

Abstract
The prognostic significance of IDH1 mutations has been demonstrated in gliomas. It is unclear whether IDH1 mutation is also a prognostic factor in gliomatosis cerebri (GC). Primary GCs can be grouped into type 1 GCs, which have the classical diffuse growth pattern without mass formation, and type 2 GCs, which form neoplastic masses in addition to classic diffuse lesions. In this study, the prognostic relevance of IDH1/2 mutations in 74 GCs (43 type 1 and 31 type 2) was evaluated. We detected 33 (44.6%) IDH1 mutations, including R132H and R132S, by bidirectional Sanger sequencing. No mutations were detected in IDH2. The percentage of 2‐year overall survival for wild‐type IDH1 patients was 46 vs. 72% for patients with IDH1‐mutated tumors. Mutations of IDH1 were strongly correlated with both increased overall survival (OS) and progression‐free survival (PFS) in patients with type 2 GCs, and IDH1 mutations were also an independent prognostic factor predicting increased OS and PFS in type 2 GC patients in multivariate analysis. However, IDH1 mutations did not correlate with survival outcomes in patients with type 1 GCs. Finally, the subgroup of GC, which has IDH1 wild‐type and additional solid component showed the worst prognosis.
Keywords: gliomatosis cerebri, IDH1, IDH2, mutations, prognostic factor
INTRODUCTION
Gliomatosis cerebri (GC) is a diffuse neoplastic glial tumor that infiltrates the brain extensively, involves at least three lobes and commonly preserves the local parenchymal architecture (6). The term GC was first coined in 1938 by Nevin (19) and refers to a clinicopathologic diagnosis, implying a heterogeneous tumor with variable microscopic phenotypes, radiologic features and clinical behavior. Most GCs have astrocytic phenotypes, but may also present as oligodendrogliomas and mixed oligoastrocytomas (6). Macroscopically, primary GCs are divided into two subtypes: type 1 and type 2 GCs 3, 13. A type 1 GC is the classical lesion that is characterized by diffuse neoplastic growth and enlargement of the involved existing structure, without the formation of a discrete tumor mass at initial clinical presentation (13). A type 2 GC is associated with the presence of obvious neoplastic masses in addition to the diffuse lesion at initial clinical presentation (13). These subgroups are referred to, but not included in, the 2007 World Health Organization (WHO) classification of tumors of the central nervous system.
Recently, a mutation affecting codon 132 of the isocitrate dehydrogenase 1 gene (IDH1), located on chromosome 2q33, was shown to be present in grade II–III gliomas and glioblastomas arising from lower‐grade gliomas (22). IDH1 encodes IDH1, which catalyzes the oxidative carboxylation of isocitrate to α‐ketoglutarate, yielding reduced nicotinamide adenine dinucleotide phosphate (NADPH). IDH1 is localized in the cytoplasm and peroxisomes, whereas the other four IDH proteins, including IDH2, are localized in mitochondria. IDH1 is one of the major enzymes responsible for the production of cytosolic NADPH that is necessary for the regeneration of reduced glutathione, which functions as the main antioxidant in mammalian cells (29). Specific mutations in the coding sequence of IDH1 can change the preference of the enzyme from isocitrate to α‐ketoglutarate, resulting in the NADPH‐dependent conversion of α‐ketoglutarate to R(‐)‐2‐hydroxyglutarate. Mutations in IDH1 may also decrease the wild‐type IDH1 enzymatic conversion of isocitrate to α‐ketoglutarate (29). Glial tumors occasionally have mutations in the corresponding codon 172 of the IDH2 gene, which codes for a mitochondrial enzyme with a similar function (29).
Numerous biomolecular markers have been explored to stratify malignant gliomas into prognostic subgroups. Recent studies have indicated that IDH1 mutations are strong predictors of a more favorable prognosis in grade II–IV gliomas (22). However, although GC is usually aggressive and its overall biologic behavior corresponds to WHO grade III (6), few large‐scale studies have specifically attempted to verify the prognostic value of IDH mutations in patients with GCs. In this study, we investigated the prognostic value of IDH mutations in GCs and examined whether the prognostic significance of the IDH mutations varied according to GC growth type.
MATERIALS AND METHODS
Patients and histological evaluation
This study was based on 74 cases of primary GC documented in the pathology files of Samsung Medical Center during the 17‐year period from 1995 to 2010. Twenty‐four of these cases were included in a previous study (20). The diagnosis of GC was based on the following criteria: (i) T2‐ or fluid‐attenuated inversion recovery (FLAIR)‐weighted magnetic resonance imaging (MRI) showing a diffuse infiltrative process involving more than three different lobes and relative preservation of the anatomical architecture and (ii) histological tissue analysis confirming glial cell proliferation, either diagnostic of or consistent with an infiltrative glioma. All magnetic resonance (MR) images of the 74 patients were reviewed by a neuroradiologist and a neurosurgeon and were classified as type 1 or type 2 GCs based on these images (Figure 1), as reported previously (20). The term “type 2 GC” was used to characterize lesions with an obvious neoplastic mass in addition to a diffuse infiltrative lesion involving more than three different lobes at the time of diagnosis.
Figure 1.
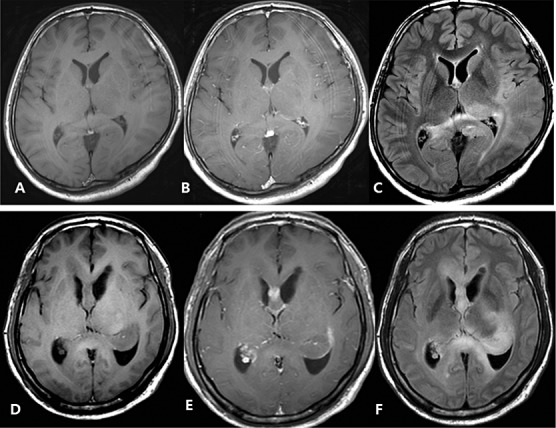
A–C. Diffuse Type 1 gliomatosis cerebri. Pre‐ (A) and postcontrast (B) images show no enhancing portion in the tumor. Flair image (C) indicates diffuse tumor infiltration in left parietotemporal lobe, splenium of corpus callosum, and right parietal lobe with no mass defect and distortion of underlying structures. D–F. Mass forming type 2 gliomatosis cerebri. Pre‐ (D) and Postcontrast (E) shows focal enhancing portion in corpus callosum. Flair image (F) shows tumor infiltrating corpus callosum, left thalamus, basal ganglia and hippocampus with mass formation at hippocampus.
All patients underwent surgical examinations for pathological assessment; 30 patients underwent craniotomy and resection of the dominant mass, one patient underwent open biopsy, one patient underwent endoscopic biopsy, 10 patients underwent navigation‐guided biopsy, and 32 patients underwent stereotactic biopsy. Histological grading and evaluation were used to classify the lesions as astrocytomas, oligodendrogliomas, mixed oligoastrocytomas or glioblastomas according to the WHO classification (6).
Clinical information including age, sex, treatment modality and survival or disease progression was analyzed using clinical records and radiological investigation results. This study was approved by the Institutional Review Board of Samsung Medical Center (Seoul, Korea).
IDH1 and IDH2 DNA sequence analyses
Genomic DNA was extracted from 10‐µm‐thick sections of 10% neutral formalin‐fixed, paraffin‐embedded (FFPE) tumor tissue blocks using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). The complete coding sequence of exon 4 of the IDH1 gene including codon 132 was obtained by overlapping polymerase chain reaction (PCR) amplification. PCR was performed in a 20‐µL volume containing 100 ng of template DNA, 10 µL PCR buffer; 0.25 mM dNTPs, 10 pmol primers, and 1.25 U Taq DNA polymerase (iNtRON, Seoul, Korea). PCR products were electrophoresed on 2% agarose gels and were purified with a QIAquick PCR purification kit (QIAGEN). Bidirectional sequencing was performed using the BigDye Terminator v1.1 kit (Applied Biosystems, Foster City, CA, USA) on an ABI 3130xl genetic analyzer (Applied Biosystems).
Sequencher version 4.10.1 (Gene Codes Corporation, Ann Arbor, MI, USA) was used along with manual chromatogram reviews for sequence analysis. Confirmatory resequencing from replicate PCR amplification reactions was performed for all sequences that were ambiguous or deviated from wild‐type, so that all abnormal sequences were confirmed at least in quadruplicate.
Immunohistochemical analysis of IDH1
IDH1‐R132H protein expression was determined by immunohistochemical staining of paraffin‐embedded tumor specimens. Histologic sections, 5 µm in thickness, were deparaffinized in xylene, rehydrated and heated at 100°C in citrate buffer (pH 6.0) for 5 minutes. Sections were incubated with the monoclonal HMab‐1 antibody that specifically recognizes IDH1‐R132H (15). The slide was incubated with HMab‐1 (5 µg/mL) at 37°C for 1 h. The DAKO LSAB kit (Dako Corp., Carpinteria, CA, USA) postprimary antibody blocker and secondary antibody were used. Color was developed using 3,3‐diaminobenzidine tetrahydrochloride (DAB) for 10 minutes, and then sections were counterstained with hematoxylin. The expression of IDH1‐R132H was determined by semiquantitatively assessing the proportion of positively‐stained tumor cells. Tumor cells that showed strong cytoplasmic staining were scored as positive for staining (2). Cases with ≥10% stained cells were considered positive, and cases with <10% stained cells were considered negative (25).
O6‐methylguanine‐DNA methyltransferase (MGMT) promoter methylation analysis by quantitative real‐time methylation‐specific PCR
DNA extracted from FFPE tissues was treated with sodium bisulfite using the EZ DNA methylation kit (Zymo Research, Orange County, CA, USA). Quantitative methylation‐specific PCR (qMSP) assays were performed in an ABI 7900HT Fast Real‐time PCR system (Applied Biosystems). Primer pairs used were as follows: MGMT forward, CGTTTCGACGTTCGTAGGT and reverse, AAAACTCCGCACTCTTCCG, with the TaqMan probe 6FAM‐AACGACCCAAACACTCACCAAATCGC‐BBQ; ACTB forward, TGGTGATGGAGGAGGTTTAGTAAGT and reverse, AACCAATAAAACCTACTCCTCCCTTAA, with the TaqMan probe 6FAM‐ACCACCACCCAACACACAATAACAAACACA‐BBQ. The housekeeping β‐actin gene (ACTB) was used for normalization of the methylation‐independent control reaction. For relative quantification, the amounts of methylated DNA (PMR, percentage of methylated reference) at an MGMT promoter region was normalized to the methylation value of the calibrator, which was defined as 100%. Universal methylated DNA (QIAGEN) was used as the calibrator. The PMR was defined as 100 × 2(sampleACTB(ct)‐sampleMGMT(ct))/2(calibratorACTB(ct)‐calibratorMGMT(ct)). The cut off value for discrimination between methylation levels was 12; samples with a methylation value (PMR) ≥ 12 were considered to be “methylated”, whereas those with a value <12 were considered “unmethylated”11, 26.
Statistical analysis
The chi‐square test was used to examine possible associations among qualitative clinicopathological variables, absence or presence of genetic alterations, and the two growth types of GCs. The association between IDH1 mutational status and age (<18 vs. ≥18 years) was determined using a two‐tailed Fisher's exact test. The relationships between growth type, patient age and the MGMT promoter methylation ratio were examined by Mann–Whitney U‐tests. Progression‐free survival (PFS) was defined as the period from the day of first surgery until tumor progression, death or the end of follow‐up. Overall survival (OS) was defined as the period from the day of first surgery until death or the end of follow‐up. Survival differences among groups were calculated using the Kaplan–Meier method with a log‐rank test. We used the Cox proportional hazards model for multivariate PFS and OS analyses. For the subgroup survival analysis, the log‐rank test with Bonferroni's correction was used. SPSS statistical software (version 18, Statistical Package for the Social Sciences, Chicago, IL, USA) and R.2.12.1 (http://www.r‐project.org) were used for all statistical analyses. P values < 0.05 were considered statistically significant.
RESULTS
Clinicopathologic demographics according to the gliomatosis cerebri growth type
The 74 patients (46 males and 28 females) examined in this study had a mean age of 45 years (Table 1). Based on the MRI findings, 43 tumors were classified as type 1 GCs and 31 tumors were classified as type 2 GCs (Figure 1). Histological evaluation of the samples revealed astrocytic, oligoastrocytic and oligodendroglial tumors with variable histological grades corresponding to WHO grades II–IV. Based on the WHO criteria, 47 (63.5%) GCs were classified as grade II, 23 (31.1%) as grade III and 4 (5.4%) as grade IV. Twenty‐one patients (28.4%) had tumors resected by debulking surgery, while 30 patients (40.5%) were treated with adjuvant chemotherapy (temozolomide; TMZ). During the follow‐up period ranging from 2–191 months (mean, 37 months), 37 patients (50%) comprising 17 patients with type 1 GCs and 20 patients with type 2 GC patients died. Statistical analyses revealed that there were no significant differences between type 1 and type 2 GCs according to sex, age, histological grade or MGMT promoter methylation status (P = 0.270, P = 0.488, P = 0.181 and P = 0.517, respectively).
Table 1.
Patient demographics and clinical characteristics.
| Characteristic | Gliomatosis cerebri | ||
|---|---|---|---|
| Type 1 | Type 2 | P | |
| n = 43 | n = 31 | ||
| Sex | 0.270 | ||
| Male | 29 | 17 | |
| Female | 14 | 14 | |
| Age (years) | |||
| Median (mean ± SD) | 49 (46.3 ± 16.3) | 45 (43.5 ± 17.6) | 0.488 |
| Range | 10–68 | 3–73 | |
| Histological grade | 0.181 | ||
| Grade II | 25 | 22 | |
| Grade III | 14 | 9 | |
| Grade IV | 4 | 0 | |
| Debulking surgery | 0.250 | ||
| Done | 10 | 11 | |
| Not done | 33 | 20 | |
| Adjuvant chemotherapy | 0.150 | ||
| Done (including TMZ) | 14 (7) | 16 (7) | |
| Not done | 29 | 15 | |
| MGMT promoter methylation status | 0.517 | ||
| Methylated | 6 | 2 | |
| Unmethylated | 30 | 22 | |
| NA | 7 | 7 | |
| qMSP ratio (mean ± SD) (mMGMT/ACTB) × 1000 | 9.2 ± 22.1 | 2.7 ± 7.1 | 0.173 |
MGMT = O6‐methylguanine‐DNA methyltransferase; NA = not applicable; qMSP = Quantitative methylation‐specific polymerase chain reaction; SD = standard deviation; TMZ = temozolomide.
IDH1/IDH2 mutation analysis and correlations with clinicopathological parameters
We detected 33 (44.6%) of 74 mutations known to occur in codon 132 of IDH1: 32 (97.0%) G395A (Arg132His) mutations and one (3.0%) C394A (Arg132Ser) mutation (Figure 2). In contrast, we detected no mutations in codon 172 of IDH2.
Figure 2.
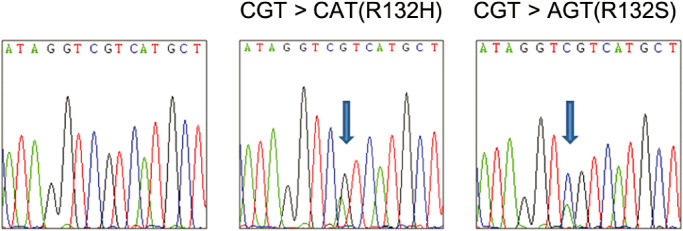
Chromaview snapshots of IDH1 wild‐type and mutant type sequences (R132H and R132S).
We next investigated if there was a correlation between IDH1 mutation status and clinicopathologic parameters (Table 2). There were no statistical differences in sex, age, growth type, histological grade, MGMT promoter methylation status, debulking surgery status, or adjuvant chemotherapy treatment status between patients with GCs with IDH1 mutations and patients with GCs with wild‐type IDH1 (P = 0.225, P = 0.725, P = 0.934, P = 0.695, P = 0.103, P = 0.742, and P = 0.511, respectively).
Table 2.
Correlations between clinicopathological characteristics and IDH1 mutation status.
| Total | IDH1 mutation | IDH1 wild type | P | |
|---|---|---|---|---|
| n = 74 | n = 33 | n = 41 | ||
| Sex | 0.225 | |||
| Male | 46 | 18 | 28 | |
| Female | 28 | 15 | 13 | |
| Age (years) | 0.725 | |||
| <18 | 8 | 3 | 5 | |
| ≥18 | 66 | 30 | 36 | |
| Growth type | 0.934 | |||
| Type 1 | 43 | 19 | 24 | |
| Type 2 | 31 | 14 | 17 | |
| Histological grade | 0.695 | |||
| Grade II | 47 | 22 | 25 | |
| Grade III | 23 | 10 | 13 | |
| Grade IV | 4 | 1 | 3 | |
| MGMT promoter methylation status | 0.103 | |||
| Methylated | 8 | 5 | 3 | |
| Unmethylated | 52 | 17 | 35 | |
| NA | 14 | 11 | 3 | |
| Debulking surgery | 0.742 | |||
| Done | 21 | 10 | 11 | |
| Not done | 53 | 23 | 30 | |
| Adjuvant chemotherapy | 0.511 | |||
| Done (including TMZ) | 30 | 12 (5) | 18 (9) | |
| Not done | 44 | 21 | 23 |
MGMT = O6‐methylguanine‐DNA methyltransferase; TMZ = temozolomide.
MGMT promoter methylation status and its correlation with OS and PFS
The methylation levels determined by qMSP are presented as ratios of the relative amount of methylated MGMT to that of ACTB. The ratios for patients with type 1 GCs ranged from 0 to 83, whereas the ratios for patients with type 2 GCs ranged from 0 to 31. MGMT methylation promoter status could be determined in 60 tumors, of which eight (13.3%) were considered methylated using the cutoff value of 12. MGMT methylation was not associated with IDH1 mutation status (P = 0.103) (Table 2).
Of the 74 patients with GCs, 14 were treated with TMZ with or without radiotherapy. Of these 14 patients, eight had an unmethylated MGMT promoter while four had a methylated MGMT promoter. The methylation status of the MGMT promoter could not be determined in the two remaining patients. MGMT promoter methylation was not correlated with prolonged OS or PFS in patients treated with TMZ compared with those patients who received no treatment (median 58 vs. 19 months, P = 0.673 and median 18 vs. 13 months, P = 0.657, respectively). In addition, there were no differences in OS or PFS between patients with a methylated MGMT promoter and those with an unmethylated MGMT promoter (median 60 vs. 27 months, P = 0.458 and median 28 vs. 15 months, P = 0.551, respectively).
Relationship between IDH1 mutation status, OS and PFS
We analyzed the prognostic relevance of IDH1 mutations and other clinicopathological parameters on the OS and PFS of patients with GCs (Table 3) (Figure 3). OS was strongly correlated with IDH1 mutation status in all GC patients. The 2‐year OS percentage for wild‐type IDH1 patients was 46% vs. 72% for patients with IDH1‐mutated tumors. Patients with tumors with IDH1 mutations had significantly better OS (mean 119 months) than those patients with IDH1 wild‐type tumors (mean 49 months; P = 0.016). However, there were no significant differences in PFS between the patients with IDH1‐mutations and those without (median 28 vs. 15 months, P = 0.121).
Table 3.
Clinicopathological and biological factors affecting overall survival (OS) and progression‐free survival (PFS) rates of patients with type 1 or type 2 gliomatosis cerebri (GC) by univariate and multivariate analyses.
| OS | PFS | |||||||
|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||
| Type 1 GCs | HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P |
| Age at diagnosis: | ||||||||
| >18 years vs. ≤18 years | 1.02 (0.22–4.64) | 0.98 | 0.71 (0.12–4.10) | 0.71 | 0.87 (0.25–3.02) | 0.83 | 1.16 (0.25–5.35) | 0.86 |
| Sex | ||||||||
| Male vs. female | 1.79 (0.58–5.50) | 0.31 | 2.41 (0.73–7.88) | 0.15 | 2.24 (0.90–5.58) | 0.08 | 3.17 (0.20–8.39) | 0.06 |
| IDH1 mutation | ||||||||
| Wild‐type vs. mutant | 0.66 (0.24–1.79) | 0.42 | 0.61 (0.22–1.72) | 0.35 | 1.20 (0.56–2.56) | 0.65 | 1.55 (0.67–3.59) | 0.31 |
| Histological grade | ||||||||
| Grade II, III vs. IV | 3.31 (1.19–9.19) | 0.02 | 2.90 (0.96–8.72) | 0.06 | 3.55 (1.58–7.98) | 0.002 | 4.45 (1.70–11.6) | 0.002 |
| Debulking surgery | ||||||||
| Not done vs. done | 1.03 (0.34–3.19) | 0.95 | 1.09 (0.34–3.46) | 0.89 | 0.99 (0.41–2.42) | 0.99 | 1.14 (0.46–2.81) | 0.78 |
| Adjuvant CTx | ||||||||
| Not done vs. done | 0.36 (0.14–0.94) | 0.04 | 0.40 (0.14–1.10) | 0.08 | 0.59 (0.27–1.26) | 0.17 | 0.79 (0.33–1.92) | 0.61 |
| Type 2 GCs | ||||||||
|---|---|---|---|---|---|---|---|---|
| Age at diagnosis: | ||||||||
| >18 years vs. ≤18 years | 1.60 (0.36–7.02) | 0.54 | 5.05 (0.83–30.8) | 0.08 | 1.21 (0.28–5.22) | 0.80 | 3.75 (0.65–21.6) | 0.14 |
| Sex | ||||||||
| Male vs. female | 1.44 (0.58–3.55) | 0.43 | 1.31 (0.45–3.87) | 0.62 | 1.15 (0.49–2.70) | 0.75 | 0.75 (0.26–2.18) | 0.60 |
| IDH1 mutation | ||||||||
| Wild‐type vs. mutant | 0.25 (0.09–0.66) | 0.01 | 0.26 (0.08–0.87) | 0.03 | 0.29 (0.11–0.73) | 0.008 | 0.24 (0.07–0.84) | 0.03 |
| Histological grade | ||||||||
| Grade II, III vs. IV | 2.79 (1.14–6.82) | 0.03 | 1.87 (0.67–5.19) | 0.23 | 4.65 (1.77–12.24) | 0.002 | 3.31 (1.18–9.26) | 0.02 |
| Debulking surgery | ||||||||
| Not done vs. done | 2.21 (0.80–6.08) | 0.13 | 2.07 (0.55–7.82) | 0.29 | 2.06 (0.80–5.30) | 0.14 | 1.18 (0.33–4.23) | 0.80 |
| Adjuvant CTx | ||||||||
| Not done vs. done | 0.88 (0.36–2.15) | 0.77 | 1.45 (0.48–4.43) | 0.51 | 1.00 (0.43–2.32) | 0.99 | 1.82 (0.62–5.29) | 0.28 |
Bold indicates statistically significant (P < 0.05).
Adjuvant CTx = adjuvant chemotherapy; CI = confidence interval; HR = hazard ration; OS = overall survival; PFS = progression‐free survival.
Figure 3.
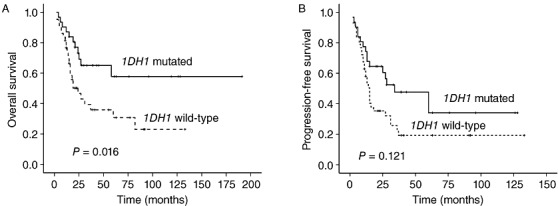
(A) Overall survival curves and (B) progression‐free survival curves in patients with gliomatosis cerebri according to IDH1 mutation status.
Correlation of IDH1 mutation status with OS and PFS according to GC growth type
We further analyzed the prognostic impact of IDH1 mutations on OS and PFS according to the growth type of the GCs (Figure 4) (Table 3). IDH1 mutations were strongly correlated with both increased OS and PFS in patients with type 2 GCs (P = 0.003 and P = 0.004, respectively), whereas IDH1 mutations were not correlated with OS or PFS in patients with type 1 GCs (P = 0.410 and P = 0.638, respectively). In patients with type 2 GCs and wild‐type IDH1, the 2‐year OS percentage was 26% vs. 79% for patients with type 2 GCs and mutated IDH1. The overall survival period of patients with IDH1‐mutated type 2 GCs (mean 82 months) was significantly better than that of patients with IDH1‐wild‐type type 2 GCs (mean 24 months). Patients with IDH1‐mutated type 2 GCs had a longer PFS period (median 34 months) than patients with IDH1 wild‐type type 2 GCs (median 12 months). Neither debulking surgery nor adjuvant chemotherapy treatment were correlated with OS (P = 0.13 and P = 0.77, respectively) or PFS (P = 0.14 and P = 0.99, respectively) in patients with type 2 GCs.
Figure 4.
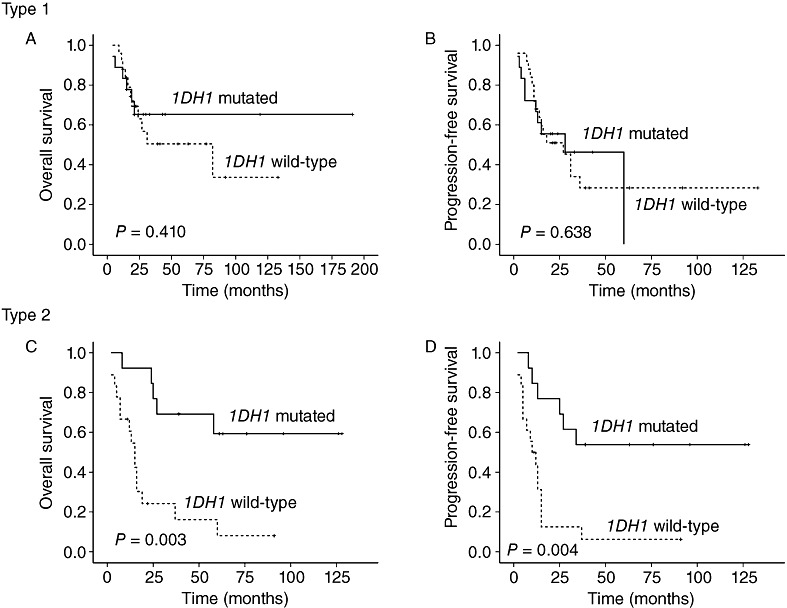
Correlations between IDH1 mutation status and overall survival (OS) and progression‐free survival (PFS) according to gliomatosis cerebri growth type. IDH1 mutations were not associated with (A) OS or (B) PFS in patients with type 1 GCs. Conversely, the presence of IDH1 mutations in type 2 GC predicted favorable (C) OS and (D) PFS.
Cox multivariate analyses with stepwise selection of type 2 GCs revealed that the presence of IDH1 mutations was the only independent prognostic factor predicting increased OS [P = 0.03, hazard ratio (HR) = 0.26, 95% confidence interval (CI), 0.08–0.87]. In addition, the presence of IDH1 mutations and grade IV tumors were independent prognostic factors associated with PFS in patients with type 2 GCs [P = 0.03, HR = 0.24, 95% CI, 0.07–0.84; P = 0.02, HR = 3.31, 95% CI, 1.18–9.26].
In patients with type 1 GCs, grade IV tumors were associated with decreased OS and PFS (P = 0.02 and P = 0.002, respectively), and histological grade was the only independent prognostic factor predicting PFS [P = 0.002, HR = 4.45, 95% CI, 1.70–11.6]. Patients treated with adjuvant chemotherapy survived for longer than those not treated with adjuvant chemotherapy (P = 0.04), but a history of adjuvant chemotherapy was not an independent prognostic factor of OS in patients with type 1 GCs [P = 0.08, HR = 0.40, 95% CI, 0.14–1.10].
Analyses of combinations of GC types and IDH1 status showed that the GC type was a significant prognostic factor of OS only in IDH1‐wild‐type cases (P = 0.014) (Figure 5). Among the four subgroups, patients with type 2 GCs and wild‐type IDH1 had a drastically worse prognosis in terms of both OS and PFS than the other three patient subgroups.
Figure 5.
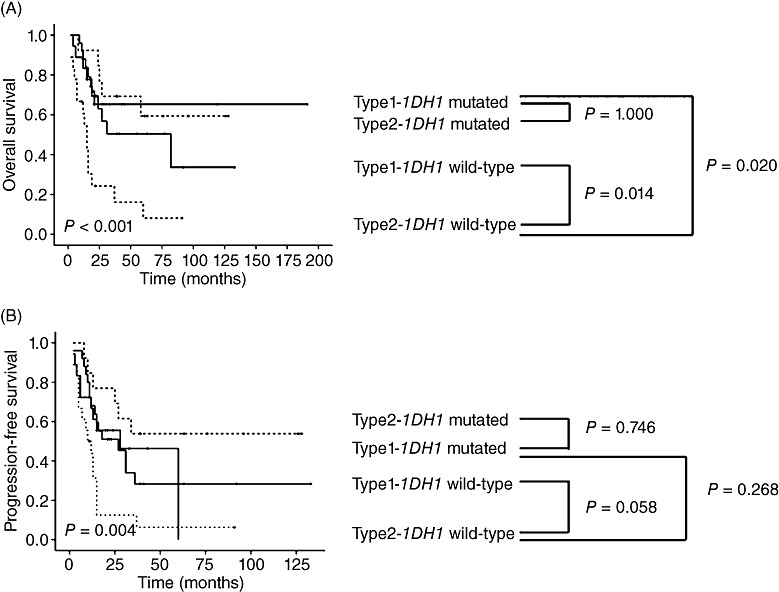
(A) Overall survival and (B) progression‐free survival in the four subgroups defined according to GC type and IDH1 mutation status. GC type was a significant prognostic factor of overall survival only in IDH1‐wild‐type cases.
Comparison of IDH1 mutation status and expression of IDH1‐R132H between primary first‐biopsy specimens and disease‐progressed second‐biopsy specimens in seven disease‐progressed cases
The disease of seven patients progressed during follow‐up monitoring, and we examined the initial and progressed tumor samples for IDH1 and IDH2 mutations (Table 4). IDH2 mutations were not detected in any of the initial or progressed specimens.
Table 4.
Comparison of IDH1 mutation status between primary first‐biopsy specimens and disease‐progressed second‐biopsy specimens in seven cases with disease progression.
| No. | Specimen | Sex | Age | Interval (days) | Treatment | IDH1 | MGMT | Growth type | Histological grade |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Initial | Male | 59 | 230 | Bx + RT | Wild‐type | NA | Type 1 | Grade II |
| Second | 60 | Wild‐type | UM | Type 1 | |||||
| 2 | Initial | Male | 46 | 915 | Bx + CCRT | p.R132H | M | Type 1 | Grade II |
| Second | 49 | p.R132H | UM | Type 1 | |||||
| 3 | Initial | Male | 57 | 237 | Bx + CCRT | Wild‐type | UM | Type 2 | Grade II |
| Second | 58 | Wild‐type | UM | Type 2 | |||||
| 4 | Initial | Male | 38 | 1060 | Bx + CCRT | p.R132H | UM | Type 2 | Grade II |
| Second | 41 | p.R132H | UM | Type 2 | |||||
| 5 | Initial | Male | 57 | 382 | Rx + RT | p.R132H | M | Type 1 | Grade III |
| Second | 58 | p.R132H | M | Type 1 | |||||
| 6 | Initial | Male | 54 | 349 | Bx + CCRT | Wild‐type | UM | Type 1 | Grade II |
| Second | 55 | Wild‐type | UM | Type 1 | |||||
| 7 | Initial | Female | 29 | 1229 | Bx + CCRT | p.R132H | UM | Type 1 | Grade II |
| Second | 32 | Wild‐type | UM | Type 1 |
MGMT = O6‐methylguanine‐DNA methyltransferase; UM = unmethylated; M = methylated; Bx = biopsy; RT = radiation therapy, Rx = resection; CCRT = concordant chemoradiation therapy.
Six cases had an identical IDH1 status in the initial and progressed specimens, but one case showed a discordant IDH1 mutation pattern when the initial and progressed specimens were compared. The discordant case was a type 1 GC with an initial histological morphology of a grade II astrocytoma with an IDH1 mutation. The patient was treated with concomitant chemoradiation therapy. The follow‐up MRI revealed progression of the lesion after 40 months and another biopsy specimen was taken at this point. The disease‐progressed biopsy specimen taken after 41 months showed similar features with minimally increased cellularity, and the IDH1 mutation was not detected (Figure 6A,B). The MGMT promoter was unmethylated in both the initial and second biopsy specimens. We performed immunohistochemical staining of the first and second biopsy specimens with an antibody that specifically recognizes IDH1‐R132H to confirm the presence of tumor cells in the specimen. Positive tumor cells were present in both the initial and progressed biopsy specimens (Figure 6C,D). This discordant case was considered to be an IDH1 mutant in further analysis. The other IDH1 mutation‐positive cases showed diffuse strong cytoplasmic staining in both the initial and progressed biopsy samples (Figure 7A,B). The wild‐type case was negative for staining in both the initial and progressed biopsy specimens (Figure 7C,D).
Figure 6.
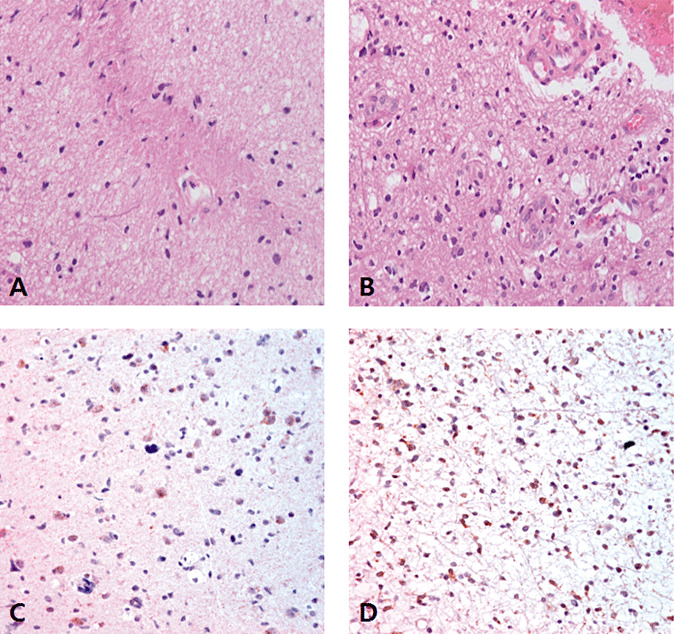
Histological features and IDH1‐R132H immunohistochemical staining of the initial (A,C) and progression (B,D) biopsy specimens of discordant case No.7 (X400). A. The initial biopsied specimen with an IDH1 mutation revealed a low cellular, mildly nuclear pleomorphic, oval or rod‐shaped tumor with an astrocytic phenotype. B. The disease‐progressed biopsy sample taken 41 months after the initial sample showed similar features with minimally increased cellularity. C,D. IDH1‐R132H immunohistochemical staining revealed scattered positive tumor cells.
Figure 7.
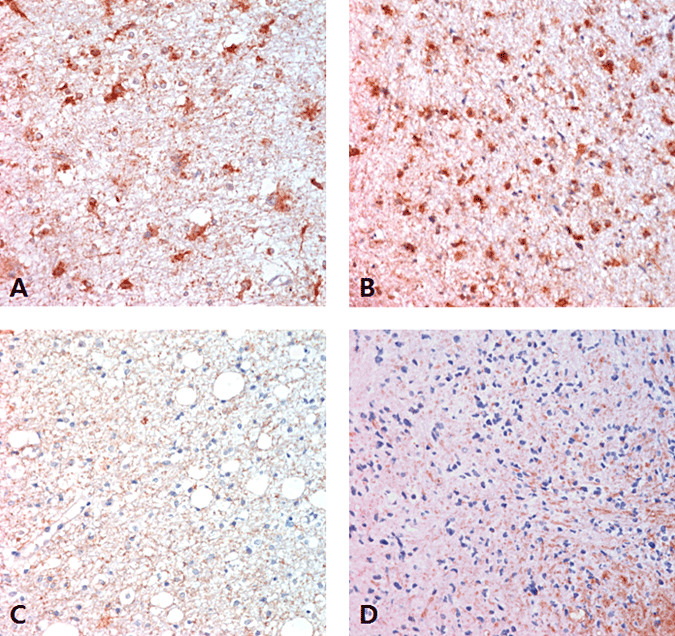
IDH1‐R132H immunohistochemistry in IDH1 mutant and wild‐type GCs. A,B. The tumor cell cytoplasm of an initial biopsy specimen was strongly positive for IDH1‐R132H (A) and the progressed biopsy sample was also strongly IDH1‐R132H positive (B) in a IDH1‐mutant case (X400). C,D. IDH1‐R132H staining was negative in the initial (C) and progressed biopsy samples (D) of a wild‐type IDH1 case (X400).
DISCUSSION
The purpose of the present study was to determine the prognostic significance of IDH1 and IDH2 mutations in different growth type GCs. IDH1 mutations were strongly correlated with both increased OS and PFS in patients with type 2 GCs, whereas IDH1 mutations were not correlated with OS and PFS in patients with type 1 GCs. Multivariate analyses revealed that the presence of IDH1 mutations was an independent prognostic factor predicting increased OS and PFS in type 2 GC patients. In contrast, grade IV tumors were associated with decreased OS and PFS in patients with type 1 GCs.
Only a few studies have investigated the prognostic significance of IDH1 mutations in GCs. Glas et al (9) demonstrated that IDH1 mutations are associated with prolonged survival in 35 GC patients. Desestret et al (4) reported that immunohistochemical staining of GC tumor specimens for the IDH1‐R132H mutant protein had prognostic value. However, the prognostic significance of IDH1 mutations according to the GC growth type has not previously been evaluated. We found that IDH1 mutations were an independent favorable prognostic factor in type 2 GCs, but not in type 1 GCs. In particular, IDH1 wild‐type type 2 GCs had the worst prognosis among the four IDH1 mutation status and growth type subgroups. Therefore, IDH1 mutation status may be an accurate prognostic factor in patients diagnosed with GCs. In addition, the strong prognostic significance of IDH1 mutations in type 2 GCs may allow prediction of clinical outcomes and informed selection of patients who would benefit from further adjuvant treatment.
Overall, 44.6% of 74 GCs had IDH1 mutations but no IDH2 mutations were detected in the present study. An IDH1 mutation was detected in 44.2% of type 1 GCs and 45.2% of type 2 GCs. Recent studies reported an IDH1 mutation rate of between 41.6% and 48% and an IDH2 mutation rate of zero in GCs 9, 18, with IDH1 mutations in 44.4% of type 1 GCs and 16.7% of type 2 GCs (18). The overall frequencies of IDH1/IDH2 mutations and the specific frequency of IDH1 mutations in type 1 GCs are similar to those observed in our study. In contrast, an earlier study reported an IDH1 mutation rate of 28.6% in 35 GCs. The authors of that study concluded that type 1 and type 2 GCs are biologically different subtypes based on the absence of IDH1 mutations in type 1 GCs, but the presence of IDH1 mutations (42%) in type 2 GCs (23). IDH2 mutations have been detected in 0.9%–6% of other WHO grade III glial neoplasms of the same grade as GCs (10); the much lower frequency of IDH2 mutations in GCs is intriguing.
To date, all reported IDH1 mutations in GCs have been R132H 4, 18, 23. In this study, we detected 33 (44.6%) mutations affecting codon 132 of IDH1, including 32 (97.0%) c.395G>A (Arg132His) mutations and one (3.0%) c.394C>A (Arg132Ser) mutation. The R132H IDH1 mutation is found in 90.5% of gliomas, with other mutations at this position occurring at far lower frequencies (R132S 4.8%, R132G 3.2%, and R132C 1.6%) (5). The R132S IDH1 mutation has also been exclusively detected in astrocytic glial tumors, such as diffuse, anaplastic astrocytomas, as well as in glioblastomas, but has not been detected in oligodendrogliomas or oligoastrocytomas (5). In our study, the GC with the R132S mutation of IDH1 was a grade II and type 2 GC with an astrocytic phenotype.
MGMT is an enzyme that removes methyl and alkyl groups from the O6 position of guanine in DNA, thereby detoxifying alkylating agents such as TMZ. MGMT methylation as well as IDH1 mutations are favorable prognostic factors associated with increased survival and chemosensitivity to TMZ in common gliomas (14). The prognostic value of MGMT methylation in patients with GCs is controversial. We found no correlations between MGMT methylation and survival outcomes or IDH1 mutation status. A recent study also found no difference in survival outcomes of GC patients with methylated MGMT promoters vs. those with nonmethylated promoters (14). However, another recent study found a positive correlation between MGMT methylation status and IDH1 mutation status and reported that MGMT methylation status was a prognostic and predictive factor in 35 GC patients treated with procarbazine and lomustine (9). The absence of reliable prognostic and predictive markers for GCs highlights the importance of assessing IDH1 mutation status and MGMT methylation status in patients with GCs.
While palliative surgery, radiotherapy and chemotherapy are used to treat patients with GCs, the optimal therapeutic strategy for GCs remains unclear. Radiotherapy has been reported to be effective in approximately half of patients with GCs and to increase overall survival (12). Chemotherapy is often given to patients with GC, but its effectiveness remains controversial. TMZ is currently widely used as a first‐line treatment for GCs because it is more tolerable and less toxic than nitrosourea‐based chemotherapy (14) and it may be effective in patients with slow‐growing low‐grade GCs (21). However, the optimal length of administration of TMZ is unknown (27). The potential for severe hematologic toxicity such as myelosuppression must be considered with prolonged use (8). Furthermore, TMZ is economically cost ineffective (7). In the present study, there was no difference in the survival rates of TMZ‐treated patients with methylated MGMT promoters and those with unmethylated promoters. Furthermore, there were no prognostic differences between patients who had received debulking surgery and those who had not. However, only a few of the patients included in our retrospective study had received TMZ treatment, thus our findings concerning TMZ in MGMT methylated GCs should not be considered conclusive.
Comparison of the IDH1 and IDH2 mutation status between the initial and progressed biopsy specimens of seven cases that showed disease progression revealed an identical IDH1 and IDH2 mutation status in six cases; only one case showed a discrepancy. That case was a type 1 GC, and an IDH1 mutation was detected in the initial tumor that had a grade II astrocytic phenotype. However, no IDH1 mutation was identified in the progressed glioma specimen. The presence of only a small proportion of tumor cells in the biopsy or resection specimens might have been responsible for this negative result. However, hematoxylin and eosin (H&E) staining slides revealed a good proportion of tumor cells in the progressed biopsy specimen, indicating that this was not the reason for the negative result. Another possibility is molecular heterogeneity within the GC. Several studies have demonstrated different molecular patterns in different areas of GCs 16, 17. Kattar et al (16) observed a nonmonoclonal pattern in a single GC patient in an analysis of X chromosomal inactivation. Mawrin et al (17) found different regional patterns of genetic TP53 alterations in GCs. Markedly different karyotypes among cells and even within established cell lines, as well as variable expression of antigenic markers, have been reported in gliomas 1, 24, 28. We performed immunohistochemical staining of the initial and progressed biopsy samples of the seven cases with disease progression using an IDH1‐R132H‐specific antibody. In the discordant case, both the initial and progressed specimens showed scattered IDH1‐R132H‐positive tumor cells. However, the intensity of staining was weaker than in the other IDH1 mutant cases. We therefore considered the result from the progressed specimen to be a false‐negative result and analyzed this case as an IDH1‐mutant case.
In conclusion, IDH1 alterations had clear prognostic significance in the overall sample of GCs examined in our study. A detailed investigation according to growth type showed that type 2 GCs exhibited different clinical courses depending on their IDH1 mutation status, whereas the prognosis of type 1 GCs was not affected by the mutation status of IDH1. Discrepancies in IDH1 results between initial and progressed specimens may be related to molecular heterogeneity within the GC.
ACKNOWLEDGMENTS
We are grateful to Joonghyun Ahn (Biostatistic Team of Samsung Biomedical Research Institute) for statistical support. This work was supported by a grant (A092255) from the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare Affairs, and by the Global Frontier (NRF‐M1AXA002‐2010‐0029795) grant of National Research Foundation funded by the Ministry of Education, Science, and Technology of Korea.
Authors' contributions
MJK (Mi Jung Kwon) designed the study, interpreted the data and wrote the manuscript; STK participated in data collection, interpreted the data and critically revised the manuscript; MJK (Mi Jeong Kwon) wrote the manuscript and critically revised the manuscript; YLC and YLS participated in study design and coordination, data analysis, data interpretation and drafting of the manuscript; SYK and JYS carried out the experiments and data acquisition; DL, SP and YK: participated in data collection and critically revised the manuscript; and DG and DHN participated in data collection. All authors read and approved the final manuscript.
REFERENCES
- 1. Bonavia R, Inda MD, Cavenee WK, Furnari FB (2011) Heterogeneity maintenance in glioblastoma: a social network. Cancer Res 71:4055–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D et al (2009) Characterization of R132H mutation‐specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Desclee P, Rommel D, Hernalsteen D, Godfraind C, de Coene B, Cosnard G (2010) Gliomatosis cerebri, imaging findings of 12 cases. J Neuroradiol 37:148–158. [DOI] [PubMed] [Google Scholar]
- 4. Desestret V, Ciccarino P, Ducray F, Criniere E, Boisselier B, Labussiere M et al (2011) Sanson M Prognostic stratification of gliomatosis cerebri by IDH1(R132H) and INA expression. J Neurooncol 105:219–224. [DOI] [PubMed] [Google Scholar]
- 5. Felsberg J, Wolter M, Seul H, Friedensdorf B, Goppert M, Sabel MC (2010) Reifenberger G Rapid and sensitive assessment of the IDH1 and IDH2 mutation status in cerebral gliomas based on DNA pyrosequencing. Acta Neuropathol 119:501–507. [DOI] [PubMed] [Google Scholar]
- 6. Fuller GN, Kros JM (2007) Gliomatosis cerebri. In: WHO Classification of Tumours of the Central Nervous System, Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 50–52. IARC Press: Lyon. [Google Scholar]
- 7. Garside R, Pitt M, Anderson R, Rogers G, Dyer M, Mealing S et al (2007) The effectiveness and cost‐effectiveness of carmustine implants and temozolomide for the treatment of newly diagnosed high‐grade glioma: a systematic review and economic evaluation. Health Technol Assess 11:iii–iiv. ix‐221. [DOI] [PubMed] [Google Scholar]
- 8. Gerber DE, Grossman SA, Zeltzman M, Parisi MA, Kleinberg L (2007) The impact of thrombocytopenia from temozolomide and radiation in newly diagnosed adults with high‐grade gliomas. Neuro Oncol 9:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Glas M, Bahr O, Felsberg J, Rasch K, Wiewrodt D, Schabet M et al (2011) NOA‐05 phase 2 trial of procarbazine and lomustine therapy in gliomatosis cerebri. Ann Neurol 70:445–453. [DOI] [PubMed] [Google Scholar]
- 10. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A et al (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118:469–474. [DOI] [PubMed] [Google Scholar]
- 11. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. [DOI] [PubMed] [Google Scholar]
- 12. Inoue T, Kumabe T, Kanamori M, Sonoda Y, Watanabe M, Tominaga T (2010) Prognostic factors for patients with gliomatosis cerebri: retrospective analysis of 17 consecutive cases. Neurosurg Rev 34:197–208. [DOI] [PubMed] [Google Scholar]
- 13. Jennings MT, Frenchman M, Shehab T, Johnson MD, Creasy J, LaPorte K, Dettbarn WD (1995) Gliomatosis cerebri presenting as intractable epilepsy during early childhood. J Child Neurol 10:37–45. [DOI] [PubMed] [Google Scholar]
- 14. Kaloshi G, Everhard S, Laigle‐Donadey F, Marie Y, Navarro S, Mokhtari K et al (2008) Genetic markers predictive of chemosensitivity and outcome in gliomatosis cerebri. Neurology 70:590–595. [DOI] [PubMed] [Google Scholar]
- 15. Kato Y, Jin G, Kuan CT, McLendon RE, Yan H, Bigner DD (2009) A monoclonal antibody IMab‐1 specifically recognizes IDH1R132H, the most common glioma‐derived mutation. Biochem Biophys Res Commun 390:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kattar MM, Kupsky WJ, Shimoyama RK, Vo TD, Olson MW, Bargar GR, Sarkar FH (1997) Clonal analysis of gliomas. Hum Pathol 28:1166–1179. [DOI] [PubMed] [Google Scholar]
- 17. Mawrin C, Kirches E, Schneider‐Stock R, Scherlach C, Vorwerk C, Von Deimling A et al (2003) Analysis of TP53 and PTEN in gliomatosis cerebri. Acta Neuropathol 105:529–536. [DOI] [PubMed] [Google Scholar]
- 18. Narasimhaiah D, Miquel C, Verhamme E, Desclee P, Cosnard G, Godfraind C (2011) IDH1 mutation, a genetic alteration associated with adult gliomatosis cerebri. Neuropathology [Epub ahead of Print]. [DOI] [PubMed] [Google Scholar]
- 19. Nevin S (1938) Gliomatosis cerebri. Brain 61:170–191. [Google Scholar]
- 20. Park S, Suh YL, Nam DH, Kim ST (2009) Gliomatosis cerebri: clinicopathologic study of 33 cases and comparison of mass forming and diffuse types. Clin Neuropathol 28:73–82. [DOI] [PubMed] [Google Scholar]
- 21. Sanson M, Cartalat‐Carel S, Taillibert S, Napolitano M, Djafari L, Cougnard J et al (2004) Initial chemotherapy in gliomatosis cerebri. Neurology 63:270–275. [DOI] [PubMed] [Google Scholar]
- 22. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F et al (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154. [DOI] [PubMed] [Google Scholar]
- 23. Seiz M, Tuettenberg J, Meyer J, Essig M, Schmieder K, Mawrin C et al (2010) Detection of IDH1 mutations in gliomatosis cerebri, but only in tumors with additional solid component: evidence for molecular subtypes. Acta Neuropathol 120:261–267. [DOI] [PubMed] [Google Scholar]
- 24. Shapiro JR, Yung WK, Shapiro WR (1981) Isolation, karyotype, and clonal growth of heterogeneous subpopulations of human malignant gliomas. Cancer Res 41:2349–2359. [PubMed] [Google Scholar]
- 25. Takano S, Tian W, Matsuda M, Yamamoto T, Ishikawa E, Kaneko MK et al (2011) Detection of IDH1 mutation in human gliomas: comparison of immunohistochemistry and sequencing. Brain Tumor Pathol 28:115–123. [DOI] [PubMed] [Google Scholar]
- 26. van Nifterik KA, van den Berg J, van der Meide WF, Ameziane N, Wedekind LE, Steenbergen RD et al (2010) Absence of the MGMT protein as well as methylation of the MGMT promoter predict the sensitivity for temozolomide. Br J Cancer 103:29– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Villano JL, Seery TE, Bressler LR (2009) Temozolomide in malignant gliomas: current use and future targets. Cancer Chemother Pharmacol 64:647–655. [DOI] [PubMed] [Google Scholar]
- 28. Wikstrand CJ, Bigner SH, Bigner DD (1983) Demonstration of complex antigenic heterogeneity in a human glioma cell line and eight derived clones by specific monoclonal antibodies. Cancer Res 43:3327–3334. [PubMed] [Google Scholar]
- 29. Yen KE, Bittinger MA, Su SM, Fantin VR (2010) Cancer‐associated IDH mutations: biomarker and therapeutic opportunities. Oncogene 29:6409–6417. [DOI] [PubMed] [Google Scholar]