

Abstract
Multiple sclerosis (MS) is an inflammatory and neurodegenerative disease characterized by myelin and axonal pathology. In a viral model of MS, we tested whether axonopathy initiation and development are based on an impaired transport of neurofilaments. Spinal cords of Theiler's murine encephalomyelitis virus (TMEV)‐infected and mock‐infected mice and TMEV infected neuroblastoma N1E‐115 cells were analyzed by microarray analysis, light microscopy and electron and laser confocal microscopy. In vivo axonal accumulation of non‐phosphorylated neurofilaments after TMEV infection revealed a temporal development caused by the impairments of the axonal traffic consisting of the downregulation of kinesin family member 5A, dynein cytoplasmic heavy chain 1, tau‐1 and β‐tubulin III expression. In addition, alterations of the protein metabolism were also noticed. In vitro, the TMEV‐infected N1E‐115 cells developed tandem‐repeated swellings similar to in vivo alterations. Furthermore, the hypothesis of an underlying axonal self‐destruction program involving nicotinamide adenine dinucleotide depletion was supported by molecular findings. The obtained data indicate that neurofilament accumulation in TME is mainly the result of dysregulation of their axonal transport machinery and impairment of neurofilament phosphorylation and protein metabolism. The present findings allow a more precise understanding of the complex interactions responsible for initiation and development of axonopathies in inflammatory degenerative diseases.
Keywords: axonopathy, multiple sclerosis, TMEV, transport defects
INTRODUCTION
Multiple sclerosis (MS) is an inflammatory neurodegenerative disease of the human central nervous system. Its precise etiology is unknown, but most likely MS occurs as a result of a combination of genetic factors, autoimmune conditions and environmental factors like infectious and non‐infectious pathogens. Clinically, MS shows a relapsing‐remitting and chronic‐progressive type with primary‐progressive, secondary‐progressive and progressive‐relapsing as subtypes (36). The majority of MS patients show a biphasic form of the disease with a relapsing‐remitting phase followed by a secondary‐progressive state (33). Animal models simulating immunologic and/or infectious features of MS are powerful tools to investigate the pathogenesis of the disease. Theiler's murine encephalomyelitis (TME) virus (TMEV)‐induced demyelination represents a well‐characterized and highly suitable virus model for MS with clinical features similar to the primary‐progressive form of MS. Both, the animal model and the human disease, share characteristic pathogenetic and mechanistic features like demyelination, inflammation and axonal injury 6, 12, 27, 32, 52 Despite decades of intensive research, it is still unknown whether MS and TME pathology follows the model of primary or secondary demyelination. In the primary model, the demyelination caused by myelino‐ and/or oligodendrogliopathies is considered to be the primary lesion that predisposes to the secondary lesion, namely the axonal injury. In the secondary scenario, the neuroaxonal damages acts as trigger for the extensive demyelination. In MS and TME it seems both events contribute substantially to disease development and progression 13, 50, 52. Similar mechanisms have been described in other naturally occurring animal models of MS like canine distemper virus (CDV) (34).
Investigations regarding the axonal pathology were promoted by the identification of amyloid precursor protein (APP) and non‐phosphorylated neurofilaments (n‐NFs) as early markers for axonal damages 6, 7, 15. Though these markers allow a precise detection of axonal damage in MS, TME and CDV lesions 10, 13, 14, 34, 40, 50, 52, 55 the molecular mechanisms of APP and n‐NF accumulation as well as their role during disease initiation and progression remain to be determined. Under physiological conditions, NFs are synthesized in the neuronal cell body as non‐phosphorylated proteins, and then they undergo a complex pattern of phosphorylation along the axon forming phosphorylated NFs (p‐NFs) (36). Phosphorylation and dephosphorylation of NFs is primarily regulated by the p35‐activated cyclin‐dependent kinase 5 (Cdk5) (25) and protein phosphatase 2A (Ppp2r2a, Ppp2r2c) (58) and their activity correlates with the NF dynamics (46). Along the axon, NFs are moved on the microtubular polymers, formed by monomeric α and β‐tubulins, in the anterograde direction after association with the kinesin family member 5A (Kif5A) (62) or in the retrograde orientation by coupling with dynein heavy chain (Dync1h1) (31). Microtubule–microtubule (MT) and MT‐motor protein interactions important for axonal transport, are controlled by tau‐1 protein 17, 60. Once arrived at the axonal terminus, NFs are degraded either by protease digestion (42) and/or by the ubiquitin‐proteasome system (28). In conclusion, NF expression may be modulated at multiple levels and abnormalities in any of these steps could cause NFs accumulation, a feature of many neurodegenerative diseases (9) including MS and TME (41).
In the present study, different processes that could be responsible for the axonal accumulation of n‐NF were investigated in vivo and in vitro to substantiate the hypothesis that the development of TMEV‐induced axonopathy is based on an impaired bidirectional axonal transport of NFs. Thus, abnormalities in the NF axonal transport were analyzed by monitoring the expression of cytoskeleton proteins such as β‐tubulin III and α‐acetylated tubulin as well as cytoskeleton‐associated proteins including tau‐1, Kif5A and Dync1h1. Possible deficiencies in NF dephosphorylation followed by the expression of Pp2ac and Pp2aa, subunits of Pp2a, and pathological modifications of the protein degradation pathway of the ubiquitin–protein conjugates and ubiquitin carboxy‐terminal hydrolase L1 expression (Uchl‐1) were investigated at the transcriptional and translational level. Data indicate that NF accumulation in TME is not only the result of specific dysregulations in their axonal transport but also the sequel of non‐specific impairments in the neuronal protein metabolism. Notably, microarray analysis of transcriptional changes in TME spinal cords support the hypothesis that the observed impairment of axonal transport is part of an axonal self‐destruction program relying on active depletion of axonal nicotinamide adenine dinucleotide (NAD) as a mechanism for axonal degeneration in demyelinating diseases like MS and TME 3, 36.
MATERIALS AND METHODS
In vivo and in vitro study design
Animal experiments were done as described before (53). Five‐week‐old female SJL/JHanHsd mice were purchased from Harlan Winkelmann (Borchen, Germany) and were inoculated under general anaesthesia into the right cerebral hemisphere with 1.63 × 106 plaque‐forming units/mouse of BeAn strain of TMEV in 20 µL Dulbecco's modified Eagle medium (PAA Laboratories, Cölbe, Germany) with 2% fetal calf serum and 50 µg/mL gentamycin. Mock‐infected mice received 20 µL of the diluent only. Placebo and infected groups were killed after 0, 4, 7, 14, 28, 42, 98 and 196 days. Each group consisted of six animals. At necropsy, the thoracic segment of the spinal cords was removed from each animal. Subsequently, tissues were fixed in 10% formalin, decalcified in 25% ethylenediaminetetraacetic acid for 48 h and then embedded in paraffin.
For differentiation into neurons, N1E‐115 murine neuroblastoma cells (ATCC, CRL‐2263) were maintained 14 days in DMEM with 2% FCS. Then, cells were incubated for 1 h at a multiplicity of infection of 10 with the BeAn strain of TMEV (23) and analyzed by immunofluorescence at 1, 3 and 7 days post‐infection.
Histochemistry and immunohistochemistry
To identify demyelination, formalin‐fixed, paraffin‐embedded thoracic spinal cord sections were stained with Luxol fast blue cresyl‐echt violet (LFB) (56). Identification of normal and pathological axonal structures was done by using a modified Bielschowsky silver stain (9). Immunohistochemistry was performed as described previously 37, 55, 56 using the primary antibodies mentioned in Table 1. The 2‐ to 3‐µm‐thick paraffin‐embedded sections were dried in an oven at 50°C for 30 minutes. Then, sections were deparaffinized and rehydrated using graded alcohols. The endogenous peroxidase was quenched in 0.5% H2O2 prepared in methanol. Antigen retrieval was done by incubation in citrate buffer for 25 minutes at 800 W or 0.25% Triton® X‐100 in phosphate‐buffered saline (PBS) for 15 minutes at room temperature (RT). To block non‐specific binding sites, sections were incubated with normal goat serum for 20 minutes at RT and then overnight with the primary antibodies (Table 1) at 4°C. For negative controls, the primary antibody was substituted by either rabbit serum or ascites from non‐immunized BALB/cJ mice. Subsequently, after washing in PBS sections were incubated with either biotinylated goat anti‐mouse or anti‐rabbit serum, respectively (Vector Laboratories, Burlingame, CA, USA). Positive antigen‐antibody reactions were visualized by incubation with 3,3′‐diaminobenzidine‐tetrahydrochloride (DAB)‐ H2O2 in 0.1 M imidazole, pH 7.08 for 10 minutes followed by slight counterstaining with Mayer's hemalaun.
Table 1.
Antibodies used for immunohistochemistry and immunofluorescence. Abbreviations: IHC = immunohistochemistry; IF = immunofluorescence; PBS = phosphate‐buffered saline; NA = not applicable; ND = not done.
| Nr | Antibody | Supplier | Catalog number | Source | Dilution | Pre‐treatment for IHC | |
|---|---|---|---|---|---|---|---|
| IHC | IF | ||||||
| 1. | Non‐phosphorylated neurofilament (n‐NF) | Sternberger Monoclonals | SMI311 | Mouse | 1:8000 | 1:1000 | Boiled in citrate buffer in the microwave |
| 2. | Phosphorylated neurofilament (p‐NF) | Sternberger Monoclonals | SMI312 | Mouse | 1:8000 | 1:1000 | Triton® X‐100 0.25% in PBS |
| 3. | Amyloid precursor protein (APP) | Chemicon | MAB348 | Mouse | 1:2000 | ND | Boiled in citrate buffer in the microwave |
| 4. | Protein phosphatase 2 subunit C (Ppp2r2c) | Cell Signaling Technology | 2038 | Rabbit | 1:50 | ND | Boiled in citrate buffer in the microwave |
| 5. | Protein phosphatase 2 subunit A (Ppp2r2a) | Cell Signaling Technology | 2041 | Rabbit | 1:50 | ND | Boiled in citrate buffer in the microwave |
| 6. | Kinesin heavy chain isoform 5A (Kif5A) | Sigma Aldrich | MAB1614 | Mouse | 1:100 | 1:60 | Boiled in citrate buffer in the microwave |
| 7. | Dynein heavy chain (Dync1h1) | Santa Cruz Biotechnology | sc‐9115 | Rabbit | 1:10 | 1:10 | Boiled in citrate buffer in the microwave |
| 8. | β‐tubulin III | Sigma Aldrich | T8660 | Mouse | 1:5000 | 1:2500 | Boiled in citrate buffer in the microwave |
| 9. | α‐Acetylated tubulin | Sigma Aldrich | T6793 | Mouse | 1:1000 | ND | Triton 0.25% in PBS |
| 10. | Tau‐1 | Chemicon | MAB3420 | Mouse | 1:2000 | 1:200 | Boiled in citrate buffer in the microwave |
| 11. | Ubiquitin | Chemicon | AB1690 | Rabbit | 1:1000 | 1:200 | Boiled in citrate buffer in the microwave |
| 12. | Ubiquitin C‐terminal hydrolase (Uchl‐1) | Chemicon | AB1761 | Rabbit | 1:1000 | NA | Boiled in citrate buffer in the microwave |
| 13. | VP1‐BeAn‐TMEV | * | NA | Rabbit | 1:2000 | 1:200 | ND |
Kummerfeld et al (23).
The obtained brown signal was evaluated quantitatively by counting the number of positive axons (number of immunoreactive axons/mm2) in the white matter. The percentage of p‐NF and Bielschowsky's silver stain‐positive thoracic axons in the ventrolateral white matter area was evaluated by digitalizing the spinal cord section with a color video camera (Color View II, 3.3 Megapixel CCD; Soft Imaging System, Münster, Germany) mounted on an Axiophot microscope (Zeiss, Oberkochen, Germany) with a 5× objective. The positive structures were measured interactively after manually outlining the total white matter area using the analysis 3.1 software package (Soft Imaging System). Data are presented as percentage of labeled axonal profiles in relation to the total white matter area.
Data obtained by the LFB stain, Bielschowsky silver stain and immunohistochemistry were subjected to statistical analysis using the program “SPSS” for Windows, version 13.0 (SPSS Inc., Chicago, IL, USA) using the Wilcoxon test as group‐wise test, as described previously (26). A P‐value of less than 0.05 was considered to be statistically significant. The correlation coefficient of p‐NF and n‐NF immunohistochemistry was calculated by using the non‐parametric Spearman's correlation test.
Immunofluorescence, electron and laser confocal microscopy
For immunostaining, N1E‐115 cells were washed with PBS and then fixed with 4% paraformaldehyde for 30 minutes at RT. After an additional washing step with PBS, cells were permeabilized with 0.25% Triton® X‐100 (Sigma‐Aldrich, Taufkirchen, Germany) in PBS (PBST) for 15 minutes. To block unspecific binding sites, cells were kept in 1% bovine serum albumin (BSA) diluted in PBST for 30 minutes. The cells were then incubated overnight at 4°C with the primary antibody diluted in PBST (Table 1). After 10 minutes washing in PBST, cells were incubated for 1 h with Cy2/Cy3 conjugated AffiniPure goat anti‐rabbit/anti‐mouse (Jackson‐Immuno Research Laboratories, West Grove PA, USA) diluted 1:200 in PBST. Then, cells were washed with and kept in PBS until embedded with Fluorescent mounting medium® (Dako Deutschland GmbH, Hamburg, Germany).
Confocal laser microscopy was performed as described previously (22) using the primary antibodies mentioned in Table 1. The labeled cells were imaged with an inverted microscope Leica DM IRE2 (Leica, Wetzlar, Germany) using UV light, 405 nm (bis‐benzimide), Ar‐Ion, 488 nm (Cy2), and HeNe, 543 nm (Cy3) lasers.
Electron microscopy was performed as described previously (55). Groups of six TMEV‐infected and control mice were killed at 14, 42, 98 and 196 dpi and spinal cords were immersed in 5% glutaraldehyde/cacodylate buffer and subsequently in 1% osmium tetroxide. Then, sections were dehydrated and embedded in epoxy resin. Ultrathin sections (70 nm) were stained with uranyl acetate and lead citrate and then analyzed by a Zeiss electron microscope EM 10 C (Zeiss).
Microarray analysis of transcriptional changes
RNA was isolated from frozen spinal cord specimens using the RNeasy Mini Kit (Qiagen, Hilden, Germany), amplified and labeled using the MessageAmp II‐Biotin Enhanced Kit (Ambion, Austin, TX, USA) and hybridized to Affymetrix mouse genome 430 2.0 arrays (Affymetrix, Santa Clara, CA, USA) as described (56). Six biological replicates were used per group and time point, except for five TMEV‐infected mice at 98 dpi. Background adjustment and quantile normalization was performed using RMAExpress (5). Further analysis focused on manually curated lists of candidate genes associated with: (i) axonal transport; and (ii) axonal self‐destruction, respectively, extracted from peer‐reviewed articles (Supporting Information Tables S1 and S2). The fold change was calculated as the ratio of the inverse‐transformed arithmetic means of the log2‐transformed expression values of TMEV‐infected vs. mock‐infected mice. Downregulations are shown as negative reciprocal values. Data were analyzed using pair‐wise Mann–Whitney U‐tests. Statistical significance was designated as P ≤ 0.05.
RESULTS
Morphological changes
The progression of viral infection, demyelination and axonal loss in TME were in accordance to previously described findings 29, 52, 55, 56. Briefly, the majority of TMEV‐positive axons in the spinal cords were detected 1 month after infection. First features of demyelination using the LFB stain were observed at 42 dpi and loss of myelin continuously increased until 196 dpi, whereas a significant axonal loss and occurrence of swollen axons termed spheroids were observed between 28 and 196 dpi (Figure 1A). Details are listed in the Supporting Information (Supporting Information Figures S1–3). To identify subtle axonal changes, the expression of APP, p‐NF and n‐NF was investigated. APP expression was confined to few glial cells in the white matter and to neuronal cell bodies in the gray matter in placebos (Figure 2A). In addition, APP immunopositive axons were observed in the ventromedial and lateral columns between 28 and 196 dpi in TMEV infected mice in addition (1, 2).
Figure 1.

Kinetic of axonal pathology in TME. The box and whisker plots illustrate, in a time‐dependent manner, the loss of nerve fibers as detected by the Bielschowsky silver stain (A) and changed immunoreactivity of amyloid precursor protein (APP) (B), phosphorylated neurofilament (p‐NF) (C), non‐phosphorylated (n‐NF) (D), kinesin family member 5A (Kif5A) (E), dynein cytoplasmic heavy chain 1 (Dync1h1) (F), β‐tubulin III (G), tau‐1 (H) and ubiquitin‐ligated proteins (I) following Theiler's murine encephalomyelitis virus (TMEV) infection. Statistical significant differences (P ≤ 0.05) were marked with asterisks (*). White box plots represent controls; gray box plots represent infected animals.
Figure 2.
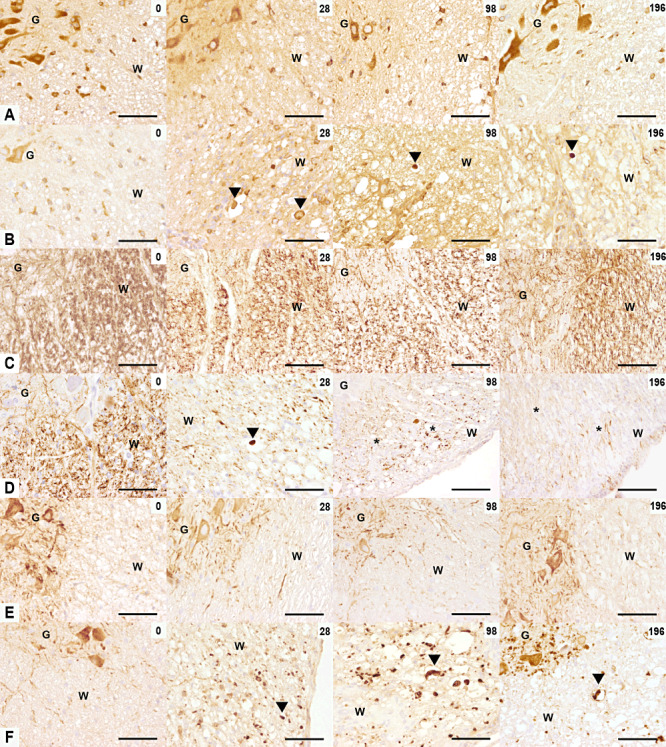
Amyloid precursor protein (APP), phosphorylated neurofilament (p‐NF), non‐phosphorylated NF (n‐NF) expression in TME. The rows illustrate APP (A,B), p‐NF (C,D), n‐NF (E,F) immunoreactivity in the ventromedial spinal cord of controls (A,C,E) and TMEV‐infected mice (B,D,F) at 0, 28, 98 and 196 dpi. (A,B) At 0 dpi, control and infected mice showed a similar pattern of APP expression in glial cells of the white matter and neurons of the gray matter. From 28 until 196 dpi, APP‐positive spheroids were observed in TMEV‐infected mice (arrowheads). (C,D) At 0 dpi, control and infected mice showed a similar pattern of p‐NF expression with p‐NF‐positive axons in the white matter and p‐NF‐negative neurons in the gray matter. Between 28 and 196 dpi a decreased p‐NF expression was observed in TME. A p‐NF‐positive spheroid is marked with arrowhead and severe axonal loss with asterisks). (E,F) At 0 dpi, control and infected mice showed the same pattern of n‐NF expression with n‐NF‐negative axons in the white matter and n‐NF‐positive neurons in the gray matter. n‐NF axonal expression, marked with an arrowhead, appeared at 28 until 196 dpi in TMEV‐infected mice only. Scale bar = 100 µm. Abbreviations: G = gray matter; TMEV = Theiler's murine encephalomyelitis virus; W = white matter.
p‐NF expression was limited to axons and not found in neuronal perikarya in controls (Figure 2C). In infected mice, loss of p‐NF expression of axons became evident at 14 dpi. A statistical significant decrease beginning at 28 dpi was observed (1, 2). In addition, few p‐NF‐positive spheroids were detected between 28 and 98 dpi (Figure 2D). n‐NF expression characterized by positive neuronal perikarya and negative axons in controls was complementary to the p‐NF immunoreactivity (Figure 2E). The first n‐NF axonal accumulations were observed at 14 dpi in TME mice. Thereafter, axonal expression of n‐NF increased significantly and reached its maximum at 98 dpi (1, 2). n‐NF positive axons were often enlarged and contained densely accumulated NFs. Evaluation of the correlation coefficient showed a highly negative association between p‐NF and n‐NF (Spearman rank correlation ρ = −0.824, P < 0.01).
For a more detailed characterization of the demyelinating process, electron microscopy was performed. In controls (Figure 3A), nerve fibers, enwrapped by myelin sheaths of various calibers were observed as expected. The earliest demyelinating changes in the spinal cord of infected mice were observed at 14 dpi and were characterized by separation of the individual lamellae at the intraperiod line, formation of fluid‐filled intramyelinic spaces and vesicles, and marked distention of the myelin sheaths (Figure 3B). At 42 dpi, morphologically altered myelin sheaths mostly displayed an invasion of their separated lamellae by interposed cellular processes containing a lysosome‐rich cytoplasm, interpreted to be of macrophage/microglial origin. Furthermore, completely demyelinated axons surrounded by macrophage/microglia and Gitter cells with abundant whirls of lamellar and/or amorphous electron‐dense debris in membrane‐bound cytoplasmic vesicles as well as reactive microglial processes were detected between 42 and 196 dpi. Notably, at 196 dpi, axons surrounded by relatively thin myelin sheaths, interpreted as remyelination were present within the lesions (Figure 3B,C).
Figure 3.
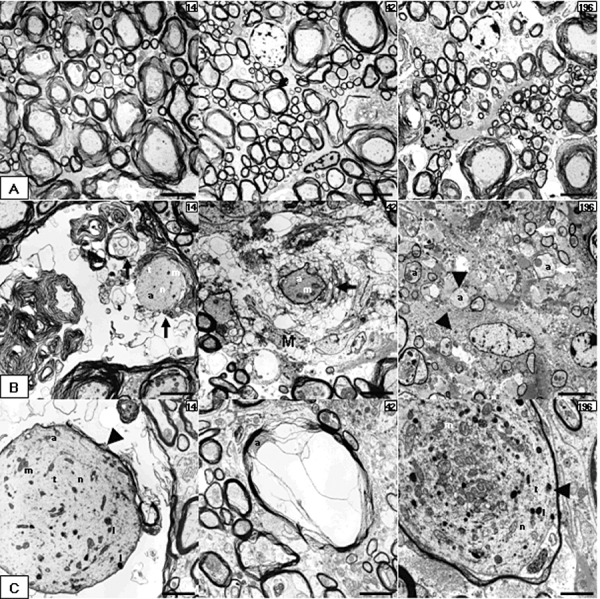
Electron microscopic features in TME. (A–C) Electron micrographs obtained from spinal cords of placebo (A) and TMEV‐infected mice (B,C) at 14, 42 and 98 dpi. (A) Intact axons at different time points. (B) Specific features of myelin pathology including myelin vesiculation and loss (black arrows, 14 and 42 dpi), naked axons (arrowheads, 196 dpi) and remyelination (white arrows, 196 dpi). (C) Particularities of axonal pathology‐like distended axons (arrowheads) with accumulation of microtubule, neurofilaments, vesiculotubular structures, mitochondria and lysosomes (14 and 196 dpi) or a compressed axon by myelin‐sheath edema (42 dpi). A and B196 scale bar = 4 µm; B14, B42, C14 and C42 scale bar = 2 µm; C196 scale bar = 1 µm. Abbreviations: a = axon; l = lysosomes; m = mitochondria; M = microglia/macrophage cytoplasm; n = neurofilaments; t = microtubules; TMEV = Theiler's murine encephalomyelitis virus.
Initiation of axonal degeneration
A series of recent publications suggests the existence of axonal self‐destruction programs, contributing to the antiviral host response in TMEV infection 35, 52. In the present study, a manually selected list of axonal self‐destruction‐related genes (Supporting Information Table S2), was individually analyzed. Twenty‐one out of these 65 axonal self‐destruction‐related probe sets, corresponding to 17 individual genes, were differentially expressed in the spinal cord of TMEV‐infected mice compared with mock‐infected mice (Table 2). Notably, a mild transcriptional downregulation of nicotinamide mononucleotide adenylyl‐transferases‐1, ‐2 (Nmnat‐1,2) and a mild upregulation of poly(ADP)ribose polymerase 1 (PARP1) were observed in the chronic demyelinating phase of the disease. Concerning the dual leucine kinase (DLK)/c‐Jun N‐terminal kinase (JNK)‐induced axonal‐degeneration pathway 13, 30, 44, 48, a mild downregulation was observed for the mitogen‐activated protein kinase kinase kinase 12 (Map3k12; synonym:DLK) at 196 dpi. Its downstream target mitogen‐activated protein kinase 8 (Mapk8; synonym: JNK) displayed a mild transcriptional upregulation at 14 dpi, followed by a mild downregulation from 42 to 196 dpi. Concerning the APP/DR6/caspase6‐induced axonal‐degeneration pathway 14, 33, a mild upregulation was observed for Caspase‐6 (Casp6) throughout the observation period. APP displayed a mild upregulation in the acute phase and a mild downregulation in the chronic phase. Brain‐derived neurotrophic factor (BDNF) was mildly downregulated from 14 to 98 dpi, and the proto‐oncogene tyrosine‐protein kinase receptor Ret (Ret) was mildly downregulated at 98 dpi. Concerning neuroaxonal–microglial interactions, moderate transcriptional upregulations were observed for multiple CD200‐receptors (CD200r) from 42 to 196 dpi, and G protein‐coupled receptor 109A (Gpr109a) from 14 to 196 dpi. The question remains: which downstream axonal changes are initiated by these activations.
Table 2.
List of differentially expressed genes related to the initiation of axonal self‐destruction in TME.
| Gene symbol | Gene title | Probe set ID† | Time post infection | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 14 | 42 | 98 | 196 | |||||||
| Fold change‡ | P‐value§ | Fold change‡ | P‐value§ | Fold change‡ | P‐value§ | Fold change‡ | P‐value§ | |||
| APP | Amyloid beta (A4) precursor protein | 1420621_a_at | 1.03 | 0.9372 | −1.06 | 0.9372 | −1.07 | 0.1255 | −1.05* | 0.0152 |
| APP | Amyloid beta (A4) precursor protein | 1427442_a_at | 1.06* | 0.0260 | −1.01* | 0.0260 | −1.03 | 0.3290 | −1.04* | 0.0260 |
| APP | Amyloid beta (A4) precursor protein | 1444705_at | 1.51* | 0.0411 | 1.04* | 0.0411 | 1.06 | 0.5368 | −1.03 | 0.6991 |
| Atg5 | Autophagy‐related 5 (yeast) | 1415684_at | −1.13 | 0.1797 | −1.11 | 0.0649 | −1.14* | 0.0087 | −1.13* | 0.0411 |
| Bdnf | Brain‐derived neurotrophic factor | 1422168_a_at | −1.19* | 0.0411 | −1.23* | 0.0411 | −1.40* | 0.0173 | −1.14 | 0.2403 |
| Casp6 | Caspase 6 | 1415995_at | 1.14* | 0.0411 | 1.15* | 0.0411 | 1.41* | 0.0303 | 1.30* | 0.0022 |
| Cd200r1 | CD200 receptor 1 | 1422191_at | 1.03 | 0.9372 | 1.27* | 0.0043 | 1.29* | 0.0087 | 1.20* | 0.0411 |
| Cd200r3 | CD200 receptor 3 | 1453813_at | 1.09 | 0.2403 | 1.26* | 0.0152 | −1.05 | 0.3290 | 1.08 | 0.3095 |
| Cd200r4 | Cd200 receptor 4 | 1435313_at | −1.01 | 0.8182 | 2.36* | 0.0022 | 4.79* | 0.0043 | 4.21* | 0.0022 |
| Gpr109a | G protein‐coupled receptor 109A | 1419721_at | 1.67* | 0.0022 | 4.00* | 0.0022 | 5.27* | 0.0043 | 3.27* | 0.0022 |
| Map3k12 | Mitogen‐activated protein kinase kinase kinase 12 | 1417115_at | −1.02 | 0.3939 | −1.06* | 0.0022 | −1.11* | 0.0173 | −1.12* | 0.0022 |
| Mapk8 | Mitogen‐activated protein kinase 8 | 1420932_at | −1.03 | 0.9372 | −1.10* | 0.0022 | −1.10* | 0.0087 | −1.03 | 0.8182 |
| Mapk8 | Mitogen‐activated protein kinase 8 | 1437045_at | 1.72* | 0.0043 | 1.11 | 0.6991 | −1.10 | 0.7922 | −1.04 | 0.8182 |
| Mapk8 | Mitogen‐activated protein kinase 8 | 1457936_at | 1.01 | 1.0000 | −1.24* | 0.0411 | −1.18 | 0.3290 | −1.12 | 0.3939 |
| Nmnat1 | Nicotinamide nucleotide adenylyltransferase 1 | 1425773_s_at | −1.06 | 0.9372 | 1.00 | 0.6991 | −1.06 | 0.4286 | −1.44* | 0.0022 |
| Nmnat2 | Nicotinamide nucleotide adenylyltransferase 2 | 1436155_at | −1.02 | 0.2403 | −1.09* | 0.0043 | −1.08* | 0.0173 | −1.08* | 0.0411 |
| Parp1 | Poly (ADP‐ribose) polymerase family, member 1 | 1422502_at | 1.01 | 0.8182 | −1.03 | 0.3939 | 1.09 | 0.1255 | 1.16* | 0.0152 |
| Pttg1 | Pituitary tumor‐transforming 1 | 1438390_s_at | −1.05 | 0.6991 | 1.20* | 0.0152 | 1.36* | 0.0087 | 1.07 | 0.2403 |
| Ret | Ret proto‐oncogene | 1421359_at | −1.19 | 0.1797 | −1.22 | 0.1797 | −1.12* | 0.0303 | 1.10 | 0.0931 |
| Ube4b | Ubiquitination factor E4B, UFD2 homolog (Saccharomyces cerevisiae) | 1432499_a_at | −1.01 | 0.9372 | −1.12* | 0.0260 | −1.12 | 0.1255 | −1.06 | 0.3939 |
| Vcp /// LOC675857 | Valosin containing protein /// similar to Transitional endoplasmic reticulum ATPase (TER ATPase) (15S Mg(2+)‐ATPase p97 subunit) (valosin‐containing protein) (VCP) | 1423030_at | 1.02 | 0.4848 | 1.02 | 0.4848 | 1.02 | 0.7922 | 1.09* | 0.0260 |
Statistically significant difference (P ≤ 0.05).
The probe set ID represents the unique probe set identifier of the Affymetrix mouse genome 430 2.0 array.
The fold change was calculated as the ratio of the inverse‐transformed arithmetic means of the log2‐transformed expression values of Theiler's murine encephalomyelitis virus (TMEV)‐infected vs. mock‐infected mice. Downregulations are shown as negative reciprocal values.
P‐value as obtained by pairwise comparison of TMEV‐infected and mock‐infected mice by Mann–Whitney U‐test.
Transcriptional changes of axon‐transport related genes
To identify possible candidate genes and to outline possible pathways causing abnormal axonal expression of p‐NF and n‐NF in TME mice, the data sets of a previously published microarray experiment were reanalyzed using an independent approach 18, 56. In the present study, a manually curated list of canonical axonal transport‐related genes (Supporting Information Table S1) was individually analyzed. Sixty‐six out of these 111 axonal transport‐related probe sets, corresponding to 59 individual genes were differentially expressed in the spinal cord of TMEV‐infected mice compared with mock‐infected mice (Table 3). There was a mild transcriptional downregulation of 15 out of 18 differentially expressed probe sets related to subunits of the kinesin molecule complex especially during the demyelinating phase of the disease between 42 and 196 dpi. Similarly, a mild transcriptional downregulation was observed for 15 out of the 18 differentially expressed probe sets related to subunits of the dynein molecule complex. Furthermore all eight out of eight differentially expressed probe sets related to subunits of protein phosphatase 2 complex (Ppp2r2a, Ppp2r2c) exhibited a mild transcriptional downregulation during the demyelinating phase of the disease between 42 and 196 dpi. Additionally, six out of seven and six out of eight differentially expressed probe sets related to various alpha and beta tubulins displayed transcriptional downregulations, respectively. Further minor transcriptional downregulations were observed for microtubule‐associated protein tau, the NF heavy polypeptide, NF light polypeptide, prion protein and Uchl‐1. The amyloid beta (A4) precursor protein (APP) displayed a more differentiated time course of transcription, with a mild upregulation at 14 dpi and a mild downregulation at 196 dpi.
Table 3.
List of axonal transport‐related differentially expressed genes.
| Gene symbol | Gene title | Probe set ID† | Time post infection | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 14 | 42 | 98 | 196 | |||||||
| Fold change‡ | P‐value§ | Fold change‡ | P‐value§ | Fold change‡ | P‐value§ | Fold change‡ | P‐value§ | |||
| APP | Amyloid beta (A4) precursor protein | 1427442_a_at | 1.06* | 0.0260 | −1.01 | 0.5887 | −1.03 | 0.3290 | −1.04* | 0.0260 |
| Dnahc2 /// Dnhd3 | Dynein, axonemal, heavy chain 2 /// dynein heavy chain domain 3 | 1438763_at | −1.18* | 0.0087 | −1.17 | 0.0931 | −1.23* | 0.0043 | −1.10 | 0.3939 |
| Dnahc5 | Dynein, axonemal, heavy chain 5 | 1421434_at | −1.00 | 0.8182 | 1.08 | 0.5887 | 1.02 | 0.9307 | −1.16* | 0.0260 |
| Dnahc8 | Dynein, axonemal, heavy chain 8 | 1425692_a_at | −1.06 | 0.3939 | 1.01 | 0.8182 | −1.16* | 0.0303 | 1.01 | 0.9372 |
| Dnahc9 | Dynein, axonemal, heavy chain 9 | 1457401_at | −1.03 | 0.6991 | −1.06 | 0.4848 | −1.02 | 0.7922 | −1.14* | 0.0260 |
| Dnaic1 | Dynein, axonemal, intermediate chain 1 | 1437094_x_at | −1.13 | 0.0649 | −1.10 | 0.2403 | −1.24* | 0.0087 | −1.13 | 0.0931 |
| Dnalc1 | Dynein, axonemal, light chain 1 | 1455630_at | −1.02 | 0.5887 | −1.13* | 0.0022 | −1.15* | 0.0087 | −1.11 | 0.0931 |
| Dnalc4 | Dynein, axonemal, light chain 4 | 1416870_at | 1.00 | 1.0000 | −1.13* | 0.0152 | −1.03 | 0.7922 | −1.02 | 0.9372 |
| Dnali1 | Dynein, axonemal, light intermediate polypeptide 1 | 1455379_at | 1.14 | 0.3939 | −1.01 | 1.0000 | −1.08 | 0.5368 | 1.17* | 0.0260 |
| Dync1i1 | Dynein cytoplasmic 1 intermediate chain 1 | 1416361_a_at | −1.10* | 0.0022 | −1.08* | 0.0152 | −1.16* | 0.0043 | −1.23* | 0.0043 |
| Dync1li1 | Dynein cytoplasmic 1 light intermediate chain 1 | 1424947_at | −1.11* | 0.0411 | −1.09 | 0.0649 | −1.20* | 0.0303 | −1.15 | 0.0931 |
| Dync2h1 | Dynein cytoplasmic 2 heavy chain 1 | 1429250_at | 1.10 | 0.2403 | −1.15* | 0.0260 | −1.12 | 0.0823 | −1.08 | 0.1320 |
| Dynll1 | Dynein light chain LC8‐type 1 | 1448682_at | −1.02 | 0.4848 | −1.09* | 0.0411 | −1.08 | 0.2468 | −1.00 | 0.5887 |
| Dynll1 /// EG627788 /// LOC637840 /// LOC672375 | Dynein light chain LC8‐type 1 /// predicted gene, EG627788 /// Similar to dynein, cytoplasmic, light peptide /// similar to dynein, cytoplasmic, light peptide | 1417339_a_at | −1.12 | 0.0649 | −1.14* | 0.0087 | −1.04 | 0.4286 | −1.10* | 0.0411 |
| Dynll2 | Dynein light chain LC8‐type 2 | 1418258_s_at | 1.03 | 0.9372 | −1.00 | 0.9372 | −1.10* | 0.0173 | −1.15* | 0.0411 |
| Dynlrb1 | Dynein light chain roadblock‐type 1 | 1428257_s_at | −1.09 | 0.0649 | −1.11* | 0.0022 | −1.06 | 0.0823 | −1.10* | 0.0022 |
| Dynlt1 | Dynein light chain Tctex‐type 1 | 1428116_a_at | 1.07 | 0.3095 | 1.11 | 0.2403 | 1.25* | 0.0303 | 1.22* | 0.0411 |
| Dynlt1 /// LOC671261 | Dynein light chain Tctex‐type 1 /// similar to Cytoplasmic dynein light chain (T‐complex testis‐specific protein 1) (TCTEX‐1) | 1453473_a_at | 1.08 | 0.3095 | 1.06 | 0.9372 | 1.28* | 0.0087 | 1.19 | 0.0649 |
| Dynlt3 | Dynein light chain Tctex‐type 3 | 1459854_s_at | −1.13* | 0.0022 | −1.09 | 0.1797 | −1.02 | 0.6623 | −1.10 | 0.0649 |
| Kif12 | Kinesin family member 12 | 1424910_at | −1.01 | 0.4848 | 1.06 | 0.2403 | −1.13* | 0.0303 | −1.01 | 0.9372 |
| Kif17 | Kinesin family member 17 | 1419827_s_at | 1.06 | 0.6991 | −1.07* | 0.0152 | −1.08 | 0.1255 | −1.15 | 0.0649 |
| Kif18a | Kinesin family member 18A | 1424107_at | −1.07 | 0.3095 | −1.03 | 0.5887 | 1.17* | 0.0087 | −1.05 | 0.3939 |
| Kif21a | Kinesin family member 21A | 1450738_at | 1.04 | 0.8182 | −1.04 | 0.9372 | −1.33* | 0.0043 | −1.22 | 0.1797 |
| Kif21b | Kinesin family member 21B | 1435772_at | 1.21* | 0.0260 | 1.10 | 0.1320 | 1.12 | 0.1255 | −1.04 | 0.4848 |
| Kif23 | Kinesin family member 23 | 1455990_at | −1.16 | 0.1320 | 1.21* | 0.0260 | 1.28* | 0.0303 | 1.02 | 0.6991 |
| Kif24 | Kinesin family member 24 | 1431937_at | −1.05 | 0.8182 | −1.14 | 0.0931 | −1.04 | 0.6623 | −1.22* | 0.0260 |
| Kif2a | Kinesin family member 2A | 1454107_a_at | 1.20 | 0.8182 | 1.19 | 0.1320 | −1.16 | 0.3290 | −1.47* | 0.0260 |
| Kif2c | Kinesin family member 2C | 1432392_at | −1.13 | 0.0931 | 1.04 | 0.8182 | 1.04 | 0.7922 | −1.24* | 0.0260 |
| Kif3b | Kinesin family member 3B | 1450074_at | −1.09 | 0.1320 | 1.05 | 0.3095 | −1.22* | 0.0173 | −1.12 | 0.3939 |
| Kif3c | Kinesin family member 3C | 1448946_at | 1.06 | 0.0649 | −1.05 | 0.1320 | −1.07* | 0.0087 | −1.10* | 0.0022 |
| Kif5a | Kinesin family member 5A | 1434670_at | 1.02 | 1.0000 | −1.10* | 0.0022 | −1.19* | 0.0043 | −1.18* | 0.0022 |
| Kif5a /// Kif5c | Kinesin family member 5A /// kinesin family member 5C | 1450249_s_at | −1.16 | 0.2403 | −1.04 | 0.3095 | −1.38* | 0.0043 | −1.33* | 0.0087 |
| Kif5b | Kinesin family member 5B | 1418431_at | 1.03 | 1.0000 | −1.09 | 0.2403 | −1.54* | 0.0303 | −1.31 | 0.0931 |
| Kif5c | Kinesin family member 5C | 1422945_a_at | −1.03 | 0.6991 | −1.04 | 0.6991 | −1.52 | 0.0519 | −1.48* | 0.0087 |
| Kifap3 | Kinesin‐associated protein 3 | 1451783_a_at | −1.04 | 0.1797 | −1.08* | 0.0260 | −1.14* | 0.0087 | −1.09* | 0.0043 |
| Klc2 | Kinesin light chain 2 | 1418214_at | −1.04 | 0.8182 | −1.09 | 0.0649 | −1.17* | 0.0043 | −1.16* | 0.0022 |
| Kns2 | Kinesin 2 | 1417005_at | −1.04 | 0.0931 | −1.03 | 0.1320 | −1.12* | 0.0087 | −1.09 | 0.0649 |
| Mapt | Microtubule‐associated protein tau | 1424719_a_at | 1.05 | 0.3095 | −1.07 | 0.1797 | −1.16* | 0.0043 | −1.06 | 0.5887 |
| Nefh | Neurofilament, heavy polypeptide | 1424847_at | −1.01 | 0.8182 | −1.07* | 0.0260 | −1.12* | 0.0303 | −1.10 | 0.0649 |
| Nefl | Neurofilament, light polypeptide | 1426255_at | −1.03 | 0.0649 | −1.02 | 0.5887 | −1.10* | 0.0173 | −1.08 | 0.0931 |
| Ppp2ca | Protein phosphatase 2 (formerly 2A), catalytic subunit, alpha isoform | 1417367_at | −1.03 | 0.1320 | −1.07* | 0.0022 | −1.07* | 0.0303 | −1.03* | 0.0043 |
| Ppp2cb | Protein phosphatase 2 (formerly 2A), catalytic subunit, beta isoform | 1421823_a_at | −1.03 | 0.6991 | −1.12* | 0.0043 | −1.16 | 0.1255 | −1.11* | 0.0087 |
| Ppp2r1a | Protein phosphatase 2 (formerly 2A), regulatory subunit A (PR 65), alpha isoform | 1438383_x_at | 1.02 | 0.5887 | 1.02 | 0.9372 | −1.06* | 0.0087 | −1.06* | 0.0152 |
| Ppp2r2a | Protein phosphatase 2 (formerly 2A), regulatory subunit B (PR 52), alpha isoform | 1429715_at | 1.04 | 0.4848 | −1.07 | 0.0649 | −1.21* | 0.0087 | −1.08 | 0.1320 |
| Ppp2r2b | Protein phosphatase 2 (formerly 2A), regulatory subunit B (PR 52), beta isoform | 1426621_a_at | −1.09 | 0.1320 | −1.05 | 0.1320 | −1.20 | 0.0519 | −1.14* | 0.0260 |
| Ppp2r2c | Protein phosphatase 2 (formerly 2A), regulatory subunit B (PR 52), gamma isoform | 1438671_at | 1.05 | 0.3095 | −1.12* | 0.0260 | −1.20* | 0.0043 | −1.23* | 0.0043 |
| Ppp2r3a | Protein phosphatase 2 (formerly 2A), regulatory subunit B, alpha | 1455198_a_at | −1.09 | 0.1320 | −1.15* | 0.0260 | −1.12 | 0.0823 | −1.05 | 0.5887 |
| Ppp2r5e | Protein phosphatase 2, regulatory subunit B (B56), epsilon isoform | 1428462_at | −1.01 | 1.0000 | −1.13* | 0.0087 | −1.15* | 0.0043 | −1.16* | 0.0043 |
| Prnp | Prion protein | 1446507_at | 1.07 | 0.4848 | 1.08 | 0.8182 | −1.02 | 1.0000 | −1.12* | 0.0152 |
| Spata6 | Spermatogenesis associated 6 | 1418650_at | −1.07 | 0.6991 | 1.02 | 0.9372 | 1.11 | 0.1255 | 1.16* | 0.0260 |
| Tuba1 | Tubulin, alpha 1 | 1418884_x_at | −1.05* | 0.0260 | −1.03 | 0.1797 | −1.02 | 0.2468 | −1.11* | 0.0022 |
| Tuba2 | Tubulin, alpha 2 | 1423846_x_at | −1.01 | 0.8182 | −1.00 | 0.9372 | −1.04* | 0.0173 | −1.04* | 0.0260 |
| Tuba3 | Tubulin, alpha 3 | 1448296_x_at | −1.10 | 0.0931 | −1.13* | 0.0152 | −1.09* | 0.0043 | −1.14* | 0.0087 |
| Tuba3 /// Tuba7 /// LOC384954 | Tubulin, alpha 3 /// tubulin, alpha 7 /// similar to tubulin, alpha 1 | 1416311_s_at | −1.14 | 0.2403 | −1.16* | 0.0411 | −1.09* | 0.0303 | −1.17* | 0.0411 |
| Tuba4 | Tubulin, alpha 4 | 1417373_a_at | −1.01 | 0.2403 | −1.05* | 0.0022 | −1.13* | 0.0043 | −1.13* | 0.0152 |
| Tuba6 /// EG626534 | Tubulin, alpha 6 /// predicted gene, EG626534 | 1448232_x_at | 1.11 | 0.3939 | 1.76* | 0.0022 | 2.12* | 0.0043 | 1.53* | 0.0022 |
| Tuba8 | Tubulin, alpha 8 | 1419518_at | 1.03 | 0.8182 | −1.01 | 0.9372 | −1.15* | 0.0303 | −1.10 | 0.3939 |
| Tubb2a | Tubulin, beta 2a | 1427838_at | −1.02 | 0.9372 | −1.04 | 0.9372 | −1.04 | 0.7922 | −1.12* | 0.0411 |
| Tubb2a /// Tubb2b /// LOC665524 /// LOC671656 | Tubulin, beta 2a /// tubulin, beta 2b /// similar to tubulin, beta 3 /// similar to tubulin, beta 3 | 1427347_s_at | −1.07 | 0.0649 | −1.05 | 0.0649 | −1.05 | 0.0519 | −1.10* | 0.0022 |
| Tubb2b | Tubulin, beta 2b | 1452679_at | −1.10 | 0.1320 | −1.03 | 0.3939 | 1.11 | 0.2468 | −1.22* | 0.0411 |
| Tubb2c | Tubulin, beta 2c | 1456470_x_at | −1.07 | 0.5887 | −1.00 | 1.0000 | −1.02 | 0.5368 | −1.20* | 0.0152 |
| Tubb3 | Tubulin, beta 3 | 1415978_at | −1.08 | 0.2403 | −1.09 | 0.1797 | −1.16* | 0.0043 | −1.00 | 0.8182 |
| Tubb4 | Tubulin, beta 4 | 1423221_at | 1.05 | 0.6991 | −1.00 | 0.9372 | −1.13 | 0.0823 | −1.24* | 0.0022 |
| Tubb5 | Tubulin, beta 5 | 1455719_at | 1.06 | 0.3939 | 1.28* | 0.0022 | 1.49* | 0.0043 | 1.33* | 0.0022 |
| Tubb6 | Tubulin, beta 6 | 1416431_at | −1.03 | 0.9372 | 1.26* | 0.0152 | 1.39* | 0.0303 | 1.39* | 0.0087 |
| Uchl1 | Ubiquitin carboxy‐terminal hydrolase L1 | 1448260_at | −1.11 | 0.2403 | −1.09* | 0.0152 | −1.24* | 0.0043 | −1.14* | 0.0411 |
Statistically significant difference (P ≤ 0.05).
The probe set ID represents the unique probe set identifier of the Affymetrix mouse genome 430 2.0 array.
The fold change was calculated as the ratio of the inverse‐transformed arithmetic means of the log2‐transformed expression values of Theiler's murine encephalomyelitis virus (TMEV)‐infected vs. mock‐infected mice. Downregulations are shown as negative reciprocal values.
P‐value as obtained by pairwise comparison of TMEV‐infected and mock‐infected mice by Mann–Whitney U‐test.
Translational changes of axonal transport‐related proteins
To further substantiate the transcriptional changes and to dissect the molecular axonopathogenesis in TME, immunohistochemistry of candidate genes was performed. Kif5A was expressed in neuronal cell bodies of the gray matter and axons of the white matter in controls (Figure 4A). At day 14, Kif5A‐positive spheroids appeared and axons showed an overall reduction of Kif5A expression in infected mice. At 98 dpi, Kif5A reached a peak of decreased expression when less than 50% of the axons showed Kif5A immunoreactivity (1, 4). Dync1h1 immunohistochemistry revealed a similar downregulation at the translational level as observed for Kif5A (1, 4). However, no dynein‐positive spheroids were detected (Figure 4D).
Figure 4.
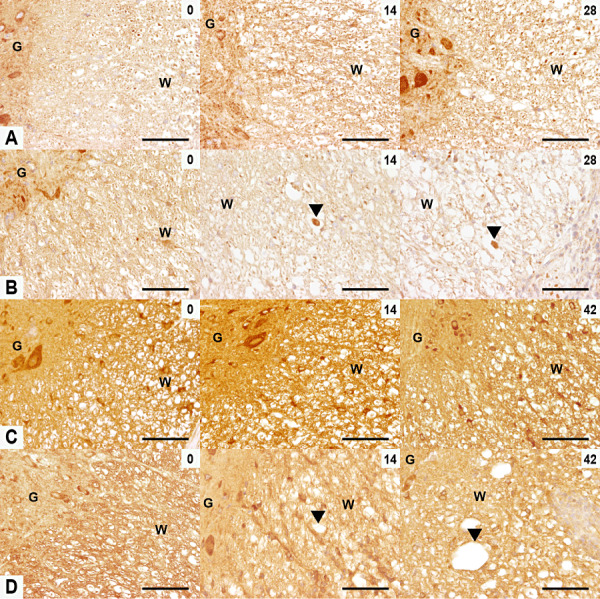
Expression of kinesin family member 5A (Kif5A) and dynein cytoplasmic heavy chain 1 (Dync1h1) in TME. Kif5A (A,B) and Dync1h1 (C,D) immunoreactivity in the ventromedial and lateral spinal cord of control (A,C) and TMEV‐infected mice (B,D) at 0, 14 and 28/42 dpi (d). (A,B) At 0 dpi control and infected mice showed the same pattern of Kif5A expression consisting of positive axons of the white matter and neurons in the gray matter. A decreased Kif5A expression and the appearance of Kif5A‐positive spheroids (arrowheads) were observed in the TMEV‐infected mice at 14 and 28 dpi. (C,D) At 0 dpi, axons in the white matter and neurons in gray matter were Dync1h1‐positive in control and infected groups. A decrease in axonal Dync1h1 expression was seen in the TMEV‐infected mice at 14 and 42 dpi (arrowheads). Scale bar = 100 µm. Abbreviations: G = gray matter; TMEV = Theiler's murine encephalomyelitis virus; W = white matter.
To assess the stability of the axonal cytoskeleton, β‐III, α‐acetylated tubulin and tau‐1 expression were analyzed. The latter is essential for the kinesin‐ and dynein‐mediated transport of NF. In controls, β‐tubulin III and α‐acetylated tubulin (data not shown) followed the Kif5A expression pattern. In TME, their expression decreased significantly at 98 dpi (Figure 1G).
tau‐1 was expressed in oligodendroglial cells of the gray and white matter and axons of the white the matter in controls. In TME, tau‐1 axonal expression was significantly reduced between 42 and 196 dpi (Figure 1H). Furthermore, based on the diametrical axonal expression of p‐NF and n‐NF it was hypothesized that the excessive presence of axonal n‐NF is the result of excessive p‐NF dephosphorylation. To analyze abnormalities in the dephosphorylation processes, the expression of Pp2ac and Pp2aa was studied. At the translational level Pp2a subunits were similarly expressed in control and TME (data not shown).
To investigate whether the abnormalities in NF, Kif5A, Dync1h1, β‐tubulin III and tau‐1 expression are the result of a dysregulated axonal protein degradation system, the axonal presence of ubiquitin–protein conjugates and Uchl‐1 was studied. Both antibodies showed similar expression patterns characterized by positive neurons in the gray matter and glial cells and axons in the white matter of controls (Figure 5A,C). In TME mice, their expression dropped significantly at 28 dpi and remained very low until 196 dpi (1, 5).
Figure 5.
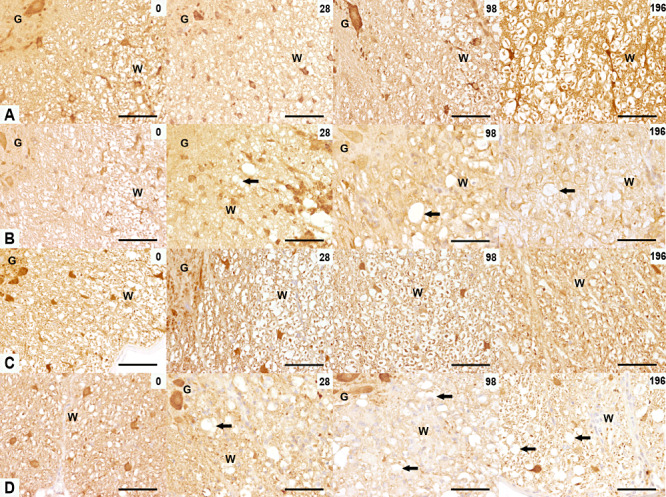
Expression of ubiquitin‐ligated proteins and ubiquitin carboxy‐terminal hydrolase ligase (Uchl‐1) in TME. Ubiquitin (A,B) and Uchl‐1 (C,D) immunoreactivity in the ventromedial spinal cord of control (A,C) and TMEV‐infected mice (B,D) at 0, 28, 98 and 196 dpi. (A,B) At 0 dpi, axonal ubiquitin‐ligated proteins were present in control and infected mice. At 28, 98 and 196 dpi, a lower expression of ubiquitin‐ligated proteins was seen in the TMEV‐infected mice (arrows). (C,D) At 0 dpi, axonal Uchl‐1 was present in the spinal cord of both control and infected mice. At 28, 98 and 196 dpi a lower expression of Uchl‐1 was seen in the TMEV‐infected mice (arrows). Scale bar = 100 µm. Abbreviations: G = gray matter; TMEV = Theiler's murine encephalomyelitis virus; W = white matter.
TMEV‐infected N1E‐115 cells
To answer the question whether TMEV alone or both the microenvironment and/or the pathogen may cause axonal changes, N1E‐115 cells were analyzed after differentiation into neurons and following infection with the BeAn strain of TMEV. The differentiated N1E‐115 cells displayed a neuron‐like morphology whereas non‐differentiated N1E‐115 cells were round and compact (Figure 6).
Figure 6.
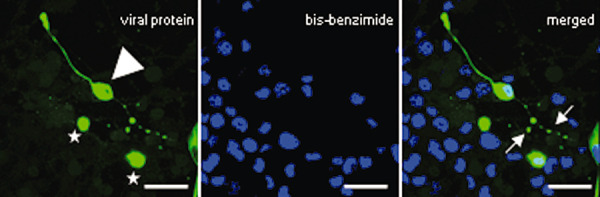
Axonal pathology in N1E‐115 Theiler's murine encephalomyelitis virus (TMEV)‐infected cells. The differentiated TMEV‐infected cells showed tandem‐repeated swellings along their axon‐like processes. N1E‐115 cells were stained with a polyclonal anti‐capsid protein VP1‐BeAn‐TMEV specific antibody and Cy2‐conjugated AffiniPure goat anti‐rabbit serum (viral protein = green) and bis‐benzimide (blue). Non‐differentiated cells are marked with stars, differentiated cells with an arrowhead, the focal swelling with arrows. Scale bars = 16 µm.
Approximately 80% of non‐differentiated and 20% of differentiated cells were infected with TMEV‐BeAn. The differentiated and infected N1E‐115 cells showed a ubiquitous expression of the TMEV antigen, and were characterized by a particular focally accentuated immunoreactivity of their processes that resembled the in vivo focal axonal swellings (Figure 6). These focal swellings were further characterized with respect to n‐NF and p‐NF expression and revealed similarities with the in vivo situation. Thus, the axonal enlargements showed higher and lower levels of n‐NF (Figure 7B) and p‐NF (Figure 7D), respectively, resembling immunoreactivity of spheroids in vivo. In contrast, axonal‐like structures of non‐infected cells displayed an uniform expression of p‐NF and a lack of n‐NF immunoreactivity (Figure 7A,C).
Figure 7.
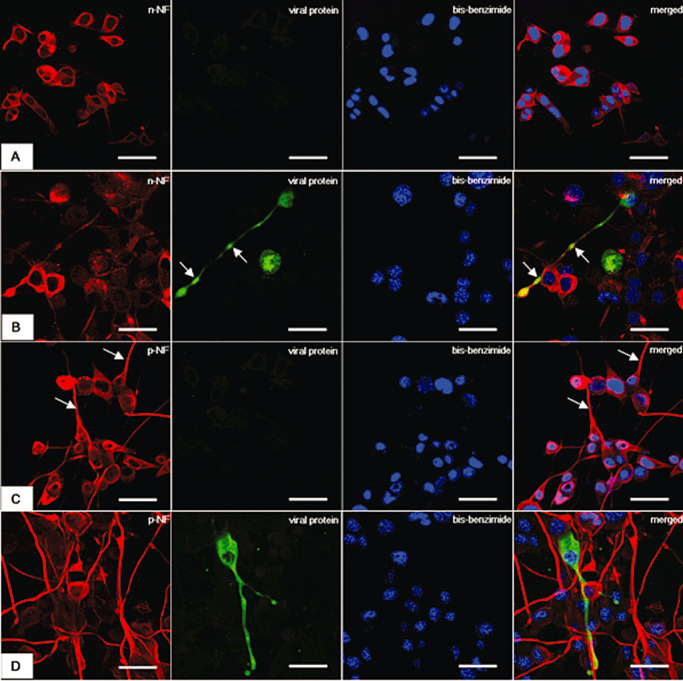
Expression of non‐phosphorylated neurofilament (n‐NF) and phosphorylated NF (p‐NF) in Theiler's murine encephalomyelitis virus (TMEV)‐infected N1E‐115 cells. The figures illustrate n‐NF (A,B) and p‐NF (C,D) immunofluorescence in controls (A,C) and TMEV‐infected cells (B,D). A. Non‐infected cells showed a n‐NF expression restricted to the cell body and lacking of TMEV expression. B. Following infection with TMEV tandem‐repeated swellings (arrows) with n‐NF increased immunoreactivity can be recognized. Simultaneous expression of viral and n‐NF proteins, especially of the swelling, can be observed in the merged picture as yellow discoloration. C. p‐NF shows a uniform expression (arrows) along the axon‐like structures in non‐infected cells. D. Following TMEV expression a decrease in p‐NF immunoreactivity can be observed (arrow) as demonstrated by weak focal yellow discoloration in the merged figure. Confocal laser scanning microscopy for the TMEV BeAn‐strain capsid‐protein VP1 with a Cy2‐coupled secondary antibody (green), combined with either n‐NF or p‐NF with a Cy3‐coupled secondary antibody (red), and nuclei with bisbenzimide (blue). Scale bars = 16 mm.
DISCUSSION
In TME, the onset of the axonopathy during the initial stage is suggested to be a virus‐induced effect (52), whereas the axonal degeneration and loss at later stages are more related to an immune‐mediated attack 11, 19, and a lack of glia‐derived trophic support, which trigger preprogramed neurodegenerative mechanisms in vulnerable, demyelinated axons (35). Both, an axonal decrease in p‐NF‐ and an increase in n‐NF‐immunoreactivity represent well‐known immunohistological markers for axonal pathology 27, 41, 49, 52, 55. In the present study, possible pathogenetic mechanisms involved in an altered axonal NF homeostasis in TME were investigated using in vivo and in vitro experiments 24, 43. Therefore, the aims were: (i) to describe and characterize the degree of axonopathies in TMEV BeAn‐infected SJL/J‐mice; (ii) to investigate possible mechanisms underlying the altered NF‐pattern focusing on molecules involved in their axonal transport and metabolism; and (iii) to investigate the transcriptional profile of a selected list of genes suggested to be involved in the initiation of axonal self‐destruction.
A detailed description of the NF expression during the progression of TME revealed an early onset decrease in p‐NF levels and an increase in the number of n‐NF‐positive axons in the ventrolateral white matter of the spinal cord of diseased animals, in concordance with previous studies 52, 55. Molecular analysis of TMEV‐infected N1E‐115 neuroblastoma cells showed similar results, with n‐NF accumulations at the level of axonal swellings after 7 dpi. Despite the fact that axonal pathology has been detected as early as 1 week after infection in the acute polioencephalomyelitis phase of TME using n‐NF immunohistology (52), significant axonal loss exhibits a different time course beginning in the chronic demyelinating disease phase at 45 dpi for large myelinated fibers and at 195–220 dpi for medium to large myelinated fibers (29). Recently it was demonstrated that a major histocompatibility complex (MHC) class I‐restricted, perforin‐mediated CD8+ T‐cell response directed against a currently unknown neuron‐derived epitope is responsible for the bulk of axonal loss and clinical deficiency in TMEV DA strain‐infected B10Q mice at 90 dpi (11). It can be concluded from the latter study that a CD8+ T cell‐mediated axonal damage and loss in chronic phase of TME depends on the precedence of demyelination and is independent from the anti‐viral immune response (11). In contrast, the description of axonal pathology preceding demyelination (52), the finding of TMEV trafficking via axons of retinal ganglion cells to the cytoplasmic channels of myelin (39) and the fact that demyelinating lesions in TME can be triggered by artificially inducing Wallerian degeneration (53) point towards the importance of virus‐induced axonal pathology for the induction of lesion development. Therefore an altered NF‐pattern, which represents an unquestioned marker for axonal pathology is associated with multiple pathogenic pathways. To further analyze pathogenetic mechanisms contributing to an altered axonal NF homeostasis, a variety of key mechanisms like NF‐transport, phosphorylation and degradation were investigated at the transcriptional and translational level. After labeling of reticulospinal, vestibulospinal and rubrospinal neurons, an impairment of the retrograde transport in TMEV‐infected mice was described by others (57). However, a precise analysis of the proteins involved in this dysregulation is still lacking. It was suggested that NF aggregates are the result of a defective NF axonal transport and/or phosphorylation in neurodegenerative disorders (43). Microarray results of the present study revealed a mild transcriptional downregulation for the members of the kinesin, dynein and protein phosphatase 2 complex, indicating an impairment of the axonal transport. Previous studies investigating NF phosphorylation by using mass spectrometry revealed no differences between amyotrophic lateral sclerosis (ALS) and control tissues 4, 8. This indicated that NF accumulation can be the result of their mislocation in different cellular compartments as it has been shown for some transgenic models of ALS 2, 3, 14. A similar mechanism has been hypothesized for NF aggregation in MS (29). To obtain more detailed information about these processes, the expression of motor and cytoskeleton proteins involved in the axonal transport of NF was further studied by immunohistochemistry in the present investigation. Thus, the specific motor protein for the anterograde transport of NF (46), Kif5A, showed a reduced expression in the spinal cord, beginning at 14 dpi. Furthermore, Dync1h1, involved in NF retrograde transport (47) and tau‐1, responsible for the MT stability (58), showed a similar downregulation as Kif5A. In addition, previous results showed a reduction of the retrograde transport in TMEV‐infected mice using retrograde labeling of reticulospinal, vestibulospinal and rubrospinal neurons (55). Furthermore the specific elimination of a subset of cytotoxic CD8+ T cells directed against the immunodominant VP2121‐130 of TMEV resulted in partial preservation of the axonal retrograde transport and improved motor function in TMEV‐susceptible H‐2b mice with genetic deletion of the interferon gamma receptor (IFNγR–/–) despite of an unaltered amount of axonal loss (19). Based on this observation, it is reasonable to suggest that the previously mentioned CD8+ T‐cell response directed against a currently unknown neuron‐derived epitope in TMEV DA‐strain infected B10Q mice (11) can also induce an interruption of axonal transport. In contrast to the motor proteins, the main components of the microtubules, α and β tubulins, showed a more stable expression at the translational levels, despite mild significant transcriptional changes. Recent data showed that MT stabilization reduces scarring and enhances axonal regeneration after spinal cord injury (18).
An impaired axonal transport could have multiple possible consequences: (i) decrease of TMEV transport as shown before for vaccinia virus (34); (ii) impairment of APP and NF axonal function; and (iii) a reduction of axonal p‐NF expression, and (iv) impaired transport of Nmat‐2 2, 16, 35, APP, a sensitive indicator of early axonal injury was detected in small amounts in axons after TMEV infection beginning at 28 dpi. Kinesin light chains transport APP along the axons (20), therefore a reduction in kinesin expression could trigger a lower APP accumulation. It still remains unclear how an impairment of the NF transport may result in increased expression of axonal n‐NF. Besides the differences in their neuronal localization and affinity for motor proteins 31, 50, 61, there is a specific association between hypophosphorylated/phosphorylated‐NF and other cytoskeleton components. Thus, unlike the extensively phosphorylated NF, only hypophosphorylated NF binds either MTs (19) or myosin with high affinity, and translocates along actin in the axon 1, 20. Moreover, the hypophosphorylated NF‐MT association is regulated by the amount of tau‐1 fragments (45). A reduction in tau‐1 expression as observed in TME could facilitate a hypophosphorylated‐NF MT‐dependent axonal transport. Thus, n‐NF can be alternatively transported and can accumulate along the axons in the absence of motor proteins. It is well known that axonal NF transport and phosphorylation are intimate processes (43). Although the amount of p‐NF and n‐NF was similar in control and ALS patients (4), active kinases like Cdk5 and p38 Map‐kinase were found predominantly in perikarya and in proximal axons. The latter represent subcellular compartments with p‐NF accumulations (4). The complementary expression of both NF forms during TME led to the assumption that a local dephosphorylation process could be an additional mechanism responsible for the conversion of the preexisting p‐NF into axonal n‐NF. Pp2a represents the main enzyme exhibiting the most abundant endogenous phosphatase activity in spinal cord extracts 38, 49. Interestingly, transcription of protein phosphatase 2 complex members was mildly downregulated and Ppp2r2a and Ppp2r2c translation was similar in TMEV‐infected and mock‐infected mice. Though this indicated phosphorylation might not represent a major pathogenetic mechanism, it cannot exclude possible changes in protein phosphatase 2a activity or the participation of other phosphatases in the axonal NF dephosphorylation process.
Either transported from the neuronal cell bodies and/or locally produced in the axon, n‐NF could represent a first step towards NF degradation. Previous studies reported that n‐NF undergoes degradation (26), whereas p‐NF was protected by phosphorylation (17). The question arose why n‐NF degradation during TME is not as efficient as in controls. Various proteases are involved in the NF degradation process. In addition, ubiquitination was described as an important mechanism of NF degradation (43). Ubiquitin‐conjugated proteins are components of the intraneuronal inclusions, and are found in several neurodegenerative diseases (43). In the present study, in contrast to other neurodegenerative disorders, the expression of ubiquitin–protein conjugates and Uchl‐1 was significantly reduced. Because ubiquitination is the first step in a non‐lysosomal degradation pathway of proteins, it was concluded that a derangement of this proteolytic pathway may occur in infected mice. The presumed direct factors involved in the axonal accumulation of n‐NF based on the present investigation are schematically summarized and displayed in Figure 8.
Figure 8.
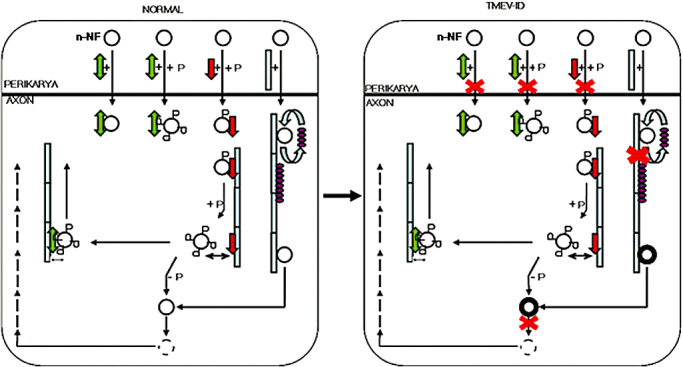
Schematic outline of the impact of the direct factors involved in alterations of neurofilament (NF) metabolism during Theiler's murine encephalomyelitis. The figure shows perikaryon‐axonal transport of neurofilaments and associated proteins. In normal animals, dynein is essential for the antero‐ and retrograde transport of non‐ and phosphorylated NFs whereas, kinesin is responsible for the anterograde transport of low phosphorylated NF. There is a continuous exchange between tau‐1 and n‐NF attached to microtubules. In axons, the phosphorylated NFs are dephosphorylated and then degraded. Following TME virus infection, impairment of these processes (marked with red crosses) that give rise to NF axonal aggregations can be observed. Non‐phosphorylated neurofilaments (○); phosphorylated neurofilaments ( ); kinesin (
); kinesin ( ); dynein (
); dynein ( ); microtubules‐MT‐ (
); microtubules‐MT‐ ( ); tau (
); tau ( ).
).
Recently, it was proposed that axonal degeneration is part of an active self‐destructive response to the TMEV infection preventing further axonal spread of the virus 51, 54. The demonstration of a downregulated transcription of the NAD‐synthesizing enzymes Nmnat‐1 and Nmnat‐2 and an upregulation of the NAD consuming PARP1 in the chronic demyelinating phase of TME is in agreement with the hypothesis that an active neuronal depletion of NAD mediates axonal self‐destruction (35). Notably, Nmat2 is a somatically produced axon survival factor, whose function depends on the anterograde transport (16). The upregulation of Mapk8 at 14 dpi is in agreement with an induced DLK/JNK‐dependent axon‐degeneration pathway during the early phase of polioencephalomyelitis (30). In contrast, the downregulation of both Map3k12 and Mapk8 at later time points indicates that this respective pathway plays a different role during the phase of chronic progressive demyelination in TME. The observed upregulation of Caspr‐6 from 14 to196 dpi suggests that the APP/DR6/Casp‐6‐dependent axon‐degeneration pathway is involved in the anti‐viral response against TMEV throughout the course of the disease (33). The observed downregulation of Bdnf within the spinal cord is a possible trigger leading to trophic deprivation of the axons, shedding of surface APP, activation of death receptor 6 (DR6, Tnfrsf21) and lastly activation of Caspr6 inducing axonal degeneration, similar to the situation encountered in MS and other neurodegenerative diseases 14, 26, 33. The moderate upregulation of CD200r points towards an activation of CD200r‐positive microglia by neuroaxonal CD200 ligands, leading to a self‐perpetuating cycle of neuroaxonal damage and microaglia activation, most likely involved in generation of secondary axonal degeneration (35). Furthermore, Gpr109a [synonyms: HM74a, protein upregulated in macrophages by IFN‐γ (PUMA‐G)], a high‐affinity nicotinic acid G‐protein‐coupled receptor involved in production of prostaglandins and activation of peroxisome proliferator‐activated receptor (PPAR)‐γ dependent transcription within macrophages, is also moderately upregulated (21).
In conclusion, TMEV‐associated axonopathy is characterized by NF accumulations caused by various impaired molecular interactions including mechanisms responsible for axonal transport, NF‐phosphorylation and protein metabolism. Thus, as shown by this study, NF accumulation in TME seems to be the result of specific dysregulations in their axonal transport (Kif5A) and also the sequel of nonspecific impairments in the neuronal protein metabolism (ubiquitin–protein conjugates). These findings suggest that axonal degeneration is induced by different mechanisms in primarily TMEV‐induced axonal damage (inside‐out) compared with secondary axonal degeneration following demyelination (outside‐in), converging on a common Nmat‐sensitive end pathway of NAD depletion, lack of ATP and finally axonal degeneration (59). Further functional studies concerning the axon self‐destruction mechanisms are needed in order to increase our understanding of their impact on disease initiation and development and design of new therapeutical strategies.
Supporting information
Figure S1. Theiler's murine encephalomyelitis virus spread in the spinal cord. A. Distribution of TMEV‐positive cells in the ventromedial spinal cord at 28 dpi. TMEV was found predominantly in microglia/macrophages (arrows). Scale bar = 200 μm. B. Inset of A which shows TMEV‐positive microglia/macrophages cells (arrows) and a positive axon (arrowhead). Scale bar = 50 μm. Abbreviations: G = gray matter; W = white matter.
Figure S2. TMEV‐induced demyelination. (A,B) Luxol fast blue cresyl‐echt violet‐stained ventromedial spinal cord of control (A) and TMEV‐infected mice (B) at 4, 42 and 98 dpi. At 4 dpi, the spinal cord of control and infected mice showed a uniformly Luxol fast blue‐stained white matter. At 42 dpi, lymphohistiocytic (arrow) and mild demyelination of the white matter (asterisks) were present. At 98 dpi, a minimal diffuse parenchymal inflammatory reaction and lymphohistiocytic meningitis (arrows) and an increased area of demyelination can be observed (asterisks). Scale bar = 100 μm. Abbreviations: G = gray matter; W = white matter.
Figure S3. Axonal loss in TME. (A,B) Results of Bielschowsky silver stain in the ventromedial spinal cord of control (A) and TMEV‐infected mice (B) at 0, 28, 98 and 196 dpi. At 0 dpi, normal staining of axons in the spinal cord of the control and TMEV‐infected mice was seen. From 28 until 196 dpi extensive pathology of the nerve fibers characterized by variation of axonal thickness, loss of axons (arrows) and presence of spheroids (arrowheads) was observed in TMEV‐infected mice compared with controls. Scale bar = 100 μm. Abbreviation: W = white matter.
Table S1. Axonal transport associated probe sets manually curated from peer‐reviewed scientific articles.
Table S2. Axonal self‐destruction associated probe sets manually curated from peer‐reviewed scientific articles.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
The authors would like to thank Petra Grünig, Bettina Buck, Danuta Waschke, Julia Schirrmeier, Kerstin Rohn, Claudia Hermann, Anuschka Unold, Martin Gamber, Thomas Feidl, Frank Seeliger, Ingo Gerhauser, Heinz Theobald and Michael Müller for their excellent technical support. The BeAn strain of TMEV was a generous gift of Dr. H.L. Lipton, Department of Microbiology‐Immunology, University of Illinois, Chicago, IL, USA. Mihaela Kreutzer was supported by a Georg‐Christoph‐Lichtenberg scholarship provided by the Ministry for Science and Culture of Lower Saxony. Frauke Seehusen received a scholarship from Bayer HealthCare, Leverkusen. Reiner Ulrich (RU) and Robert Kreutzer (RK) received scholarship from the Center for Systems Neuroscience, Hannover, Germany (RU) and the University for Veterinary Medicine Foundation, Hannover, Germany (RK). This study was, in part, supported by the German Research Foundation (DFG, Research Unit: Neurodegeneration and Regeneration in Canine CNS Diseases; BA 815/10‐1).
REFERENCES
- 1. Ahmad FJ, Echeverri CJ, Vallee RB, Baas PW (1998) Cytoplasmic dynein and dynactin are required for the transport of microtubules into the axon. J Cell Biol 140:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA (2010) NAD+ depletion is necessary and sufficient for poly(ADP‐ribose) polymerase‐1‐mediated neuronal death. J Neurosci 30:2967–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Araki T, Sasaki Y, Milbrandt J (2004) Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305:1010–1013. [DOI] [PubMed] [Google Scholar]
- 4. Bajaj NP, al‐Sarraj ST, Leigh PN, Anderson V, Miller CC (1999) Cyclin dependent kinase‐5 (CDK‐5) phosphorylates neurofilament heavy (NF‐H) chain to generate epitopes for antibodies that label neurofilament accumulations in amyotrophic lateral sclerosis (ALS) and is present in affected motor neurones in ALS. Prog Neuropsychopharmacol Biol Psychiatry 23:833–850. [DOI] [PubMed] [Google Scholar]
- 5. Bolstad BM, Irizarry RA, Astrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193. [DOI] [PubMed] [Google Scholar]
- 6. Coleman M (2005) Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 6:889–898. [DOI] [PubMed] [Google Scholar]
- 7. Collard JF, Cote F, Julien JP (1995) Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 375:61–64. [DOI] [PubMed] [Google Scholar]
- 8. Conforti L, Adalbert R, Coleman MP (2007) Neuronal death: where does the end begin? Trends Neurosci 30:159–166. [DOI] [PubMed] [Google Scholar]
- 9. Czasch S, Paul S, Baumgartner W (2006) A comparison of immunohistochemical and silver staining methods for the detection of diffuse plaques in the aged canine brain. Neurobiol Aging 27:293–305. [DOI] [PubMed] [Google Scholar]
- 10. Deb C, Lafrance‐Corey RG, Zoecklein L, Papke L, Rodriguez M, Howe CL (2009) Demyelinated axons and motor function are protected by genetic deletion of perforin in a mouse model of multiple sclerosis. J Neuropathol Exp Neurol 68:1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deb C, Lafrance‐Corey RG, Schmalstieg WF, Sauer BM, Wang H, German CL et al (2010) CD8+ T cells cause disability and axon loss in a mouse model of multiple sclerosis. PLoS ONE 5:e12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dziedzic T, Metz I, Dallenga T, Konig FB, Muller S, Stadelmann C, Bruck W (2010) Wallerian degeneration: a major component of early axonal pathology in multiple sclerosis. Brain Pathol 20:976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferguson B, Matyszak MK, Esiri MM, Perry VH (1997) Axonal damage in acute multiple sclerosis lesions. Brain 120(Pt 3):393–399. [DOI] [PubMed] [Google Scholar]
- 14. Gehrmann J, Banati RB, Cuzner ML, Kreutzberg GW, Newcombe J (1995) Amyloid precursor protein (APP) expression in multiple sclerosis lesions. Glia 15:141–151. [DOI] [PubMed] [Google Scholar]
- 15. Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW (1993) Beta‐amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett 160:139–144. [DOI] [PubMed] [Google Scholar]
- 16. Gilley J, Coleman MP (2010) Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 8:e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gotow T (2000) Neurofilaments in health and disease. Med Electron Microsc 33:173–199. [DOI] [PubMed] [Google Scholar]
- 18. Haist VUR, Kalkuhl A, Deschl U, Baumgärtner W (2011) Mild transcriptional changes of extracellular matrix but extensive matrix accumulation in a distinct spatio‐temporal pattern within chronic demyelinated spinal cord lesions in Theiler's murine encephalomyelitis. Brain Pathol doi:10.1111/j.1750‐3639.2011.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Howe CL, Ure D, Adelson JD, LaFrance‐Corey R, Johnson A, Rodriguez M (2007) CD8+ T cells directed against a viral peptide contribute to loss of motor function by disrupting axonal transport in a viral model of fulminant demyelination. J Neuroimmunol 188:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamal A, Almenar‐Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001) Kinesin‐mediated axonal transport of a membrane compartment containing beta‐secretase and presenilin‐1 requires APP. Nature 414:643–648. [DOI] [PubMed] [Google Scholar]
- 21. Knowles HJ, te Poele RH, Workman P, Harris AL (2006) Niacin induces PPARgamma expression and transcriptional activation in macrophages via HM74 and HM74a‐mediated induction of prostaglandin synthesis pathways. Biochem Pharmacol 71:646–656. [DOI] [PubMed] [Google Scholar]
- 22. Kreutzer R, Kreutzer M, Propsting MJ, Sewell AC, Leeb T, Naim HY, Baumgartner W (2008) Insights into post‐translational processing of beta‐galactosidase in an animal model resembling late infantile human G‐gangliosidosis. J Cell Mol Med 12:1661–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kummerfeld M, Meens J, Haas L, Baumgartner W, Beineke A (2009) Generation and characterization of a polyclonal antibody for the detection of Theiler's murine encephalomyelitis virus by light and electron microscopy. J Virol Methods 160:185–188. [DOI] [PubMed] [Google Scholar]
- 24. Lee VM, Carden MJ, Schlaepfer WW, Trojanowski JQ (1987) Monoclonal antibodies distinguish several differentially phosphorylated states of the two largest rat neurofilament subunits (NF‐H and NF‐M) and demonstrate their existence in the normal nervous system of adult rats. J Neurosci 7:3474–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lew J, Winkfein RJ, Paudel HK, Wang JH (1992) Brain proline‐directed protein kinase is a neurofilament kinase which displays high sequence homology to p34cdc2. J Biol Chem 267:25922–25926. [PubMed] [Google Scholar]
- 26. Lindquist S, Hassinger S, Lindquist JA, Sailer M (2011) The balance of pro‐inflammatory and trophic factors in multiple sclerosis patients: effects of acute relapse and immunomodulatory treatment. Mult Scler 17:851–866. [DOI] [PubMed] [Google Scholar]
- 27. Mancardi G, Hart B, Roccatagliata L, Brok H, Giunti D, Bontrop R et al (2001) Demyelination and axonal damage in a non‐human primate model of multiple sclerosis. J Neurol Sci 184:41–49. [DOI] [PubMed] [Google Scholar]
- 28. Masaki R, Saito T, Yamada K, Ohtani‐Kaneko R (2000) Accumulation of phosphorylated neurofilaments and increase in apoptosis‐specific protein and phosphorylated c‐Jun induced by proteasome inhibitors. J Neurosci Res 62:75–83. [DOI] [PubMed] [Google Scholar]
- 29. McGavern DB, Murray PD, Rodriguez M (1999) Quantitation of spinal cord demyelination, remyelination, atrophy, and axonal loss in a model of progressive neurologic injury. J Neurosci Res 58:492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A (2009) A dual leucine kinase‐dependent axon self‐destruction program promotes Wallerian degeneration. Nat Neurosci 12:387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Motil J, Chan WK, Dubey M, Chaudhury P, Pimenta A, Chylinski TM et al (2006) Dynein mediates retrograde neurofilament transport within axons and anterograde delivery of NFs from perikarya into axons: regulation by multiple phosphorylation events. Cell Motil Cytoskeleton 63:266–286. [DOI] [PubMed] [Google Scholar]
- 32. Murray PD, Pavelko KD, Leibowitz J, Lin X, Rodriguez M (1998) CD4(+) and CD8(+) T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J Virol 72:7320–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nikolaev A, McLaughlin T, O'Leary DD, Tessier‐Lavigne M (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG (2000) Multiple sclerosis. N Engl J Med 343:938–952. [DOI] [PubMed] [Google Scholar]
- 35. Penberthy WT, Tsunoda I (2009) The importance of NAD in multiple sclerosis. Curr Pharm Des 15:64–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perrot R, Berges R, Bocquet A, Eyer J (2008) Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol Neurobiol 38:27–65. [DOI] [PubMed] [Google Scholar]
- 37. Pringproa K, Rohn K, Kummerfeld M, Wewetzer K, Baumgartner W (2010) Theiler's murine encephalomyelitis virus preferentially infects immature stages of the murine oligodendrocyte precursor cell line BO‐1 and blocks oligodendrocytic differentiation in vitro . Brain Res 1327:24–37. [DOI] [PubMed] [Google Scholar]
- 38. Qian W, Shi J, Yin X, Iqbal K, Grundke‐Iqbal I, Gong CX, Liu F (2010) PP2A regulates tau phosphorylation directly and also indirectly via activating GSK‐3beta. J Alzheimers Dis 19:1221–1229. [DOI] [PubMed] [Google Scholar]
- 39. Roussarie JP, Ruffie C, Brahic M (2007) The role of myelin in Theiler's virus persistence in the central nervous system. PLoS Pathog 3:e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sato F, Omura S, Martinez NE, Tsunoda I (2011) Animal model for multiple sclerosis. In: Neuroinflammation, Minagar A (ed.), pp. 55–79. Elsevier Books: Burlington, MA. [Google Scholar]
- 41. Schirmer L, Antel JP, Bruck W, Stadelmann C (2011) Axonal loss and neurofilament phosphorylation changes accompany lesion development and clinical progression in multiple sclerosis. Brain Pathol 21:428–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schlaepfer WW, Zimmerman UJ (1985) Mechanisms underlying the neuronal response to ischemic injury. Calcium‐activated proteolysis of neurofilaments. Prog Brain Res 63:185–196. [DOI] [PubMed] [Google Scholar]
- 43. Seehusen F, Baumgartner W (2010) Axonal pathology and loss precede demyelination and accompany chronic lesions in a spontaneously occurring animal model of multiple sclerosis. Brain Pathol 20:551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Selvaraj V, Soundarapandian MM, Chechneva O, Williams AJ, Sidorov MK, Soulika AM et al (2009) PARP‐1 deficiency increases the severity of disease in a mouse model of multiple sclerosis. J Biol Chem 284:26070–26084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shah JV, Flanagan LA, Janmey PA, Leterrier JF (2000) Bidirectional translocation of neurofilaments along microtubules mediated in part by dynein/dynactin. Mol Biol Cell 11:3495–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shea TB, Chan WK (2008) Regulation of neurofilament dynamics by phosphorylation. Eur J Neurosci 27:1893–1901. [DOI] [PubMed] [Google Scholar]
- 47. Sobottka B, Harrer MD, Ziegler U, Fischer K, Wiendl H, Hunig T et al (2009) Collateral bystander damage by myelin‐directed CD8+ T cells causes axonal loss. Am J Pathol 175:1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D (2009) Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci 29:14965–14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Strack S, Westphal RS, Colbran RJ, Ebner FF, Wadzinski BE (1997) Protein serine/threonine phosphatase 1 and 2A associate with and dephosphorylate neurofilaments. Brain Res Mol Brain Res 49:15–28. [DOI] [PubMed] [Google Scholar]
- 50. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285. [DOI] [PubMed] [Google Scholar]
- 51. Tsunoda I (2008) Axonal degeneration as a self‐destructive defense mechanism against neurotropic virus infection. Future Virol 3:579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tsunoda I, Kuang LQ, Libbey JE, Fujinami RS (2003) Axonal injury heralds virus‐induced demyelination. Am J Pathol 162:1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tsunoda I, Tanaka T, Saijoh Y, Fujinami RS (2007) Targeting inflammatory demyelinating lesions to sites of Wallerian degeneration. Am J Pathol 171:1563–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tsunoda I, Tanaka T, Terry EJ, Fujinami RS (2007) Contrasting roles for axonal degeneration in an autoimmune versus viral model of multiple sclerosis: when can axonal injury be beneficial? Am J Pathol 170:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ulrich R, Seeliger F, Kreutzer M, Germann PG, Baumgartner W (2008) Limited remyelination in Theiler's murine encephalomyelitis due to insufficient oligodendroglial differentiation of nerve/glial antigen 2 (NG2)‐positive putative oligodendroglial progenitor cells. Neuropathol Appl Neurobiol 34:603–620. [DOI] [PubMed] [Google Scholar]
- 56. Ulrich R, Kalkuhl A, Deschl U, Baumgartner W (2010) Machine learning approach identifies new pathways associated with demyelination in a viral model of multiple sclerosis. J Cell Mol Med 14:434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ure D, Rodriguez M (2000) Extensive injury of descending neurons demonstrated by retrograde labeling in a virus‐induced murine model of chronic inflammatory demyelination. J Neuropathol Exp Neurol 59:664–678. [DOI] [PubMed] [Google Scholar]
- 58. Veeranna SKT, Link WT, Jaffe H, Wang J, Pant HC (1995) Neuronal cyclin‐dependent kinase‐5 phosphorylation sites in neurofilament protein (NF‐H) are dephosphorylated by protein phosphatase 2A. J Neurochem 64:2681–2690. [DOI] [PubMed] [Google Scholar]
- 59. Vohra BP, Sasaki Y, Miller BR, Chang J, DiAntonio A, Milbrandt J (2010) Amyloid precursor protein cleavage‐dependent and ‐independent axonal degeneration programs share a common nicotinamide mononucleotide adenylyltransferase 1‐sensitive pathway. J Neurosci 30:13729–13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72:1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xia CH, Roberts EA, Her LS, Liu X, Williams DS, Cleveland DW, Goldstein LS (2003) Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol 161:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yabe JT, Pimenta A, Shea TB (1999) Kinesin‐mediated transport of neurofilament protein oligomers in growing axons. J Cell Sci 112:3799–3814. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Theiler's murine encephalomyelitis virus spread in the spinal cord. A. Distribution of TMEV‐positive cells in the ventromedial spinal cord at 28 dpi. TMEV was found predominantly in microglia/macrophages (arrows). Scale bar = 200 μm. B. Inset of A which shows TMEV‐positive microglia/macrophages cells (arrows) and a positive axon (arrowhead). Scale bar = 50 μm. Abbreviations: G = gray matter; W = white matter.
Figure S2. TMEV‐induced demyelination. (A,B) Luxol fast blue cresyl‐echt violet‐stained ventromedial spinal cord of control (A) and TMEV‐infected mice (B) at 4, 42 and 98 dpi. At 4 dpi, the spinal cord of control and infected mice showed a uniformly Luxol fast blue‐stained white matter. At 42 dpi, lymphohistiocytic (arrow) and mild demyelination of the white matter (asterisks) were present. At 98 dpi, a minimal diffuse parenchymal inflammatory reaction and lymphohistiocytic meningitis (arrows) and an increased area of demyelination can be observed (asterisks). Scale bar = 100 μm. Abbreviations: G = gray matter; W = white matter.
Figure S3. Axonal loss in TME. (A,B) Results of Bielschowsky silver stain in the ventromedial spinal cord of control (A) and TMEV‐infected mice (B) at 0, 28, 98 and 196 dpi. At 0 dpi, normal staining of axons in the spinal cord of the control and TMEV‐infected mice was seen. From 28 until 196 dpi extensive pathology of the nerve fibers characterized by variation of axonal thickness, loss of axons (arrows) and presence of spheroids (arrowheads) was observed in TMEV‐infected mice compared with controls. Scale bar = 100 μm. Abbreviation: W = white matter.
Table S1. Axonal transport associated probe sets manually curated from peer‐reviewed scientific articles.
Table S2. Axonal self‐destruction associated probe sets manually curated from peer‐reviewed scientific articles.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item