

Abstract
The assumption in the twin model that genotypic and environmental variables are uncorrelated is primarily made to ensure parameter identification, not because researchers necessarily think that these variables are uncorrelated. Although the biasing effects of such correlations are well understood, a method to estimate these parameters in the twin model would be useful. Here we explore the possibility of relaxing this assumption by adding polygenic scores to the (univariate) twin model. We demonstrate that this extension renders the additive genetic (A)—common environmental (C) covariance (σAC) identified. We study the statistical power to reject σAC = 0 in the ACE model and present the results of simulations.
Supplementary Information
The online version of this article (10.1007/s10519-020-10035-7) contains supplementary material, which is available to authorized users.
Keywords: Classical twin design, Polygenic risk scores, A–C covariance, Identification, Statistical power
Introduction
The classical twin design (CTD; Eaves et al. 1978; Jinks and Fulker 1970) has been one of the most productive genetically informative designs in the study of human traits (Polderman et al. 2015). Twin studies have contributed greatly to our knowledge concerning genetic and environmental contributions to individual differences in psychological and medical traits, disease phenotypes and 'omics' variables (van Dongen et al. 2012). Multivariate and longitudinal extensions of the CTD have provided insights into the etiology of comorbidity and stability of traits and disorders. It is well understood that the correct interpretation of results based on the CTD depend on the tenability of the model assumptions (Eaves et al. 1977; Jinks and Fulker 1970; Plomin et al. 2016). The main assumptions of the CTD concern genotype-environment covariance (assumed to be absent), genotype-environment interaction (assumed to be absent), the equal environment assumption (environment does not cause larger resemblance in MZ than in DZ twins), and parental mating (assumed to be random, or that parental resemblance is due to social homogamy rather than phenotypic assortment). Given these assumptions, the results from the CTD can provide unbiased estimates of additive genetic (A), unshared environmental (E), and common environmental (C) and dominance (D) variance components. The effect of violations of these assumptions are well understood (Verhulst and Hatemi 2013; Purcell 2002; Keller et al. 2010), so that estimates of variance components obtained in the twin model may be interpreted in the light of possible model violations.
Many papers have been devoted to the detection and accommodation of model violations, either within the CTD (e.g., Purcell 2002; Molenaar et al. 2012; Eaves and Erkanli 2003; Carey 1986; Dolan et al. 2014; Beam and Turkheimer 2013), or in extended designs (e.g., Plomin et al. 1985; Narusyte et al. 2008; Neale and Fulker 1984; Fulker 1988; D’Onofrio et al. 2003; Keller et al. 2009; Heath et al. 1985; Maes et al. 2006). The aim of the present paper is to demonstrate that the incorporation of polygenic risk scores (PRSs) in the classical twin design allows one to estimate the covariance between A and C (σAC). In the study of childhood intelligence, σAC > 0 is considered plausible, stemming from a process of cultural transmission (Keller et al. 2009; Fulker 1988), which gives rise to passive genotype-environment covariance in children (Plomin et al. 1977; Scarr and McCartney 1983, Knafo and Jaffee 2013; Kendler 2011; Rutter and Silberg 2002).
Measured genetic variables have been incorporated in genetically informative designs with various aims, apart from the gene finding of traditional linkage or combined linkage-association analysis (e.g., Fulker et al. 1999; Neale 2000). For instance, van den Oord and Snieder (2002) presented an extended twin model with measured genetic variables to test association in the presence of population stratification and to test causal relationships. Neale et al. (2000) partitioned variation in serum APOE levels into that associated with the APOE locus and residual genetic variance. In a study of attention problems, van Beijsterveldt et al. (2011) incorporated measured candidate gene information on SNPs in the serotonergic, dopaminergic system and the BDNF gene. The effect of SNPs was tested on a latent factor that summarized multiple assessments of attention problems across childhood. Minică et al. (2018) presented an integration of the CTD and Mendelian randomization method, in which PRSs feature as genetic instruments.
The use of measured genetic information specifically to study genotype-environment covariance is relatively new. Bates et al. (2018) and Kong et al. (2018) proposed the use of polygenic scores based on transmitted and non-transmitted alleles from parents to offspring to detect the effects of non-transmitted alleles on phenotype outcomes in their children. Warrington et al. (2018) used structural equation modeling to determine the fetal and maternal effects of measured genetic variants on birthweight, thus revealing genotype-environment covariance. Cheesman et al. (2020) applied PRSs in an adoption design to detect (passive) gene-environment correlation in educational attainment. Selzam et al. (2019) used PRSs measured in DZ twin pairs to demonstrate the presence of gene-environment correlation for cognitive abilities, and the mediating role of social economic status therein. Wertz et al. (2018) used PRSs in a parent and offspring design to demonstrate the gene-environment correlation originating in parental behavior.
The present aim is to incorporate PRSs in the twin design with the aim of estimating A–C covariance. This approach allows us to determine the presence of A–C covariance, but sheds no light on the process that gave rise to the A–C covariance. For instance, the A–C covariance may be due to active (e.g., niche picking), passive (e.g., cultural transmission) or evocative processes (Plomin et al. 1977). The outline of this paper is as follows. First, we present the classical twin model, and the model extended with PRSs. Second, given the model for PRSs in MZ and DZ twins, we address the issues of identification and statistical power. Third, we present the results of a small simulation to determine the effects of using estimated weights in calculating PRSs (i.e., the standard procedure) in comparison to exact known weights.
The Twin Model with Polygenic Risk Scores: A–C Covariance
Let Ph denote the phenotype of interest, and let GVk denote the k-th genetic variant (GV) contributing to the variance of Ph, where k = 1…K, and K is the number of GVs. We limit our presentation to diallelic GVs (e.g., SNPs) with additive effects (additively coded, e.g., 0, 1, or 2). The phenotype Ph is modeled as follows:
where b0 is the intercept, bk is the k-th regression coefficient, subscript i denotes person, and E and C represent unshared and shared environmental factor scores of individual i. In the classical twin model, under the assumptions mentioned in the introduction, the variance of Ph is decomposed, in the ACE model, into the components σ2A, σ2C, and σ2E:
The additive genetic variance equals the sum of the contributions of the individual GVs and their covariances (σGVk,GVl) attributable to linkage disequilibrium:
Given σAC ≠ 0, we have (assuming no AE covariance)
where σAC is the covariance of A and C. In this model, the parameter σAC is not identified. If we assume σAC = 0, while in truth σAC > 0, the variance σ2C is biased in the twin model, as σAC acts as C, thus inflating the estimate of σ2C in the standard ACE model (see Purcell 2002; Verhulst and Hatemi 2013). If in truth, σAC < 0, the MZ and DZ twin correlations suggest the presence of dominance variance (D).
Given estimates of the regression coefficients (bk) obtained in independent genome-wide association studies (GWASs), the PRS can be calculated GVli (Purcell et al. 2009; Evans et al. 2009; Dudbridge 2013), where the set of L GVs is a subset of the K GVs. The set of L GVs may be chosen on the basis of the p value of the individual GVs or other considerations. Let the PRS equal p*Ap, with variance p2*σ2Ap. The scaling parameter p accommodates the fact that the phenotype Ph and the PRS are not measured on the same scale. Let Aq denote the residual additive genetic variable. The model is now:
The variance decomposition of the additive genetic variable A and the phenotype Ph are:
where σAC = σApC + σAqC. The parameters σApC and σAqC are the covariances of Ap and C and of Aq and C, respectively. The parameter σApAq is the covariance of the additive variables Ap and Aq. We parameterize the covariance terms σApC and σAqC as a function of the single covariance term σAC as follows. We derive the coefficient γp by tracing from C to A (C ↔ A with coefficient σAC), and then from A to Ap (A → Ap) where γp is the regression coefficient in the regression of Ap on A. We do the same with Aq using γq. The path diagram is shown in Fig. 1.
Fig. 1.

The covariance between C and Ap and Aq are derived as σACγp and σACγq, respectively, where γp = σ2Ap/σ2A and γq = σ2Aq/σ2A
We thus obtain the constraints:
where
| 1 |
| 2 |
Note that σAC = σApC + σAqC, as γ1 + γ2 = 1. At this point two comments are in order. First, it is not possible to estimate both the scaling parameter p and the parameter σApAq. We therefore set σApAq to equal zero. Given this identifying constraint, γp = σ2Ap/σ2A and γq = σ2Aq/σ2A. We demonstrate below that the constraint σApAq = 0 has no bearing on the likelihood ratio test of σAC = 0, or on the maximum likelihood estimates of σAC, σ2A, and σ2C. Second, we recognize that if σAC ≠ 0, the PRS weights (bl) obtained in meta-analyses of the results of independent GWASs, which are used to calculate the PRS, will be upwardly (downwardly) biased given σAC > 0 (σAC < 0). This raises the question of whether this has any effect on the estimate of σAC. We address this question below. The model is depicted in Fig. 2. We have parameterized this model in terms of variance components, i.e., we fixed the path coefficients terminating in the phenotype to one, and estimated the variance components (σ2Ap, σ2Aq, σ2C, σ2E, along with the parameter p). One may also consider fixing the variance components to one, and estimating the path coefficients. However, this parameterization may complicate statistical tests of the variance components (see Verhulst et al. 2019, for details).
Fig. 2.

ACE twin model with PRSs, including A–C covariances σApC and σAqC (dashed double headed arrows). This is the model in DZ twins (i.e., rz = 0.5). The covariance between Ap and Aq is fixed to zero, but, as demonstrated in the text, this has no bearing on the derived estimate of the total A, C covarianc (σA,C)
Simulation I: Power
In this model, the observed statistics are the 3 × 3 MZ (PRS, phenotype twin 1, phenotype twin 2) and the 4 × 4 DZ (PRS1, PRS2, phenotype twin 1, phenotype twin 2) covariance matrices (ΣMZ and ΣDZ, respectively), and the 3- and 4-dimensional mean vectors. The MZ covariance matrix ΣMZ is 3 × 3, as MZ twins, being genetically identical, have identical polygenic scores. We do not consider the mean structure of the phenotype data beyond noting that we adopt the standard (testable) assumptions that the means are equal over twins within a pair and over zygosity. Let the vector θ contain the six parameters of the covariance structure model:
The vector θ does not include σApC and σAqC explicitly, because these parameters depend on the parameters σ2Ap, σ2Aq, and the total covariance σAC, as shown above. We evaluated local identification numerically using the OpenMx function mxCheckIdentification (written by Michael Hunter). The model is locally identified if the Jacobian matrix, J(θ), is full column rank (Bekker et al. 1994). The Jacobian matrix contains the first-order derivatives of the non-redundant elements in the matrices ΣMZ(θ) and ΣDZ(θ) with respect to the parameters in θ. Given 6 + 10 elements in ΣMZ(θ) and ΣDZ(θ), and 6 parameters J(θ) is a 16 × 6 matrix. The mxCheckIdentification function is convenient as it does the necessary calculations automatically, and can be applied directly to the OpenMx script that one uses to fit the model.
Having established local identification, we proceeded to address the question of resolution by considering the statistical power to reject σAC = 0 given various parameter settings. We used exact data simulation (van der Sluis, et al. 2008), which is equivalent to analyzing the expected (true) covariance matrices. We used normal theory maximum likelihood estimation throughout, and based our power calculations on the non-centrality parameter (NCP) associated with the (non-central) chi-square distribution (Martin et al. 1978). Given exact data simulation, the NCP equals the loglikelihood ratio (LLR) test of σAC = 0. We used the OpenMx library (Boker et al. 2011; Neale et al. 2016) in the R program (R Core Team 2018), and we used R for data simulation, and power calculations. In the power analyses, we set the MZ and DZ sample sizes equal (Nmz = 1000, Ndz = 1000), and we report the power given Nmz = Ndz = 1000, and the required sample sizes to achieve a power of 0.80, given α = 0.05.
Results I
The numerical check of model identification demonstrated that the model is locally identified, bearing in mind that we have set σApAq = 0. That is, given the 3 × 3 MZ and the 4 × 4 DZ phenotypic covariance matrices, ΣMZ(θ) and ΣDZ(θ), we can obtain unique estimates of the six parameters p, σ2Ap, σ2Aq, σ2C, σ2E, and σAC. From the perspective of the path model (Fig. 2), the key to the identification is the covariance between the phenotype and the PRS, which does not depend on zygosity. This covariance equals p*σAp2 + p*σApC or p*γ1*(σA2 + σAC), where p, γ1 and σA2 are identified based on the phenotypic and PRS MZ and DZ twin covariances. Table 1 contains the results of the power study. This table includes the 16 parameter settings and the power to reject σAC = 0.
Table 1.
Statistical power to reject the null hypothesis that A–C covariance is zero (alpha = 0.05)
| prPRS | prPh | σ2A | σ2C | σ2E | σAC | rAC | σ2Ph | pr2σAC | Power | N (0.80) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.2 | 0.054 | 0.3 (0.27) | 0.2 (0.18) | 0.5 (0.45) | 0.048 | 0.2 | 1.09 | 0.088 | 0.216 | 11,420 |
| 2 | 0.2 | 0.052 | 0.3 (0.26) | 0.2 (0.17) | 0.5 (0.43) | 0.073 | 0.3 | 1.14 | 0.128 | 0.415 | 5158 |
| 3 | 0.2 | 0.053 | 0.3 (0.27) | 0.3 (0.26) | 0.4 (0.36) | 0.060 | 0.2 | 1.12 | 0.107 | 0.323 | 6971 |
| 4 | 0.2 | 0.050 | 0.3 (0.25) | 0.3 (0.25) | 0.4 (0.34) | 0.090 | 0.3 | 1.18 | 0.153 | 0.606 | 3161 |
| 5 | 0.4 | 0.109 | 0.3 (0.27) | 0.2 (0.18) | 0.5 (0.45) | 0.048 | 0.2 | 1.09 | 0.088 | 0.402 | 5352 |
| 6 | 0.4 | 0.104 | 0.3 (0.26) | 0.2 (0.17) | 0.5 (0.43) | 0.073 | 0.3 | 1.14 | 0.128 | 0.724 | 2408 |
| 7 | 0.4 | 0.107 | 0.3 (0.27) | 0.3 (0.26) | 0.4 (0.36) | 0.060 | 0.2 | 1.12 | 0.107 | 0.595 | 3241 |
| 8 | 0.4 | 0.101 | 0.3 (0.25) | 0.3 (0.25) | 0.4 (0.34) | 0.090 | 0.3 | 1.18 | 0.153 | 0.906 | 1462 |
| 9 | 0.2 | 0.088 | 0.5 (0.44) | 0.2 (0.18) | 0.3 (0.27) | 0.063 | 0.2 | 1.12 | 0.112 | 0.238 | 10,107 |
| 10 | 0.2 | 0.084 | 0.5 (0.42) | 0.2 (0.17) | 0.3 (0.25) | 0.094 | 0.3 | 1.18 | 0.159 | 0.455 | 4594 |
| 11 | 0.2 | 0.086 | 0.5 (0.43) | 0.3 (0.26) | 0.2 (0.17) | 0.077 | 0.2 | 1.15 | 0.134 | 0.362 | 6084 |
| 12 | 0.2 | 0.081 | 0.5 (0.40) | 0.3 (0.24) | 0.2 (0.16) | 0.116 | 0.3 | 1.23 | 0.189 | 0.661 | 2784 |
| 13 | 0.4 | 0.177 | 0.5 (0.44) | 0.2 (0.18) | 0.3 (0.26) | 0.063 | 0.2 | 1.12 | 0.112 | 0.465 | 4481 |
| 14 | 0.4 | 0.168 | 0.5 (0.42) | 0.2 (0.17) | 0.3 (0.25) | 0.094 | 0.3 | 1.18 | 0.158 | 0.794 | 2028 |
| 15 | 0.4 | 0.173 | 0.5 (0.43) | 0.3 (0.26) | 0.2 (0.17) | 0.077 | 0.2 | 1.15 | 0.133 | 0.682 | 2650 |
| 16 | 0.4 | 0.162 | 0.5 (0.41) | 0.3 (0.24) | 0.2 (0.16) | 0.116 | 0.3 | 1.23 | 0.188 | 0.950 | 1206 |
Given σ2A = σ2Ap + σ2Aq, prPRS equals σ2Ap/σ2A, i.e., the proportion of additive genetic variance attributable to the PRS, and prPh is the proportion of phenotypic variance attributable to the PRS, σ2Ap/σ2Ph; rAC and σAC are the correlation and covariance of A and C, σ2Ph is the phenotypic variance; pr2*σAC is the proportion of phenotypic variance due to 2*σAC
The standardized A, C, E variance components are given in parentheses. For instance, in setting 16, the raw variance is 0.5 + 0.3 + 0.2 + 0.116*2 = 1.23, and the standardized variance is 0.41 + 0.24 + 0.16 + 0.188 = ~ 1
The power is given for Nmz = Ndz = 1000, given α = 0.05; N(0.80) is the sample size (N = Nmz + Ndz, where Nmz = Ndz) associated with a power of 0.80, given α = 0.05
Table 1 contains the proportions of genetic and phenotypic variance explained by the PRS (prPRS and prPh in Table 1). These range from 0.2 to 0.4 (prPRS) and 0.050 to 0.177 (prPh). The correlation between A and C (rAC) was chosen to equal 0.2 or 0.3. In addition to this correlation, we express the σAC effect size as the proportion (2σAC)/σ2Ph (i.e., pr2σAC in Table 1), where σ2Ph = σ2A + σ2C + σ2E + 2σAC. This proportion ranges from 0.088 to 0.189. The proportion of A variance is ~ 0.26 or ~ 0.40; the proportion of C variance is ~ 0.18 or ~ 0.25, and the proportion of E variance varies between 0.09 and 0.44). The settings, which are limited, were chosen merely to identify some circumstance in which the power to reject σAC = 0 is acceptable, given the present sample sizes.
The greatest power is obtained in settings 8, 14, and 16: 0.906 (8), 0.793 (14), and 0.950 (16). Here, the PRS accounts for 10.1% (8), 16.8% (14), and 16.2% (16) of the phenotypic variance, and 2σAC accounts for 15.3% (8), 15.9% (14), and 18.9% (16) of the phenotypic variance. We see the lowest power given settings 1 (0.216) and 9 (0.238). Unsurprisingly, these are associated with relative low values of pr2σAC (8.8% and 11.2%) and prPh (5.4% and 8.8%). The relative contributions of pr2σAC and prPh to the power are apparent in the correlations of these with the power: 0.71 and 0.53, respectively. Regressing the power on these parameters, we found that they explain 65% of the variance in power (β pr2σAC: 0.625 and β prPh 0.400). Both are important, but the contribution of pr2σAc to the power is greater. The comparison of the σ2A = 0.3 and the σ2A = 0.5 conditions show that the magnitude of σ2A does not greatly influence the power. In terms the ratio of the N (power = 0.80) are about 1.1–1.2 (favoring the σ2A = 0.5 conditions). Finally, to determine the effect of the sign of σAC, we repeated the power analysis with rAC set to equal − 0.2 or − 0.3, and all other parameters unchanged. Figure 3 displays the plot of the power given positive and negative rAC. We note that the difference in power is small suggesting that the sign of rAC is unimportant in calculating power.
Fig. 3.
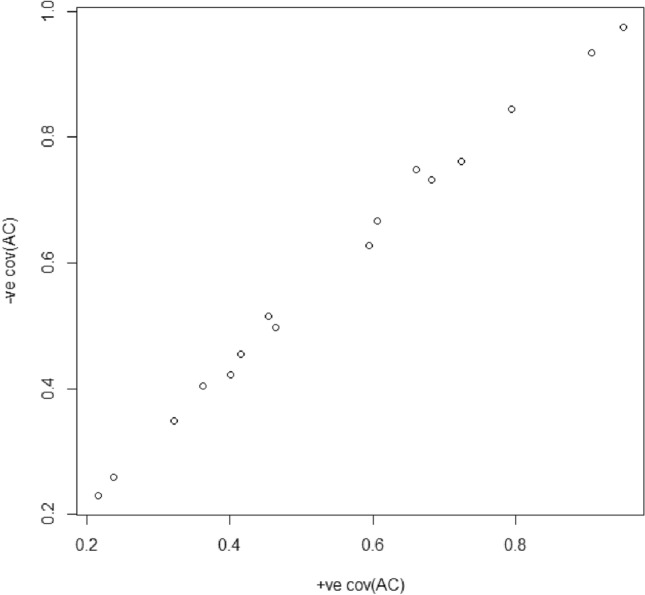
Power of the LLR test to reject σAC = 0 given positive and negative σAC. The parameter settings are given in Table 1. The only difference is the sign of σAC. The power to reject σAC = 0 given positive σAC is given in Table 1
Simulation II: Bias and Type I Error Rate
As noted above, the weights (bl) used to calculate the PRS are expected be biased upwards if σAC > 0. We investigated the effects of this in a simulation study. Specifically, we simulated data according to the nuclear twin family (NTF) design (Fulker 1988; Keller et al. 2009), which comprises MZ and DZ twins and their parents. This model includes cultural transmission, i.e., the direct contribution of the parental phenotype to the twin's environment (parameter m in the notation of Keller et al. 2009 notation). This contribution gives rise to a shared environmental variable (F in the notation of Keller et al. 2009), and to covariance between this variable (F) and the additive genetic factor (A), σAF. In addition to the shared variance due to F, the model includes a shared environmental variance term, due to shared influences other than cultural transmission. We denote this C* to distinguish it from the C in the standard ACE model. The original NTF model accommodates phenotypic assortative mating. However, here we assume that mating is random. The MZ and DZ expected phenotypic covariance matrices are shown in Table 2. It is not possible to resolve F and C* in the absence of parental phenotypes. Thus, in the standard ACE model and in the model with PRSs, we estimate a single shared environmental variance, σ2C which actually equals σ2C* + σ2F.
Table 2.
The expected covariance matrices in simulations 1–3
| MZ 1 | MZ 2 | |
|---|---|---|
| MZ 1 | σ2A + σ2C* + σ2F + 2σAF + σ2E | σ2A + σ2C* + σ2F + 2σAF |
| MZ 2 | σ2A + σ2C* + σ2F + 2σAF | σ2A + σ2C* + σ2F + 2σAF + σ2E |
| DZ 1 | DZ 2 | |
| DZ 1 | σ2A + σ2C* + σ2F + 2σAF + σ2E | ½σ2A + σ2C* + σ2F + 2σAF |
| DZ 2 | ½σ2A + σ2C* + σ2F + 2σAF | σ2A + σ2C* + σ2F + 2σAF + σ2E |
| σF2 = 2m2(σ2A + σ2C* + σ2F + 2σAF+ σ2E) = 2m2σ2Ph | ||
| σAF = (mσA)/(1−σF m) | ||
Parameter m is the regression coefficient in regression of parental phenotype on F in twins (shared environmental factor attributable to cultural transmission)
σ2F is shared environmental variance due to cultural transmission (see Keller et al. 2009, for the derivation)
σ2C* is shared environmental variance, not due to cultural transmission
σ2E is unshared environmental variance
σ2A additive genetic variance
σAF covariance of A and F (see Keller et al. 2009, for the derivation)
Note in fitting the model we estimate σ2C (i.e., σ2F + σ2C*)
First, we considered the model without cultural transmission, i.e., cultural transmission parameter (m) was zero. This implies that σ2F and σAF are zero, as the parameter m is the source of the A–C covariance. However, we included σ2C* > 0. Thus, there is shared (by the twins) environmental variance, but it is not due to cultural transmission. This model allows us to determine the Type I error rate associated with the test of σAC = 0. As an aside, in this simulation, the inclusion of σ2C* > 0 also allows us to establish the power to detect C variance in a twin model with PRS, in addition to checking the Type I error rate in test of σAC = 0. Second, we considered a model with cultural transmission, with m > 0, so that σAF > 0 and σ2F > 0. In this model we set σ2C* to zero, so that there are no shared environmental influences other than those stemming from the cultural transmission. Third, we considered a model with cultural transmission (m > 0), so that σAF > 0 and σ2F > 0, and σ2C* > 0. As mentioned, we cannot resolve σ2F and σ2C*, so we fitted a single shared environmental factor, representing C and F. In summary, given σ2C = σ2F + σ2C*, we have the following settings. Simulation 1: m = 0, σ2F = 0, σ2C* > 0, σ2C = σ2C*, σAC = σAF = 0. Simulation 2: m > 0, σ2F > 0, σ2C* = 0, σ2C = σ2F, σAC = σAF and σAF > 0. Simulation 3: m > 0, σ2F > 0, σ2C* > 0, σ2C = σ2F + σ2C*, σAC = σAF and σAF > 0.
In each simulation study based on these three models, we carried out 500 replications. Each data set comprised genotypic and phenotypic data in parents and twins. The parental data were discarded, and the twin data were used to fit the model. The additive genetic variable comprised 100 uncorrelated diallelic genetic variants, of which 40 were used to calculate the PRS. We carried out the simulation twice: once with exact, unbiased PRS weights (i.e., the parameters b in the expression PRSi = GVli), and once with estimated weights b. We estimated the weights in independent data (not the data used to fit the actual model) by regressing the phenotype on the genetic variants. Given the absence of A–C covariance, the estimated weights are unbiased. In each replication the sample sizes were N = 2000 to estimate the parameter weights b, and Nmz = 1000 and Ndz = 1000 (in total 2000 pairs) to fit the actual model. Parameter values and effect sizes are given in Table 3. In simulations 1–3, the true values of prPh are 0.2, 0.141, and 0.147, and the true values of pr2σA,C*+F are 0.0, 0.353, and 0.368, respectively.
Table 3.
Means and standard deviation of parameter estimates in simulation 1–3 based on 500 replications (Nmz = 1000; Ndz = 1000)
| b est |
σ2Ap | σ2Aq | σ2C* | σ2F | σ2C = σ2C* + σ2F |
σ2E | σA,C | |
|---|---|---|---|---|---|---|---|---|
| Simulation 1 | ||||||||
| True | 0.20 | 0.30 | 0.20 | 0 | 0.20 | 0.30 | 0.00 | |
| Mean | No | 0.199 | 0.298 | 0.197 | – | – | 0.301 | 0.003 |
| s.d. | 0.026 | 0.045 | 0.058 | – | – | 0.013 | 0.033 | |
| s.e.(mean) | 0.0012 | 0.0020 | 0.0026 | 0.0006 | 0.0015 | |||
| Mean | Yes | 0.184* | 0.316* | 0.199 | – | – | 0.300 | 0.001 |
| s.d. | 0.026 | 0.047 | 0.063 | – | – | 0.013 | 0.036 | |
| s.e.(mean) | 0.0012 | 0.0021 | 0.0028 | 0.0006 | 0.0016 | |||
| Simulation 2 | ||||||||
| True | 0.20 | 0.30 | 0 | 0.091 | 0.091 | 0.30 | 0.125 | |
| Mean | No | 0.200 | 0.300 | – | 0.087 | – | 0.301 | 0.126 |
| s.d. | 0.026 | 0.047 | – | 0.078 | – | 0.013 | 0.037 | |
| s.e.(mean) | 0.0012 | 0.0021 | 0.0035 | 0.0006 | 0.0017 | |||
| Mean 2 | Yes | 0.189* | 0.315* | – | 0.090 | – | 0.300 | 0.124 |
| s.d. | 0.025 | 0.046 | – | 0.079 | – | 0.013 | 0.038 | |
| s.e.(mean) | 0.0011 | 0.0021 | 0.0035 | 0.0006 | 0.0017 | |||
| Simulation 3 | ||||||||
| True | 0.20 | 0.30 | 0.20 | 0.108 | 0.308 | 0.30 | 0.125 | |
| Mean | No | 0.200 | 0.302 | – | – | 0.302 | 0.300 | 0.126 |
| s.d. | 0.026 | 0.045 | – | – | 0.077 | 0.013 | 0.039 | |
| s.e.(mean) | 0.0012 | 0.0020 | 0.0034 | 0.0006 | 0.0017 | |||
| Mean | Yes | 0.185* | 0.320* | – | – | 0.304 | 0.299 | 0.125 |
| s.d. | 0.026 | 0.050 | – | – | 0.087 | 0.013 | 0.041 | |
| s.e.(mean) | 0.0012 | 0.0022 | 0.0039 | 0.0006 | 0.0018 | |||
| Simulation 2a | ||||||||
| True | 0.20 | 0.30 | 0 | 0.091 | 0.091 | 0.30 | 0.125 | |
| Mean | Yes | 0.189* | 0.314* | – | 0.095 | – | 0.299 | 0.122 |
| s.d. | 0.025 | 0.045 | – | 0.061 | – | 0.013 | 0.032 | |
| s.e.(mean) | 0.0011 | 0.0020 | 0.0027 | 0.0006 | 0.0014 |
Values shown in bold are the true parameter values
Simulation 2a: subject to constraints of positive definiteness of the Ap–C and Aq–C covariance matrices
b est: weights for PRS estimated (yes), or fixed to true values (no)
Simulation 1: r(A,F + C) = 0; σ2Ph = 0.20 + 0.30 + 0.20 + 0.30 = 1; r(MZ) = 0.70 & r(DZ) = 0.45; prPH = 0.2; pr2σAC = 0.0
Simulation 2: r(A,F + C) = 0.125/sqrt(0.5*0.091) = 0.586; σ2Ph = 1.141; r(MZ) = 0.74 & r(DZ) = 0.52; prPH = 0.141; pr2σAC = 0.353
Simulation 3: r(A,F + C) = 0.125/sqrt(0.5*0.308) = 0.318; σ2Ph = 1.358; r(MZ) = 0.78 & r(DZ) = 0.59; prPH = 0.147; pr2σAC = 0.368
*Deviation from true value is significant given α = 0.01
Note in fitting the model we estimated the single variance term σ2C, which equals σ2F + σ2C*. In simulations 1, σ2F is zero and σAC = 0; in simulation 2 σ2C* is zero, σAC > 0; in simulation 3, σ2F > 0, σ2C* > 0, and σAC > 0
In summary, the aims were (1) to establish that the Type 1 error rate was correct and (2) to investigate the effects, if any, of biased weights on the Type I error rate and the parameter estimates (bias), (3) to determine whether the presence of PRS, given zero A–C covariance, increases the power to detect C variance.
Results II
We first discuss the parameter estimates and then the loglikelihood ratio (LLR) test statistics. Table 3 contains the true parameter values and the mean and standard deviation of the parameter estimates based on the 500 replications. In simulation 1, given exact PRS weights, the parameter estimates are unbiased, as expected. Given estimated PRS weights, the estimates of σ2Ap and σ2Aq are biased: the mean values are 0.184 (underestimated; true 0.2) and 0.316 (overestimated; true: 0.3), respectively. The estimate of the covariance term σAC is unbiased: the mean value is 0.001 (true: 0.0). In simulation two, given exact weights, the parameter estimates are again unbiased, and given estimated PRS weights, the estimates of σ2Ap and σ2Aq are again biased: the mean values are 0.189 (underestimated; true 0.2) and 0.315 (overestimated; true: 0.3), respectively. The estimate of σAC is unbiased: mean value 0.124 (true 0.125). Simulation three produced the same results as simulation two, in terms of parameter bias stemming from using estimated PRS weight. The main finding is that using estimated PRS weights results in biased estimates of the variance components σ2Ap and σ2Aq, but has little effect on the estimate of the covariance term σAC.
Table 4 contains the results of the LLR tests. We tested the hypotheses σA,C = 0, and σ2C = 0 given σA,F+C = 0 in the twin model with the PRSs. In addition, we tested the hypothesis σC2 = 0 in the standard univariate ACE model. The test of σ2C = 0 is of interest in simulation 1, as the comparison of σ2C = 0 given σA,F+C = 0 (in the full model) and hypothesis σ2C = 0 in the standard univariate ACE model tell us whether the presence of PRSs helps to resolve C. The LLR statistic associated with the test of σA,F+C = 0 in simulation 1, where in truth σA,F+C = 0, should follow central chi2(1) distribution, which is characterized by a mean of 1 and a standard deviation of √(2) = 1.414. Given exact PRS weights, the mean and standard deviation of the LLR statistic are 0.970 and 1.423 (see Table 4). These do not differ from the expected values of 1 and √2 (LLR test: 0.27, df = 2, p = 0.87). The Type I error rate equaled 0.049 (CI95: 0.032–0.072). Given estimated weights, the values are 1.041 and 1.515. While these do not appear to deviate from the expected values (LLR test: 5.18, df = 2, p = 0.075), the variance is larger (1.515 vs. 1.414), as is the Type I error rate: 0.063 (CI95: 0.043–0.088). In terms of the mean LLR test, we note that the test of σC2 = 0 is more powerful in the full model (given σA,C = 0) than in the standard univariate ACE twin model. With exact PRS weights, the mean LLR test statistics equal 22.2 (with PRSs) and 16.06 (standard ACE model), and with estimated PRS weight, 21.5 and 15.6.
Table 4.
Means and standard deviation of loglikelihood ratio tests simulation 1–3 based on 500 replications (Nmz = 1000; Ndz = 1000)
| b est | σA,C = 0 | σ2C = 0 given σA,C = 0 |
σ2C = 0 in ACE model |
|
|---|---|---|---|---|
| Simulation 1 | ||||
| Mean | No | 0.970a | 22.2 | 16.06 |
| sd | 1.423 | 8.88 | 7.344 | |
| Mean | Yes | 1.041a | 21.52 | 15.62 |
| sd | 1.515 | 8.94 | 7.49 | |
| Simulation 2 | ||||
| Mean | No | 15.11 | 25.12 | 37.16 |
| sd | 7.47 | 10.13 | 11.23 | |
| Mean | Yes | 13.51 | 25.99 | 36.63 |
| sd | 7.38 | 9.66 | 11.25 | |
| Simulation 3 | ||||
| Mean | No | 13.48 | 78.87 | 78.98 |
| sd | 7.16 | 15.40 | 15.96 | |
| Mean | Yes | 11.96 | 74.59 | 78.43 |
| sd | 6.65 | 16.93 | 17.05 | |
| Simulation 2b | ||||
| Mean | Yes | 13.92 | 25.54 | 36.66 |
| sd | 7.49 | 9.92 | 12.11 |
Means are the mean of the 1-df likelihood ratio test
b est: weights for PRS estimated (yes), or fixed to true values (no)
Simulation 1: r(A,C) = 0; σ2Ph = 0.20 + 0.30 + 0.20 + 0.30 = 1; r(MZ) = 0.70 & r(DZ) = 0.45; prPH = 0.2; pr2σAC = 0.0
Simulation 2: r(A, C) = 0.125/sqrt(0.5*0.091) = 0.586; σ2Ph = 1.141; r(MZ) = 0.74 & r(DZ) = 0.52; prPH = 0.141; pr2σAC = 0.353
Simulation 3: r(A,C) = 0.125/sqrt(0.5*0.308) = 0.318; σ2Ph = 1.358; r(MZ) = 0.78 & r(DZ) = 0.59; prPH = 0.147; pr2σAC = 0.368
aExpected mean value = 1, expected stdev = √2 = 1.414
bSubject to constraints of positive definiteness of the Ap–C and Aq–C covariance matrices
The results of simulation 2 and 3 are comparable. The test of σAC = 0 suffers slightly given estimated PRS weights: the mean LLR test statistics equal 15.10 and 13.5 (simulation 2) and 13.48 and 11.96 (simulation 3). The effect of the test of σ2C = 0 given σAC = 0 is slight (in simulation 3: 78.8 vs 74.6). We note that the test of C in the standard twin model appears to be more powerful. However, given σAC > 0, this is a combined test of σC2 = 0 and σAC = 0.
In simulation 2, we set σ2C* = 0 and σ2F = 0.091 (6.4% of the phenotypic variance). This relatively low value does not rule out a considerable contribution of σAC to the phenotypic variance (35%). The true twin correlations in the simulation 2 (rMZ = 0.74, rDZ = 0.52) suggest a considerable contribution of C (~ 30%). This implies that (1) substantial C in the classical twin model may be mainly due to A–C covariance, while (2) the C variance, which in simulation 2 comprised only σ2F, may be small. Given that this variance may be small, its estimate may, given the present variance component parameterization, assume negative values. Indeed, in simulation 2, we encountered a negative variance component estimate in about 11% (exact PRS weights) and 13% (estimated weights) of the replications (we did not remove these cases in calculating the results in Tables 3 and 4 to avoid the bias caused by truncating the distribution of the parameters to the admissible solutions). The problem of negative variance can be solved by imposing the constraint that the 2 × 2 covariance matrix of Ap and C and the 2 × 2 covariance matrix of Aq and C be positive definite. This means that the eigenvalues of the covariance matrices are constrained to be larger than zero. This PD constraint is simple to implement in OpenMx using an mxAlgebra statement, as OpenMx includes a function to calculate eigenvalues (see the OpenMx script). Only constraining σ2C to be greater than zero is insufficient, as this by itself does not ensure that the covariance matrix of Ap (or Aq) and C is positive definite. In addition, if σ2C were to hit the lower bound, the parameter σAC would no longer be defined. We repeated simulation 2 with these PD constraints to gauge the effects on the parameters. The results, as obtained with estimated PRS weights, are included in Tables 3 and 4. These results are largely consistent with those obtained without the PD constraints. We do note that the PD constraints affect the distribution of the estimates of σ2C positively skewed, as shown in Fig. 4. Without the positive definiteness constraints, this distribution is normal. In contrast, the distribution of the estimates of σAC appear to be quite normal, which suggests that, at least in the present scenario, the LLR test of σAC = 0 will not be affected by the PD constraints.
Fig. 4.

Distribution of estimates of σ2F (left) and σAF (right) given positive definiteness constraints (simulation 2). The true values are σ2F = 0.091 and σAF = 0.125
The Identifying Constraint σApAq = 0
As mentioned above, we have constrained σApAq to equal 0, because it is not possible to estimate the scaling parameter p (in σ2PRS = p2σ2Ap) and the covariance simultaneously. This raises the question how the results are affected if in fact σApAq > 0, as σApAq = 0 is implausible given linkage disequilibrium. In fact, the constraint σApAq = 0 has no effect of the estimates of σAC, σ2A, and σ2C. To demonstrate this, we used simulation exact data with σApAq > 0, and fitted the model twice. Once with σApAq fixed to its true value, and once σApAq fixed to zero. Specifically, we chose the parameter values shown in Table 5. Fitting the model with σApAq fixed to equal its true value (σApAq = 0.1224; correlation: ρApAq = 0.5), we recovered the parameter values, including σAC = 0.077, and the total additive genetic variance 0.2 + 0.3 + 2*0.1224 = ~ 0.745 (i.e., σ2Ap + σ2Aq + 2σApAq). The power to reject σAC = 0 equals 0.742 (α = 0.05). Fixing σApAq = 0, we obtain identical results, except for the values of σ2Ap and σ2Aq. The total genetic variance is now composed as follows 0.5199 + 0.2250 = 0.745 (i.e., σ2Ap + σ2Aq). We checked this result with a wide variety of parameter values. So in principle, one can constrain σApAq to equal any sensible value. However, while the estimates and tests of σAC, σ2A, and σ2C are unaffected, we note that the values of σ2Ap and σ2Aq do depend on this sensible value.
Table 5.
Results with σApAq fixed to its true value (row A), and σApAq fixed to equal zero (row B)
| σ2Ap | σ2Aq | σApAq | σ2A | σ2C | σ2E | σAC | LLR | Power | |
|---|---|---|---|---|---|---|---|---|---|
| A | 0.20 | 0.30 | 0.1224* | 0.745a | 0.2 | 0.3 | 0.077 | 6.81 | 0.742 |
| B | 0.5199 | 0.2250 | 0* | 0.745b | 0.2 | 0.3 | 0.077 | 6.81 | 0.742 |
σApAq = 0.122 corresponds to a correlation of 0.1224/sqrt(0.20*0.30) = 0.5
σAC = 0.077 corresponds to a correlation of 0.077/sqrt(0.745*0.2) = 0.2
Power: to reject σAC = 0 is given α = 0.05, and Nmz = 1000, Ndz = 1000, based on the LLR statistic
*Fixed parameters
aσ2A = 0.745 = 0.30 + 0.30 + 2*0.1224
bσ2A = 0.745 = 0.5199 + 0.2250
Discussion
The present aim was to estimate A–C covariance in the classical twin model with PRS. To this end, we proposed the model depicted in Fig. 1, in which the covariance between Ap (PRS) and Aq and C are modeled as a function of the single covariance of A (Ap + Aq) and C. We found that the power to reject σAC = 0 depends mainly on the proportion of phenotypic variance due to the covariance term (σAC) and the PRS, where the former is more important than the latter. We investigated the influence of using estimated PRS weights. The use of estimated weights resulted in downwards bias of σAp and an upwards bias of σAq. However, the estimate of σAC was not affected. The use of estimated weights had a small effect of the Type I error rate in the test of σAC = 0.
In the most favorable settings qua power (8, 14, and 16 in Table 1), the proportions pr2σAC (phenotypic variance due to σAC) equaled 0.153, 0.159, and 0.189, and the proportions prPh (phenotype variance due to the PRS) equaled 0.101, 0.168 and 0.162. We consider these values (prPh) to be generally large by today's standards, but note that, while pr2σAC is given, the proportion prPh is likely to increase with the ongoing progress of GWAS meta analyses of many phenotypes. For instance, at present PRSs explain ~ 15% of the variance of educational attainment and ~ 11% of the variance of IQ (Allegrini et al. 2019).
The results of the power study and simulations shed some light on the viability of the model. But the results of simulation 2 also demonstrated that positive cultural transmission can result in large C in the standard ACE model, while most of this C variance is due to σAC. The actual C variance (without σAC) can be quite small. It is therefore advisable to fit the model with the positive definiteness constraint, outlined above. In this connection, we note that the finding that C variance in cognitive abilities is large in young children, but declines quickly in magnitude as children grow older (Haworth et al. 2010; Tucker-Drob and Bates 2016) may well be due to a decline in the magnitude of cultural transmission, in combination with an increase in genetic variance. This may be testing by extending the present model to include age as a moderator in the manner of Purcell (2002). This is relatively simple to do in OpenMx. In this connection, we also note that the estimate of σAC obtained using the present model may tell us that σAC is present. It does not, unlike other models, reveal the source of the σAC. For instance, in the NTF design, cultural transmission is the source the covariance between A and F, where the distinction is made between F (shared environmental effects due to cultural transmission) and residual C, which we denoted C* above.
We considered negative σAC in the power study, and found that the power to reject σAC = 0 was about the same regardless of the sign of σAC. We note that negative σAC (e.g., originating in negative cultural transmission) tends to produce twin correlations, which are suggestive of an ADE model (2*rDZ < rMZ). This is to be expected as − σAC lowers the MZ and DZ correlations to the same extent. Finally, the present results demonstrated that the addition of PRSs to the ACE model increases the power to detect C variance, assuming σAC = 0. This may be of interest, as in the classical twin model, the power to detect C variance is known to be generally poor (Visscher et al. 2008; Martin et al. 1978).
In closing, we note the following limitations. We have assumed that dominance variance (D) is absent, and acknowledge that the twin univariate design is limited to ACE or ADE. As demonstrated by Keller et al. (2010), a well fitting ACE model does not rule out the represent of D. It is possible that the addition of PRSs may aid in resolving D (in an ACDE) model, but we consider this beyond the present scope. Boomsma et al (2020; see this issue) showed that it is possible to estimate all four variance components (A, C, D, and E) in special cases of the multivariate twin model. Second as mentioned above, the settings of the power study and the simulations are limited in scope. In addition, we considered only equal MZ and DZ sample sizes (Nmz = Ndz = 1000). The ratio Nmz/Ndz has a general bearing on the power in the twin design (Visscher 2004). However, power calculations with unequal MZ and DZ sample sizes are simple to carry out. Third, the simulations that we carried out to gauge the effect of using estimated PRS weights, involved only a small number of associated GVs with relatively large effects. Simulation studies with more realistic designs will provide additional information concerning the effects of using estimated PRS weights. Fourth, we assumed that phenotypic mating is random. However, we note that the PRSs in the DZ twins offer the means to tests this, as the correlation between the additive genetic PRSs will equal 0.5 given phenotypic random mating. In addition, the present model may be extended to include parental data to accommodate phenotypic assortative mating as outlined in Keller et al. (2009). Fifth, we have not considered the effect of violations of other standard twin design assumptions on the estimate of σAC (Eaves et al. 1977; Purcell 2002; Keller et al 2010). Genotype—unshared environment covariance (σAE) is not identified in the present model. Unmodeled (positive) σAE and A × C interaction contribute to A. We do not see how either could result in spurious σAC. A × E and C × E interaction contribute to E, which has no bearing on A, C, or σAC. Sixth, we have not considered the possibility that the σAC is due to stratification. We know that spatial (geographical) allele frequency gradients may given rise to spurious C variance in the classical twin design (Tamimy et al. 2020; see this issue). A positive spatial correlation between C effects and allele frequencies, may given rise to A–C covariance. One way to detect this by including as fixed covariates principal components that reflect the allele frequency gradient (Price et al. 2006). If this kind of stratification is an issue, then the size of the C variance (see Tamimy et al. 2020) and the size of the A–C covariance should decline following the introduction of these covariates. Finally, we have assumed that the PRSs weights were obtained from GWASs of the phenotype of interest. Whether the present approach can be adapted to handle PRSs weights based on a genetically correlated phenotype (i.e., correlated with the phenotype of interest) remains to be seen.
Supplementary information
Below is the link to the electronic supplementary material.
Acknowledgements
CVD acknowledges NWO top-talent grant 406-12-124 (awarded to the late Janneke de Kort); DIB acknowledges KNAW Academy Professor Award (PAH/6635); MCN, CVD, DIB and CCM were supported by National Institute on Drug Abuse grants (DA049867 & DA018673; PI: MCN). CCM acknowledges grant 5R37 MH107649. We thank the editor David Evans, and the reviewers Matt Keller, Yongkang Kim, and Brad Verhulst for their helpful comments.
Compliance with ethical standards
Conflict of interest
Conor V. Dolan, Roel C. A. Huijskens, Camelia C. Minică , Michael C. Neale, & Dorret I. Boomsma declare that they have no conflicts of interest related to the publication of this article.
Ethical approval
This article does not contain any studies with human participants or animal subjects performed by any of the authors.
Footnotes
Edited by David Evans.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Allegrini AG, Selzam S, Rimfeld K, von Stumm S, Pingault JB, Plomin R. Genomic prediction of cognitive traits in childhood and adolescence. Mol Psychiatry. 2019;24(6):819–827. doi: 10.1038/s41380-019-0394-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates TC, Maher BS, Medland SE, et al. The nature of nurture: using a virtual-parent design to test parenting effects on children's educational attainment in genotyped families. Twin Res Hum Genet. 2018;21(2):73–83. doi: 10.1017/thg.2018.11. [DOI] [PubMed] [Google Scholar]
- Beam CR, Turkheimer E. Phenotype–environment correlations in longitudinal twin models. Dev Psychopathol. 2013;25:7–16. doi: 10.1017/S0954579412000867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker PA, Merckens A, Wansbeek TJ. Identification, equivalent models, and computer algebra: statistical modeling and decision science. New York: Academic Press; 1994. [Google Scholar]
- Boker SM, Neale MC, Hermine H, Maes HH, Wilde MJ, Spiegel M, Brick TR, Spies J, Estabrook R, Kenny S, Bates TC, Mehta P, Fox J. OpenMx: an open source extended structural equation modeling framework. Psychometrika. 2011 doi: 10.1007/s11336-010-9200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomsma DI, van Beijsterveldt CEM, Odintsova VV, Neale MC, Dolan CV (2020) Genetically informed regression analysis: application to aggression prediction by inattention and hyperactivity in children and adults. Behav Genet. 10.1007/s10519-020-10025-9. (Online ahead of print). [DOI] [PMC free article] [PubMed]
- Carey G. Sibling imitation and contrast effects. Behav Genet. 1986;16:319–341. doi: 10.1007/BF01071314. [DOI] [PubMed] [Google Scholar]
- Cheesman R, Hunjan A, Coleman JRI, et al. Comparison of adopted and nonadopted individuals reveals gene-environment interplay for education in the UK Biobank. Psychol Sci. 2020;31(5):582–591. doi: 10.1177/0956797620904450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Onofrio BM, Turkheimer EN, Eaves LJ, Corey LA, Berg K, Solaas MH, et al. The role of the children of twins design in elucidating causal relations between parent characteristics and child outcomes. J Child Psychol Psychiatry. 2003;44(8):1130–1144. doi: 10.1111/1469-7610.00196. [DOI] [PubMed] [Google Scholar]
- Dolan CV, de Kort JM, van Beijsterveldt TC, Bartels M, Boomsma DI. GE covariance through phenotype to environment transmission: an assessment in longitudinal twin data and application to childhood anxiety. Behav Genet. 2014;44(3):240–253. doi: 10.1007/s10519-014-9659-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudbridge F (2013) Power and predictive accuracy of polygenic risk scores. PLoS Genet 9(3):e1003348. 10.1371/journal.pgen.1003348. Epub 2013 Mar 21. Erratum in: PLoS Genet 2013 Apr;9(4) [DOI] [PMC free article] [PubMed]
- Eaves LJE, Erkanli A. Markov Chain Monte Carlo Approaches to Analysis of Genetic and Environmental Components of Human Developmental Change and G × E Interaction. Behav Genet. 2003;33:279–299. doi: 10.1023/A:1023446524917. [DOI] [PubMed] [Google Scholar]
- Eaves LJ, Last K, Martin NG, Jinks JL. A progressive approach to non-additivity and genotype-environmental covariance in the analysis of human differences. Br J Math Stat Psychol. 1977;30(1):1–42. doi: 10.1111/j.2044-8317.1977.tb00722.x. [DOI] [Google Scholar]
- Eaves LJ, Last KA, Young PA, Martin NG. Model-fitting approaches to the analysis of human behavior. Heredity. 1978;41:249–320. doi: 10.1038/hdy.1978.101. [DOI] [PubMed] [Google Scholar]
- Evans DM, Visscher PM, Wray NR. Harnessing the information contained within genome-wide association studies to improve individual prediction of complex disease risk. Hum Mol Genet. 2009;18:3525–3531. doi: 10.1093/hmg/ddp295. [DOI] [PubMed] [Google Scholar]
- Fulker DW (1988) Genetic and cultural transmission in human behavior. In: Weir BS, Eisen EJ, Goodman MM, Namkoong G (eds) Proceedings of the second international conference on quantitative genetics. Sinauer, Sunderland, MA, pp 318–340
- Fulker W, Cherny SS, Sham PC, Hewitt JKR. Combined linkage and association sib-pair analysis for quantitative traits. Am J Hum Genet. 1999;64(1):259–267. doi: 10.1086/302193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth CM, Wright MJ, Luciano M, Martin NG, de Geus EJ, van Beijsterveldt CE, Bartels M, Posthuma D, Boomsma DI, Davis OS, Kovas Y, Corley RP, Defries JC, Hewitt JK, Olson RK, Rhea SA, Wadsworth SJ, Iacono WG, McGue M, Thompson LA, Hart SA, Petrill SA, Lubinski D, Plomin R. The heritability of general cognitive ability increases linearly from childhood to young adulthood. Mol Psychiatry. 2010;15(11):1112–1120. doi: 10.1038/mp.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath AC, Kendler KS, Eaves LJ, Markell D. The resolution of cultural and biological inheritance: informativeness of different relationships. Behav Genet. 1985;15(5):439–465. doi: 10.1007/BF01066238. [DOI] [PubMed] [Google Scholar]
- Jinks JL, Fulker DW. Comparison of the biometrical genetical, MAVA, and classical approaches to the analysis of the human behavior. Psychol Bull. 1970;73(5):311–349. doi: 10.1037/h0029135. [DOI] [PubMed] [Google Scholar]
- Keller MC, Medland SE, Duncan LE. Are extended twin family designs worth the trouble? A comparison of the bias, precision, and accuracy of parameters estimated in four twin family models. Behav Genet. 2010;40(3):377–393. doi: 10.1007/s10519-009-9320-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MC, Medland SE, Duncan LE, Hatemi PK, Neale MC, Maes HHM, Eaves LJ. Modeling extended twin family data I: description of the cascade model. Twin Res Hum Genet. 2009;12(1):8–18. doi: 10.1375/twin.12.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS. A conceptual overview of gene–environment interaction and correlation in a developmental context. In: Kendler KS, Jaffee SR, Romer D, editors. The dynamic genome and mental health: the role of genes and environments in youth development. New York: Oxford University Press; 2011. pp. 5–28. [Google Scholar]
- Knafo A, Jaffee SR. Gene–environment correlation in developmental psychopathology. Dev Psychopathol. 2013;25:1–6. doi: 10.1017/S0954579412000855. [DOI] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Frigge ML, Vilhjalmsson BJ, Young AI, Thorgeirsson TE, Benonisdottir S, Oddsson A, Halldorsson BV, Masson G, Gudbjartsson DF. The nature of nurture: effects of parental genotypes. Science. 2018;359:424–428. doi: 10.1126/science.aan6877. [DOI] [PubMed] [Google Scholar]
- Maes HH, Neale MC, Kendler KS, Martin NG, Heath AC, Eaves LJ. Genetic and cultural transmission of smoking initiation: an extended twin kinship model. Behav Genet. 2006;36(6):795–808. doi: 10.1007/s10519-006-9085-4. [DOI] [PubMed] [Google Scholar]
- Martin NG, Eaves LJ, Kersey MJ, Davies P. The power of the classical twin study. Heredity. 1978;40:97–116. doi: 10.1038/hdy.1978.10. [DOI] [PubMed] [Google Scholar]
- Minică CC, Dolan CV, Boomsma DI, de Geus E, Neale MC. Extending causality tests with genetic instruments: an integration of Mendelian randomization with the classical twin design. Behav Genet. 2018;48:337–349. doi: 10.1007/s10519-018-9904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narusyte J, Neiderhiser JM, D’Onofrio BM, Reiss D, Spotts EL, Ganiban J, Lichtenstein P. Testing different types of genotype-environment correlation: an extended children-of-twins model. Dev Psychol. 2008;44(6):1591–1603. doi: 10.1037/a0013911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MC. Flexible QTL mapping with Mx. In: Spector T, Snieder H, MacGregor A, editors. Advances in twin and sib pair analysis. London: Greenwich Medical Media; 2000. pp. 219–243. [Google Scholar]
- Neale MC, Fulker DW. A bivariate path analysis of fear data on twins and their parents. Acta Genet Med Gemellol. 1984;33:273–286. doi: 10.1017/S0001566000007327. [DOI] [PubMed] [Google Scholar]
- Neale MC, de Knijff P, Havekes LM, Boomsma DI. ApoE polymorphism accounts for only part of the genetic variation in quantitative ApoE levels. Genet Epidemiol. 2000;18:331–340. doi: 10.1002/(SICI)1098-2272(200004)18:4<331::AID-GEPI6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Neale MC, Hunter MD, Pritikin JN, Zahery M, Brick TR, Kirkpatrick RM, Estabrook R, Bates TC, Maes HH, Boker SM. OpenMx 2.0: extended structural equation and statistical modeling. Psychometrika. 2016;81(2):535–49. doi: 10.1007/s11336-014-9435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, DeFries JC, Loehlin JC. Genotype-environment interaction and correlation in the analysis of human behavior. Psychol Bull. 1977;84:309–322. doi: 10.1037/0033-2909.84.2.309. [DOI] [PubMed] [Google Scholar]
- Plomin R, Loehlin JC, DeFries JC. Genetic and environmental components of "environmental" influences. Dev Psychol. 1985;21(3):391–402. doi: 10.1037/0012-1649.21.3.391. [DOI] [Google Scholar]
- Plomin R, DeFries JC, Knopik VS, Neiderhise JM. Top 10 replicated findings from behavioral genetics. Perspect Psychol Sci. 2016;11(1):3–23. doi: 10.1177/1745691615617439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polderman TJC, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, Posthuma D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47:702–709. doi: 10.1038/ng.3285. [DOI] [PubMed] [Google Scholar]
- Price A, Patterson N, Plenge R, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Purcell S. Variance components models for gene–environment interaction in twin analysis. Twin Res. 2002;5:554–571. doi: 10.1375/136905202762342026. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
- Rutter M, Silberg J. Gene–environment interplay in relation to emotional and behavioral disturbance. Annu Rev Psychol. 2002;53:463–490. doi: 10.1146/annurev.psych.53.100901.135223. [DOI] [PubMed] [Google Scholar]
- Scarr S, McCartney K. How people make their own environments: a theory of genotype → environment effects. Child Dev. 1983;54:424–435. doi: 10.1111/j.1467-8624.1983.tb03884.x. [DOI] [PubMed] [Google Scholar]
- Selzam S, Ritchie SJ, Pingault J-B, Reynolds CA, O’Reilly PF, Plomin R. Comparing within- and between-family polygenic score prediction. Am J Hum Genet. 2019;105:351–363. doi: 10.1016/j.ajhg.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamimy Z, Kevenaar ST, Hottenga JJ, Hunter MD, de Zeeuw EL, Neale MC, van Beijsterveldt CEM, Dolan CV, van Bergen E, Boomsma DI (2020) Multilevel twin models: geographical region as a third level variable. Behav Genet. bioRxiv. 10.1101/2020.11.11.377820. [DOI] [PMC free article] [PubMed]
- Tucker-Drob EM, Bates TC. Large cross-national differences in gene × socioeconomic status interaction on intelligence. Psychol Sci. 2016;27(2):138–149. doi: 10.1177/0956797615612727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beijsterveldt CEM, Middeldorp CM, Slof-Op’t Landt MCT, Bartels M, Hottenga JJ, Eka H, Suchiman D, Slagboom PE, Boomsma DI Influence of candidate genes on attention problems in children: a longitudinal study. Behav Genet. 2011;41:155–164. doi: 10.1007/s10519-010-9406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Oord EJCG, Snieder H. Including measured genotypes in statistical models to study the interplay of multiple factors affecting complex traits. Behav Genet. 2002;32(1):1–22. doi: 10.1023/A:1014474711118. [DOI] [PubMed] [Google Scholar]
- van der Molenaar D, Sluis S, Boomsma DI, Dolan CV. Detecting specific genotype by environment interactions using marginal maximum likelihood estimation in the classical twin design. Behav Genet. 2012;42:483–499. doi: 10.1007/s10519-011-9522-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluis S, Dolan CV, Neale MC, Posthuma D. Power calculations using exact data simulation: a useful tool for genetic study designs. Behav Genet. 2008;38:202–211. doi: 10.1007/s10519-007-9184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen J, Slagboom P, Draisma H, et al. The continuing value of twin studies in the omics era. Nat Rev Genet. 2012;13:640–653. doi: 10.1038/nrg3243. [DOI] [PubMed] [Google Scholar]
- Verhulst B, Hatemi PK. Gene-environment interplay in twin models. Polit Anal. 2013;21(3):368–389. doi: 10.1093/pan/mpt005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhulst B, Prom-Wormley E, Keller M, Medland S, Neale MC. Type I error rates and parameter bias in multivariate behavioral genetic models. Behav Genet. 2019;49:99–111. doi: 10.1007/s10519-018-9942-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher P. Power of the classical twin design revisited. Twin Res. 2004;7(5):505–512. doi: 10.1375/twin.7.5.505. [DOI] [PubMed] [Google Scholar]
- Visscher M, Gordon S, Neale MC. Power of the classical twin design revisited: II detection of common environmental variance. Twin Res Hum Genet. 2008;11(1):48–54. doi: 10.1375/twin.11.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington NM, Freathy RM, Neale MC, Evans DM. Using structural equation modelling to jointly estimate maternal and fetal effects on birthweight in the UK Biobank. Int J Epidemiol. 2018;47(4):1229–1241. doi: 10.1093/ije/dyy015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz J, Moffitt TE, Agnew-Blais J, Arseneault L, Belsky DW, Corcoran DL, Houts R, Matthews T, Prinz JA, Richmond-Rakerd LS, Sugden K, Caspi A. Using DNA from mothers and children to study parental investment in children’s educational attainment. Child Dev. 2018 doi: 10.1111/cdev.13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.