

Abstract
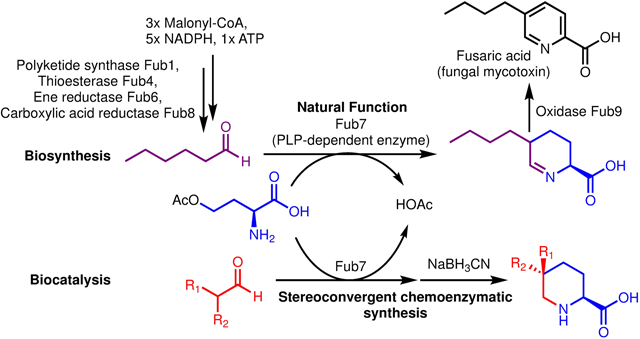
Fusaric acid (FA) is a well-known mycotoxin that plays an important role in plant pathology. The biosynthetic gene cluster for FA has been identified but the biosynthetic pathway remains unclarified. Here, we elucidated the biosynthesis of FA, which features a two-enzyme catalytic cascade, a pyridoxal 5’-phosphate (PLP)-dependent enzyme (Fub7) and a flavin mononucleotide (FMN)-dependent oxidase (Fub9) in synthesizing the picolinic acid scaffold. FA biosynthesis also involves an off-line collaboration between a highly reducing polyketide synthase (HRPKS, Fub1) and a nonribosomal peptide synthetase (NRPS)-like carboxylic acid reductase (Fub8) in making an aliphatic α,β-unsaturated aldehyde. By harnessing the stereoselective C-C bond forming activity of Fub7, we established a chemoenzymatic route for stereoconvergent synthesis of a series of 5-alkyl, 5,5-dialkyl, and 5,5,6-trialkyl-L-pipecolic acids of high diastereomeric ratio.
Graphical Abstract
INTRODUCTION
Picolinic acids and their derivatives (including the ring hydrogenated derivatives, pipecolic acids) are privileged scaffolds frequently used in pharmaceuticals and agrochemicals, and also common moieties found in many natural products (Figure 1).1,2 As a bidentate chelator, picolinic acids are also important metal-binding pharmacophores (MBPs) that show activity against several metalloenzymes.2c Fusaric acid (FA), a picolinic acid-based fungal natural product, is a classic mycotoxin isolated from numerous Fusarium species (Figure 1).3 It exhibits strong phytotoxicity and causes wilt symptoms in a variety of plants, and has long been regarded as a virulence factor of pathogenic Fusarium species.4 FA also shows mild toxicity to animals and is found to be an uncompetitive inhibitor of mammalian dopamine β-hydroxylase.5a Consequently, the amidated derivative of FA, bupicomide (5-butyl-2-pyridinecarboxamide), was clinically used as a vasodilator and experimentally as a β-blocker.5b Furthermore, the hydrogenated derivative of FA, trans-5-butyl-DL-pipecolic acid mixtures (Figure 1) was also shown to exhibit antihypertensive activity.6
Figure 1.
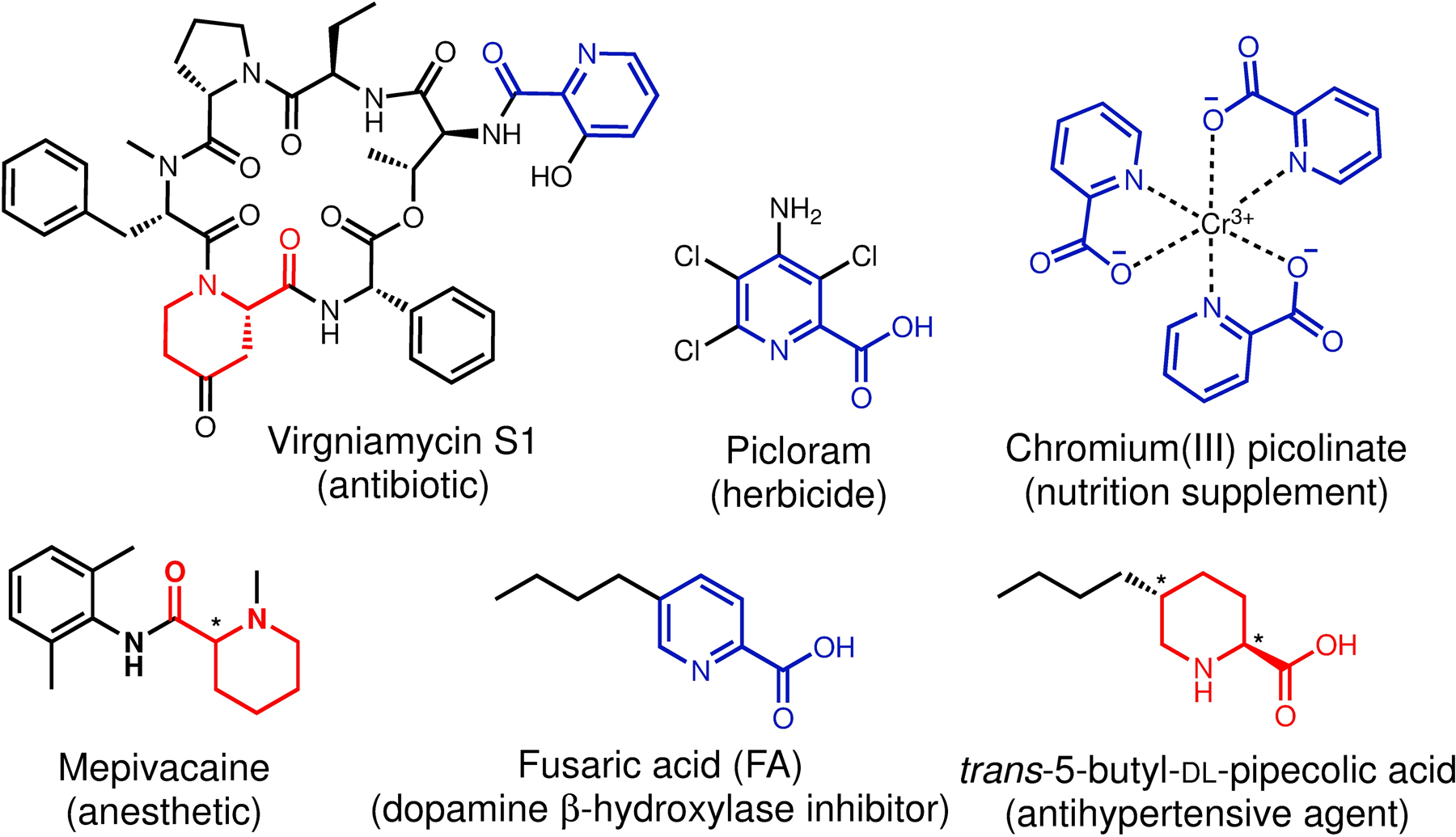
Representative picolinic acids and pipecolic acids-containing natural products, pharmaceuticals, and agrochemicals. The racemate stereocenters are indicated with asterisks.
Despite the importance of FA in plant pathology, fungal ecology and food safety,7 the biochemical basis for FA biosynthesis has not been established. Early isotope-labeled precursor feeding experiments revealed that FA was derived from aspartate (or oxaloacetate) and three acetate units (Figure 2A).8 Recently, the well-conserved biosynthetic gene cluster for FA in multiple Fusarium species has been identified via transcriptomics and comparative genomics approaches (Figure 2B).9 Gene inactivation studies showed that five genes (fub1, fub4, and fub6–8) were essential for FA biosynthesis, which encode a highly-reducing polyketide synthase (HRPKS, fub1), an α/β hydrolase (fub4), a medium-chain dehydrogenase/reductase (MDR, fub6), a PLP-dependent enzyme (fub7), and a nonribosomal peptide synthetase (NRPS)-like carboxylic acid reductase (fub8). Additionally, a volatile compound trans-2-hexenal was identified in both Δfub6 and Δfub7 mutants, but was absent in Δfub1, Δfub4, or Δfub8 mutants. Chemical complementation of Δfub1 mutant with feeding of trans-2-hexenal restored production of FA, implicating its intermediacy in FA biosynthesis as well as its polyketide origin. Based on these evidences, Studt et al proposed a biosynthetic pathway for FA featuring a 6π electrocyclization step to synthesize the picolinic acid scaffold (Figure 2C).9d Several steps in the proposed pathway, however, are not consistent with the predicted functions of the biosynthetic enzymes, including: (1) the α/β-hydrolase (Fub4) acts as a thioester reductase to offload trans-2-hexenal from an HRPKS (Fub1); (2) the predicted PLP-dependent enzyme (Fub7) catalyzes a γ-elimination reaction on an imine substrate that lacks a free α-amino group to form the PLP-aldimine, a key intermediate involved in all PLP-dependent enzymes except glycogen phosphorylase.10
Figure 2.
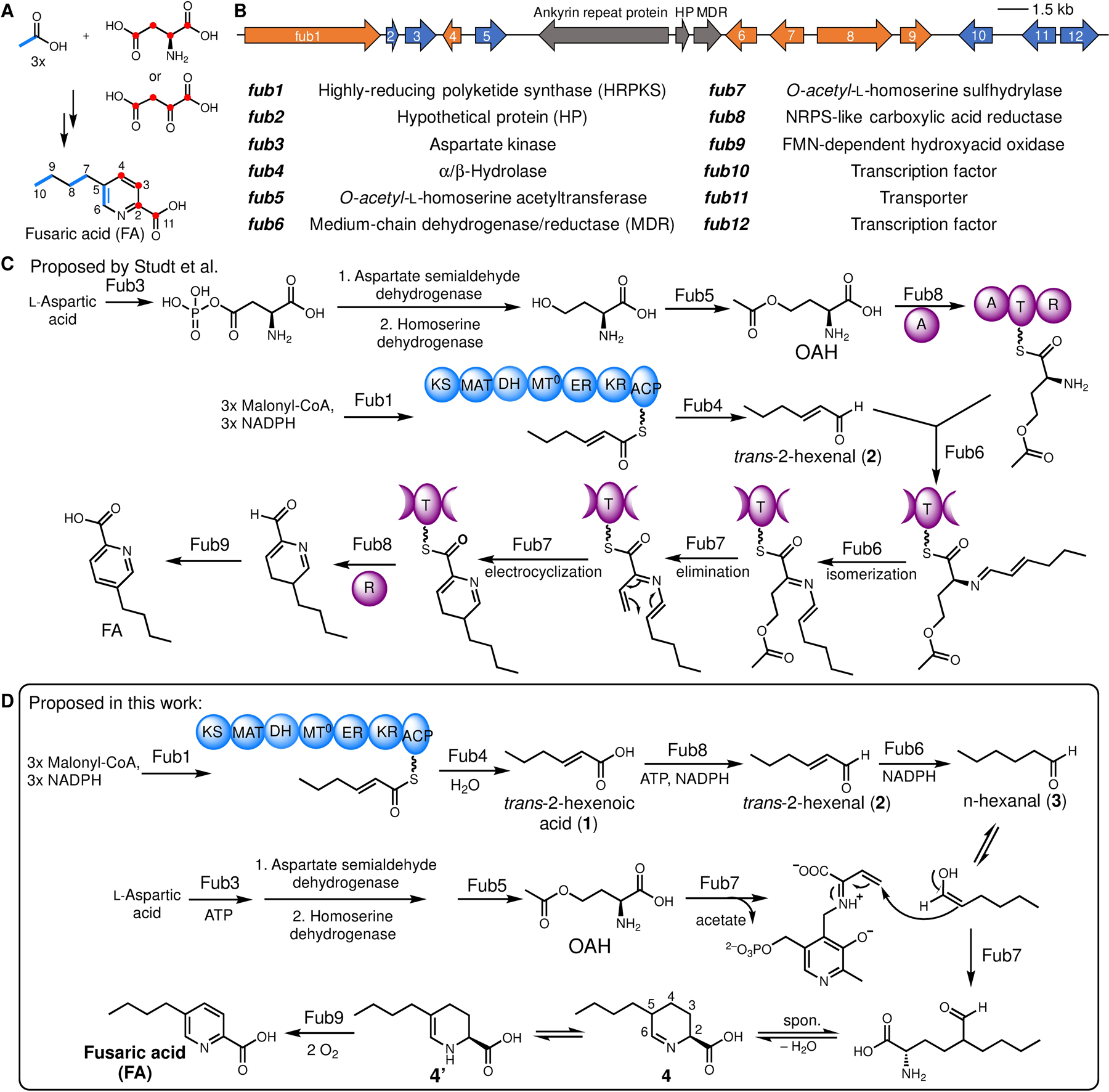
Biosynthesis of fusaric acid. (A) Feeding experiment summary.8 (B) Reported gene cluster fub in Fusarium fujikuroi.9b (C) Previously proposed biosynthetic pathway of FA by Studt et al.9d Domain abbreviations: KS, keto-synthase, MAT, malonyl acyltransferase; DH, dehydratase; MT0, degenerate methyltransferase domain; ER, enoyl-reductase; KR, keto-reductase; ACP, acyl-carrier protein; A, adenylation; T, thiolation; R, thioester reductase. (D) Biosynthetic pathway established in this study.
Motivated by these unresolved problems, we studied the biosynthesis of FA through biochemical characterization of these Fub enzymes. Results from our studies enabled the proposal of a biosynthetic route to the substituted picolinic acid as shown in Figure 2D. Using the key C-C bond forming enzyme Fub7 as a biocatalyst, we developed a facile method to prepare a panel of 5-alkyl, 5,5-dialkyl, and 5,5,6-trialkyl-L-pipecolic acids. The synthetic utility of Fub7 complements the recently discovered PLP-dependent enzyme CndF, which was used to prepare a suite of 6-alkyl-L-pipecolic acids.11 Together with CndF, these PLP-dependent enzymes can be useful biocatalytic tools for the synthesis of an assortment of nitrogen containing heterocycles.
RESULTS AND DISCUSSIONS
Reconstitution of FA biosynthesis
To elucidate FA biosynthetic pathway, we first attempted to reconstitute FA production using the Aspergillus nidulans ΔEM strain as a heterologous host.12 We initially expressed only the essential genes (fub1, 4, 6–8) as suggested by the knockout studies (ref. 9d), and relied on the endogenous metabolic pathway in A. nidulans to supply the putative amino acid precursor O-acetyl-l-homoserine (OAH).13 However, we did not observe production of FA. Instead, a new compound 4 (m/z of 184 for [M+H]+) emerged, of which the molecular weight is in accord with the proposed tetrahydrofusaric acid intermediate (Figure 3A). Derivatizing this compound with 2-aminobenzaldehyde (2AB) gave the expected dihydroquinazolinium adduct (2AB-4, m/z of 287 [M+H]+ and λmax at 460 nm) suggesting that this compound indeed bears an imino functional group (Fig. S1). Chemical reduction of 4 by NaBH3CN afforded 5 and 6 with a diastereomeric ratio (d.r.) of 18:82 (Figure 3B), which were isolated and determined to be cis-5-butyl-L-pipeoclic acid and trans-5-butyl-L-pipeoclic acid, respectively (Table S1, S2). Characterization of 5 and 6 as the reduction product of 4 not only confirmed that 4 is tetrahydrofusaric acid, but also implied that 4 is epimerized at C5 (pKa ≈ 8, ref. 14) presumably through facile tautomerization between the imine tautomer and the enamine tautomer 4’ (Figure 2D). Interestingly, we also observed 5 and 6 as shunt products from the same A. nidulans transformant that produced 4 when it was cultured on solid medium. The diastereomeric ratio (93:7) between 5 and 6 formed in vivo is completely reversed as compared to the ones formed by chemical reduction, which suggests that a yet-unknown endogenous imine reductase (IRED) from A. nidulans may preferentially catalyze reduction on the thermodynamically less favored cis-diastereomer of 4 (cis-4) leading to 5 as the dominant shunt product (Figure S2).
Figure 3.
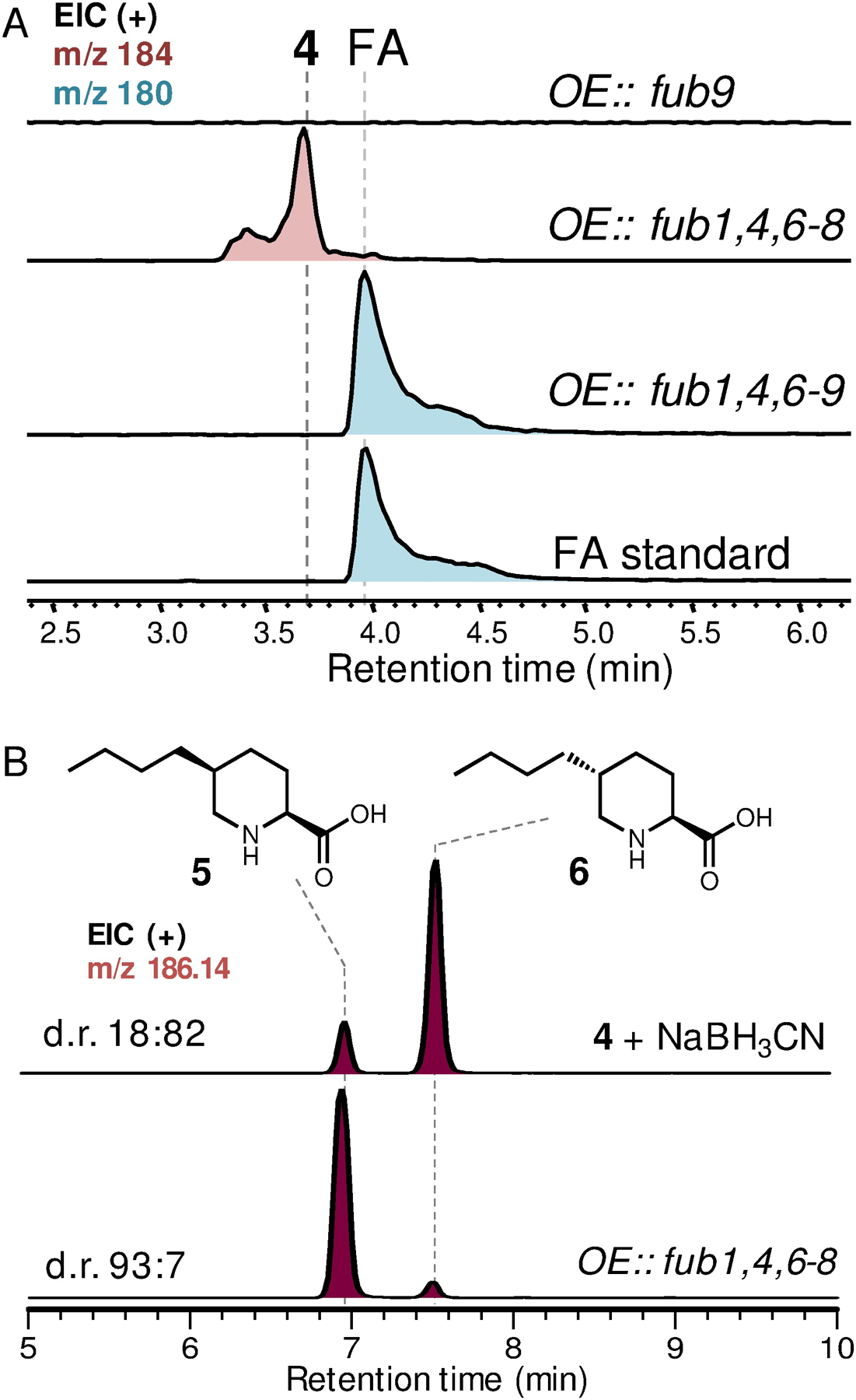
Heterologous production of fusaric acid and 5-butyl-L-pipecolic acids. (A) Reconstitution of FA biosynthesis in A. nidulans. OE, overexpressing. (B) Reduction of 4 led to pipecolic acid diastereomer 5 and 6. Traces shown here are extracted ion chromatograms (EIC).
Nevertheless, observation of 4 instead of FA in our initial reconstitution indicates the enzyme responsible for the last 4-electron oxidation step converting 4 to FA was missing in our heterologous expression experiment. We reasoned that one of the remaining genes, fub9, which encodes an FMN-dependent oxidase, could be a likely candidate responsible for this oxidation event. Indeed, including fub9 in our reconstitution successfully yielded FA (Figure 3A). The discrepancy between our result that Fub9 is involved in FA biosynthesis and the previous gene-inactivation study (ref. 9d), which showed fub9 was unnecessary for FA formation in Fusarium verticillioides, suggests there must be a redundant pathway from 4 to FA in F. verticillioides. We have hence now defined the minimal set of genes required to synthesize FA in a heterologous host and set the stage for detailed biochemical investigation of these enzymes.
Fub7 is a PLP-dependent enzyme catalyzing C-C bond formation
The cyclic Schiff base 4 is in equilibrium with the (2S)-2-amino-5-formylnonanoic acid (Figure 2D). Retrobiosynthetic dissection of the γ−δ C-C bond in (2S)-2-amino-5-formylnonanoic acid revealed this compound could be synthesized from OAH and n-hexanal (3) through an apparent γ-substitution reaction. Such reactions can be catalyzed by PLP-dependent enzymes,11 of which Fub7 is predicted to be (Figure 2D). To test this hypothesis, we purified recombinant Fub7 from E. coli and characterized its function in vitro (Figure S3). As-isolated Fub7 was yellow and exhibited maximal absorbance at ~425 nm (Figure S4a), indicating the presence of PLP cofactor that is covalently bound as an internal aldimine and exists in the form of ketoenamine.15 Fub7 (1 μM) readily afforded 4 in the presence of OAH (1 mM) and 3 (1 mM) as substrates (Figure 4a), and the activity was abolished and no OAH consumption was observed when the enzyme was pre-treated with hydroxylamine (1 mM), a common inhibitor for PLP-dependent enzymes. When OAH was titrated into Fub7 solution, the absorbance peak at 425 nm disappeared indicating the internal PLP aldimine reacted with OAH. The peak can be gradually recovered after cosubstrate 3 was added to the reaction mixture (Figure S4a). O-succinyl-L-homoserine could also serve as the amino acid donor, albeit with much lower efficiency. Other O-substituted homoserines or S-substituted homocysteines/methionines are not substrates for Fub7 (Figure S4b). Unlike the previously characterized C-C bond forming PLP-dependent enzyme CndF,11 Fub7 does not show any metal ion dependence (Figure S4c). Rather, some divalent metal ions can strongly inhibit the enzyme activity, such as Cu2+ and Zn2+. The origin of the inhibition is currently unknown. We next used a Fub9-peroxidase-coupled assay (vide infra) to study the steady-state kinetics of Fub7-catalyzed C-C bond formation between 3 and OAH (Figure S4d). The reaction has an apparent KM of 1.8 mM for 3 and an apparent KM of 15 μM for OAH, with an overall kcat of 0.14 s−1.
Figure 4.
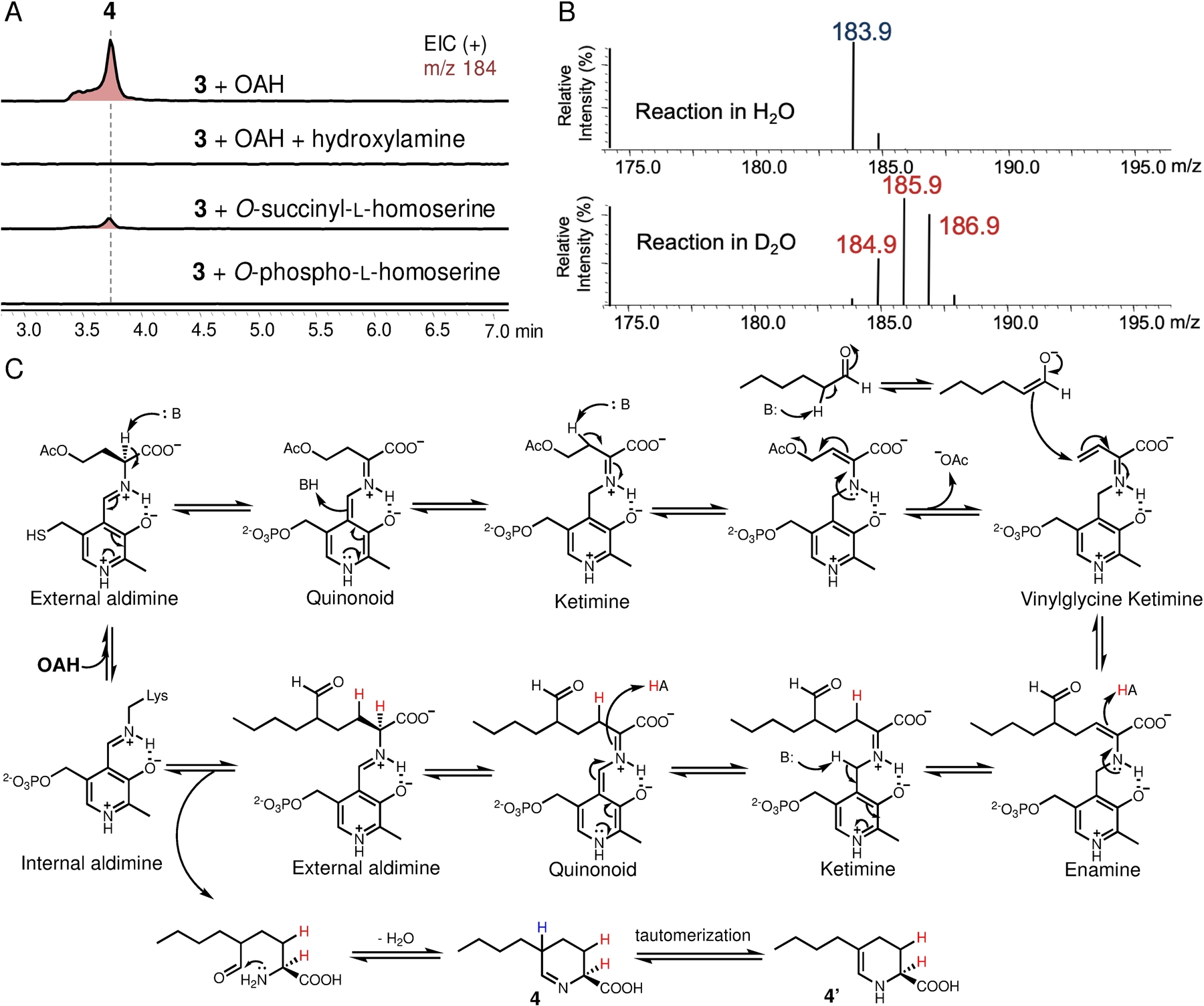
Characterization of Fub7 function. (A) In vitro biochemical assay of Fub7. (B) MS analysis of Fub7 catalyzed reaction in D2O (95% v/v in H2O). A +3 Da mass shift of product 4 is shown. (C) Proposed catalytic mechanism of Fub7. The proton exchanged with solvent due to spontaneous epimerization is colored in blue, whereas the protons incorporated through enzyme catalyzed protonation are colored in red. All three protons can be exchanged with deuterium when the reaction is performed in D2O.
To test whether OAH and 3 undergo a direct displacement reaction (SN2), or the reaction follow an elimination-addition mechanism, we repeated the enzymatic assay in D2O. If the reaction proceeds via a SN2 pathway, only one deuterium atom (blue H in Figure 4C) should be “washed in” as a result of the nonenzymatic epimerization at C5 (+1 Da mass shift of 4). Alternatively, two additional deuterium atoms (red H in Figure 4C) would be incorporated if the reaction proceeds through an elimination-addition mechanism.16 As shown in Figure 4B, a +3 Da mass shift of 4 was observed when the reaction was performed in D2O, which clearly rules out the SN2 mechanism and supports a γ-elimination-Michael addition pathway analogous to that of cystathionine γ-synthase (Figure 4C).16 Briefly, Fub7 first generates a vinylglycine intermediate (ketimine form) from OAH, which then undergoes conjugate addition by attack from the enolate tautomer of 3 to give an enamine adduct. Protonation at Cβ (where deuterium is incorporated) followed by quinonoid formation, and protonation at Cα (where the second deuterium is incorporated) yields the final external aldimine adduct. Offloading the product and regeneration of the internal aldimine set the enzyme ready for another catalytic cycle. Released (2S)-2-amino-5-formylnonanoic acid can undergo intramolecular cyclization and dehydration to afford 4.
PLP-dependent aldolases generally use aldehyde substrates as electrophiles, such as threonine aldolase and serine hydroxymethyltransferase.17 To our knowledge, Fub7 is the first PLP-dependent enzyme shown to use aldehyde substrates as nucleophiles. We next examined whether Fub7 can accept other nucleophiles (S, O, or N) to form carbon-heteroatom bond.18 In the presence of hydrogen sulfide and OAH, Fub7 can synthesize L-homocysteine. Given 1-dodecanethiol as the nucleophile, Fub7 can synthesize S-dodecyl-L-homocysteine from OAH (Figure S5). These results demonstrate Fub7 has sulfhydrylase activity. In addition, Fub7 can also accept aliphatic amines (e.g. amylamine, 3-phenylpropylamine, etc) as nucleophiles to form different Nδ-alkyl-L-norornithine (Figure S6). Furthermore, with aliphatic alcohols as nucleophiles, Fub7 could also afford an array of O-alkyl-L-homoserines (Figure S7); while using aliphatic carboxylic acids as the nucleophiles, Fub7 formally acts as a transacylase to convert OAH into corresponding O-acyl-L-homoserines (Figure S8a). These results together suggest Fub7 has relaxed substrate specificity towards different nucleophiles. Besides, the “unnatural” O-acyl-L-homoserines synthesized by the transacylase activity could also undergo γ-elimination and act as amino acid donor in synthesizing 4 (Figure S8b). This result not only suggest above reactions all go through the elimination/addition mechanism via the same key vinylglycine ketimine intermediate, but also indicate the transacylation reaction is fully reversible and thermodynamically controlled (i.e. in equilibrium).
Fub9 is an FMN-dependent oxidase catalyzing dehydrogenative aromatization.
Preparation of 4 by Fub7 in vitro allowed us to further verify the function of Fub9. Purified recombinant Fub9 appeared yellow, which is attributed to the enzyme-bound cofactor FMNox (Figure S9). When we added Fub9 into Fub7 reaction mixture using OAH and 3 as substrates, oxidation of 4 into FA was observed (Figure 5A). By employing a horseradish peroxidase-based fluorometric assay, we also detected formation of H2O2. The stoichiometry between product FA and H2O2 was determined to be 1:2 (Figure 5B), which clearly indicates that two equivalents of molecular oxygen are reduced to hydrogen peroxide accompanying each 4-electron oxidation of 4. This results also demonstrates that Fub9 is a bona fide oxidase and 4 is an on-pathway intermediate en route to FA.
Figure 5.
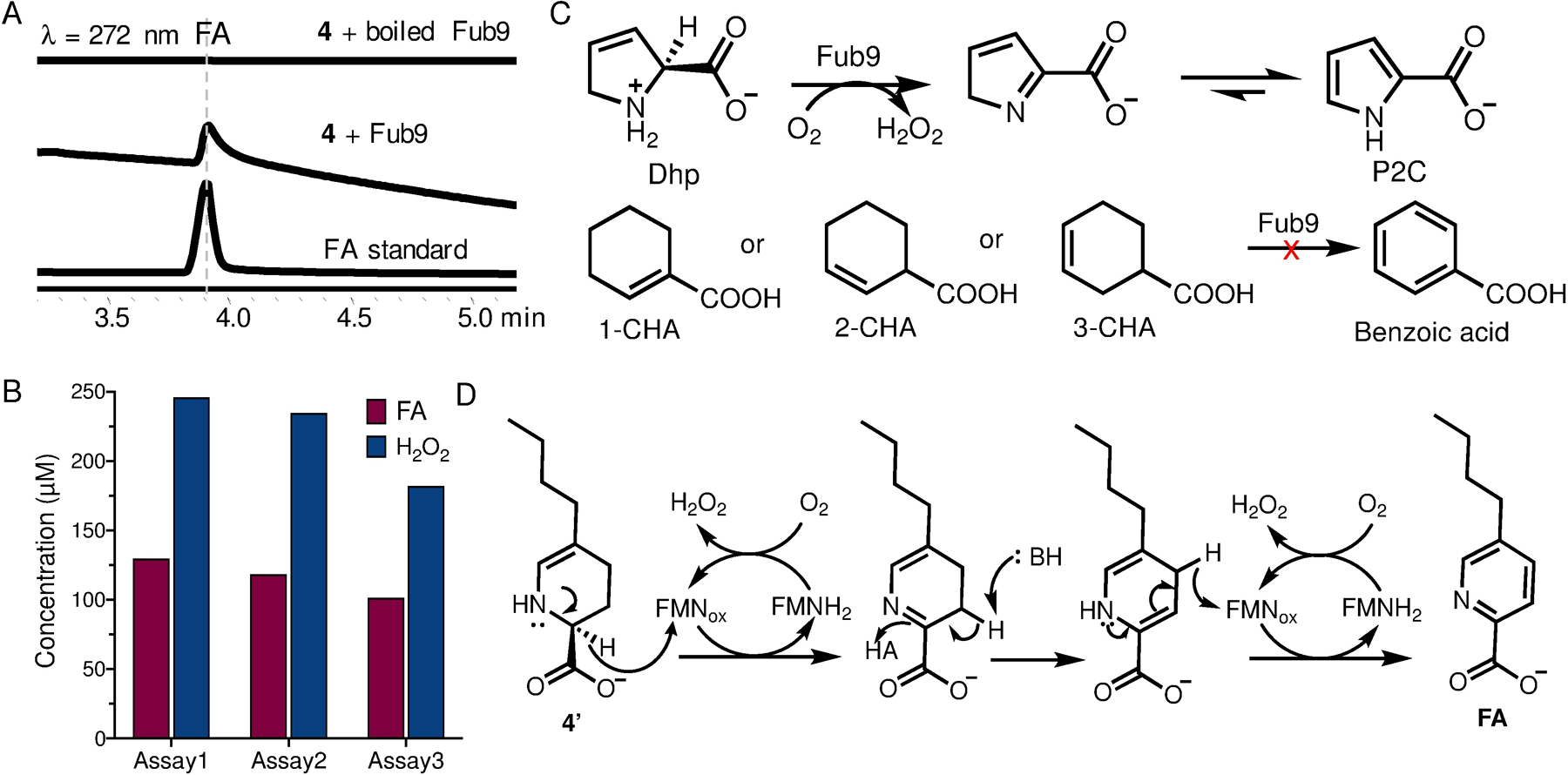
Characterization of Fub9. (A) In vitro biochemical assay of Fub9. (B) Determining the stoichiometry of FA and H2O2 by quantifying the product concentration. Three independent assays were performed. (C) Substrate analogues tested for Fub9. (D) Proposed catalytic mechanism of Fub9.
To gain more insight into Fub9 function, we tested several substrate analogues (Figure 5C). The five-membered ring analogue 3,4-dehydro-L-proline (Dhp), which mimics 4’ instead of 4, can be oxidized to pyrrole-2-carboxylate (P2C) by Fub9. The steady-state kinetics was determined based on the rate of H2O2 formation. The initial catalytic rate was not saturated even up to 10 mM Dhp (app. Km > 50 mM) and an apparent kcat/Km of 421 M−1s−1 was obtained (Figure S10). The poor affinity to Dhp is perhaps due to the lack of alkyl substitution group on the substrate. The Fub9-catalyzed oxidation of Dhp suggests that Fub9 specifically oxidizes C-N bond instead of C-C bond, which is reminiscent of the well-characterized flavin adenine dinucleotide (FAD)-dependent oxidase NikD involved in nikkomycin biosynthesis.19 Similar to NikD, we propose the reactive substrate for Fub9 is the enamine 4’ instead of imine 4. Upon the first hydride transfer from C2 to FMNox, Fub9 catalyzes tautomerization to yield another enamine, dihydrofusaric acid, which then transfers hydride from C4 to a regenerated FMNox to complete the dehydrogenative aromatization (Figure 5D). Consistent with this proposal, neither 2-cyclohexene-1-carboxylate (2-CHA) nor 3-cyclohexene-1-carboxylate (3-CHA) was a substrate for Fub9, which mimic imine 4 and enamine 4’ respectively, (Figure S10).
Biosynthesis of n-hexanal (3) involves a collaboration between an HRPKS and an NRPS-like enzyme
Having elucidated the biosynthetic mechanism for picolinic acid formation, we then focused on the biogenesis of the alkyl building block n-hexanal (3). Since trans-2-hexenal (2) accumulated in both Δfub6 and Δfub7 mutants was confirmed to be an on-pathway intermediate (ref. 9d), we reasoned that the MDR Fub6 may catalyze ene-reduction of 2 to 3. Indeed, addition of purified recombinant Fub6 into solution containing 2 and NADPH (but not NADH) led to rapid decrease of absorbance at 340 nm, indicating oxidation of NADPH (Figure S11). Analyzing the reaction mixture by GC-MS revealed that 2 was reduced to 3 exclusively, without formation of n-hexanol (7) or trans-2-hexenol (8). Therefore, Fub6 is a strict ene-reductase and does not possess any aldehyde reductase activity (Figure 6A). Fub6 does not show any metal ion dependence and the reaction has a KM(app) of 60 μM for 2 and a kcat of 3.1 s−1 (Figure S11). Low apparent KM implies that the intracellular concentration of the good Michael-acceptor 2 is maintained at a low level to avoid undesired nonenzymatic reactions. Other α/β-unsaturated aldehydes, such as crotonaldehyde and cinnamaldehyde, are also substrates for Fub6, but the catalytic efficiencies are more than 10-fold lower than that for 2 (Table S16). Like other ene-reductases,20 Fub6 can only reduce activated C=C double bonds, as the non-activated trans-2-hexenol (8) and the weakly-activated trans-2-hexenoic acid (1) are not substrates for Fub6 (Figure S11).
Figure 6.
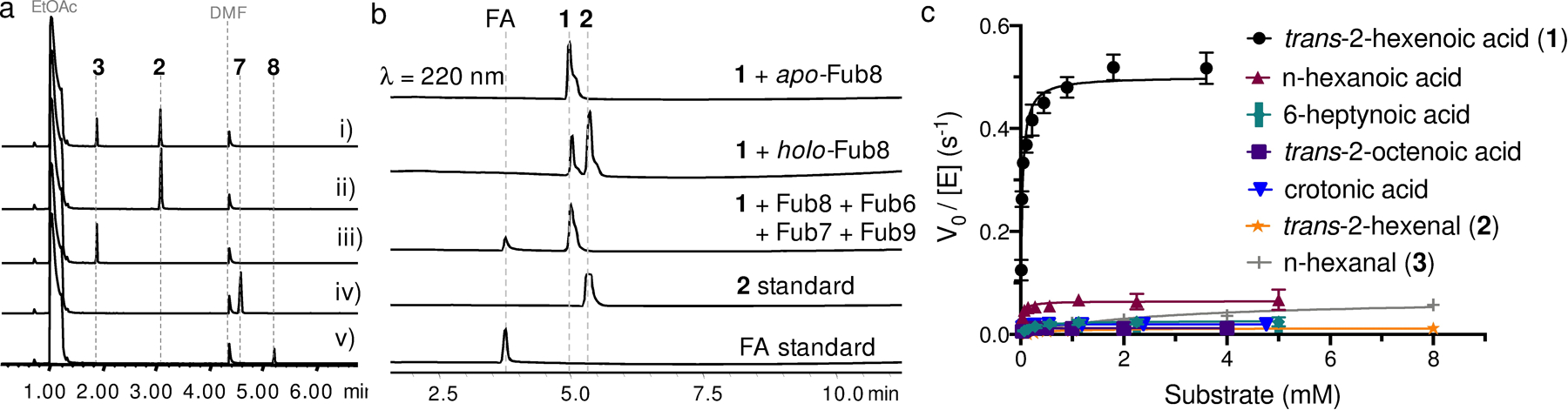
Characterization of Fub6 and Fub8. (A) GC-MS analysis of Fub6-catalyzed ene-reduction of 2 in vitro: i) reaction extract (2); ii) 2 standard; iii) n-hexanal (3) standard, iv) n-hexanol (7) standard, v) trans-2-hexenol (8) standard. (B) HPLC analysis of Fub8-catalyzed ATP-dependent reduction of 1. (C) Substrate specificity of Fub8 determined through steady-state kinetics analysis.
The remaining essential enzymes for FA biosynthesis, which include Fub1 (HRPKS), Fub4 (α,β-hydrolase), and Fub8 (NRPS-like carboxylic acid reductase), must be responsible for synthesizing trans-2-hexenal (2). We propose that Fub1 and Fub4 synthesize and hydrolytically release the triketide trans-2-hexenoic acid (1), which is then activated through adenylation and reduced by Fub8 to give 2 (Figure 2D). To test this proposal, we purified recombinant Fub8 from E. coli (Figure S12). Apo-Fub8 was enzymatically converted to the holo-form by using phosphopantetheinyl transferase NpgA as described previously.21 In the presence of ATP, NADPH, and MgCl2, 1 can be reduced to 2 by holo-Fub8, but not apo-Fub8 (Figure 6B). Although prolonged incubation of Fub8 with 1 in the presence of excess amount of NADPH led to formation of over-reduced alcohol product 8 (Figure S12), the catalytic efficiency of Fub8 aldehyde reductase activity (8 as substrate, app. kcat/KM = 19 M−1s−1) is three orders of magnitude lower than that of its carboxylic acid reductase activity (1 as substrate, app. kcat/KM = 17,000 M−1s−1, Table S16). Thus, like many other NRPS-like carboxylic acid reductases,22 Fub8 does not have processive 2+2 electron reduction activities under physiological conditions. Successfully reconstituting Fub8 activity also allowed us to synthesize FA from 1 and OAH in one-pot using a four-enzyme cocktail (Figure 6B).
Furthermore, Fub8 is shown to be highly specific to 1 in terms of both chain length and functional groups (Figure 6C). Reduction of the saturated substrate analogue n-hexanoic acid is 10-fold slower than that of 1, and neither the 2-carbon shorter crotonoic acid (kcat = 0.02 s−1) or the 2-carbon longer trans-2-octenoic acid (kcat = 0.01 s−1) is a good substrate for Fub8. Such stringent substrate specificity of Fub8 suggests a dedicated role of Fub8 in reducing the triketide precursor 1, proposed to be synthesized by Fub1 and Fub4. Although we are not able to reconstitute the activity of the HRPKS Fub1 and Fub4 in vitro, such combination of HRPKS and hydrolase in producing polyketides is well-documented.23 Therefore, biosynthesis of 2 requires the collaborative effort between two multi-domain enzymes: an HRPKS (Fub1) and an NRPS-like carboxylic acid reductase (Fub8).
The revised biosynthetic pathway to FA is shown in Figure 2D. Unexpectedly, it takes a village (i.e. four enzymes) to biosynthesize 3, a seemingly simple, saturated C6 aldehyde. This biosynthetic logic is intriguing, since one would not design such an atomically non-economical pathway. For instance, Fub6 could be bypassed if the HRPKS system can afford the fully saturated n-hexanoic acid, which is well within the biosynthetic capability of HRPKSs. Moreover, the HPRKS Fub1 could directly release the polyketide chain to yield 3 using a reductase (R) release domain as seen in the biosynthesis of betaenone B.24 These strategies are all energetically more efficient as compared to the current pathway. One possible explanation why Fusarium species adopt the current pathway is that the free intermediates 1 and 2 may serve other purposes, such as participating in other yet-unknown metabolic pathways, or act as signaling or regulatory molecules. Future studies are required to understand this logic.
Biocatalytic utility of Fub7
Our biochemical characterization established the biosynthetic pathway of FA, which features a four-enzyme combination to make the short-chain fully saturated aliphatic aldehyde (3), a PLP-dependent enzyme (Fub7) to connect 3 with an amino acid primary metabolite O-acetyl-L-homoserine to generate the nitrogen-containing heterocycle 4, and an oxidase to furnish the aromatic pyridine ring at last. Many of these enzymes characterized here can be useful reagents in biocatalysis and synthetic biology.20 Among them, the C-C bond forming activity of Fub7 intrigued us to further explore its biocatalytic potential.
We first tested Fub7 substrate specificity using different linear aldehyde substrates with chain length varying from C3 to C8. OAH was used as the limiting reagent in each reaction. To facilitate product detection and quantification, we coupled this assay to Fub9 enabling oxidation of any cyclic imine products into chromophoric picolinic acids. As shown in Figure 7, HPLC analysis indicates a series of 5-alkyl picolinic acids (9–16) can be synthesized using this coupled enzymatic system starting from OAH and aldehydes. Five compounds (10, 11, 14–16) were isolated from scaled-up, over-night reactions (volume varies between 10 to 30 mL depending on the yield) for structural characterization. Analysis of the reactions by LC-MS did not reveal any accumulated tetrahydropicolinic acid intermediates, therefore the yield of each product is likely limited by the substrate specificity of Fub7. Aldehydes bearing bulky groups at Cβ position are not favored by Fub7 as 2-phenylacetaldehyde gave low yield (product 15) and 2-cyclohexylacetaldehyde was completely inactive (vide infra), presumably due to increased steric hinderance impeding Fub7-catalyzed enolization of aldehydes. Overall, Fub7 exhibits somewhat relaxed substrate specificity towards linear aliphatic aldehydes and prefers substrate chain length C4-C7.
Figure 7.
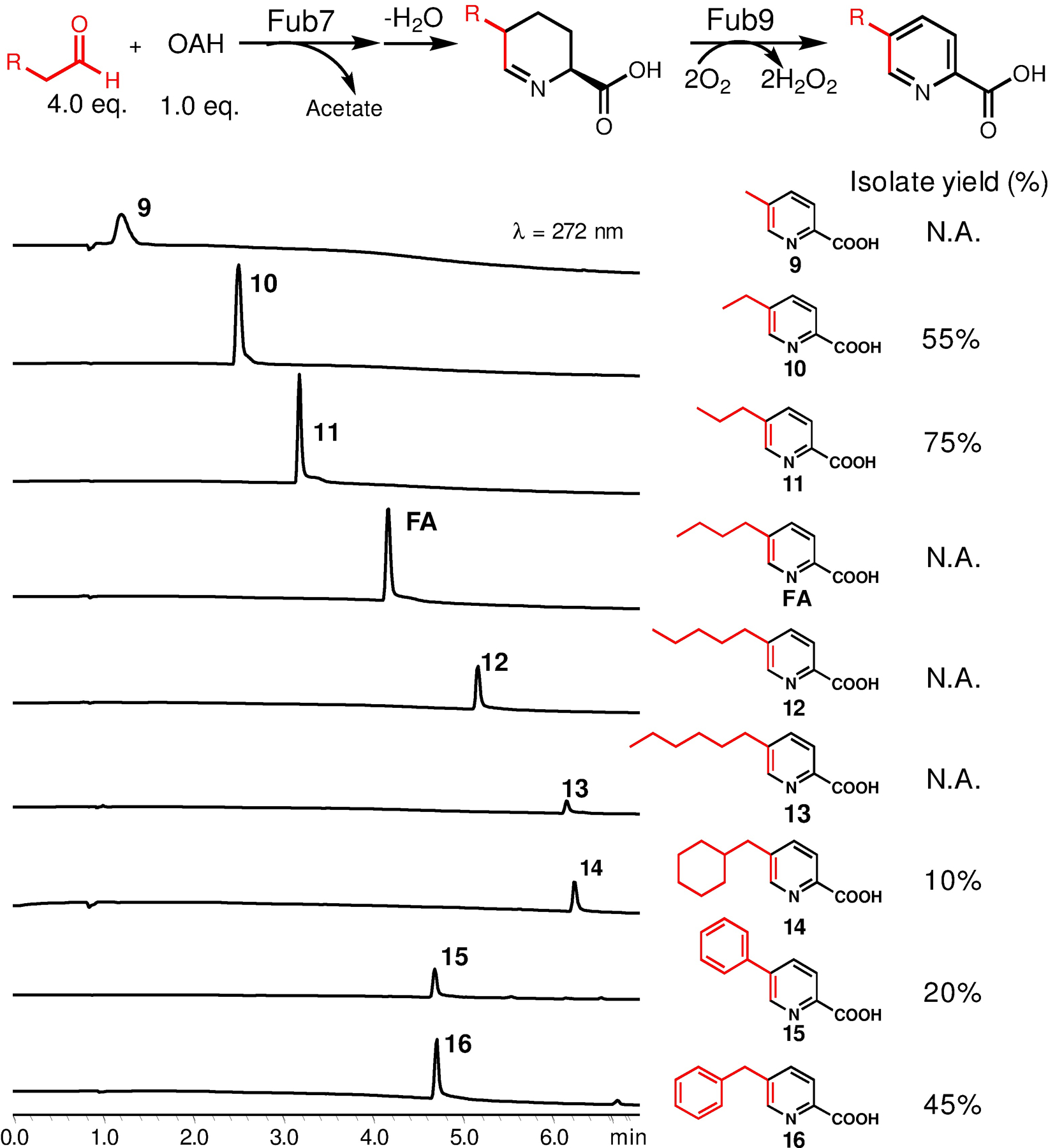
Biocatalytic synthesis of 5-alkyl picolinic acid derivatives. Analytical HPLC chromatograms of each reactions at small scale (200 μL). Assay conditions: 2 mM OAH, 10 μM Fub7, 5 μM Fub9, 0.1 μM catalase, and 8 mM aldehydes mixed in phosphate buffer [50 mM K2HPO4 (pH 7.5), 100 mM NaCl, 3% (v/v) DMSO] and incubated at 20 °C. Five compounds (10, 11, 14–16) were isolated for structural characterization. N.D., not determined.
This relaxed substrate specificity of Fub7 encouraged us to repurpose its activity for the synthesis of 5-alkyl- and 5-dialkyl-L-pipecolic acids by intercepting Fub7-synthesized cyclic imine products through chemical reduction using NaBH3CN. Compared to the reported method in which 5-alkyl-DL-pipcolic acids were prepared through hydrogenation of 5-alkyl-picolinic acids,6 our chemoenzymatic route is advantageous in that it preserves the stereocenter at C2 position (L-configuration) originated from OAH. Furthermore, we reasoned that C-C bond formation catalyzed by Fub7 should be stereoselective, which can be leveraged to make L-pipecolic acids bearing a quaternary stereocenter at C5 position by starting from asymmetric α-branched aldehydes.
As shown in Figure 8, a library of 5-substituted-L-pipecolic acids can be prepared chemoenzymatically. When the yield is sufficiently high, the final products were purified and structurally characterized by NMR as indicated in the figure. Diastereomeric ratios were determined by either NMR or by LCMS when the compounds can be separated. As expected, for linear aliphatic aldehydes substrates (entry 1–4), a pair of 2,5-diastereomers (cis and trans) were formed with the trans-isomers being the major products (Figure S13). For α-branched aldehyde substrates (entry 5–14), Fub7 could accept a series of α-methyl aldehydes with carbon chain length C4-C7 preferred (products 20–24), but did not tolerate aldehydes with ethyl or any other larger substitution groups at Cα, which is also likely due to steric clash with enzyme active site residues. It is noteworthy to mention that one of the products, 5,5-dimethyl-L-pipecolic acid (20), is a natural metabolite involved in flavunoidine biosynthesis.25 It was proposed that 20 could be synthesized from OAH and α-keto-isovalerate by the PLP-dependent enzyme FlvA and imine reductase FlvB. Our result shown here shed more light on the biosynthetic mechanism of 20 that the carbon nucleophile for FlvA could be isobutylaldehyde instead of α-keto-isovalerate.
Figure 8.
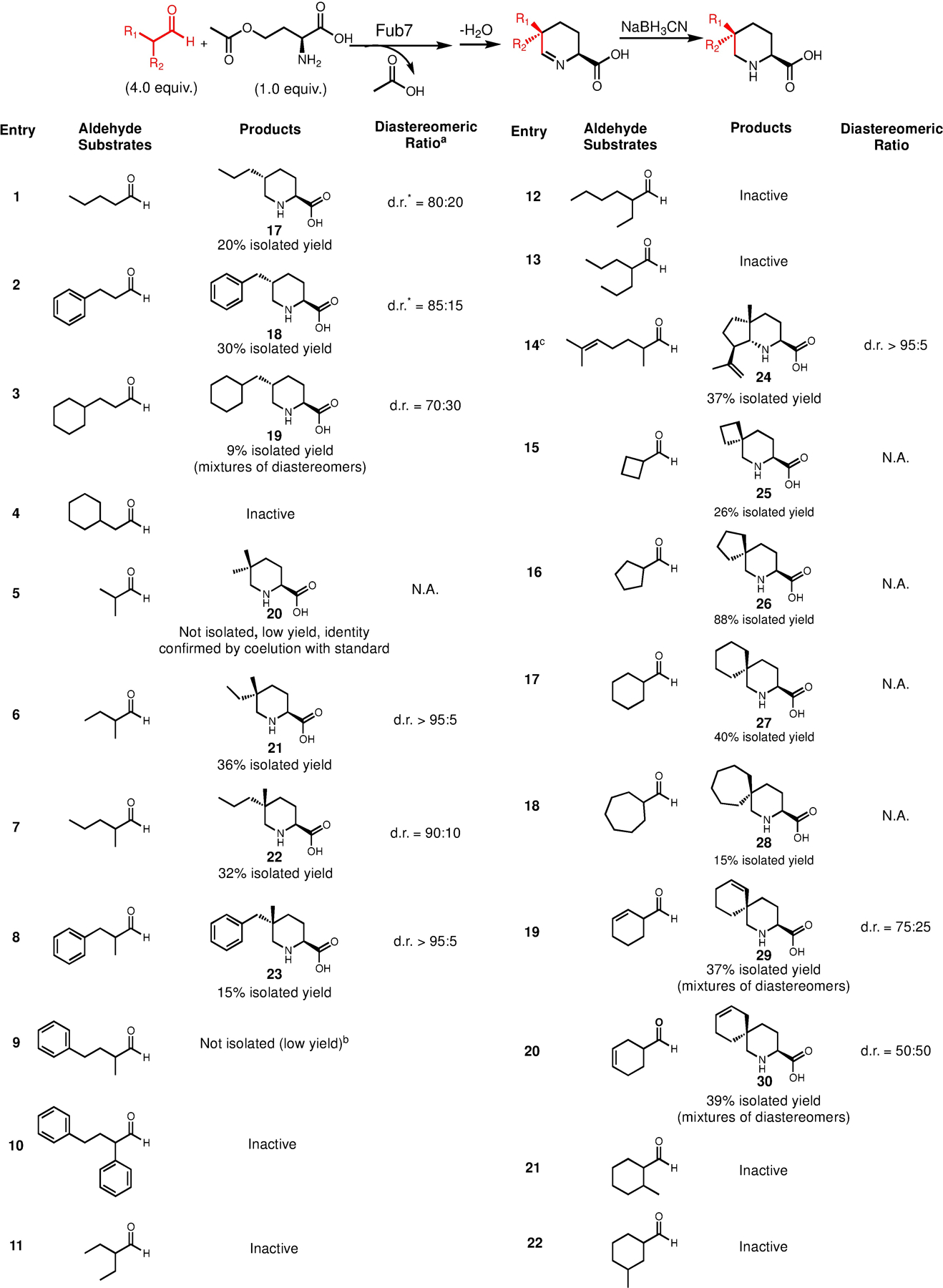
Substrate scope of Fub7 in making 5-alkyl-l-pipecolic acids and 5,5-dialkyl-l-pipecolic acids. Reaction conditions: 2 mM OAH, 10 μM Fub7, and 8 mM aldehydes mixed in phosphate buffer [50 mM K2HPO4 (pH 7.5), 100 mM NaCl, 3% (v/v) DMSO] and incubated at 20 °C. Reaction volume varies between 20 to 40 mL depending on the yield. N.A., not applicable. aThe d.r. values were analyzed by NMR or LC-MS when the diastereomers can be separated by HPLC. bAttempts to purify the target product from a 40 mL reaction did not yield enough materials for NMR characterization (<0.5 mg). cNaBH3CN was not used in this reaction.
Moreover, reactions with asymmetric racemate α-methyl aldehyde substrates (products 21–24) all convergently yielded trans-5-alkyl-5-methyl-L-pipecolic acids. Because this C5 quaternary stereocenter can no longer epimerize, this result strongly supports our hypothesis that Fub7 stereoselectively catalyzes C-C bond formation between aldehyde-derived enolate and PLP-bound vinylglycine ketimine. Notably, product 24 is a 5,5,6-trialkyl-L-pipecolic acid with three contiguous stereocenters, which are set through a cascade reaction triggered by Fub7: stereoselective C-C bond formation between 2,6-dimethyl-5-heptenal and OAH, followed by cyclization and dehydration; the newly-formed imine intermediate then undergoes a Pictet-Spengler-like reaction to give the final bicyclic scaffold. Synthesis of 24 also demonstrates that the reactive Fub7-synthesized imine products can be leveraged to generate new scaffolds which could open up new opportunities in constructing more complex L-pipecolic acids.
Last but not least, Fub7 can also accept various α-branched cyclic aldehydes (entry 15–20) leading to pipecolic acids derived spirocycles (product 25-30). The low diastereomeric ratios of product 29 and 30 indicate that significant substrate desymmetrization (appending substitution groups) will be required for Fub7 to achieve diastereoselectivity, as Fub7 does not distinguish the re-face and si-face of the asymmetric enolate substrates (entry 19–20) very well. However, additional methyl substitution could not be accommodated (entry 21–22), which further suggests that the enzyme active site is naturally tailored to best suit linear aliphatic aldehyde, such as its natural substrate 3. Enlarging the active site by protein engineering on Fub7 may be required to recognize more bulky substrates.
Summary
In this work, we elucidated the biosynthetic pathway for fusaric acid and characterized the enzymatic activity of four key enzymes, Fub6–9. Among these, we demonstrated the synthetic potential of Fub7 in synthesizing either 5-alkyl-picolinic acids when coupled with Fub9 or substituted pipecolic acids via a chemoenzymatic route. In particular, the stereoselective C-C bond formation catalyzed by Fub7 can be harnessed to stereoconvergently access 5-alkyl-5-methyl-L-pipecolic acids with high diastereomeric ratio. Fub7-enabled cascade reaction synthesizing 24 indicates the asymmetrically synthesized imine products from Fub7-catalyzed reaction can be potentially borrowed by many other downstream pathways leading to more complex structural scaffolds. Our study also demonstrated how biosynthesis study could facilitate biocatalytic innovations. Given the vast genetic potential of microbial genomes, more PLP-dependent biosynthetic enzyme will be uncovered and hold promise in biocatalysis and synthetic biology.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH 1R35GM118056 to YT. YH was a Life Sciences Research Foundation fellow sponsored by the Mark Foundation for Cancer Research. AH was supported by the Lorraine H. and Masuo Toji Summer Research fellowship. YH also acknowledges University of California Santa Barbara start-up funds for support of this work.
Footnotes
Supporting Information.
This material is available free of charge via the Internet at http://pubs.acs.org.
Experimental procedures, and spectroscopic data.
No competing financial interests have been declared.
REFERENCES
- (1).(a) Majumdar KC and Chattopadhyay SK Heterocycles in Natural Product Synthesis. Wiley-VCH, Weinheim, 2011, pp 267–297. [Google Scholar]; (b) O’Hagan D Pyrrole, pyrrolidine, pyridine, piperidine and tropane alkaloids. Nat. Prod. Rep 2000, 17, 435–446. [DOI] [PubMed] [Google Scholar]; (c) Dewick PM Medicinal natural products: A biosynthetic approach. 3rd ed. John Wiley & Sons Ltd., 2009. [Google Scholar]
- (2).(a) Taylor R; MacCoss M; and Lawson ADG Rings in drugs. J. Med. Chem 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; and Njardarson JT Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U. S. FDA approved pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (c) Jacobsen JA; Fullagar JL; Miller MT; and Cohen SM Identifying chelators for metalloprotein inhibitors using a fragment-based approach. J. Med. Chem 2011, 54, 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yabuta T; Kambe T; and Hayashi T Biochemical studies of the ‘bakanae’ fungus of rice. I. Fusarinic acid, a new product of the ‘Bakanae’ fungus. J. Agric. Chem. Soc. Jpn 1934, 10, 1059–1068. [Google Scholar]
- (4).(a) Bacon CW; Porter JK; Norred WP; and Leslie JF Production of fusaric acid by Fusarium species. Appl. Environ. Microbiol 1996, 62, 4039–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bani M; Rispail N; Evidente A; Rubiales D; and Cimmino A Identification of the main toxins isolated from Fusarium oxysporum f. sp. pisi Race 2 and their relation with isolates’ pathogenicity. J. Agric. Food Chem 2014, 64, 2574–2580. [DOI] [PubMed] [Google Scholar]; (c) Singh VK; Singh HB; and Upadhyay RS Role of fusaric acid in the development of ‘Fusarium wilt’ symptoms in tomato: Physiological, biochemical and proteomic perspective. Plant Physiol. Biochem 2017, 320–332. [DOI] [PubMed] [Google Scholar]
- (5).(a) Nagatsu T; Hidaka H; Kuzuya H; Takeya K; and Umezawa H Inhibition of dopamine beta-hydroxylase by fusaric acid (5-butylpicolinic acid) in vitro and in vivo. Biochem. Pharmacol 1970, 19, 35–44. [DOI] [PubMed] [Google Scholar]; (b) Velasco M, Gilbert CA; Rutledge CO; and McNay JL Antihypertensive effect of a dopamine beta hydroxylase inhibitor, bupicomide: a comparison with hydralazine. Clin. Pharmacol. Ther 1975, 18, 145–153. [DOI] [PubMed] [Google Scholar]
- (6).Jones PH and Somani P 5-Alkylpipecolic acids as antihypertensive agents. US Patent 3840663 A 1974. (b)
- (7).(a) Munkvold GP Fusarium species and their associated mycotoxins. Methods. Mol. Biol 2007, 1542, 51–106. [DOI] [PubMed] [Google Scholar]; (b) Gruber-Dorninger C; Novak B; Nagl V; and Berthiller F Emerging mycotoxins: beyond traditionally determined food contaminants. J. Agric. Food Chem 2017, 65, 7052–7070. [DOI] [PubMed] [Google Scholar]
- (8).(a) Dobson TA; Desaty D; Brewer D; and Vining LC Biosynthesis of fusaric acid in cultures of Fusarium oxysporum Schlecht. Can. J. Biochem 1967, 45, 809–823. [DOI] [PubMed] [Google Scholar]; (b) Stipanovic RD; Wheeler MH; Puckhaber LS; Liu J; Bell AA; Williams HJ Nuclear magnetic resonance (NMR) studies on the biosynthesis of fusaric acid from Fusarium oxysporum f. sp. vasinfectum. J. Agric. Food Chem 2011, 59, 5351–5356. [DOI] [PubMed] [Google Scholar]
- (9).(a) Brown DW; Butchko RA; Busman M; and Proctor RH Identification of gene clusters associated with fusaric acid, fusarin, and perithecial pigment production in Fusarium verticillioides. Fungal. Genet. Biol 2012, 49, 521–532. [DOI] [PubMed] [Google Scholar]; (b) Niehaus EM; von Bargen KW; Espino JJ; Pfannmuller A; Humpf HU; and Tudzynski B Characterization of the fusaric acid gene cluster in Fusarium fujikuroi. Appl. Microbio. Biotechnol 2014, 98, 1749–1762. [DOI] [PubMed] [Google Scholar]; (c) Brown DW; Lee SH; Kim LH; Ryu JG; Lee S; Seo Y; Kim YH; Busman M; Yun SH; Proctor RH; Lee T Identification of a 12-gene fusaric acid biosynthetic gene cluster in Fusarium species through comparative and functional genomics. Mol. Plant Microbe. Interact 2015, 28, 319–332. [DOI] [PubMed] [Google Scholar]; (d) Studt L; Janevska S; Niehaus EM; Burkhardt I; Arndt B; Sieber CM; Humpf HU; Dickschat JS; Tudzynski B Two separate key enzymes and two pathway-specific transcription factors are involved in fusaric acid biosynthesis in Fusarium fujikuroi. Environ. Microbiol 2016, 18, 936–956. [DOI] [PubMed] [Google Scholar]
- (10).Palm D; Klein HW; Schinzel R; Buehner M; and Helmreich EJM The role of pyridoxal 5’-phosphate in glycogen phosphorylase catalysis. Biochemistry 1990, 29, 1099–1107. [DOI] [PubMed] [Google Scholar]
- (11).Chen M; Liu C-T; and Tang Y Discovery and application of a PLP-dependent amino acid γ-substitution enzyme that catalyzes C−C bond formation. J. Am. Chem. Soc 2020, 142, 10506–10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liu N; Hung Y-S; Gao S-S; Hang L; Zou Y; Chooi Y-H; and Tang Y Identification and heterologous production of a benzoyl-primed tricarboxylic acid polyketide intermediate form the zaragozic acid A biosynthetic pathway. Org. Lett 2017, 19, 13, 3560–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pieniazek A and Grabski J Studies on beta cystathionase and acetylhomoserine sulfhydrylase as the enzymes of alternative methionine biosynthetic pathways in Aspergillus nidulans. Acta. Biochim. Pol 1973, 20, 159–168. [PubMed] [Google Scholar]
- (14).Nishina Y; Sato K; and Shiga K (1991) Isomerization of Δ−1-piperideine-2-carboxylate to Δ−2-piperideine-2-carboxylate on complexation with the flavoprotein D-amino acid oxidase. J. Biochem 1991, 109, 705–710. [DOI] [PubMed] [Google Scholar]
- (15).Griswold WR; Toney MD Role of the pyridine nitrogen in pyridoxal 5’-phosphae catalysis: activity of three classes of PLP enzymes reconstituted with deazapyridoxal 5’-phosphate. J. Am. Chem. Soc 2011, 133, 14823–14830. [DOI] [PubMed] [Google Scholar]
- (16).Brzovic P; Holbrook EL; Greene RC; and Dunn MF Reaction mechanism of Escherichia coli cystathionine γ-synthase: direct evidence for a pyridoxamine derivative of vinylglyoxylate as a key intermediate in pyridoxal phosphate dependent γ-elimination and γ-replacement reactions. Biochemistry 1990, 29, 442–451. [DOI] [PubMed] [Google Scholar]
- (17).(a) Franz SE and Stewart JD Threonine aldolases. Adv. Appl. Microbiol 2014, 142, 710–714. [DOI] [PubMed] [Google Scholar]; (b) Florio R di Salvo ML Vivoli M; Contestabile R Serine hydroxymethyltransferase: a model enzyme for mechanistic, structural and evolutionary studies. Biochim. Biophys. Acta 2011, 1814, 1489–1496. [DOI] [PubMed] [Google Scholar]
- (18).Cui Z; Overbay J; Wang X; Liu X; Zhang Y; Bhardwaj M; Lemke A; Wiegmann D; Niro G; Thorson JS; Ducho C; Van Lanen SG Pyridoxal-5’-phosphate-dependent alkyl transfer in nucleoside antibiotic biosynthesis. Nat. Chem. Biol 2020, 16, 904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bruckner BC and Jorns MS Spectral and kinetic characterization of intermediates in the aromatization reaction catalyzed by NikD, an unusual amino acid oxidase. Biochemistry. 2009, 48, 4455–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Toogood HS and Scrutton NS Discovery, characterization, engineering, and applications of ene-reductases for industrial biocatalysis. ACS Catal. 2018, 8, 3532–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hai Y; Huang AM; and Tang Y Structure-guided function discovery of an NRPS-like glycine betaine reductase for choline biosynthesis in fungi. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 10348–10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gahloth D; Dunstan MS; Quaglia D; Klumbys E; Lockhart-Clairns MP; Hill AM; Derrington SR; Scrutton NS; Turner NJ; and Leys D Structures of carboxylic acid reductase reveal domain dynamics underlying catalysis. Nat. Chem. Biol 2017, 13, 975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Xu W; Chooi Y-H; Choi JW; Li S; Vederas JC; Da Silva NA; and Tang Y LovG: the thioesterase required for dihydromonacolin L release and lovastatin nonaketide synthase turnover in lovastatin biosynthesis. Angew. Chem. Biol 2017, 13, 975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ugai T; Minami A; Fuji R; Tanaka M; Oguri H; Gomi K; Oikawa H Heterologous expression of highly reducing polyketide synthase involved in betaenone biosynthesis. Chem. Commun 2015, 51, 1878–1881. [DOI] [PubMed] [Google Scholar]
- (25).Yee DA; Kakule TB; Cheng W; Chen M; Chong CTY; Hai Y; Hang LF; Hung Y-S; Liu N; Ohashi M; Okorafor IC; Song Y; Tang M; Zhang Z; Tang Y, Genome Mining of Alkaloidal Terpenoids from a Hybrid Terpene and Nonribosomal Peptide Biosynthetic Pathway. J. Am. Chem. Soc 2020, 142, 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.