

Abstract
Epigenetic regulation of gene expression by DNA methylation and histone modification is frequently altered in human cancers including gliomas, the most common primary brain tumors. In diffuse astrocytic and oligodendroglial gliomas, epigenetic changes often present as aberrant hypermethylation of 5′‐cytosine‐guanine (CpG)‐rich regulatory sequences in a large variety of genes, a phenomenon referred to as glioma CpG island methylator phenotype (G‐CIMP). G‐CIMP is particularly common but not restricted to gliomas with isocitrate dehydrogenase 1 (IDH1) or 2 (IDH2) mutation. Recent studies provided a mechanistic link between these genetic mutations and the associated widespread epigenetic modifications. Specifically, 2‐hydroxyglutarate, the oncometabolite produced by mutant IDH1 and IDH2 proteins, has been shown to function as a competitive inhibitor of various α‐ketoglutarate (α‐KG)‐dependent dioxygenases, including histone demethylases and members of the ten‐eleven‐translocation (TET) family of 5‐methylcytosine (5mC) hydroxylases. In this review article, we briefly address (i) the basic principles of epigenetic control of gene expression; (ii) the most important methods to analyze focal and global epigenetic alterations in cells and tissues; and (iii) the involvement of epigenetic alterations in the molecular pathogenesis of gliomas. Moreover, we discuss the promising roles of epigenetic alterations as molecular diagnostic markers and novel therapeutic targets, and highlight future perspectives toward unraveling the “glioma epigenome.”
Keywords: DNA methylation, glioma, histone modification, IDH1, MGMT
INTRODUCTION
Cancer development and progression are driven by complex aberrations involving structural genetic alterations, such as point mutations, deletions, insertions, translocations, amplifications and other genetic rearrangements, as well as various types of epigenetic changes resulting in aberrant gene expression because of altered patterns of DNA methylation and post‐translational modification of histone proteins [for recent reviews see 71, 117]. The molecular mechanisms underlying the transforming and growth‐promoting effects of cancer‐associated genetic and epigenetic changes are still poorly understood in their entire complexity, as is the mechanistic interplay between both types of alterations in cancer cells. However, recent data have provided intriguing new insights by the identification of genetic changes affecting genes directly functioning as epigenetic regulators, for example, DNA and histone methyltransferases, or by linking mutations in genes encoding the isocitrate dehydrogenase 1 (IDH1) and 2 (IDH2) with specific metabolic changes that influence the function and activity of various epigenetic regulators, thereby leading to global alterations in DNA methylation and chromatin structure. Interestingly, the molecular characterization of epigenetic alterations in cancer has not only improved our understanding of disease‐driving molecular pathomechanisms but has also resulted in the identification of valuable novel biomarkers for improved tumor diagnostics, as well as a better prediction of therapy response and prognosis. Moreover, as alterations in DNA methylation and histone modifications can be reversed by treatment with specific drugs, such epigenetic changes represent promising molecular targets for novel therapeutic approaches.
In this review article, we will (i) briefly summarize the basic principles of epigenetic control of gene expression with a particular focus on DNA methylation and histone modification; (ii) introduce the most commonly used methods to investigate such epigenetic alterations at the level of individual genes up to the entire genome; and (iii) focus on recent progress concerning the pathomechanistic roles and clinical significance of epigenetic alterations in primary brain tumors, in particular gliomas.
BASIC PRINCIPLES OF EPIGENETIC CONTROL OF GENE EXPRESSION
“An epigenetic trait is a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence”(12). DNA methylation and post‐translational modification of histone proteins are the best‐characterized epigenetic mechanisms that determine the access to genetic information as required for DNA replication, transcription and repair. Physiologically, epigenetic control plays a crucial role in X chromosome inactivation (64), genomic imprinting (77), silencing of retrotransposons (4), telomere length control (15), centromere functionality (120) and recently was also shown to impact alternative splicing of pre‐mRNAs (85).
DNA methylation
DNA may be methylated at the 5′‐position of cytosine usually within cytosine‐guanine (CpG) dinucleotides. Approximately 70%–80% percent of all CpG dinucleotides are methylated in human somatic cells. A concentration of CpG sites is seen in the so‐called CpG islands, which are usually short CpG‐rich DNA regions located in more than half of all human gene promoters, and in so‐called CpG island shores, which are located upstream of promoter‐associated CpG islands or in regions of large repetitive sequences, such as centromeres and retrotransposon elements 19, 28, 138. There are three main DNA methyltransferases that catalyze cytosine methylation in CpG dinucleotides. Acting as de novo methyltransferases, DNMT3A and DNMT3B target unmethylated CpGs. In contrast, DNMT1 targets hemimethylated DNA to maintain the methylation pattern following DNA replication (19). The effects of DNA methylation depend on its localization in the genome. For example, CpG methylation of repetitive sequences has been associated with increased genome stability. Hypermethylation of promoter‐associated CpG‐rich sequences may repress transcription and, hence, gene expression, whereas methylated CpGs located in the genes themselves are associated with active genes (123). The molecular mechanisms by which CpG methylation influences the regulation of gene expression are not fully understood. First models suggested that CpG methylation in promoter regions may directly block the binding of transcription factors to their target sequences in the DNA (58). However, more recent studies demonstrated that promoter methylation inhibits the binding of the transcriptional machinery by recruiting methylated CpG‐binding proteins and modifying chromatin structure via further protein interactions 87, 136.
5‐methylcytosine (5mC) generated by DNA methyltransferases may be further modified by hydroxylation to 5‐hydroxymethylcytosine, a modification that is mediated by the ten‐eleven‐translocation (TET) family of α‐ketoglutarate (α‐KG)‐ and Fe(II)‐dependent dioxygenases (124). In Purkinje neurons, for example, 5‐hydroxymethylcytosine is approximately 40% as abundant as 5mC, suggesting an important biological function (70). However, the precise functional roles of 5mC hydroxylation are still poorly understood and a matter of active research 26, 147. It has been suggested that 5‐hydroxymethylcytosine induces passive demethylation by preventing DNMT1‐mediated maintenance methylation and inducing active demethylation by DNA repair mechanisms. Very recent data in fact show that oxidation of 5mC and 5‐hydroxymethylcytosine by TET proteins, followed by thymine‐DNA glycosylase (TDG)‐mediated base excision of the resulting 5‐carboxylcytosine, constitutes an important pathway for active DNA demethylation (53). Moreover, several methyl‐CpG‐binding proteins display reduced affinity for 5‐hydroxymethylcytosine, which, in turn, may have effects on transcriptional regulation.
Post‐translational histone modifications
Chromatin is not just an inert structure required for the proper package of DNA within the cell nucleus but represents a highly dynamic and instructive DNA scaffold (9). The histones H2A, H2B, H3 and H4 associate as octamers with DNA to form a nucleosome, the fundamental repeating unit in chromatin (3). Histones may be post‐translationally modified, mainly in their N‐terminal tails. The list of such modifications is constantly growing and comprises acetylation (ac), methylation (me), phosphorylation (ph), sumoylation (sumo), ubiquitination (ub) and others. Histone modifications influence the overall structure of chromatin and positively or negatively modulate the binding of effector molecules (9). Thereby, several enzymes that add or remove histone modifications interact to produce a chromatin structure that has a more activating or a more repressive effect on gene transcription. Histone acetylation, for example, acetylation of lysine 56 on histone H3 (abbreviated H3K56ac) and acetylation of lysine 16 on histone H4 (H4K16ac), seems to be generally associated with activation of transcription. On the other hand, histone methylation may either repress gene expression (H3K9me2, H3K27me3) or activate transcription (eg H3K4me2, H3K4me3), depending on the specific sites of methylation (78). The consequences of histone modifications depend on the genomic context. For instance, H3K6ac and H3K6me are both associated with active genes, the former being enriched in promoter sequences, the latter being enriched in the bodies of active genes (95).
Interplay between DNA methylation and histone modifications
Different types of histone modifications are strongly interconnected with each other in the regulation of gene expression. Histone modifications may crosstalk in situ (at the same residue), in cis (on the same histone) and in trans (on different histones) (95). For instance, H3K9 methylation, related to gene repression, blocks H3K9 acetylation that occurs on promoters of active genes and vice versa (100). As an example of a histone crosstalk in cis, H3S10ph is required for H3K14 acetylation (82). On the other hand, H3S10ph inhibits H3K9 methylation and vice versa (110). H3K4me seems to crosstalk with H4K16 acetylation in trans (29). Moreover, bidirectional complex and as yet poorly understood molecular interactions are taking place between histone modifications and DNA methylation. For example, H3K9 trimethylation is required for DNMT3B‐dependent DNA methylation of pericentromeric repeats (76). On the other hand, H3K4 methylation was shown to be mutually exclusive to de novo DNA methylation by DNMT3A/B 89, 92, 104. The ubiquitin‐like protein containing plant homeo domain (PHD) and ring finger domains 1 (UHRF1) protein recognizes H3K9me marks and, after binding to hemimethylated DNA, recruits DNMT1 to allow for maintenance of DNA methylation in heterochromatin following replication (118). In addition, UHRF1 induces histone deacetylation and transcriptional repression by recruiting histone deacetylase 1 (HDAC1) after binding to methylated CpG (132). These are only some examples of the complex epigenetic interplay between histone modifications and DNA methylation in the regulation of gene expression (for a more comprehensive overview see (95)).
COMMONLY USED METHODS FOR THE DETECTION OF EPIGENETIC DNA AND HISTONE ALTERATIONS
Methods for the detection of 5mC
The investigation of methylated cytosines in genomic DNA relies mainly on the following three principles: (i) sodium bisulfite conversion of nonmethylated cytosines to uracil; (ii) use of methylation‐sensitive and nonsensitive isoschizomeric restriction enzymes; and (iii) immunoprecipitation of methylated DNA by antibodies against 5mC or affinity‐binding to methylated DNA‐binding enzymes. These pretreatment methods can be combined with different analytical platforms, including microarrays and high‐throughput sequencing to enable genome‐wide DNA methylation analysis (Figure 1, Table 1).
Figure 1.
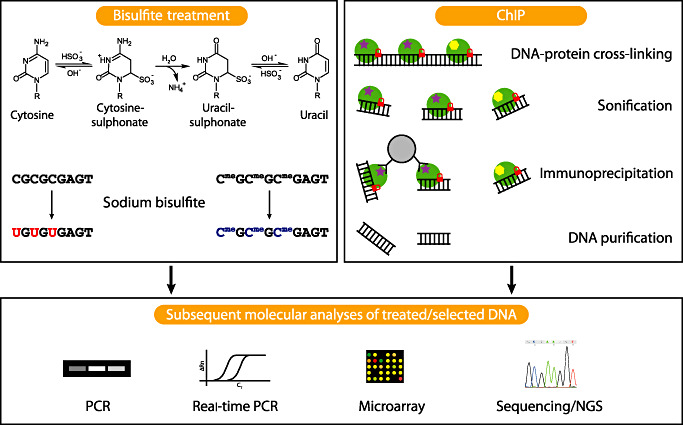
Commonly used methods to analyze epigenetic alterations. Sodium bisulfite treatment, which is frequently used for the investigation of DNA methylation, converts cytosine to uracil, while 5‐methylcytosine (5mC) and 5‐hydroxymethylcytosine are protected against this conversion. Subsequent analyses of the bisulfite‐treated DNA using PCR [methylation‐specific PCR (MSP)], real‐time PCR (MethyLight), microarrays, different sequencing approaches or mass spectrometry (MassARRAY EpiTyper assay, not depicted) allow to distinguish cytosines converted to uracil from unaltered cytosines. Chromatin immunoprecipation (ChIP) is frequently used to investigate specific histone modifications. Therefore, histones are stably cross‐linked to DNA by formaldehyde, in vivo. After cell lysis, DNA is fragmented by sonification. Antibodies against the modified histone of interest are added, bind to the modified histone and are afterwards pulled down by, for example, protein A/G coated agarose beads (protein A/G is capable of binding the Fc fragments of the specific antibodies). Thereafter, DNA protein cross‐links are reversed, and the DNA that was bound to the modified histone is purified for subsequent analyses. PCR and real‐time PCR focus on specific DNA regions, whereas microarray based methods (ChIP‐on‐chip) or next generation sequencing methods (ChIP‐seq) enable whole genome coverage. Histones are symbolized by green circles; two different histone modifications are symbolized by a purple star and a yellow hexagon, respectively; DNA‐protein cross‐linking is indicated by a red lock; the grey circle represents an agarose bead with protein A/G coating.
Table 1.
Summary of commonly used methods for the detection of methylated DNA.
| Bisulfite conversion | Methylation‐sensitive restriction enzymes | Immunoprecipitation or affinity‐binding of methylated DNA | |
|---|---|---|---|
| Locus‐specific DNA methylation analyses | MSP | Digest followed by PCR or Southern Blot | Immunoprecipitation or affinity‐binding followed by PCR |
| MethyLight | MS‐MLPA | ||
| COBRA | |||
| Sanger‐Sequencing | |||
| Pyrosequencing | |||
| Genome‐wide DNA methylation analyses | NGS | RLGS | MeDIP |
| DMH | MIRA | ||
| MCA | NGS | ||
| NGS |
COBRA = combined bisulfite restriction analysis; DMH = differential methylation hybridization; MS‐MLPA = methylation‐specific multiplex ligation‐dependent probe amplification; MCA = methylated CpG island amplification; MeDIP = methylated DNA immunoprecipitation; MIRA = methylated CpG island recovery assay; NGS = next‐generation sequencing; MSP = methylation‐specific PCR; RLGS = restriction landmark genomic scanning.
Methods used for the targeted detection of promoter methylation of individual genes
Several of the most commonly used methods for locus‐specific DNA methylation analysis are based on the treatment of DNA with sodium bisulfite, resulting in a conversion of nonmethylated cytosines to uracil, whereas methylated cytosines remain unaltered. Modified DNA is used as a template for amplification in the methylation‐specific PCR (MSP) technique, a rapid and very sensitive method for the detection of methylated DNA (55). MSP is the most frequently used technique for the diagnostic determination of promoter hypermethylation of the O6‐methylguanine‐DNA methyltransferase gene (MGMT) in glioblastomas (142). PCR is performed using primer sets designed to distinguish methylated from unmethylated DNA. This method is even useful for the investigation of DNA methylation in formalin‐fixed paraffin embedded tissues (FFPE).
MethyLight is a fluorescent‐based real‐time PCR method used to detect and quantify DNA methylation in bisulfite‐converted DNA (31). The method is highly sensitive and no post‐PCR manipulations, such as gel electrophoresis, are required. However, similar to MSP, this method generates no information about the precise regional methylation pattern of the genomic DNA at the individual CpG sites investigated.
Combined bisulfite restriction analysis (COBRA) is a quantitative technique for methylation detection that also depends on bisulfite‐converted DNA (148). The genomic region of interest is amplified with primers not discriminating methylated from nonmethylated DNA but encompassing a restriction enzyme site, which is affected by the bisulfite treatment. The PCR product is digested with the respective enzyme and quantified using gel electrophoresis and densitometry. COBRA is compatible with DNA extracted from FFPE tissue but provides data only for the specific restriction enzyme recognition site.
The gold standard for mapping of methylated CpG sites is sequencing of sodium bisulfite‐modified DNA (44) either by chain termination sequencing (“Sanger sequencing”) or pyrosequencing, which is a sequencing‐by‐synthesis method 21, 128. Both sequencing methods rely on the sodium bisulfite conversion of genomic DNA and PCR amplification of the region of interest using primers specific for bisulfite‐converted DNA but without covering CpG sites. Both methods generate a complete picture of the methylation pattern of the region of interest. In addition, pyrosequencing allows for the quantification of multiple CpG methylation sites in one sequencing run. In contrast to conventional sequencing, however, only short sequences can be read efficiently, therefore limiting the number of CpG sites that may be analyzed in one read.
The EpiTYPERTM assay (Sequenom, San Diego, CA, USA) involves PCR amplification from bisulfite‐converted DNA, followed by in vitro transcription into RNA from the reverse strand and base‐specific cleavage of the transcribed RNAs. The cleavage products result in distinct fragments for methylated and non‐methylated DNA that are detected by MALDI‐TOF mass spectrometry (MassARRAYTM system; Sequenom). This method allows for the rapid, sensitive and quantitative detection of methylation patterns of multiple CpG‐containing amplicons of up to 600 base pairs and is ideally suited for the screening of defined gene sets in larger sample numbers. However, it requires access to a specialized mass spectrometry system that is not available at all places.
Several nonbisulfite methods for the detection of methylated cytosines in specific DNA sequences rely on the digestion of DNA with methylation‐sensitive enzymes followed by different detection methods of the restriction fragments, for example, by PCR or Southern blot analysis. Another method often used for the analysis of methylated cytosines is based on multiplex ligation‐dependent probe amplification (MLPA) in combination with methylation‐sensitive restriction enzymes (methylation‐specific MLPA, MS‐MLPA). In MS‐MLPA the ligation of the MLPA probe oligonucleotides is combined with digestion of the genomic DNA‐probe hybrid complex by methylation‐sensitive restriction enzymes (102). This method is also being used for the semiquantitative detection of MGMT promoter hypermethylation in the routine diagnostic assessment of gliomas (61).
Methods used for large‐scale DNA methylation analysis
One of the earliest methods used for genome‐wide, non‐microarray‐based DNA methylation analysis is restriction landmark genomic scanning (RLGS) (52). RLGS is a two‐dimensional DNA gel electrophoresis technique that allows for the quantification of gene copy number and methylation status at the same time. A drawback of the RLGS method is its limited sensitivity for the detection of methylated CpG sites.
Another method based on the use of methylation‐sensitive restriction enzymes is differential methylation hybridization (DMH) that allows for the determination of the methylation levels of a large number of CpG‐rich loci at the same time (57). DMH is an array‐based method employing CpG island fragments gridded on high‐density arrays. The genomic DNA of interest is digested with methylation‐sensitive enzymes, and the digestion products are used as templates for PCR after linker ligation. The amplification products are labeled and used as probes that are hybridized to the CpG island arrays. Neither further cloning nor sequencing of the fragments of interest is necessary. A major disadvantage of the methods using restriction enzymes is the reliance on specific restriction enzyme‐cutting sites and incomplete digestion that can result in false‐positive results. The methylated CpG island amplification (MCA) method addresses the problem of false‐positive results generated by RLGS or DMH by using methylation‐sensitive and insensitive isoschizomeres followed by adaptor ligation and PCR amplification (129). Thereby, methylated CpG‐rich sequences are preferentially amplified, labeled and hybridized to microarrays. This method generates less frequently false‐positive results than DMH, but the genome coverage is still limited.
The third basic method for DNA methylation analysis is the enrichment of methylated genomic sequences by affinity‐binding of methylated sequences to 5mC antibodies (methylated DNA immunoprecipitation; MeDIP) (141) or to columns that contain methyl‐CpG‐binding domain proteins, like MBD2 (methylated CpG island recovery assay; MIRA) (109). The enriched methylated sequences can be used as templates either for locus‐specific amplification or for genome‐wide methylation analysis using microarrays. MeDIP and MIRA allow for the rapid identification of multiple CpG sites in the genomic DNA of interest. However, they depend on the availability of specific and efficiently working antibodies and methyl‐CpG‐binding domain proteins, and, like the other microarray‐based, genome‐wide DNA methylation detection methods, the genome coverage is still limited because the entire human genome is not represented on the microarrays. This problem is circumvented by the use of high‐throughput genomic sequencing approaches. On next‐generation sequencing platforms, bisulfite converted DNA and methylated DNA enriched by using methylation‐sensitive restriction enzymes, as well as affinity binding of methylated sequences to 5mC antibodies or to methylated DNA binding proteins can be used to assess millions of DNA fragments, thus allowing for detection of DNA methylation across the entire genome, including interspersed repeat sequences that are inaccessible when using microarrays (42).
Methods to distinguish 5mC from 5‐hydroxymethylcytosine
The discovery of 5‐hydroxymethylcytosine, which is highly abundant in the human brain (70), raised the question of how to specifically identify 5‐hydroxymethylcytosine and how to distinguish it from 5mC. Techniques based on bisulfite conversion of DNA are incapable of distinguishing 5mC from 5‐hydroxymethylcytosine as both bases are not altered by sodium bisulfite and are recognized as “cytosine” in downstream detection methods. Methylation‐sensitive restriction enzymes are also strongly inhibited by 5‐hydroxymethylcytosine, while techniques based on immunoprecipitation with antibodies against methylcytosine or methyl‐CpG‐binding domain proteins are specific for 5mC and do not detect 5‐hydroxymethylcytosine (63). Recently, specific antibodies against 5‐hydroxymethylcytosine have become available as well as assays that enable the relative quantitation of 5mC and 5‐hydroxymethylcytosine by the addition of glucose to the hydroxyl group of 5‐hydroxymethylcytosine via an enzymatic reaction utilizing T4 β‐glycosyltransferase, thereby converting a cleavable MspI site to a noncleavable site.
Detection of histone modifications
The gold standard for the detection of post‐translational modifications of histone proteins is mass spectrometry (34). It is the most accurate method for identifying histone modifications but requires a high degree of technical expertise and is difficult to apply genome‐wide. More commonly, the chromatin immunoprecipitation assay (ChIP‐assay) (90) is used for linking histone modifications to particular DNA sequences. Chromatin immunoprecipitation is performed with antibodies against specific histone modifications, for example, trimethylation of histone H3 at lysine 4 (H3K4me3), a mark associated with active chromatin (19). The immunoprecipitated DNA is then typically analyzed by PCR with specific primers to investigate a candidate gene region of interest. To identify histone modifications at a genome‐wide level, the immunoprecipitated DNA can be hybridized to microarrays (ChIP‐on‐chip) (113) or can be applied to massively parallel sequencing techniques (ChIP‐seq) (10).
FREQUENT ALTERATIONS OF CpG METHYLATION AND HISTONE MODIFICATION IN CANCER CELLS
Cancer cells often show a variety of epigenetic aberrations including altered DNA methylation, as well as changes in histone methylation and acetylation (117) (summarized in Figure 2). Interestingly, such epigenetic changes may already be present in early tumor stages before characteristic mutations of tumor suppressor genes or proto‐oncogenes are detectable 39, 43. In general, malignant tumor cells typically demonstrate genome‐wide hypomethylation, site‐specific CpG island hypermethylation (117), as well as differential methylation of CpG island shores (59). DNA hypomethylation of repetitive elements, retrotransposons and introns has been linked to increased genomic instability (32), whereas focal hypermethylation of CpG island in promoter regions may cause transcriptional silencing of tumor suppressor genes and other genes (117). Promoter hypomethylation, on the other hand, may activate the transcription of proto‐oncogenes (140). Moreover, the histone code, that is, the pattern of post‐translational modifications of core histones, is commonly altered in cancer cells. For instance, levels of H4K20me3 and H4K16ac are often reduced at hypomethylated DNA repetitive sequences in cancer (43).
Figure 2.
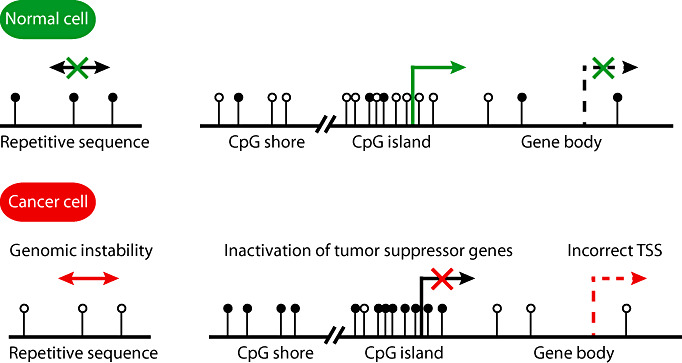
Common epigenetic alterations in cancer. Epigenetic changes are observed in many cancer types. Hypermethylation of gene promoters decreases the expression of proteins or non‐coding RNAs that have antiproliferative or proapoptotic effects, or mediate DNA repair, or inhibit invasion or angiogenesis. In some cases, hypomethylation of the promoters of proto‐oncogenes was demonstrated (not depicted). In certain cancers, for example, colorectal carcinomas, differential methylation was found mainly in so‐called CpG island shores that reside upstream of promoter‐associated CpG islands. As an example, CpG island shore hypermethylation is depicted resulting in reduced transcription. Hypomethylation of the gene body may unblock alternative transcription start sites. Moreover, hypomethylation of repetitive sequences is associated with genomic instability. The cancer‐associated changes in DNA methylation pattern are accompanied by further “epimutations”, such as histone modifications that alter the chromatin structure (not depicted). Methyl groups are symbolized by black circles; green and red crosses symbolize the block of gene expression or transposition; TSS: transcriptional start site.
EPIGENETIC ALTERATIONS IN GLIOMA—TOWARD UNRAVELING THE GLIOMA EPIGENOME
Glioma‐associated histone modifications
Several studies have provided evidence for a deregulation of genes encoding proteins involved in the regulation of histone modifications in gliomas. In particular, large‐scale sequencing of glioblastoma samples detected several mutations in genes encoding HDAC (HDAC2, HDAC9), histone demethylases (JMJD1A JMJD1B), histone methyltransferases (SET7, SETDB2, MLL, MLL3, MLL4) and the methyl‐CpG‐binding domain protein 1 (MBD1) (106). In medulloblastoma, mutations in the histone‐lysine N‐methyltransferase genes MLL2 or MLL3 were recently identified in 16% of the cases (107). The nuclear receptor SET domain containing protein‐1 (NSD1) gene, which encodes a histone methyltransferase involved in chromatin regulation, has been shown to undergo CpG island methylation and subsequent transcriptional downregulation in neuroblastomas and glioblastomas, thereby demonstrating an interesting crosstalk between different layers of epigenetic regulation, that is, the epigenetic inactivation of NSD1 by DNA methylation leads to reduced methylation of the histone lysine residues H4‐K20 and H3‐K36, respectively (11). Other authors reported that transcript levels of various histone deacetylases (HDAC5‐11) are decreased in glioblastoma when compared with low‐grade astrocytoma and normal brain tissue, a finding that was associated with higher levels of histone H3 acetylation in glioblastoma (84). Immunohistochemical analysis of glioma tissue microarrays showed strong nuclear expression of HDAC1, HDAC2 and the nuclear receptor corepressor 2 (NCOR2) in glioma cells, whereas NCOR1 and HDAC3 were sparsely detected in the cytoplasm and nuclei of tumor cells (18). HDAC3 expression inversely correlated with tumor grade and increased HDAC3 expression was a marker of better prognosis. On the other hand, expression of HDAC1 and HDAC2 increased during tumor recurrence and progression (18). Another immunohistochemical study investigated the expression of various histone H3 and H4 modifications in tumor tissue samples of 230 glioma patients and identified prognostically distinct patient subgroups based on the expression of different sets of histone modifications (80).
In addition to these global aberrations at the histone level, individual histone modifications and their downstream effects in gliomas have been studied. For example, the candidate tumor suppressor gene RRP22 was shown to be transcriptionally repressed in gliomas by 5′‐CpG island hypermethylation and/or heterochromatinization as evidenced by increased levels of H3K9me3 and decreased pan‐Ac‐H3 bound to this gene (119). HDAC4 was reported to suppress expression of the cell cycle regulator p21waf1/Cip1 in cancer cells by reducing histone H3 acetylation at the Sp1/Sp3‐binding site‐rich proximal promoter of the CDKN1A gene (94). Silencing of HDAC4 induced p21WAF1/Cip1 and reduced tumor growth of glioblastoma cells, suggesting HDAC4 inhibition as a possible therapeutic target (94). Other authors identified a mechanism by which the oncogenic PI3K‐AKT pathway, which is frequently altered in malignant gliomas, upregulates the expression of HOXA9 through histone modifications, which could possibly be initiated by AKT‐induced EZH2 histone methyltransferase activity (24). HOXA9 is a pro‐proliferative, antiapoptotic transcription factor and associated with poor prognosis in gliomas (24). Despite these interesting studies, current knowledge on the functional roles of histone modifications in gliomas is still limited, and the relevant set of genes epigenetically regulated by post‐translational histone changes remains to be characterized. However, a growing number of studies provides evidence that drugs resulting in global modifications of the histone code, in particular HDAC inhibitors, may have therapeutic effects on glioma growth by reducing proliferation and inducing differentiation (see later discussion).
Alterations of DNA methylation in gliomas
Global DNA hypomethylation and focal hypermethylation of CpG‐rich promoter‐associated sequences are common findings in malignant gliomas (86). Hypomethylation has been detected at Sat2 pericentromeric DNA, the subtelomeric repeat sequence D4Z4 and interspersed Alu elements (17). Moreover, hypomethylation was associated with copy number changes of the adjacent euchromatin and induced the reactivation of the cancer‐testis antigen MAGEA1, indicating that global hypomethylation may promote glioma growth by increasing genomic instability and the disinhibition of proto‐oncogenes (17). Interestingly, global DNA hypomethylation, as evidenced by reduced levels of LINE‐1 methylation, has been associated with less favorable outcome in glioma patients (103). Another recent study found that overexpression of DNMT1 and DNMT3B in glioma was mediated by histone modification and promoter hypomethylation, respectively, and that inhibition of these DNA‐methyltransferases resulted in re‐expression of tumor suppressor genes (108).
The list of genes reported as being epigenetically silenced in gliomas by aberrant promoter methylation is steadily growing and includes genes involved in a variety of cellular processes linked to glioma development and progression. For example, focused methylation analyses of candidate genes in primary glioma tissues revealed promoter methylation of cell cycle regulatory genes, for example, RB1 (98), CDKN2A/p16INK4a, p14ARF and CDKN2B/p15INK4b 25, 97, 146; apoptosis‐regulating genes, for example, PYCARD (121); DNA repair genes, for example, CHK2 (139) and MGMT (see later discussion); migration and invasion related genes, for example, TIMP3 (35), CDH1 (30), PCDH‐gamma‐A11 (137) and SOCS3 (79), as well as various established or putative tumor suppressor genes, such as PTEN (6), RASSF1A (56), DIRAS3 (114), EMP3 (1), CITED4 (127), BLU (83), NDRG2 (126), CTMP (68), ECRG4 (48) and different Wnt pathway regulatory genes (49). Accelerated by recent large‐scale DNA methylation analyses, many more genes have been identified as being aberrantly methylated in gliomas 74, 101, with certain types of gliomas showing concurrent hypermethylation of multiple genes. This phenomenon has been referred to as glioma‐CpG island methylator phenotype (G‐CIMP) (101), analogous to the CIMP observed in certain epithelial cancers, in particular colorectal carcinomas (60). However, promoter hypermethylation was found to be inconsistently associated with transcriptional downregulation 74, 101, and the individual roles of the hypermethylated genes in promoting glioma growth remain to be clarified.
Patterns and frequency of promoter hypermethylation depend on glioma type and grade
Evidence is increasing that not only the patterns of genetic aberrations but also the epigenetic profiles in gliomas differ between different tumor types and malignancy grades. For example, secondary glioblastomas were shown to have a higher frequency of promoter methylation of p14ARF, p16INK4a (97), RB1 (98), MGMT (96) , TIMP3 (99), EMP3 (73), and the Wnt inhibitor DKK1 (49) when compared with primary glioblastomas. On the other hand, promoter hypermethylation and transcriptional downregulation of NDRG2 (126), SFRP1, SFRP2, and NKD2 (49) is detectable in at least 40% of primary glioblastomas but only in low fractions of secondary glioblastomas.
Oligodendrogliomas often show aberrant promoter methylation of multiple genes 2, 146, which has recently been found to correspond to the G‐CIMP phenomenon (101). In fact, many of the genes found to be commonly hypermethylated in oligodendroglial tumors are also hypermethylated in diffuse astrocytic gliomas and secondary glioblastomas but not in primary glioblastomas. Thus, gliomas may be stratified into two major groups according to their epigenomic profiles, that is, one group consisting mostly of diffuse gliomas and secondary glioblastomas with concurrent hypermethylation of multiple gene promoters, which largely overlaps with the group of IDH1 mutant gliomas, and another group consisting mostly of primary glioblastomas with global DNA hypomethylation and less frequent promoter hypermethylation, which largely overlaps with IDH1 wild‐type gliomas 74, 101. Within the group of diffuse gliomas with multiple hypermethylated genes, tumors with or without 1p/19q deletion have been reported to display evidence for distinct methylation profiles (74). Similarly, a study investigating the methylation status at CpG islands associated with 15 distinct gene loci in 139 gliomas demonstrated characteristic methylation profiles in different glioma subtypes, with pilocytic astrocytomas rarely showing hypermethylation at the investigated loci (131). Interestingly, global methylation patterns of individual gliomas seem to remain strikingly stable upon tumor progression and recurrence 74, 101. This fact may indicate that altered DNA methylation is usually an early event in glioma development. In line with these findings, the MGMT promoter methylation status remains stable between primary and recurrent tumors in the vast majority of glioblastoma patients (40).
A mechanistic link between IDH1 mutation and the altered epigenome of gliomas
Noushmehr and colleagues performed an unsupervised hierarchical clustering of the glioblastoma samples included in The Cancer Genome Atlas project based upon methylation profiles (101). They found a small cluster of glioblastomas (8.8%) that was characterized by concerted hypermethylation of many gene loci (G‐CIMP). Further analyses revealed that G‐CIMP was common in secondary and recurrent glioblastomas but also in diffuse gliomas, in particular oligodendroglial tumors (53/56 cases). G‐CIMP was associated with proneural gene expression profiles (135) in glioblastomas and with prolonged survival (101). Among the differentially hypermethylated genes within the proneural G‐CIMP‐positive tumors, the expression of several genes, for example, FABP5, PDPN, CHI3L1 and LGALS3, was also downregulated (101), while high expression of these four genes was associated with poor prognosis (22). It has been speculated that patients with G‐CIMP gliomas may benefit from therapeutic modification of DNA methylation (20).
Interestingly, G‐CIMP was strongly associated with mutation of the IDH1 gene, which represents an early and common event in the pathogenesis of diffuse astrocytic and oligodendroglial tumors, as well as secondary glioblastomas, but are rare in primary glioblastomas and all other types of brain tumors 8, 106, 115, 150. In the series of Noushmehr et al, one of four secondary glioblastomas was G‐CIMP negative. This particular tumor did not carry an IDH1 mutation. Among the investigated primary (de novo) glioblastomas, the majority of tumors lacked IDH1 mutations and were G‐CIMP negative (101). Two independent studies confirmed that a hypermethylation phenotype was common in diffuse gliomas, including secondary glioblastomas, and tightly associated with IDH1 mutation 20, 74.
Recent studies provided a mechanistic explanation for the striking association between mutation of IDH1 or IDH2, that is, enzymes that take part in the tricarboxylic acid cycle, and the observed large‐scale epigenetic changes in glioma and acute myeloid leukemia cells (Figure 3). The NADP+‐dependent isocitrate dehydrogenase 1 normally catalyzes the oxidative decarboxylation of isocitrate to α‐KG (synonym: 2‐oxoglutarate) (111). IDH1 mutations, however, reduce IDH1 activity as compared with the wild‐type enzyme, which results in reduced α‐KG levels (153). Moreover, the mutant IDH1 enzyme gains a neomorphic enzymatic activity and catalyses the NADPH‐dependent reduction of α‐KG to R(‐)‐2‐hydroxyglutarate (2‐HG). Therefore, IDH1 mutant tumors, including gliomas and acute myeloic leukemias, demonstrate elevated levels of the neometabolite 2‐HG 27, 41. In addition, IDH1 and IDH2 mutations decrease NADPH production (150), probably predisposing cells to oxidative stress (111). Profiling of more than 200 metabolites revealed altered levels of amino acids, glutathione metabolites, choline derivatives and tricarboxylic acid cycle intermediates in cells expressing mutant IDH1 (112), which demonstrates an extensive impact of IDH1 mutations on the cellular metabolome.
Figure 3.
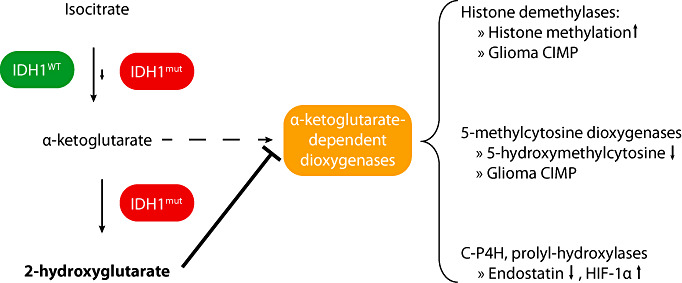
Impact of IDH1 mutation on epigenetic control. Isocitrate dehydrogenase 1 (IDH1) catalyzes the oxidative decarboxylation of isocitrate to α‐ketoglutarate (α‐KG), which is coupled to the reduction of NADP+ to NADPH + H+. IDH1 (eg, R132H mutation) results in a neomorphic IDH1 activity that catalyzes the NADPH consuming reduction of α‐KG to 2‐hydroxyglutarate (2‐HG). Thereby, α‐KG levels are reduced, and 2‐HG levels are strongly increased in IDH1 mutant tumor cells. 2‐HG is a competitive inhibitor of a large number of α‐KG dependent dioxygenases, including histone demethylases and 5‐methylcytosine hydroxylases of the TET protein family. Thereby, IDH1 mutation leads to altered methylation at different histone residues and decreased levels of 5‐hydroxymethylcytosine. Together, both epigenetic alterations are supposed to result in widespread changes in DNA methylation and the histone code, namely hypermethylation of multiple promoter‐associated CpG islands (G‐CIMP) and more compact chromatin. In addition, 2‐HG produced by mutant IDH1 also inhibits collagen‐prolyl‐4‐hydroxylase (C‐P4H) and prolyl‐hydroxylases, which may lead to tumor promotion by stabilization of HIF‐1α.
Importantly, recent studies revealed that 2‐HG competitively inhibits the activity of various α‐KG‐dependent dioxygenases, including histone demethylases and the TET family of 5mC hydroxylases 41, 149. Xu and collaborators showed that ectopic expression of IDH1 R132H in U‐87 cells decreased α‐KG levels by 60% and increased 2‐HG levels more than 20‐fold resulting in an increase of different histone methylations (H3K4me, H3K27me2, H3K4me3, H3K9me3, H3K79me2). Other authors also observed altered histone methylation in gliomas with IDH1 mutation (20). Moreover, 2‐hydroxyglutarate was shown to inhibit the activity of 5mC hydroxylases of the TET family, in particular TET2, and thereby lower the levels of 5‐hydroxymethylcytosine in IDH1 mutant leukemias and gliomas 41, 149. In hematopoietic malignancies, TET2 mutations were mutually exclusive with IDH1 mutations and similarly linked to a DNA hypermethylation phenotype, which fueled the hypothesis that reduced DNA demethylation because lowered 5‐hydroxymethylcytosine levels eventually results in hypermethylation of multiple CpG islands (41). In low‐grade gliomas lacking IDH1 and IDH2 mutation, only a small proportion of tumors showed TET2 promoter hypermethylation, while no TET2 mutations were detected (67). Taken together, the finding that 2‐HG, which is aberrantly produced by mutant IDH1 and IDH2 proteins, inhibits α‐KG‐dependent enzymes involved in the regulation of DNA, and histone methylation provides a mechanistic link between mutations in either of these genes, altered glioma cell metabolism and widespread epigenetic changes, in particular, aberrant methylation of multiple genes and global histone modifications.
Are there genetic changes other than IDH1 mutation that may alter the glioma epigenome?
Several recent studies have identified mutations in different genes directly involved in the regulation of DNA methylation and chromatin remodeling. In myeloid malignancies, for example, somatic mutations in different epigenetic regulators have been identified, including the de novo methyltransferase gene DNMT3A, the 5mC hydroxylase gene TET2, as well as the polycomb protein and histone methylase gene EZH2 in subsets of acute and chronic myeloid leukemias [for review, see (38)]. Interestingly, these mutations appear to occur mutually exclusive to each other and to mutations in IDH1 or IDH2.
Additional potential targets for cancer‐associated mutations leading to widespread epigenetic changes include adenosine triphosphate (ATP)‐dependent chromatin remodeling enzymes 69, 145, histone variants (125), histone chaperones (5) and histone readers (152). In fact, recent large‐scale sequencing studies have identified frequent mutations in various chromatin remodeling genes in different types of cancer. For example, mutations in UTX, MLL‐MLL3, CREBBP‐EP300, NCOR1, ARID1A and CHD6 have been detected in the majority of transitional cell carcinomas of the bladder (50). In renal cell carcinomas, 41% of the cases carried mutations in the switch/sucrose non‐fermentable (SWI/SNF) chromatin remodeling complex gene PBRM1, with mutations in the histone demethylase genes UTX and JARID1C or the histone methylase SETD2 being each restricted to smaller subsets of cases (134). Among non‐Hodgkin lymphomas, 32% of diffuse large B‐cell lymphomas and 89% of follicular lymphomas were found to carry somatic mutations in the histone methyltransferase gene MLL2, while 10%–15% of these lymphomas had mutations in MEF2B, a calcium‐regulated gene involved in histone acetylation (93). Moreover, somatic mutations in the chromatin remodeling enzyme genes ATRX and DAXX are frequent in pancreatic neuroendocrine tumors and have been associated with altered telomeres and increased genomic instability (62). The histone variant macroH2A suppresses melanoma progression in vitro and in vivo (65). ASF1B was identified as a histone chaperone isoform that is nescessary for proliferation and whose mRNA overexpression is associated with outcome in breast cancer patients (23). Aberrant expression of the histone reader TRIM24, which binds unmethylated H3K4, H3K23ac and estrogen receptor to induce estrogen‐dependent genes, also correlates with the survival of breast cancer patients (130).
Among pediatric malignant brain tumors, mutations in the ATP‐dependent SWI/SNF chromatin‐remodeling complex gene SMARCB1 (INI1) and, less commonly, SMARCA4 (BRG1) are characteristic features of atypical teratoid rhabdoid tumors (51). In addition, pediatric medulloblastomas carry inactivating mutations in the histone methyltransferase genes MLL2 or MLL3 in 16% of the cases (107). In contrast, little is known about alterations of any of these genes in gliomas to date. Nevertheless, as these alterations appear to be so common in various types of cancers, it may be possible that genetic mutations or epigenetic changes of any of these genes, or in yet other epigenetic regulators, are involved in glioma pathogenesis, for example, in those gliomas lacking IDH1 or IDH2 mutation but demonstrating widespread epigenomic alterations.
CLINICAL RELEVANCE OF EPIGENETIC MARKERS IN GLIOMAS
The paradigm of MGMT promoter methylation in malignant gliomas
The impressive progress in unraveling epigenetic alterations in cancer has not only greatly advanced our understanding of tumor pathogenesis but has already achieved clinical relevance by providing valuable molecular biomarkers, as well as promising novel therapeutic targets. In particular, the promoter methylation status of the MGMT gene has become an important prognostic and predictive biomarker in the diagnostic assessment of patients with high‐grade malignant gliomas [for recent reviews see 116, 142]. Based on a subpopulation of glioblastoma patients from the EORTC/NCIC 22981/26981 trial (122), Hegi and colleagues found a significantly better survival of patients treated with radiotherapy combined with concurrent and adjuvant temozolomide when their tumors demonstrated a hypermethylated MGMT promoter (54). In contrast, patients with MGMT‐unmethylated glioblastomas did not appear to derive a significant survival benefit from the addition of temozolomide to radiotherapy, suggesting that the MGMT promoter methylation status serves as a predictive marker for response to alkylating chemotherapy in glioblastoma patients. Mechanistically, the predictive function of MGMT promoter methylation is explained by the fact that MGMT functions as a repair enzyme that counteracts the DNA damage induced by alkylating chemotherapeutic agents. These drugs cause an alkylation on the O6 position of guanine that conveys most of the cytotoxic effects (46). Tumor cells with high MGMT protein levels can repair this therapy‐induced damage and, thereby, poorly respond to the treatment. In turn, MGMT promoter methylation, as seen in about 40% of primary glioblastomas, leads to low or absent MGMT expression and, thereby, inability to counteract the therapy‐induced DNA damage and consequently better therapy response.
In anaplastic gliomas, MGMT promoter methylation has been linked to more favorable outcome not only in patients treated with temozolomide but also in patients treated with radiotherapy only 133, 144. One potential explanation for this surprising observation may again be related to the frequent presence of G‐CIMP in anaplastic gliomas. In addition to MGMT promoter methylation, these tumors may in fact carry epigenetically silenced genes whose gene products normally confer radiotherapy resistance. Further studies addressing this hypothesis by using genome‐wide methylation analyses are needed to identify and characterize such candidate genes.
Diagnostic testing for MGMT promoter methylation has already become a standard in clinical trials involving patients with high‐grade gliomas. In fact, several trials are ongoing that specifically recruit only patients with either MGMT promoter methylated or unmethylated glioblastomas. Concerning the routine diagnostic setting, however, MGMT testing has not yet become a standard of care, mainly because of the still limited therapeutic alternatives that can be offered to glioblastoma patients. Nevertheless, it is foreseeable that clinical MGMT testing will gain increasing importance also in the routine diagnostic assessment of malignant glioma patients.
Various methods are being used to detect MGMT promoter methylation in clinical specimens, including a number of the techniques discussed above, such as MSP analysis 54, 55, methylation‐specific pyrosequencing (91), the COBRA method (91), as well as techniques that do not require bisulfite conversion of DNA, such as methylation‐quantification of endonuclease‐resistant DNA (Methyl‐QESD) (14) and MS‐MLPA (61). The use of various methods and thresholds for the detection of MGMT promoter methylation, as well as the testings of variable numbers and locations of CpG sites within the promoter region, may give rise to a substantial degree of interlaboratory variation in testing results. Thus, centralized MGMT methylation testing is required in clinical trials. Moreover, application of MGMT promoter methylation testing in the routine diagnostic setting should be subjected to standardization, for example, by the definition of standard operating procedures and the implementation of interlaboratory quality control measures.
Role of other epigenetic markers in gliomas
A number of studies have reported on individual genes whose epigenetic silencing by promoter methylation was found to correlate with clinical outcome of glioma patients. Moreover, recent data even suggest the global DNA methylation status determined by LINE1 methylation analysis as a powerful prognostic marker (103). However, none of these molecular alterations has as yet been promoted to the level of a clinically relevant marker that would be comparable with the MGMT status. In fact, many of the other reported prognostically relevant genes are preferentially hypermethylated in 1p/19q‐deleted and/or IDH1 mutant gliomas, thus, are likely members of the large group of genes concurrently hypermethylated in G‐CIMP‐positive gliomas (see above). In these tumors, the prognostic impact of each individual hypermethylated gene is difficult to assess. Moreover, it is not entirely clear whether the clinically less aggressive course of IDH1 mutant and G‐CIMP‐positive gliomas is primarily because of the widespread epigenomic alterations or possibly related to other, yet to be characterized metabolic effect of the IDH1 mutation. Interestingly, both oligodendroglial and diffuse astrocytic gliomas share frequent IDH1 mutation, while the clinical outcome in anaplastic glioma patients appears to be significantly better in patients with oligodendroglial tumors (144). This suggests that genetic and/or epigenetic aberrations in addition to IDH1 mutation/G‐CIMP are of prognostic relevance in diffuse gliomas, for example, the 1p/19q status [for review, see (115)]. In this respect, the functional and prognostic roles of the recently identified candidate genes on 1p and 19q that are frequently mutated in oligodendrogliomas, namely FUBP1 and CIC (13), remain to be investigated, in particular as to how they may synergize with IDH1 mutation and G‐CIMP in oligodendrogliomas.
Interestingly, glioma‐associated epigenetic DNA modifications can not only be demonstrated in resected tumor tissue specimens but may also be detectable in tumor DNA circulating in body fluids, such as serum and cerebrospinal fluid (CSF). Several studies reported on the diagnostic utility of detecting promoter methylation of various genes, including MGMT, CDKN2A, THBS1, TIMP3 and RASFF1 in these body fluids 7, 75, 81. These studies found a high concordance between methylation of these genes detected in serum or CSF in relation to the corresponding tumor tissue, suggesting a potential role of serum‐based DNA methylation profiling in the diagnostic and prognostic assessment of glioma patients.
CONCLUSIONS AND PERSPECTIVES
During the last decades, a large number of epigenetic changes have been discovered in gliomas that contribute to the inactivation of tumor suppressor genes and increase genomic instability of these tumors. With MGMT promoter methylation, one “epimutation” has reached clinical significance as a molecular biomarker of predictive and prognostic relevance in malignant gliomas. Moreover, the identification of a mechanistic link between IDH1 or IDH2 mutation and widespread epigenetic alterations manifesting as G‐CIMP via the inhibition of α‐KG‐dependent epigenetic regulators represents a major step forward in the understanding of glioma pathogenesis. The recent technical advancements concerning genome‐wide sequencing and large‐scale epigenetic profiling methods will certainly provide important new insights into the complex interplay between genetic and epigenetic alterations in these tumors, thereby leading to a deeper understanding of the genomes and epigenomes of various glioma subtypes and malignancy grades.
Perspectives concerning “epigenetic therapy” of gliomas
More detailed knowledge of glioma‐associated mutations and epimutations will also be necessary to further characterize those genes and pathways that essentially drive glioma growth and thus represent promising novel targets for pathogenesis‐based therapeutic approaches. In contrast to genetic alterations, pharmacological repair of epigenetic changes may be easier to accomplish, although targeted reversal of specific epimutations without globally changing the epigenetic landscape is still challenging. To date, “epigenetic therapies” mainly consist in a rather broadly acting inhibition of the epigenetic machinery, in particular, inhibition of DNA methyltransferases and HDACs. However, the arsenal of epigenetic drugs is constantly growing, and the issue of more precisely modulating distinct epigenetic players is intensively being investigated [for review, see 16, 117]. Concerning glioma treatment, preclinical data suggested efficacy of HDAC inhibition in experimental glioma models when given as single therapy 36, 151 or in combination with radiotherapy (33). Preliminary clinical results suggested low toxicity and moderate activity of treatment with HDAC inhibitors in pediatric malignant glioma patients (88), as well as adult patients with recurrent glioblastoma (45). In addition, prolonged survival has been noted in a subset of glioblastoma patients of the EORTC/NCIC trial who had been treated with valproic acid, an antiepileptic drug that has HDAC inhibitory activity (143). Several other trials evaluating HDAC inhibitors in the treatment of malignant glioma patients are ongoing to evaluate potential synergistic effects in combination with radiotherapy and various types of chemotherapy (see http://www.clinicaltrials.gov/).
Another aspect of potential clinical relevance concerns the effects of cytotoxic therapy, in particular, alkylating chemotherapy, on the glioma epigenome and its potential role in acquired therapy resistance. While the MGMT promoter methylation status usually remains stable between primary and recurrent glioblastomas after radiochemotherapy (40), experimental studies revealed that temozolomide and carmustine may cause large‐scale heterochromatin reorganization in glioma cells by decreasing global levels of histone H3 acetylation and increasing levels of histone H3 trimethylated on lysine 9 (105). In fact, these epigenetic effects of DNA alkylators may be counteracted by concurrent treatment with HDAC inhibitors, thereby possibly providing one explanation for a synergistic role of both treatment approaches.
Glioma epigenetics—role of noncoding RNAs
Finally, it has to be acknowledged that an additional level of complexity in cancer epigenetics is emerging by recent insights into the role of noncoding RNAs as modifiers of DNA methylation and histone modification. This includes a variety of distinct types of noncoding RNAs, as well as different molecular mechanisms, such as double‐stranded RNA (dsRNA)‐mediated activation of gene expression associated with histone demethylation, endogenous antisense RNA‐mediated CpG demethylation, putatively miRNA‐mediated paramutation, piwi‐RNA‐mediated de novo DNA methylation of transposons, endogenous antisense RNA‐mediated imprinting, long noncoding RNA‐ and xiRNA‐mediated X inactivation, and other mechanisms [for review, see (154)]. Interestingly, mammalian microRNAs (miRNAs) that usually regulate gene expression post‐transcriptionally by binding to target mRNAs in their 3′‐UTR also may directly affect transcriptional gene silencing. For example, miR‐320a is encoded on the minus strand within the promoter region of POLR3D. By inducing the association of AGO1, the polycomb group member EZH2, and H3K27me3 with the POLR3D promoter, miR‐320a is able to directly repress the transcription of POLR3D (66). Moreover, miRNAs may change the epigenome of tumor cells by inhibiting DNA methyltransferases and other epigenetic regulators, as exemplified by the function of the miR‐29 family in the post‐transcriptional regulation of DNMT3A expression. Overexpression of miR‐29 in lung cancer cell lines restored normal patterns of DNA methylation, induced re‐expression of methylation‐silenced tumor suppressor genes, and inhibited tumorigenicity (37). In gliomas, miR‐128 has been shown to regulate the chromatin‐modifying protein Bmi‐1, thereby increasing H3K27 trimethylation in addition to Akt phosphorylation and upregulation of p21waf1 (47). On the other hand, expression of many miRNAs appears to be epigenetically modified by aberrant DNA methylation in cancer cells. A recent meta‐analysis revealed a total of 122 miRNAs that were reported to be epigenetically regulated in 23 different cancer types (72). Hypermethylated miRNAs often are located in or have associated 5′‐CpG islands and appear to cluster in certain genomic regions mapping to chromosome arms 1q, 7q, 11q, 14q and 19q, respectively (72).
Taken together, there is still a long way to go to unravel the glioma epigenome in its entire complexity. However, the tools are available to comprehensively characterize the various levels of epigenetic aberrations in these tumors, and novel discoveries are to be expected in the near future in this rapidly advancing field of molecular neuro‐oncology. These will hopefully result in the identification of additional clinically useful biomarkers and promising novel targets for innovative epigenetic therapy approaches.
REFERENCES
- 1. Alaminos M, Dávalos V, Ropero S, Setién F, Paz MF, Herranz M et al (2005) EMP3, a myelin‐related gene located in the critical 19q13.3 region, is epigenetically silenced and exhibits features of a candidate tumor suppressor in glioma and neuroblastoma. Cancer Res 65:2565–2571. [DOI] [PubMed] [Google Scholar]
- 2. Alonso ME, Bello MJ, Gonzalez‐Gomez P, Arjona D, Lomas J, de Campos JM et al (2003) Aberrant promoter methylation of multiple genes in oligodendrogliomas and ependymomas. Cancer Genet Cytogenet 144:134–142. [DOI] [PubMed] [Google Scholar]
- 3. Andrews AJ, Luger K (2011) Nucleosome structure(s) and stability: variations on a theme. Annu Rev Biophys 40:99–117. [DOI] [PubMed] [Google Scholar]
- 4. Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF et al (2008) A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31:785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avvakumov N, Nourani A, Côté J (2011) Histone chaperones: modulators of chromatin marks. Mol Cell 41:502–514. [DOI] [PubMed] [Google Scholar]
- 6. Baeza N, Weller M, Yonekawa Y, Kleihues P, Ohgaki H (2003) PTEN methylation and expression in glioblastomas. Acta Neuropathol 106:479–485. [DOI] [PubMed] [Google Scholar]
- 7. Balaña C, Ramirez JL, Taron M, Roussos Y, Ariza A, Ballester R et al (2003) O6‐methyl‐guanine‐DNA methyltransferase methylation in serum and tumor DNA predicts response to 1,3‐bis(2‐chloroethyl)‐1‐nitrosourea but not to temozolamide plus cisplatin in glioblastoma multiforme. Clin Cancer Res 9:1461–1468. [PubMed] [Google Scholar]
- 8. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602. [DOI] [PubMed] [Google Scholar]
- 9. Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21:381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barski A, Cuddapah S, Cui K, Roh T, Schones DE, Wang Z et al (2007) High‐resolution profiling of histone methylations in the human genome. Cell 129:823–837. [DOI] [PubMed] [Google Scholar]
- 11. Berdasco M, Ropero S, Setien F, Fraga MF, Lapunzina P, Losson R et al (2009) Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc Natl Acad Sci USA 106:21830–21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A (2009) An operational definition of epigenetics. Genes Dev 23:781–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH et al (2011) Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 333:1453–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bettstetter M, Dechant S, Ruemmele P, Vogel C, Kurz K, Morak M et al (2008) MethyQESD, a robust and fast method for quantitative methylation analyses in HNPCC diagnostics using formalin‐fixed and paraffin‐embedded tissue samples. Lab Invest 88:1367–1375. [DOI] [PubMed] [Google Scholar]
- 15. Blasco MA (2007) The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8:299–309. [DOI] [PubMed] [Google Scholar]
- 16. Boumber Y, Issa JJ (2011) Epigenetics in cancer: what's the future? Oncology (Williston Park) 25:220–226. 228. [PubMed] [Google Scholar]
- 17. Cadieux B, Ching T, Vandenberg SR, Costello JF (2006) Genome‐wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res 66:8469–8476. [DOI] [PubMed] [Google Scholar]
- 18. Campos B, Bermejo JL, Han L, Felsberg J, Ahmadi R, Grabe N et al (2011) Expression of nuclear receptor corepressors and class I histone deacetylases in astrocytic gliomas. Cancer Sci 102:387–392. [DOI] [PubMed] [Google Scholar]
- 19. Chen Z, Riggs AD (2011) DNA methylation and demethylation in mammals. J Biol Chem 286:18347–18353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Christensen BC, Smith AA, Zheng S, Koestler DC, Houseman EA, Marsit CJ et al (2011) DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J Natl Cancer Inst 103:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Colella S, Shen L, Baggerly KA, Issa JP, Krahe R (2003) Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. BioTechniques 35:146–150. [DOI] [PubMed] [Google Scholar]
- 22. Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, Rivera A et al (2010) A multigene predictor of outcome in glioblastoma. Neuro-Oncol 12:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Corpet A, Koning L, Toedling J, Savignoni A, Berger F, Lemaître C et al (2011) Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J 30:480–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Costa BM, Smith JS, Chen Y, Chen J, Phillips HS, Aldape KD et al (2010) Reversing HOXA9 oncogene activation by PI3K inhibition: epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res 70:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Costello JF, Berger MS, Huang HS, Cavenee WK (1996) Silencing of p16/CDKN2 expression in human gliomas by methylation and chromatin condensation. Cancer Res 56:2405–2410. [PubMed] [Google Scholar]
- 26. Dahl C, Grønbæk K, Guldberg P (2011) Advances in DNA methylation: 5‐hydroxymethylcytosine revisited. Clin Chim Acta 412:831–836. [DOI] [PubMed] [Google Scholar]
- 27. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al (2009) Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature 462:739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doi A, Park I, Wen B, Murakami P, Aryee MJ, Irizarry R et al (2009) Differential methylation of tissue‐ and cancer‐specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 41:1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J et al (2005) Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121:873–885. [DOI] [PubMed] [Google Scholar]
- 30. D'Urso PI, D'Urso OF, Storelli C, Catapano G, Gianfreda CD, Montinaro A et al (2010) Retrospective protein expression and epigenetic inactivation studies of CDH1 in patients affected by low‐grade glioma. J Neurooncol 104:113–118. [DOI] [PubMed] [Google Scholar]
- 31. Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D et al (2000) MethyLight: a high‐throughput assay to measure DNA methylation. Nucleic Acids Res 28:E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eden A, Gaudet F, Waghmare A, Jaenisch R (2003) Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300:455. [DOI] [PubMed] [Google Scholar]
- 33. Entin‐Meer M, Yang X, Vandenberg SR, Lamborn KR, Nudelman A, Rephaeli A et al (2007) In vivo efficacy of a novel histone deacetylase inhibitor in combination with radiation for the treatment of gliomas. Neuro-Oncol 9:82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Esteller M (2007) Cancer epigenomics: DNA methylomes and histone‐modification maps. Nat Rev Genet 8:286–298. [DOI] [PubMed] [Google Scholar]
- 35. Esteller M, Corn PG, Baylin SB, Herman JG (2001) A gene hypermethylation profile of human cancer. Cancer Res 61:3225–3229. [PubMed] [Google Scholar]
- 36. Eyüpoglu IY, Hahnen E, Buslei R, Siebzehnrübl FA, Savaskan NE, Lüders M et al (2005) Suberoylanilide hydroxamic acid (SAHA) has potent anti‐glioma properties in vitro, ex vivo and in vivo . J Neurochem 93:992–999. [DOI] [PubMed] [Google Scholar]
- 37. Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E et al (2007) MicroRNA‐29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 104:15805–15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fathi AT, Abdel‐Wahab O (2012) Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic‐targeted therapy, Advances in Hematology [Epub ahead of print; doi:10.1155/2012/469592]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feinberg AP (2005) Cancer epigenetics is no Mickey Mouse. Cancer Cell 8:267–268. [DOI] [PubMed] [Google Scholar]
- 40. Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M et al (2011) Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer 129:659–670. [DOI] [PubMed] [Google Scholar]
- 41. Figueroa ME, Abdel‐Wahab O, Lu C, Ward PS, Patel J, Shih A et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fouse SD, Nagarajan RP, Costello JF (2010) Genome‐scale DNA methylation analysis. Epigenomics 2:105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fraga MF, Ballestar E, Villar‐Garea A, Boix‐Chornet M, Espada J, Schotta G et al (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37:391–400. [DOI] [PubMed] [Google Scholar]
- 44. Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW et al (1992) A genomic sequencing protocol that yields a positive display of 5‐methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA 89:1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS et al (2009) Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol 27:2052–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gerson SL (2004) MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer 4:296–307. [DOI] [PubMed] [Google Scholar]
- 47. Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G et al (2008) Targeting of the Bmi‐1 oncogene/stem cell renewal factor by microRNA‐128 inhibits glioma proliferation and self‐renewal. Cancer Res 68:9125–9130. [DOI] [PubMed] [Google Scholar]
- 48. Götze S, Feldhaus V, Traska T, Wolter M, Reifenberger G, Tannapfel A et al (2009) ECRG4 is a candidate tumor suppressor gene frequently hypermethylated in colorectal carcinoma and glioma. BMC Cancer 9:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Götze S, Wolter M, Reifenberger G, Müller O, Sievers S (2010) Frequent promoter hypermethylation of Wnt pathway inhibitor genes in malignant astrocytic gliomas. Int J Cancer 126:2584–2593. [DOI] [PubMed] [Google Scholar]
- 50. Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S et al (2011) Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet 43:875–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F et al (2011) Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35:933–935. [DOI] [PubMed] [Google Scholar]
- 52. Hayashizaki Y, Hirotsune S, Okazaki Y, Hatada I, Shibata H, Kawai J et al (1993) Restriction landmark genomic scanning method and its various applications. Electrophoresis 14:251–258. [DOI] [PubMed] [Google Scholar]
- 53. He Y, Li B, Li Z, Liu P, Wang Y, Tang Q et al (2011) Tet‐mediated formation of 5‐carboxylcytosine and its excision by TDG in mammalian DNA. Science 333:1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hegi ME, Diserens A, Gorlia T, Hamou M, de Tribolet N, Weller M et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. [DOI] [PubMed] [Google Scholar]
- 55. Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (1996) Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 93:9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Horiguchi K, Tomizawa Y, Tosaka M, Ishiuchi S, Kurihara H, Mori M et al (2003) Epigenetic inactivation of RASSF1A candidate tumor suppressor gene at 3p21.3 in brain tumors. Oncogene 22:7862–7865. [DOI] [PubMed] [Google Scholar]
- 57. Huang TH, Perry MR, Laux DE (1999) Methylation profiling of CpG islands in human breast cancer cells. Hum Mol Genet 8:459–470. [DOI] [PubMed] [Google Scholar]
- 58. Iguchi‐Ariga SM, Schaffner W (1989) CpG methylation of the cAMP‐responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev 3:612–619. [DOI] [PubMed] [Google Scholar]
- 59. Irizarry RA, Ladd‐Acosta C, Wen B, Wu Z, Montano C, Onyango P et al (2009) The human colon cancer methylome shows similar hypo‐ and hypermethylation at conserved tissue‐specific CpG island shores. Nat Genet 41:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Issa J (2004) CpG island methylator phenotype in cancer. Nat Rev Cancer 4:988–993. [DOI] [PubMed] [Google Scholar]
- 61. Jeuken JWM, Cornelissen SJB, Vriezen M, Dekkers MMG, Errami A, Sijben A et al (2007) MS‐MLPA: an attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab Invest 87:1055–1065. [DOI] [PubMed] [Google Scholar]
- 62. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A et al (2011) DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331:1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jin S, Kadam S, Pfeifer GP (2010) Examination of the specificity of DNA methylation profiling techniques towards 5‐methylcytosine and 5‐hydroxymethylcytosine. Nucleic Acids Res 38:e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kalantry S (2011) Recent advances in X‐chromosome inactivation. J Cell Physiol 226:1714–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO et al (2010) The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468:1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim DH, Saetrom P, Snøve O, Rossi JJ (2008) MicroRNA‐directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci USA 105:16230–16235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kim Y, Pierscianek D, Mittelbronn M, Vital A, Mariani L, Hasselblatt M et al (2011) TET2 promoter methylation in low‐grade diffuse gliomas lacking IDH1/2 mutations. J Clin Pathol 64:850–852. [DOI] [PubMed] [Google Scholar]
- 68. Knobbe CB, Reifenberger J, Blaschke B, Reifenberger G (2004) Hypermethylation and transcriptional downregulation of the carboxyl‐terminal modulator protein gene in glioblastomas. J Natl Cancer Inst 96:483–486. [DOI] [PubMed] [Google Scholar]
- 69. Korber P, Becker PB (2010) Nucleosome dynamics and epigenetic stability. Essays Biochem 48:63–74. [DOI] [PubMed] [Google Scholar]
- 70. Kriaucionis S, Heintz N (2009) The nuclear DNA base 5‐hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324:929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kulis M, Esteller M (2010) DNA methylation and cancer. Adv Genet 70:27–56. [DOI] [PubMed] [Google Scholar]
- 72. Kunej T, Godnic I, Ferdin J, Horvat S, Dovc P, Calin GA (2011) Epigenetic regulation of microRNAs in cancer: an integrated review of literature. Mutat Res [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 73. Kunitz A, Wolter M, van den Boom J, Felsberg J, Tews B, Hahn M et al (2007) DNA hypermethylation and aberrant expression of the EMP3 gene at 19q13.3 in Human Gliomas. Brain Pathol 17:363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Laffaire J, Everhard S, Idbaih A, Crinière E, Marie Y, Reyniès A et al (2011) Methylation profiling identifies 2 groups of gliomas according to their tumorigenesis. Neuro-Oncol 13:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lavon I, Refael M, Zelikovitch B, Shalom E, Siegal T (2010) Serum DNA can define tumor‐specific genetic and epigenetic markers in gliomas of various grades. Neuro-Oncol 12:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lehnertz B, Ueda Y, Derijck AAHA, Braunschweig U, Perez‐Burgos L, Kubicek S et al (2003) Suv39h‐mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13:1192–1200. [DOI] [PubMed] [Google Scholar]
- 77. Li Y, Sasaki H (2011) Genomic imprinting in mammals: its life cycle, molecular mechanisms and reprogramming. Cell Res 21:466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128:707–719. [DOI] [PubMed] [Google Scholar]
- 79. Lindemann C, Hackmann O, Delic S, Schmidt N, Reifenberger G, Riemenschneider MJ (2011) SOCS3 promoter methylation is mutually exclusive to EGFR amplification in gliomas and promotes glioma cell invasion through STAT3 and FAK activation. Acta Neuropathol 122(2):241–251. [DOI] [PubMed] [Google Scholar]
- 80. Liu B, Cheng J, Zhang X, Wang R, Zhang W, Lin H et al (2010) Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol Biomarkers Prev 19:2888–2896. [DOI] [PubMed] [Google Scholar]
- 81. Liu B, Cheng J, Zhang W, Zhang X, Wang R, Lin H et al (2010) Quantitative detection of multiple gene promoter hypermethylation in tumor tissue, serum, and cerebrospinal fluid predicts prognosis of malignant gliomas. Neuro-Oncol 12:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lo WS, Duggan L, Emre NC, Belotserkovskya R, Lane WS, Shiekhattar R et al (2001) Snf1—a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 293:1142–1146. [DOI] [PubMed] [Google Scholar]
- 83. Lorente A, Mueller W, Urdangarín E, Lázcoz P, Lass U, von Deimling A et al (2009) RASSF1A, BLU, NORE1A, PTEN and MGMT expression and promoter methylation in gliomas and glioma cell lines and evidence of deregulated expression of de novo DNMTs. Brain Pathol 19:279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lucio‐Eterovic AKB, Cortez MAA, Valera ET, Motta FJN, Queiroz RGP, Machado HR et al (2008) Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer 8:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T (2011) Epigenetics in alternative pre‐mRNA splicing. Cell 144:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Martinez R, Esteller M (2010) The DNA methylome of glioblastoma multiforme. Neurobiol Dis 39:40–46. [DOI] [PubMed] [Google Scholar]
- 87. Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y et al (2003) DNA methylation‐related chromatin remodeling in activity‐dependent BDNF gene regulation. Science 302:890–893. [DOI] [PubMed] [Google Scholar]
- 88. Masoudi A, Elopre M, Amini E, Nagel ME, Ater JL, Gopalakrishnan V et al (2008) Influence of valproic acid on outcome of high‐grade gliomas in children. Anticancer Res 28:2437–2442. [PubMed] [Google Scholar]
- 89. Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A et al (2008) Genome‐scale DNA methylation maps of pluripotent and differentiated cells. Nature 454:766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Meluh PB, Koshland D (1997) Budding yeast centromere composition and assembly as revealed by in vivo cross‐linking. Genes Dev 11:3401–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mikeska T, Bock C, El‐Maarri O, Hübner A, Ehrentraut D, Schramm J et al (2007) Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. J Mol Diagn 9:368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB et al (2008) Lineage‐specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30:755–766. [DOI] [PubMed] [Google Scholar]
- 93. Morin RD, Mendez‐Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD et al (2011) Frequent mutation of histone‐modifying genes in non‐Hodgkin lymphoma. Nature 476:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mottet D, Pirotte S, Lamour V, Hagedorn M, Javerzat S, Bikfalvi A et al (2009) HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1‐dependent, p53‐independent mechanism. Oncogene 28:243–256. [DOI] [PubMed] [Google Scholar]
- 95. Murr R (2010) Interplay between different epigenetic modifications and mechanisms. Adv Genet 70:101–141. [DOI] [PubMed] [Google Scholar]
- 96. Nakamura M, Watanabe T, Yonekawa Y, Kleihues P, Ohgaki H (2001) Promoter methylation of the DNA repair gene MGMT in astrocytomas is frequently associated with G:C – A:T mutations of the TP53 tumor suppressor gene. Carcinogenesis 22:1715–1719. [DOI] [PubMed] [Google Scholar]
- 97. Nakamura M, Watanabe T, Klangby U, Asker C, Wiman K, Yonekawa Y et al (2001) p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol 11:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H (2001) Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest 81:77–82. [DOI] [PubMed] [Google Scholar]
- 99. Nakamura M, Ishida E, Shimada K, Kishi M, Nakase H, Sakaki T et al (2005) Frequent LOH on 22q12.3 and TIMP‐3 inactivation occur in the progression to secondary glioblastomas. Lab Invest 85:165–175. [DOI] [PubMed] [Google Scholar]
- 100. Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292:110–113. [DOI] [PubMed] [Google Scholar]
- 101. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nygren AOH, Ameziane N, Duarte HMB, Vijzelaar RNCP, Waisfisz Q, Hess CJ et al (2005) Methylation‐specific MLPA (MS‐MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res 33:e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ohka F, Natsume A, Motomura K, Kishida Y, Kondo Y, Abe T et al (2011) The Global DNA Methylation Surrogate LINE‐1 Methylation Is Correlated with MGMT Promoter Methylation and Is a Better Prognostic Factor for Glioma. PLoS ONE 6:e23332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z et al (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448:714–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Papait R, Magrassi L, Rigamonti D, Cattaneo E (2009) Temozolomide and carmustine cause large‐scale heterochromatin reorganization in glioma cells. Biochem Biophys Res Commun 379:434–439. [DOI] [PubMed] [Google Scholar]
- 106. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC et al (2011) The genetic landscape of the childhood cancer medulloblastoma. Science 331:435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Rajendran G, Shanmuganandam K, Bendre A, Mujumdar D, Goel A, Shiras A (2011) Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J Neurooncol 104:483–494. [DOI] [PubMed] [Google Scholar]
- 109. Rauch T, Pfeifer GP (2005) Methylated‐CpG island recovery assay: a new technique for the rapid detection of methylated‐CpG islands in cancer. Lab Invest 85:1172–1180. [DOI] [PubMed] [Google Scholar]
- 110. Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M et al (2000) Regulation of chromatin structure by site‐specific histone H3 methyltransferases. Nature 406:593–599. [DOI] [PubMed] [Google Scholar]
- 111. Reitman ZJ, Yan H (2010) Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst 102:932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW et al (2011) Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci USA 108:3270–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I et al (2000) Genome‐wide location and function of DNA binding proteins. Science 290:2306–2309. [DOI] [PubMed] [Google Scholar]
- 114. Riemenschneider MJ, Reifenberger J, Reifenberger G (2008) Frequent biallelic inactivation and transcriptional silencing of the DIRAS3 gene at 1p31 in oligodendroglial tumors with 1p loss. Int J Cancer 122:2503–2510. [DOI] [PubMed] [Google Scholar]
- 115. Riemenschneider MJ, Jeuken JWM, Wesseling P, Reifenberger G (2010) Molecular diagnostics of gliomas: state of the art. Acta Neuropathol 120:567–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Riemenschneider MJ, Hegi ME, Reifenberger G (2010) MGMT promoter methylation in malignant gliomas. Target Oncol 5:161–165. [DOI] [PubMed] [Google Scholar]
- 117. Rodríguez‐Paredes M, Esteller M (2011) Cancer epigenetics reaches mainstream oncology. Nat Med 17:330–339. [DOI] [PubMed] [Google Scholar]
- 118. Rottach A, Frauer C, Pichler G, Bonapace IM, Spada F, Leonhardt H (2010) The multi‐domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res 38:1796–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Schmidt N, Windmann S, Reifenberger G, Riemenschneider MJ (2011) DNA hypermethylation and histone modifications downregulate the candidate tumor suppressor gene RRP22 on 22q12 in human gliomas. Brain pathology (Zurich, Switzerland). [DOI] [PMC free article] [PubMed]
- 120. Stimpson KM, Sullivan BA (2010) Epigenomics of centromere assembly and function. Curr Opin Cell Biol 22:772–780. [DOI] [PubMed] [Google Scholar]
- 121. Stone AR, Bobo W, Brat DJ, Devi NS, van Meir EG, Vertino PM (2004) Aberrant methylation and down‐regulation of TMS1/ASC in human glioblastoma. Am J Pathol 165:1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. [DOI] [PubMed] [Google Scholar]
- 123. Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9:465–476. [DOI] [PubMed] [Google Scholar]
- 124. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al (2009) Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Talbert PB, Henikoff S (2010) Histone variants–ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 11:264–275. [DOI] [PubMed] [Google Scholar]
- 126. Tepel M, Roerig P, Wolter M, Gutmann DH, Perry A, Reifenberger G et al (2008) Frequent promoter hypermethylation and transcriptional downregulation of the NDRG2 gene at 14q11.2 in primary glioblastoma. Int J Cancer 123:2080–2086. [DOI] [PubMed] [Google Scholar]
- 127. Tews B, Roerig P, Hartmann C, Hahn M, Felsberg J, Blaschke B et al (2007) Hypermethylation and transcriptional downregulation of the CITED4 gene at 1p34.2 in oligodendroglial tumours with allelic losses on 1p and 19q. Oncogene 26:5010–5016. [DOI] [PubMed] [Google Scholar]
- 128. Tost J, Dunker J, Gut IG (2003) Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. BioTechniques 35:152–156. [DOI] [PubMed] [Google Scholar]
- 129. Toyota M, Ho C, Ahuja N, Jair KW, Li Q, Ohe‐Toyota M et al (1999) Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res 59:2307–2312. [PubMed] [Google Scholar]
- 130. Tsai W, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S et al (2010) TRIM24 links a non‐canonical histone signature to breast cancer. Nature 468:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Uhlmann K, Rohde K, Zeller C, Szymas J, Vogel S, Marczinek K et al (2003) Distinct methylation profiles of glioma subtypes. Int J Cancer 106:52–59. [DOI] [PubMed] [Google Scholar]
- 132. Unoki M, Nishidate T, Nakamura Y (2004) ICBP90, an E2F‐1 target, recruits HDAC1 and binds to methyl‐CpG through its SRA domain. Oncogene 23:7601–7610. [DOI] [PubMed] [Google Scholar]
- 133. van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJB, Wesseling P et al (2010) IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res 16:1597–1604. [DOI] [PubMed] [Google Scholar]
- 134. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P et al (2011) Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wade PA (2001) Methyl CpG binding proteins: coupling chromatin architecture to gene regulation. Oncogene 20:3166–3173. [DOI] [PubMed] [Google Scholar]
- 137. Waha A, Güntner S, Huang TH, Yan PS, Arslan B, Pietsch T et al (2005) Epigenetic silencing of the protocadherin family member PCDH‐gamma‐A11 in astrocytomas. Neoplasia 7:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wang Y, Leung FCC (2004) An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics 20:1170–1177. [DOI] [PubMed] [Google Scholar]
- 139. Wang H, Wang S, Shen L, Chen Y, Zhang X, Zhou J et al (2010) Chk2 down‐regulation by promoter hypermethylation in human bulk gliomas. Life Sci 86:185–191. [DOI] [PubMed] [Google Scholar]
- 140. Watt PM, Kumar R, Kees UR (2000) Promoter demethylation accompanies reactivation of the HOX11 proto‐oncogene in leukemia. Genes Chromosomes Cancer 29:371–377. [DOI] [PubMed] [Google Scholar]
- 141. Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL et al (2005) Chromosome‐wide and promoter‐specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37:853–862. [DOI] [PubMed] [Google Scholar]
- 142. Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W et al (2010) MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol 6:39–51. [DOI] [PubMed] [Google Scholar]
- 143. Weller M, Gorlia T, Cairncross JG, van den Bent MJ, Mason W, Belanger K et al (2011) Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 77:1156–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F et al (2009) NOA‐04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27:5874–5880. [DOI] [PubMed] [Google Scholar]
- 145. Wilson BG, Roberts CWM (2011) SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11:481–492. [DOI] [PubMed] [Google Scholar]
- 146. Wolter M, Reifenberger J, Blaschke B, Ichimura K, Schmidt EE, Collins VP et al (2001) Oligodendroglial tumors frequently demonstrate hypermethylation of the CDKN2A (MTS1, p16INK4a), p14ARF, and CDKN2B (MTS2, p15INK4b) tumor suppressor genes. J Neuropathol Exp Neurol 60:1170–1180. [DOI] [PubMed] [Google Scholar]
- 147. Wu SC, Zhang Y (2010) Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 11:607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Xiong Z, Laird PW (1997) COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res 25:2532–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S et al (2011) Oncometabolite 2‐hydroxyglutarate is a competitive inhibitor of α‐ketoglutarate‐dependent dioxygenases. Cancer Cell 19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Yin D, Ong JM, Hu J, Desmond JC, Kawamata N, Konda BM et al (2007) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo . Clin Cancer Res 13:1045–1052. [DOI] [PubMed] [Google Scholar]
- 152. Yun M, Wu J, Workman JL, Li B (2011) Readers of histone modifications. Cell Res 21:564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P et al (2009) Glioma‐derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF‐1alpha. Science 324:261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Zhou H, Hu H, Lai M (2010) Non‐coding RNAs and their epigenetic regulatory mechanisms. Biol Cell 102:645–655. [DOI] [PubMed] [Google Scholar]