

Abstract
We recently reported that the vast majority (>90%) of low‐grade diffuse gliomas (diffuse astrocytoma, oligoastrocytoma and oligodendroglioma) carry at least one of the following genetic alterations: IDH1/2 mutation, TP53 mutation or 1p/19q loss. Only 7% of cases were triple‐negative (ie, lacking any of these alterations). In the present study, array comparative genomic hybridization (CGH) in 15 triple‐negative WHO grade II gliomas (eight diffuse astrocytomas and seven oligodendrogliomas) showed loss at 9p21 (p14ARF, p15INK4b, p16INK4a loci) and 13q14–13q32 (containing the RB1 locus) in three and two cases, respectively. Further analyses in 31 triple‐negative cases as well as a total of 160 non‐triple‐negative cases revealed that alterations in the RB1 pathway (homozygous deletion and promoter methylation of the p15INK4b, p16INK4a and RB1 genes) were significantly more frequent in triple‐negative (26%) than in non‐triple‐negative cases (11%; P = 0.0371). Multivariate analysis after adjustment for age, histology and treatment showed that RB1 pathway alterations were significantly associated with unfavorable outcome for patients with low‐grade diffuse glioma [hazard ratio, 3.024 (1.279–6.631); P = 0.0057]. These results suggest that a fraction of low‐grade diffuse gliomas lacking common genetic alterations may develop through a distinct genetic pathway, which may include loss of cell‐cycle control regulated by the RB1 pathway.
Keywords: diffuse glioma, IDH1, IDH2, p14ARF, p15INK4b, p16INK4a, RB1, TP53
INTRODUCTION
The present World Health Organization (WHO) classification recognizes three main histologic types of WHO grade II low‐grade diffuse glioma: diffuse astrocytoma; oligoastrocytoma; and oligodendroglioma (14). We have recently reported that at least one of the following alterations—IDH1/2 mutation, TP53 mutation or 1p/19q loss—is present in the vast majority (93%) of low‐grade diffuse gliomas; the most frequent combinations were IDH1/2 mutations plus TP53 mutations, IDH1/2 mutations plus 1p/19q loss or IDH1/2 mutations only (11). As a means to predict patient survival, molecular classification based on these alterations showed similar power to histological classification (11). In 7% of all cases, none of these genetic alterations were detected (triple‐negative cases) (11), suggesting the possibility of alternative genetic pathway(s) in the development of low‐grade diffuse glioma.
In the present study, we used array comparative genomic hybridization (aCGH) to screen 15 triple‐negative cases in order to identify chromosomal loci associated with their development. As the results showed loss at 9p21 (p14ARF, p15INK4b, p16INK4a loci) and 13q14–13q32 (containing the RB1 locus), we then screened for alterations in the RB1 pathway (homozygous deletion and promoter methylation of the p15INK4b, p16INK4a and RB1 genes) and alterations in the p14ARF gene (homozygous deletion and promoter methylation), a member of the TP53 pathway in a larger number of triple‐negative (31 cases) and non‐triple‐negative (160 cases) low‐grade diffuse gliomas.
MATERIALS AND METHODS
Tumor samples
Tumor samples were obtained from the Department of Neuropathology, University Hospital Zurich, Switzerland (88 cases); the Department of Neuropathology, University Hospital Frankfurt, Germany (45 cases); the Institute of Neuropathology and Department of Neurosurgery, University Hospital Muenster, Germany (24 cases); the Department of Neuropathology and Neurosurgery, University Hospital Essen (16 cases), Germany; the Department of Pathology, Gunma University, Japan (six cases); the Institute of Neuroscience, Bordeaux, France (six cases); and the Department of Neurosurgery, University Hospital Bern, Switzerland (six cases). We collected clinical data, including age and sex of the patient, location of tumors, histological diagnosis, date of surgical resection, extent of surgery, other treatment (radiotherapy, chemotherapy), date of last follow‐up or last contact and date of death. Patients who died within 4 months after surgery were excluded from the survival analysis in order to eliminate cases in which death was attributable to surgery and surgical complications. Before genetic analyses, histology review was carried out as previously described (11). All tumors were subjected to gross total or subtotal resection, without therapy prior to surgery. Needle‐biopsy cases were not included in this study.
Analyses showed that 31 tumors were triple‐negative, that is, lacked IDH1/2 mutations, TP53 mutations and 1p/19q loss (11). Histologically, these tumors were classified as diffuse astrocytoma WHO grade II (16 cases), oligoastrocytoma WHO grade II (2 cases) and oligodendroglioma WHO grade II (13 cases). The clinical and histological features of these triple‐negative low‐grade diffuse gliomas are shown in Table 1. These triple‐negative cases, as well as a total of 160 non‐triple‐negative cases (50 cases with IDH1/2 mutation only, 54 cases with IDH1/2 plus TP53 mutations, and 56 cases with IDH1/2 mutation plus 1p/19q loss; or, as classified histologically, 77 diffuse astrocytomas, 43 oligoastrocytomas, 40 oligodendrogliomas) published previously (11), were screened for alterations in the RB1 and TP53 pathways.
Table 1.
Clinical and histological features of triple negative low‐grade diffuse gliomas with and without alterations in the RB1 pathway.
| Case | Age | Sex | Histology | Tumor location | Ki‐67 index | Survival (months) | Alterations in the RB1 pathway |
|---|---|---|---|---|---|---|---|
| WP31 | 28 | M | Diffuse astrocytoma* | − | 7% | 14 | + |
| PB677 | 38 | F | Diffuse astrocytoma* | Left temporal | <1% | 12.7 | + |
| PB718 | 54 | F | Diffuse astrocytoma* | Left temporal and parietal | <1% | 20.1 | + |
| PB678 | 61 | M | Diffuse astrocytoma | Left temporal | <1% | 31.3 | + |
| M01 | 61 | F | Diffuse astrocytoma | Right frontal | 2% | − | + |
| PB659 | 63 | M | Diffuse astrocytoma* | Left frontal | <1% | 4.4 | + |
| M54 | 7 | F | Oligodendroglioma | Left frontal | 1% | 47 | + |
| AV13 | 56 | F | Oligodendroglioma | Temporal | 12% | − | + |
| M44 | 5 | M | Diffuse astrocytoma | Right thalamus | <5% | 56 | − |
| M08 | 24 | F | Diffuse astrocytoma | Left frontal | <1% | 85 | − |
| PB773 | 38 | F | Diffuse astrocytoma | Left frontal | <1% | 120 | − |
| DBB404 | 41 | F | Diffuse astrocytoma | Crus cerebri / Pons area | <1% | 13.8 | − |
| BA39 | 43 | M | Diffuse astrocytoma | − | nd | 63 | − |
| M38 | 49 | M | Diffuse astrocytoma | Left temporal | <1% | 42 | − |
| M43 | 49 | M | Diffuse astrocytoma | Left insular | <1% | 40 | − |
| M33 | 52 | M | Diffuse astrocytoma | Right temporal and parietal | <1% | 19 | − |
| PB653 | 58 | F | Diffuse astrocytoma | Left frontal and temporal | <1% | 9.2 | − |
| BA53 | 73 | M | Diffuse astrocytoma | − | nd | 90 | − |
| M83 | 69 | M | Oigoastrocytoma | Right temporal | 5% | 4 | − |
| DBB247 | − | M | Oigoastrocytoma | Basal gaglia | <1% | 43.3 | − |
| PB760 | 3 | M | Oigodendroglioma | Right temporal, thalamus, insular | <1% | 52.4 | − |
| M49 | 4 | F | Oigodendroglioma | Right frontal | 1% | − | − |
| DBB4951 | 5 | F | Oigodendroglioma | Left temporal | 6% | 214.5 | − |
| PB777 | 14 | M | Oigodendroglioma | Right insular | <1% | 149.5 | − |
| M15 | 21 | M | Oligodendroglioma | Right parietal | 1% | 21 | − |
| PB745 | 22 | F | Oligodendroglioma | Thalamus 4/5–7/8 | <1% | 25.5 | − |
| PB1002 | 29 | M | Oligodendroglioma | Left temporal | nd | 190.3 | − |
| M57 | 30 | F | Oligodendroglioma | Left temporal | <1% | 32 | − |
| M51 | 31 | M | Oligodendroglioma | Cerebellum | 2% | 96 | − |
| PB778 | 41 | M | Oligodendroglioma | Left frontal | <1% | 144.6 | − |
| DBB524 | 48 | F | Oligodendroglioma | Temporal and parietal | <1% | 124.5 | − |
With gemistocytic components.
nd = not determined.
DNA extraction
DNA was extracted from formalin‐fixed paraffin‐embedded histological sections as previously described (9). Briefly, tumor areas were scraped from the histological slide and were deparaffinized in xylene for 15 minutes and then in 100% ethanol for 10 minutes. The pellets were dried in acetone and washed with 0.4% Tween 20 solution and then with phosphate‐buffered saline (PBS; pH 7.4), before being dissolved in 400 µL 1 M NaSCN solution. After overnight incubation at 37°C, samples were suspended in 400 µL of DNA extraction buffer, composed of 360 µL of ATL buffer and 40 µL of proteinase K (DNeasy Mini kit, Qiagen, Valencia, CA, USA), and were incubated overnight at 55°C. Additional proteinase K (40 µL) was added 12 and 24 h later, with a total incubation time of 60 h. After incubation with 8 µL RNase (100 mg/mL) for 10 minutes at room temperature (RT), 420 µL ATL buffer was added and the samples were separated into two parts (each 450 µL). Each part was mixed with 450 µL AL buffer and 450 µL 100% ethanol, and incubated at RT for 5 minutes. The samples were loaded into DNeasy Mini spin columns (Qiagen). After washing the columns with AW1 buffer and drying the column membrane with 80% ethanol, the purified genomic DNA was eluted with 25 µL nuclease‐free H2O. The DNA concentration was determined by spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Absorption was measured at 230, 260 and 280 nm and the DNA quality was evaluated by A260/A230 and A260/A280 ratios.
Array comparative genomic hybridization (CGH)
Array CGH (aCGH) analysis was carried out as described previously (9). Briefly, the genomic profile changes of paired DNA samples were compared using a 2 × 105‐K CGH oligonucleotide microarray (Agilent Technologies, Santa Clara, CA, USA; 15.0‐Kb average probe resolution) according to the manufacturer's instructions. Briefly, the sample (1 µg) and the sex‐matched reference DNA were chemically labeled with ULS‐Cy5 and ULS‐Cy3, respectively, at 85°C for 30 minutes using an Oligo aCGH labeling kit for FFPE samples (Agilent). The labeled samples were purified with the genomic DNA purification module (Agilent), combined, mixed with human Cot‐1 DNA, denatured at 95°C using a Oligo aCGH hybridization kit (Agilent), and were applied to microarrays. After hybridization at 65°C for 40 h, microarrays were washed in Oligo aCGH wash buffer 1 at RT for 5 minutes and in wash buffer 2 at 37°C for 1 minute. After drying, the microarrays were scanned with a DNA microarray scanner G2565BA (Agilent) and data (log2) were extracted from the raw microarray image files using Feature Extraction software version 9. Data were analyzed by DNA Analytics software (version 3.5) with default filter settings. The aberration detection method 2 (ADM2) algorithm with fuzzy zero correction was used to define aberrant intervals. The log2 ratio of <−1.0 at the region of interest was considered to represent homozygous loss, and values of −1.0 to −0.2 were considered to represent heterozygous loss (24).
Differential PCR
The analysis for homozygous deletion of the p14ARF, p15INK4b, p16INK4a (at 9p21) and the RB1 (at 13q14) genes were carried out by using differential PCR, as previously reported (20). The CF gene sequence was used as a reference (25). For the detection of homozygous deletion of the p14ARF, p15INK4b, p16INK4a and RB1 genes, the following primers were used: 5′‐ACC CCG CTT TCG TAG TTT‐3′ (sense) and 5′‐AAA TGG ACA TTT ACG GTA GTG G‐3′ (antisense) for p14ARF and p16INK4a (PCR product, 101 bp), 5′‐AAT TTT TGG AAC AAA GAT AAT GGA A −3′ (sense) and 5′‐CCT CTA ATG ATT GAG TGC TTA AGT GA‐3′ (antisense) for p15INK4b (PCR product, 100 bp), 5′‐AAA ACT GTA CAT TTA AAA TTG CTA TG‐3′ (sense) and 5′‐CAC AAC ATC AGA CCA TTA AGA CTC‐3′ (antisense) for RB1 (PCR product, 104 bp) and 5′‐GGC ACC ATT AAA GAA AAT ATC ATC TT‐3′ (sense) and 5′‐GTT GGC ATG CTT TGA TGA CGC TTC‐3′ (antisense) for CF (PCR product, 79 bp). PCR was carried out with 28 cycles, with an annealing temperature at 55°C. The PCR products were loaded on 8% acrylamide gels and stained with ethidium bromide. The mean ratio of signal of p14ARF, p15INK4b, p16INK4a and RB1 to CF of normal DNA (14 samples of normal tissue) was approximately 1.0. Samples with a ratio of ≤0.2 were considered to present deletions (27).
Methylation‐specific PCR
Screening with methylation‐specific PCR for promoter methylation of p14ARF, p15INK4b, p16INK4a and RB1 was performed as described previously 16, 17. Briefly, approximately 300 ng of DNA extracted from paraffin sections was modified with sodium bisulfite using an EZ DNA Methylation KitTM (Zymo Research, Orange, CA, USA). Briefly, DNA was denatured with dilution buffer at 37°C for 15 minutes, incubated with CT conversion reagent at 50°C for 16 h, and cleaned up and desulfonated by using columns. The primer sequences of p14ARF, p15INK4b, p16INK4a and RB1 for the methylated and unmethylated PCR have been reported previously 16, 17. DNA extracted from normal blood samples were used as negative controls, and CpGenomeTM Universal methylated DNA (Chemicon International Inc., Temecula, CA, USA) was used as a positive control for methylated DNA, as previously described 16, 17.
Statistical analyses
The χ2 test or the Fisher's exact test was conducted to analyze the significance of the association between triple‐negative status and alterations in the RB1 and TP53 pathways. Statistical analysis was performed with Stat‐View for Windows 5.01 software® (SAS Institute Inc., Cary, NC, USA). For survival analyses, the Kaplan–Meier method was used. Multivariate analyses were performed with adjustment for age, sex and treatment (surgery and radiotherapy, surgery only).
RESULTS
Array CGH
Array CGH was carried out for 15 triple‐negative low‐grade diffuse gliomas. Chromosomal imbalances observed in at least two cases and confirmed by differential PCR were loss at 9p21, containing the p14ARF, p15INK4b and p16INK4a loci (three cases), and loss at 13q14–13q32, containing the RB1 locus (two cases) (Figure 1). In one case, array CGH showed loss at 10p13 (C10orf30), which was confirmed by differential PCR (data not shown).
Figure 1.
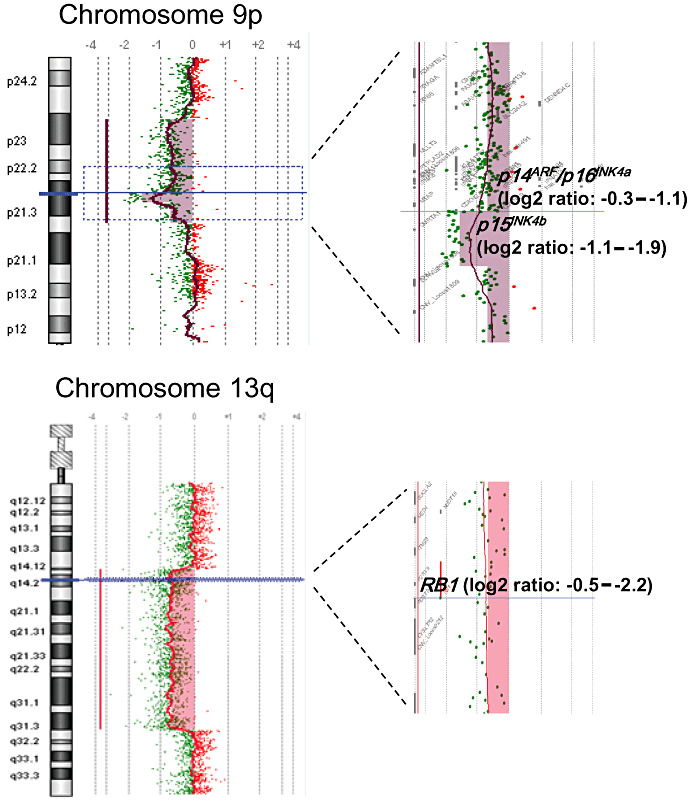
Array comparative genomic hybridization (CGH) showing chromosomal loss at 9p21 (p14ARF, p15INK4b, p16INK4a loci) in a “triple‐negative” diffuse astrocytoma and 13q14–13q32 (containing the RB1 locus) in a “triple‐negative” oligodendroglioma.
Alterations in the p14ARF, p15INK4b, p16INK4a and RB1 genes
Homozygous deletion and promoter methylation of the p14ARF, p15INK4b, p16INK4a and RB1 genes were assessed in 31 triple‐negative and 160 non‐triple‐negative low‐grade diffuse gliomas. Alterations in the RB1 pathway were more frequent in triple‐negative than in non‐triple‐negative cases (26% vs. 11%; P = 0.0371; Table 2). In particular, homozygous deletion of p15INK4b was significantly more frequent in triple‐negative than in non‐triple‐negative cases (13% vs. 1%; P = 0.0069; Table 2).
Table 2.
Alterations in the RB1 and TP53 signalling pathways in low‐grade diffuse gliomas.
| No. of cases with | Triple negative* (n = 31) | Non‐triple negative** (n = 160) | P value |
|---|---|---|---|
| At least one alteration in the RB1 pathway | 8 (26%) *** | 17 (11%) | 0.0371 |
| p15INK4b homozygous deletion | 4 (13%) *** | 2 (1%) | 0.0069 |
| p15INK4b promoter methylation | 0 | 7 (4%) | ns |
| p16INK4a homozygous deletion | 1 (3%) | 2 (1%) | ns |
| p16INK4a promoter methylation | 2 (6%) | 4 (3%) | ns |
| RB1 homozygous deletion | 2 (6%) | 5 (3%) | ns |
| RB1 promoter methylation | 0 | 0 | ns |
| At least one alteration in the TP53 pathway | 2 (6%) | 79 (49%) *** | 0.0001 |
| p14ARF homozygous deletion | 1 (3%) | 2 (1%) | ns |
| p14ARF promoter methylation | 1 (3%) | 34 (21%) *** | 0.0199 |
| TP53 mutations | 0 | 54 (34%) | 0.0001 |
| No alterations | 23 (74%) | 72 (45%) | 0.0032 |
Cases without any of IDH1/IDH2 mutation, TP53 mutation, or 1p/19q loss.
Non‐triple negative cases included 54 cases (IDH1/2 plus TP53 mutations), 56 cases (IDH1/2 mutation plus 1p/19q loss) and 50 cases (IDH1/2 mutation only).
Bold letters indicate statistical significance.
ns, not significant.
Among triple‐negative cases, 6 diffuse astrocytomas and 2 oligodendrogliomas showed alterations in the RB1 pathway, whereas 10 diffuse astrocytomas, 2 oligoastrocytomas and 11 oligodendrogliomas lacked alterations in the RB1 pathway (Table 1). Gemistocytic components were observed in 4 out of 6 triple‐negative diffuse astrocytomas (67%) with an alteration in the RB1 pathway, although the fraction of gemistocytes did not reach >20% to fulfil the diagnostic criteria of gemistocytic astrocytomas (14). In contrast, gemistocytic components were not recognized in any of the 10 triple‐negative diffuse astrocytomas without alterations in the RB1 pathway (Table 1) and in only 5 out of 77 (6%) non‐triple negative diffuse astrocytomas.
Promoter methylation of the p14ARF was significantly less frequent in triple‐negative than in non‐triple‐negative tumors (3% vs. 21%; P = 0.0199; Table 2). Overall, alterations in the TP53 pathway were also significantly less frequent in triple‐negative than in non‐triple‐negative cases (6% vs. 49%; P = 0.0001; Table 2). This tendency was consistent when we analyzed each histologic type separately, but a significant difference was observed only in diffuse astrocytoma (data not shown).
Clinical outcome
The mean survival of patients with triple‐negative and non‐triple‐negative tumors was not markedly different (63.0 ± 57.9 months vs. 67.6 ± 48.8 months; P = 0.6603). Among all low‐grade diffuse gliomas, multivariate analysis after adjusting for age, sex, histology and treatment showed that alterations in the RB1 pathway were significantly associated with shorter survival [hazard ratio, 3.024 (1.279–6.631); P = 0.0057].
Among triple‐negative tumors, median survival of patients with diffuse astrocytoma with an alteration in the RB1 pathway was 14.0 months, significantly shorter than that of patients with diffuse astrocytoma lacking RB1‐pathway alterations (49 months, P = 0.0187; Figure 2). Among triple‐negative cases without alterations in the RB1 pathway, patients with oligodendroglioma or oligoastrocytoma had a significant longer survival than those with diffuse astrocytoma (median, 74 vs. 49 months; P = 0.0475; Figure 2).
Figure 2.
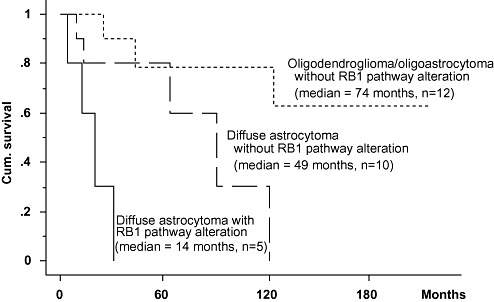
Median survival of diffuse astrocytoma patients with RB1 pathway alteration was 14 months, significantly shorter than that of diffuse astrocytoma patients lacking RB1 pathway alteration (49 months, P = 0.0187). Note that among triple‐negative cases without RB1 pathway alteration, oligodendroglioma and oligoastrocytoma patients have significant longer survival than those with diffuse astrocytoma (median = 74 vs. 49 months; P = 0.0475).
DISCUSSION
The cyclin D‐CDK4,CDK6/INK4/RB1/E2F pathway plays a key role in controlling cell growth by integrating multiple mitogenic and anti‐mitogenic stimuli (19). The CDK4‐cyclin D1 complex phosphorylates the RB1 protein, thereby inducing release of the transcription factor E2F, which activates genes involved in the G1 → S transition 3, 23. The members of the INK4 family (p16INK4a, p15INK4b, p18INK4c and p19INK4d) block the progression of the cell cycle by binding to either CDK4 or CDK6 and inhibiting the action of cyclin D (5). In addition to their capacity to arrest cells in the G1 phase of the cell cycle, the INK4 proteins participate in a number of cellular processes, including senescence, apoptosis and DNA repair (5). As the RB1 signaling pathway is deregulated by genetic and epigenetic alterations in a variety of human tumors 7, 18, certain molecules in the pathway are being considered as targets for cancer therapy (6).
In contrast to glioblastoma, in which frequent alterations in the RB1 pathway (40–70% of cases) have been reported 4, 18, there are few such reports for low‐grade diffuse glioma. Alterations in the RB1 pathway have been reported in 6 out of 46 (13%) diffuse astrocytomas (RB1 methylation in one case, p16INK4a methylation in three cases, p15INK4b methylation or homozygous deletion in three cases) (29). We have previously shown that alterations in the RB1 pathway are rare in oligodendrogliomas (WHO grade II) (4%), but frequent (65%) in anaplastic oligodendrogliomas (WHO grade III) (28).
In order to identify novel genetic pathways to triple‐negative low‐grade diffuse gliomas, which lack IDH1/2 mutation, TP53 mutation or 1p/19q loss, we first carried out array CGH analyses. Chromosomal imbalances that were observed in at least two cases were loss at 9p21 (p14AR F, p15INK4b, p16INK4a loci) and loss at 13q14–13q32 (containing the RB1 locus). As these two chromosomal loci contain genes involved in the RB1 signaling pathway (p15INK4b, p16INK4a, RB1) (5), we further assessed alterations in these genes in triple‐negative as well as non‐triple negative low‐grade diffuse gliomas. We found that approximately one quarter (26%) of triple‐negative low‐grade diffuse gliomas carry alterations in the RB1 pathway, as represented by p15INK4b homozygous deletion, homozygous deletion or promoter methylation of the p16INK4a, or RB1 homozygous deletion. Such alterations were significantly more frequent than in non‐triple negative cases (17/160; 11%).
By definition, triple‐negative cases do not have TP53 mutations. In the present study, we also noted that promoter methylation of the p14ARF gene, another component of the TP53 pathway was significantly less frequent in triple‐negative than in non‐triple‐negative low‐grade diffuse gliomas (3% vs. 21%; P = 0.0199). Such infrequent alterations in the TP53 pathway thus appear to be typical of triple‐negative low‐grade diffuse gliomas.
Alterations in RB1 pathway‐related genes have been reported to be associated with shorter survival (median survival, 1.4 vs. 5.8 years; P = 0.0009) in patients with anaplastic astrocytoma (2). Abnormalities in the p15INK4b, p16INK4a, RB1 or CDK4 genes are also associated with shorter survival in patients with glioblastoma (P = 0.0002) (1). However, the prognostic value of alterations in the RB1 signaling pathway has not been studied in low‐grade diffuse gliomas.
In the present study, among all low‐grade diffuse gliomas, multivariate analysis after adjusting for age, histology and treatment showed that alterations in the RB1 pathway were significantly associated with shorter survival [hazard ratio, 3.024 (1.279–6.631); P = 0.0057]. Among triple‐negative diffuse astrocytomas, survival of patients with tumours carrying RB1 pathway alterations was significantly shorter than that of patients with tumours without RB1 pathway alterations (Figure 2). It was of interest to note that 4 out of 6 triple‐negative diffuse astrocytomas with RB1 pathway alterations but none of 10 cases lacking RB1 pathway alterations showed gemistocytic components (Table 1). This may at least in part explain the poorer outcome of patients with RB1 pathway alterations, as gemistocytic astrocytomas or astrocytomas containing a significant fraction of gemistocytes are characterized by a tendency for rapid recurrence and malignant progression 12, 13, 15, 22, 26 and shorter patient survival 13, 26.
It is well established that oligodendrogliomas with 1p/19q loss have a favorable prognosis 8, 10, 14, 21. In the present study, among triple‐negative tumors without alterations in the RB1 pathway, survival of patients with oligodendrogliomas or oligoastrocytomas was significantly longer than those with diffuse astrocytoma, further presenting evidence that, even without 1p/19q loss, outcome for patients with oligodendroglial tumors was significantly better than for patients with diffuse astrocytomas (Figure 2).
In summary, we present evidence that alterations in the RB1 pathway are common in triple‐negative low‐grade diffuse gliomas, and that genetic alterations in the RB1 pathway are associated with unfavorable outcome for the patient. These results suggest that a fraction of low‐grade diffuse gliomas lacking common genetic alterations (IDH1/2 mutation, TP53 mutation and 1p/19q loss) may develop through a distinct genetic pathway, which may include loss of cell‐cycle control regulated by the RB1 pathway.
REFERENCES
- 1. Backlund LM, Nilsson BR, Goike HM, Schmidt EE, Liu L, Ichimura K, Collins VP (2003) Short postoperative survival for glioblastoma patients with a dysfunctional Rb1 pathway in combination with no wild‐type PTEN. Clin Cancer Res 9:4151–4158. [PubMed] [Google Scholar]
- 2. Backlund LM, Nilsson BR, Liu L, Ichimura K, Collins VP (2005) Mutations in Rb1 pathway‐related genes are associated with poor prognosis in anaplastic astrocytomas. Br J Cancer 93:124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bogler O, Huang HJ, Kleihues P, Cavenee WK (1995) The p53 gene and its role in human brain tumors. Glia 15:308–327. [DOI] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Canepa ET, Scassa ME, Ceruti JM, Marazita MC, Carcagno AL, Sirkin PF, Ogara MF (2007) INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life 59:419–426. [DOI] [PubMed] [Google Scholar]
- 6. Chano T, Ikebuchi K, Ochi Y, Tameno H, Tomita Y, Jin Y et al (2010) RB1CC1 activates RB1 pathway and inhibits proliferation and cologenic survival in human cancer. Plos ONE 5:e11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collins VP (2004) Brain tumours: classification and genes. J Neurol Neurosurg Psychiatry 75(Suppl. 2):ii2–i11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Felsberg J, Erkwoh A, Sabel MC, Kirsch L, Fimmers R, Blaschke B et al (2004) Oligodendroglial tumors: refinement of candidate regions on chromosome arm 1p and correlation of 1p/19q status with survival. Brain Pathol 14:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang J, Pang J, Watanabe T, Ng HK, Ohgaki H (2009) Whole genome amplification for array comparative genomic hybridization using DNA extracted from formalin‐fixed, paraffin‐embedded histological sections. J Mol Diagn 11:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kanner AA, Staugaitis SM, Castilla EA, Chernova O, Prayson RA, Vogelbaum MA et al (2006) The impact of genotype on outcome in oligodendroglioma: validation of the loss of chromosome arm 1p as an important factor in clinical decision making. J Neurosurg 104:542–550. [DOI] [PubMed] [Google Scholar]
- 11. Kim YH, Nobusawa S, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K et al (2010) Molecular classification of low‐grade diffuse gliomas. Am J Pathol 177:2708–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kleihues P, Davis RL, Ohgaki H, Burger PC, Westphal MM, Cavenee WK (2000) Diffuse astrocytoma. In: Pathology and Genetics of Tumours of the Nervous System, Kleihues P, Cavenee WK (eds), Chapter pp. 22–26. IARC Press: Lyon. [Google Scholar]
- 13. Krouwer HG, Davis RL, Silver P, Prados M (1991) Gemistocytic astrocytomas: a reappraisal. J Neurosurg 74:399–406. [DOI] [PubMed] [Google Scholar]
- 14. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007) WHO Classification of Tumours of the Central Nervous System. IARC: Lyon. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McLendon RE, Enterline DS, Tien RD, Thorstad WL, Bruner JM (1998) Tumors of central neuroepithelial origin. In: Russell and Rubinstein's Pathology of Tumors of the Nervous System, Bigner DD, McLendon RE, Bruner JM (eds), Chapter 9 pp. 307–571. Arnold: London, Sydney, Auckland. [Google Scholar]
- 16. Nakamura M, Watanabe T, Klangby U, Asker CE, Wiman KG, Yonekawa Y et al (2001) P14Arf deletion and methylation in genetic pathways to glioblastomas. Brain Pathol 11:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H (2001) Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest 81:77–82. [DOI] [PubMed] [Google Scholar]
- 18. Ohgaki H, Kleihues P (2009) Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci 100:2235–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ortega S, Malumbres M, Barbacid M (2002) Cyclin d‐dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta 1602:73–87. [DOI] [PubMed] [Google Scholar]
- 20. Rollbrocker B, Waha A, Louis DN, Wiestler OD, von Deimling A (1996) Amplification of the cyclin‐dependent kinase 4 (CDK4) gene is associated with high cdk4 protein levels in glioblastoma multiforme. Acta Neuropathol 92:70–74. [DOI] [PubMed] [Google Scholar]
- 21. Sasaki H, Zlatescu MC, Betensky RA, Johnk LB, Cutone AN, Cairncross JG, Louis DN (2002) Histopathological‐molecular genetic correlations in referral pathologist‐diagnosed low‐grade “oligodendroglioma”. J Neuropathol Exp Neurol 61:58–63. [DOI] [PubMed] [Google Scholar]
- 22. Shaw EG, Daumas Duport C, Scheithauer BW, Gilbertson DT, O'Fallon JR, Earle JD et al (1989) Radiation therapy in the management of low‐grade supratentorial astrocytomas. J Neurosurg 70:853–861. [DOI] [PubMed] [Google Scholar]
- 23. Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev 13:1501–1512. [DOI] [PubMed] [Google Scholar]
- 24. Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A et al (2005) Genome‐wide array‐based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 24:1348–1358. [DOI] [PubMed] [Google Scholar]
- 25. Tohma Y, Gratas C, Biernat W, Peraud A, Fukuda M, Yonekawa Y et al (1998) PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol 57:684–689. [DOI] [PubMed] [Google Scholar]
- 26. Watanabe K, Tachibana O, Yonekawa Y, Kleihues P, Ohgaki H (1997) Role of gemistocytes in astrocytoma progression. Lab Invest 76:277–284. [PubMed] [Google Scholar]
- 27. Watanabe T, Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H (2001) Promoter hypermethylation and homozygous deletion of the p14ARF and p16INK4a genes in oligodendrogliomas. Acta Neuropathol 101:185–189. [DOI] [PubMed] [Google Scholar]
- 28. Watanabe T, Yokoo H, Yokoo M, Yonekawa Y, Kleihues P, Ohgaki H (2001) Concurrent inactivation of RB1 and TP53 pathways in anaplastic oligodendrogliomas. J Neuropathol Exp Neurol 60:1181–1189. [DOI] [PubMed] [Google Scholar]
- 29. Watanabe T, Katayama Y, Yoshino A, Komine C, Yokoyama T (2003) Deregulation of the TP53/p14ARF tumor suppressor pathway in low‐grade diffuse astrocytomas and its influence on clinical course. Clin Cancer Res 9:4884–4890. [PubMed] [Google Scholar]