

Abstract
Background:
Cerebral vasospasm is a major contributor to disability and mortality after aneurysmal subarachnoid hemorrhage. Oxidation of cell-free hemoglobin plays an integral role in neuroinflammation and is a suggested source of tissue injury after aneurysm rupture. This study sought to determine if patients with subarachnoid hemorrhage and cerebral vasospasm were more likely to have been exposed to early hyperoxemia than those without vasospasm.
Methods:
This single-center retrospective cohort study included adult patients presenting with aneurysmal subarachnoid hemorrhage to Vanderbilt University Medical Center between January 2007 and December 2017. Patients with an ICD-9/10 diagnosis of aneurysmal subarachnoid hemorrhage were initially identified (N=441) and subsequently excluded if they did not have intracranial imaging, arterial PaO2 values, or died within 96 hours post-rupture (N=96). The final cohort was 345 subjects. The degree of hyperoxemia was defined by the highest PaO2 measured within 72 hours after aneurysmal rupture. The primary outcome was development of cerebral vasospasm, which included asymptomatic vasospasm and delayed cerebral ischemia (DCI). Secondary outcomes were mortality and modified Rankin Scale.
Results:
345 patients met inclusion criteria; 218 patients (63%) developed vasospasm. Of those that developed vasospasm, 85 were diagnosed with delayed cerebral ischemia (DCI, 39%). The average patient age of the cohort was 55±13 years, and 68% were female. Ninety percent presented with Fisher Grade 3 or 4 hemorrhage (N=310) while 42% presented as Hunt-Hess Grade 4 or 5 (N=146). In univariable analysis, patients exposed to higher levels of PaO2 by quintile of exposure had a higher mortality rate and were more likely to develop vasospasm in a dose-dependent fashion (P=0.015 and P=0.019, respectively). There were no statistically significant predictors that differentiated asymptomatic vasospasm from DCI and no significant difference in maximum PaO2 between these two groups. In multivariable analysis, early hyperoxemia was independently associated with vasospasm (OR=1.15 per 50 mmHg increase in PaO2 [1.03, 1.28]; P=0.013), but not mortality (OR=1.10 [0.97, 1.25]; P=0.147) following subarachnoid hemorrhage.
Conclusions:
Hyperoxemia within 72 hours post-aneurysmal rupture is an independent predictor of cerebral vasospasm, but not mortality in subarachnoid hemorrhage. Hyperoxemia is a variable that can be readily controlled by adjusting the delivered FiO2 and may represent a modifiable risk factor for vasospasm.
Keywords: hyperoxemia, vasospasm, subarachnoid hemorrhage, aneurysm, oxidative stress
Introduction
Subarachnoid hemorrhage (SAH) due to a ruptured intracranial aneurysm accounts for 5% of all strokes in North America1. Although the mortality rate associated with aneurysmal SAH has declined over the past few decades, it remains greater than 30%2. A major contributor to death and disability after SAH is the development of cerebral vasospasm. In fact, it is estimated that half of the deaths in patients surviving treatment after SAH are attributable to vasospasm. Vasospasm has a spectrum of severity, spanning from asymptomatic patients to those that develop delayed cerebral ischemia (DCI) and infarction. The consequences of vasospasm contribute to high rates of long-term complications and poor quality of life due to significant neurocognitive impairment and loss of independence3.
Currently there is no effective preventative therapy for cerebral vasospasm. The discovery that nimodipine significantly reduces the occurrence of severe neurologic deficits from vasospasm after SAH was made over 36 years ago and changed practice4. However, despite the fact that nearly all SAH patients receive nimodipine, the burden of disability and death from vasospasm persists3. In addition to nimodipine, pharmacologic measures to optimize cerebral perfusion by inducing hypertension are routinely utilized after the aneurysm has been secured to ameliorate the effects of vasospasm5. Once DCI develops, additional endovascular approaches are commonly employed to aid in delivery of vasodilators or provide restoration of flow in narrowed arterial vessels through cerebral angioplasty6,7. These therapies, while important and effective, are not preventative. New insights into the pathophysiology of cerebral vasospasm after SAH are necessary to develop preventative therapies for this devastating condition.
The holy grail of understanding vasospasm pathophysiology has been the identification of a “spasmogen”8. The proximate triggering event of vasospasm is the initial deposition of blood in the subarachnoid space9. The Fisher scale illustrates a dose-dependent relationship between the amount and pattern of blood and development of vasospasm; however, clot removal or fibrinolysis has not proven to prevent vasospasm10,11,12. As such, current research has focused on understanding the ‘spasmogenic’ nature of the hemorrhage. During clot resolution, extravascular cell-free hemoglobin (CFH) levels rise with the blood breakdown13. CFH is a known mediator of inflammation and oxidative tissue injury, which makes it a good candidate “spasmogen”14,15. Hyperoxemia increases oxidation of CFH, which has known spasmogenic effects in the pulmonary vasculature, but the effects of hyperoxemia and resultant oxidation of CFH on the cerebral vasculature remains uncertain16,17,18. The authors’ goal was to test the hypothesis that there was a higher incidence of cerebral vasospasm and mortality for patients with aneurysmal SAH who were exposed to hyperoxemia within 72 hours post-aneurysm rupture compared to those who were not exposed to hyperoxemia.
Methods
Data Source
Patients greater than or equal to 18 years of age with non-traumatic subarachnoid hemorrhage secondary to ruptured aneurysm who presented to Vanderbilt University Medical Center between 2007 and 2017 were identified. Records were obtained from the Synthetic Derivative (SD), which is a de-identified database containing clinical information from the hospital electronic medical record19. The SD is not linked to the original medical record and is modified to ensure no identifiers can be traced back to an individual patient. Given the de-identified data source, the study was determined to be exempt by the Vanderbilt Institutional Review Board.
Inclusion and Exclusion Criteria
Patients (N=1,391) were identified by International Classification of Diseases (ICD) codes for nontraumatic subarachnoid hemorrhage. The specific codes were 430 and I60.XX for ICD-9 and ICD-10, respectively. Aneurysmal SAH was confirmed by manual review of each SD record for a computed tomography angiography (CTA) scan and/or a catheter-based angiography procedure which indicated the presence of an aneurysm. Patients who did not have at least one CT of the head or an arterial blood gas in the first 72 hours were excluded. The 72-hour window was selected as vasospasm usually begins on or after post-hemorrhage day three. Time zero was measured from the onset of patient symptoms. Additionally, those who died in the first 96 hours after aneurysmal rupture were excluded from the study as they did not survive long enough to develop vasospasm and thus were unlikely to have a clinical outcome modified by early hyperoxemia. There were 345 patients included in the study (Supplemental Figure I).
Data Collection
Clinical and demographic covariates were identified on the basis of commonly reported risk factors for vasospasm or mortality secondary to aneurysm rupture including age, gender, ethnicity, tobacco use, and Fisher and Hunt-Hess grades20–22. Study data were stored in a Research Electronic Data Capture (REDCap) database, which is a secure, web-based application designed to manage clinical data for research studies23.
Outcome Measures
The primary outcome of this study was the development of cerebral vasospasm, which included both asymptomatic and symptomatic patients. Secondary outcomes studied were mortality and modified Rankin Scale at hospital discharge24. Cerebral vasospasm was defined as vasospasm occurring greater than 72 hours after the time of aneurysmal rupture. Vasospasm was defined by an elevated Lindegaard (Li) ratio in the anterior (>3.0) or posterior circulation (>2.0) on transcranial doppler, a report of vasospasm on CTA, MRI with evidence of ischemic stroke, and/or the need for direct intra-arterial therapy for vasospasm in the operating room. Asymptomatic vasospasm was defined by positive transcranial dopplers or CTA but no need for emergent endovascular therapy and no MRI with evidence of infarct. Symptomatic patients, or those with DCI, were defined by the need for emergent intra-arterial therapy and/or MRI with evidence of ischemic stroke. Measures of mortality were confined to in-hospital deaths after the first 96 hours post-aneurysm rupture for reasons previously stated. The primary exposure variable was early hyperoxemia as captured by arterial blood gas analyses completed for clinical reasons. All arterial blood gas results from the first 72 hours after aneurysm rupture were recorded for each patient. The mean number of arterial blood gas analyses in the first 72 hours after aneurysm rupture was 4 ± 3 per patient. The timing of aneurysm rupture was defined by patient-reported symptom onset. PaO2 was a continuous variable and its maximum value within 72 hours post-aneurysm rupture was used in our analyses. For some analyses, quintiles of maximum PaO2 in the first 72 hours were compared across the cohort.
Statistical Analysis
Patient characteristics were analyzed by the Kruskal-Wallis test for continuous variables and Fisher’s exact test for categorical variables. Multivariable logistic regression was performed to determine whether PaO2 was independently associated with development of vasospasm or mortality. Prespecified potential confounders, including age, gender, ethnicity, tobacco abuse, Fisher grade10, Hunt Hess grade25, and minimum PaCO2 within 72 hours of aneurysmal rupture were adjusted in all models. Fisher and Hunt-Hess grades were dichotomized into more and less severe groups. The “more severe” groups were Fisher grades 3 and 4 and Hunt-Hess grades 4 and 5, which is consistent with prior vasospasm studies26. Multiple imputation with predictive mean matching was used to impute missing values of covariates in regression models27. Among variables used in the multivariable model, there was a negative correlation between minimum PaCO2 and maximum PaO2. There was a positive correlation between Fisher and Hunt-Hess grades and maximum PaO2. Although there was correlation among these variables, there was no evidence of multicollinearity when the variance inflation factor was used, indicating that the models are statistically valid. Two-sided P values less than 0.05 were deemed statistically significant. Statistical analysis and graphics were completed using R software Version 3.
Results
Demographics and Clinical Characteristics
There were 345 patients who met inclusion criteria. In total, 218 patients or 63% developed cerebral vasospasm. Overall patient characteristics and a comparative analysis between those with and without vasospasm are found in Table 1. 68% of patients were female (N=234). By univariable analysis, patients who developed vasospasm were younger in age at 52.9±11.9 years versus 57.2±15.2 years (P=0.004). Patients with higher Fisher and Hunt-Hess grades were more likely to develop vasospasm (P=0.003 and 0.035, respectively). There was no significant difference in gender (66% versus 72% female; P=0.325) or tobacco use (61% vs. 50%; P=0.062) between the vasospasm and no vasospasm groups, respectively. Ninety-nine percent of all patients with or without vasospasm received nimodipine therapy (N=342; P=0.788). There was no association between vasospasm and use of open versus endovascular treatment to secure the aneurysm (P=0.283).
Table 1.
Patient demographics and clinical characteristics (N=345).
| All patients (N = 345) | No vasospasm (N = 127) | Vasospasm (N = 218) | p-value | |
|---|---|---|---|---|
| Age in years (mean (SD)) | 54.5 (13.4) | 57.2 (15.2) | 52.9 (11.9) | 0.004 |
| Sex (n (%)) | 0.325 | |||
| Female | 234 (68) | 91 (72) | 143 (66) | |
| Male | 111 (32) | 36 (28) | 75 (34) | |
| Race (n (%)) | ||||
| White | 269 (78) | 92 (72) | 177 (81) | 0.203 |
| Black | 50 (15) | 23 (18) | 27 (12) | |
| Other | 26 (8) | 12 (10) | 14 (7) | |
| Tobacco use (n (%)) | 0.062 | |||
| Yes | 195 (57) | 63 (50) | 132 (61) | |
| No | 150 (43) | 64 (50) | 86 (39) | |
| Fisher grade (n (%)) | 0.003 | |||
| 1 | 6 (2) | 5 (4) | 1 (1) | |
| 2 | 29 (8) | 13 (10) | 16 (7) | |
| 3 | 57 (17) | 29 (23) | 28 (13) | |
| 4 | 253 (73) | 80 (63) | 173 (79) | |
| Hunt-Hess grade (n (%)) | 0.035 | |||
| 1 | 24 (7) | 10 (8) | 14 (6) | |
| 2 | 81 (24) | 40 (32) | 41 (19) | |
| 3 | 94 (27) | 35 (28) | 59 (27) | |
| 4 | 94 (27) | 25 (20) | 69 (32) | |
| 5 | 52 (15) | 17 (13) | 35 (16) | |
| Initial treatment (n (%)) | 0.283 | |||
| Endovascular | 277 (81) | 107 (85) | 170 (78) | |
| Open surgery | 59 (17) | 16 (13) | 43 (20) | |
| Other | 7 (2) | 3 (2) | 4 (2) | |
| Nimodipine use (n (%)) | 0.788 | |||
| Yes | 342 (99) | 126 (99) | 216 (99) | |
| No | 3 (1) | 1 (1) | 2 (1) | |
| Maximum PaO2 (mean (SD)) | 218.8 (117.3) | 195.4 (101.0) | 232.4 (124.1) | 0.005 |
| Minimum PaCO2 (mean (SD)) | 34.1 (6.5) | 34.8 (6.7) | 33.7 (6.3) | 0.100 |
Of the 218 patients with vasospasm, 143 met the criteria for asymptomatic vasospasm while 85 (39%) developed delayed cerebral ischemia, as shown in Table 2. There were no statistically significant predictors that differentiated these two groups and no significant difference in maximum PaO2 between them. Therefore, the primary outcome of cerebral vasospasm included patients who developed DCI in addition to those who developed asymptomatic vasospasm.
Table 2.
Vasospasm subgroup analysis: asymptomatic versus delayed cerebral ischemia
| Total N = 218 | Asymptomatic Vasospasm (n= 143) | Delayed Cerebral Ischemia (n=85) | p-value |
|---|---|---|---|
| Age in years (mean (SD)) | 52.6 (12.3) | 53.9 (12.1) | 0.438 |
| Race | 0.614 | ||
| White | 83 (71) | 113 (79) | |
| Black | 23 (20) | 19 (13) | |
| Other | 11 (9) | 11 (8) | |
| Sex (n (%)) | 0.445 | ||
| Female | 97 (68) | 52 (62) | |
| Male | 146 (32) | 33 (38) | |
| Tobacco use (n (%)) | 0.565 | ||
| Yes | 84 (59) | 54 (64) | |
| No | 59 (41) | 31 (36) | |
| Fisher grade (n (%)) | 0.636 | ||
| 1 | 0 (0) | 1 (1) | |
| 2 | 10 (7) | 6 (7) | |
| 3 | 18 (13) | 11 (13) | |
| 4 | 115 (80) | 67 (79) | |
| Hunt-Hess grade (n (%)) | 0.217 | ||
| 1 | 9 (6) | 6 (7) | |
| 2 | 32 (22) | 10 (12) | |
| 3 | 40 (28) | 21 (25) | |
| 4 | 42 (29) | 30 (35) | |
| 5 | 20 (14) | 18 (21) | |
| Initial treatment (n (%)) | 0.560 | ||
| Endovascular | 110 (78) | 68 (80) | |
| Open surgery | 30 (21) | 15 (18) | |
| Other | 2 (1) | 2 (1) | |
| Nimodipine use (n (%)) | -- | ||
| Yes | 142 (100) | 84 (100) | |
| No | 0 (0) | 0 (0) | |
| Maximum PaO2 (mean (SD)) | 240.5 (126.9) | 213.1 (113.2) | 0.102 |
| Minimum PaCO2 (mean (SD)) | 33.5 (6.4) | 33.9 (6.1) | 0.659 |
Vasospasm and Maximum PaO2
The highest PaO2 value in the first 72 hours after aneurysm rupture was significantly higher in patients who went on to develop vasospasm compared to those who did not (232 ± 124 mmHg versus 195 ± 101 mmHg; P=0.012; (Figure 1A). There was an apparent dose-dependent relationship between degree of hyperoxemia as measured by quintile of exposure and risk of vasospasm (Figure 1B). In a multivariable logistic regression for vasospasm controlling for potential confounders (Table 3), higher PaO2 was associated with a higher risk of vasospasm (OR=1.15 per 50 mmHg increase in PaO2 [1.03, 1.28]; P=0.013). Variables that were associated with a lower risk of development of vasospasm included non-Caucasian ethnicity (OR=0.51 [0.28, 0.92]; P=0.026), older age (OR=0.97 [0.96, 0.99]; P=0.007), and lower Hunt-Hess grade (OR=0.59 [0.35, 0.98]; P=0.043). All other variables, including Fisher grade, were not significant. (Figure 2)
Figures 1A and 1B. Maximum PaO2 and vasospasm.
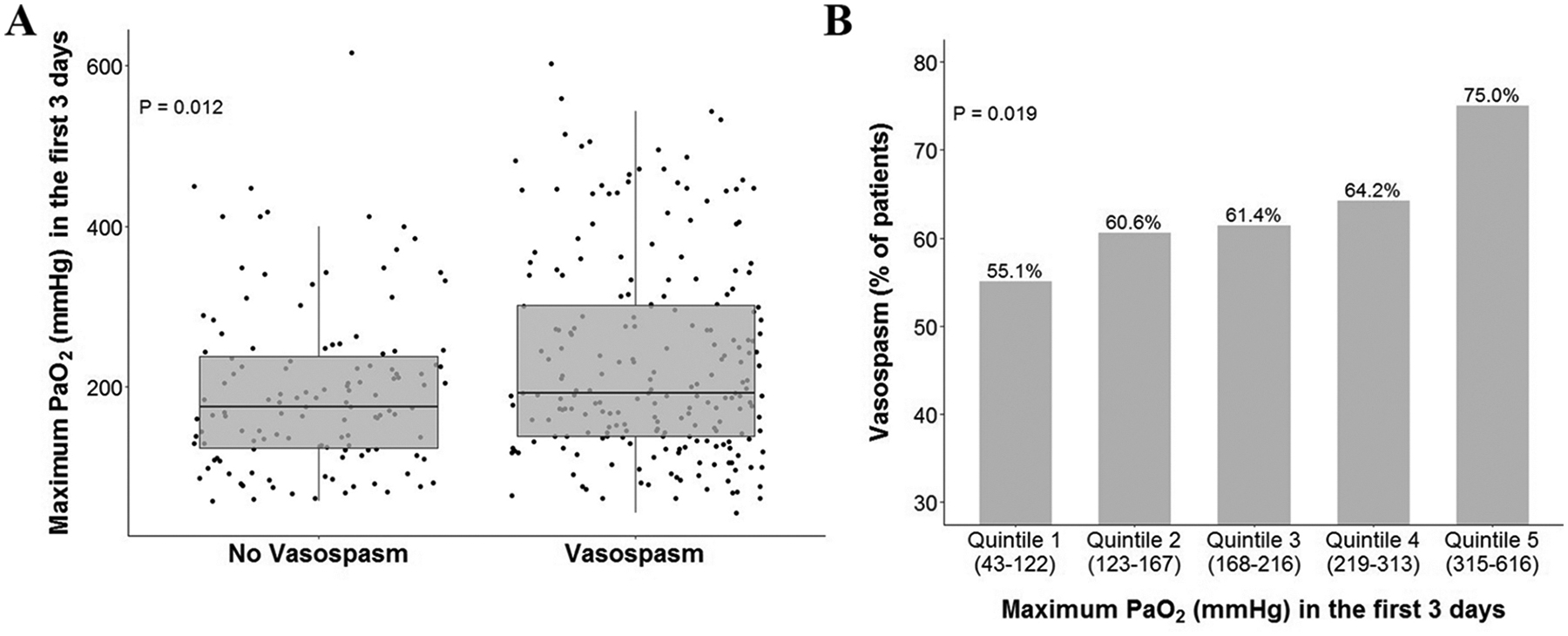
(A) Maximum PaO2 is significantly higher in patients who subsequently develop cerebral vasospasm in unadjusted analysis (P=0.012 by Kruskal-Wallis test). (B) Maximum PaO2 exhibited behavior similar to a dose-dependent effect on vasospasm. (P = 0.019 by linear-by-linear test).
Table 3.
Multivariable analyses for vasospasm and mortality.
| Vasospasm | ||
|---|---|---|
| Variable | Odds Ratio [2.5%, 97.5% CI] | p-value |
| Sex (female) | 0.85 [0.51, 1.41] | 0.538 |
| Age (years) | 0.97 [0.96, 0.99] | 0.007 |
| Tobacco use | 1.41 [0.88, 2.27] | 0.153 |
| Ethnicity (Non-Caucasian) | 0.51 [0.28, 0.92] | 0.026 |
| Fisher grade (low) | 0.58 [0.27, 1.23] | 0.159 |
| Hunt-Hess grade (low) | 0.59 [0.35, 0.98] | 0.043 |
| Minimum PaCO2 (per 10mmHg increase) | 0.99 [0.67, 1.45] | 0.951 |
| Maximum PaO2 (per 50mmHg increase) | 1.15 [1.03, 1.28] | 0.013 |
| Mortality | ||
| Sex (female) | 0.90 [0.44, 1.81] | 0.762 |
| Age (years) | 1.04 [1.01, 1.06] | 0.002 |
| Tobacco use | 0.57 [0.30, 1.08] | 0.084 |
| Ethnicity (Non-Caucasian) | 0.67 [0.26, 1.71] | 0.406 |
| Fisher grade (low) | 2.58 [0.88, 7.51] | 0.083 |
| Hunt-Hess grade (low) | 0.58 [0.29, 1.16] | 0.125 |
| Minimum PaCO2 (per 10mmHg increase) | 0.39 [0.22, 0.69] | 0.001 |
| Maximum PaO2 (per 50mmHg increase) | 1.10 [0.97, 1.25] | 0.147 |
Figure 2. Multivariable analysis for development of vasospasm.
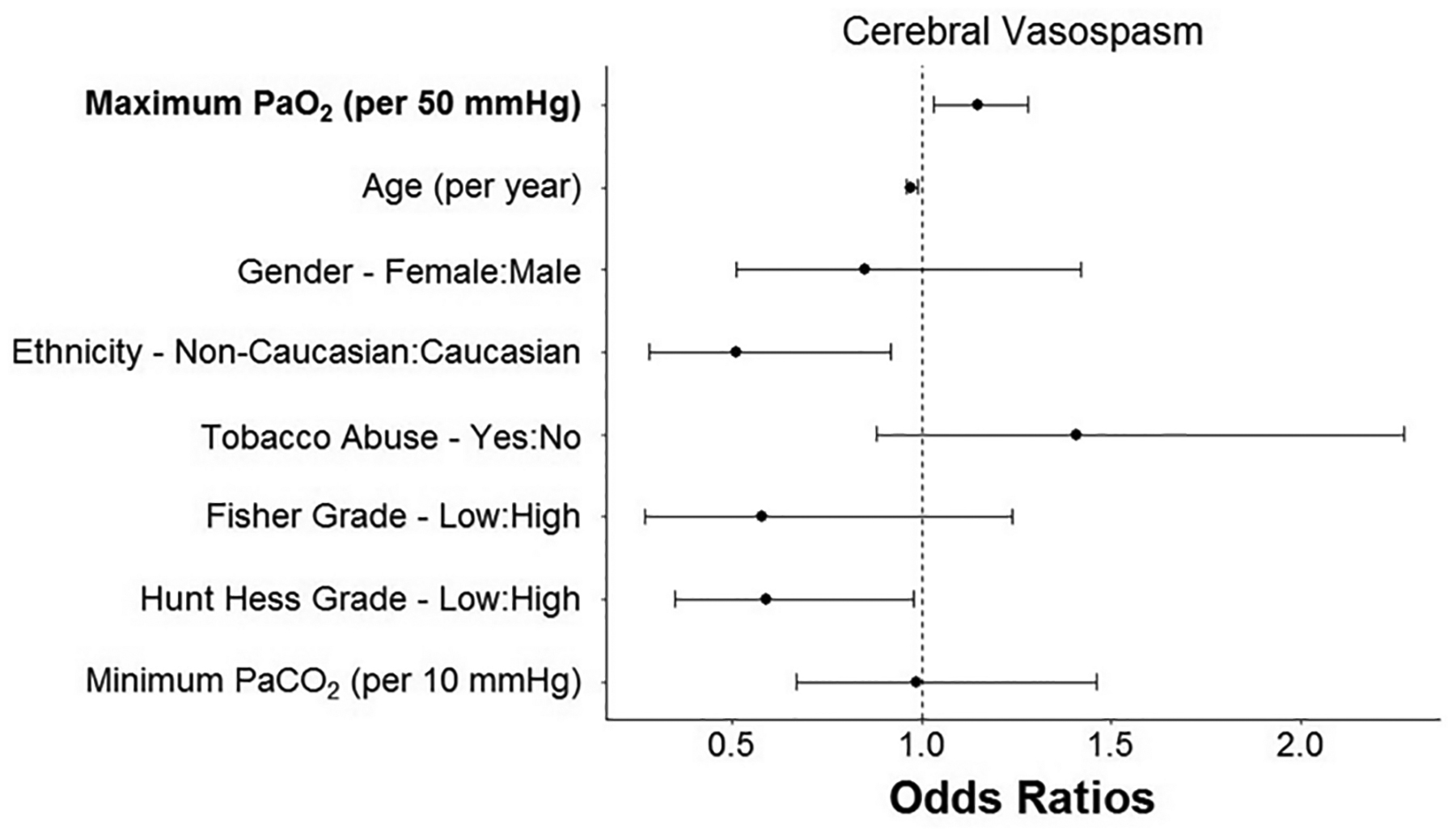
Early hyperoxia was significantly associated with vasospasm controlling for potential confounders.
Mortality and Maximum PaO2
The hospital mortality rate in the included patient population was 15% (N=52). In the multivariable analysis for mortality (Table 2), older age (OR=1.04 per year [1.01, 1.06]; P=0.002) was significantly associated with mortality. Conversely, a higher minimum PaCO2 was associated with better outcomes (OR=0.39 [0.22, 0.69]; P=0.001). Although in univariable analysis maximum PaO2 was higher in patients who died in hospital (P=0.017, Figure 3A) and there was a dose response relationship between degree of hyperoxemia and risk of death (Figure 3B), maximum PaO2 was not a significant predictor of mortality in multivariable analysis after adjusting for potential confounders (OR=1.10 per 50 mmHg [0.97, 1.25]; P=0.147).
Figures 3A and 3B. Maximum PaO2 and mortality.
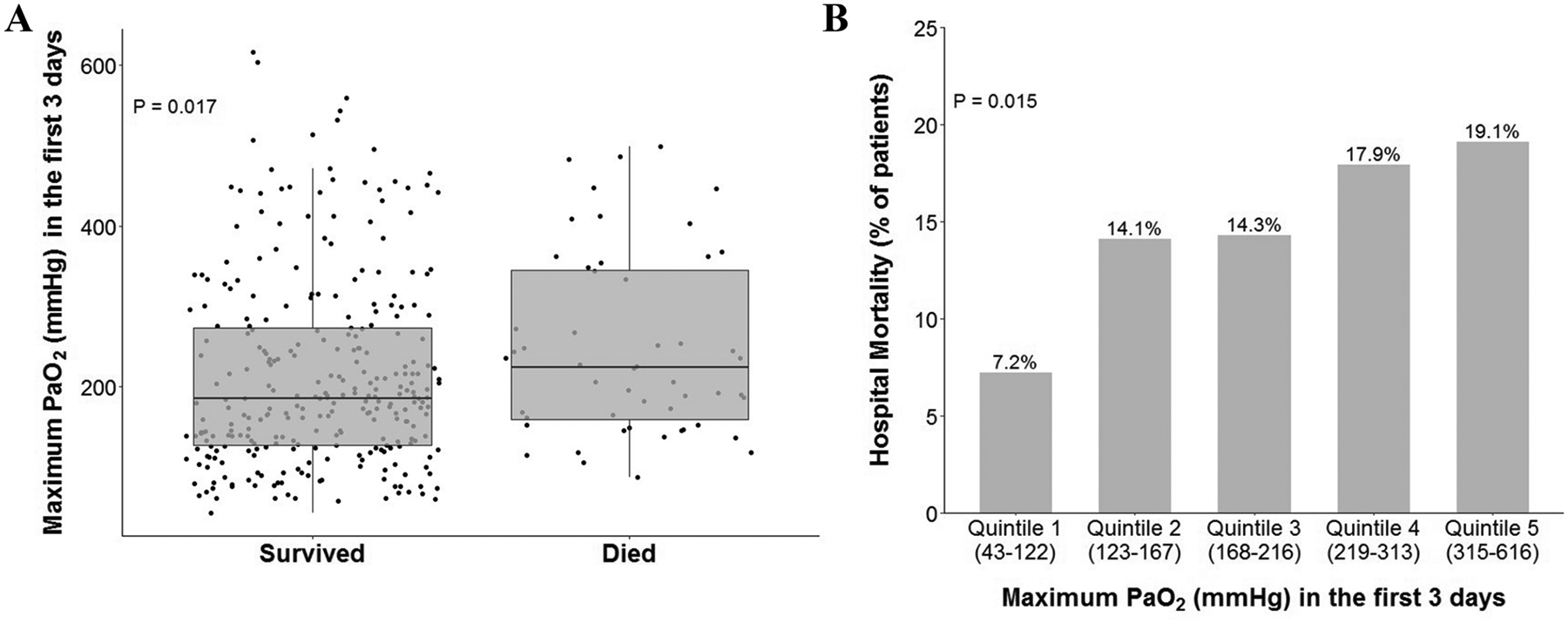
(A) Maximum PaO2 is significantly higher in patients who died in hospital in unadjusted analysis (P=0.017 by Kruskal-Wallis test). (B) Maximum PaO2 exhibited behavior similar to a dose-dependent effect on hospital mortality (P = 0.015 by linear-by-linear test).
Modified Rankin Scale and Maximum PaO2
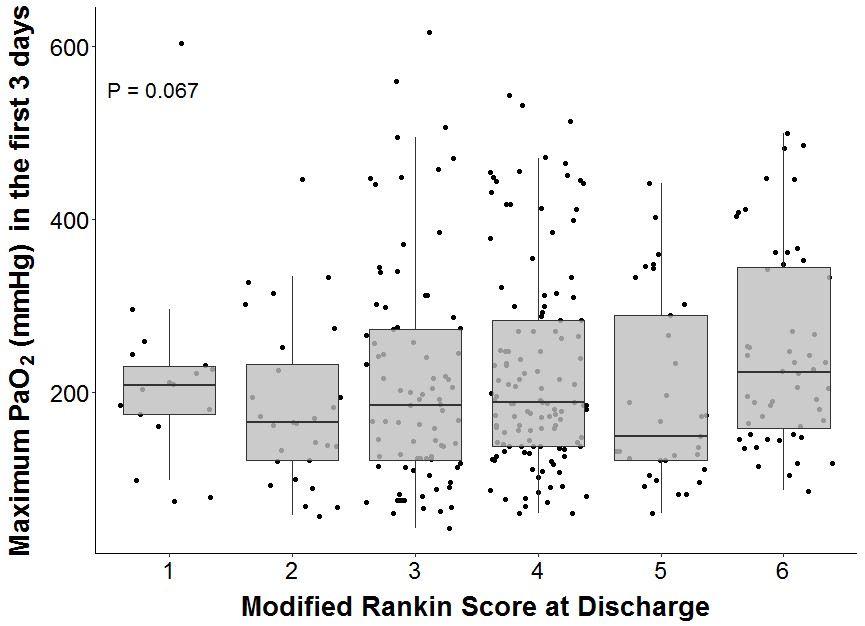
Although there was a trend towards an association between higher maximum PaO2 and lower modified Rankin Scale at discharge, this was not statistically significant (P=0.067, see Supplemental Figure II).
Discussion
Summary of Findings
Our results identify a novel and potentially modifiable risk factor for cerebral vasospasm after subarachnoid hemorrhage. We demonstrate that early hyperoxemia, as measured by maximum PaO2 within the first 72 hours after aneurysmal rupture, is associated with a higher incidence of cerebral vasospasm. Notably, this finding persisted across the range of severity of SAH and vasospasm. Additionally, the overall degree of hyperoxemia in our study was only moderate, with a mean maximum PaO2 of 219 mmHg in the study population, consistent with the use of low to moderate levels of supplemental oxygen. In addition to hyperoxemia, ethnicity, age and Hunt-Hess grade were independently associated with cerebral vasospasm, which is consistent with some of the existing literature28. Hyperoxemia was also associated with higher hospital mortality in a dose-dependent fashion in univariable analysis, although this association did not remain significant after adjustment for potential confounders.
Hyperoxemia is Common after Aneurysmal Rupture
Ideally, patients with aneurysmal subarachnoid hemorrhage are admitted to a neurologic intensive care unit where a multidisciplinary team can best manage their complex care. The initial focus is to secure the aneurysm by surgical or endovascular means to prevent re-bleeding. As a result, patients are often managed in multiple environments including the operating room, endovascular suite and intensive care unit and are managed by many teams including neuro-intensivists, neuro-anesthesiologists, stroke neurologists and cerebrovascular neurosurgeons. In concert with securing the aneurysm, there is intense attention paid to the key physiological imperative of maximizing cerebral perfusion through careful blood pressure management, often with the institution of induced hypertension using vasoactive support. In addition, the PaCO2 is carefully calibrated through adjustment of the minute ventilation to optimize cerebral perfusion and minimize cerebral ischemia. While blood pressure and PaCO2 are thus monitored closely by teams across all care environments, comparatively little attention has been paid to the PaO2. Indeed, in this study of over 345 aneurysmal SAH patients, there was wide variability in the maximum PaO2 measured in the first 72 hours after aneurysm rupture. In fact, patient arterial oxygen levels in our study ranged from approximately 60 to 600mmHg, confirming that PaO2 is not a parameter that is tightly titrated after SAH. This variability is not unique to aneurysmal SAH and is common across different diagnoses and various intensive care unit settings29.
Proposed Underlying Mechanism of Hyperoxemia and Vasospasm
Hyperoxemia is defined by excess oxygen content in the arterial blood. In hospital settings, hyperoxemia is caused by administration of excessive supplemental oxygen. While oxygen is critical to survival, supra-physiologic levels can drive damaging cellular responses through generation of reactive oxygen species leading to proinflammatory effects. Reactive oxygen species have been implicated in multiple deleterious processes including cell death30, cancer31, and aging32. We hypothesize that hyperoxemia in SAH promotes the upregulation of reactive oxygen species through the oxidation of cell-free hemoglobin in the subarachnoid space33. Oxidized extracellular hemoglobin subsequently drives lipid and protein oxidation to cause neuronal apoptosis and brain injury34. The excess free radicals further stimulate the hypersensitive arterial system to cause vasospasm35. In addition to effects on cell-free hemoglobin in the subarachnoid space, other postulated mechanisms of hyperoxemia-induced vasospasm include disrupted mitochondrial respiration, uncoupling of nitric oxide synthase, and disruption of intrinsic oxidative signaling pathways17,36,37.
Hyperoxemia Associates with Neurologic Morbidity
Hyperoxemia has been associated with adverse clinical outcomes in other clinical conditions. For example, in animal models, hyperoxemia can precipitate acute lung injury when the fraction of inspired oxygen (FIO2) exceeds 0.7 through generation of high levels of reactive oxygen species that overwhelm natural antioxidant defenses in the lung and cause destruction of cellular structures38. Hyperoxemia in premature infants with exposure to mean PaO2 values of 107.3 ± 59.3 mmHg has been correlated with compromised long-term neurodevelopmental outcomes39. In post-cardiac arrest patients, hyperoxemia is associated with higher mortality rate and worse Cerebral Performance Category Scale at hospital discharge among survivors40, while studies of hyperoxemia in the setting of stroke and traumatic brain injury studies have yielded mixed results41,42. Several recent clinical studies have attempted to examine whether any association exists between blood oxygen levels and outcomes following SAH, with varying results. A study by Yokoyama et al suggested that hyperoxemia, defined categorically as PaO2 > 120 mmHg, within the first 24 hours in an intensive care unit is associated with worse neurological disability defined as modified Rankin Scale at hospital discharge in patients with mild-to-moderate SAH43. Another study by Lång et al similarly examined arterial oxygen levels during the first 24 hours in an intensive care unit; this study did not demonstrate any association between oxygen levels and Glasgow Outcome Scale or mortality44. Both studies were potentially limited by their short observation windows since vasospasm most commonly occurs at least 72 hours post-SAH. A study by Jeon et al suggested an association between sustained hyperoxemia and delayed cerebral ischemia along with poor 3-month neurological outcomes in long-term mechanically ventilated patients; however, this publication was biased toward a more severely ill patient population than our study18. To our knowledge, this is the first study to demonstrate an independent association between early hyperoxemia and cerebral vasospasm in patients presenting with SAH of varying degrees of severity.
Study Limitations
The principal limitations of this study are attributable to its retrospective nature at a single academic center. Further prospective human studies at multiple institutions are required. The inclusion and exclusion criteria may limit the generalizability of the findings. Patients who died within the first 96 hours were excluded from this study since early death would preclude development of cerebral vasospasm, potentially confounding the analysis. Although exclusion of early deaths may have shifted the analysis to include less severely ill patients, the spectrum of Hunt-Hess grades was widely distributed across the cohort, thereby including patients of all hemorrhage severities. The requirement for an arterial blood gas analysis in the first 72 hours may have excluded less severely ill patients whose respiratory status could be managed without blood gas monitoring. Less severely ill patients may also have presented later than 72 hours after initial symptom onset, thereby excluding them from the study since no PaO2 values would have been obtained prior to hospital presentation. Conversely, since hyperoxemia is an iatrogenic phenomenon, the study inclusion criteria may introduce bias toward the more severely ill, mechanically ventilated patients. In attempts to mitigate this fact, hemorrhage severity scores and PaCO2 values were controlled for in the multivariable analyses. Lastly, we elected to use the highest PaO2 value in the first three days after hemorrhage as an index of hyperoxemia, but this reflects a single data point in time and is not representative of duration of time spent at this elevated value. All arterial blood gases were obtained for clinical reasons and thus the timing and number of blood gases was not standardized. A continuous measurement of oxygenation including time spent at each elevated value would provide a more accurate measurement of hyperoxemia, but this data was not available retrospectively. Despite these limitations, the study includes a diverse patient population across varying degrees of hemorrhage severity and multiple PaO2 measurements, which are reflective of a “real-world” sampling pattern.
Conclusions
In patients with aneurysmal subarachnoid hemorrhage, hyperoxemia is common within the first 72 hours after aneurysm rupture and is an independent predictor of cerebral vasospasm. Since hyperoxemia is readily measured and can be eliminated by titration of the level of supplemental oxygen, it may be an easily modifiable risk factor for cerebral vasospasm. Larger prospective studies are needed to validate the clinical significance of this finding.
Supplementary Material
Supplemental Figure II. Maximum PaO2 and modified Rankin Scale. Boxplot summary of maximum PaO2 in the first 3 days by modified Rankin Scale at discharge among survivors (n=336). 9 patients without available modified Rankin Scales were excluded. P = 0.067 by Kruskal-Wallis test.
{kind=link}
Supplemental Figure I. Flow chart of study population.
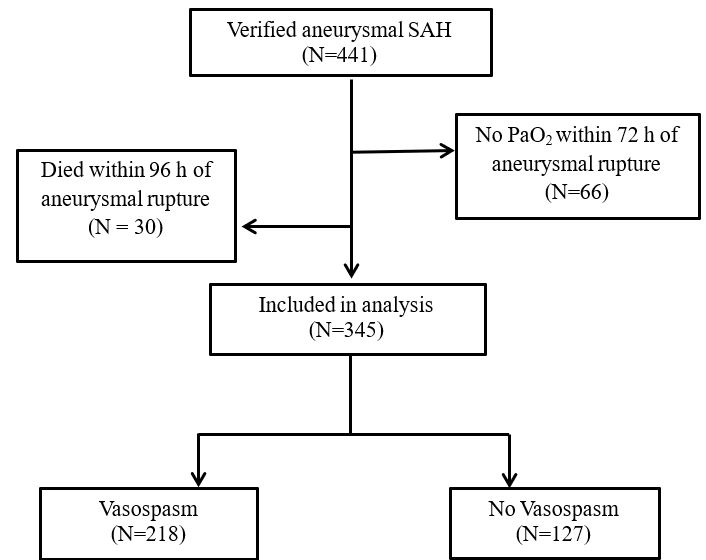{kind=link}
Acknowledgements
The authors would like to thank the REDCap team for assistance with secure data storage.
Sources of Funding
Dr. Ware was funded by NIH HL103836. Drs. Ware and Bastarache are funded by HL135849.
Financial support for this project included NIH grants HL103836 and HL135849.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Disclosures
Author LB Ware has received research support from Global Blood Therapeutics, CSL Behring, and Boehringer Ingelheim in the past and currently receives research support from Genentech. She also has received consulting fees from Citius, Foresee, Boehringer Ingelheim, Quark, CSL Behring and Merck. The authors have no other conflicts of interest concerning the materials or methods used in this study or the findings specified in this paper.
The content of this paper has not been presented or published previously.
References
- 1.Etminan N, Chang HS, Hackenberg K, et al. Worldwide Incidence of Aneurysmal Subarachnoid Hemorrhage According to Region, Time Period, Blood Pressure, and Smoking Prevalence in the Population: A Systematic Review and Meta-analysis. JAMA Neurol. 2019;76(5):588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vergouwen MD, Jong-Tjien-Fa AV, Algra A, et al. Time trends in causes of death after aneurysmal subarachnoid hemorrhage: A hospital-based study. Neurology. 2016;86(1):59–63. [DOI] [PubMed] [Google Scholar]
- 3.Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke. 2010;41(8):e519–536. [DOI] [PubMed] [Google Scholar]
- 4.Allen GS. Role of calcium antagonists in cerebral arterial spasm. Am J Cardiol. 1985;55(3):149b–153b. [DOI] [PubMed] [Google Scholar]
- 5.Francoeur CL, Mayer SA. Management of delayed cerebral ischemia after subarachnoid hemorrhage. Crit Care. 2016;20(1):277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel AS, Griessenauer CJ, Gupta R, et al. Safety and Efficacy of Noncompliant Balloon Angioplasty for the Treatment of Subarachnoid Hemorrhage-Induced Vasospasm: A Multicenter Study. World Neurosurg. 2017;98:189–197. [DOI] [PubMed] [Google Scholar]
- 7.Kwon HJ, Lim JW, Koh HS, et al. Stent-Retriever Angioplasty for Recurrent Post-Subarachnoid Hemorrhagic Vasospasm - A Single Center Experience with Long-Term Follow-Up. Clin Neuroradiol. 2018. [DOI] [PubMed] [Google Scholar]
- 8.Zimmermann M, Seifert V. Endothelin and subarachnoid hemorrhage: an overview. Neurosurgery. 1998;43(4):863–875; discussion 875–866. [DOI] [PubMed] [Google Scholar]
- 9.Pasqualin A. Epidemiology and pathophysiology of cerebral vasospasm following subarachnoid hemorrhage. J Neurosurg Sci. 1998;42(1 Suppl 1):15–21. [PubMed] [Google Scholar]
- 10.Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery. 1980;6(1):1–9. [DOI] [PubMed] [Google Scholar]
- 11.Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified fisher scale. Neurosurgery. 2006;59(1):21–27; discussion 21–27. [DOI] [PubMed] [Google Scholar]
- 12.Hosoda K, Fujita S, Kawaguchi T, et al. Effect of clot removal and surgical manipulation on regional cerebral blood flow and delayed vasospasm in early aneurysm surgery for subarachnoid hemorrhage. Surg Neurol. 1999;51(1):81–88. [DOI] [PubMed] [Google Scholar]
- 13.Macdonald RL, Weir BK. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke. 1991;22(8):971–982. [DOI] [PubMed] [Google Scholar]
- 14.Hugelshofer M, Sikorski CM, Seule M, et al. Cell-Free Oxyhemoglobin in Cerebrospinal Fluid After Aneurysmal Subarachnoid Hemorrhage: Biomarker and Potential Therapeutic Target. World Neurosurg. 2018;120:e660–e666. [DOI] [PubMed] [Google Scholar]
- 15.Bulters D, Gaastra B, Zolnourian A, et al. Haemoglobin scavenging in intracranial bleeding: biology and clinical implications. Nat Rev Neurol. 2018;14(7):416–432. [DOI] [PubMed] [Google Scholar]
- 16.Shaver CM, Wickersham N, McNeil JB, et al. Cell-free hemoglobin promotes primary graft dysfunction through oxidative lung endothelial injury. JCI Insight. 2018;3(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Chen S, Zhang JM. The Updated Role of Oxidative Stress in Subarachnoid Hemorrhage. Curr Drug Deliv. 2017;14(6):832–842. [DOI] [PubMed] [Google Scholar]
- 18.Jeon SB, Choi HA, Badjatia N, et al. Hyperoxia may be related to delayed cerebral ischemia and poor outcome after subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 2014;85(12):1301–1307. [DOI] [PubMed] [Google Scholar]
- 19.Roden DM, Pulley JM, Basford MA, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84(3):362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Marchis GM, Schaad C, Fung C, et al. Gender-related differences in aneurysmal subarachnoid hemorrhage: A hospital based study. Clin Neurol Neurosurg. 2017;157:82–87. [DOI] [PubMed] [Google Scholar]
- 21.Krishnamurthy S, Kelleher JP, Lehman EB, et al. Effects of tobacco dose and length of exposure on delayed neurological deterioration and overall clinical outcome after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2007;61(3):475–480; discussion 480–471. [DOI] [PubMed] [Google Scholar]
- 22.Lantigua H, Ortega-Gutierrez S, Schmidt JM, et al. Subarachnoid hemorrhage: who dies, and why? Crit Care. 2015;19:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Swieten JC, Koudstaal PJ, Visser MC, et al. Interobserver agreement for the assessment of handicap in stroke patients. Stroke. 1988;19(5):604–607. [DOI] [PubMed] [Google Scholar]
- 25.Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg. 1968;28(1):14–20. [DOI] [PubMed] [Google Scholar]
- 26.van der Steen WE, Leemans EL, van den Berg R, et al. Radiological scales predicting delayed cerebral ischemia in subarachnoid hemorrhage: systematic review and meta-analysis. Neuroradiology. 2019;61(3):247–256. [DOI] [PubMed] [Google Scholar]
- 27.Little R, Rubin D. Statistical Analysis with Missing Data. New York: John Wiley & Sons; 1987. [Google Scholar]
- 28.Torbey MT, Hauser TK, Bhardwaj A, et al. Effect of age on cerebral blood flow velocity and incidence of vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 2001;32(9):2005–2011. [DOI] [PubMed] [Google Scholar]
- 29.Helmerhorst HJ, Arts DL, Schultz MJ, et al. Metrics of Arterial Hyperoxia and Associated Outcomes in Critical Care. Crit Care Med. 2017;45(2):187–195. [DOI] [PubMed] [Google Scholar]
- 30.Peixoto MS, de Oliveira Galvao MF, Batistuzzo de Medeiros SR. Cell death pathways of particulate matter toxicity. Chemosphere. 2017;188:32–48. [DOI] [PubMed] [Google Scholar]
- 31.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931–947. [DOI] [PubMed] [Google Scholar]
- 32.Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med. 2013;60:1–4. [DOI] [PubMed] [Google Scholar]
- 33.Lucke-Wold BP, Logsdon AF, Manoranjan B, et al. Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review. Int J Mol Sci. 2016;17(4):497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaetani P, Lombardi D. Brain damage following subarachnoid hemorrhage: the imbalance between anti-oxidant systems and lipid peroxidative processes. J Neurosurg Sci. 1992;36(1):1–10. [PubMed] [Google Scholar]
- 35.Nishihashi T, Trandafir CC, Wang A, et al. Hypersensitivity to hydroxyl radicals in rat basilar artery after subarachnoid hemorrhage. J Pharmacol Sci. 2006;100(3):234–236. [DOI] [PubMed] [Google Scholar]
- 36.Pyne-Geithman GJ, Caudell DN, Prakash P, et al. Glutathione peroxidase and subarachnoid hemorrhage: implications for the role of oxidative stress in cerebral vasospasm. Neurol Res. 2009;31(2):195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciurea AV, Palade C, Voinescu D, et al. Subarachnoid hemorrhage and cerebral vasospasm - literature review. J Med Life. 2013;6(2):120–125. [PMC free article] [PubMed] [Google Scholar]
- 38.Kallet RH, Matthay MA. Hyperoxic acute lung injury. Respir Care. 2013;58(1):123–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deuber C, Terhaar M. Hyperoxia in very preterm infants: a systematic review of the literature. J Perinat Neonatal Nurs. 2011;25(3):268–274. [DOI] [PubMed] [Google Scholar]
- 40.Janz DR, Hollenbeck RD, Pollock JS, et al. Hyperoxia is associated with increased mortality in patients treated with mild therapeutic hypothermia after sudden cardiac arrest. Crit Care Med. 2012;40(12):3135–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Damiani E, Adrario E, Girardis M, et al. Arterial hyperoxia and mortality in critically ill patients: a systematic review and meta-analysis. Crit Care. 2014;18(6):711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujita M, Oda Y, Yamashita S, et al. Early-Stage Hyperoxia Is Associated with Favorable Neurological Outcomes and Survival after Severe Traumatic Brain Injury: A Post-Hoc Analysis of the Brain Hypothermia Study. J Neurotrauma. 2017. [DOI] [PubMed] [Google Scholar]
- 43.Yokoyama S, Hifumi T, Kawakita K, et al. Early Hyperoxia in The Intensive Care Unit is Significantly Associated With Unfavorable Neurological Outcomes in Patients With Mild-to-Moderate Aneurysmal Subarachnoid Hemorrhage. Shock. 2019;51(5):593–598. [DOI] [PubMed] [Google Scholar]
- 44.Lang M, Raj R, Skrifvars MB, et al. Early Moderate Hyperoxemia Does Not Predict Outcome After Aneurysmal Subarachnoid Hemorrhage. Neurosurgery. 2016;78(4):540–545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure II. Maximum PaO2 and modified Rankin Scale. Boxplot summary of maximum PaO2 in the first 3 days by modified Rankin Scale at discharge among survivors (n=336). 9 patients without available modified Rankin Scales were excluded. P = 0.067 by Kruskal-Wallis test.
Supplemental Figure I. Flow chart of study population.