

Abstract
Membrane proteins are incredibly important biomolecules because they mediate interactions between a cell’s external and internal environment. Obtaining information about membrane protein structure and interactions is thus important for understanding these essential biomolecules. Compared to the analyses of water-soluble proteins, the structural analysis of membrane proteins is more challenging owing to their unique chemical properties and the presence of lipid components that are necessary to solubilize them. The combination of covalent labeling (CL) and mass spectrometry (MS) has recently been applied with great success to study membrane protein structure and interactions. These studies have demonstrated the many advantages that CL-MS methods have over other traditional biophysical techniques. In this review, we discuss both amino-acid specific and non-specific labeling approaches and the special considerations needed to address the unique challenges associated with interrogating membrane proteins. This review highlights the aspects of this approach that require special care to be applied correctly and provides a comprehensive review of the membrane protein systems that have been studied by CL-MS.
Keywords: Covalent Labeling, Diethylpyrocarbonate, Hydroxyl Radical, Mass Spectrometry, Membrane Proteins, Membrane Protein Structural Analysis, Higher-order Structure, Membrane Protein Interactions, Fast Photochemical Oxidation of Proteins
I. INTRODUCTION
Membrane proteins, comprising 30% of the proteome, perform numerous cellular functions in living organisms, such as cell signaling, transport, cell adhesion, catalysis and more. They are incredibly important biomolecules because they mediate interactions between a cell’s external and internal environment. Their importance is further emphasized by the fact that more than 60% of FDA-approved drugs target membrane proteins (Yin & Flynn, 2016), with a remarkable 19% of these drugs directed toward ion channels (Santos et al., 2017). However, compared to water-soluble proteins, it is difficult to characterize their higher order structures and to study their binding interactions owing to their partially hydrophobic surfaces, flexibility and lack of stability (Carpenter, Beis, Cameron, & Iwata, 2008). Moreover, when the natural lipid bilayer is removed, the exposure of hydrophobic surfaces usually results in protein denaturation and/or aggregation. Several approaches have been developed to overcome these hurdles. One common approach is to study only the soluble domain of the protein. This method enables the use of all the tools that have been developed for soluble proteins, but it limits structural information and does not represent the full three-dimensional context of the protein or its membrane environment. An alternative method is to embed the protein into an artificial membrane, such as micelles, bicelles, amphipols, or lipid nanodiscs, which mimic cell membrane environments. This approach allows the full membrane protein to be studied in the context of a lipid environment, even though it does not always fully account for diversity of components that are present in real cell membranes.
Techniques for membrane protein structure investigations include X-ray crystallography (McPherson, 2004), cryo-electron microscopy (cryo-EM) (Thonghin, Kargas, Clews, & Ford, 2018), solution or solid-state nuclear magnetic resonance (NMR) (Liang & Tamm, 2016), and single-molecule tracking with fluorescence spectroscopy (Alcor, Gouzer, & Triller, 2009; Christie, Shi, & Smith, 2020; García-Sáez & Schwille, 2007). However, despite these techniques, the number of membrane protein with known structures is 2488 or 1.5% of the 165422 structures in Protein Data Bank. This relatively low number of structures is due to the significant challenges associated with applying traditional structural analysis methods to membrane proteins. For example, co-crystallization of membrane protein with artificial membranes is difficult, limiting the use of X-ray crystallography. NMR is typically more applicable, but molecular weight restrictions and difficulties with analyzing mixtures limits its broad usage for membrane proteins. Cryo-EM has great promise for membrane protein structural analysis, although its resolution for intact cell studies is currently limited.
The popularity of utilizing mass spectrometry (MS) to study membrane proteins is on the rise because of its high sensitivity, good structural resolution, relatively high-throughput, and ability to study proteins in complex mixtures (Calabrese & Radford, 2018). In native MS, protein non-covalent interactions can be maintained upon ionization so that it can provide protein structural information and binding stoichiometries. Amphipols, nanodiscs, and bicelles are often used to directly analyze membrane proteins in native MS because they solubilize the full protein and are compatible with electrospray ionization. Ion mobility (IM)-MS, coupled with collision-induced dissociation (CID), collision-induced unfolding (CIU), or surface-induced dissociation (SID) can then report protein structural information, albeit with low resolution. Hydrogen/deuterium exchange (HDX)-MS can provide more highly resolved information for membrane proteins and yields insight into solvent accessible regions of the protein main chain and backbone dynamics that can reflect conformational changes upon interactions with ligands (Hebling et al., 2010; Reading et al., 2017; Vahidi, Bi, Dunn, & Konermann, 2016). Similarly, chemical cross-linking (XL)-MS provides structural information at the residue level for protein regions that are adjacent to one another. XL-MS also can be applied to study subunit connectivity of membrane protein complexes and protein interactions (Komolov et al., 2017; Liu et al., 2013).
Covalent labeling (CL)-MS is also emerging as a complementary technique to HDX and has some advantages over XL-MS in terms of measurement simplicity. CL-MS has been used study membrane protein structure and interactions due to some of its inherent advantages. In CL-MS, a reagent covalently modifies solvent exposed side chains on a protein or protein complex. The resulting modification is typically very stable, allowing the use of optimized proteomics techniques to facilitate its localization. Because the reagent leads to essentially a ‘dead-end’ modification, analysis of the resulting MS and MS/MS data is simpler than in XL-MS methods. A variety of labeling chemistries are available, and they fall into two general categories – residue non-specific reagents (e.g. hydroxyl radical) and residue specific reagents (e.g. N-ethylmaleimide for Cys residues) (Mendoza & Vachet, 2009; Xu & Chance, 2007). When CL-MS methods are used to study membrane proteins, the covalent modification readily withstands the purification steps necessary to facilitate membrane protein analysis. The typical steps involved are illustrated in Figure 1. After the labeling reaction, the membrane components are removed, the proteins are digested, and the resulting fragments are separated by liquid chromatography (LC) and analyzed by tandem mass spectrometry (MS/MS) to identify the modified residues. Comparing modification percentages of a given protein under one condition (e.g. without ligand) to that of the same protein under another condition (e.g. with ligand) can reveal conformational changes, ligand binding sites, and protein-protein interfaces often with residue-level resolution. CL-MS is not subject to back exchange and label scrambling, and data interpretation is typically more straightforward.
Figure 1.

In covalent labeling-mass spectrometry (CL-MS) techniques, a chemical reagent modifies solvent accessible side chains of membrane proteins, followed by lipid removal, enzymatic digestion, and LC-MS/MS analysis to determine modification sites. Comparing modification levels under one condition (e.g. without ligand) to another (e.g. with ligand) can reveal interactions sites or conformational changes.
While several excellent reviews on hydroxyl radical footprinting and CL-MS of soluble proteins have appeared elsewhere (Chance, Farquhar, Yang, Lodowski, & Kiselar, 2020; Kiselar & Chance, 2018; Limpikirati, Liu, & Vachet, 2018; Liu, Zhang, & Gross, 2020; Mendoza & Vachet, 2009; Niu & Gross, 2019), no review solely focused on CL-MS of membrane proteins has appeared recently. In this review, we focus on the chemistry and methods that are unique to membrane proteins. In particular, we highlight the technical challenges and innovations associated with conducting CL experiments on membrane protein both in vitro and in vivo. We also describe several ways in which CL-MS has been applied to understand better membrane protein structures as well as membrane protein-ligand or protein-protein interactions. Proximity-dependent labeling methods (Chen & Perrimon, 2017; Roux, Kim, & Burke, 2013; Samavarchi-Tehrani, Samson, & Gingras, 2020), such as those that use horseradish peroxidase (HRP), ascorbate peroxidase (APEX), or biotin ligase (BioID), are not included in this review because we focus on direct labeling techniques that do not require molecular biology on the proteins of interest.
II. RESIDUE NON-SPECIFIC LABELING
A. Labeling Reagents
Residue non-specific reagents are able to simultaneously modify a wide variety of amino acid side chains in CL experiments. Among those reagents for membrane protein studies, hydroxyl radical is the most commonly used because they can label almost all amino acid side chains in proteins. Hydroxyl radicals (•OH) can be generated through synchrotron water radiolysis or UV laser photolysis of H2O2 (248 nm or 266 nm) (Li, Shi, & Gross, 2018; Maleknia, Brenowitz, & Chance, 1999; Zhang, Cheng, Rempel, & Gross, 2018), and when used to oxidatively modify proteins, the process has been termed synchrotron-based hydroxyl radical footprinting (HRF) or fast photochemical oxidation of proteins (FPOP), respectively (Figure 2). The labeling reactions usually occur on the ~ μs to ms timescale (Gau, Sharp, Rempel, & Gross, 2009; Hambly & Gross, 2005; Takamoto & Chance, 2006), which helps avoid labeling-induced structural changes during the course of the experiment, as proteins are labeled faster than they can typically undergo structural changes. Although the reactions of proteins with hydroxyl radical produce over 50 different product types, which can complicate MS analyses (Xu & Chance, 2007), the predominant products are the addition of a hydroxyl group with a mass shift of +16 Da or a carbonyl group with a mass shift of +14 Da (Xu & Chance, 2005). Sulfur-containing residues and aromatic residues have the highest intrinsic reactivity. Because the amount of •OH generated in solution during HRF or FPOP is in large excess, the extent of oxidative labeling tends to correlate with the solvent accessible surface area (SASA) of amino acid side chains, which can provide insight about protein structure and their interactions (Deperalta et al., 2013; Konermann, Stocks, Pan, & Tong, 2010; Liu, Rempel, & Gross, 2019). In contrast to synchrotron HRF and FPOP, which produce hydroxyl radical everywhere in solution, •OH can also be produced site specifically at a transition metal center via Fenton and Fenton-like chemistry. These metal-catalyzed oxidation (MCO) reactions selectively oxidize residue side chains that are bound or near the metal center (Bridgewater, Lim, & Vachet, 2006; Bridgewater & Vachet, 2005; Lim & Vachet, 2003).
Figure 2.

(a) Scheme illustrating the main reactions during synchrotron-based hydroxyl radical footprinting (HRF). Activated water (H2O*), hydrated electrons (eaq−) and •OH radical are generated via radiolysis of bulk water. Then, •OH radical can be quenched by buffer, self-associate to form peroxide, or label protein surface residues. (b) Scheme illustrating an FPOP platform. Figures used with permission from Liu et al., 2020, DOI: 10.1021/acs.chemrev.9b00815.
Carbenes are another residue non-specific labeling reagent that has been used for membrane protein analysis. Carbenes are generated from the photolysis of diazirine derivatives using near-UV light (~ 350 nm). This labeling reagent can irreversibly insert into any X-H bond (X stands for C, O, N, or S) and generate a variety of products, which can lead to some difficulties in getting residue-level information (Limpikirati et al., 2018; Manzi et al., 2017). The challenge of pinpointing carbene labeling sites requires a different approach to data analysis than is typically used in HRF or FPOP (Jumper, Bomgarden, Rogers, Etienne, & Schriemer, 2012; Ziemianowicz, Sarpe, & Schriemer, 2019). Most recently, labeling methods based on trifluoromethyl radicals (•CF3) have been reported. Trifluoromethyl radicals can be generated by laser or synchrotron irradiation of triflinate anions (Cheng et al., 2020), and these radicals can insert into X-H bonds of amino acid side chains. Trifluoromethyl radicals tend to label all amino acid residues, except Met and Cys, which makes the reagent complementary to hydroxyl radical which has the highest reactivity with these sulfur-containing residues (Liu et al., 2020).
B. Technical Challenges of Residue Non-specific Labeling Experiments on Membrane Proteins
Covalent labeling of membrane proteins relies on many of the same approaches used for soluble proteins, but additional considerations and steps in the digestion and separation procedures are typically necessary due to inherent properties of membrane proteins. Moreover, the presence of lipids, which are necessary to solubilize and stabilize membrane proteins, can affect the experimental procedures and labeling results. Below we indicate the technical challenges associated with non-specific labeling reagents, but it should be recognized that many of these same challenges are present when using residue specific labeling reagents. For example, the same challenges associated with the choice of the artificial membrane system, artificial membrane removal, and membrane protein digestion are common to both residue non-specific labeling experiments and residue specific labeling experiments.
1. Choice of artificial membrane
Artificial membranes are a common way to solubilize, enclose and stabilize membrane proteins in aqueous solutions (Figure 3). Detergents that form micelles when used at concentrations above their critical micelle concentration (CMC) are commonly used artificial membranes. In principle, detergent micelles mimic natural lipid bilayers and ensure the native conformations of both the membrane-embedded and soluble domains of the protein, while at the same time allowing the protein to interact with other molecules. However, in practice, micelles do not have ideal shapes to match the proper curvature of real cell membranes, and their sizes can vary even in the same sample (Lu et al., 2016). Moreover, if a given membrane protein has two soluble domains with an intervening transmembrane domain, micelles cannot properly recapitulate the structure of such membrane proteins. Another option is amphipols, which are amphiphilic polymers capable of embedding membranes proteins. They are more stable than simple detergents but have some of the same limitations as micelles. In bicelles, membrane proteins can be encapsulated by a lipid bilayer in a manner that better mimics the native cell-membrane environment, especially for proteins with transmembrane domains (Cavagnero, Dyson, & Wright, 1999). Other important membrane components, such as cholesterol and cardiolipin, can be doped into bicelles to further mimic the diversity of biological membranes. However, one challenge with bicelles is that there morphology can change at low lipid concentrations (Lu et al., 2012), which can be limiting in some applications. Nanodiscs have grown in popularity recently for their ability to solubilize membrane proteins. They use not only a lipid bilayer to stabilize the membrane protein but also membrane scaffold proteins to encircle the bilayer. Nanodiscs are well defined in size and more stable than micelles and bicelles at low concentrations (Roos et al., 2014). Liposomes can also be used to reconstitute and solubilize membrane proteins. They have a lipid bilayer that can form a spherical shape that is morphologically analogous to cells, thereby allowing proteins with transmembrane domains to be properly structured. A challenge, however, is that most membrane proteins have a proper orientation, and ensuring that each soluble domain presents itself on the appropriate side of the liposome typically proves difficult.
Figure 3.

Scheme illustrating some artificial membranes that are used to solubilize and stabilize membrane proteins. a) micelles, b) amphiphols, c) bicelles, d) liposomes, e) nanodiscs.
2. Choice of labeling reagents
An important characteristic of membrane proteins is the presence of large hydrophobic domains, which typically embed or span the membrane, and large soluble domains that orient outside or inside the cell. A unique feature of CL, as opposed to HDX, is the ability to use reagents that can target one domain or the other. Depending on the goals of the experiment, water-soluble reagents are used to probe solvent exposed side chains, whereas lipophilic reagents are used to probe residues that are buried in membrane regions. For example, hydroxyl radical has been used to label soluble domains of the membrane protein while some carbenes can modify residues in hydrophobic transmembrane regions due to their amphipathic properties (Manzi et al., 2017). Although trifluoromethyl radicals are hydrophobic, they do not readily diffuse into the membrane and thus only modify extra-membrane domains (Cheng, Zhang, Cui, & Gross, 2017). This inability to penetrate the membrane may result from the well-known poor interactions between fluorocarbons and hydrocarbons.
3. Reagent concentrations and reaction time optimization
Optimizing reaction conditions is necessary to ensure detectable and reproducible levels of labeling while at the same time maintaining a protein’s conformation integrity during the CL experiments. Structural perturbations are minimized when a protein’s exposure to the residue non-specific reagents (e.g. •OH) is kept on the μs timescale. In FPOP, this is achieved by tuning sample flow rate, laser pulse width, and irradiation frequency, so that proteins in solution are only illuminated once. Radical scavengers such as the amino acid Glu or adenine are also added to minimize the •OH lifetime, which not only prevents over-labeling but improves reproducibility (Liu et al., 2020). In HRF, dose-response plots that measure labeling kinetics are often used to provide additional assurance that a protein’s structural integrity has been maintained. Reproducible labeling in both FPOP and HRF is necessary to properly compare proteins under different conditions (e.g. with ligand vs. without ligand), and dosimetry methods have been described to maximize method reproducibility. Typically, reproducible labeling in FPOP is accomplished by measuring the effective hydroxyl radical concentrations via the UV absorbance of adenine at 265 nm upon oxidation (Sharp, Misra, Persoff, Egan, & Weinberger, 2018). In synchrotron-based HRF, the dose of •OH is reported by monitoring the radiolytic degradation of the Alexa 488 fluorophore (Gupta, Sullivan, Toomey, Kiselar, & Chance, 2007). As long as the dose of hydroxyl radical is controlled properly, it appears then that membrane proteins can be studied by HRF and FPOP in a manner analogous to soluble proteins.
4. Scavenging effects of membrane components and other molecules
One concern that could arise when studying membrane proteins is the potential for the membrane components and other molecules to react with the labeling reagents. Often, the decreased modification levels caused by sample components can be addressed by simply increasing the concentration of the labeling reagent. For example, to compensate for the •OH scavenging effects of artificial membranes, the X-ray exposure time in HRF of membrane proteins is increased, while still controlling •OH levels by radical dosimetry approaches (Angel, Gupta, Jastrzebska, Palczewski, & Chance, 2009). Because hydroxyl radical is usually present in excess to the protein and membrane components in FPOP and HRF experiments, the presence of these additional components are not expected to significantly affect overall reactivity as long as the proper radical levels are maintained. Similarly, the reaction of carbenes with detergents decreases the modification events at residue side chains, and the diazirine precursor reagent concentrations need to be increased accordingly (Manzi et al., 2017). A more challenging situation arises when other molecules are added to change the structure or conformation of the membrane protein. For example, Chance and co-workers used HRF to study the open and closed states of a potassium channel, and used EDTA, a well-known radical scavenger, to achieve the open state (Gupta et al., 2010). By conducting the radical dosimetry approaches, the authors found that 1 mM EDTA reduces the hydroxyl radical dose by a factor of 2.5. Consequently, they normalized the measured residue rate constants in the open state by a factor of 2.5 to allow a better comparison to the closed state. While this normalization factor seems reasonable, this simple approach may not work for every labeling reagent, and so such a correction should be approached with caution.
In addition, the lipid components of the membrane system can be oxidized, and lipid oxidation is a very complicated process. Lipid oxidation starts with the reaction of double bonds in polyunsaturated fatty acids with free radicals (e.g. hydroxyl radical). The resulting lipid hydroperoxides that are generated decompose to form multiple secondary compounds such as alcohols, aldehydes, and ketones (Domínguez et al., 2019). Several studies have pointed out that lipid oxidation has a significant impact on membrane physical properties. In brief, lipids will reorganize their packing due to the change of their structures after oxidation. For example, Jacob et al. and Ayuyan et al. have found the formation of large cholesterol rafts that decrease the width of lipid bilayers (Ayuyan & Cohen, 2006; Jacob & Mason, 2005). Bonneau and coworkers observed the shape transition of giant unilamellar vesicles after oxidation (Heuvingh & Bonneau, 2009). These changes influence membrane fluidity, permeability, its thermodynamics properties, and how membrane proteins are embedded in lipids. Hence, HRF approaches could oxidize lipids, causing the deformation of the lipid bilayer and subsequently influencing membrane protein structures. Typically HRF experiments are conducted in such a way that oxidation only occurs on the micro- to millisecond timescale, which should minimize changes in membrane properties that could influence protein conformation, but possible membrane changes should not be ignored. Moreover, radicals could propagate from oxidized lipids to membrane proteins on a longer timescale (> ms) via free-radical mediated chain reactions (Porter, Caldwell, & Mills, 1995; Pratt, Tallman, & Porter, 2011), giving rise to the possibility of indirect modifications to the protein of interest. While we are not aware that such a phenomenon has been described during HRF experiments, it is possible that researchers have not investigated this possibility. If such radical propagation reactions do occur, the correlation of modification extent with residue solvent accessibility would likely decrease, as membrane embedded regions could be oxidized.
5. Artificial membrane removal
After labeling experiments, the sample components that solubilize the membrane protein (e.g. lipids in the artificial membrane) usually must be removed to facilitate the subsequent digestion and LC-MS/MS analysis. Several approaches are used to achieve this purification. In one approach a mixture of methanol, water, and chloroform are used to separate the proteins from the lipids. In this approach, proteins precipitate and remain at the interface of the aqueous and organic layers, while the lipids dissolve in the organic phase. In another approach, trichloroacetic acid (TCA) and acetone are used for precipitation. In this solvent system, lipids dissolve in acetone but proteins do not. Then, the lipids are removed easily through centrifugation. More sophisticated approaches rely on expressing membrane protein with affinity tags that bind to spin columns, while lipids are washed away before later eluting the bound proteins. Lipids can also be separated from proteins/peptides via reverse phase (RP) liquid chromatography columns either before enzymatic digestion or during LC/MS analyses due to the differences in the retention of lipids and proteins.
6. Membrane protein digestion
In most cases, membrane proteins can be digested by commonly used proteolytic enzymes, such as trypsin, chymotrypsin, Lys-C, etc. These enzymes usually provide high sequence coverage for the soluble domains of the membrane protein after protein denaturation; however, it can be difficult to digest hydrophobic transmembrane and membrane-embedded domains owing to their hydrophobicity and lack of residues recognized by proteases. Using multiple enzymes to digest the protein is one way to overcome this challenge. A second method is to perform in-gel protease digestions. In this approach, the protein bands of interest are excised, disulfide bonds are reduced and alkylated, the protein is destained and then enzymatically digested all in the gel where the protein had been previously solubilized by sodium dodecyl sulfate (SDS) (Shevchenko, Tomas, Havli, Olsen, & Mann, 2006). Another common challenge in analyzing membrane proteins is the poor sequence coverage that results in LC/MS analyses because of the poor LC separation of highly hydrophobic peptides. Often, mobile phase and gradient conditions that are normally used for soluble proteins are unsuitable. A common solution is to use a shallower gradient of the organic mobile phase to better separate the many hydrophobic peptides that can be produced during enzymatic digestion.
C. Applications of Residue Non-specific Labeling to Membrane Proteins
1. Structural characterization
Because the extent of labeling is related to the solvent accessibility of amino acid side chains, CL-MS techniques can provide information about a protein’s higher-order structure. An early example of using non-specific labeling reagents to study membrane proteins was by Konermann and co-workers, who utilized FPOP to characterize the structure of bacteriorhodopsin (BR) in Halobacterium salinarum purple membrane suspensions (Pan, Stocks, Brown, & Konermann, 2009). Only Met residues in the protein were labeled owing to their high intrinsic reactivity and the relative high percentage of this residue in BR. Three Met residues in solvent-exposed loops were found to have higher extents of modification than the remaining six Met residues that are in transmembrane helices, indicating that FPOP can identify residues in the soluble domain of membrane proteins. The conformational changes of BR under semi-denaturing conditions (low pH, SDS exposure, and heat) were studied as well (Pan, Brown, & Konermann, 2009). Surprisingly, the labeling pattern of BR at low pH is similar to the protein in native state, indicating that unlike other proteins, BR does not have large conformational changes at low pH. When BR was heated or treated with SDS, two additional Met residues are labeled, indicating that partial unfolding of some helices is necessary to oxidize these residues.
The refolding of SDS-denatured BR in the presence of bicelles and free retinal molecules was also studied by producing •OH in ~1 μs increments at various time points after mixing with the artificial membranes (Pan, Brown, & Konermann, 2011). The fraction of unmodified M20 in transmembrane helix A increases significantly from 0 to 20 ms, showing the residues in this region are protected by the refolding of helix A. In this early stage of folding, BR forms an intermediate, I1, and then it refolds to the intermediates I2 and IR after binding with the retinal chromophore. The interaction between the retinal chromophore and the protein reduces the solvent accessibility of M118, as revealed by the declining oxidation level of M118 on helix D from 0.5 s until 4 s. Finally, BR folds into IR* at 4 s, IR** at 10 s, and native state. These early FPOP experiments on a well-studied membrane protein demonstrated the great potential of oxidative labeling for understanding membrane protein structure and folding.
When structural changes are significant enough, HRF can be used to identify physiologically relevant conformational changes in membrane proteins. One of the first examples of such studies was work by Chance and co-workers in which the open and closed states of the potassium channel KirBac3.1 in the detergent n-tridecyl-b-D-maltopyranoside were investigated with millisecond HRF (Gupta et al., 2010). From modification rate constant measurements of residues in the open and closed states, it was found that L122 and L124 on one of the transmembrane helices, M94 near the pore cavity, and T84, D85, F88, P107, and N110 at the selectivity filter react faster with hydroxyl radical during channel opening (Figure 4). The residues near the selectivity filter that have increased labeling rates were consistent with prior postulations that an additional gating mechanism could exist. Importantly, changes in modification rates for some residues at the slide-helix/N-terminal linker region and G-loop/CD-loop region were some of the first direct experimental evidence for the role of the G-loop as a gate in the cytoplasmic domain of the protein.
Figure 4.

Crystal structure of potassium channel KirBac3.1 in the closed state. Oxidatively modified residues are shown as sticks. The magnitudes of the changes in modification rate constants from the closed to open state are color-coded. Figure used with permission from Gupta et al., 2010, DOI: 10.1016/j.str.2010.04.012
Synchrotron HRF has also been used to study solvent exposed regions in the photosystem II (PS II) membrane protein, including presumed water channels (Frankel et al., 2013). The PS II protein consists of four subunits (CP47, CP43, D1, and D2), and two other subunits from cytochrome b559. Around 75% of the residues that are oxidatively modified sit on the surface of PS II. More interesting is the fact that some residues located in the interior of the protein complex were labeled as well, including M331, E333, R334, and E323, which map to a putative water channel. These experimental results complemented prior computational studies by identifying residues involved in transporting water to the active metal center that then convert water to oxygen. The HRF results in this study also led the authors to posit that another channel existed that functions as an O2 or ROS exit pathway. The ability to identify water channels or structural water molecules in transmembrane regions appears to be a unique capability of synchrotron HRF because it can dissociate water molecules into hydroxyl radical that directly oxidize Phe, Cys, Met, and Tyr residues, which are often found in transmembrane segments.
An example of HRF being used to examine the role of structurally important water molecules in integral membrane proteins is the work on the G protein-coupled receptor (GPCR) rhodopsin (Rho) by Chance and co-workers (Angel et al., 2009). Rho, which is a light-activated membrane protein essential for vision, was studied by HRF in its ground state, photo-activated state, and inactive state to understand the role of crystallographically observed water molecules on the structure and function of this important protein. By rapidly mixing H218O into the samples and performing HRF, the researchers were able to show that residues in the transmembrane helical bundle were labeled with 16O from localized structural H216O that had limited exchange with bulk H218O, whereas residues in the soluble domain were modified with 18O from bulk H218O. The 16O labeling, and lack of 18O labeling, only in the transmembrane regions was independent of the protein state, indicating that the structural waters in the transmembrane segments do not exchange with bulk solvent even upon activation or inactivation. Instead, Rho activation changed the sites of oxidation in the transmembrane helical bundle, suggesting that protein and water dynamics are an important part of the mechanism controlling Rho activity. Overall, the structural and dynamic information obtainable by HRF provides a valuable complement to X-ray crystallography and NMR studies of membrane proteins.
Since the level of covalent labeling reflects the solvent accessibility and intrinsic reactivity of modifiable residue side chains, oxidative modification by hydroxyl radical can be used to assess the accuracy of computational protein structure prediction (Pan, Ruan, Valvano, & Konermann, 2012). WaaL, a membrane-bound O-antigen ligase from E. coli with unknown higher order structure, was studied by oxidative footprinting. High modification extents on M19, M291, and M316 and the lack of modification on other Met and Cys residues were consistent with an algorithm that predicts transmembrane helices in protein sequences. However, one Met residue, M151, that was predicted to reside in a transmembrane helix was modified as well. This result led to a refined structural model for WaaL that placed this residue closer to the periplasmic interface.
Complementary to hydroxyl radical, •CF3, which can label 18 of the 20 common amino acid residues, has been used to investigate membrane proteins. Gross and co-workers used this reagent to characterize the structure of vitamin K epoxide reductase (VKOR) (Cheng et al., 2017). Ten residues on the extra-membrane regions were found to be labeled, including Ala and Gly residues, which show low reactivity towards •OH. Surprisingly, •CF3 did not modify any transmembrane regions despite its hydrophobicity. We speculate that the poor interactions between fluorocarbons and hydrocarbons prevent the diffusion of trifluoromethyl radicals into the membrane.
Covalent labeling with hydroxyl radical can also be used in a complementary fashion with HDX/MS to obtain greater insight into the conformational changes undergone by membrane proteins. As an example, the glycerol facilitator (GF) from E. coli, which is a homotetramer where each monomer contains a transmembrane channel allowing the selective permeation of glycerol and water, was interrogated by HDX-MS and FPOP (Pan, Piyadasa, O'Neil, & Konermann, 2012). Each monomeric unit in the protein consists of six helices and two half-helices that cross the membrane bilayer and two extra-membrane helices (EM1 and 2) that make up the soluble domain. Upon performing FPOP experiments on n-dodecyl-D-maltoside (DDM)-solubilized GF, Konermann and co-workers monitored the labeling extents of 13 Met, Cys, and Trp residues. They observed that the completely or partially solvent exposed residues were primarily labeled (red in Figure 5), and the buried residues were generally not labeled (blue in Figure 5). Surprisingly, C134 and M237, which are found buried in the crystal structure of GF, were modified by •OH, suggesting that the half-helical transmembrane segment in which these residues are found is only marginally stable. HDX-MS results were consistent with this observation, showing moderate conformational dynamics in this region. From these results, the authors postulated that the flexibility of this transmembrane helix is necessary to strike a balance between selectivity and efficient transport of glycerol and water.
Figure 5.

Structure of the glycerol facilitator (GF). The transmembrane segment, TM7, is shown in orange. Modified residues are shown in red while unmodified ones are shown in blue. The dashed line represents the region of the protein near the N-terminus that is not resolved in the crystal structure. Figure used with permission from Pan, Piyadasa, O'Neil, & Konermann, 2012, DOI: 10.1016/j.jmb.2011.12.052
2. Protein-ligand and protein-protein interactions
The interactions between proteins and ligands and proteins with other proteins lead to buried surfaces and conformational changes, both of which lead to changes in residue solvent accessibility. Thus, CL-MS can be a powerful way to map binding interfaces and discover structural changes by comparing the differences in labeling extent with and without ligand/protein. A common class of membrane proteins that undergo conformational changes upon protein or ligand binding are GPCRs, and they have been the focus of several CL-MS studies. In addition to using HRF to study the role of structural waters in the GPCR Rho, Chance and co-workers examined the conformational changes associated with photoactivation of Rho and the subsequent interactions of this activated state with the G protein transducin (Gt) (Orban et al., 2012). By comparing the modification rate constants of residues in the ground state, photo-activated state, and the complex of activated Rho and Gt, it was found that Gt binding causes the formation of a structural state in Rho that is different from the ground state or photoactivated uncomplexed state. Specifically, the researchers found that the oxidation rates of residues A346, M86, P303, and M288 significantly change in Rho-Gt complex compared to Rho and activated Rho, allowing some molecular details about this new structural state to be revealed.
Du et al. have demonstrated the merits of time-resolved HRF together with rapid mixing in their CL-MS studies of the β2 adrenergic receptor (β2AR), which is another GPCR (Du et al., 2019). In this work, the early conformational changes of β2AR forming a complex with the G protein Gαsβ1γ2 (Gs) were studied by pulse-labeling of hydroxyl radical at various time points ranging from 20 ms to 30 s after mixing (Figure 6). The duration of X-ray exposure (~ 50 μs) and the inherently fast reactivity of hydroxyl radical allowed the researchers to capture early conformational changes. These conformational changes revealed transient intermediates that seem to be important as selectivity filters and are states that are not evident from crystal and cryo-EM structures. The residue-specific information possible with HRF pinpointed the conformational changes to specific α-helices and β strands that likely explain GDP release, which is important for eventual GTP binding and activation.
Figure 6.

A) The oxidation levels of ICL2 on β2AR decrease over time because this region forms an α-helix from an unstructured loop upon binding. B) The change of labeling extent of M386 reveals that the interactions of C-terminus of Gαs with β2AR exposes its side chain and then forms a stabilized α-helix. C) The rise in oxidative modifications of M221 and V375/F376 is explained by the conformational rearrangement after GDP release. When these residues have new interactions with other hydrophobic residues, the labeling level drops. Figure used with permission from Du et al., 2019, DOI: 10.1016/j.cell.2019.04.022
A proton-coupled Zn(II) transporter YiiP was labeled by hydroxyl radical in both apo- and Zn- form to unravel the structural differences in two conformations as a way to reveal the mechanism of Zn(II) transport (Gupta et al., 2014). Again, the fast reactivity of hydroxyl radical and the ability to do time-resolved experiments yielded insight inaccessible to other methods. In these experiments, the labeling rate of the region around V48/D49/I50 drops after Zn(II) binding because D49 coordinates the ion. Furthermore, the energy released from the binding leads to the reorientation of the transmembrane helix TM5, which is reflected by a drop in the labeling rates of L152 and M197 and the increase in labeling of M151, M159, and M160. Time-resolved CL experiments were carried out by rapid mixing of apo-YiiP with Zn(II) followed by pulsed •OH labeling to capture structural dynamics in millisecond range. Averaging the labeling rates of four modified residues on TM5 provided the approximate rate of TM5 motion, which is on the same order of the macroscopic transport rate.
HRF can also be used to provide structural insight into the conformational changes caused by ligand binding to a membrane protein in the absence of any crystallographic information. As an example, the binding of one type of serotonergic receptor 5-HT4, with one of its antagonists, GR125487, was characterized by HRF and compared to homology models (Padayatti et al., 2013). Upon binding of the antagonist, changes in the oxidation rates of several residues provide insight into the ligand-binding site, structural changes caused by antagonist binding, and the presence and movement of several structural waters that are likely located in transmembrane regions as was seen in other GPCRs. Even though no high-resolution structural data existed to compare to the HRF data, confidence in the results came from the fact that most of the changes in oxidative labeling matched docking calculations, and the data shared ligand-binding characteristics that are typical of other GPCRs in the same class. Moreover, most of the labeled residues were found to be located either at the solvent accessible sites or in transmembrane regions known to have integral structural water molecules. The study demonstrates the power of CL-MS in revealing binding sites and structural changes after ligand binding to membrane proteins with no other direct structural data.
The sensitivity of HRF rates to the solvent accessibility of protein sites makes the technique very valuable for studying protein-protein interactions in membrane protein complexes. The interactions of photosystem II (PS II) with PsbP and PsbQ, which are two water-soluble extrinsic proteins in higher plants that play essential roles in oxygen evolution under physiological conditions, is a good example (Mummadisetti et al., 2014). The organization of the 20-protein PS II system, particularly the locations of PsbP and PsbQ, is unclear, even though individual high-resolution structures of these water-soluble proteins exist. In studies by Bricker and co-workers, HRF revealed that regions on the surfaces of these proteins were resistant to oxidative modification when in contact with components of PS II, indicating that these two proteins define buried regions within the larger PS II system. While relatively few HRF studies of membrane protein-protein interactions in artificial systems have been reported, the large buried interfaces associated with protein-protein binding would seem to make CL-MS a powerful tool for mapping such sites.
While HRF can provide insight into membrane protein binding, it primarily reports on soluble domain sites unless the transmembrane segments already have structural waters or water channels. Manzi et al. have investigated the capability of photoactivatable aryl diazirines, which are precursors to highly reactive carbenes, as a complementary means of labeling transmembrane regions of a protein (Manzi et al., 2017). Using an aryl diazirine (Figure 7), these researchers were able to label the micelle-embedded regions of the outer membrane porin, OmpF, a homotrimeric membrane protein. In contrast to hydroxyl radical, the aryl diazirine can insert into the micelles, and upon laser irradiation at 349 nm, carbenes can be generated that modify hydrophobic transmembrane regions. Interestingly, most of the extracellular regions of the protein were unlabeled, suggesting this diazirine precursor is particularly suited for membrane-embedded regions. Moreover, most of the modified residues in the artificial membrane are located on one side of the β-barrel structure, revealing the other side to be the trimeric interface of OmpF. More water-soluble diazirines, such as photoleucine (Figure 7) were unable to label membrane-embedded regions of OmpF. Not surprisingly, the carbenes generated by the aryl diazirine react extensively with the detergent used to solubilize OmpF. This observation means higher concentrations of labeling reagent are needed. These necessary higher concentrations also raise concerns about how the diazirine precursors might influence membrane structure and fluidity, thereby also negatively affecting membrane protein structure. Assuming the precursor concentrations are kept as low as possible, then membrane perturbation is probably not a large concern.
Figure 7.
Structures of aryl diazirine (left) and photoleucine (right).
3. Impact of different lipids on proteins
CL-MS has also been used to examine the interactions between different lipids and membrane proteins. Gross and colleagues investigated the different impact of the detergent DDM and nanodiscs on the topology of light-harvesting complex 2 (LHC 2) from B850 bacteriochlorophyll a using FPOP (Lu et al., 2016). To compare the extent of oxidation at the residue level in the two membrane environments, leucine enkephalin was added as a reporter peptide to normalize LHC 2 reactivity due to the differences in reactivity of DDM and nanodiscs with hydroxyl radical. The researchers found that LHC 2 residues were oxidized to a greater extent in DDM, perhaps due to the higher mobility of this detergent. However, nanodiscs provided better protection for the membrane-embedded regions of the proteins suggesting that the protein higher-order structure in the nanodiscs were more native like than in the simple detergent. In addition to providing some insight into how different detergents affect protein reactivity with hydroxyl radical, this study also indicates that nanodiscs might be a better artificial membrane system for some proteins than the commonly used DDM detergent.
In an analogous study, Watkinson et al. studied the interactions of DDM and amphipols (A8-35) with the membrane protein OmpT via FPOP (Watkinson, Calabrese, Ault, Radford, & Ashcroft, 2017). Some previous studies had shown that polymeric amphipols can be more effective than detergent micelles at maintaining the native structure of membrane proteins, and FPOP was used to compare the solvent accessibility of OmpT in these two artificial membrane systems. The group found that oxidation levels were different for key residues when DDM and amphipols were used, suggesting that the artificial membranes interact differently with OmpT. Moreover, the site-specific differences in the modification extents indicate greater intermolecular contacts between the amphipols and OmpT, providing a molecular-level rationale for the greater stability of OmpT in amphipols.
D. Residue non-specific labeling in cells and in vivo
The integration of CL with MS to investigate membrane proteins in cells or in vivo is a relatively new and exciting area of research. In contrast to CL-MS of proteins in vitro, experiments conducted in cells or in vivo seek to study native proteins in their normal cellular environment. Studying proteins under these conditions provides a more relevant setting in which to understand a membrane protein’s structure and interaction. For example, in a cellular context, membrane proteins may have competing interactions with multiple different molecules and the presence of clusters of lipids, ions, and small and large molecules, which could all influence their structure. Although in cell and in vivo labeling are relatively new areas that have primarily used FPOP, researchers have made a lot of promising progress in the last five years. There are numerous technical issues to consider when using non-specific reagents to conduct CL-MS experiments on living cells or organisms, including the following.
1. Reagent toxicity
An important aspect to consider when performing CL-MS experiments on cells or in vivo is the effect of the reagent on cell or organism viability. To minimize toxicity, optimization of reagent concentrations and reaction time are necessary. Often, the viability of cells are reported by measuring ATP concentrations (Espino, Mali, & Jones, 2015) while fluorescence from propidium iodide, which inserts into the DNA of dead cells, is used to indirectly reflect the viability of organisms, such as C. elegans, at different reagent concentrations (Espino & Jones, 2019).
2. Chemical penetration enhancers (CPEs)
Reagent access to the cells or organisms is one of the more challenging aspects of CL-MS experiments, yet it is essential to control as it largely determines the labeling efficiency, especially for the intracellular side of membrane proteins and for multicellular organisms. Jones and co-workers have found that the addition of CPEs can increase the uptake of labeling reagents via mild disruption to the lipid bilayer (Espino, Zhang, & Jones, 2020). These CPEs should not be toxic to the cell or organism within the working concentrations and ideally should not interfere with the labeling reagent. The addition of 1% 1-dodecylazacycloheptan-2-one, known by its registered trademark name Azone®, for FPOP experiments has been found to increase the number of modified proteins and their oxidation extents in the nematode C. elegans. Azone is capable of increasing the penetration of the H2O2, which is needed for FPOP experiments, while exhibiting low toxicity itself. CPE use is far from optimized in such labeling experiments, so searching for other CPE compounds that can be compatible with reagents and facilitate even higher labeling efficiencies is still required.
3. Choice of quench/scavenger reagents
Not only is it important to consider the penetration of the labeling reagent, cell permeability of the quenching/scavenging reagent is vital as well. Because cells have high concentrations of proteins, the radical scavengers that are used in hydroxyl radical labeling experiments (i.e. Gln or Phe) are typically not required in cell labeling experiments. Quenching reagents are still needed to consume H2O2, and Jones and co-workers have found that N’-dimethyl-thiourea (DMTU) and N-tert-butyl-α-phenylnitrone (PBN) are better than catalase and methionine owing to their good cell permeability properties (Espino et al., 2015).
4. Labeling platform
Labeling cells and organisms introduces additional logistical challenges that are absent in simple solution-based experiments. Consequently, Jones and co-workers have developed a novel microflow system for IC-FPOP to increase labeling efficiency and to improve modification reproducibility during the experiments (Rinas, Mali, Espino, & Jones, 2016). By adding two sheath flows besides the central cell flows with relatively high velocity, optimal laser irradiation of single cells at a time allows for increased oxidation extents (Figure 8a). To increase the throughput of their initial system, they designed a Platform Incubator with XY movement (PIXY) in which FPOP in cell or in vivo (i.e. C. elegans) can be done on an optical bench (Figure 8b) (Johnson, Punshon-Smith, Espino, Gershenson, & Jones, 2020). Four pumps control the flow of reagents or buffers automatically while laser pulses occur at each well. By utilizing this improved platform, the number of modified proteins further increases, and some proteins have more modified residues compared to their initial microflow system. Precursor isotopic labeling with isobaric tagging is also integrated into their FPOP system to greatly reduce analysis time and to increase biological reproducibility in vivo (Espino, King, Jones, & Robinson, 2020).
Figure 8.

a) Scheme illustrating the IC-FPOP microflow system created by Jones and co-workers. b) Scheme illustrating the Platform Incubator with XY movement. Figure used with permission from Rinas, Mali, Espino, & Jones, 2016, DOI: 10.1021/acs.analchem.6b02357 and Johnson, Punshon-Smith, Espino, Gershenson, & Jones, 2020, DOI: 10.1021/acs.analchem.9b04933.
5. Protein purification and enrichment
Just as with artificial membrane systems, non-protein molecules are removed after the modification experiment by precipitation, SDS-PAGE, and/or affinity columns, as indicated earlier. Given the complexity of the samples, however, the use of affinity tags linked to the proteins of interest often needs to be considered to improve detectability, especially for membrane proteins that are underrepresented in proteomics experiments on cells and tissues. An important consideration is that the affinity tag should ideally not react with the labeling reagents, thereby reducing enrichment efficiency. The broad reactivity of •OH makes affinity enrichment for challenging when FPOP or synchrotron HRF is used.
In cell FPOP (IC-FPOP) has been applied to study protein structure (Espino et al., 2015). After optimizing the H2O2 concentrations and cell culture volumes to ensure moderate cell viability, many proteins in different cellular compartments, including membrane proteins, with a variety of abundances can be modified. MS measurements show that 17 different types of residues can be labeled, with Met, Glu, and Asp being the most frequently detected. In one of the first examples of IC-FPOP on cells, the extent of modification of residues in the protein actin were found to have the same trend as when the protein is labeled in vitro. The in-cell labeling showed a stronger correlation with the open state of actin as compared to the close state, indicating most of the actin was in open state in Vero cells during the FPOP experiments.
The conformational changes of epidermal growth factor receptor (EGFR) upon binding with EGF were characterized by nanosecond laser photolysis protein footprinting in HEK 293-K cells (Zhu et al., 2017). The majority of the modified residues that were detected are solvent accessible with and without EGF activation. Upon EGF binding, five peptides showed increased levels of oxidation, whereas only two peptides showed decreased oxidation. These regions that increase in labeling span across some subdomains in the extracellular regions, demonstrating that the binding of EGF triggers these regions to be more extended. The decreasing oxidation of two peptides reflects that they are more buried causing by the conformational changes after EGF binding.
An exciting recent development by the Jones group is the application of FPOP to investigate protein structure in vivo (Espino & Jones, 2019). The nematode C. elegans was mixed with 200 mM H2O2 immediately before photolysis, and then the UV laser was fired at 50 Hz, resulting in 545 modified proteins from the nervous, alimentary, muscle, and epithelial systems. Among them, membrane proteins from the transmembrane 9 superfamily, the TWiK family of potassium channels, the KvQLT family of potassium channels, and olfactory channels were found to be oxidized. The authors found that the oxidation levels of the modified residues from the heat shock protein 90 (Hsp90) correlate with their SASA values acquired from the protein’s crystal structure. Likewise, the oxidation levels of the protein actin were found to be consistent with its oxidation levels in cells. The ability to perform FPOP on live organisms opens up many possibilities for characterizing membrane protein structures and interactions in vivo.
Oxidative labeling of membrane proteins in live cells was also conducted by Zhu et al. to study the structure and dynamics of trimeric OmpF under different physiological conditions (Zhu et al., 2009). In this case, •OH were generated through an in situ Fenton reactions rather than laser photolysis of H2O2 because the E. coli culture medium interfered with the transmission of light. The modification extent of the residues in loops L1, L3 and L6, which are at or close to extracellular domains, are higher than the oxidation levels of residues in the internal β-barrels that comprise the transmembrane regions. There were no modifications of loops L2 and L4 located at the trimer interfaces. Interestingly, when OmpF is subjected to low pH, low ionic strength, or both, the oxidation extent of residues in the internal β strands significantly drops. This lower oxidation extent was interpreted as being caused by the closure of OmpF channel preventing Fe(II) ions from initiating the Fenton reactions inside the channel.
Metal-catalyzed oxidation (MCO) reactions have also been used to identify proteins binding to cell surface glycoproteins in three cell lines (PNT2, Caco-2, and A549) (Li, Xie, Xu, & Lebrilla, 2019). In this work, azido groups were incorporated into sialylated glycoproteins followed by click chemistry to link Fe(II) complexes to the glycoproteins (Figure 9). The addition of H2O2 to the samples catalyzed the production of hydroxyl radical at the Fe center, causing proteins that were close to the functionalized sialic acids to be oxidatively modified (Figure 9). MS was then used to identify the oxidized proteins, and they were found to fall into three categories: 1) sialylated glycoproteins having transmembrane domains, 2) non-sialylated membrane proteins having interactions with sialic acid, and 3) extracellular proteins without transmembrane domains but having interactions with sialic acid.
Figure 9.

Scheme illustrating protein oxidation near cell surface glycoproteins as generated by Fenton chemistry. Fe(II) complexes are incorporated into glycoproteins via click chemistry, and then proteins are oxidized through metal-catalyzed oxidation reactions. Figure used with permission from Li, Xie, Xu, & Lebrilla, 2019, DOI: 10.1039/C9SC01360A.
Oxidative labeling of membrane protein labeling in cells can also been conducted using H2O2 alone without the addition of metals or irradiation with light. In such cases, the extent of labeling is often too low to be confidently identified, but some structural insight can be obtained by quantifying the levels of unmodified peptides, as a way to reveal indirectly which solvent exposed sites are oxidized. As an example, Yao and co-workers used such an approach to study the cystic fibrosis transmembrane conductance regulator (CFTR), which is a chloride ion channel on the surface of epithelial cells (Farrokhi et al., 2015). To improve the quantification accuracy of the unmodified peptides, internal standards were added, and measurements were conducted using multiple reaction monitoring (MRM). The researchers found that unmodified peptides from the loop regions on CFTR decreased in abundances as H2O2 concentrations were increased, while unmodified peptides from transmembrane regions changed less.
III. RESIDUE SPECIFIC LABELING
A. Labeling reagents
In contrast to non-specific reagents that probe the topology of a variety of residues altogether, residue-specific reagents only modify one or a few residue side chains in a given protein. Residue-specific reagents are easy to use, as they are typically just added to solution, and their limited modification scope actually simplifies the resulting MS analysis. However, they generally provide lower structural resolution, and some prior structural information about the protein of interest is often needed to apply them most effectively. A wide variety of residue-specific labeling reagents have been created and used in CL-MS experiments (Liu et al., 2020; Mendoza & Vachet, 2009), but the ones most commonly used for membrane protein studies are: the combination of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) and glycine ethyl ester (GEE) for Asp/Glu labeling, butane-2,3-dione (BD) for Arg, diethylpyrocarbonate (DEPC) for nucleophilic residues such as Lys, His, Tyr, Ser, and Thr, N-ethylmaleimide (NEM) for Cys, and sulfosuccinimidyl acetate for Lys. The polar residues that are targeted by these reagents are often either located on the extra- or intra-cellular segment of membrane proteins, and they often are involved in protein-ligand and protein-protein interactions. Thus, residue specific labeling can unravel the important roles of these residues in regulating protein structures or mediating protein interactions. Moreover, as alluded to earlier, experiments that use these reagents can be conducted without a complex setup (e.g. laser), and the data interpretation is much simpler than with oxidative labeling. An additional aspect of residue-specific labels is that these reagents can be isotopically encoded. By analyzing together proteins that are labeled with a ‘light’ isotopically encoded reagent under one condition and proteins that are labeled with a ‘heavy’ reagent under another condition, detection time can be dropped in half. In addition, the quantification accuracy is improved because the isotopically labeled peptides from the two conditions are detected at the same time and have the same ionization efficiency (Shen et al., 2018).
B. Technical Challenges of Residue-Specific Labeling Experiments
Many of the technical challenges for membrane protein analysis that were described in section II.B. for non-specific labeling reagents are also present when using residue specific labeling reagents. These include the choice of the artificial membrane, the removal of the membrane components before analysis, and protein digestion. In the process of using residue-specific labeling reagents, one must also consider a protein’s structural integrity during the labeling reaction. In sharp contrast to non-specific reagents such as hydroxyl radical, residue-specific reagent react more slowly, and it is possible that proteins change their structure during the longer reaction times (i.e. seconds to minutes) that are required for a protein to obtain adequate modification extents. To avoid these structural changes, reagent concentrations and reaction times must be optimized to avoid overlabeling the protein. In addition, often a complementary technique such as circular dichroism (CD), fluorescence spectroscopy or activity assays must be used to confirm that the protein’s structure has not been significantly changed. However, all of these techniques are only able to detect significant conformational changes and are not sensitive to local structural perturbations (Limpikirati et al., 2018). A more reliable, but more experimentally involved way, to monitor the structural integrity of the protein during these labeling reactions is to measure the labeling reaction kinetics, as deviations in kinetic plots are a very sensitive way to identify structural perturbations. Alternatively, restricting the extent of labeling to one label per protein on average is a safe way to obtain reliable information, but this approach also limits the number of labeled peptides that will be detected, leading to less structural information.
C. Applications of Residue Specific Labeling to Membrane Proteins
1. Structural characterization
Like CL with non-specific reagents, CL with residue-specific reagents is affected by the solvent accessibility of residue’s side chains, and so it can be used to monitor the topology of membrane proteins. As an example, Schmidt et al. used DEPC labeling data and computational modeling to study an F-type ATP synthase (cATPase) (Schmidt et al., 2017). The structure information about this assembly is limited, and CL-MS was able to provide solvent accessibility information of residue side chains to facilitate structure prediction. The protein was subjected to DEPC labeling, and modified residues were identified through LC-MS/MS analysis. Using the DEPC labeling results as modeling constraints and applying a customized scoring algorithm together with native MS and cross-linking data, the authors were able to assemble a structural model for cATPase. Based on this model, they were able to conduct MD simulations to investigate the dynamics of this nanomotor.
2. Protein-ligand and protein-protein interactions
Residue-specific reagents have been more commonly used with MS to study membrane protein-ligand or protein-protein interactions. One example was work by Knapp and colleagues, who studied photo-activated Rho (Rho*) and its binding interactions with α subunit of the G-protein transducin (Gtα) in the detergent DDM (Wang, Kim, Ablonczy, Crouch, & Knapp, 2004). Ground-state Rho, Rho*, Rho with the Gtα peptide 340-350, and Rho* with the Gtα peptide 340-350 were separately incubated with the Lys-specific reagent sulfosuccinimidyl acetate followed by enzymatic digestion and LC-MS/MS measurements. Most of the residues labeled by this reagent were found to be on the cytoplasmic side of the protein. The acetylation levels of K66/K67 on one soluble loop and K141 on another soluble loop slightly dropped after light activation, while the reactivity of K325 at the C-terminus declined dramatically, providing some insight into the structural changes induced by light. After binding with the Gtα peptide fragment, the extents of modification of K66/K67 and K311 on another soluble loop decreased due to interactions with the peptide, while K141 was less impacted by Gtα peptide binding. This work represents one of the earliest examples of CL-MS confirming structural changes of Rho after photoactivation and Gtα peptide binding.
The value of lysine specific reagents to study membrane proteins is also seen in the work on N-methyl-D-aspartate receptors (NMDARs), which are glutamate receptors and ion channel proteins in the central nervous system (CNS) (Zhou et al., 2019). Binding regions of three ligands (Ro25-6981, gavestinel and UBP710) and associated conformational changes of NMDARs were studied using dimethyl labeling of Lys residues using formaldehyde followed by sodium cyanoborohydride (NaBH3CN) reduction. According to the researchers, this CL method is highly efficient and does not deactivate the membrane protein complex. Ro25-6981 and gavestinel were found to bind to two different regions on the receptor based on labeling decreases at Lys residues in different domains, whereas UBP170 shielded some of the same Lys residues in both regions, indicating that this molecule binds at the dimer interface created by these two different regions.
Residue-specific labeling reagents have also been used to reveal the conformational changes of β2-adrenergic receptor (β2AR) after binding with nine different ligands separately in DDM (Kahsai et al., 2011). The nine ligands included full agonists, partial agonists, weak partial agonists, antagonists, and inverse agonists. Lefkowitz and coworkers used isotopically-encoded NEM and succinic anhydride to modify Cys and Lys residues at various time points after the addition of ligands (Kahsai, Rajagopal, Sun, & Xiao, 2014). The reactivity of each modified residue in bound state versus unbound state were used to compare with the classical Gα activation mechanism. The researchers found that two Cys residues (C77 and C327) correlated with expected mechanism, whereas the reactivity of other residues were quite variable, indicating that the conformational changes of β2AR that are induced by binding are ligand dependent.
Even though free Cys residues appear much less frequently in proteins than Lys residues, Cys-specific labeling reagents can still be used to provide useful binding information for membrane proteins. One example is from studies of the D-galactose-H+ symport protein (GalP) and its structural changes upon binding D-glucose (Jones, Baldwin, Henderson, & Ashcroft, 2010). NEM was used to modify the three Cys residues in this protein. Two of the Cys residues decreased in labeling, while one residue’s reactivity remained unchanged. The decreased labeling of one of the residues (C374) could be explained by previous information about the glucose-binding site, but the unexpected decrease in C19 labeling could only be attributed to protein conformational changes. Analogous insight into conformational changes were obtained for Mhp1, which is a sodium dependent transport protein for 5-aryl-substituted hydantoins (Calabrese et al., 2017). One Cys residue, C327, was found to exhibit high reactivity in the inward-facing state of this membrane protein (Figure 10). Upon incubation with Na+ and 5-benzyl-L-hydantoin (L-BH) together, Mhp1 switches from the more predominant inward-facing state to an outward-facing state in which C327 is mostly buried, which lowers the NEM modification percentage of this residue (Figure 10). Upon titrating L-BH concentrations and measuring changes in the modification levels of this Cys residue at different Na+ concentrations, Michaelis-like constant (Km) values could be acquired for both Na+ and L-BH.
Figure 10.

Cartoon illustrating the Mhp1 transport mechanism between outward-facing and inward-facing conformations. Yellow stars are the residue C327; green dots represent Na+, and red dots represent the L-BH molecule. Figure used with permission from Calabrese et al., 2017, DOI: 10.1021/acs.analchem.7b01310.
Wen et al. used EDC/GEE to identify the binding interactions of the membrane-bound Fenna-Matthews-Olson (FMO) antenna protein, which links the large peripheral chlorosome complex to the reaction center in photosynthetic bacteria (Wen, Zhang, Gross, & Blankenship, 2009). The isolated FMO protein, FMO from chlorosome-depleted membranes of Chlorobaculum tepidum, and FMO from native membrane isolates were reacted with EDC/GEE to discover the binding interfaces (Figure 11). A comparison of the labeling sites and extents from these three constructs were able to reveal the orientation of the FMO protein in the larger photosystem complex. The researchers noted that their results were consistent with theoretical predictions about the architecture of the photosystem, but noted that the relatively small size of the reagent, and presumably its relatively slow reactivity, might cause it to miss some more weakly interacting sites.
Figure 11.

Experimental workflow for discovering the interactions of FMO with chlorosome and the reaction center. FMO alone, chlorosome-depleted complex, and the complex are labeled by EDC/GEE followed by enzymatic digestion and LC-MS/MS analysis to compare the modification extent of peptides in three cases. Figure used with permission from Wen, Zhang, Gross, & Blankenship, 2009, DOI: 10.1073/pnas.0901691106.
The binding interfaces of substrates with lactose permease (LacY), which is a membrane protein that facilitates lactose transport across cell membranes, were also investigated using carbodiimide chemistry (Weinglass et al., 2003) and the arginine specific reagent BD (Weinglass, Whitelegge, Faull, & Kaback, 2004). The studies using carbodiimides are particularly important for demonstrating the value of residue-specific reagents. In the presence of p-nitro-phenyl-α-D-galactopyranoside (p-NPGal), which is a high-affinity ligand, the modification level of E269 in LacY was dramatically reduced, indicating this Glu as a key residue that binds the substrate. Combining the modification profile of LacY with other p-NPGal derivatives that have deoxy and methoxy functional groups at different positions on the galactopyranosyl ring, Weinglass et al. proposed that E269 interacts with the C-3 OH of the ring. These observations are intriguing, as they suggest that proper experimental design can provide even higher resolution information than would be predicted based on the size and reactivity profile of the reagent. Moreover, the higher observed reactivity of E269 towards more hydrophobic carbodiimide labeling reagents suggested a more hydrophobic environment around the binding sites. This latter observation highlights another potential advantage of residue-specific labeling reagents. They can be sensitive to the local microenvironment in ways that non-specific reagents are not, allowing them to provide deeper molecular-level information (Limpikirati, Pan, & Vachet, 2019).
D. Residue specific labeling in vivo
Like non-specific CL coupled with MS to study membrane proteins in vivo, the combination of residue-specific CL with MS can provide insight into protein structure and interactions in cellular environments. Despite some technical challenges that must be considered, the past few years have witnessed several successful studies of membrane protein topology and their interactions with ligands by residue-specific CL techniques.
Technical Challenges of Residue Specific Labeling Experiments on Membrane Proteins in vivo
1. Reagent permeability
We have mentioned some challenges of CL labeling in vivo such as reagent toxicity, the choice of quench reagent, labeling platform and protein enrichment. For residue specific CL, we need to pay special attention to reagent permeability. Given the variability in their sizes, hydrophobicities, and charges at physiological pH, different reagents will have different abilities to pass through the cell membrane and thus different abilities to provide structural information for extra-cellular or intra-cellular regions of membrane proteins. In general, small molecules with no charge or hydrophobic molecules can pass through cell membrane whereas large polar or ionic molecules cannot.
2. CPEs
CPEs have been applied to increase the cellular uptake of residue-specific reagents. Digitonin, a non-ionic detergent, is often used to facilitate the delivery of small molecules into cells by permeabilizing cell membrane. Li et al. pre-treated mammalian cells with digitonin to increase the labeling efficiency of EDC/GEE without affecting functional integrity. Other CPEs, such as Azone®, could conceivably be used as well (Li, 2018).
3. Peptide abundance
Membrane proteins consist of some hydrophobic transmembrane segments that have low solubility and poor ionization efficiencies. In addition, membrane proteins and their proteolytic fragments are often underrepresented in traditional proteomics experiments, further affecting the detection of covalently labeled proteins and influencing the accuracy of their quantitation. One advantage of residue-specific labeling reagents is that functional groups such as biotin groups can be added to the reagent to facilitate enrichment of the modified peptides. Some researchers have also mixed ‘light’ and ‘heavy’ versions of labeling reagents as a means of enhancing relative quantitation during MS detection (Li, 2018).
Only a few examples of using residue-specific labeling reagents for CL-MS of membrane protein in cells have appeared in the literature. In one example, EDC/GEE labeling of Asp and Glu residues in human glucose transporters (hGLUTs) was used to identify key salt bridge interactions in two conformations (Li, 2018). When hGLUT1 is not bound to a ligand, it has an equilibrium between outward-facing and inward-facing conformations. The binding of D-glucose or D-maltose shifts the equilibrium towards outward-facing conformation in which E329 forms a salt bridge with R333, and decreased modification of E329 allowed this to be determined. In contrast, when the mycotoxin cytochalasin B is bound, hGLUT1 favors an inward-facing conformation. Labeling with EDC/GEE identified that E299 decreases in labeling under this condition because of the formation of a salt bridge with K38. Key salt bridges in different conformations of another transporter, GLUT5, which is a fructose specific binding protein, have been characterized by EDC/GEE-based CL-MS as well.
In another example, a two-step modification procedure involving EDC/N-hydroxysulfosuccinimide activation and a biotinyl cystamine reaction to label Asp/Glu residues has been used to identify soluble regions in the extracellular domains of membrane proteins in human HL60 cells (Figure 12), and this labeling data was used to help refine computational models of membrane proteins from this organism (Müller et al., 2019). The use of a biotinylated reagent along with an avidin-based enrichment column enhanced the detection of membrane protein proteolytic fragments after cell lysis and protein digestion. In total, 135 residues from 38 different transmembrane proteins were modified, and the majority of the labels were found in predicted extracellular regions, meaning that CL-MS is likely to be more broadly applicable for improving the accuracy of membrane protein topology predictions.
Figure 12.
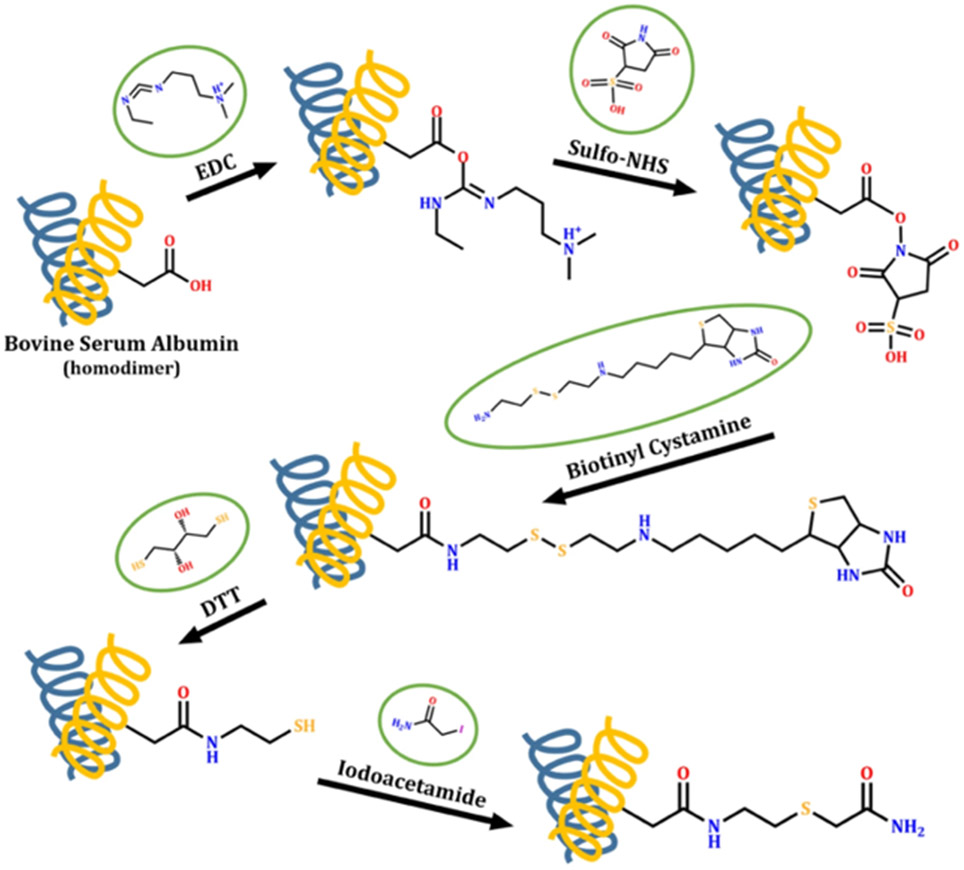
Scheme illustrating covalent labeling of Asp and Glu residues by EDC/sulfo-NHS activation and the subsequent biotinyl cystamine reaction. After cell lysis and protein digestion, peptides from membrane proteins are enriched via the binding between biotinylated Asp/Glu and neutravidin column. The addition of dithiothreitol (DTT) was applied to elute the peptides followed by iodoacetamide (IAM) alkylation. Figure used with permission from Müller et al., 2019, DOI: 10.1038/s41598-019-52188-4.
Alkylation of Cys residues by NEM has been also applied for investigating the higher order structure and binding interactions of human vitamin K epoxide (hVKOR), an enzyme in the endoplasmic reticulum (ER) membrane in HEK 293 cell line (Shen et al., 2016; Shen et al., 2018). To monitor the redox status of multiple Cys residues in hVKOR, the reduced form was first labeled by NEM in cells. After cell lysis, the oxidized form was reduced and labeled by d5-NEM. By calculating the apparent oxidized fraction from the ratio of NEM labeling extent with the light and heavy reagent, the researchers were able to identify which Cys residues formed disulfide bonds and which did not. When hVKOR was labeled in the presence of warfarin, a known binder of this protein, it was also demonstrated that certain disulfide bonds and a free Cys were crucial for warfarin binding, facilitating the construction of warfarin-hVKOR model from molecular dynamics (MD) simulations.
IV. OUTLOOK
The past 10 years have witnessed the rapid development of CL-MS techniques for membrane protein structural analysis. Since membrane proteins are common drug targets, knowledge about their higher order structures and interactions can facilitate the research and development of new therapeutics. We predict that more researchers will begin to use CL-MS techniques to study the wide range of membrane protein systems given their immense importance. Moreover, because there is a dearth of structural information about membrane proteins in live cells, CL-MS methods have the potential to have a huge impact on the field in the future. From a technique standpoint, one need is the development of new reagents with different chemical properties that can separately react with exposed or membrane-buried region of the protein. For studies of intact cells or organisms, the cell penetration efficiency of reagents will also need to be further enhanced by identifying reagents that can pass through cell membranes or by finding better chemical penetration enhancers. The effect of the lipid membrane itself and components in real cell membranes need more study as well. Improvements in the analysis of proteins by LC-MS/MS are always emerging, and these improvements will undoubtedly increase the structural resolution possible in CL-MS experiments, as better detection efficiency is achieved. However, approaches to selectively enrich membrane proteins will also likely be needed to access the full potential of the method. As CL-MS methods continued to be used more broadly, they will likely be combined more and more with computational modeling to predict and refine membrane protein structures. Overall, we feel that CL-MS is poised to improve our understanding of cellular biochemistry by revealing membrane structure and interactions that are currently not accessible by other techniques.
V. ACKNOWLEDGEMENTS
This work is supported by the National Institutes of Health (NIH) under Grant R01 GM075092.
VI. REFERENCES
- Alcor D, Gouzer G, & Triller A (2009). Single-particle tracking methods for the study of membrane receptors dynamics. Eur J Neurosci, 30(6), 987–997. doi: 10.1111/j.1460-9568.2009.06927.x [DOI] [PubMed] [Google Scholar]
- Angel TE, Gupta S, Jastrzebska B, Palczewski K, & Chance MR (2009). Structural waters define a functional channel mediating activation of the GPCR, rhodopsin. Proc Natl Acad Sci U S A, 106(34), 14367–14372. doi: 10.1073/pnas.0901074106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuyan AG, & Cohen FS (2006). Lipid peroxides promote large rafts: effects of excitation of probes in fluorescence microscopy and electrochemical reactions during vesicle formation. Biophys J, 91(6), 2172–2183. doi: 10.1529/biophysj.106.087387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgewater JD, Lim J, & Vachet RW (2006). Using metal-catalyzed oxidation reactions and mass spectrometry to identify amino acid residues within 10 Å of the metal in Cu-binding proteins. J Am Soc Mass Spectrom, 17(11), 1552–1559. doi: 10.1016/j.jasms.2006.06.003 [DOI] [PubMed] [Google Scholar]
- Bridgewater JD, & Vachet RW (2005). Metal-catalyzed oxidation reactions and mass spectrometry: the roles of ascorbate and different oxidizing agents in determining Cu-protein-binding sites. Anal Biochem, 341(1), 122–130. doi: 10.1016/j.ab.2005.02.034 [DOI] [PubMed] [Google Scholar]
- Calabrese AN, Jackson SM, Jones LN, Beckstein O, Heinkel F, Gsponer J, Henderson PJF, et al. (2017). Topological Dissection of the Membrane Transport Protein Mhp1 Derived from Cysteine Accessibility and Mass Spectrometry. Anal Chem, 89(17), 8844–8852. doi: 10.1021/acs.analchem.7b01310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese AN, & Radford SE (2018). Mass spectrometry-enabled structural biology of membrane proteins. Methods, 147, 187–205. doi: 10.1016/j.ymeth.2018.02.020 [DOI] [PubMed] [Google Scholar]
- Carpenter EP, Beis K, Cameron AD, & Iwata S (2008). Overcoming the challenges of membrane protein crystallography. Curr Opin Struct Biol, 18(5), 581–586. doi: 10.1016/j.sbi.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavagnero S, Dyson HJ, & Wright PE (1999). Improved low pH bicelle system for orienting macromolecules over a wide temperature range. J Biomol NMR, 13(4), 387–391. doi: 10.1023/A:1008360022444 [DOI] [PubMed] [Google Scholar]
- Chance MR, Farquhar ER, Yang S, Lodowski DT, & Kiselar J (2020). Protein Footprinting: Auxiliary Engine to Power the Structural Biology Revolution. J Mol Biol, 432(9), 2973–2984. doi: 10.1016/j.jmb.2020.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-L, & Perrimon N (2017). Proximity-dependent labeling methods for proteomic profiling in living cells. Wiley interdisciplinary reviews. Developmental biology, 6(4). doi: 10.1002/wdev.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Asuru A, Kiselar J, Mathai G, Chance MR, & Gross ML (2020). Fast Protein Footprinting by X-ray Mediated Radical Trifluoromethylation. J Am Soc Mass Spectrom, 31(5), 1019–1024. doi: 10.1021/jasms.0c00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Zhang B, Cui W, & Gross ML (2017). Laser-Initiated Radical Trifluoromethylation of Peptides and Proteins: Application to Mass-Spectrometry-Based Protein Footprinting. Angew Chem Int Ed Engl, 56(45), 14007–14010. doi: 10.1002/anie.201706697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie S, Shi X, & Smith AW (2020). Resolving Membrane Protein–Protein Interactions in Live Cells with Pulsed Interleaved Excitation Fluorescence Cross-Correlation Spectroscopy. Acc Chem Res, 53(4), 792–799. doi: 10.1021/acs.accounts.9b00625 [DOI] [PubMed] [Google Scholar]
- Deperalta G, Alvarez M, Bechtel C, Dong K, McDonald R, & Ling V (2013). Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. mAbs, 5(1), 86–101. doi: 10.4161/mabs.22964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez R, Pateiro M, Gagaoua M, Barba FJ, Zhang W, & Lorenzo JM (2019). A Comprehensive Review on Lipid Oxidation in Meat and Meat Products. Antioxidants, 8(10), 429. doi: 10.3390/antiox8100429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Duc NM, Rasmussen SGF, Hilger D, Kubiak X, Wang L, Chung KY et al. (2019). Assembly of a GPCR-G Protein Complex. Cell, 177(5), 1232–1242. doi: 10.1016/j.cell.2019.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espino JA, & Jones LM (2019). Illuminating Biological Interactions with in Vivo Protein Footprinting. Anal Chem, 91(10), 6577–6584. doi: 10.1021/acs.analchem.9b00244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espino JA, King CD, Jones LM, & Robinson RAS (2020). In Vivo Fast Photochemical Oxidation of Proteins Using Enhanced Multiplexing Proteomics. Anal Chem, 92(11), 7596–7603. doi: 10.1021/acs.analchem.0c00174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espino JA, Mali VS, & Jones LM (2015). In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Anal Chem, 87(15), 7971–7978. doi: 10.1021/acs.analchem.5b01888 [DOI] [PubMed] [Google Scholar]
- Espino JA, Zhang Z, & Jones LM (2020). Chemical Penetration Enhancers Increase Hydrogen Peroxide Uptake in C. elegans for In Vivo Fast Photochemical Oxidation of Proteins. J Proteome Res. doi: 10.1021/acs.jproteome.0c00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrokhi V, Bajrami B, Nemati R, McShane AJ, Rueckert F, Wells B, & Yao X (2015). Development of Structural Marker Peptides for Cystic Fibrosis Transmembrane Conductance Regulator in Cell Plasma Membrane by Reversed-Footprinting Mass Spectrometry. Anal Chem, 87(17), 8603–8607. doi: 10.1021/acs.analchem.5b01962 [DOI] [PubMed] [Google Scholar]
- Frankel LK, Sallans L, Bellamy H, Goettert JS, Limbach PA, & Bricker TM (2013). Radiolytic mapping of solvent-contact surfaces in Photosystem II of higher plants: experimental identification of putative water channels within the photosystem. J Biol Chem, 288(32), 23565–23572. doi: 10.1074/jbc.M113.487033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sáez AJ, & Schwille P (2007). Single molecule techniques for the study of membrane proteins. Appl Microbiol Biotechnol, 76(2), 257–266. doi: 10.1007/s00253-007-1007-8 [DOI] [PubMed] [Google Scholar]
- Gau BC, Sharp JS, Rempel DL, & Gross ML (2009). Fast photochemical oxidation of proteins footprints faster than protein unfolding. Anal Chem, 81(16), 6563–6571. doi: 10.1021/ac901054w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Bavro VN, D'Mello R, Tucker SJ, Vénien-Bryan C, & Chance MR (2010). Conformational changes during the gating of a potassium channel revealed by structural mass spectrometry. Structure, 18(7), 839–846. doi: 10.1016/j.str.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Chai J, Cheng J, D'Mello R, Chance MR, & Fu D (2014). Visualizing the kinetic power stroke that drives proton-coupled zinc(II) transport. Nature, 512(7512), 101–104. doi: 10.1038/nature13382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Sullivan M, Toomey J, Kiselar J, & Chance MR (2007). The Beamline X28C of the Center for Synchrotron Biosciences: a National Resource for Biomolecular Structure and Dynamics Experiments Using Synchrotron Footprinting. J Synchrotron Radiat, 14(3), 233–243. doi: 10.1107/S0909049507013118 [DOI] [PubMed] [Google Scholar]
- Hambly DM, & Gross ML (2005). Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom, 16(12), 2057–2063. doi: 10.1016/j.jasms.2005.09.008 [DOI] [PubMed] [Google Scholar]
- Hebling CM, Morgan CR, Stafford DW, Jorgenson JW, Rand KD, & Engen JR (2010). Conformational analysis of membrane proteins in phospholipid bilayer nanodiscs by hydrogen exchange mass spectrometry. Anal Chem, 82(13), 5415–5419. doi: 10.1021/ac100962c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuvingh J, & Bonneau S (2009). Asymmetric oxidation of giant vesicles triggers curvature-associated shape transition and permeabilization. Biophys J, 97(11), 2904–2912. doi: 10.1016/j.bpj.2009.08.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob RF, & Mason RP (2005). Lipid peroxidation induces cholesterol domain formation in model membranes. J Biol Chem, 280(47), 39380–39387. doi: 10.1074/jbc.M507587200 [DOI] [PubMed] [Google Scholar]
- Johnson DT, Punshon-Smith B, Espino JA, Gershenson A, & Jones LM (2020). Implementing In-Cell Fast Photochemical Oxidation of Proteins in a Platform Incubator with a Movable XY Stage. Anal Chem, 92(2), 1691–1696. doi: 10.1021/acs.analchem.9b04933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LN, Baldwin SA, Henderson PJF, & Ashcroft AE (2010). Defining topological features of membrane proteins by nanoelectrospray ionisation mass spectrometry. Rapid Commun Mass Spectrom, 24(3), 276–284. doi: 10.1002/rcm.4387 [DOI] [PubMed] [Google Scholar]
- Jumper CC, Bomgarden R, Rogers J, Etienne C, & Schriemer DC (2012). High-Resolution Mapping of Carbene-Based Protein Footprints. Anal Chem, 84(10), 4411–4418. doi: 10.1021/ac300120z [DOI] [PubMed] [Google Scholar]
- Kahsai AW, Rajagopal S, Sun J, & Xiao K (2014). Monitoring protein conformational changes and dynamics using stable-isotope labeling and mass spectrometry. Nat Protoc, 9(6), 1301–1319. doi: 10.1038/nprot.2014.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, Sun J, Lefkowitz RJ, et al. (2011). Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat Chem Biol, 7(10), 692–700. doi: 10.1038/nchembio.634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselar J, & Chance MR (2018). High-Resolution Hydroxyl Radical Protein Footprinting: Biophysics Tool for Drug Discovery. Annu Rev Biophys, 47(1), 315–333. doi: 10.1146/annurev-biophys-070317-033123 [DOI] [PubMed] [Google Scholar]
- Komolov KE, Du Y, Duc NM, Betz RM, Rodrigues J, Leib RD, Benovic JL et al. (2017). Structural and Functional Analysis of a β(2)-Adrenergic Receptor Complex with GRK5. Cell, 169(3), 407–421. doi: 10.1016/j.cell.2017.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L, Stocks BB, Pan Y, & Tong X (2010). Mass spectrometry combined with oxidative labeling for exploring protein structure and folding. Mass Spectrom Rev, 29(4), 651–667. doi: 10.1002/mas.20256 [DOI] [PubMed] [Google Scholar]
- Li K, (2018). Mass Spectrometry-based Strategies for Protein Characterization: Amyloid Formation, Protein-Ligand Interactions and Structures of Membrane Proteins in Live Cells. Arts & Sciences Electronic Theses and Dissertations, 1556. doi:https://openscholarship.wustl.edu/art_sci_etds/1556 [Google Scholar]
- Li K, Shi L, & Gross ML (2018). Mass spectrometry-based fast photochemical oxidation of proteins (FPOP) for higher order structure characterization. Acc Chem Res, 51(3), 736–744. doi: 10.1021/acs.accounts.7b00593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Xie Y, Xu G, & Lebrilla CB (2019). Identification of potential sialic acid binding proteins on cell membranes by proximity chemical labeling. Chem Sci, 10(24), 6199–6209. doi: 10.1039/C9SC01360A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B, & Tamm LK (2016). NMR as a tool to investigate the structure, dynamics and function of membrane proteins. Nat Struct Mol Biol, 23(6), 468–474. doi: 10.1038/nsmb.3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, & Vachet RW (2003). Development of a methodology based on metal-catalyzed oxidation reactions and mass spectrometry to determine the metal binding sites in copper metalloproteins. Anal Chem, 75(5), 1164–1172. doi: 10.1021/ac026206v [DOI] [PubMed] [Google Scholar]
- Limpikirati P, Liu T, & Vachet RW (2018). Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods, 144, 79–93. doi: 10.1016/j.ymeth.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpikirati P, Pan X, & Vachet RW (2019). Covalent Labeling with Diethylpyrocarbonate: Sensitive to the Residue Microenvironment, Providing Improved Analysis of Protein Higher Order Structure by Mass Spectrometry. Anal Chem, 91(13), 8516–8523. doi: 10.1021/acs.analchem.9b01732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XR, Rempel DL, & Gross ML (2019). Composite conformational changes of signaling proteins upon ligand binding revealed by a single approach: calcium-calmodulin study. Anal Chem, 91(19), 12560–12567. doi: 10.1021/acs.analchem.9b03491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang H, Niedzwiedzki DM, Prado M, He G, Gross ML, & Blankenship RE (2013). Phycobilisomes Supply Excitations to Both Photosystems in a Megacomplex in Cyanobacteria. Science, 342(6162), 1104–1107. doi: 10.1126/science.1242321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XR, Zhang MM, & Gross ML (2020). Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chem Rev, 120(10), 4355–4454. doi: 10.1021/acs.chemrev.9b00815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Van Horn WD, Chen J, Mathew S, Zent R, & Sanders CR (2012). Bicelles at Low Concentrations. Mol Pharm, 9(4), 752–761. doi: 10.1021/mp2004687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang H, Niedzwiedzki DM, Jiang J, Blankenship RE, & Gross ML (2016). Fast Photochemical Oxidation of Proteins Maps the Topology of Intrinsic Membrane Proteins: Light-Harvesting Complex 2 in a Nanodisc. Anal Chem, 88(17), 8827–8834. doi: 10.1021/acs.analchem.6b01945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A, Langó T, Turiák L, Ács A, Várady G, Kucsma N, Tusnády GE, et al. (2019). Covalently modified carboxyl side chains on cell surface leads to a novel method toward topology analysis of transmembrane proteins. Sci Rep, 9(1), 15729. doi: 10.1038/s41598-019-52188-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleknia SD, Brenowitz M, & Chance MR (1999). Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Anal Chem, 71(18), 3965–3973. doi: 10.1021/ac990500e [DOI] [PubMed] [Google Scholar]
- Manzi L, Barrow AS, Hopper JTS, Kaminska R, Kleanthous C, Robinson CV, Oldham NJ, et al. (2017). Carbene Footprinting Reveals Binding Interfaces of a Multimeric Membrane-Spanning Protein. Angew Chem Int Ed, 56(47), 14873–14877. doi: 10.1002/anie.201708254 [DOI] [PubMed] [Google Scholar]
- McPherson A (2004). Introduction to protein crystallization. Methods, 34(3), 254–265. doi: 10.1016/j.ymeth.2004.03.019 [DOI] [PubMed] [Google Scholar]
- Mendoza VL, & Vachet RW (2009). Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev, 28(5), 785–815. doi: 10.1002/mas.20203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummadisetti MP, Frankel LK, Bellamy HD, Sallans L, Goettert JS, Brylinski M, Bricker TM, et al. (2014). Use of protein cross-linking and radiolytic footprinting to elucidate PsbP and PsbQ interactions within higher plant Photosystem II. Proc Natl Acad Sci U S A, 111(45), 16178–16183. doi: 10.1073/pnas.1415165111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu B, & Gross M (2019). MS -Based Hydroxyl Radical Footprinting: Methodology and Application of Fast Photochemical Oxidation of Proteins ( FPOP ) In Mass Spectrometry-Based Chemical Proteomics, 363–416 [Google Scholar]
- Orban T, Jastrzebska B, Gupta S, Wang B, Miyagi M, Chance Mark R., & Palczewski K (2012). Conformational Dynamics of Activation for the Pentameric Complex of Dimeric G Protein-Coupled Receptor and Heterotrimeric G Protein. Structure, 20(5), 826–840. doi: 10.1016/j.str.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padayatti PS, Wang L, Gupta S, Orban T, Sun W, Salom D, Chance MR, et al. (2013). A Hybrid Structural Approach to Analyze Ligand Binding by the Serotonin Type 4 Receptor (5-HT4). Mol Cell Proteomics, 12(5), 1259–1271. doi: 10.1074/mcp.M112.025536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Brown L, & Konermann L (2009). Mapping the Structure of an Integral Membrane Protein under Semi-Denaturing Conditions by Laser-Induced Oxidative Labeling and Mass Spectrometry. J Mol Biol, 394(5), 968–981. doi: 10.1016/j.jmb.2009.09.063 [DOI] [PubMed] [Google Scholar]
- Pan Y, Brown L, & Konermann L (2011). Kinetic Folding Mechanism of an Integral Membrane Protein Examined by Pulsed Oxidative Labeling and Mass Spectrometry. J Mol Biol, 410(1), 146–158. doi: 10.1016/j.jmb.2011.04.074 [DOI] [PubMed] [Google Scholar]
- Pan Y, Piyadasa H, O'Neil JD, & Konermann L (2012). Conformational Dynamics of a Membrane Transport Protein Probed by H/D Exchange and Covalent Labeling: The Glycerol Facilitator. J Mol Biol, 416(3), 400–413. doi: 10.1016/j.jmb.2011.12.052 [DOI] [PubMed] [Google Scholar]
- Pan Y, Ruan X, Valvano MA, & Konermann L (2012). Validation of Membrane Protein Topology Models by Oxidative Labeling and Mass Spectrometry. J Am Soc Mass Spectrom, 23(5), 889–898. doi: 10.1021/jasms.8b04279 [DOI] [PubMed] [Google Scholar]
- Pan Y, Stocks BB, Brown L, & Konermann L (2009). Structural Characterization of an Integral Membrane Protein in Its Natural Lipid Environment by Oxidative Methionine Labeling and Mass Spectrometry. Anal Chem, 81(1), 28–35. doi: 10.1021/ac8020449 [DOI] [PubMed] [Google Scholar]
- Porter NA, Caldwell SE, & Mills KA (1995). Mechanisms of free radical oxidation of unsaturated lipids. Lipids, 30(4), 277–290. doi: 10.1007/bf02536034 [DOI] [PubMed] [Google Scholar]
- Pratt DA, Tallman KA, & Porter NA (2011). Free Radical Oxidation of Polyunsaturated Lipids: New Mechanistic Insights and the Development of Peroxyl Radical Clocks. Acc Chem Res, 44(6), 458–467. doi: 10.1021/ar200024c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading E, Hall Z, Martens C, Haghighi T, Findlay H, Ahdash Z, Booth PJ, et al. (2017). Interrogating Membrane Protein Conformational Dynamics within Native Lipid Compositions. Angew Chem Int Ed Engl, 56(49), 15654–15657. doi: 10.1002/anie.201709657 [DOI] [PubMed] [Google Scholar]
- Rinas A, Mali VS, Espino JA, & Jones LM (2016). Development of a Microflow System for In-Cell Footprinting Coupled with Mass Spectrometry. Anal Chem, 88(20), 10052–10058. doi: 10.1021/acs.analchem.6b02357 [DOI] [PubMed] [Google Scholar]
- Roos C, Kai L, Haberstock S, Proverbio D, Ghoshdastider U, Ma Y, Bernhard F, et al. (2014). High-level cell-free production of membrane proteins with nanodiscs. Methods Mol Biol, 1118, 109–130. doi: 10.1007/978-1-62703-782-2_7 [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, & Burke B (2013). BioID: A Screen for Protein-Protein Interactions. Curr Protoc Protein Sci, 74(1), 19.23.11–19.23.14. doi: 10.1002/0471140864.ps1923s74 [DOI] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P, Samson R, & Gingras A-C (2020). Proximity dependent biotinylation: key enzymes and adaptation to proteomics approaches. Mol Cell Proteomics. doi: 10.1074/mcp.R120.001941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Overington JP, et al. (2017). A comprehensive map of molecular drug targets. Nature reviews. Drug discovery, 16(1), 19–34. doi: 10.1038/nrd.2016.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Macpherson JA, Lau AM, Tan KW, Fraternali F, & Politis A (2017). Surface accessibility and dynamics of macromolecular assemblies probed by covalent labeling mass spectrometry and integrative modeling. Anal Chem, 89(3), 1459–1468. doi: 10.1021/acs.analchem.6b02875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp J, Misra S, Persoff J, Egan R, & Weinberger S (2018). Real Time Normalization of Fast Photochemical Oxidation of Proteins Experiments by Inline Adenine Radical Dosimetry. Anal Chem, 90, 12625–12630. doi: 10.1021/acs.analchem.8b02787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen G, Cui W, Zhang H, Zhou F, Huang W, Liu Q, Li W, et al. (2016). Warfarin traps human vitamin K epoxide reductase in an intermediate state during electron transfer. Nat Struct Mol Biol, 24(1), 69–76. doi: 10.1038/nsmb.3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen G, Li S, Cui W, Liu S, Yang Y, Gross M, & Li W (2018). Membrane Protein Structure in Live Cells: Methodology for Studying Drug Interaction by Mass Spectrometry-Based Footprinting. Biochemistry, 57(3), 286–294. doi: 10.1021/acs.biochem.7b00874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havli J, Olsen JV, & Mann M (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc, 1(6), 2856–2860. doi: 10.1038/nprot.2006.468 [DOI] [PubMed] [Google Scholar]
- Takamoto K, & Chance MR (2006). Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu Rev Biophys, 35(1), 251–276. doi: 10.1146/annurev.biophys.35.040405.102050 [DOI] [PubMed] [Google Scholar]
- Thonghin N, Kargas V, Clews J, & Ford RC (2018). Cryo-electron microscopy of membrane proteins. Methods, 147, 176–186. doi: 10.1016/j.ymeth.2018.04.018 [DOI] [PubMed] [Google Scholar]
- Vahidi S, Bi Y, Dunn SD, & Konermann L (2016). Load-dependent destabilization of the γ-rotor shaft in FOF1 ATP synthase revealed by hydrogen/deuterium-exchange mass spectrometry. Proc Natl Acad Sci U S A, 113(9), 2412–2417. doi: 10.1073/pnas.1520464113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Kim S-H, Ablonczy Z, Crouch RK, & Knapp DR (2004). Probing Rhodopsin–Transducin Interactions by Surface Modification and Mass Spectrometry. Biochemistry, 43(35), 11153–11162. doi: 10.1021/bi049642f [DOI] [PubMed] [Google Scholar]
- Watkinson TG, Calabrese AN, Ault JR, Radford SE, & Ashcroft AE (2017). FPOP-LC-MS/MS Suggests Differences in Interaction Sites of Amphipols and Detergents with Outer Membrane Proteins. J Am Soc Mass Spectrom, 28(1), 50–55. doi: 10.1007/s13361-016-1421-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinglass A, Whitelegge JP, Faull KF, & Kaback HR (2004). Monitoring conformational rearrangements in the substrate-binding site of a membrane transport protein by mass spectrometry. J Biol Chem, 279(40), 41858–41865. doi: 10.1074/jbc.M407555200 [DOI] [PubMed] [Google Scholar]
- Weinglass A, Whitelegge JP, Hu Y, Verner GE, Faull KF, & Kaback HR (2003). Elucidation of substrate binding interactions in a membrane transport protein by mass spectrometry. The EMBO Journal, 22(7), 1467–1477. doi: 10.1093/emboj/cdg145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Zhang H, Gross ML, & Blankenship RE (2009). Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proc Natl Acad Sci U S A, 106(15), 6134–6139. doi: 10.1073/pnas.0901691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, & Chance MR (2005). Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal Chem, 77(14), 4549–4555. doi: 10.1021/ac050299+ [DOI] [PubMed] [Google Scholar]
- Xu G, & Chance MR (2007). Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chemical Rev, 107(8), 3514–3543. doi: 10.1021/cr0682047 [DOI] [PubMed] [Google Scholar]
- Yin H, & Flynn AD (2016). Drugging Membrane Protein Interactions. Annu Rev Biomed Eng, 18, 51–76. doi: 10.1146/annurev-bioeng-092115-025322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Cheng M, Rempel D, & Gross ML (2018). Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology. Methods, 144, 94–103. doi: 10.1016/j.ymeth.2018.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu Z, Zhang J, Dou T, Chen J, Ge G, Wang F et al. (2019). Prediction of ligand modulation patterns on membrane receptors via lysine reactivity profiling. Chem Commun, 55(30), 4311–4314. doi: 10.1039/c9cc00520j [DOI] [PubMed] [Google Scholar]
- Zhu Y, Guo T, Park JE, Li X, Meng W, Datta A, Sze SK, et al. (2009). Elucidating in Vivo Structural Dynamics in Integral Membrane Protein by Hydroxyl Radical Footprinting. Mol Cell Proteomics, 8(8), 1999–2010. doi: 10.1074/mcp.M900081-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Serra A, Guo T, Park JE, Zhong Q, & Sze SK (2017). Application of Nanosecond Laser Photolysis Protein Footprinting to Study EGFR Activation by EGF in Cells. J Proteome Res, 16(6), 2282–2293. doi: 10.1021/acs.jproteome.7b00154 [DOI] [PubMed] [Google Scholar]
- Ziemianowicz DS, Sarpe V, & Schriemer DC (2019). Quantitative Analysis of Protein Covalent Labeling Mass Spectrometry Data in the Mass Spec Studio. Anal Chem, 91(13), 8492–8499. doi: 10.1021/acs.analchem.9b01625 [DOI] [PubMed] [Google Scholar]