

Abstract
Introduction
Quantifying associations between genetic mutations and loss of ambulation (LoA) among males diagnosed with childhood-onset dystrophinopathy is important for understanding variation in disease progression and may be useful in clinical trial design.
Methods
Genetic and clinical data from the Muscular Dystrophy Surveillance, Tracking, and Research Network for 358 males born and diagnosed from 1982–2011 were analyzed. LoA was defined as the age at which independent ambulation ceased. Genetic mutations were defined by overall type (deletion/duplication/point mutation) and among deletions, those amenable to exon-skipping therapy (exons 8, 20, 44–46, 51–53) and another group. Cox proportional hazards regression modeling was used to estimate hazards ratios (HR) and 95% confidence intervals (CI).
Results
Mutation type did not predict time to LoA. Controlling for corticosteroids, Exons 8 (HR=0.22; 95% CI=0.08,0.63) and 44 (HR=0.30; 95% CI=0.12,0.78) were associated with delayed LOA compared to other exon deletions.
Discussion
Delayed LoA in males with mutations amenable to exon-skipping therapy is consistent with previous studies. These findings suggest that clinical trials including exon 8 and 44 skippable males should consider mutation information prior to randomization.
Keywords: Duchenne muscular dystrophy, exon skipping, loss of ambulation, MD STARnet, natural history study
1. Introduction
The dystrophinopathies, Duchenne and Becker muscular dystrophies (DMD and BMD, respectively), are X-linked muscle disorders caused by mutations in the DMD gene that produce a spectrum of severity and progression.1 Individuals with DMD have earlier onset of symptoms and faster disease progression with loss of ambulation (LoA) between 10 and 12 years of age, whereas individuals with BMD usually maintain ambulation into adulthood.2 LoA is an important marker of disease progression.3 Glucocorticoids may delay LoA and are now considered standard of care.4–8
Many factors contribute to the rate of progression of the dystrophinopathies including genetic and environmental modifiers.9–16 Variability in progression is partly attributed to the specific mutation in the DMD gene. Most often, DMD is caused by out-of-frame deletions of one or more exons, whereas in-frame deletions are usually associated with BMD.17 This observation led to the development of therapeutic approaches that induce exon-skipping to restore an intact reading-frame.18 With the development of genetic therapies, a better understanding of mutation-associated variance in disease progression is important for clinical trial design (e.g. selection of a control group) and can have an effect on interpretation of results.
Prior clinic-based natural history studies suggest differences in the decline of ambulation or age at LoA in patients with DMD mutations amenable to exon skipping therapies.19–21 Describing age at LoA by mutation type in a population-based cohort may help determine whether this association is seen on a population level. The purpose of this study was to quantify the association of age at LoA by DMD mutation class in males with childhood-onset dystrophinopathies from a population-based cohort in the United States.
Methods
The Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) is a population-based, longitudinal surveillance program for childhood-onset dystrophinopathies. The MD STARnet retrospectively identified and longitudinally followed all individuals diagnosed with a dystrophinopathy who were born on or after January 1, 1982 and on or before December 31, 2011 and were diagnosed by age 21 living in defined surveillance areas. Surveillance was initiated in Arizona, Colorado, Iowa, and western New York State in 2004, and Georgia and Hawaii in 2006 and 2010, respectively.
Case ascertainment occurred using multiple sources including healthcare facilities (e.g., neuromuscular clinics, physical medicine and rehabilitation clinics, emergency departments, hospitals) and administrative data (e.g., birth defects surveillance, state vital records, hospital discharge). Details on case identification and data collection were described elsewhere.22 A committee of neuromuscular clinicians reviewed each case and assigned a diagnostic case status of definite, probable, possible, manifesting female, or asymptomatic, based on clinical trajectory and laboratory and genetic test results.23 Medical record abstraction was conducted annually through December 2011. For individuals identified during September 2011 through December 2011, medical record abstraction was conducted through December 2012.
The MD STARnet identified 1054 affected individuals. Exclusion criteria outlined in Supplemental Figure 1 were applied and data from 358 individuals were available for analysis.
Ethics Statement
All sites had public health authority and IRB approval or exemption to abstract data from medical records of individuals diagnosed with childhood-onset dystrophinopathy.
Outcome
Loss of ambulation
Ambulation status (independent or ceased) and use of a wheelchair (manual or power, part- or full-time) were entered into the abstraction form annually or when a change in mobility status (e.g., wheelchair use began) was noted in the medical record. The date (or age when date was missing) of the mobility status was also entered.
LoA was defined as full-time use of a wheelchair or indication that independent ambulation had ceased. Age at LoA was calculated using date (or age if date was not documented) of first full-time wheelchair use or when ambulation ceased. The algorithm included record review to verify consistency of mobility-related data. For time-to-event analyses, the last known age ambulating, which was identified by the last entry in the mobility table, was used for censored cases.
Predictor
Genetic mutations
The MD STARnet collected genetic test results. Available results were entered into standardized forms specific to mutation type (deletion, duplication, or point mutation). For duplications and deletions, information about which exons were deleted or duplicated and the reading frame prediction was entered. For point mutations, the predicted effect (missense, nonsense, frameshift) and affected exon, nucleotide start/stop locations, and protein and codon changes were included. Additionally, verbatim text from laboratory test results and physician notes were entered into the form for further description of the genetic findings. Genetic mutations were classified into type (deletion, duplication, or point mutation; Table 1). Deletions were further classified into subgroups comprised of mutations potentially treatable (via restoration of the reading frame) by skipping of exons 8, 20, 44, 45, 46, 51, 52, or 53, with the remaining deletions being grouped in an “Other” category (refer to footnotes of Table 1 for details). When deletions were deemed skippable at more than one exon, they were included in the subgroup that would give the highest frequency count to increase the power in that subgroup.
Table 1:
Summary of loss of ambulation (LoA) information for mutation type groups. Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) 1982–2012.
| No. | No. LoA (%) | Mean Age at LoA (years) | Median Age at LoA (years) | ||
| Mutation Type | Deletion | 259 | 138 (53.3%) | 11.14 | 10.96 |
| Duplication | 34 | 18 (52.9%) | 12.09 | 10.71 | |
| Point Mutation | 65 | 31 (47.7%) | 11.02 | 10.63 | |
| Total | 358 | 187 (52.2%) | 11.21 | 10.83 | |
| No. | No. LoA (%) | Mean Age at LoA (years) | Median Age at LoA (years) | ||
| Exon Skippable subgroup | Exon 8 Skippablea | 13 | 4 (30.8%) | 13.54 | 13.22 |
| Exon 20 Skippableb | 7 | 4 (57.1%) | 11.06 | 11.12 | |
| Exon 44 Skippablec | 12 | 6 (50.0%) | 13.75 | 13.03 | |
| Exon 45 Skippabled | 31 | 20 (64.5%) | 10.93 | 11.21 | |
| Exon 46 Skippablee | 16 | 8 (50.0%) | 11.68 | 11.58 | |
| Exon 51 Skippablef | 46 | 26 (56.5%) | 10.78 | 10.59 | |
| Exon 52 Skippableg | 17 | 8 (47.1%) | 10.71 | 9.82 | |
| Exon 53 Skippableh | 38 | 20 (52.6%) | 11.06 | 11.16 | |
| Otheri | 79 | 42 (53.2%) | 10.88 | 10.39 |
Abbreviations: LoA: loss of ambulation
Deletions of exons 3–7 (11), 5–7 (1), or 6–7 (1).
Deletions of exons 3–19 (1), 17–19 (2), 19 alone (3), or 21 alone (1).
Deletions of exons 13–43 (1), 30–43 (1), 35–43 (1), 38–43 (1), 42–43 (1), 43 alone (2), 45–54 (4), or 45–56 (1).
Deletions of exons 12–44 (2), 18–44 (2), 44 alone (9), 46–47 (8), 46–48 (3), 46–49 (1), 46–51 (3), 46–55 (2), or 46–59 (1).
Deletions of exons 26–45 (1), 43–45 (1), or 45 alone (14).
Deletions of exons 45–50 (17), 47–50 (2), 48–50 (10), 49–50 (12), 50 alone (5).
Deletions of exons 51 alone (12) or 53–55 (5).
Deletions of exons 34–52 (1), 45–52 (11), 47–52 (2), 48–52 (9), 49–52 (4), 50–52 (4), or 52 alone (7).
Comprised of all males with exon deletions not falling into one of the groups as defined in a-h.
Covariates
Corticosteroid use
Abstractors annually entered start and stop dates of corticosteroid use, type of corticosteroid used (prednisone or deflazacort), dosage, and reasons for discontinuing use if corticosteroids had been stopped. To account for variability in continuity of use and length of follow-up, the start and stop dates were used to construct corticosteroid use as a time-varying covariate.
Age at symptom onset
Clinical signs and symptoms were entered into the abstraction form using a drop-down menu until the time at which the MD STARnet clinical review committee assigned a case definition. The age at the first documented motor symptom (Gower sign, trouble walking/running/jumping, frequent falling, inability to keep up with peers, abnormal gait, loss of motor skills, gross motor delay, muscle weakness) was derived from an algorithm developed by MD STARnet researchers.12
Race and ethnicity
Parental race and ethnicity were collected from birth certificates, where available. Race was included as a categorical variable with categories White, Black or African American, Native American or American Indian or Alaska Native, Asian or Hawaiian or Pacific Islander, Multiple, Other, and Unknown. Ethnicity was included as a categorical variable with categories of not Hispanic or Latino, Hispanic or Latino, and Unknown.
Statistical Analysis
Observed mean ages at LoA among males who lost ambulation for each mutation group were compared using one-way analysis of variance (ANOVA). A single F-test was carried out to simultaneously test if any of the mutation group means differed from any other. If statistically significant (p < 0.05), post-hoc pairwise differences were reported, along with 95% confidence intervals (CIs). Pairwise differences were adjusted for multiple comparisons by calculating CIs based on the studentized range distribution using the Tukey-Kramer method to account for differences in group sizes.24 Kaplan-Meier curve estimation was used to depict the probability of ambulation by age, mutation type (deletion, duplication, point mutation) and exon skipping subgroups (each exon subgroup versus all other subgroups combined). Cox proportional hazard modeling was used to estimate annual risk of LoA by mutation type and exon subgroups. Three models were used to compare risk of LoA by mutation type (or exon skippable subgroup): Model I included mutation type (or exon skippable subgroup) and corticosteroid use; Model II included mutation type (or exon skippable subgroup), corticosteroid steroid use, race, and ethnicity; and Model III included mutation type (or exon skippable subgroup), corticosteroid use, and age at onset of symptoms. Although corticosteroid use varies substantially based on a variety of factors, corticosteroids remain the standard therapeutic treatment for males with DMD.25 Thus, we retained corticosteroid use as a time varying covariate in all models. For each model, pairwise hazard ratios (HRs) and 95% confidence intervals (CIs) were estimated and corrected for multiple comparisons using the Tukey-Kramer method. Sensitivity analyses that examined (1) the impact of a single outlier with late age at onset of symptoms and (2) reclassification of 14 males with exon 45 deletions from the exon 46 to exon 44 skippable group were also conducted, since such individuals can utilize therapy targeted at skipping either exon 44 or 45 making their classification in one group somewhat arbitrary. Analyses were carried out using R26, version 3.4.1, using the survival package27, version 3.1.8, and the multcomp package28, version 1.4.10.
Results
Mutation Type
Cases came from five states: Arizona (n = 74), Colorado (n = 92), Georgia (n = 94), Iowa (n = 53), and New York (n = 45). The overall average duration of corticosteroid use was 3.10 years (SD=2.84); 83% of cases still walking and 90% of those with LoA used corticosteroids for at least 6 months. Except for the duplication group, the observed median ages at LoA for the different mutation groups were comparable to the mean ages and were similar across groups (Table 1). Among those with duplications, the mean age was larger than the median, suggesting outliers with later ages of LoA. Testing for mean differences in age of LoA among deletion, duplication, and point mutation types using the ANOVA test was not statistically significant (F-value (df=1,180) = 1.26, p = 0.29) (data not shown). The log-rank test for the Kaplan-Meier curve estimation was also not statistically significant for mutation type (χ2 (2) = 0.89) (Figure 1) suggesting no differences in survival time by mutation type. Finally, the adjusted annual risk of LoA among those with duplication or point mutations did not differ from those with deletions (Table 2); most estimates approximated 1.00 and the 95% CIs contained the null. Further, the magnitude of the HRs did not vary substantively across the three models.
Figure 1:

Kaplan-Meier curves for mutation type groups. Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) 1982–2012. Kaplan-Meier curves comparing probability of ambulation as a function of age in years for the three major mutation groups: deletion, duplication, and point mutation.
Table 2:
Hazard ratios (HR) for Cox regression models I, II, and III for comparing loss of ambulation across mutation type groups. Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) 1982–2012.
| No. (%) | Model I | Model II | Model III | ||
|---|---|---|---|---|---|
| Factor | Level | HR (95% CI) | HR (95% CI) | HR (95% CI) | |
| Mutation Type | Deletion | 259 (72.3%) | 1a | 1a | 1a |
| Duplication | 34 (9.5%) | 0.95 (0.58, 1.58) | 1.01 (0.60, 1.69) | 1.00 (0.60, 1.66) | |
| Point Mutation | 65 (18.2%) | 0.92 (0.62, 1.36) | 1.08 (0.72, 1.63) | 0.84 (0.57, 1.24) | |
| Corticosteroid Useb | 226 (63.1%) | 0.75 (0.56, 1.00) | 0.72* (0.53, 0.97) | 0.71* (0.53, 0.95) | |
| Race | White | 274 (76.5%) | - | 1a | - |
| Asianc | † | - | 0.68 (0.22, 2.16) | - | |
| Black or AA | 21 (5.9%) | - | 0.91 (0.49, 1.67) | - | |
| Native Americand | † | - | 2.03 (0.28, 14.84) | - | |
| Multiple | 11 (3.1%) | - | 0.69 (0.25, 1.90) | - | |
| Other | 12 (3.4%) | - | 0.70 (0.30, 1.65) | - | |
| Unknown | 27 (7.5%) | - | 0.54 (0.22, 1.30) | - | |
| Ethnicity | Not Hispanic or Latino | 253 (70.7%) | - | 1a | - |
| Hispanic or Latino | 60 (16.8%) | - | 1.92* (1.30, 2.84) | - | |
| Unknown | 45 (12.6%) | - | 0.75 (0.41, 1.37) | - | |
| Age at Onset | - | - | - | 0.89* (0.84, 0.95) |
Abbreviations: HR = hazard ratio, CI = confidence interval, AA = African American. - = covariate not included in model.
Reference categories have a HR of 1 and no CI.
Reference group.
For Cox regression models in this table, corticosteroid use was included as a time-varying covariate.
Also includes Hawaiian and Pacific Islander.
Also includes American Indian and Alaska Native.
Frequency too low to report.
Statistically significant (p < 0.05)
Exon skippable subgroups
Observed median ages at LoA were similar to mean ages for most exon skippable subgroups (Table 1); the overall test for differences in observed ages across exon skippable subgroups was not statistically significant (F-value (df=8,125) = 1.72, p = 0.10) (data not shown). The curves estimated from Kaplan-Meier analyses comparing exon skippable subgroups to all other subgroups combined are presented in Figure 2. The overall log-rank tests were statistically significant for exon 8 skippable and exon 44 skippable subgroups (see Figure 2 legend) when compared to the other subgroups, respectively. Longer times to LoA were found among specific exon skippable subgroups: median LoA survival ages were 14.8 years for the exon 8 skippable subgroup and 13.8 years for the exon 44 subgroup (data not shown). The comparison group had a median LoA survival age of 11.6 for both models. No other comparisons were significantly different. Coded as a categorical variable, the results of the Cox proportional hazards model showed a statistically significant overall effect for exon skippable subgroup (likelihood ratio χ2 (9) = 30.32, p < 0.001). The pattern of risk varied across exon skippable subgroups. Compared to those with ‘other’ exon skippable mutations, reduced adjusted annual risks were observed for LoA among individuals with exon 8 skippable mutations (HR=0.22, 95% CI=0.08, 0.63) and those with exon 44 skippable mutations (HR = 0.30, 95% CI = 0.12, 0.78) (Table 3). Reduced adjusted annual risk was also found for exon 46 skippable mutations compared to other skippable exons, but the CI contained the null. Elevated adjusted annual risks of LoA were found for exon 45 skippable and exon 52 skippable mutations, compared to those with other skippable exons, but the CI contained the null. The pattern of findings was again consistent across the three models.
Figure 2:

Kaplan-Meier curves for exon skippable subgroups. Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) 1982–2012. Kaplan-Meier curves comparing age (in years) at loss of ambulation for (A) exon 8 skippable deletions, log-rank χ2 (1) = 9.68, p < 0.01; (B) exon 20 skippable deletions, log-rank χ2 (1) = 0.18, p > 0.05; (C) exon 44 skippable deletions, log-rank χ2 (1) = 5.05, p < 0.05; (D) exon 45 skippable deletions, log-rank χ2 (1) = 3.51, p > 0.05; (E) exon 46 skippable deletions, log-rank χ2 (1) = 0.54, p > 0.05; (F) exon 51 skippable deletions, log-rank χ2 (1) = 2.45, p > 0.05; (G) exon 52 skippable deletions, log-rank χ2 (1) = 1.26, p > 0.05; and (H) exon 53 skippable deletions, log-rank χ2 (1) =1.47, p > 0.05. In all subplots, the comparison group is comprised of all males classified as having a deletion genetic type not in the indicated exon skippable subgroup.
Table 3:
Hazard ratios (HR) for Cox regression models I, II, and III for comparing loss of ambulation across exon skippable subgroups. Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) 1982–2012.
| No. (%) | Model I | Model II | Model III | ||
|---|---|---|---|---|---|
| Factor | Level | HR (95% CI) | HR (95% CI) | HR (95% CI) | |
| Mutation Type | Other | 79 (30.5%) | 1a | 1a | 1a |
| Exon 8 Skippable | 13 (5.0%) | 0.22* (0.08, 0.63) | 0.21* (0.08, 0.60) | 0.23* (0.08, 0.65) | |
| Exon 20 Skippable | 7 (2.7%) | 0.96 (0.34, 2.69) | 0.93 (0.33, 2.65) | 0.85 (0.30, 2.39) | |
| Exon 44 Skippable | 12 (4.6%) | 0.30* (0.12, 0.78) | 0.33* (0.12, 0.88) | 0.37* (0.14, 0.96) | |
| Exon 45 Skippable | 31 (12.0%) | 1.30 (0.76, 2.21) | 1.54 (0.88, 2.71) | 1.15 (0.67, 1.97) | |
| Exon 46 Skippable | 16 (6.2%) | 0.62 (0.29, 1.34) | 0.54 (0.25, 1.17) | 0.58 (0.27, 1.25) | |
| Exon 51 Skippable | 46 (17.8%) | 1.18 (0.72, 1.93) | 1.03 (0.61, 1.71) | 1.06 (0.65, 1.75) | |
| Exon 52 Skippable | 17 (6.6%) | 1.34 (0.62, 2.87) | 1.21 (0.55, 2.68) | 1.29 (0.60, 2.78) | |
| Exon 53 Skippable | 38 (14.7%) | 1.10 (0.65, 1.89) | 1.04 (0.60, 1.79) | 1.07 (0.62, 1.83) | |
| Steroid Useb | 156 (60.2%) | 0.69* (0.49, 0.98) | 0.66* (0.46, 0.95) | 0.65* (0.46, 0.92) | |
| Race | White | 203 (78.4%) | - | 1a (−) | - |
| Asianc | † | - | 0.15 (0.02, 1.12) | - | |
| Black or AA | 15 (5.8%) | - | 0.87 (0.40, 1.89) | - | |
| Native Americand | † | - | 3.20 (0.43, 23.88) | - | |
| Multiple | † | - | 0.41 (0.05, 3.23) | - | |
| Other | 11 (4.2%) | - | 0.77 (0.31, 1.95) | - | |
| Unknown | 17 (6.6%) | - | 0.59 (0.22, 1.59) | - | |
| Ethnicity | Not Hispanic or Latino | 187 (72.2%) | - | 1a (−) | - |
| Hispanic or Latino | 49 (18.9%) | - | 1.59* (1.01, 2.51) | - | |
| Unknown | 23 (8.9%) | - | 0.75 (0.35, 1.63) | - | |
| Age at Onset | - | - | - | 0.91* (0.85, 0.98) |
Abbreviations: HR = hazard ratio, CI = confidence interval, AA = African American. - = covariate not included in model.
Reference categories have a HR of 1 and no CI.
Reference category.
For Cox regression models in this table, steroid use was included as a time-varying covariate.
Also includes Hawaiian and Pacific Islander.
Also includes American Indian and Alaska Native.
Frequency too low to report.
Statistically significant (p < 0.05)
Additional pairwise comparisons of all exon skippable subgroups using Model I are shown in Figure 3. Decreased adjusted annual risk of LoA was found among those with exon 8 skippable mutations compared to the other skippable subgroup, but only the risk for the pairwise comparison between the exon 8 and exon 45 and 51 skippable subgroups remained statistically significant after adjusting for multiple comparisons (Exon 45: HR = 5.80; 95% CI = 1.07, 31.41; Exon 51: HR = 5.28; 95% CI = 1.01, 27.66 for Model I). Similarly, the exon 44 skippable subgroup showed decreased adjusted annual risk of LoA compared to the other exon skipping subgroup; however, no pairwise HRs were statistically significant after adjusting for multiple comparisons. The HR for the comparison of the exon 8 and exon 44 skippable subgroups was near unity, as were the HRs for the remaining comparisons, with all CIs containing the null.
Figure 3:
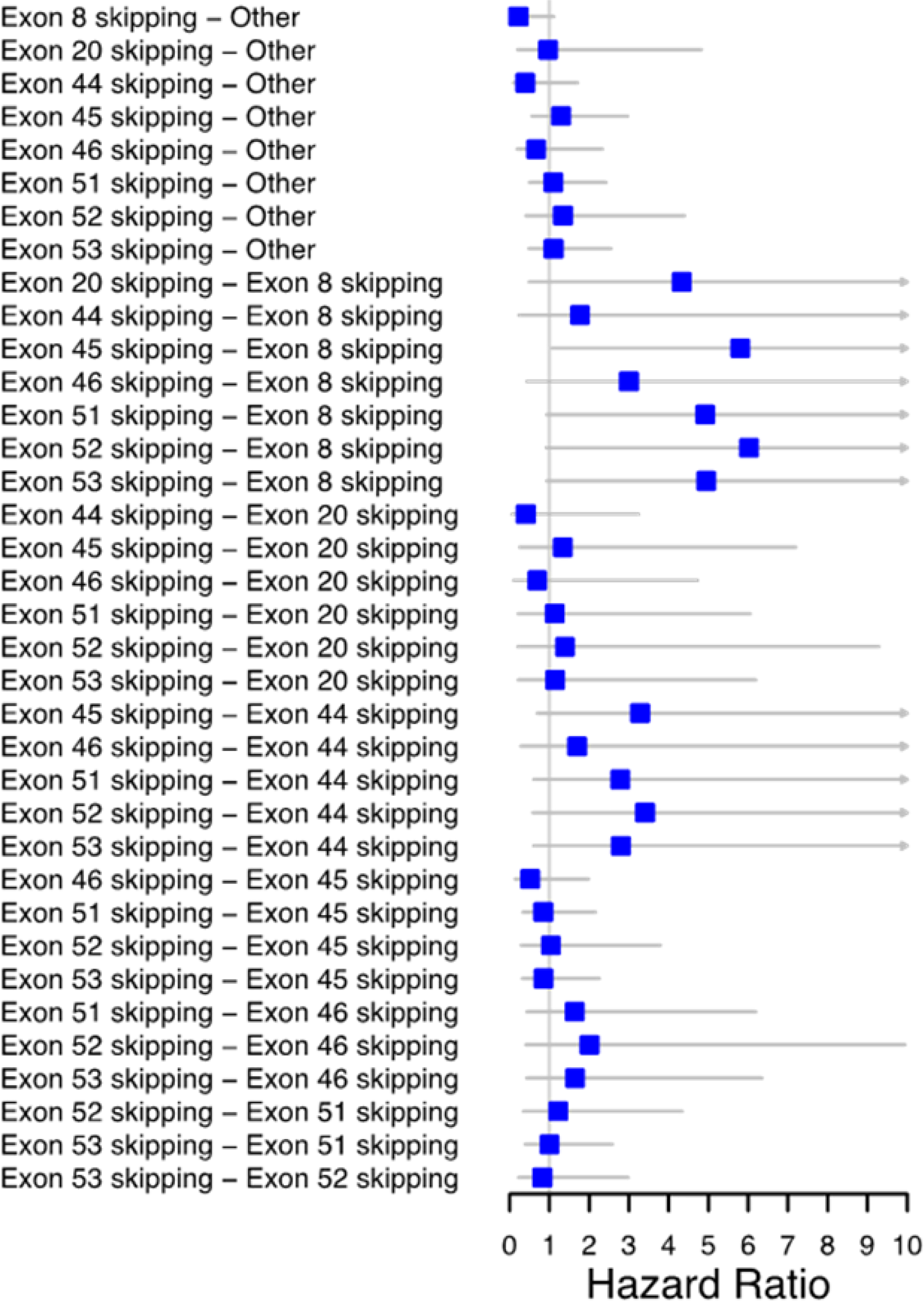
Pairwise hazard ratios for exon skippable subgroups based on Model I. Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) 1982–2012. Pairwise hazard ratio (HR) estimates for each exon skippable subgroup when controlling for steroid use. A HR estimate greater than one indicates a younger age for loss of ambulation for the group on the left hand side relative to the group listed on the right hand side. Right facing arrows indicate an estimated HR larger than 10. Confidence intervals are corrected for multiple comparisons using the Tukey-Kramer method.
Sensitivity analyses
As a sensitivity analysis, Models I and II were fit omitting a single outlier, a male from the exon 44 skippable subgroup that had a very late age of onset of symptoms (>18 years old). For both models, this omission resulted in a predictably large increase in the estimated HR for the exon 44 skippable subgroup (Model I: HR = 0.39; 95% CI = 0.15, 1.01; Model II: HR = 0.48; 95% CI = 0.18, 1.28) compared with the results in Table 3. In particular, the HRs were no longer statistically significant when compared with the other exon skippable group.
To further study the exon 44 skippable subgroup, an additional analysis was carried out in which 14 males with a single exon 45 deletion were reclassified from the exon 46 to the exon 44 skippable subgroup. For this analysis, the two remaining males in the exon 46 skippable subgroup (with exon 26–45 and exon 43–45 deletions, respectively) were moved to the “other” group. For all three models, this resulted in a modest increase in the estimated HR for the exon 44 skippable subgroup compared to the results in Table 3, while remaining statistically significantly smaller than one (HR = 0.47; 95% CI = 0.25, 0.89 for Model I). This provides further evidence for a lower annual risk of LoA among males with DMD and exon deletions amenable to exon 44 skipping.
4. Discussion
In this analysis, we examined two primary questions regarding the prognostic impact of genetic mutations on progression to LoA in males with DMD. First, we considered the question of whether there were any differences in this progression among the three broad mutation types: deletions, duplications, and point mutations. For this question, our analysis showed no differences by mutation type groups among males with dystrophinopathy. This indicates that classification of a male with dystrophinopathy based solely on one of these mutation groups was insufficient to predict any difference in disease progression.
Second, we restricted our analysis to individuals with deletions amenable to exon skipping therapies. This issue is particularly important as various therapies in development target specific exons for skipping, raising questions on how to properly construct a control group from a limited patient pool. Regarding this issue, our analyses demonstrated differences among exon skippable subgroups, although specific differences varied across models depending on which characteristics were controlled for. The most consistent finding from these analyses is that the exon 8 and 44 skippable subgroups showed lower annual risks of LoA relative to other amenable subgroups, although specific estimates must take into account the limited sample sizes. Our findings are consistent with those from the CINRG natural history study,19 which showed a significantly longer time to LoA in males with deletions of exons 3–7, a subgroup of the exon 8 skippable subgroup considered here. The CINRG cohort also observed that males with deletions amenable to exon 44 skipping had longer time to LoA when compared to a group of males with DMD who had out-of-frame deletions not amenable to skipping of exons 44, 45, 51, or 53. The differences in LOA and the exon 3–7 and exon 44 skippable groups are corroborated by previous phenotyping and may be explained at a molecular level. In our group, eleven of thirteen mutations in the exon 8 skippable group were composed of exon 3–7 deletions, which have previously been reported with milder phenotype and may be an exception to the reading frame rule.29 Moreover, various other exon 44 skippable mutations (e.g. exon 45 deletions) have also previously been associated with milder phenotypes and potential for endogenous exon skipping.30 It should be noted that in the CINRG dataset, LoA was defined as patient-or-caregiver-reported age at continuous wheelchair use, approximated to the nearest month, and verified as unable to perform the 10 m run/walk assessment by a CINRG-trained clinical evaluator. In our analysis, LoA was defined as continuous wheelchair use only. Although the differences for the exon 44 group disappeared when adjusting for multiple comparisons, these results should be considered in light of the conservative nature of such methods and the very small sample sizes available for the exon 44 skippable subgroup.
Strengths of this study include utilizing a large cohort of males with dystrophinopathy identified through a population-based surveillance system. This study has limitations. MD STARnet retrospectively identified cases using medical records. Medical record abstraction started in 2004 and some medical records were up to 20 years old; we may have not identified all cases who died before medical record abstraction started. Some cases may have received care outside the surveillance area and this information may not have been abstracted. Incomplete information in the medical records may lead to misclassification of variables. During the surveillance period, diagnostic testing patterns changed over time and point mutation testing in this cohort was low.31 Among the definite cases identified in the surveillance system, we excluded approximately a quarter of individuals because they did not have genetic information available in their clinic record. Results from this analysis may not be generalizable to all males living with dystrophinopathy in the United States.
In conclusion, we investigated the association of mutation class, including mutations amenable to exon skipping therapies, with age at LoA in males with dystrophinopathy. The finding of prolonged time to LoA in males with mutations amenable to skipping of exons 8 or 44 is consistent with previous studies. By better characterizing the patient populations to be enrolled, our findings may be informative for clinical trials that seek to find treatments that are specific to subpopulations with dystrophinopathies.
Supplementary Material
{kind=link}
Acknowledgments:
We acknowledge the contributions made to the MD STARnet by the many staff members at each MD STARnet site. Author (Venkatesh Atul Bhattaram) would like to acknowledge help from Sharonjit Sagoo (Executive Program and Project Management staff, Office of Clinical Pharmacology, CDER, FDA) for her help with project management.
Funding and Disclaimer:
This publication was supported by the Cooperative Agreement numbers, DD000187, DD000189, DD000190, DD000191, DD000392, DD001108, DD001119, DD001123, DD001126, DD001116, DD001117, funded by the Centers for Disease Control and Prevention. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Diseases Control and Prevention or the Department of Health and Human Services. This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
This project was supported in part by an appointment to the Research Fellowship Program at the Office of Translational Sciences/Office of Clinical Pharmacology/Center for Drug Evaluation and Research, US Food and Drug Administration, administered by the Oak Ridge Institute for Science and Engineering through an interagency agreement between US Department of Energy and FDA.
Financial Disclosures:
Dr. Haber reports no disclosures.
Dr. Conway reports no disclosures.
Dr. Paramsothy reports no disclosures.
Dr. Roy reports no disclosures.
Dr. Rogers reports no disclosures.
Dr. Ling reports no disclosures.
Dr. Kozauer reports no disclosures.
Ms. Street reports no disclosures.
Dr. Romitti reports no disclosures.
Ms. Fox reports no disclosures.
Dr. Phan is a principal investigator for Sarepta, Wave, Fibgrogen, Takeda, Shire, Catabasis, Mallinckrodt, Roche, Santhera, Teva, and Pfizer. She has served on scientific advisory board for Wave, Sarepta, and Mallinckrodt.
Dr. Mathews reports no disclosures.
Dr. Ciafaloni has received personal compensation for serving on advisory boards and/or as a consultant for Avexis, Inc, Biogen, Medscape, Pfizer, PTC Therapeutics, Sarepta Therapeutics, Ra pharma, Wave, and Strongbridge Biopharma plc. Dr. Ciafaloni has received personal compensation for serving on a speaker’s bureau for Biogen. Dr. Ciafaloni has received research and/or grant support from the Centers for Disease Control and Prevention, CureSMA, Muscular Dystrophy Association, National Institutes of Health, the Patient-Centered Outcomes Research Institute, Parent Project Muscular Dystrophy, PTC Therapeutics, Santhera, Sarepta Therapeutics, Orphazyme, and the US Food and Drug Administration.
Dr. Ciafaloni has received royalties from Oxford University Press and compensation from Medlink for editorial duties.
Dr. Oleszek reports no disclosures.
Dr. James reports no disclosures.
Ms. Galindo reports no disclosures.
Dr. Whitehead reports no disclosures.
Dr. Johnson has received grant funding from NINDS (4K23NS091511; R01NS104010), CDC (DD19-002) and the FDA (7R01FD006071-02). He receives royalties from the CCMDHI and the CMTHI. He receives research funds from Dyne, AveXis, CSL Behring, Vertex Pharmaceuticals, Fulcrum Therapeutics, ML Bio, Sarepta, and Acceleron Pharma. He has provided consultation for AveXis, AMO Pharma, Strongbridge BioPharma, Acceleron Pharma, Fulcrum Therapeutics, Dyne, Avidity, and Vertex Pharmaceuticals.
Dr. Butterfield is receiving funding via contracts for clinical trials from PTC Therapeutics, Sarepta Therapeutics, Pfizer, Biogen, Carpricor, and Catabasis. He serves on scientific advisory boards for Sarepta Therapeutics, Biogen, and Pfizer.
Dr. Pandya reports no disclosures.
Dr. Venkatesh reports no disclosures.
Dr. Bhattaram reports no disclosures.
Abbreviations:
- BMD
Becker muscular dystrophy
- CI
confidence interval
- DMD
Duchenne muscular dystrophy
- HR
hazard ratio
- LoA
loss of ambulation
- MD STARnet
Muscular Dystrophy Surveillance, Tracking, and Research Network
Appendix I.
| Author | Degree | Affiliation | Role | Contribution |
|---|---|---|---|---|
| Gregory Haber | PhD | Division of Cancer Epidemiology and Genetics (DCEG), National Cancer Institute (NCI) | Author | Contributed to study design; analyzed data; drafted and revised manuscript |
| Kristin Conway | PhD | University of Iowa, Department of Epidemiology | Author | Analysis or interpretation of the data |
| Pangaja Paramsothy | PhD, MPH | National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention (CDC) | Author | Contributed to the design of the study, discussion of analysis/interpretation of results, and revising manuscript |
| Anindya Roy | PhD | Department of Mathematics and Statistics, University of Maryland – Baltimore County (UMBC) | Author | Contributed to the statistical analysis plan, data analysis and interpretation; revised the manuscript |
| Hobart Rogers | PharmD, PhD | Office of Clinical Pharmacology (OCP), Center for Drug Evaluation and Research (CDER), Food & Drug Administration (FDA) | Author | Design of conceptualized study; interpretation of data; drafted manuscript |
| Xiang Ling | PhD | Division of Biometrics 1, Office of Biostatistics (OB), CDER, FDA | Author | Contributed to the statistical analysis plan, data analysis and interpretation; revised the manuscript |
| Nicholas Kozauer | MD | Office of Neuroscience, CDER, FDA | Author | Co-investigator, interpretation of findings and checked the manuscript for intellectual content |
| Natalie Street | MS | National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention (CDC) | Author | Data collection manuscript review and revision |
| Paul A Romitti | MS, PhD | Department of Epidemiology, University of Iowa, Iowa City, IA | Author | Major role in the acquisition of data; interpretation of data; and revision of manuscript |
| Deborah J. Fox | MPH | Bureau of Environmental and Occupational Epidemiology, New York State Department of Health | Author | Interpreted the data; revised the manuscript for intellectual content |
| Han Phan | MD | CDC | Author | Manuscript review |
| Dennis Matthews | MD | Department of Physical Medicine and Rehabilitation, Children’s Hospital Colorado, Aurora, CO | Author | Revised manuscript |
| Emma Ciafaloni | MD, FAAN | University of Rochester, Department of Neurology | Author | Manuscript review |
| Joyce Olesek | MD | Department of Rehabilitation, University of Colorado and Children’s Hospital Colorado | Author | Drafted and revised manuscript for intellectual content |
| Katherine A. James | PhD, MSPH, MS | University of Colorado School of Public Health University of Colorado Anschutz Medical Campus | Author | Analysis or interpretation of the data |
| Maureen Galindo | MS, RN | University of Arizona, Department of Pediatrics | Author | Data collection; Revised manuscript for intellectual content and internal processes. |
| Nedra Whitehead | MS, PhD | Biostatistics and Epidemiology Environmental and Health Sciences RTI International |
Author | Data collection manuscript review |
| Nick Johnson | MD, MSCI | Department of Neurology, Virginia Commonwealth University | Author | Acquisition and analysis of data |
| Russell J Butterfield | MD, PhD | Department of Pediatrics and Neurology, University of Utah | Author | Interpretation of results and revision of manuscript for intellectual content. |
| Shree Pandya | PT, DPT | Department of Neurology, School of Medicine and Dentistry, University of Rochester, Rochester, NY | Author | Revised the manuscript for intellectual content |
| Swamy Venkatesh | MD | Department of Neurology, University of South Carolina, School of Medicine, Columbia, SC | Author | Manuscript review |
| Venkatesh Atul Bhattaram | PhD | Office of Clinical Pharmacology, CDER, FDA | Author | Contributed to study design; analyzed data; drafted and revised manuscript |
Footnotes
Ethical publication statement:
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Contributor Information
Gregory Haber, Division of Cancer Epidemiology and Genetics, National Cancer Institute.
Kristin M. Conway, Department of Epidemiology, University of Iowa.
Pangaja Paramsothy, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention.
Anindya Roy, Department of Mathematics and Statistics, University of Maryland, Baltimore County.
Hobart Rogers, Center for Drug Evaluation and Research, Food & Drug Administration.
Xiang Ling, Center for Drug Evaluation and Research, Food & Drug Administration.
Nicholas Kozauer, Center for Drug Evaluation and Research, Food & Drug Administration.
Natalie Street, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention.
Paul A. Romitti, Department of Epidemiology, University of Iowa.
Deborah J. Fox, Bureau of Environmental and Occupational Epidemiology, New York State Department of Health.
Han C. Phan, Department of Pediatrics, Division of Neurology, University of Alabama, Birmingham.
Dennis Matthews, Department of Physical Medicine and Rehabilitation, Children’s Hospital Colorado.
Emma Ciafaloni, Department of Neurology, University of Rochester.
Joyce Oleszek, Department of Physical Medicine and Rehabilitation, Children’s Hospital Colorado.
Katherine A. James, School of Public Health, University of Colorado.
Maureen Galindo, Department of Pediatrics, University of Arizona.
Nedra Whitehead, Research Triangle Institute International.
Nicholas Johnson, Department of Neurology, Virginia Commonwealth University.
Russell J. Butterfield, Department of Pediatrics and Neurology, University of Utah.
Shree Pandya, Department of Neurology, University of Rochester.
Swamy Venkatesh, Department of Neurology, University of South Carolina.
Venkatesh Atul Bhattaram, Center for Drug Evaluation and Research, Food & Drug Administration.
References
- 1.Kunkel LM, Hejtmancik JF, Caskey CT, et al. Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature. 1986;322(6074):73–77. [DOI] [PubMed] [Google Scholar]
- 2.Magri F, Govoni A, D’Angelo MG, et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol. 2011;258(9):1610–1623. [DOI] [PubMed] [Google Scholar]
- 3.Humbertclaude V, Hamroun D, Bezzou K, et al. Motor and respiratory heterogeneity in Duchenne patients: Implication for clinical trials. European Journal of Paediatric Neurology. 2012;16(2):149–160. [DOI] [PubMed] [Google Scholar]
- 4.Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goemans N, van den Hauwe M, Wilson R, van Impe A, Klingels K, Buyse G. Ambulatory capacity and disease progression as measured by the 6-minute-walk-distance in Duchenne muscular dystrophy subjects on daily corticosteroids. Neuromuscular Disorders. 2013;23(8):618–623. [DOI] [PubMed] [Google Scholar]
- 6.Henricson EK, Abresch RT, Cnaan A, et al. The cooperative international neuromuscular research group Duchenne natural history study: Glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle & Nerve. 2013;48(1):55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim S, Campbell KA, Fox DJ, Matthews DJ, Valdez R, the MDS. Corticosteroid Treatments in Males With Duchenne Muscular Dystrophy: Treatment Duration and Time to Loss of Ambulation. Journal of Child Neurology. 2015;30(10):1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald CM, Henricson EK, Abresch RT, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. 2018;391(10119):451–461. [DOI] [PubMed] [Google Scholar]
- 9.Bello L, Flanigan KM, Weiss RB, et al. Association Study of Exon Variants in the NF-kappaB and TGFbeta Pathways Identifies CD40 as a Modifier of Duchenne Muscular Dystrophy. Am J Hum Genet. 2016;99(5):1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bello L, Kesari A, Gordish‐Dressman H, et al. Genetic modifiers of ambulation in the cooperative international Neuromuscular research group Duchenne natural history study. Annals of Neurology. 2015;77(4):684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bello L, Pegoraro E. The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. J Clin Med. 2019;8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciafaloni E, Kumar A, Liu K, et al. Age at onset of first signs or symptoms predicts age at loss of ambulation in Duchenne and Becker Muscular Dystrophy: Data from the MD STARnet. J Pediatr Rehabil Med. 2016;9(1):5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flanigan KM, Ceco E, Lamar K-M, et al. LTBP4 genotype predicts age of ambulatory loss in duchenne muscular dystrophy. Annals of Neurology. 2013;73(4):481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hogarth MW, Houweling PJ, Thomas KC, et al. Evidence for ACTN3 as a genetic modifier of Duchenne muscular dystrophy. Nat Commun. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pegoraro E, Hoffman EP, Piva L, et al. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology. 2011;76(3):219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss RB, Vieland VJ, Dunn DM, Kaminoh Y, Flanigan KM, United Dystrophinopathy P. Long-range genomic regulators of THBS1 and LTBP4 modify disease severity in duchenne muscular dystrophy. Ann Neurol. 2018;84(2):234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. 1989;45(4):498–506. [PMC free article] [PubMed] [Google Scholar]
- 18.Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. 2016;79(2):257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bello L, Morgenroth LP, Gordish-Dressman H, Hoffman EP, McDonald CM, Cirak S. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology. 2016;87(4):401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brogna C, Coratti G, Pane M, et al. Long-term natural history data in Duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. PLOS ONE. 2019;14(6):e0218683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang RT, Barthelemy F, Martin AS, et al. DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat. 2018;39(9):1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller LA, Romitti PA, Cunniff C, et al. The muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): surveillance methodology. Birth Defects Res A Clin Mol Teratol. 2006;76(11):793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathews KD, Cunniff C, Kantamneni JR, et al. Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): Case Definition in Surveillance for Childhood-Onset Duchenne/Becker Muscular Dystrophy. Journal of Child Neurology. 2010;25(9):1098–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kramer CY. Extension of Multiple Range Tests to Group Means with Unequal Numbers of Replications. Biometrics. 1956;12(3):307–310. [Google Scholar]
- 25.Fox DJ, Kumar A, West NA, et al. Trends With Corticosteroid Use in Males With Duchenne Muscular Dystrophy Born 1982–2001. Journal of Child Neurology. 2015;30(1):21–26. [DOI] [PubMed] [Google Scholar]
- 26.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, 2016. [Google Scholar]
- 27.Therneau T A Package for Survival Analysis in R. Version 3.1–12, 2020.
- 28.Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50(3):346–363. [DOI] [PubMed] [Google Scholar]
- 29.Muntoni F, Gobbi P, Sewry C, et al. Deletions in the 5’ region of dystrophin and resulting phenotypes. J Med Genet. 1994;31(11):843–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dwianingsih EK, Malueka RG, Nishida A, et al. A novel splicing silencer generated by DMD exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J Hum Genet. 2014;59(8):423–429. [DOI] [PubMed] [Google Scholar]
- 31.Cunniff C, Andrews J, Meaney FJ, et al. Mutation analysis in a population-based cohort of boys with Duchenne or Becker muscular dystrophy. J Child Neurol. 2009;24(4):425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.