

Our body responds to environmental stress by changing the expression levels of a series of cytoprotective enzymes/proteins through multilayered regulatory mechanisms, including the KEAP1-NRF2 system. While NRF2 upregulates the expression of many cytoprotective genes, there are fundamental limitations in short-read RNA sequencing (RNA-Seq), resulting in confusion regarding interpreting the effectiveness of cytoprotective gene induction at the transcript level. To precisely delineate isoform usage in the stress response, we conducted independent full-length transcriptome profiling (isoform sequencing; Iso-Seq) analyses of lymphoblastoid cells from three volunteers under normal and electrophilic stress-induced conditions.
KEYWORDS: KEAP1, NRF2, oxidative stress, transcription, transcriptome
ABSTRACT
Our body responds to environmental stress by changing the expression levels of a series of cytoprotective enzymes/proteins through multilayered regulatory mechanisms, including the KEAP1-NRF2 system. While NRF2 upregulates the expression of many cytoprotective genes, there are fundamental limitations in short-read RNA sequencing (RNA-Seq), resulting in confusion regarding interpreting the effectiveness of cytoprotective gene induction at the transcript level. To precisely delineate isoform usage in the stress response, we conducted independent full-length transcriptome profiling (isoform sequencing; Iso-Seq) analyses of lymphoblastoid cells from three volunteers under normal and electrophilic stress-induced conditions. We first determined the first exon usage in KEAP1 and NFE2L2 (encoding NRF2) and found the presence of transcript diversity. We then examined changes in isoform usage of NRF2 target genes under stress conditions and identified a few isoforms dominantly expressed in the majority of NRF2 target genes. The expression levels of isoforms determined by Iso-Seq analyses showed striking differences from those determined by short-read RNA-Seq; the latter could be misleading concerning the abundance of transcripts. These results support that transcript usage is tightly regulated to produce functional proteins under electrophilic stress. Our present study strongly argues that there are important benefits that can be achieved by long-read transcriptome sequencing.
INTRODUCTION
The KEAP1-NRF2 system is a master regulatory system regulating the cytoprotective response upon exposure to oxidative or electrophilic stress (1). Nuclear factor erythroid 2-related factor 2 (NRF2) is a cap ’n’ collar (CNC) family transcription factor that regulates inducible expression of an array of cytoprotective genes (2, 3). Under the normal (unstressed) condition, the NRF2 protein is constitutively trapped by Kelch-like ECH-associated protein 1 (KEAP1) and is degraded through the proteasome pathway in the cytoplasm (4). Oxidative and electrophilic stresses inactivate KEAP1 and stabilize NRF2, which leads to the accumulation of the NRF2 protein in the nucleus and upregulation of target genes by forming a heterodimer with small Maf proteins (2, 5, 6). The target genes of NRF2 encode various cytoprotective enzymes or proteins involved in detoxification, elimination of reactive oxygen species (ROS), drug exclusion, and NADPH production (7–12).
The ability to produce multiple transcripts through the use of an alternative transcription start site (TSS), alternative transcription termination site (TTS), or alternative splicing is an essential regulatory mechanism in eukaryotes and enables a cell to increase the transcript diversity from a single gene (13). The expression of alternative 5′ structures is often coupled to promoter choice. The promoters that contain different regulatory elements corresponding to tissue-specific and/or developmental stage-specific regulation allow diverse regulation of gene expression in various environments (14–16). Structural changes in mRNA give rise to modifications of the reading frame, resulting in multiple protein isoforms with diverse functions, and affect gene expression by altering the efficiency of mRNA translation. One salient example of this phenomenon is known in the KEAP1-NRF2 system; in lung cancers and head-and-neck cancers, aberrant NFE2L2 (NRF2) transcript variants missing exon 2, which encode NRF2 protein isoforms lacking the KEAP1 interacting domain, have been identified (17). This exon skipping results in aberrant NRF2 stabilization and accumulation, leading to the aberrant induction of NRF2 target genes.
To date, the sequences of over 80,000 protein-coding transcripts expressed from approximately 20,000 protein-coding genes have been deposited in public databases, such as GENCODE (18). It has been estimated that more than 90% of multiexon genes undergo alternative splicing, and approximately 60% of genes in humans have at least one alternative TSS (19–21). However, while there are ample lines of evidence for the expression of multiple transcripts from a single gene, it is less clear whether these transcripts are expressed equally abundantly or one or a few transcripts are expressed predominantly from the gene. For instance, human KEAP1 has been reported to have 10 transcript isoforms, but the usage of the isoforms remains to be clarified (21).
This question becomes more important with the growth in numbers of alternative transcripts annotated in databases following the development and expansion of high-throughput technologies, including RNA sequencing (RNA-Seq). In combination with the development of whole-genome sequencing analysis, RNA-Seq analysis brought about the delineation of precise gene structures, and RNA-Seq analysis further realized good quantifications of gene expression across the genome. To date, RNA-Seq analysis using short-read sequencers (short-read RNA-Seq) has become a standard method to detect and quantify gene expression; short-read RNA-Seq generates comprehensive and high-quality data to evaluate the expression levels of genes. However, it is important to note that there is a fundamental limitation in short-read RNA-Seq analysis of transcript levels, as accurate allocation of sequence reads to specific isoforms and precise estimation of transcript abundance are challenging by short-read RNA-Seq analysis (22).
Recent developments in several long-read sequencing technologies have given rise to a new approach for transcriptome analysis. One is single-molecule real-time (SMRT) sequencing, which is also referred to as isoform sequencing (Iso-Seq), and was generated by Pacific Biosciences (PacBio) (23). The other is nanopore sequencing, which was introduced by Oxford Nanopore Technologies (24, 25). Both technologies are expected to provide promising breakthroughs in transcriptome analysis, especially to solve the bottleneck problem of short-read RNA-Seq. These technologies enable the production of much longer sequence reads than those produced by short-read RNA-Seq. Maximum lengths of the reads will reach over tens to hundreds of thousands of base pairs (23, 25). We surmise that this progress will realize accurate assignments of transcript isoforms as the long lead sequence ensures the determination of full-length transcripts as a single read.
NRF2 regulates a subset of cytoprotective genes in an inducible manner. The expression of NRF2 target genes increases substantially upon exposure to oxidative or electrophilic stress (26). Therefore, we hypothesize that there are alternative uses of exons or generated transcript isoforms, which produce diverse transcript isoforms that are utilized specifically in various stress-mediated contexts. To the best of our knowledge, while numerous studies have been executed to characterize many NRF2 target genes, investigations focused on the molecular basis for producing transcriptional diversity as well as posttranscriptional regulation, including transcript isoform usage of the NRF2 target genes, have not been conducted.
Therefore, in this study, we decided to apply Iso-Seq technology to analyze the transcriptome of human cells to determine the transcript isoforms of NRF2-regulated cytoprotective genes expressed in response to stress. We found that the transcription of KEAP1 and NFE2L2 mRNA was initiated from two distinct first exons. We also found, through the evaluation of the expression levels of NRF2 target gene transcripts, that a few isoforms are dominantly expressed from the genes regulated by NRF2, suggesting the presence of posttranscriptional regulatory mechanisms to express specific isoforms in response to stress. Of note, we found evident discrepancies in the assignment of isoform abundance between Iso-Seq and short-read RNA-Seq. There is a possibility that on certain occasions, the estimation of the expression level of transcript isoforms by short-read RNA-Seq produces misleading information on the abundance of isoforms. We conclude with the benefits of long-read sequencing to address the inherent limitations of short-read RNA-Seq.
RESULTS
Inducible expression of NRF2 target genes in response to electrophilic stress in LCLs.
To analyze the transcript usage of KEAP1 and NFE2L2 (encoding NRF2) genes and NRF2 target genes in the electrophilic stress response, in this study, we utilized Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines (LCLs) as a model system. Our concurrent (65) and previous studies on the environmental stress response in human cells (27, 28) have revealed that LCLs retain their response to electrophilic stress. We independently established LCLs from three healthy volunteers and treated the cells with an electrophilic agent, diethylmaleate (DEM), to induce the activation of the NRF2 pathway (Fig. 1A). Showing very good agreement with a previous study (27), immunoblot analysis revealed that under the DEM-treated (stress) condition, the NRF2 protein accumulated comparably in the nucleus of LCLs from all three participants (Fig. 1B), indicating that the stress responsiveness of each LCL is comparable in terms of intercellular NRF2 localization and abundance.
FIG 1.

DEM treatment-activated NRF2 target genes in LCLs established from three healthy donors. (A) Scheme for the establishment of LCLs and the induction of the electrophilic response. The LCLs were established from three healthy volunteers (named no. 1, no. 2, and no. 3) and treated with DEM to induce electrophilic stress. (B) The accumulation of the NRF2 protein in the nucleus of LCLs under normal (N) and stress (S) conditions. Lamin B was used as a loading control. (C) MA plot representing differentially expressed genes. The x axis represents the mean read count among all samples, and the y axis represents the log2(fold change) in gene expression under the stress condition compared to that under the normal condition. Each dot represents one gene, with red, blue, or brown dots representing genes with significantly upregulated expression values, those with downregulated expression values, or other genes, respectively. (D) GSEA enrichment plot of NRF2 target genes. The normalized enrichment score (NES) and the false discovery rate (FDR) are shown. (E) Heat map representing the expression level of NRF2-related genes. The genes were significantly upregulated in the NFE2L2.V2 gene set. The values are shown by the z-score of normalized counts. The values of each sample (no. 1, no. 2, and no. 3 shown in panel A) are shown. Prototype NRF2 target genes are indicated in green.
We next conducted RNA-Seq analysis using a short-read sequencer to estimate transcriptional changes between normal and DEM-treated (stress) conditions. We found that the expression levels of 4,100 genes were significantly changed upon DEM stimulation in LCLs, in which 2,013 genes were upregulated and 2,087 genes were downregulated among the 58,721 genes in the GENCODE database (Fig. 1C). Since the NRF2 protein was induced by the DEM challenge, in this analysis, we examined how NRF2 target genes were upregulated. Gene set enrichment analysis (GSEA) showed that the gene set including the canonical target genes of NRF2 was significantly more enriched under the stress condition than under the normal condition, and the majority of the genes in this category were upregulated by DEM treatment (Fig. 1D). Of the upregulated genes, we found the expression levels of prototype NRF2 target genes, which are shown in green characters in Fig. 1E. This group of genes includes those related to heme metabolism (heme oxygenase 1 [HMOX1], ferritin heavy chain 1 and light chain [FTH1 and FTL, respectively], and ferrochelatase [FECH]), quinone detoxification [NAD(P)H quinone oxidoreductase 1 and 2 (NQO1 and NQO2, respectively)], glutathione metabolism (glutamate-cysteine ligase modifier subunit and catalytic subunit [GCLM and GCLC, respectively] and glutathione-disulfide reductase [GSR]), transcription factor (v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog G [MAFG]), autophagy (sequestosome 1 [SQSTM1]), and pentose phosphate pathway (transaldolase 1 [TALDO1], malic enzyme 1 [ME1], transketolase [TKT], and phosphogluconate dehydrogenase [PGD]) (9, 29–32). These results demonstrate that the NRF2 pathway was successfully induced by the DEM treatment in LCLs.
Detection and quantification of transcript isoforms using Iso-Seq.
Currently, in transcriptome analysis, quantification of the read abundance for individual transcript isoforms relies on counting of sequence reads that overlap known isoforms defined by transcript annotation, such as GENCODE. As shown in Fig. 2A, one of the limitations inherent to this approach is that, in the case of short-read RNA-Seq, it is difficult to accurately allocate a read to a specific isoform (22). Substantial proportions of the reads appear to be mapped ambiguously, as an exon or an exon-exon junction are often shared by multiple isoforms (see isoforms A and B in Fig. 2A). Therefore, to overcome this difficulty, we decided to adopt an approach utilizing long-read RNA-Seq, as this approach has the potential to identify and quantify isoforms by determining the end-to-end cDNA sequence. To this end, in this study, we conducted Iso-Seq analysis developed for the PacBio sequencer and quantified the expression levels of each isoform in LCLs under normal and/or oxidative stress conditions.
FIG 2.

Assessment of expression at the isoform level using Iso-Seq. (A) Isoform detection by short-read RNA-Seq or Iso-Seq technologies. A gene structure is represented with black boxes and lines representing exons and introns, respectively, and structures of transcript isoforms expressed from the gene are shown (isoform A and isoform B are in red and blue, respectively). While the sequence reads ambiguously annotated to isoforms A and B are shown in brown, those unambiguously annotated to isoform A or B are shown in red or blue under the transcript models. (B) The workflow of data analysis of Iso-Seq data. Software used in the analysis is shown in parentheses. (C) Number of reads annotated to each novelty category of transcripts under normal conditions. The criteria used for the classification of the reads into the four categories (FSM, red; ISM, blue; NIC, green; and NNC, navy) are provided in the main text. The results of each sample (no. 1, no. 2, and no. 3 shown in Fig. 1A) under normal conditions are represented. (D) Saturation curve for the number of genes (top) and transcripts (bottom) identified using Iso-Seq analysis under normal conditions. The numbers of FSM reads are on the x axis, and the numbers of genes and transcripts detected are on the y axis. The results of each sample (no. 1, no. 2, and no. 3 shown in Fig. 1A) under normal conditions are shown. (E) Venn diagram showing the common annotated isoform detected by short-read RNA-Seq and Iso-Seq. The transcripts in the GENCODE database with a read under any condition for short-read RNA-Seq and with a read in any sample for Iso-Seq were considered to be the detected transcripts. (F) Box plot showing the expression level of the unique isoforms in short-read RNA-Seq (short-read RNA-Seq only; red) and the common isoforms (short-read RNA-Seq and Iso-Seq; green) under the normal condition. The expression levels were estimated by the log2(TPM value) of short-read RNA-Seq. (G) Box plot showing the expression level of the unique isoforms in Iso-Seq (Iso-Seq only; blue) and the common isoforms (short-read RNA-Seq and Iso-Seq; green) under the normal condition. The expression levels were estimated by the log2(count value) of Iso-Seq. (H) Scatterplot showing the expression level of each isoform estimated by short-read RNA-Seq [x axis; mean log2(TPM value)] and Iso-Seq [y axis; mean log2(count value)] under normal conditions.
As shown in Fig. 2B, the Iso-Seq protocol includes constructing a library, selecting the cDNA size, sequencing the cDNA, and processing the data. One of the critical points is the size selection of cDNAs to avoid any loading biases. Therefore, during these processes, cDNAs were fractionated based on their length to produce four libraries in size ranges of 1 to 2 kbp, 2 to 3 kbp, 3 to 5 kbp, and 5 to 10 kbp. We prepared libraries from LCLs of three individuals with and without DEM treatment (i.e., a total of six sets of 4 size-selected libraries). Each library was sequenced independently by the PacBio RSII system, and 2,184,241 ± 106,718 (means ± standard deviations [SD]) circular consensus sequence (CCS) reads were obtained (Table 1). We further selected the CCS reads to 1,607,689 ± 98,355 full-length reads, which had both 5′ and 3′ barcoded primers and poly(A) tails. We then employed the LoRDEC algorithm (33) to correct errors by employing short-read RNA-Seq data that were independently determined for each sample. Following mapping of the long-read sequence to the reference genome, reference-based error correction was conducted to remove microinsertions, microdeletions, mismatches, and noncanonical splice junctions (34, 35).
TABLE 1.
Number of reads obtained for Iso-Seq analysis
| Sample and condition | Library size at: |
||||
|---|---|---|---|---|---|
| 1–2 kb | 2–3 kb | 3–5 kb | 5–10 kb | Total | |
| CCS reads | |||||
| 1 | |||||
| Normal | 694,795 | 655,375 | 558,807 | 329,294 | 2,238,271 |
| Stress | 622,425 | 431,778 | 530,784 | 410,958 | 1,995,945 |
| 2 | |||||
| Normal | 643,199 | 610,078 | 553,821 | 333,271 | 2,140,369 |
| Stress | 687,545 | 659,969 | 504,990 | 301,612 | 2,154,116 |
| 3 | |||||
| Normal | 731,989 | 641,458 | 504,138 | 358,635 | 2,236,220 |
| Stress | 722,501 | 631,056 | 509,231 | 477,734 | 2,340,522 |
| Full-length reads | |||||
| 1 | |||||
| Normal | 559,255 | 509,878 | 425,131 | 233,893 | 1,728,157 |
| Stress | 513,991 | 326,149 | 402,070 | 244,230 | 1,486,440 |
| 2 | |||||
| Normal | 427,826 | 416,115 | 407,569 | 220,104 | 1,471,614 |
| Stress | 537,162 | 487,284 | 361,156 | 218,634 | 1,604,236 |
| 3 | |||||
| Normal | 546,215 | 476,120 | 396,482 | 243,908 | 1,662,725 |
| Stress | 523,360 | 462,530 | 381,939 | 325,133 | 1,692,962 |
To obtain sequence reads that are unambiguously annotated to unique isoforms, we annotated and quantified the mapped sequence reads to GENCODE version 29 reference transcripts using the TALON pipeline, which is an official ENCODE pipeline for long-read data analysis (36). This pipeline annotates full-length reads to known or novel isoforms, as shown on the right of Fig. 2C, and reports the abundance of isoforms. If a query read had a set of splice junctions that perfectly matched the reference annotation, the read was categorized as full splice match (FSM). The FSM reads can be recognized as reads unambiguously assigned to unique isoforms. In cases where a read matched a subsection of a known transcript model and had a novel putative TSS or TTS, it was categorized as incomplete splice match (ISM). ISM reads can be a combination of potentially real shorter versions of long reference transcripts as well as partial fragments resulting from artifacts reflecting mRNA decay. The transcripts with known splice donors and acceptors but new combinations between them were categorized as novel in catalog (NIC), and transcripts that contained at least one novel splice donor or acceptor were referred to as novel not in catalog (NNC). Following these categorizations, we found that between 790,758 and 1,087,493 reads obtained from each sample from the three volunteers under the normal condition were categorized as FSM, while a relatively small proportion of reads (from 204,535 to 216,257) were categorized as ISM (Fig. 2C, left). Three samples with the DEM treatment showed almost similar results (data not shown). Thus, these results demonstrate that our Iso-Seq analysis successfully captured full-length transcripts.
To assess how many genes and transcripts expressed in LCLs were detected by our Iso-Seq analysis, we next plotted the number of individual genes and transcripts as a function of the sum of FSM reads (Fig. 2D). We found that the number of genes reached a plateau of approximately 10,000 detected genes at approximately 300,000 reads. Interestingly, whereas the number of detected transcripts started saturating, we still observed a steady increase in detected transcripts as the sequence depth increased to a million FSM reads. These results suggest that deeper sequencing is required for a comprehensive viewing of the transcripts, especially for detecting rare transcripts.
Of the annotated transcripts obtained from the Iso-Seq data, we found 42,736 transcripts that were expressed one or more times in any of the six samples. Comparing this number of Iso-Seq transcripts to those detected in short-read RNA-Seq (56,959 transcripts), we found that 25,714 transcripts were detected by both short-read RNA-Seq and Iso-Seq, 31,245 transcripts were detected only by short-read RNA-Seq, and 17,022 transcripts were detected only by Iso-Seq (Fig. 2E).
It should be noted that the expression levels of transcripts uniquely detected by short-read RNA-Seq were lower than those of transcripts commonly detected by short-read RNA-Seq and Iso-Seq (Fig. 2F), and this correlation was similar for those of transcripts detected by Iso-Seq (Fig. 2G), indicating that in both techniques, it was difficult to precisely detect the transcripts with lower expression levels. To further address how widely the expression level of each transcript can be quantified by Iso-Seq and whether the extent correlates with that quantified by short-read RNA-Seq, we compared the expression levels of each transcript quantified using Iso-Seq to those using short-read RNA-Seq. While there was a limited correlation between the results of two techniques in the transcripts with very low expression levels, transcripts with moderate to high expression levels showed substantial levels of correlations (Fig. 2H). These results support our belief that the Iso-Seq approach provides high-quality full-length transcripts with a reasonable level of quantitative assessment.
Multiple first-exon usage in NFE2L2 and KEAP1 genes encoding regulators of stress response.
We next evaluated the transcript isoform usage of electrophilic stress response and antioxidant genes, along with NFE2L2 and KEAP1 genes encoding their regulators, by employing Iso-Seq data. We first examined NFE2L2 and KEAP1 genes by aligning the Iso-Seq reads (Fig. 3A and C, middle) to the reference genome and by annotating them using the reference transcripts in the GENCODE database (Fig. 3A and C, top). These alignments revealed that the NFE2L2 and KEAP1 loci were mostly fully covered with single continuous Iso-Seq reads, indicating that our Iso-Seq strategy successfully captured the full-length transcripts of the NFE2L2 and KEAP1 genes.
FIG 3.

Identification of alternative TSS usage in NFE2L2 and KEAP1 genes. (A and C) Representative Iso-Seq reads aligned to NFE2L2 (A) and KEAP1 (C) genes. The transcript structures in the GENCODE database are represented with black (GENCODE transcripts). The reads of Iso-Seq analysis are shown in purple (Iso-Seq reads). The transcript models made from Iso-Seq data are shown in green (Iso-Seq transcripts). The distal and proximal first exons are highlighted in red and blue, respectively. The ensemble IDs (with prefix of ENST) for transcripts annotated in the GENCODE database or the temporary IDs (with prefix of TALONT) for transcripts not annotated in the database are shown on the transcript models. (B and D) Expression levels of transcript isoforms of NFE2L2 (B) and KEAP1 (D) genes detected by Iso-Seq (left) and/or short-read-based RNA-Seq (right). The expression levels under normal and stress conditions are represented. The values are shown as the means ± SD.
Closer inspection of the data revealed that among 13 transcript isoforms of the NFE2L2 gene that appeared in the GENCODE database, the Iso-Seq reads showed only five corresponding isoforms based on the positions of the splice junctions, namely, ENST00000397062, ENST00000397063, ENST00000448782, ENST00000421929, and ENST00000423513. While the latter four transcripts shared a common sequence at the 3′ portion of the first exon, the first transcript (ENST00000397062) used an alternative first exon, which was transcribed from an area upstream of that used for the other transcripts.
To determine the most abundant isoform transcribed from the NFE2L2 genes, we quantified the expression of these transcript isoforms using the read count data of Iso-Seq. We found that ENST00000397062 and ENST00000397063 were the isoforms with the highest and second highest expression levels, respectively, in the transcript isoforms of the NFE2L2 gene (Fig. 3B, left). ENST00000393062 and ENST00000397063 utilize alternative first exons (highlighted in red and blue in Fig. 3A) but commonly share four downstream exons (exons 2 to 5). The rest of the transcript isoforms showed much lower expression levels than those of these two isoforms. These expression levels of the NFE2L2 transcript isoforms did not change significantly when challenged with the DEM-treated stress (blue and red bars in Fig. 3B).
To validate the observation in Iso-Seq analysis, short-read RNA-Seq data were used to quantify the abundance of the transcript isoforms. Showing very good agreement, the short-read RNA-Seq data revealed the ENST00000397062 and ENST00000397063 transcripts as the primary and secondary transcript isoforms, respectively (Fig. 3B, right). There were some minor discrepancies between the quantification using Iso-Seq and short-read RNA-Seq data (for instance, the detection of the ENST00000464747 isoform). Nonetheless, these results indicate that Iso-Seq analysis successfully captured full-length transcripts and determined the primary transcript isoform expressed in LCLs under both normal and DEM-treated stress conditions.
In the case of the KEAP1 gene, Iso-Seq analysis identified ENST00000393623 and ENST00000171111 as the transcript isoforms with the highest and second highest expression, respectively. ENST00000393623 and ENST00000171111 shared five exons (exons 2 to 6) but possessed alternative first exons (highlighted in red and blue in Fig. 3C). While both transcript isoforms were expressed under the normal and stress conditions, ENST00000393623 was significantly upregulated under the DEM-treated stress condition compared to expression under the normal condition (Fig. 3D). The relative abundances of the KEAP1 transcript isoforms and their order were not changed between the normal and the stressed conditions. This increase may be because the KEAP1 gene is under NRF2 regulation (37). We conclude based on these results that the quantification of isoform expression using Iso-Seq is a promising strategy for determining the transcript isoforms dominantly expressed under normal and stress-induced conditions. In this regard, we found one ISM isoform that was not annotated in GENCODE, so we named it TALONT000863285. We surmise that TALONT000863285 is a novel shorter version of the KEAP1 transcript.
Expression of isoform transcripts of the NQO1 gene quantified by Iso-Seq and short-read RNA-Seq.
Since we successfully determined the primary transcript isoforms of the NFE2L2 and KEAP1 genes by Iso-Seq analysis, we extended the analysis to evaluate the isoform usage of NRF2 target genes. To this end, we first focused on the NQO1 gene, one of the prototypical target genes of NRF2 (38). As seven transcript isoforms expressed from the NQO1 gene by alternative first exons or alternative splicing are annotated in the GENCODE catalog (Fig. 4A, top), we considered that the NQO1 gene may be a good target for transcriptome analysis using Iso-Seq. The Iso-Seq analysis identified five transcript isoforms of the NQO1 gene expressed in the LCLs: ENST00000320623, ENST00000379047, ENST00000379046, ENST00000439109, and ENST00000569118 (Fig. 4A, middle and bottom). Inspection of the locus indicated that ENST00000320623 was a full-length isoform of the NQO1 gene, but ENST00000379047, ENST00000379046, and ENST00000439109 were alternative splicing isoforms in which exon 4, exon 5, or both of them were skipped (Fig. 4A, bottom).
FIG 4.

Estimation of transcript isoforms of the NQO1 gene. (A) Representative Iso-Seq reads aligned to the NQO1 gene. The transcript structures in the GENCODE database are represented with black (GENCODE transcripts). The reads of Iso-Seq analysis are shown in purple (Iso-Seq reads). The transcript models made from Iso-Seq data are shown in green (Iso-Seq transcripts). The ensemble IDs are shown on the transcript models. (B) Expression levels of transcript isoforms of the NQO1 gene detected by Iso-Seq (left) and short-read-based RNA-Seq (right). The expression levels under normal (blue) and stress (red) conditions are shown. Data represent the means ± SD. (C) Representative sequence reads aligned to the NQO1 gene. The transcript structures in the GENCODE database are represented with black boxes and lines representing exons and introns, respectively. The short reads aligned to the NQO1 locus are shown with pink and purple, indicating reads aligned to the plus strand and the minus strand, respectively. The ensemble IDs are shown on the transcript models. (D) Mapping of skipped exons on the protein structure of NQO1. Exons 4 and 5 are shown in purple and green, respectively. (E) RT-qPCR validation of transcript isoforms of the NQO1 gene identified by the Iso-Seq analysis. The full-length transcript and transcript isoforms with exon skipping of exon 4, exon 5, or both are shown. RT-qPCR analysis for the detection of NQO1 isoforms was performed using primers in exon 3 and exon 6 (triangles). The expected amplicon sizes obtained from each transcript isoform are shown in parentheses. (F) The relative abundance of the NQO1 isoforms. Representative electrophoresis image obtained from the RT-qPCR analysis is shown on the left. Relative abundance of the isoforms under normal and stress conditions is shown on the right. Data represent the means ± SD. (G) Quantification of transcript isoforms of the NQO1 gene using StringTie, Kallisto, and Salmon software under normal and stress conditions. The values are shown as the means ± SD.
We next quantified the abundances of these isoforms using Iso-Seq reads and determined that ENST00000320623 was the most abundant isoform transcript expressed from NQO1 gene transcripts under both normal and stress conditions and was significantly upregulated under the stress condition compared to that under the normal condition (Fig. 4B). In stark contrast, quantification of short-read RNA-Seq data using the RSEM algorithm showed that the ENST00000564043 isoform, but not the ENST00000320623 isoform, was the most abundant isoform. Iso-Seq reads were not annotated for ENST00000564043. The ENST00000320623 and ENST00000564043 isoforms were generated by alternative first-exon usage (Fig. 4C). To delineate the details, we further examined the short-read RNA-Seq data, and, interestingly, we observed that although the short reads were successfully aligned to the splice donor junction of the first exon of ENST00000320623, the short-read RNA-Seq data incorrectly assigned the transcript reads for ENST00000564043.
We also directly determined that three alternative splicing isoforms, ENST00000379047, ENST00000379046, and ENST00000439109, were expressed from the NQO1 gene using Iso-Seq analysis, and all of them were induced in an electrophilic stress-dependent manner (Fig. 4B). Since the domains encoded by exon 4 (residues 102 to 139 of the full-length protein) and exon 5 (residues 140 to 173) are the binding sites of FAD and NAD(P)H, respectively (Fig. 4D), skipping of these exons destroys the enzymatic activity of the NQO1 protein (39, 40). The Iso-Seq data indicated that the transcript isoforms lacking exon 4 and/or exon 5 were minor transcripts whose expression levels were less than 10% of the expression level of the full-length transcript isoform (ENST00000320623) (Fig. 4B), which is consistent with the results of previous biochemical assays (41). To validate the relative abundance of the transcript isoforms, we next conducted a reverse transcription-quantitative PCR (RT-qPCR) analysis. We designed PCR primers on exon 3 and exon 6 to detect full-length isoforms (the expected amplicon size of 271 bp) and splicing isoforms with exon skipping of exon 5 (ΔE5; 169 bp), exon 4 (ΔE4; 157 bp), or exon 4 and exon 5 (ΔE4 and E5; 55 bp) (Fig. 4E). Showing very good agreement with the Iso-Seq data, we found that the full-length isoform was expressed abundantly, but the latter isoforms with exon skipping were the minor components compared with the full-length isoform under the normal and stress conditions (Fig. 4F).
To address the discrepancy in the quantification of isoform abundance between Iso-Seq and short-read RNA-Seq in more detail, we exploited three transcript abundance estimation algorithms for short-read RNA-Seq, StringTie, Kallisto, and Salmon (42–44), in addition to RSEM (45). While the assessment by Kallisto and Salmon (Fig. 4G) showed a tendency similar to that by RSEM, in which ENST00000564043 was identified as the primary isoform, StringTie identified ENST00000439109, ENST00000379047, and ENST00000320623 as the abundantly expressed isoforms. Importantly, none of the algorithms for short-read RNA-Seq identified ENST0000032623 as the most abundantly expressed isoform, even though the short reads aligned to the position of the first exon of ENST0000032623 (Fig. 4C and G). These findings clearly demonstrate that accurate quantification of transcript isoforms by a short-read transcriptome approach is still a challenging project despite continuous improvements in the computational tools.
Identification of the primary isoforms of NRF2 target genes.
We next extended the Iso-Seq-based approach of evaluating the transcript usage to other NRF2 target genes. To this end, we selected several NRF2 target genes, including multiple transcript isoforms cataloged in the GENCODE database; we selected FTH1, TXNRD1, SQSTM1, and TALDO1 (Fig. 5). The FTH1 gene is a canonical NRF2 target gene to which 12 transcript isoforms are annotated in the database. Among them, Iso-Seq identified ENST00000273550 as the primarily expressed transcript isoform that was upregulated under the stress condition (Fig. 5A). In contrast, short-read RNA-Seq reported ENST000000620041 (by RSEM) and ENST00000534719 (by StringTie) generated by alternative splicing in the 5′ untranslated region and by retention of the third intron, respectively, as the most abundant transcripts. All of the algorithm software for short-read RNA-Seq, including Kallisto and Salmon, failed to detect ENST00000273550 as the primary isoform (Fig. 5A).
FIG 5.

Identification and quantification of primary isoforms of NRF2 target genes using Iso-Seq and short-read RNA-Seq data. Quantification of transcript isoforms of the FTH1 (A), SQSTM1 (B), TXNRD1 (C), and TALDO1 (D) genes using Iso-Seq and short-read RNA-Seq, including RSEM, StringTie, Kallisto, and Salmon software. Data represent the means ± SD. The transcript models are shown in black. The ensemble IDs (with prefix of ENST) for transcripts annotated in the GENCODE database or the temporary IDs (with prefix of TALONT) for transcripts not annotated in the database are also shown.
The SQSTM1 gene encodes the SQSTM1 protein (also referred to as p62), which is an important autophagy chaperone protein (11, 32). Iso-Seq analysis and short-read RNA-Seq analysis using StringTie concordantly identified ENST00000389805 as the most abundant isoform. However, the other algorithms for short-read RNA-Seq estimated that the isoforms with an alternative first exon (ENST00000360718), a fragmented transcript (ENST00000626660), and the seventh exon skipped (ENST00000510187) abundantly exist in LCLs (Fig. 5B).
In contrast, the assignment of the primary transcript isoform of TXNRD1 (Fig. 5C) and TALDO1, which encode thioredoxin reductase 1 (46) and transaldolase 1 (9), respectively, by Iso-Seq and short-read RNA-Seq showed good agreement (Fig. 5D). Even in these cases, short-read RNA-Seq showed that the expression levels of ENST00000526691 of TXNRD1 and ENST00000528097 of TALDO1, which contain an alternative first exon and alternative donor site in intron 6, respectively, existed substantially in LCLs. It seems that the latter transcript of each gene may not be abundant within LCLs. These results support our contention that Iso-Seq analysis is powerful in determining the primary transcript isoform of the cytoprotective genes that are dominantly expressed within cells under normal and stress conditions.
Relative transcript usage of NRF2 target genes under normal and stress conditions.
To address how environmental stress influences transcript isoform utilization and whether there is a selective usage of specific isoforms in response to the electrophilic stress, we extended our examination of transcript isoform usage in response to stress. For this purpose, we examined the relative abundance of transcript isoforms under normal and stress conditions. We categorized transcript isoforms into three groups: primary transcript (the most abundantly expressed), secondary transcript (the second most abundantly expressed), and other transcripts, based on the expression levels under the normal condition (Fig. 6A).
FIG 6.

Relative transcript usage of NRF2 target genes under normal and stress conditions. (A) The relative abundance of transcript isoforms under normal and stress conditions. The relative expression levels of the primary transcript with the highest expression in the gene under normal conditions (primary transcript), the second highest expression (secondary transcript), or the other transcripts are shown. (B) Schematic model of transcript isoforms expressed under normal (left) and stress (right) conditions. The primary and secondary isoforms expressed under the normal condition are shown. Although the transcript level of each isoform is upregulated under the stress condition, the proportion of the transcript isoforms is not changed.
We found that the expression levels of the primary transcript accounted for the majority of transcripts expressed from a single gene under both normal and stress conditions (Fig. 6A). These results indicate that in the electrophilic stress response, a specific transcript isoform (or a few varieties of transcript isoforms) of NRF2 target genes that are expressed under the normal condition are upregulated under the stress condition and expressed dominantly without any switching of the isoform usage (Fig. 6B). In some genes, the secondary and other transcripts were expressed substantially under the normal condition, but the proportions of these transcripts did not change much in response to the challenge with electrophilic stress (Fig. 6A).
In conclusion, in this study, we determined the dominantly expressed transcript isoforms of cytoprotective genes, including the multiple first exon usage in KEAP1 and NFE2L2 genes. The expression of the primary isoform is tightly regulated under normal and electrophilic stress conditions, and we have clarified the limitation of short-read RNA-Seq technology and benefits of long-read technology in the quantification of transcript isoform abundance.
DISCUSSION
While hundreds of NRF2 target genes have been identified and multiple transcript isoforms expressed from these genes have also been identified, it is uncertain whether these genes regulated by NRF2 express multiple transcript isoforms with almost equal abundance per gene or whether a single transcript is produced predominantly per gene. We surmise that this is due to technical limitations inherent to the short-read RNA-Seq technology, in which precisely allocating a read to a specific transcript is technically not feasible. Therefore, to address this issue utilizing more advanced technology, we conducted transcriptome analysis by means of long-read technology and examined transcript isoforms expressed under electrophilic stress response. As summarized in Fig. 7, in this study, we determined primary transcript isoforms of cytoprotective genes, including NFE2L2, KEAP1, NQO1, FTH1, TALDO1, TXNRD1, and SQSTM1. We found through this analysis that in all cases, a single or a few transcript isoforms per gene were expressed predominantly, while expression levels of the other isoforms are negligible or much lower than that of the primary isoform. In addition, we found that short-read RNA-Seq tends to overestimate the expression levels of the alternative isoforms. We conclude, based on these results, that transcript usage is tightly regulated to produce functional proteins from cytoprotective genes. There are substantial benefits of long-read transcriptome analysis in the identification of transcript isoforms and quantification of their expression levels.
FIG 7.
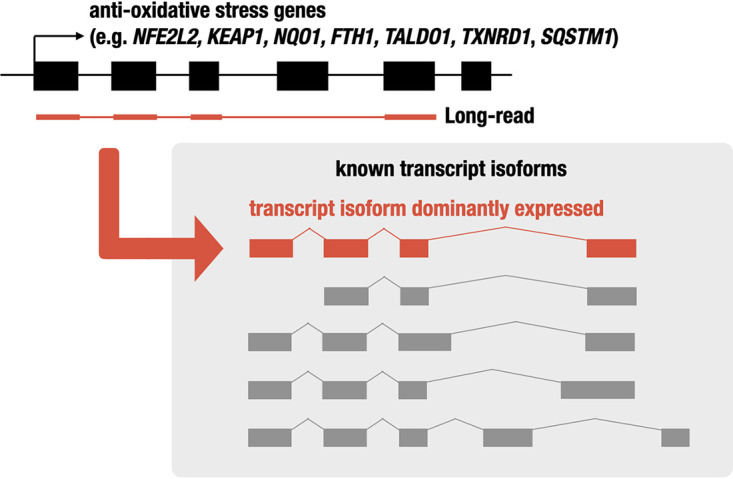
Schematic model of transcript isoform usage in the electrophilic stress response. Our study identified the transcript isoform that is dominantly or alternatively expressed in the electrophilic stress response (red or gray, respectively) using long-read RNA-Seq analysis, which can identify and quantify isoforms by sequencing cDNA molecules end to end. Our assignment indicates that in the NRF2 target genes, a few isoforms are expressed dominantly, and the others are alternatives.
Many eukaryote genes have multiple transcript isoforms (19–21). These isoforms are produced from different TSSs and TTSs or as a consequence of alternative splicing. These transcriptional or posttranscriptional regulatory mechanisms of gene expression are able to change the abundance, function, cellular localization, and stability of the corresponding RNA and protein products. Therefore, differential usages of transcript isoforms under different conditions, which are often referred to as isoform switching, seem to have a substantial biological impact (47–49). As a salient example for the transcript isoform usage responsible for the specialized expression of genes in unique cells, we have identified the utilization of multiple TSSs of GATA1 and GATA2 genes, i.e., first exons IT (testis) and IE (erythroid) for the GATA1 gene (16) and first exons IS (specific) and IG (general) for the GATA2 gene (15). Regulatory elements in the proximity of these exons are responsible for cell type-specific expression of these genes (50). Similarly, we hypothesized that there are specific first exons for the expression of inducible genes that respond to environmental stress. To address this hypothesis, in this study, we selected the KEAP1-NRF2 system as a model system and examined KEAP1 and NFE2L2 genes along with a number of NRF2 target genes. We identified two first exons in the KEAP1 and NFE2L2 genes and the inducible expression of the KEAP1 gene under electrophilic stimulus. However, the utilization of these distinct transcript isoforms does not change during the inducible expression. The other genes examined in this study unfortunately do not harbor the alternative first exon system, so this hypothesis remains to be clarified in future studies.
Recent developments in high-throughput technologies, including RNA-Seq, have accelerated the identification of transcript isoforms annotated in databases. Under this circumstance, it becomes important to determine the primary transcript isoform expressed from the gene in a cell lineage or a condition-specific manner. In this study, Iso-Seq analysis revealed that cytoprotective genes under NRF2 regulation often express one or a few dominant isoforms under stable conditions, which remain dominant under stress conditions. Showing very good agreement, a recent proteome analysis suggests that a multi-isoform gene expresses only one predominant isoform at the protein level in a given tissue (51). We surmise that the transcriptional and posttranscriptional regulatory mechanisms act to converge the expression of stress-responsive genes into the expression of proper transcripts that produce functional proteins to effectively counteract stress conditions.
To our surprise, there are notable discrepancies in the quantification of the transcript expression between short-read RNA-Seq and Iso-Seq. There are substantial differences, even among the algorithms developed for the transcript quantification of the short-read RNA-Seq. We have used a typical design for short-read RNA-Seq analysis, in which 76 bp are sequenced from both ends of the libraries, but we surmise that an analysis using longer reads (e.g., 150 bp) will improve the accuracy of the estimation (52). We feel that accurate transcript reconstruction and estimation of transcript abundance remain challenging for short-read RNA-Seq despite the development of sophisticated algorithms (53). Whereas the long-read transcriptome brings about accurate transcript isoform information, the read counts obtained from the Iso-Seq experiment are limiting compared to those obtained from the short-read RNA-Seq experiment, partly because of the sequencing cost. Therefore, hybrid approaches that use long reads to define the isoforms expressed in the samples and short reads to obtain enough counts for well-powered differential expression seem to be necessary for the increase of the long-read analysis throughput and the decreases of its sequencing cost.
In summary, in this study, we challenged the determination of transcript isoform usage in electrophilic stress genes regulated by NRF2 using long-read technology. The results demonstrate that there are strict regulatory mechanisms over transcript isoform usage in stress-responsive cytoprotective genes to warrant the efficient production of functional proteins. The study also revealed important benefits of long-read sequencing in the field of gene expression research.
MATERIALS AND METHODS
Establishment of human LCLs.
Three adult male Japanese volunteers, who are the same individuals as jg1a, jg1b, and jg1c reported by Takayama et al. (54), were recruited and participated in this study with written informed consent. They were self-reportedly healthy without genetic diseases. LCLs were established from the volunteers and cultured as previously described (55, 56).
Immunoblot analyses.
Nuclear lysates for immunoblotting were prepared from LCLs treated with DEM for 6 h using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific). The nuclear lysate was subjected to immunoblotting using anti-NRF2 (57) and anti-lamin B (D4Q4Z) (number 12586S; Cell Signaling Technology) antibodies.
Short-read RNA-Seq library preparation and sequencing.
For transcriptome analysis, the LCLs were treated with 100 μM DEM for 8 h. Total RNA was prepared by using an RNeasy minikit (Qiagen). Isolation of poly(A)-tailed RNA and library construction were performed using the SureSelect strand-specific RNA sample preparation kit (Agilent Technologies). The libraries were sequenced using HiSeq 2500 (Illumina) for 76 cycles of paired-end reads, and more than 40 million reads were generated for each sample.
Short-read RNA-Seq data analysis.
The sequence reads were aligned to the human genome (GRCh38) using STAR (version 2.6.1) (58). Quantification of genes and transcripts annotated in the GENCODE comprehensive annotation (version 29) was performed using RSEM (version 1.3.1) (45) with default parameters. The DESeq2 package (version 1.22.2) (59) was used for differential expression analysis. An adjusted P value cutoff of 0.05 was applied to identify the differentially expressed genes. For comparison analyses, StringTie (version 2.1.1) (43), Kallisto (version 0.46.1) (42), and Salmon (version 1.2.1) (44) were used to quantify expression levels with default parameters. Gene set analysis was conducted using GSEA software (60, 61). The gene set of NFE2L2.V2 (M2870) was downloaded from the Molecular Signatures Database (MSigDB; https://www.gsea-msigdb.org/gsea/msigdb/cards/NFE2L2.V2).
PacBio Iso-Seq library preparation and sequencing.
The same RNA aliquots prepared for short-read RNA-Seq analysis were subjected to library preparation for Iso-Seq analysis. The Iso-Seq library was prepared according to the Iso-Seq protocol using the Clontech SMARTer PCR cDNA synthesis kit and the BluePippin size selection system protocol as described by PacBio. Briefly, 2 μg of total RNA was used for input into the Clontech SMARTer reaction. Amplification of cDNA was performed using the KAPA HiFi HotStart ReadyMix PCR kit (KAPA Biosystems). The amplicons were then size selected using BluePippin (Sage Science) with the following bins for each sample: 1 to 2 kb, 2 to 3 kb, 3 to 6 kb, and 5 to 10 kb. After size selection, large-scale PCR was performed using the eluted DNA from the previous step to generate more double-stranded cDNA for SMRTbell library construction. The amplified and size-selected cDNA products were subjected to SMRTbell template libraries according to the Iso-Seq protocol. The libraries were prepared for sequencing by annealing a sequencing primer and binding polymerase to the primer-annealed template. Each library was sequenced independently using 234 SMRT cells of a PacBio RSII system.
Iso-Seq data analysis.
We used the SMRT Link 6.0.0 software provided by PacBio to obtain the CCS reads from Iso-Seq raw reads. Next, we determined full-length transcripts using the poly(A) tail and 5′/3′ primer information from the cDNA kit and provided them for downstream analysis. To improve the accuracy of the CCS reads, we performed error correction using LoRDEC (33). The corrected full-length transcripts were mapped to the human reference genome (GRCh38) using minimap2 (version 2.7–654) with “-ax splice -uf –secondary=no -C5 -O6,24 -B4” options by following the manufacturer’s recommendations. By following the ENCODE guidelines, we then performed reference-based error correction using TranscriptClean software (35) and the quantification of transcripts in the GENCODE comprehensive annotation (version 29) using the TALON (version 5.0) pipeline (36). For transcript-level quantification, post-TALON filtering was performed to remove novel transcripts that were not reproducibly detected across biological replicates.
Protein structure analysis.
Structural analyses of NQO1 were performed by using PDB (62) entry ID4A (chain A). Isoform sequences were taken from UniProt (NQO1_HUMAN, full-length, P15559; isoform-2, P15559-2; and isoform-3, P15559-3) (63), and missing regions were identified. Visualization was performed by using jV (64). The missing region in isoform-2 (139-172 in ID4A) and that in isoform-3 (101-138 in ID4A) are colored green and purple, respectively.
RT-qPCR analysis.
RNA was reverse transcribed with a SuperScript VILO cDNA synthesis kit (Thermo Fisher Scientific). RT-qPCR analysis for the detection of NQO1 isoforms was performed using a sense primer (GCC GCA GAC CTT GTG ATA TT) in exon 3 and an antisense primer (GAA GCC ACA GAA ATG CAG AA) in exon 6. A TapeStation gel electrophoresis system (Agilent Technologies) was used for the measurement of amplicon size and quantification of their molarity.
Data availability.
The transcriptome data are available at the jMorp website: https://jmorp.megabank.tohoku.ac.jp. The raw sequence data are available upon request after approval of the Ethical Committee and the Materials and Information Distribution Review Committee of Tohoku Medical Megabank Organization.
ACKNOWLEDGMENTS
We thank Keiko Tateno, Nozomi Hatanaka, and Noriko Takahashi for technical support. We appreciate all the volunteers who participated in the TMM Project.
This work was supported by the Tohoku Medical Megabank (TMM) Project from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the Japan Agency for Medical Research and Development (AMED; grant numbers JP19km0105001 and JP19km0105002) and by the Advanced Genome Research and Bioinformatics Study to Facilitate Medical Innovation (GRIFIN) project of Platform Program for Promotion of Genome Medicine from AMED (grant number JP20km0405203). All computational resources were provided by the Tohoku University Tohoku Medical Megabank Organization supercomputer system (http://sc.megabank.tohoku.ac.jp/en), which is supported by the Facilitation of R&D Platform for AMED Genome Medicine Support conducted by AMED (grant number JP20km0405001). This work was also supported in part by JSPS KAKENHI (grant number 19H05649 to M.Y. and JP19K16511 to A.O.).
REFERENCES
- 1.Yamamoto M, Kensler TW, Motohashi H. 2018. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 98:1169–1203. doi: 10.1152/physrev.00023.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 3.Otsuki A, Yamamoto M. 2020. Cis-element architecture of Nrf2-sMaf heterodimer binding sites and its relation to diseases. Arch Pharm Res 43:275–285. doi: 10.1007/s12272-019-01193-2. [DOI] [PubMed] [Google Scholar]
- 4.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. 1999. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motohashi H, Katsuoka F, Engel JD, Yamamoto M. 2004. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc Natl Acad Sci U S A 101:6379–6384. doi: 10.1073/pnas.0305902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katsuoka F, Otsuki A, Takahashi M, Ito S, Yamamoto M. 2019. Direct and specific functional evaluation of the Nrf2 and MafG heterodimer by introducing a tethered dimer into small Maf-deficient cells. Mol Cell Biol 39:e00273-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayes JD, McMahon M. 2009. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci 34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 8.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. 2010. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal 13:1713–1748. doi: 10.1089/ars.2010.3221. [DOI] [PubMed] [Google Scholar]
- 9.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. 2012. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Uruno A, Yagishita Y, Katsuoka F, Kitajima Y, Nunomiya A, Nagatomi R, Pi J, Biswal SS, Yamamoto M. 2016. Nrf2-mediated regulation of skeletal muscle glycogen metabolism. Mol Cell Biol 36:1655–1672. doi: 10.1128/MCB.01095-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. 2010. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otsuki A, Suzuki M, Katsuoka F, Tsuchida K, Suda H, Morita M, Shimizu R, Yamamoto M. 2016. Unique cistrome defined as CsMBE is strictly required for Nrf2-sMaf heterodimer function in cytoprotection. Free Radic Biol Med 91:45–57. doi: 10.1016/j.freeradbiomed.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert W. 1978. Why genes in pieces? Nature 271:501. doi: 10.1038/271501a0. [DOI] [PubMed] [Google Scholar]
- 14.Kalsotra A, Cooper TA. 2011. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet 12:715–729. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minegishi N, Ohta J, Suwabe N, Nakauchi H, Ishihara H, Hayashi N, Yamamoto M. 1998. Alternative promoters regulate transcription of the mouse GATA-2 gene. J Biol Chem 273:3625–3634. doi: 10.1074/jbc.273.6.3625. [DOI] [PubMed] [Google Scholar]
- 16.Ito E, Toki T, Ishihara H, Ohtani H, Gu L, Yokoyama M, Engel JD, Yamamoto M. 1993. Erythroid transcription factor GATA-1 is abundantly transcribed in mouse testis. Nature 362:466–468. doi: 10.1038/362466a0. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein Leonard D, Lee J, Gnad F, Klijn C, Schaub A, Reeder J, Daemen A, Bakalarski Corey E, Holcomb T, Shames David S, Hartmaier Ryan J, Chmielecki J, Seshagiri S, Gentleman R, Stokoe D. 2016. Recurrent loss of NFE2L2 exon 2 is a mechanism for Nrf2 pathway activation in human cancers. Cell Rep 16:2605–2617. doi: 10.1016/j.celrep.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, Barnes I, Berry A, Bignell A, Carbonell Sala S, Chrast J, Cunningham F, Di Domenico T, Donaldson S, Fiddes IT, Garcia Giron C, Gonzalez JM, Grego T, Hardy M, Hourlier T, Hunt T, Izuogu OG, Lagarde J, Martin FJ, Martinez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Ruffier M, Schmitt BM, Stapleton E, Suner MM, Sycheva I, Uszczynska-Ratajczak B, Xu J, Yates A, Zerbino D, Zhang Y, Aken B, Choudhary JS, Gerstein M, Guigo R, Hubbard TJP, et al. 2019. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, Forrest AR, Alkema WB, Tan SL, Plessy C, Kodzius R, Ravasi T, Kasukawa T, Fukuda S, Kanamori-Katayama M, Kitazume Y, Kawaji H, Kai C, Nakamura M, Konno H, Nakano K, Mottagui-Tabar S, Arner P, Chesi A, Gustincich S, Persichetti F, Suzuki H, Grimmond SM, Wells CA, Orlando V, Wahlestedt C, Liu ET, Harbers M, Kawai J, Bajic VB, Hume DA, Hayashizaki Y. 2006. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 21.Encode Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stark R, Grzelak M, Hadfield J. 2019. RNA sequencing: the teenage years. Nat Rev Genet 20:631–656. doi: 10.1038/s41576-019-0150-2. [DOI] [PubMed] [Google Scholar]
- 23.Wang B, Tseng E, Regulski M, Clark TA, Hon T, Jiao Y, Lu Z, Olson A, Stein JC, Ware D. 2016. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat Commun 7:11708. doi: 10.1038/ncomms11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Workman RE, Tang AD, Tang PS, Jain M, Tyson JR, Razaghi R, Zuzarte PC, Gilpatrick T, Payne A, Quick J, Sadowski N, Holmes N, de Jesus JG, Jones KL, Soulette CM, Snutch TP, Loman N, Paten B, Loose M, Simpson JT, Olsen HE, Brooks AN, Akeson M, Timp W. 2019. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat Methods 16:1297–1305. doi: 10.1038/s41592-019-0617-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sessegolo C, Cruaud C, Da Silva C, Cologne A, Dubarry M, Derrien T, Lacroix V, Aury JM. 2019. Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Sci Rep 9:14908. doi: 10.1038/s41598-019-51470-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. 2000. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 27.Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, Xue P, Pi J, Kleeberger SR, Bell DA. 2012. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res 40:7416–7429. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Campbell MR, Lacher SE, Cho HY, Wan M, Crowl CL, Chorley BN, Bond GL, Kleeberger SR, Slattery M, Bell DA. 2016. A polymorphic antioxidant response element links NRF2/sMAF binding to enhanced MAPT expression and reduced risk of Parkinsonian disorders. Cell Rep 15:830–842. doi: 10.1016/j.celrep.2016.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katsuoka F, Motohashi H, Engel JD, Yamamoto M. 2005. Nrf2 transcriptionally activates the mafG gene through an antioxidant response element. J Biol Chem 280:4483–4490. doi: 10.1074/jbc.M411451200. [DOI] [PubMed] [Google Scholar]
- 30.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. 2004. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A 101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwak M-K, Itoh K, Yamamoto M, Sutter TR, Kensler TW. 2001. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dithiole-3-thione. Mol Med 7:135–145. doi: 10.1007/BF03401947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. 2010. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 33.Salmela L, Rivals E. 2014. LoRDEC: accurate and efficient long read error correction. Bioinformatics 30:3506–3514. doi: 10.1093/bioinformatics/btu538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wyman D, Mortazavi A. 2019. TranscriptClean: variant-aware correction of indels, mismatches and splice junctions in long-read transcripts. Bioinformatics 35:340–342. doi: 10.1093/bioinformatics/bty483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wyman D, Balderrama-Gutierrez G, Reese F, Jiang S, Rahmanian S, Forner S, Matheos D, Zeng W, Williams B, Trout D, England W, Chu S-H, Spitale RC, Tenner AJ, Wold BJ, Mortazavi A. 2020. A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification. bioRxiv doi: 10.1101/672931. [DOI]
- 37.Lee OH, Jain AK, Papusha V, Jaiswal AK. 2007. An auto-regulatory loop between stress sensors INrf2 and Nrf2 controls their cellular abundance. J Biol Chem 282:36412–36420. doi: 10.1074/jbc.M706517200. [DOI] [PubMed] [Google Scholar]
- 38.Favreau LV, Pickett CB. 1993. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Characterization of a DNA-protein interaction at the antioxidant responsive element and induction by 12-O-tetradecanoylphorbol 13-acetate. J Biol Chem 268:19875–19881. [PubMed] [Google Scholar]
- 39.Gasdaska PY, Fisher H, Powis G. 1995. An alternatively spliced form of NQO1 (DT-diaphorase) messenger RNA lacking the putative quinone substrate binding site is present in human normal and tumor tissues. Cancer Res 55:2542–2547. [PubMed] [Google Scholar]
- 40.Lienhart WD, Strandback E, Gudipati V, Koch K, Binter A, Uhl MK, Rantasa DM, Bourgeois B, Madl T, Zangger K, Gruber K, Macheroux P. 2017. Catalytic competence, structure and stability of the cancer-associated R139W variant of the human NAD(P)H:quinone oxidoreductase 1 (NQO1). FEBS J 284:1233–1245. doi: 10.1111/febs.14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato M, Takagi M, Mizutani S. 2010. Irradiation-induced p53 expression is attenuated in cells with NQO1 C465T polymorphism. J Med Dent Sci 57:139–145. [PubMed] [Google Scholar]
- 42.Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 43.Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, Pertea M. 2019. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20:278. doi: 10.1186/s13059-019-1910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakurai A, Nishimoto M, Himeno S, Imura N, Tsujimoto M, Kunimoto M, Hara S. 2005. Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. J Cell Physiol 203:529–537. doi: 10.1002/jcp.20246. [DOI] [PubMed] [Google Scholar]
- 47.Vitting-Seerup K, Sandelin A. 2017. The landscape of isoform switches in human cancers. Mol Cancer Res 15:1206–1220. doi: 10.1158/1541-7786.MCR-16-0459. [DOI] [PubMed] [Google Scholar]
- 48.Mitra M, Lee HN, Coller HA. 2020. Splicing busts a move: isoform switching regulates migration. Trends Cell Biol 30:74–85. doi: 10.1016/j.tcb.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L, Yao Y, Sun L, Zhou J, Miao M, Luo S, Deng G, Li J, Wang J, Tang J. 2017. Snail driving alternative splicing of CD44 by ESRP1 enhances invasion and migration in epithelial ovarian cancer. Cell Physiol Biochem 43:2489–2504. doi: 10.1159/000484458. [DOI] [PubMed] [Google Scholar]
- 50.Tan JS, Mohandas N, Conboy JG. 2006. High frequency of alternative first exons in erythroid genes suggests a critical role in regulating gene function. Blood 107:2557–2561. doi: 10.1182/blood-2005-07-2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ezkurdia I, Rodriguez JM, Carrillo-de Santa Pau E, Vazquez J, Valencia A, Tress ML. 2015. Most highly expressed protein-coding genes have a single dominant isoform. J Proteome Res 14:1880–1887. doi: 10.1021/pr501286b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chhangawala S, Rudy G, Mason CE, Rosenfeld JA. 2015. The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biol 16:131. doi: 10.1186/s13059-015-0697-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A. 2016. A survey of best practices for RNA-seq data analysis. Genome Biol 17:13. doi: 10.1186/s13059-016-0881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takayama J, Tadaka S, Yano K, Katsuoka F, Gocho C, Funayama T, Makino S, Okamura Y, Kikuchi A, Kawashima J, Otsuki A, Yasuda J, Kure S, Kinoshita K, Yamamoto M, Tamiya G. 2019. Construction and integration of three de novo Japanese human genome assemblies toward a population-specific reference. bioRxiv doi: 10.1101/861658. [DOI] [PMC free article] [PubMed]
- 55.Minegishi N, Nishijima I, Nobukuni T, Kudo H, Ishida N, Terakawa T, Kumada K, Yamashita R, Katsuoka F, Ogishima S, Suzuki K, Sasaki M, Satoh M, Tohoku Medical Megabank Project Study Group, Yamamoto M. 2019. Biobank establishment and sample management in the Tohoku Medical Megabank Project. Tohoku J Exp Med 248:45–55. doi: 10.1620/tjem.248.45. [DOI] [PubMed] [Google Scholar]
- 56.Neitzel H. 1986. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet 73:320–326. doi: 10.1007/BF00279094. [DOI] [PubMed] [Google Scholar]
- 57.Maruyama A, Tsukamoto S, Nishikawa K, Yoshida A, Harada N, Motojima K, Ishii T, Nakane A, Yamamoto M, Itoh K. 2008. Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch Biochem Biophys 477:139–145. doi: 10.1016/j.abb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 58.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. 2003. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 61.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burley SK, Berman HM, Kleywegt GJ, Markley JL, Nakamura H, Velankar S. 2017. Protein Data Bank (PDB): the single global macromolecular structure archive. Methods Mol Biol 1607:627–641. doi: 10.1007/978-1-4939-7000-1_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.UniProt Consortium Team. 2018. UniProt: the universal protein knowledgebase. Nucleic Acids Res 46:2699. doi: 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kinoshita K, Nakamura H. 2004. eF-site and PDBjViewer: database and viewer for protein functional sites. Bioinformatics 20:1329–1330. doi: 10.1093/bioinformatics/bth073. [DOI] [PubMed] [Google Scholar]
- 65.Ishida N, Aoki Y, Katsuoka F, Nishijima I, Nobukuni T, Anzawa H, Bin L, Tsuda M, Kumada K, Kudo H, Terakawa T, Otsuki A, Kinoshita K, Yamashita R, Minegishi N, Yamamoto M. 2020. Landscape of electrophilic and inflammatory stress-mediated gene regulation in human lymphoblastoid cell lines. Free Radic Biol Med 161:71–83. doi: 10.1016/j.freeradbiomed.2020.09.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The transcriptome data are available at the jMorp website: https://jmorp.megabank.tohoku.ac.jp. The raw sequence data are available upon request after approval of the Ethical Committee and the Materials and Information Distribution Review Committee of Tohoku Medical Megabank Organization.