

Abstract
Sudden unexpected death in epilepsy (SUDEP) is the most common epilepsy‐related cause of death, yet the cause is unknown. Our previous studies suggest a role for arrhythmia‐related ion channel genes in the pathogenesis of SUDEP. Hyperpolarization‐activated cyclic nucleotide‐gated cation (HCN1–4) channels are ion channels involved in generating spontaneous rhythmic activity in cardiac pacemaker and neuronal cells. This study sought to determine the role of pathogenic DNA variants in the HCN1–4 genes in a large SUDEP cohort collected from 1993 to 2009. Post‐mortem DNA samples were amplified and analyzed for each HCN exon. Genetic analysis in 48 SUDEP cases (age range 12–82 years) identified six novel and three previously reported nonsynonymous (amino acid changing) variants in HCN1 (n = 1), HCN2 (n = 2), HCN3 (n = 2) and HCN4 (n = 4). The Phe738Cys and Pro802Ser variants in HCN2, and Gly973Arg in HCN4 were absent in control alleles and affecting highly conserved residues in the carboxyl‐cytoplasmic tail region. Our results support a pathogenic link between the heart and brain in SUDEP, mediated by the HCN neuro‐cardiac ion channel genes.
Keywords: arrhythmias, genetics, HCN gene, sudden unexpected death in epilepsy
INTRODUCTION
Sudden unexpected death in epilepsy (SUDEP) is the most common epilepsy‐related cause of death (21) and accounts for up to 17% of all deaths in epilepsy patients 9, 10, 37. SUDEP is characterized by a sudden, unexpected, witnessed or unwitnessed, nontraumatic and nondrowning death in a patient with epilepsy, where the post‐mortem does not reveal a cause of death. Despite the increased awareness of SUDEP and its associated risk factors, the underlying cause has yet to be clearly defined. Evidence suggests seizure‐related abnormalities of cardiac and respiratory function may have an integral role in the pathogenesis of SUDEP (30). A commonly suggested terminal event in SUDEP is a cardiac arrhythmia, and notably, post‐mortem examination of SUDEP cases shows no evidence of structural cardiac abnormalities to suggest a pathological cause of death (ie, negative post‐mortem) (32). As cardiac electrical disorders, such as long QT syndrome (LQTS), also result in a negative post‐mortem, this supports a possible mechanistic link between SUDEP and cardiac arrhythmias. To support this emerging hypothesis, we recently reported the presence of potentially pathogenic variants in the three most common LQTS genes [ie, KCNQ1, KCNH2 (HERG) and SCN5A] in our SUDEP cohort (35). In addition, studies by other groups have also identified potentially pathogenic variants in the cardiac ion channel genes SCN5A and RYR2 1, 13, further supporting the notion that genetic variants associated with cardiac arrhythmias may play a pathogenic role in SUDEP.
While variants in cardiac ion channel genes are likely to play a significant role in the predisposition to sudden death in SUDEP, ion channels which are co‐expressed in both the heart and brain are also attractive potential candidates. There is growing evidence for the role of hyperpolarization‐activated cyclic nucleotide‐gated cation (HCN) channels in both idiopathic 19, 31 and acquired epilepsy 2, 3, 4, 14, 25. HCN channels are voltage‐gated ion channels primarily involved in the generation of spontaneous rhythmic activity in both cardiac pacemaker and neuronal cells 7, 23. These channels are dually activated by voltage hyperpolarization and direct binding of cAMP, and are permeable to both K+ and Na+ ions 8, 11, 24. HCN channels are encoded by a gene family that consists of four members: HCN1, HCN2, HCN3 and HCN4, and unlike the cardiac‐specific LQTS genes, HCN genes are expressed at varying levels in both the heart and brain 12, 17, 18, 26, 28. Of most significance is the recent association of HCN channels with cardiac rhythm disturbances and epileptic seizures, which are clinical features common to epilepsy patients. Specifically, genetic variants in the HCN1, HCN2 and HCN4 genes have been reported to account for familial sinus bradycardia 15, 20, 22, 27, 36 and familial epilepsy syndromes 6, 33. Hence, HCN channels represent ideal candidates for genetic screening in SUDEP cases because of their association with human disease, and expression in the heart and brain.
We propose that the pathogenic mechanism underlying SUDEP is caused by rhythm disturbances in the heart and brain caused by pathogenic variants in the cardiac‐ and neuron‐expressed HCN ion channels, giving rise to epileptic seizures and a predisposition to cardiac arrhythmias. This study sought to identify the presence of pathogenic variants in HCN genes that may predispose epilepsy patients to an increased susceptibility of cardiac arrhythmia and sudden death in SUDEP.
METHODS
Study cohort
Autopsy reports between 1993 and 2009 inclusive at the Department of Forensic Medicine, Sydney, Australia were reviewed in detail, as previously described (35). Specifically, cases of SUDEP were identified, and all available demographic, clinical and autopsy data were collected for each SUDEP case that fulfilled the following criteria: died suddenly and unexpectedly, had a history of epilepsy reported in the medical history and/or evidence provided of anti‐epileptic drug (AED) therapy, and the post‐mortem examination revealed no structural, noncardiac or toxicological cause of death (35). This study was approved by the Office of the NSW State Coroner and performed in accordance with institutional human ethics guidelines.
Genetic analysis
For SUDEP cases in which a post‐mortem sample was available (n = 48), genomic DNA was extracted from post‐mortem blood as previously described 5, 35 and used in the genetic screening of the coding regions and intron/exon boundaries of the four known HCN genes (ie, HCN1, HCN2, HCN3 and HCN4). Although HCN3 expression has not been detected in the heart, this isoform was also screened for potential neuron‐expressed genetic variants. Primers flanking the intron/exon boundaries were designed to amplify protein‐encoding exons of each HCN gene, in conjunction with previously published HCN2 and HCN4 primers 27, 33. All gene segments were amplified by PCR, visualized on a 2% agarose gel, DNA sequenced, then analyzed for sequence variants using Sequencher® v4.8 (Gene Codes Corporation, Ann Arbor, MI, USA). Primer sequences and PCR conditions are available on request. Despite numerous attempts, HCN2 exon 1 with a GC content of 84% could not be successfully amplified and DNA sequenced.
Nonsynonymous variants were genotyped in at least 320 control ethnicity‐matched alleles by DNA sequencing or restriction digestion, and assessed for possible disease pathogenicity using the following criteria: (i) absence of the variant in at least 320 healthy control alleles, in dbSNP, an online database of single nucleotide polymorphisms (SNPs) (http://www.ncbi.nlm.nih.gov/snp), and in 1000 Genomes Project, a catalog of human genetic variation in 1258 control alleles; (ii) conservation level of the amino acid residue among orthologous proteins; and (iii) prediction of pathogenicity using the online resource PolyPhen, which predicts the possible impact of an amino acid substitution on the structure and function of a human protein on the basis of three‐dimensional structure and multiple alignment of homologous sequences (http://genetics.bwh.harvard.edu/pph/).
RESULTS
Cohort characteristics
The SUDEP cohort consisted of 48 cases from a single major forensic center in Sydney, Australia, which we have reported on recently (35). The age range of SUDEP cases was 12–82 years (mean 40 ± 15) with a male predominance of 2.4:1. Of these 48 cases, 16 (33%) were on no AED therapy, 23 (48%) were on one AED, 8 (17%) were on two AEDs and 1 (2%) was on three AEDs. These AEDs included carbamazepine, lamotrigine, oxcarbazepine, phenytoin and sodium valproate. Forty‐five cases were unwitnessed events with more than half the cases found deceased in bed (51%). The specific cause of epilepsy was known in three cases and attributed to a motor vehicle accident or head injury. The cardiac findings in all cases were considered normal or noncontributory to the cause of death.
Genetic analysis
We screened 48 SUDEP cases for DNA variants in the coding regions and splice signal sequences of the ion channel genes HCN1–4, and identified 22 synonymous variants in HCN2 (n = 14), HCN3 (n = 1) and HCN4 (n = 7), of which six were novel (Table 1). Of most interest were nine nonsynonymous variants in HCN1 (n = 1), HCN2 (n = 2), HCN3 (n = 2) and HCN4 (n = 4), of which six were novel (Table 2). Each nonsynonymous variant was genotyped in at least 320 ethnicity‐matched control alleles, and allele frequencies of each variant were also sought in dbSNP. Of the nine nonsynonymous variants, three were absent in both our control samples and in dbSNP, and were further assessed for their potential pathogenicity. These three variants were HCN2 Phe738Cys, HCN2 Pro802Ser and HCN4 Gly973Arg, each detected once in each case (Table 2).
Table 1.
Synonymous variants identified in HCN genes.
| Gene | SNP | Exon | Amino acid change | MAF (%) | |
|---|---|---|---|---|---|
| SUDEP (n ≥ 47) | dbSNP European controls* | ||||
| HCN2 | rs56131056 | 2 | Thr241Thr | 13.5 | 13.0–15.2 |
| rs55659726 | 2 | Tyr286Tyr | 13.5 | 9.7–10.8 | |
| rs56170955 | 2 | Phe305Phe | 13.5 | 12.0–15.9 | |
| rs56180027 | 2 | IIe307IIe | 6.3 | 12.0–12.2 | |
| rs55780677 | 2 | Arg321Arg | 13.5 | 12.0–15.9 | |
| rs56279131 | 3 | Ser362Ser | 1.1 | 5.9 | |
| rs55839339 | 3 | Ala363Ala | 3.2 | 5.9 | |
| rs12981860 | 3 | Pro389Pro | 39.4 | 31.7 | |
| rs3752158 | 4 | Leu413Leu | 4.3 | 3.4–4.5 | |
| rs34397648 | 5 | Glu484Glu | 10.6 | 11.8 | |
| rs2301778 | 6 | Ala548Ala | 33.7 | 23.3 | |
| rs1054786 | 7 | Ala624Ala | 31.3 | 44.2 | |
| Novel | 8 | Ala743Ala | 1.3 | NA | |
| rs55691080 | 8 | Ala751Ala | 25.0 | NA | |
| HCN3 | Novel | 7 | IIe547IIe | 1.0 | NA |
| HCN4 | Novel | 1 | Leu12Leu | 1.0 | NA |
| Novel | 3 | Ser452Ser | 1.0 | NA | |
| rs12909882 | 4 | Leu520Leu | 5.2 | 5.0 | |
| rs117819825 | 8 | Pro852Pro | 9.6 | 1.7 | |
| Novel | 8 | Gly973Gly | 1.0 | NA | |
| Novel | 8 | Pro1016Pro | 1.1 | NA | |
| rs529004 | 8 | Pro1200Pro | 46.9 | 3.3–5.8 | |
Bold indicates novel variants. MAF = minor allele frequency.
Multiple European control groups are reported in dbSNP. MAF shows the frequency range (ie, the lowest and highest frequency for each variant). NA = MAF is not available in a European control population.
Table 2.
Nonsynonymous variants identified in HCN genes.
| Gene | SNP | Exon | Amino acid change | MAF (%) | ||
|---|---|---|---|---|---|---|
| SUDEP (n ≥ 47) | Controls (n ≥ 160) | dbSNP* | ||||
| HCN1 | Novel | 1 | Gly46Val | 2.1 | 0.6 | NA |
| HCN2 | Novel | 8 | Phe738Cys | 1.2 | 0 | NA |
| Novel | 8 | Pro802Ser | 1.2 | 0 | NA | |
| HCN3 | rs61812063 | 1 | Lys69Arg | 3.1 | 1.5 | NA |
| rs35001694 | 8 | Pro630Leu | 1.0 | 4.0 | 8.0 | |
| HCN4 | Novel | 1 | Gly36Glu | 7.3 | 8.4 | NA |
| rs62641689 | 8 | Val759IIe | 2.1 | 0.3 | 1.3 | |
| Novel | 8 | Gly973Arg | 1.0 | 0 | NA | |
| Novel | 8 | Arg1044Trp | 1.0 | 2.1 | NA | |
Bold indicates novel variants which were absent in controls; MAF = minor allele frequency.
No genotype frequencies are available in dbSNP except rs35001694 and rs62641689.
Significant nonsynonymous variants
HCN2 Phe738Cys is a substitution of a nonpolar and neutral phenylalanine residue for a cysteine residue with a large physicochemical difference (Grantham score of 205), and is predicted to be possibly damaging by PolyPhen. The Phe738 residue is located in the carboxyl‐cytoplasmic tail of the HCN2 protein and is highly conserved among mammals, birds and fish (Figure 1). HCN2 Pro802Ser is a substitution of a nonpolar and neutral proline residue for a polar and neutral serine residue with a moderate physicochemical difference (Grantham score of 74) at a poorly conserved residue in the carboxyl‐cytoplasmic tail, and is predicted to be benign by PolyPhen (Figure 1). HCN4 Gly973Arg is a substitution of a nonpolar and neutral glycine residue for a polar and positively charged arginine residue with a moderate physicochemical difference (Grantham score of 125) in the carboxyl‐cytoplasmic tail at a highly conserved residue in mammals, and is predicted to be benign as the corresponding residues are absent in nonmammalian orthologues (Figure 2).
Figure 1.
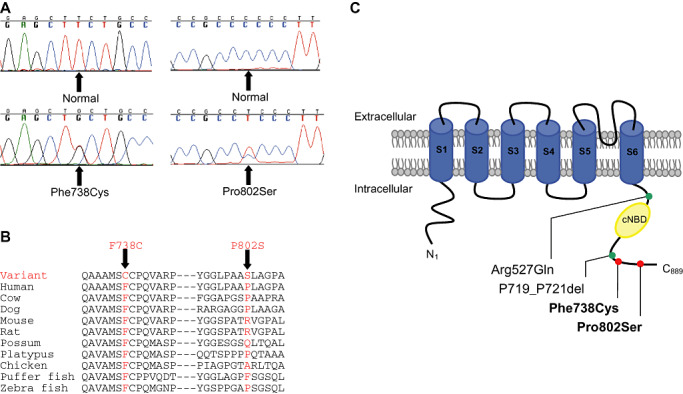
HCN2 Phe738Cys and Pro802Ser mutation in sudden unexpected death in epilepsy (SUDEP). (A) Sequence chromatograms and (B) amino acid conservation across species. (C) Schematic topology of the HCN2 channel with six transmembrane segments (S1–S6) and the location of HCN2 variants. cNBD = cyclic nucleotide‐binding domain; red circle = novel HCN2 variant identified in SUDEP; green circle = previously identified HCN2 variant in familial epilepsy syndromes.
Figure 2.
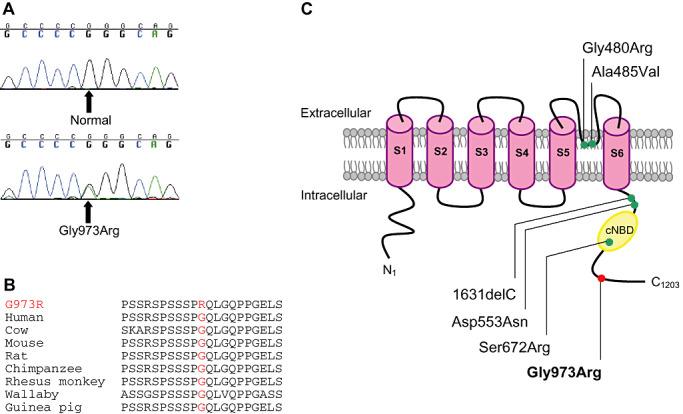
HCN4 Gly973Arg mutation in sudden unexpected death in epilepsy (SUDEP). (A) Sequence chromatograms and (B) amino acid conservation of Gly973Arg across species. (C) Schematic topology of the HCN4 channel with six transmembrane segments (S1–S6) and the location of HCN4 variants. cNBD = cyclic nucleotide‐binding domain; red circle = novel HCN4 variant identified in SUDEP; green circle = previously identified HCN4 variant in familial sinus bradycardia.
DISCUSSION
This study reports the genetic analysis of cardiac‐ and neuron‐expressed HCN ion channel variants in a large, unique cohort of SUDEP cases. A total of 31 HCN variants were detected, of which 12 were novel. Importantly, nine protein‐changing variants were found in exon 1 and exon 8, corresponding to the amino‐ and carboxyl‐cytoplasmic tail of the HCN protein respectively, both of which harbor previously identified nonsynonymous variants. Of most significance was the identification of three potential pathogenic variants in HCN2 (Phe738Cys, Pro802Leu) and HCN4 (Gly973Arg), which may have contributed to the genetic predisposition of these epilepsy individuals to seizures and cardiac arrhythmia, resulting in sudden death. This study proposes the possibility of an underlying genetic link between the heart and brain in epilepsy patients susceptible to SUDEP.
SUDEP is a devastating complication in patients with epilepsy. The underlying mechanisms predisposing to the risk of sudden death remain unclear. A number of risk factors have been identified which appear to be associated with SUDEP, including poor seizure control, lack of compliance with AEDs, polytherapy and early‐onset epilepsy 32, 34. Of note, the absence of a specific cause of death at post‐mortem, including the presence of an anatomically normal heart, supports the possibility that sudden death in SUDEP may be primarily arrhythmogenic, and therefore may involve alterations in ion channels, as observed in other arrythmogenic syndromes such as familial LQTS.
Cardiac ion channels harboring genetic variants can lead to altered ion channel function, causing an increased risk of cardiac arrhythmia and sudden death. Recently, our group and others have revealed a likely role for genetic variants in cardiac ion channel genes [KCNQ1, KCNH2 (HERG), SCN5A and RYR2] in the susceptibility of epilepsy patients to cardiac arrhythmia and sudden death 1, 13, 35. Furthermore, Le Gal et al reported a susceptibility to SUDEP in a Dravet syndrome patient with a frameshifting duplication mutation in the neuron ion channel gene, SCN1A (16). Collectively, these findings provide a basis for further investigation of the role of ion channel genes in SUDEP. Our current study identified three particular DNA variants of pathogenic interest.
HCN2 Phe738Cys
The HCN2 Phe738Cys variant was detected in a 52‐year‐old male. The death was unwitnessed and the deceased was found slumped over the bath in the evening. The post‐mortem was negative; however, it showed brain changes consistent with previous seizures. No excess fluid was found in the lungs and no lacerations were visible on the tongue. The primary cause of death was unascertained, which is consistent with SUDEP. The deceased had a known history of epilepsy since age 16 years, but was noncompliant with AEDs. He had regular seizures, the last of which was 18 months prior to death.
Previous screening of the three most common LQTS genes detected the KCNH2 Arg1047Leu variant (35). Interestingly, Phe738Cys is located in the carboxyl‐cytoplasmic tail of the HCN2 protein close to previously identified HCN2 variants, Arg527Gln and P719_P721del, which have been associated with familial epilepsy syndromes (Figure 1) 6, 33. Functional studies found that the HCN2 Arg527Gln variant demonstrated a trend toward a decreased slope of the conductance–voltage relation, while currents generated by HCN2 channels with the P719_P721 deletion were 35% greater than those of wild‐type HCN2 channels 6, 33. Hence, the possibility of altered HCN2 channel function caused by the Phe738Cys variant needs to be considered. Overall, the Phe738Cys variant may have caused neuronal and cardiac rhythm disturbances, leading to regular seizures and sudden death, which was exacerbated by noncompliance with AED therapy. Moreover, the KCNH2 Arg1047Leu variant could have also contributed to a cardiac arrhythmia in a combined effect.
HCN2 Pro802Ser
The HCN2 Pro802Ser variant was detected in a 43‐year‐old male. The deceased had a seizure at night, and death occurred in bed and was witnessed. The post‐mortem was negative, no brain lesions or tongue lacerations were detected, while the heart showed mild coronary artery disease and generalized atheroma. The primary cause of death was undetermined; however, the possibility of SUDEP was noted, although the deceased had not had previous AED therapy. Although Pro802Ser is predicted to be benign and the proline residue is poorly conserved across species, this variant may be a contributing factor in the seizure episode prior to sudden death. This could be further investigated by studying the functional effect of Pro802Ser on the HCN2 ion channel. In the same case, our initial genetic studies revealed a protein‐changing variant in the SCN5A gene, Pro2006Ala, and the effect of multiple nonsynonymous variants may have led to a more severe risk of cardiac arrhythmia and sudden death in this epilepsy patient (35).
HCN4 Gly973Arg
The HCN4 Gly973Arg variant was detected in a 44‐year‐old male. Death was unwitnessed and the deceased was found on the lounge room floor. The post‐mortem was negative, no brain lesions, heart fibrosis or tongue lacerations were found; however, excess fluid was present in the lungs. The primary cause of death was undetermined; however, the possibility of SUDEP was noted, as the deceased was reported as having regular seizures prior to death, and at post‐mortem subtherapeutic levels of lamotrigine and oxcarbazepine were detected. Previous screening of the three most common LQTS genes revealed no genetic variants (35). A potential role of HCN4 variants in the genetic predisposition to cardiac arrhythmia was highlighted with the discovery of HCN4 variants (Gly480Arg, Ala485Val, 1631delC, Asp553Asn and Ser672Arg) in patients with familial sinus bradycardia 15, 20, 22, 27, 36. These variants are distributed in the HCN4 carboxyl‐cytoplasmic tail and the loop between transmembrane domains S5 and S6 (Figure 2). Functional expression of each mutated channel showed greater activation at more negative voltages than wild‐type HCN4 channels, indicating reduced HCN4 ion channel activity. Thus, it is possible the Gly973Arg variant could affect channel function by altering channel activation in response to voltage hyperpolarization. The single variant combined with seizures and AED therapy, which are both risk factors for SUDEP, could have predisposed this epilepsy patient to epileptic seizures and cardiac arrhythmia leading to sudden death.
All of the nonsynonymous variants identified, and particularly HCN2 Phe738Cys, and Pro802Leu and HCN4 Gly973Arg, may have altered the function of each respective HCN ion channel, causing frequent seizure episodes reported in each SUDEP case. These genetic factors combined with multiple susceptibility factors, including a second protein‐changing genetic variant, frequent seizures, noncompliance with AED therapy may have exacerbated the risk already present in the epilepsy patient to cardiac arrhythmia and ultimately sudden death. Interestingly, the location of these nonsynonymous variants was primarily at the carboxyl‐terminal end of the channel. It is tempting to speculate that the hitherto absence of nonsynonymous variants in transmembrane domains, which form the ion channel pore, may be caused by the strict functional requirement of intact HCN channel pores for the generation of pacemaker activity in the heart and brain. Variants in transmembrane domains could potentially be lethal as the hearts of HCN4‐deficient mouse embryos contract significantly slower than wild type and are unable to be stimulated by cAMP, with embryonic lethality between 9.5 and 11.5 days, highlighting the essential role of HCN channels for the proper generation of pacemaker activity (29).
To further investigate these SUDEP cases, evaluation of first‐degree relatives would shed further light on the potential genetic/inherited basis of epilepsy and sudden death. Unfortunately, we were unable to contact surviving family members of SUDEP cases in order to assess co‐inheritance of the identified variants with disease. All cases were studied in a de‐identified manner as per ethical guidelines, hence there was no available ECG data in the deceased or in family members, and a family history of sudden death and epilepsy could not be obtained.
CONCLUSIONS
SUDEP is a tragic event, and the underlying pathogenic mechanism predisposing epilepsy patients to sudden death is not clearly defined. The combined effect of potential ion channel pathogenic variants and multiple susceptibility factors may play a contributing role in SUDEP. We report the role of HCN genetic variants involved in the genetic predisposition of epilepsy patients to seizures, cardiac arrhythmia and sudden death, supporting a pathogenic link between the brain and heart in cases of SUDEP. Further prospective studies in larger cohorts are required to further define the genetic predisposition to SUDEP.
DISCLOSURES
Nil.
ACKNOWLEDGMENTS
C.S. is the recipient of a National Health and Medical Research Council (NHMRC) Practitioner Fellowship. E.T. is the recipient of a National Heart Foundation of Australia Fellowship. The study is supported by research grants from NHMRC and the Thrasher Research Fund.
No conflicts of interest
REFERENCES
- 1. Aurlien D, Leren TP, Tauboll E, Gjerstad L (2009) New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure 18:158–160. [DOI] [PubMed] [Google Scholar]
- 2. Bender RA, Soleymani SV, Brewster AL, Nguyen ST, Beck H, Mathern GW, Baram TZ (2003) Enhanced expression of a specific hyperpolarization‐activated cyclic nucleotide‐gated cation channel (HCN) in surviving dentate gyrus granule cells of human and experimental epileptic hippocampus. J Neurosci 23:6826–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brewster A, Bender RA, Chen Y, Dube C, Eghbal‐Ahmadi M, Baram TZ (2002) Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization‐activated channels in an isoform‐ and cell‐specific manner. J Neurosci 22:4591–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen K, Aradi I, Thon N, Eghbal‐Ahmadi M, Baram TZ, Soltesz I (2001) Persistently modified h‐channels after complex febrile seizures convert the seizure‐induced enhancement of inhibition to hyperexcitability. Nat Med 7:331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiu C, Tebo M, Ingles J, Yeates L, Arthur JW, Lind JM, Semsarian C (2007) Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 43:337–343. [DOI] [PubMed] [Google Scholar]
- 6. Dibbens LM, Reid CA, Hodgson B, Thomas EA, Phillips AM, Gazina E et al (2010) Augmented currents of an HCN2 variant in patients with febrile seizure syndromes. Ann Neurol 67:542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DiFrancesco D (1993) Pacemaker mechanisms in cardiac tissue. Annu Rev Physiol 55:455–472. [DOI] [PubMed] [Google Scholar]
- 8. DiFrancesco D, Tortora P (1991) Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351:145–147. [DOI] [PubMed] [Google Scholar]
- 9. Ficker DM (2000) Sudden unexplained death and injury in epilepsy. Epilepsia 41(Suppl. 2):S7–12. [DOI] [PubMed] [Google Scholar]
- 10. Ficker DM, So EL, Shen WK, Annegers JF, O'Brien PC, Cascino GD, Belau PG (1998) Population‐based study of the incidence of sudden unexplained death in epilepsy. Neurology 51:1270–1274. [DOI] [PubMed] [Google Scholar]
- 11. Ingram SL, Williams JT (1996) Modulation of the hyperpolarization‐activated current (Ih) by cyclic nucleotides in guinea‐pig primary afferent neurons. J Physiol 492:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ishii TM, Takano M, Xie LH, Noma A, Ohmori H (1999) Molecular characterization of the hyperpolarization‐activated cation channel in rabbit heart sinoatrial node. J Biol Chem 274:12835–12839. [DOI] [PubMed] [Google Scholar]
- 13. Johnson JN, Tester DJ, Bass NE, Ackerman MJ (2010) Cardiac channel molecular autopsy for sudden unexpected death in epilepsy. J Child Neurol 25:916–921. [DOI] [PubMed] [Google Scholar]
- 14. Jung S, Jones TD, Lugo JN, Jr , Sheerin AH, Miller JW, D'Ambrosio R et al (2007) Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci 27:13012–13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laish‐Farkash A, Glikson M, Brass D, Marek‐Yagel D, Pras E, Dascal N et al (2010) A novel mutation in the HCN4 gene causes symptomatic sinus bradycardia in Moroccan Jews. J Cardiovasc Electrophysiol 21:1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le Gal F, Korff CM, Monso‐Hinard C, Mund MT, Morris M, Malafosse A, Schmitt‐Mechelke T (2010) A case of SUDEP in a patient with Dravet syndrome with SCN1A mutation. Epilepsia 51:1915–1918. [DOI] [PubMed] [Google Scholar]
- 17. Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M (1998) A family of hyperpolarization‐activated mammalian cation channels. Nature 393:587–591. [DOI] [PubMed] [Google Scholar]
- 18. Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, Biel M (1999) Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J 18:2323–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K et al (2003) Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J 22:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Milanesi R, Baruscotti M, Gnecchi‐Ruscone T, DiFrancesco D (2006) Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med 354:151–157. [DOI] [PubMed] [Google Scholar]
- 21. Nashef L (1997) Sudden unexpected death in epilepsy: terminology and definitions. Epilepsia 38(Suppl. 11):S6–S8. [DOI] [PubMed] [Google Scholar]
- 22. Nof E, Luria D, Brass D, Marek D, Lahat H, Reznik‐Wolf H et al (2007) Point mutation in the HCN4 cardiac ion channel pore affecting synthesis, trafficking, and functional expression is associated with familial asymptomatic sinus bradycardia. Circulation 116:463–470. [DOI] [PubMed] [Google Scholar]
- 23. Pape HC (1996) Queer current and pacemaker: the hyperpolarization‐activated cation current in neurons. Annu Rev Physiol 58:299–327. [DOI] [PubMed] [Google Scholar]
- 24. Pedarzani P, Storm JF (1995) Protein kinase A‐independent modulation of ion channels in the brain by cyclic AMP. Proc Natl Acad Sci USA 92:11716–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Powell KL, Ng C, O'Brien TJ, Xu SH, Williams DA, Foote SJ, Reid CA (2008) Decreases in HCN mRNA expression in the hippocampus after kindling and status epilepticus in adult rats. Epilepsia 49:1686–1695. [DOI] [PubMed] [Google Scholar]
- 26. Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR (1998) Identification of a gene encoding a hyperpolarization‐activated pacemaker channel of brain. Cell 93:717–729. [DOI] [PubMed] [Google Scholar]
- 27. Schulze‐Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O, Isbrandt D (2003) Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest 111:1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seifert R, Scholten A, Gauss R, Mincheva A, Lichter P, Kaupp UB (1999) Molecular characterization of a slowly gating human hyperpolarization‐activated channel predominantly expressed in thalamus, heart, and testis. Proc Natl Acad Sci USA 96:9391–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stieber J, Herrmann S, Feil S, Loster J, Feil R, Biel M et al (2003) The hyperpolarization‐activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc Natl Acad Sci USA 100:15235–15240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stollberger C, Finsterer J (2004) Cardiorespiratory findings in sudden unexplained/unexpected death in epilepsy (SUDEP). Epilepsy Res 59:51–60. [DOI] [PubMed] [Google Scholar]
- 31. Strauss U, Kole MH, Brauer AU, Pahnke J, Bajorat R, Rolfs A et al (2004) An impaired neocortical Ih is associated with enhanced excitability and absence epilepsy. Eur J Neurosci 19:3048–3058. [DOI] [PubMed] [Google Scholar]
- 32. Surges R, Thijs RD, Tan HL, Sander JW (2009) Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol 5:492–504. [DOI] [PubMed] [Google Scholar]
- 33. Tang B, Sander T, Craven KB, Hempelmann A, Escayg A (2008) Mutation analysis of the hyperpolarization‐activated cyclic nucleotide‐gated channels HCN1 and HCN2 in idiopathic generalized epilepsy. Neurobiol Dis 29:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tomson T, Nashef L, Ryvlin P (2008) Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol 7:1021–1031. [DOI] [PubMed] [Google Scholar]
- 35. Tu E, Bagnall RD, Duflou J, Semsarian C (2011) Post‐mortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol 21:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ueda K, Nakamura K, Hayashi T, Inagaki N, Takahashi M, Arimura T et al (2004) Functional characterization of a trafficking‐defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem 279:27194–27198. [DOI] [PubMed] [Google Scholar]
- 37. Walczak TS, Leppik IE, D'Amelio M, Rarick J, So E, Ahman P et al (2001) Incidence and risk factors in sudden unexpected death in epilepsy: a prospective cohort study. Neurology 56:519–525. [DOI] [PubMed] [Google Scholar]