

Abstract
Mutations in the X-linked gene MECP2 cause Rett syndrome, a progressive neurological disorder in which children develop normally for the first one or two years of life before experiencing profound motor and cognitive decline1,2,3. Currently, there are no effective treatments for Rett syndrome, but we hypothesized that using the period of normal development to strengthen motor and memory skills might confer some benefit. We found that intensive training beginning in the presymptomatic period dramatically improved the performance of specific motor and memory tasks in a mouse model of Rett syndrome and significantly delayed the onset of symptoms. These benefits were not observed when the training was initiated only after symptom onset. Markers of neuronal activity and chemogenetic manipulation revealed that task-specific neurons that are repeatedly activated during training develop more dendritic arbors and have better neurophysiological responses, thereby enhancing their functionality and delaying symptom onset. These results provide a rationale for newborn genetic screening for Rett syndrome, as they suggest that presymptomatic intervention might mitigate symptoms or delay their onset. Similar strategies should be studied for other childhood neurological disorders.
Rett syndrome is one of the most prevalent neurodevelopmental disorders affecting females4. The natural history of Rett syndrome is unusual, however, in that its onset is postnatal. Development appears normal until around two years, when the child loses acquired motor, cognitive, social, and linguistic milestones and develops neurological symptoms such as tremors, stereotypies, and seizures3,5. Female mice lacking one Mecp2 allele reproduce key features of Rett syndrome, with early normal development giving way to widespread neurological dysfunction6–8. Restoring Mecp2 expression in adult Rett mice rescues the phenotype9,10, however, which demonstrates that the nervous system is sufficiently intact to support recovery. Although gene therapy would, in principle, be an ideal treatment11, the brain’s sensitivity to the quantity of MeCP2 poses a particular challenge12 in that too much of the protein causes MECP2 duplication syndrome3,13,14. Because MECP2 expression in females is mosaic15, delivering additional copies of MECP2 would overload neurons expressing the normal MECP2 allele. There has therefore been considerable interest in alternative approaches to treatment. One non-pharmacological approach, forniceal deep brain stimulation (DBS), improves learning and memory in Rett mice by stimulating the hypoactive hippocampal circuits16,17. We therefore asked whether there might be ways of mimicking the effects of consistent stimulation without the drawbacks of DBS18 and turned our attention to behavioral training, which is non-invasive and improves motor and cognitive skills in young children with autism19. We decided to test whether training can enhance circuit activity and mitigate behavioral deficits in female Mecp2 heterozygous mice (hereafter referred to as Rett mice).
Early training delays symptom onset
Female Rett mice develop motor incoordination around 12 weeks of age and then deteriorate. To ascertain the effects of training before and after symptom onset, we compared the performance of three groups of 24-week-old mice on the rotating rod apparatus20,21: a control group of wild-type (WT) and Rett mice that had no rotarod training (the naive group), an early-trained group that was trained starting at 8 weeks of age, and a late-trained group that was trained for the same number of sessions but starting at 22 weeks (Fig. 1a; Extended Data Fig 1a). As expected, naive Rett mice performed worse than naive WT mice (Fig. 1b). Late-trained Rett mice performed slightly better than naive Rett mice, but the early-trained Rett mice performed significantly better than both naive and late-trained Rett mice (Fig. 1b; Extended Data Fig 1c). To determine if presymptomatic training bestowed lasting benefits on rotarod performance, we continued to train early-trained mice beyond 24 weeks of age using the same regimen (4 times per day, bi-weekly). At 32 weeks of age, the performance of early-trained Rett mice was comparable to that of naive 12-week-old Rett mice (Fig. 1c). Postsymptomatic training did not provide benefit even if it was begun earlier, at 16 weeks (Fig. 1c). Notably, early training on the rotarod did not mitigate other behavioral deficits (Extended Data Fig. 1b, 1d–o).
Fig. 1 |. Presymptomatic training improves motor performance on the rotarod in Rett mice.

a, Training regimen for naive, late-trained, and early-trained mice; each line represents 4 trials/day. b, Average motor performance on the rotarod. WT naive (n = 19), WT late-trained (n = 19), WT early-trained (n = 18), Rett naive (n = 19), Rett late-trained (n = 18), and Rett early-trained (n = 19) mice were tested across 4 days at 24 weeks of age. c, A subset of these mice underwent additional behavioral tests (see Extended Data Fig. 1) and the rest of the early-trained mice were followed for rotarod performance until 32 weeks of age. Early training delayed the onset of motor symptoms in Rett mice (n = 7) until after 22 weeks, with some benefit still apparent at 32 weeks; comparisons are WT (n = 7) at 32 weeks of age and naive Rett mice (n = 12) at 12 weeks of age. In another group of Rett mice (n = 8), we delayed training initiation until 16 weeks, following the same early-training protocol, but this yielded no benefit. The sample size (n) corresponds to the number of biologically independent mice. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), ** (p<0.01), *** (p<0.001), **** (p<0.0001).
To determine whether presymptomatic training improves cognition in Rett mice22, we trained mice in the Morris water maze23,24. The animals were either untrained, late-trained (at 11 weeks of age), or early-trained (at 4 weeks of age) (Fig. 2a; Extended Data Fig. 2a–c). At 12 weeks, early-trained Rett mice outperformed both naive and late-trained Rett mice (Fig. 2b–d; Extended Data Fig 2d). Both timing and consistency of training were important, as either delaying or skipping training sessions erased the benefits (Fig. 2e; Extended Data Fig. 2e). To determine whether presymptomatic training bestowed lasting benefits, we continued to train early-trained mice (4 times per week, monthly). Early-trained Rett mice did not show memory deficits comparable to those of naive Rett mice until they reached 24 weeks of age, though their performance began to decline after 16 weeks of age compared to early-trained WT mice (Fig. 2e). Water maze-training did not affect contextual fear memory or other behavioral deficits (Fig. 2f–g; Extended Data Fig 2f–o).
Fig. 2 |. Presymptomatic training improves spatial memory in the Morris water maze task in Rett mice.

a, Training regimen for naive, late-trained, and early-trained mice; each line represents 8 trials/day. Early training started at 4 weeks of age; late training started at 11 weeks of age. Late- and early-trained mice received the same number of training trials. b-d, Spatial memory performance in the water maze. (b) WT naive (n = 18), WT late-trained (n = 18), WT early-trained (n = 19), Rett naive (n = 19), Rett late-trained (n = 19), and Rett early-trained (n = 18) mice were tested over 4 consecutive days at 12 weeks of age. A probe trial on day 5 measured the time spent in the target quadrant (c) and number of platform area crossings (d). e, Early-trained WT (n = 7) and Rett mice (n = 7) mice were tested until 24 weeks of age and then compared to naive Rett mice (n = 12) at 12 weeks of age. In a subset of Rett mice (n = 8), training was delayed until 8 weeks of age. f-g, Conditioned fear memory in water maze-trained mice. Contextual (f) and cued memory (g) were assessed in WT naive (n = 11), WT late-trained (n = 11), WT early-trained (n = 12), Rett naive (n = 12), Rett late-trained (n = 12), and Rett early-trained (n= 11) mice. The sample size (n) corresponds to the number of biologically independent mice. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), ** (p<0.01), **** (p<0.0001).
Engagement of task-specific neurons
Water maze training activates a subset of neurons that encode task-related information25. These neurons express immediate early genes such as cFos upon training, re-express them upon additional training, and mediate synaptic plasticity and memory formation26,27. Because cortical and hippocampal dysfunction underlies the learning and memory abnormalities of Rett mice16,17,28, we asked whether task-specific neurons in these regions confer the benefits of presymptomatic training.
We used cFos-targeted recombination in active populations (FosTRAP) to label task-specific neurons activated during water maze training29,30. FosTRAP utilizes the genetically encoded cFosCreER to drive expression of a Cre-dependent reporter, in this case tdTomato, in activated neurons. Administering 4-hydroxytamoxifen immediately following training ensures that only neurons activated during training are labeled (Extended Data Fig. 3a–b). WT and Rett mice had an equivalent number of labeled, tdTomato+ neurons in the cortex and hippocampus, regions important for spatial memory31,32 (Extended Data Fig. 3c–f). In WT mice, all tdTomato+ neurons expressed MeCP2, but in Rett mice only half of the tdTomato+ neurons expressed MeCP2 (Extended Data Fig. 3g–l), indicating that FosTRAP induction is not affected by MeCP2 loss. If a labeled neuron was task-specific, cFos would be reactivated in tdTomato+ neurons after retesting mice in the water maze but not after testing mice in a novel memory assay. We trained WT and Rett mice in the water maze, performed FosTRAP labeling, and divided mice into two groups: one to be retested in the water maze, another to be tested in the fear-conditioning assay (Extended Data Fig. 4a). The number of tdTomato+ and cFos+ neurons was equivalent among early- and late-trained WT and Rett mice in both groups, but there were more tdTomato+cFos+ neurons in mice that were retested in the water maze (Extended Data Fig. 4b–j)—these neurons were task-specific because they were reactivated only in the water maze.
To determine if the activity of task-specific neurons was required for the benefits of presymptomatic training, we expressed Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) in task-specific neurons of trained mice and manipulated neural activity with clozapine-N-oxide (CNO)33. In the presence of CNO, hM4Di DREADD receptors silence, while hM3Dq DREADD receptors stimulate, neuronal activity34. We packaged Cre-dependent DREADD receptors into adeno-associated viruses (AAVs) and delivered them into cFosCreER mice by intraventricular injection at postnatal day 035. The AAV transgene is expressed in task-specific neurons only after training and cFosCreER induction29,30. A Cre-dependent mCherry labeled the same neurons as an endogenous Cre-dependent GFP in the Rosa26 locus (Extended Data Fig. 5a–c). Importantly, infected neurons in WT mice expressed MeCP2, and infected neurons in Rett mice were MeCP2+ or MeCP2− in equal numbers (Extended Data Fig. 5d–i). These results validated our method to virally label task-specific neurons.
If task-specific neurons are necessary for the benefits of presymptomatic training, preventing their reactivation would impair spatial memory. We therefore injected an AAV encoding hM4Di-mCherry or mCherry into WT and Rett mice that also expressed cFosCreER. All groups began training at 4 weeks of age and were retested in the water maze in the presence of CNO or vehicle at 13 weeks (Fig. 3a). Neurons expressing mCherry expressed cFos, but those expressing hM4Di-mCherry did not, indicating that CNO prevented the reactivation of task-specific neurons (Extended Data Fig. 6a–d). CNO impaired the spatial memory of WT and Rett mice expressing hM4Di-mCherry (Fig. 3b–d). In contrast, spatial memory remained intact in WT and Rett mice expressing mCherry and injected with CNO or expressing hM4Di-mCherry and injected with vehicle (Fig. 3b–d). Although spatial memory was initially impaired in WT mice expressing hM4Di-mCherry and treated with CNO, additional training improved their performance. Additional training did not improve the performance of Rett mice expressing hM4Di-mCherry, but if training was resumed in the absence of CNO, the benefits of presymptomatic training returned (Extended Data Fig. 6e). The activation of task-specific neurons is therefore necessary for presymptomatic training to benefit Rett mice. But is it sufficient? If so, then promoting reactivation in a neutral context (in the absence of additional water maze training) would preserve spatial memory.
Fig. 3 |. Repeated activation of task-specific neurons mediates the beneficial effects of presymptomatic training in Rett mice.

a, Training timeline for mice expressing hM4Di-mCherry or mCherry at 4, 8, and 12 weeks of age. b-d, Morris water maze performance in early-trained mice. (b) WT mCherry + CNO (n = 7), Rett mCherry + CNO (n = 6), WT hM4Di + Vehicle (n = 6), Rett hM4Di + Vehicle (n = 5), WT hM4Di + CNO (n = 7), and Rett hM4Di + CNO (n = 7) mice were tested at 13 weeks of age. A probe trial on day 6 measured the time spent in the target quadrant (c) and the number of platform area crossings (d). e, Training timeline for mice expressing hM3Dq-mCherry or mCherry at 4, 8, and 12 weeks of age. f-g, Morris water maze performance in early-trained mice. A probe trial was performed on WT mCherry + CNO (n = 7), Rett mCherry + CNO (n = 8), WT hM3Dq + Vehicle (n = 5), Rett hM3Dq + Vehicle (n = 5), WT hM3Dq + CNO (n = 7), and Rett hM3Dq + CNO (n = 10) mice at 13 weeks of age and measured the time spent in the target quadrant (f) and the number of platform area crossings (g). The sample size (n) corresponds to the number of biologically independent mice. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), * (p<0.05), ** (p<0.01), **** (p<0.0001).
We therefore injected an AAV encoding hM3Dq-mCherry or mCherry into WT and Rett mice that expressed cFosCreER. We labeled task-specific neurons in 4-week-old mice after one four-day training session in the water maze, then, instead of subjecting the mice to additional training sessions, we administered CNO or vehicle in the neutral context of their homecage (Fig. 3e). Neurons expressing hM3Dq-mCherry expressed cFos, but those expressing only mCherry did not, indicating that CNO reactivated task-specific neurons (Extended Data Fig. 7a–d). CNO preserved the spatial memory of WT and Rett mice expressing hM3Dq-mCherry, who had undergone only one four-day training session early in life (Fig. 3f–g). In contrast, spatial memory faded over time in WT and Rett mice expressing mCherry and injected with CNO or expressing hM3Dq-mCherry and injected with vehicle (Fig. 3f–g). Inhibiting or activating a random subpopulation of neurons did not affect the memory of WT or Rett mice expressing Camk2aCreER (Extended Data Fig. 8a–l). Activation of task-specific neurons is thus necessary and sufficient to confer benefits of training in Rett mice.
Early training improves neural function
Strategies that reverse Rett-like phenotypes in mice produce corresponding improvements in neuronal morphology and electrophysiology9,16,17,36–38. We therefore compared the morphology of task-specific hippocampal CA1 neurons from 13-week-old late- and early-trained mice in the water maze. Presymptomatic training enhanced dendritic complexity and spine density of MeCP2− CA1 neurons in Rett mice but did not affect the soma or nuclear area (Fig. 4a–d). The morphology of MeCP2− neurons that were not task-specific in early-trained mice was indistinguishable from that of neurons in late-trained Rett mice; the morphological changes thus were not due to nonspecific improvements across the hippocampus (Extended Data Fig. 9a–e). Presymptomatic training similarly improved the morphology of MeCP2− hippocampal granule neurons and layer 5 cortical neurons in Rett mice (Extended Data Fig. 10a–h).
Fig. 4 |. Presymptomatic training improves morphological and electrophysiological defects in neurons of Rett mice.

a-d, Morphological analysis of hippocampal CA1 task-specific neurons in late- and early-trained mice. Sholl analysis (a), spine density (b), soma area (c), and nuclear area (d) were measured in neurons of WT late-trained (n = 5), WT early-trained (n = 5), Rett late-trained (n = 5), and Rett early-trained (n = 5) mice at 13 weeks of age. e-i, Electrophysiological recordings from task-specific hippocampal CA1 neurons in late- and early-trained mice. e, Representative image of a task-specific neuron (magenta) injected with biocytin (green) during recording and immunostained to determine the MeCP2 status (yellow). Frequency (f) and amplitude (g) of sIPSCs and frequency (h) and amplitude (i) of sEPSCs were measured in WT late-trained (n = 10), WT early-trained (n = 10), Rett late-trained (n = 10), Rett early-trained (n = 10) mice at 13 weeks of age. The sample size (n) corresponds to the number of biologically independent mice. Data are represented as mean ± s.e.m. Statistical significance was determined using a one-way (b-d, f-i) or two-way (a) ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), * (p<0.05), ** (p<0.01), *** (p<0.001), **** (p<0.0001).
To assess synaptic function, we recorded spontaneous inhibitory postsynaptic currents (sIPSCs) and spontaneous excitatory postsynaptic currents (sEPSCs) in task-specific hippocampal CA1 neurons from 13-week-old mice (Fig. 4e). Presymptomatic training improved the sIPSC and sEPSC frequencies of MeCP2− neurons in Rett mice (Fig. 4f,h) but not the amplitudes (Fig. 4g,i). The sIPSC and sEPSC frequencies of MeCP2− neurons that were not task-specific were indistinguishable between late-trained and early-trained Rett mice, again ruling out nonspecific improvements across the hippocampus (Extended Data Fig. 9f–j). The underlying benefits of presymptomatic training appear to act at the synaptic and/or circuit level, perhaps as a result of MeCP2’s function in organizing chromatin architecture37,38.
Discussion
The discovery that presymptomatic training leads to behavioral improvements in a physiologically relevant model of Rett syndrome has practical implications for Rett patients. Above all, it suggests that early behavioral training could improve functionality and delay the onset of specific symptoms. It is worth noting that an environmental enrichment approach in young symptomatic Rett patients (average age of 3 years) led to some improvements in gross motor skills39; our data suggest that presymptomatic diagnosis and earlier intervention would exert greater effect. Why did late training not benefit Rett mice, then, when it does benefit patients? At the age of 3 years Rett children are still in the early stages of the disease and sufficiently ambulatory to benefit from treadmill training. Our Rett mice in this study represent more advanced stages of Rett syndrome, and they cannot handle much physical training after a certain point.
Previous studies have shown that activation of task-specific neurons causes morphological and electrophysiological changes that promote memory formation40,41. It is interesting that inhibiting task-specific neurons erased the benefits of early training in Rett mice but not WT mice. We speculate that in Rett mice, neurons that were newly recruited to the relevant ensemble had not, by definition, been engaged during early training. They therefore would be equivalent to neurons in older, training-naive Rett mice, with the same abnormal morphology and physiological responses.
Although treatments are becoming a reality for some inherited neurological diseases42, newborn genetic testing for childhood neurological disorders remains uncommon43–45. We propose that newborn genetic testing for Rett syndrome followed by prompt intensive training in behavioral domains that will be affected (e.g., gait/balance, manual dexterity, and communication) could help patients retain specific milestones and delay symptom onset. Early diagnosis and intensive training may also augment the efficacy of future therapies, whether they are pharmacologic or nucleic acid-based. The fact that early intensive training is safe, non-invasive, and enhances circuit function in multiple behavioral domains makes it an appealing treatment, even for infants and young children46. Though the genetic heterogeneity of neurodevelopmental disorders poses a significant challenge for developing therapies47, the neural circuit dysfunction underlying disease phenotypes48 could represent a shared therapeutic entry point for early training. Presymptomatic diagnosis and early training might benefit not only patients with Rett syndrome but perhaps those with other neurological conditions.
METHODS
Animals
Mice were maintained on a C57BL/6J background on a 12 hr light:dark cycle at 68–72 °F and 30–70% humidity with standard mouse chow and water ad libitum. Up to five mice were housed per cage. Wild-type male mice were bred to Mecp2+/− female mice6 to generate F1 wild-type and Rett female mice for behavioral experiments. Fostm2.1(iCreERT2)Luo/J (cFosCreER; JAX stock: 030323), B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (R26lsl-tdTomato; JAX stock: 007914), B6;129S6-Tg(Camk2a-cre/ERT2)/1Aibs/J (Camk2aCreER; JAX stock: 012362) and B6;129S4-Gt(ROSA)26Sortm9(EGFP/Rpl10a)Amc/J (R26lsl-EGFP-L10a; JAX stock: 024750) mice were obtained from the Jackson Laboratories and maintained as homozygotes. Male R26lsl-tdTomato and R26lsl-EGFP-L10a mice were bred to female Mecp2+/− mice to establish a breeding colony. To generate animals for labeling experiments, male cFosCreER or Camk2aCreER mice were bred to female Mecp2+/−, Mecp2+/−;R26lsl-tdTomato, or Mecp2+/−;R26lsl-EGFP-L10a mice. Behavioral, histological, and electrophysiological analyses were performed blind to genotypes. Mice were randomly assigned to training groups. The Baylor College of Medicine Institutional Animal Care and Use Committee approved all research and animal care procedures.
Behavioral Tests
For each test, mice were habituated in the testing room for 30 min. A light intensity of 150 lx and background white noise of 60 dB was presented during habituation and throughout the testing periods. All assays were performed at the same time of day.
Accelerating Rotarod.
Mice were placed on an accelerating rotarod apparatus (Ugo Basile) and allowed to freely move as the cylinder increased from 5 r.p.m. to 40 r.p.m. over a 5-min period. Latency to fall was measured when the mouse fell off the rod or rode the cylinder for two consecutive revolutions without regaining control. Each training day consisted of four attempts with a 30 min rest in between each trial. To control for the potential effects of excessive handling, mice that did not receive early training were handled in the test room and returned to their cage.
Morris Water Maze.
The pool (1.2 m in diameter) was filled with water (60 cm deep, 22–24 °C) and made opaque with non-toxic white paint. Visual clues were placed on the wall of the testing room approximately 1 m from the pool edge. The 10 × 10 cm escape platform was placed in the southeast quadrant and submerged 2 cm under the water. Mice were placed in individual cages with ample paper towels for the duration of training. Each of the four consecutive training days consisted of eight attempts separated into two blocks. A training trial was completed when the mouse mounted and remained on the platform for 2 s or spent 60 s in the pool. After each trial, the mouse remained on the platform for an additional 30 sec. Mice were analyzed in groups of five before returning to their homecage. The starting locations were changed each day but were consistent for all mice tested. During the probe trail on day five, the platform was removed, and each animal was given 60 s to navigate the pool. ANY-maze (Stoelting) was used to track, record, and analyze swimming. In the hM4Di experiments, mice were trained for an additional day in order to detect any potential delays in relearning that occurred following CNO administration. To control for the potential effects of excessive handling, mice that did not receive early training were handled in the test room and returned to their cage.
Fear Conditioning.
On the first day, mice were placed in a holding room and delivered to the testing room in a temporary cage. Mice were trained in a fear-conditioning chamber (Med Associates, Inc.) that delivers an electric shock paired with a tone. This device was located inside a soundproof box that contained a digital camera and loudspeaker. Each mouse was placed individually in the chamber and left undisturbed for 2 min. A tone (80 dB, 5 kHz, 30 s) coincided with a foot-shock (2 s, 0.7 mA) and was repeated after 1 min. The apparatus was cleaned with isopropanol. The mouse was returned to the temporary cage after an additional minute and returned the homecage in the holding room. Fear memory was assessed after 1 day of training. To test contextual fear memory, mice were placed in the original environment for 5 min without a tone or foot-shock. Mice were returned to their homecage in the holding room. To test cued fear memory, mice were returned to the testing room and placed in the chamber, which was modified to distinguish it from the original context. The chamber was made triangular with the addition of white panels, cleaned with 70% ethanol, and scented with a cup of vanilla extract under the floor. The mouse was allowed to explore the novel environment for 3 min, after which the original tone (80 dB, 5 kHz, 3 min) was presented. Mouse movement was recorded and analyzed using ANY-maze (Stoelting). Freezing was scored only if the animal was immobile for at least 1 s.
Elevated Plus Maze.
Animals were placed in the center of a maze containing two arms (25 × 7.5 cm) surrounded by 15 cm high walls and two open arms with a raised 0.5 cm lip around the edges. The maze was elevated 50 cm above the ground with the arms equidistant from the center platform. The movement and position of the mouse was recorded over 10 min and analyzed with ANY-maze (Stoelting).
Open Field.
Mice were placed in a clear, open Plexiglas box (40 × 40 × 30 cm, Stoelting) with an overhead camera and photo beams to record horizontal and vertical movements. Activity was measured over 10 min and quantified using ANY-maze (Stoelting).
Three-Chamber Interaction.
During the habituation phase, mice were placed in the middle of the three-chamber apparatus (Ugo Basile) containing two empty barred cages in the right and left chambers for 10 min. During the social interaction phase, an age-matched C57BL/6J wild-type female mouse was placed in one cage and a black Lego block of similar size was placed in the other. Partner mice were habituated to the chamber for 1 hr per day for two consecutive days before testing. The test mouse was returned to the middle zone and allowed to explore the chamber for 10 min. Mouse movement was recorded and analyzed using ANY-maze (Stoelting).
Footslip.
Animals were placed individually into the center of a wire grid laid within an open-field chamber (Accuscan) for 10 min. The number of paw slips through the wire grid was recorded and analyzed using ANY-maze (Stoelting). The number of footslips was normalized to the total distance traveled.
Acoustic Startle Response and Prepulse Inhibition.
Mice were placed in a Plexiglas tube and allowed to habituate for 5 min with a 70 dB background noise. The test sessions consisted of six trials of each sound stimuli lasting 20 ms: no stimulus, a 120 dB sound burst, or a 120 dB sound burst with a 74 dB, 78 dB, or 82 dB pre-pulse stimuli presented 100 ms before the startle stimulus. The maximum startle response was recorded and analyzed during the 65 ms period following the onset of the startle stimulus (SR-Lab). Pre-pulse inhibition was calculated as 1 - (startle response with pre-pulse stimulus/startle response only) x 100).
Drug Preparation and Injection
For the FosTRAP experiments, 4-hydroxytamoxifen (4-HT; Cat# H6278, Sigma-Aldrich) was dissolved in ethanol at a concentration of 20 mg/mL and incubated at 37 °C for 10 min with shaking and stored at −20 °C. Before use, 4-HT was dissolved in a 1:4 mixture of castor oil:sunflower seed oil (Cat# 259853 and S5007, Sigma-Aldrich) at a concentration of 10 mg/mL and incubated at 37 °C for 15 min with shaking. The ethanol was evaporated by vacuum centrifugation for 1 hr at room temperature. 4-HT was delivered via intraperitoneal injection at a dose of 25 mg/kg at the end of the last training session. The final solution was always used on the day of preparation. Tissue was collected one week after injection. For the DREADD experiments, clozapine-N-oxide (Cat# 4936, Tocris) was dissolved in 0.9% NaCl at a concentration of 0.5 mg/mL and delivered via intraperitoneal injection at a dose of 5 mg/kg 30 min before the start of the first training trial or in the homecage.
Virus Preparation and Delivery
pAAV:hSyn-DIO-mCherry (Cat# 50459, Addgene), pAAV:hSyn-DIO-hM3D(Gq)-mCherry (Cat# 44361, Addgene), and pAAV:hSyn-DIO-hM4D(Gi)-mCherry (Cat# 44362, Addgene) were used for the DREADD experiments. All AAVs were generated by the Gene Vector Core at Baylor College of Medicine and packaged with serotype 8. For P0 injections, neonatal mice (<8 hr after birth) were collected from their cage and anesthetized on ice. AAV was diluted in sterile 0.1 M phosphate-buffered saline (PBS) containing 0.05% trypan blue and injected bilaterally into the ventricles using a 10 μl syringe (Cat# 7653–01, Hamilton) and 32-gauge needle (Cat# 7803–04, Hamilton) as previously described35. Mice that expressed cFosCreER were injected with 2 × 1010 genome copies per ventricle of pAAV:hSyn-DIO-mCherry, pAAV:hSyn-DIO-hM3D(Gq)-mCherry, and pAAV:hSyn-DIO-hM4D(Gi)-mCherry to achieve widespread distribution of the virus. At this dose, >95% of excitatory neurons in the neocortex are infected35. For sparse labeling of DREADDs in Camk2aCreER mice and morphological analysis of tdTomato- cells in cFosCreER;R26lsl-tdTomato mice, mice were injected with 5 × 107 genome copies per ventricle of pAAV:hSyn-DIO-mCherry, pAAV:hSyn-DIO-hM3D(Gq)-mCherry, pAAV:hSyn-DIO-hM4D(Gi)-mCherry, or pAAV:CBA-3xYFP35,49. Mice were left on a heating pad before returning to their mother. Viral delivery was confirmed by immunofluorescence.
Immunofluorescence Staining and Histology
Animals were transcardially perfused with 50 mL ice-cold 4% paraformaldehyde in 0.1 M PBS. Brains were dissected, post-fixed overnight at 4 °C, washed with 0.1 M PBS, and placed in 30% sucrose in 0.1 M PBS for 24 hr at 4 °C. Brains were embedded in Tissue-Tek Optimum Cutting Temperature Compound (Cat# 4583, Sakura) and stored at −80 °C. 50 μm floating sections were cut using a cryostat (Cat# CM3050S, Leica) and collected in 0.1 M PBS. For neuronal morphology analysis, 100 μm sections were obtained and collected in 0.1 M PBS. Sections were incubated in blocking solution (0.3% Triton X-100, 5% normal goat serum in 0.1 M PBS) for 1 hr at room temperature followed by primary antibody in blocking solution for 24 hr at 4 °C. The following primary antibodies were used: rabbit anti-cFos (1:500, Cat# ABE457, Millipore), rabbit anti-MeCP2 (1:1000, Cat# 3456, Cell Signaling Technologies), mouse anti-NeuN (1:250, Cat# MAB377, Millipore). Sections were washed with 0.1 M PBS and incubated in secondary antibody for 2 hr at room temperature. The following secondary antibodies were used: goat anti-mouse IgG Alexa Fluor 488 (1:500, Cat# A-11001, Thermo Fisher), goat anti-rabbit IgG Alexa Fluor 647 (1:500, Cat# A-21244, Thermo Fisher), and Streptavidin Alexa Fluor 488 (1:500, Cat# S32354, Thermo Fisher). Sections were washed with 0.1 M PBS, counterstained with 1 mM DAPI (Cat# D1306, Thermo Fisher) for 5 min, and mounted onto Superfrost Plus microscope slides (Cat# 12–550-15, Thermo Fisher) with ProLong Gold Antifade mounting medium (Cat# P10144, Thermo Fisher). 3–4 sections were analyzed per mouse.
Imaging and Quantification
Confocal images were captured with the TCS SP8 X microscope (Leica) using a 10X or 63X objective. Z-stack images were acquired at 10 μm steps, or 0.5 μm steps for morphological analysis. Laser settings were set above background levels based on the signal intensity of tissue stained only with the secondary antibody and kept consistent across samples in each experiment. Images were counted using ImageJ-Fiji50. For morphological analysis, task-specific neurons were identified by the presence of tdTomato in cFosCreER;R26lsl-tdTomato animals. Neuronal tracing of dendritic branches and spines were performed automatically from Z-stack images using 3D in Neurolucida 360 (MBF Bioscience) with the default settings as previously described51. Sholl analysis and spine density quantification of the traced images for 10–15 neurons per mouse were performed using Neurolucida 360 Explorer (MBF Bioscience). Soma and nuclear areas were quantified for 50–100 neurons per mouse using ImageJ-Fiji50.
Slice Preparation and Whole-Cell Recording
Animals were decapitated after anesthetization with 3% isoflurane at a 1L/min flow rate. The brain was placed into ice-cold oxygenated NMDG cutting solution containing (mM) 93 NMDG, 93 HCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 Thiourea, 3 sodium pyruvate, 10 MgSO4 and 0.5 CaCl2, pH 7.35. Coronal slices (300 μm) were cut using a vibratome (Cat# VT1200S, Leica) in a chamber filled with ice-cold NMDG cutting solution. Slices were transferred to 34 °C cutting solution for 10 min. They were then incubated in artificial cerebrospinal fluid (ACSF) (125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 12.5 mM Glucose, 1 mM MgCl2, 2 mM CaCl2, pH 7.4) at 34 °C for 1 hr. Slices were transferred to recording chamber and perfused with ASCF at 25±1 °C with the help of an automatic temperature controller (TC-324C, Warner Instruments) at a rate of 3 mL/min. All solutions were bubbled with 95 % O2/5 % CO2. Whole-cell recordings were made using a patch clamp amplifier (Multiclamp 700B, Molecular Devices) under a water immersion 40x objective using infrared differential interference contrast optics. Labeled neurons were visualized using a fluorescence microscope (Cat# BX51 W1, Olympus) connected with X-cite 120 LED (Lumen Dynamics). Data was acquired using a digitizer (DigiData 1440A, Molecular Devices) and analyzed with pClamp (Molecular Devices) and Minianalysis (Synaptosoft). Microelectrodes were made from borosilicate glass capillaries with a resistance of 5–7 MΩ. Spontaneous excitatory postsynaptic currents were recorded at −70 mV in voltage-clamp mode and spontaneous inhibitory postsynaptic currents were recorded at +10 mV in voltage-clamp mode. For these experiments, the microelectrode was filled with an internal solution containing 121 mM Cs-methanesulfonate, 1.5 mM MgCl2, 10 mM HEPES, 10 mM EGTA, 4 mM Mg-ATP, 0.3 mM Na-GTP, 10 mM Na2-phosphocreatine, 2 mM QX314 (Cat# 2313, Tocris), and 0.2% biocytin (Cat# B4261, Sigma-Aldrich) (295 mOsml, pH 7.35). Data were discarded when the series resistance changed by >20% during the course of the recording. After recording, slices were fixed with cold 4% PFA overnight, washed with PBS, and stained with streptavidin Alexa Fluor 488 (1:500, Cat# S32354, Thermo Fisher) and rabbit anti-MeCP2 (1:1000, Cat# 3456, Cell Signaling Technologies) to identify the MeCP2 status of the recorded neuron. 1–3 neurons were analyzed per mouse.
Statistical Analysis
Data are displayed as mean ± s.e.m. with a significance threshold of ⍺=0.05 (ns, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Sample sizes were determined based on prior statistics and data characterizing the phenotypes of Rett mice8,16. Statistical analysis was performed using Prism (Graphpad). Statistical significance was determined using a two-tailed, unpaired student’s t-test, one-way ANOVA with Tukey’s multiple comparisons test, or two-way ANOVA with Tukey’s multiple comparisons test. Data were analyzed with the experimenters blinded to genotype.
Extended Data
Extended Data Fig. 1 |. Presymptomatic motor training on the rotarod does not improve other behavioral deficits in Rett mice.

a, Motor performance on the rotarod. WT (n = 12) and Rett (n = 10) mice were tested over 4 days beginning at 8 weeks of age. b, Timeline for additional behavioral assays in rotarod-trained mice. c, Performance curves of rotarod-trained Rett naive (n = 19), Rett late-trained (n = 18), and Rett early-trained (n = 19) mice across training and test days. d-o, After rotarod testing at 24 weeks of age, WT naive (n = 12), Rett naive (n = 12), WT late-trained (n = 12), Rett late-trained (n = 11), WT early-trained (n = 11), Rett early-trained (n = 12) mice were tested on a variety of behavioral assays. Motor function was tested in the footslip (d) and open field assays (e). Spatial memory was tested in the Morris water maze (f-h). Social behavior was tested in the 3-chamber assay (i). Anxiety was tested in the elevated plus maze (j). Sensorimotor gating was tested in the acoustic startle (k) and pre-pulse inhibition assays (l). Contextual (m) and cued (n) memory were assessed in the fear conditioning assays. Body weight was measured (o). The sample size (n) corresponds to the number of biologically independent mice. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), **** (p<0.0001).
Extended Data Fig. 2 |. Presymptomatic memory training in the Morris water maze does not improve other behavioral deficits in Rett mice.

a-c, Spatial memory performance in the Morris water maze. WT (n = 8) and Rett (n = 8) mice were tested over 4 days beginning at 4 weeks of age (a). A probe trial on day 5 revealed the time spent in the target quadrant (b) and the number of platform area crossings (c). d, Performance curves of Morris water maze-trained Rett naive (n = 19), Rett late-trained (n = 19), and Rett early-trained (n = 18) mice across training and test days. e, Spatial memory in the Morris water maze over time in early-trained Rett mice (n = 7) and Rett mice (n = 8) that missed a training session. f, Swim speed in the Morris water maze of WT naive (n = 18), WT late-trained (n = 18), WT early-trained (n = 19), Rett naive (n = 19), Rett late-trained (n = 19), and Rett early-trained (n = 18) mice. g, Timeline for additional behavioral assays in Morris water maze-trained mice. h-o, After Morris water maze testing at 12 weeks of age, WT naive (n = 11), Rett naive (n = 12), WT late-trained (n = 11), Rett late-trained (n = 12), WT early-trained (n = 12), Rett early-trained (n = 11) mice were tested on a variety of behavioral assays. Motor function was tested in rotarod (h), footslip (i), and open field assays (j). Body weight was measured (k). Social behavior was tested in the 3-chamber assay (l). Anxiety was tested in the elevated plus maze (m). Sensorimotor gating was tested in the acoustic startle (n) and pre-pulse inhibition (o) assays. The sample size (n) corresponds to the number of biologically independent mice. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-tailed, unpaired student’s t-test (b-c) or two-way ANOVA with Tukey’s multiple comparisons test (a, d-o); ns (p>0.05), **** (p<0.0001).
Extended Data Fig. 3 |. FosTRAP labels task-specific neurons, including MeCP2+ and MeCP2− neurons in Rett mice.
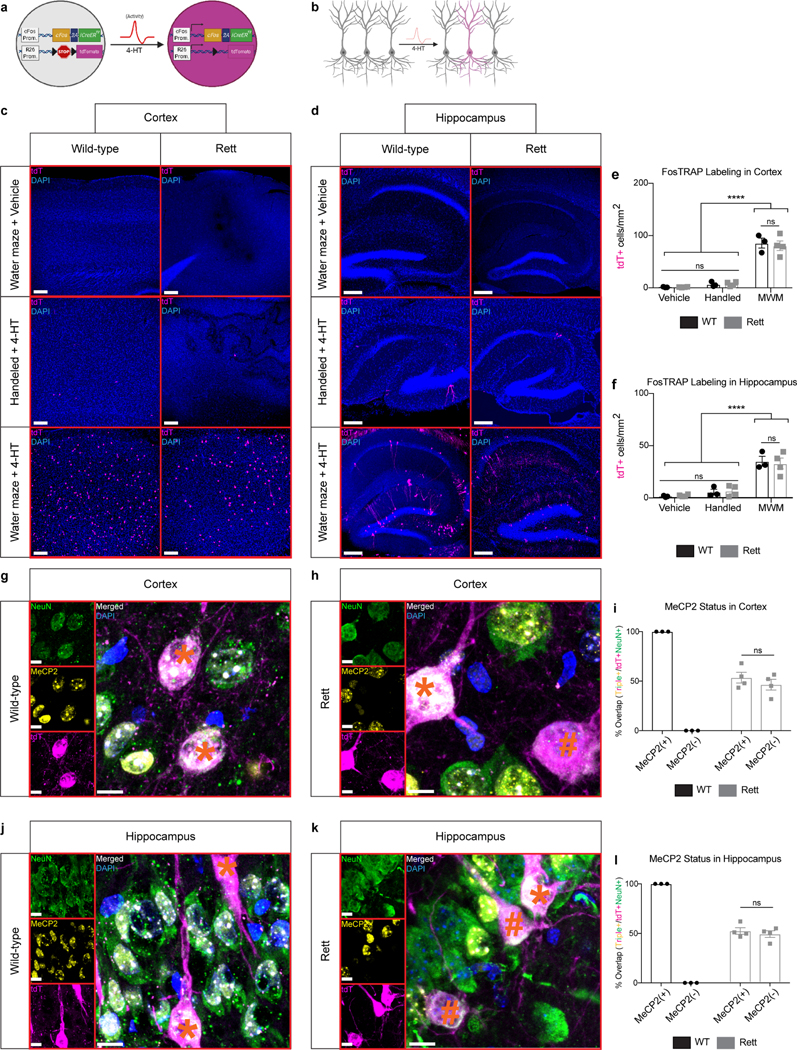
a, Schematic of FosTRAP. In the presence of 4-hydroxytamoxifen (4-HT), neural activity activates cFos expression and subsequent expression of a Cre-dependent reporter, tdTomato. b, FosTRAP labels a subset of neurons following 4-HT administration. c-f, FosTRAP labeling during the Morris water maze labels neurons in multiple brain regions. Labeled neurons in the cortex (c) and hippocampus (d) from 13-week-old mice that were trained in the Morris water maze and injected with vehicle or 4-HT as well as mice that were handled and injected with 4-HT. Quantification in the cortex (e) and hippocampus (f) of WT (n = 3) and Rett (n = 4) mice. Scale bar, 200 μm. g-l, FosTRAP labeling in MeCP2+ and MeCP2− neurons. FosTRAP labels NeuN+/MeCP2+ neurons in the cortex (g) and hippocampus (j) of WT mice. FosTRAP labels NeuN+/MeCP2+ and NeuN+/MeCP2− in the cortex (h) and hippocampus (k) of Rett mice. Quantification in the cortex (i) and hippocampus (l) of WT (n = 3) and Rett (n = 4) mice. (*) denotes a MeCP2+ neuron. (#) denotes a MeCP2− neuron. Scale bar, 20 μm. The sample size (n) corresponds to the number of biologically independent mice. 3–4 sections were analyzed per mouse. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), **** (p<0.0001).
Extended Data Fig. 4 |. Morris water maze-associated neurons are reactivated during the Morris water maze but not during fear conditioning.
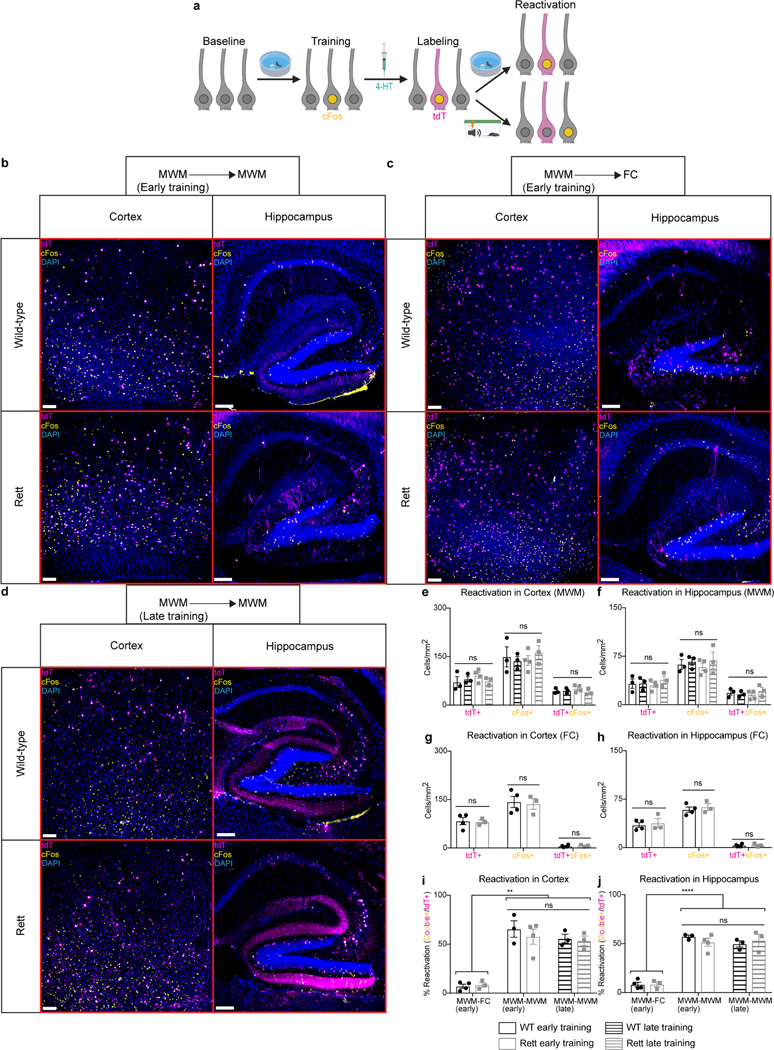
a, FosTRAP labeling of Morris water maze-associated neurons, with subsequent reactivation of task-specific neurons during retesting in the Morris water maze or fear conditioning assay. b-j, cFos reactivation in Morris water maze (MWM)-associated neurons of early-trained mice retested in the Morris water maze (b), early-trained mice tested in a novel fear conditioning (FC) assay (c), and late-trained mice retested in the Morris water maze (d) at 13 weeks of age. The number of tdTomato+ (tdT+), cFos+, and tdT+cFos+ neurons were quantified in the cortex (e) and hippocampus (f) of early-trained WT (n = 3) and Rett (n = 4) mice and late-trained WT (n = 3) and Rett (n = 3) mice after retesting in the Morris water maze. The number of tdTomato+ (tdT+), cFos+, and tdT+cFos+ neurons were quantified in the cortex (g) and hippocampus (h) of early-trained WT (n = 4) and Rett (n = 3) mice after fear conditioning. The reactivation percentage, defined as the percentage of tdT+ neurons that were also cFos+, were quantified in the cortex (i) and hippocampus (j) of Morris water maze-trained mice after retesting in the Morris water maze or the fear conditioning assay. Scale bar, 200 μm. The sample size (n) corresponds to the number of biologically independent mice. 3–4 sections were analyzed per mouse. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), ** (p<0.01), **** (p<0.0001).
Extended Data Fig. 5 |. Viral delivery of DREADD-containing AAVs labels task-specific neurons, including MeCP2+ and MeCP2− neurons in Rett mice.

a, P0 AAV delivery of Cre-dependent mCherry into cFosCreER mice that also contain a Cre-dependent GFP in the Rosa26 locus. Expression of mCherry in GFP+ neurons demonstrates that the AAVs are reactivated in task-specific neurons. b-c, Overlap between injected AAV (mCherry) and endogenous reporter (GFP) in early-trained mice at 13 weeks of age. Quantification of GFP+ cells, mCherry+ cells, and overlap in the cortex and hippocampus of WT (n = 3) and Rett (n = 4) mice (c). Scale bar, 200 μm. d-i, FosTRAP labeling in MeCP2+ and MeCP2− neurons from AAV-injected mice trained in the Morris water maze. FosTRAP labels MeCP2+ neurons in the cortex (d) and hippocampus (g) of WT mice. FosTRAP labels MeCP2+ and MeCP2− neurons in the cortex (e) and hippocampus (h) of Rett mice. Quantification in the cortex (f) and hippocampus (i) of WT (n = 3) and Rett (n = 4) mice. (*) denotes a MeCP2+ neuron. (#) denotes a MeCP2− neuron. Scale bar, 20 μm. The sample size (n) corresponds to the number of biologically independent mice. 3–4 sections were analyzed per mouse. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05).
Extended Data Fig. 6 |. CNO prevents reactivation of hM4Di-expressing neurons.
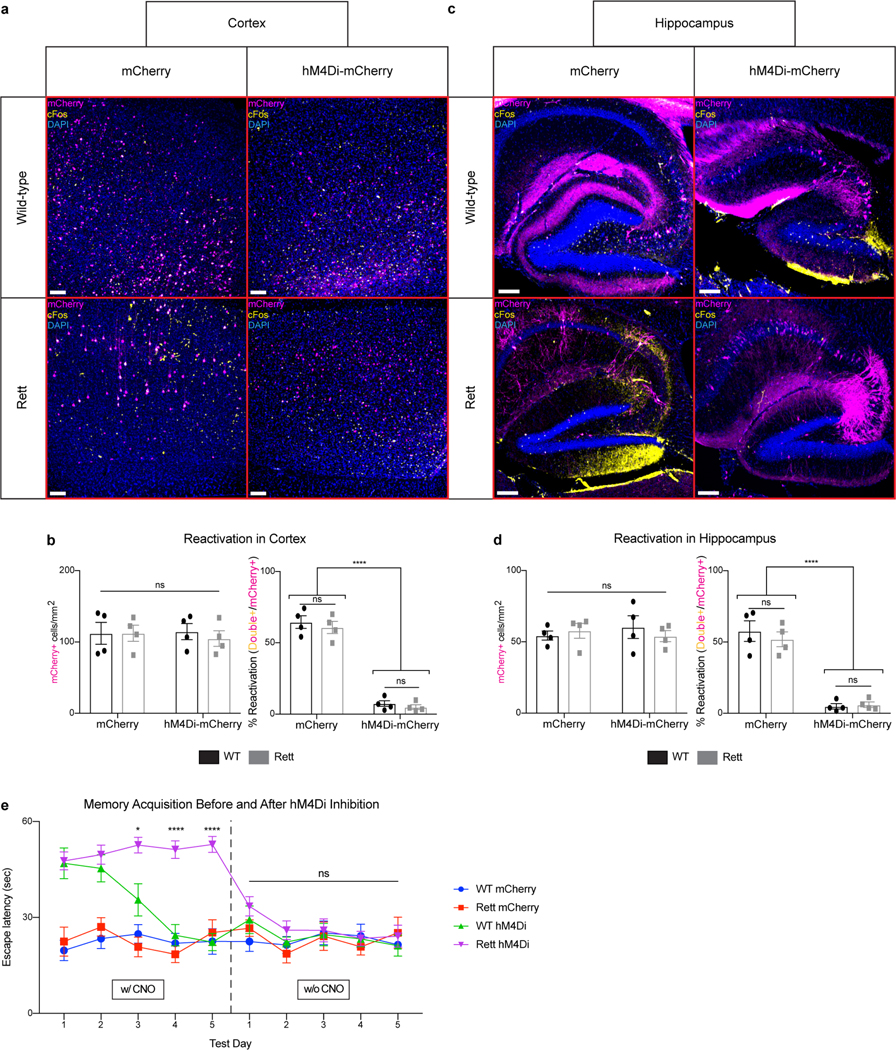
a-d, cFos reactivation in task-specific neurons expressing mCherry or hM4Di-mCherry. Overlap between cFos and mCherry in the cortex (a) and hippocampus (c) of mice injected with CNO during testing in the Morris water maze at 13 weeks of age. Quantification of mCherry+ cells and reactivation from cortex (b) and hippocampus (d) in WT (n = 4) and Rett (n = 4) mice. Scale bar, 200 μm. e, The effect of removing CNO on Morris water maze performance in WT mCherry (n = 6), Rett mCherry (n = 5), WT hM4Di (n = 5), and Rett hM4Di (n = 6) mice at 13 weeks of age. The sample size (n) corresponds to the number of biologically independent mice. 3–4 sections were analyzed per mouse. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), * (p<0.05), **** (p<0.0001).
Extended Data Fig. 7 |. CNO reactivates hM3Dq-expressing neurons.

a-d, cFos reactivation in task-specific neurons expressing mCherry or hM3Dq-mCherry. Overlap between cFos and mCherry in the cortex (a) and hippocampus (c) of mice injected with CNO in the homecage at 13 weeks of age. Quantification of mCherry+ cells and reactivation from cortex (b) and hippocampus (d) in WT (n = 4) and Rett (n = 4) mice. Scale bar, 200 μm. The sample size (n) corresponds to the number of biologically independent mice. 3–4 sections were analyzed per mouse. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), **** (p<0.0001).
Extended Data Fig. 8 |. Repeated activation of a random subset of neurons does not alter the performance of early-trained mice.

a, AAV injection paradigm to express DREADDs in a random subset of neurons and manipulate their activity with CNO. High-titer AAV is delivered to cFosCreER mice, which express Cre in task-specific neurons following 4-hydroxytamoxifen administration. Low-titer AAV is delivered to Camk2aCreER mice, which express Cre ubiquitously in excitatory neurons following 4-hydroxytamoxifen administration. cFosCreER and Camk2aCreER mice both express the same number of labeled neurons, but those in cFosCreER mice are task-specific. b-d, Overlap between injected AAV (mCherry) and endogenous reporter (GFP) in early-trained mice at 13 weeks of age. Quantification of mCherry+ cells in the cortex (c) and hippocampus (d) of WT (n = 3) and Rett (n = 4) mice expressing cFosCreER and WT (n = 4) and Rett (n = 4) expressing Camk2aCreER. Scale bar, 200 μm. e-f, cFos reactivation in Camk2aCreER mice expressing mCherry. Quantification of reactivation in the cortex and hippocampus of WT (n = 4) and Rett (n = 3) mice after Morris water maze training at 13 weeks of age (f). g-i, Morris water maze performance in early-trained WT and Rett mice expressing Camk2aCreER and hM4Di-mCherry or mCherry. WT mCherry (n = 7), Rett mCherry (n = 8), WT hM4Di (n = 8), and Rett hM4Di (n = 6) mice were tested at 13 weeks of age and injected with CNO during testing (g). A probe trial on day 6 measured the time spent in target quadrant (h) and the number of platform area crossings (i). j-k, Morris water maze performance in early-trained WT and Rett mice expressing Camk2aCreER and hM3Dq-mCherry or mCherry. A probe trial on WT mCherry (n = 7), Rett mCherry (n = 8), WT hM3Dq (n = 7), and Rett hM3Dq (n = 7) measured the time spent in the target quadrant (j) and the number of platform area crossings (k). The sample size (n) corresponds to the number of biologically independent mice. Data are represented as mean ± s.e.m. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test; ns (p>0.05).
Extended Data Fig. 9 |. Morphological and electrophysiological benefits are evident in task-specific neurons after presymptomatic training.

a, P0 AAV delivery of YFP to assess the morphology of neurons that are not task-specific (tdT-). b-e, Morphological analysis of MeCP2− hippocampal CA1 neurons that are task-specific (tdT+) and not task-specific (tdT-/YFP+) in trained Rett mice at 13 weeks of age. Sholl analysis (b), spine density (c), soma area (d), and nuclear area (e) were measured in MeCP2− neurons of late-trained (n = 5) and early-trained (n = 5) Rett mice. f-j, Electrophysiological recordings of MeCP2− CA1 neurons in late- and early-trained Rett mice at 13 weeks of age. f, Representative image of neurons that are task-specific (magenta) and not task-specific (no magenta), both of which were injected with biocytin (green) during recording and immunostained to determine the MeCP2 status (yellow). Frequency (g) and amplitude (h) of sIPSCs and frequency (i) and amplitude (j) of sEPSCs were measured in MeCP2− neurons from late-trained (n = 10) and early-trained (n = 10) Rett mice. The sample size (n) corresponds to the number of biologically independent mice. For b-c, 10–15 neurons were analyzed per mouse. For d-e, 50–100 neurons were analyzed per mouse. For g-j, 1–3 neurons were analyzed per mouse. Data are represented as mean ± s.e.m. Statistical significance was determined using a one-way (b) or two-way (c-e, g-j) ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), ** (p<0.01), **** (p<0.0001).
Extended Data Fig. 10 |. Presymptomatic training improves dendritic morphology of task-specific hippocampal granule and cortical neurons in Rett mice.

a-d, Morphological analysis of task-specific hippocampal granule neurons in late- and early-trained WT and Rett mice at 13 weeks of age. Sholl analysis (a), spine density (b), soma area (c), and nuclear area (d) were measured in neurons of WT late-trained (n = 5), WT early-trained (n = 5), Rett late-trained MeCP2− (n = 5), Rett early-trained MeCP2+ (n = 5), and Rett early-trained MeCP2− (n = 5) neurons. e-h, Morphological analysis of layer 5 cortical task-specific neurons in late- and early-trained WT and Rett mice. Sholl analysis (e), spine density (f), soma area (g), and nuclear area (h) were measured in neurons of WT late-trained (n = 5), WT early-trained (n = 5), Rett late-trained MeCP2+ (n = 5), Rett late-trained MeCP2− (n = 5), Rett early-trained MeCP2+ (n = 5), and Rett early-trained MeCP2− (n = 5) neurons. The sample size (n) corresponds to the number of biologically independent mice. For a-b and e-g, 10–15 neurons were analyzed per mouse. For c-d and g-h, 50–100 neurons were analyzed per mouse. Data are represented as mean ± s.e.m. Statistical significance was determined using a one-way (b-d, f-h) and two-way (a, e) ANOVA with Tukey’s multiple comparisons test; ns (p>0.05), * (p<0.05), ** (p<0.01), *** (p<0.001), **** (p<0.0001).
Supplementary Material
Acknowledgments
We thank the Neurovisualization and Neurobehavioral Cores at the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital and the BCM-IDDRC (U.S. NIH Grant U54HD083092). This project was supported by NIH/NINDS 5R01NS057819–13 (H.Y.Z.), NIH/NICHD F30HD097871–01 (N.P.A.), and the Henry Engel Fund. We thank the Baylor College of Medicine Center for Comparative Medicine for mouse colony management, Surabi Veeraragavan for mouse behavior assistance, Dinghui Yu for microscopy assistance, and the Baylor College of Medicine Gene Vector core for AAV production. We thank members of the Zoghbi lab for discussions and comments on the manuscript, and Vicky Brandt and Callison Alcott for helpful editorial input. Figure diagrams were created with BioRender.com.
Footnotes
Data Availability
No datasets were generated or analyzed during the current study. Source data for all figures are provided with the paper.
Competing interests The authors declare no competing interests.
Additional information
Supplementary information is available for this paper.
Reprints and permission information is available at www.nature.com/reprints.
References
- 1.Amir RE et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23, 185–188 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Hagburg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol 14, 471–479 (1983). [DOI] [PubMed] [Google Scholar]
- 3.Sandweiss AJ, Brandt VL, Zoghbi HY Advances in understanding of Rett syndrome and MECP2 duplication syndrome: prospects for future therapies. Lancet Neurol 19, 689–698 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Laurvick CL et al. Rett syndrome in Australia: a review of the epidemiology. J Pediatr 148, 347–353 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Neul JL et al. Developmental delay in Rett syndrome: data from the natural history study. J Neurodevelop Disord 6, 20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27, 322–326 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Katz DM et al. Preclinical research in Rett syndrome: setting the foundation for translational success. Dis Model Mech 5, 733–745 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samaco RC et al. Female Mecp2(+/−) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum Mol Genet 22, 96–109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garg SK et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci 33, 13612–13620 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hocquemiller M, Giersch L, Audrain M, Parker S, Cartier N. Adeno-associated virus-based gene therapy for CNS diseases. Hum Gene Ther 27, 478–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clarke AJ, Abdala Sheikh AP A perspective on a “cure” for Rett syndrome. Orphanet J Rare Dis 13, 44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Esch H. et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 77, 442–453 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins AL et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 13, 2679–2689 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Braunschweig D, Simcox T, Samaco RC, LaSalle JM X-chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet 13, 1275–1286 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Hao S. et al. Forniceal deep brain stimulation rescues hippocampal memory in Rett mice. Nature 526, 430–434 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu H. et al. Loss and gain of MeCP2 cause similar hippocampal circuit dysfunction that is rescued by deep brain stimulation in a Rett syndrome mouse model. Neuron 91, 739–747 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lozano AM et al. Deep brain stimulation: current challenges and future directions. Nat Rev Neurol 15:148–160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dawson G. et al. Randomized, controlled trial of an intervention for toddlers with autism: the Early Start Denver Model. Pediatrics 125, e17–23 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaevtiz LR, Gómez NB, Zhen DP, Berger-Sweeney JE MeCP2 R168X male and female mutant mice exhibit Rett-like behavioral deficits. Gene Brain Behav 12, 732–740 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Deacon RM Measuring motor coordination in mice. J Vis Exp 75, e2609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kee SE, Mou X, Zoghbi HY, Ji D. Impaired spatial memory codes in a mouse model of Rett syndrome. Elife 7, e31451 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11, 47–60 (1984). [DOI] [PubMed] [Google Scholar]
- 24.Vorhees CV, Williams MT Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1, 848–858 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chowdhury A, Caroni P. Time units for learning involve maintenance of system-wide cFos expression in neuronal assemblies. Nat Commun 9, 4122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallo FT, Katche C, Morici JF, Medina JH, Weisstaub NV Immediate early genes, memory, and psychiatric disorders: focus on c-Fos, Egr1 and Arc. Front Behav Neurosci 12, 79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attardo A. et al. Long-term consolidation of ensemble neural plasticity patterns in hippocampal area CA1. Cell Rep 25, 640–650.e2 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Lau BYB, Krishan K, Haung J, Shea SD Maternal experience-dependent cortical plasticity in mice is circuit- and stimulus-specific and requires MECP2. J Neurosci 40, 1514–1526 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guenthner CJ Miyamichi K, Yang HH, Heller HC, Luo L. Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 78, 773–784 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeNardo LA et al. Temporal evolution of cortical ensembles promoting remote memory retrieval. Nat Neurosci 22, 460–469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunsaker MR, Kesner RP Evaluating the different roles of the dorsal dentate gyrus, dorsal CA3, and dorsal CA1 during temporal ordering for spatial locations task. Hippocampus 18, 955–964 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guise KG, Shapiro ML Medial prefrontal cortex reduces memory interference by modifying hippocampal encoding. Neuron 94, 183–192.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexander GM et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roth BL DREADDs for neuroscientists. Neuron 89, 683–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JY et al. Viral transduction of the neonatal brain delivers controllable genetic mosaicism for visualizing and manipulating neuronal circuits in vivo. Eur J Neurosci 37, 1203–1220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rietveld L, Stuss DP, McPhee D, Delaney KR Genotype-specific effects of Mecp2 loss-of-function on morphology of layer V pyramidal neurons in heterozygous female Rett syndrome model mice. Front Cell Neurosci 9, 145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Connolly DR, Zhou Z. Genomic insights into MeCP2 function: a role for the maintenance of chromatin architecture. Curr Opin Neurobiol 59, 174–179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linhoff MW, Garg SK, Mandel G. A high-resolution imaging approach to investigate chromatin architecture in complex tissues. Cell 163, 246–255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Downs J. et al. Environmental enrichment intervention in Rett syndrome: an individually randomised stepped wedge trial. Orphanet J Rare Dis 13, 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryan TJ, Roy DS, Pignatelli M, Arons A, Tonegawa S. Engram cells retain memory under retrograde amnesia. Science 348, 1007–1013 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pignatelli M, et al. Engram cell excitability state determines the efficacy of memory retrieval. Neuron 101, 274–284.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Finkel RS et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Kayton A. Newborn screening: a literature review. Neonatal Netw 26, 85–95 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Valente EM, Ferraris A, Dallapiccola B. Genetic testing for paediatric neurological disorders. Lancet Neurol 7, 1113–1126 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Pitt JJ Newborn screening. Clin Biochem Rev 31, 57–68 (2010). [PMC free article] [PubMed] [Google Scholar]
- 46.Landa RJ Efficacy of early interventions for infants and young children with, and at risk for, autism spectrum disorders. Int Rev Psychiatry 30, 25–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiurazzi P, Pirozzi F. Advances in understanding – genetic basis of intellectual disability. F1000Res 5, F1000 Faculty Rev-599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kroon T, Sierksma MC, Meredith RM Investigating mechanisms underlying neurodevelopmental phenotypes of autistic and intellectual disability disorders: a perspective. Front Syst Neurosci 7, 75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu HC et al. Disruption of the ATXN1-CIC complex causes a spectrum of neurobehavioral phenotypes in mice and humans. Nat Genet 49, 527–536 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schindelin J. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dickstein DL et al. Automatic dendritic spine quantification from confocal data with Neurolucida 360. Curr Protoc Neurosci 77,1.27.1–1.27.21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.