

Abstract
Formal Cu(III) complexes bearing an oxygen-based auxiliary ligand ([CuOR]2+, R = H or CH2CF3) were stabilized by modulating the donor character of supporting ligand LY (LY = 4-Y, N,N′-bis(2,6-diisopropylphenyl)-2,6-pyridinedicarboxamide, Y = H or OMe) and/or the basicity of the auxiliary ligand, enabling the first characterization of these typically highly reactive cores by NMR spectroscopy and X-ray crystallography. Enhanced lifetimes in solution and slowed rates of PCET with a phenol substrate were observed. NMR spectra corroborate the S = 0 ground states of the complexes, and X-ray structures reveal shortened Cu–ligand bond distances that match well with theory.
Understanding the molecular structures, spectroscopic properties, and reactivity of copper–oxygen complexes1–5 is important for gaining insight into the mechanisms by which copper enzymes and other catalysts function.6,7 Among the various complexes studied, those comprising the [CuOH]2+ core supported by dicarboxamide ligands (Figure 1)8–14 are notably reactive, attacking C–H and O–H bonds via proton-coupled electron transfer (PCET) processes and undergoing electron transfer at high rates (cf. the rate constant for the reaction of LHCuOH with 1,2-dihydroanthracene 50 M−1 s−1 at −25 °C;9 electron-transfer self-exchange rate constant ~104 M−1 s−1 at −88 °C13). The formulations of the complexes are supported by UV–vis spectroscopy (diagnostic absorptions with ligand-to-metal charge-transfer (LMCT) character), EPR silence, resonance Raman spectroscopy (νCu–O ≈ 630 cm−1),13 EXAFS (avg Cu–N,O ≈ 0.1 Å shorter than for the [CuIIOH]+ precursor),8 and theory.8,15
Figure 1.
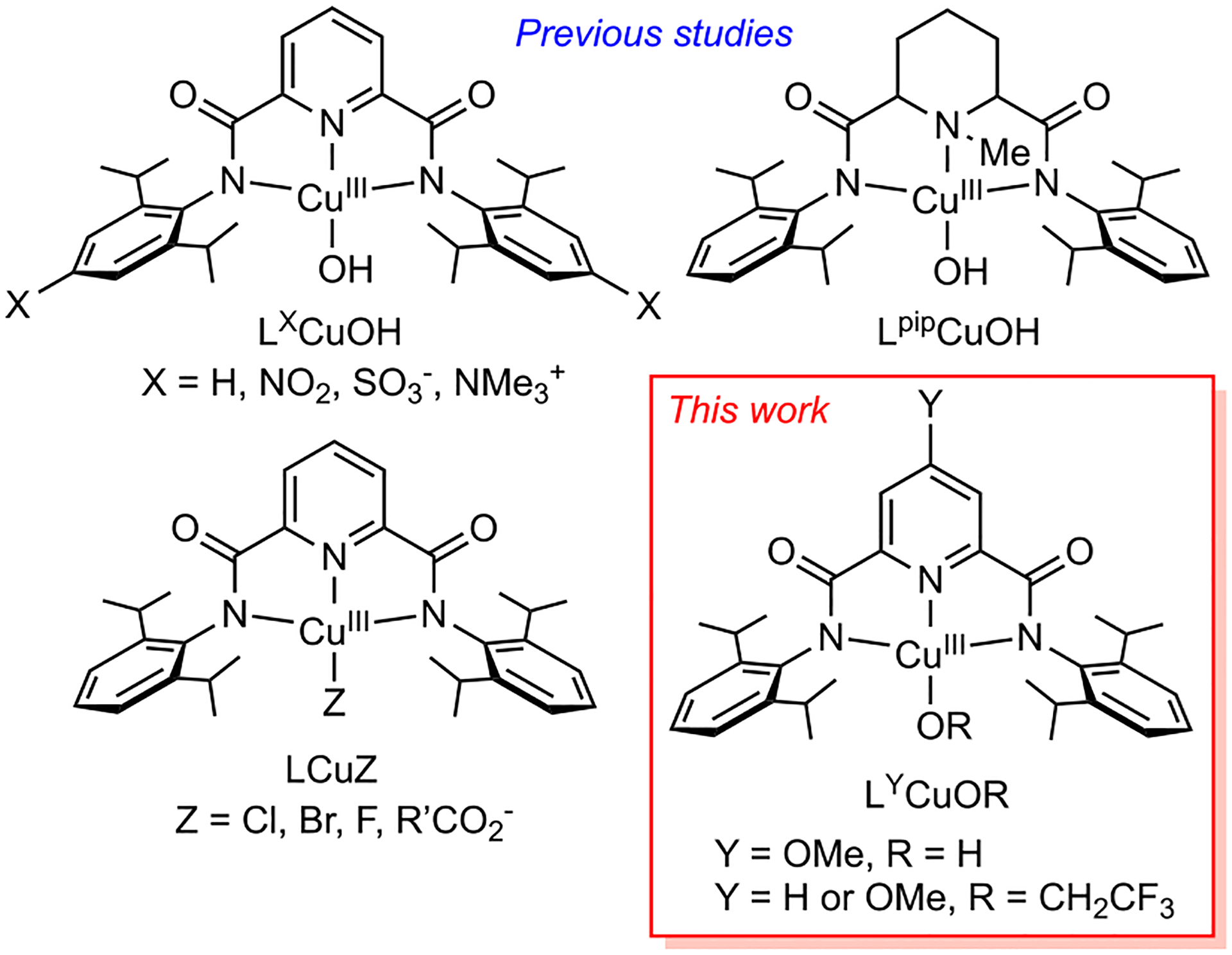
Complexes studied previously and the compounds that comprise the focus of this work. R′ = Me or aryl groups.
Perturbations of the [CuOH]2+ core have been effected by the installation of remote substituents on the flanking aryl rings (LXCuOH, Figure 1).11,12 These perturbations are reflected by shifts in the LMCT energies (lower), redox potentials (higher), basicities (lower), and PCET reaction rates (faster) that may be rationalized by electron withdrawal by the X groups. Changing the pyridyl group to a piperidine (LpipCuOH) has the opposite effects, attributed to greater electron donation by the amine donor. In no case has a complex with the [CuOH]2+ core been structurally defined by X-ray crystallography, in large part due to its high reactivity and poor thermal stability (t1/2 ≈ minutes at −80 °C in THF). These characteristics and the fact that decomposition yields paramagnetic Cu(II) species have also inhibited efforts to obtain NMR spectra that would be useful in confirming the proposed S = 0 ground state. Significantly slower reactions and greater stability were observed for the LCuZ derivatives (Z = halide8,16 or carboxylate17) featuring a less-basic X-type reactive moiety, of which the halide complexes were characterized recently by X-ray diffraction and NMR spectroscopy.16 Inspired by that success, we hypothesized that more complete characterization of the [CuOH]2+ core might be attained if its stability could be enhanced. Toward this end, we targeted two modifications: the placement of an electron-donating para-methoxy pyridyl group (Y = OMe; LOMe) and the use of a less-basic alkoxide moiety (CF3CH2O−). On the basis of previous work,9,11,12 we hypothesized that these changes would enhance the solution stability, thereby facilitating handling and characterization. We now report the confirmation of these hypotheses through the synthesis of the targeted derivatives and their characterization by X-ray crystallography and NMR spectroscopy, which with comparison to results from theory provides new insights into the molecular and electronic structures of complexes with [CuOH]2+ and [CuOCH2CF3]2+ cores.
The reaction of proligand H2LOMe with Cu(OTf)2 in the presence of NaOMe and CH3CN led to the isolation of key precursor LOMeCu(CH3CN) (77%), which upon treatment with NBu4OH led to [NBu4][LOMeCuOH] (76%). [NBu4]-[LYCuOCH2CF3] (Y = H or OMe) complexes were obtained from the respective hydroxide precursors via protonolysis with HOCH2CF3. The complexes LOMeCu(CH3CN), [NBu4]-[LOMeCuOH], and [NBu4][LYCuOCH2CF3] (Y = H or OMe) were characterized by UV–vis and X-band EPR spectroscopy, CHN analysis, and X-ray crystallography (SI). The complexes feature a slightly distorted square-planar geometry (τ4 = 0.15–0.22)18 with Cu–N,O bond distances typical for such Cu(II) species (Figures S43–S45; Table 1) and characteristic S = 1/2 rhombic signals with Cu and N hyperfine patterns in EPR spectra (Figures S7–S9).
Table 1.
Bond Distances (Å) Obtained by X-ray Crystallography for [CuOR]2+ (CuIII) and [CuOR]+ (CuII) Species and by DFT Geometry Optimization for [CuOR]2+ (Theory)a
| bond | species | LOMe, OH | LH, OCH2CF3 | LOMe, OCH2CF3 |
|---|---|---|---|---|
| Cu-N2 | CuIII | 1.841(3) | 1.864(13) | 1.856(3) |
| theory | 1.845 | 1.875 | 1.866 | |
| CuII | 1.927(2) | 1.925(4) | 1.927(1) | |
| CuIII−CuII | −0.086 | −0.061 | −0.071 | |
| Cu-N1,3 | CuIII | 1.900(3) | 1.950(12) | 1.948(3) |
| theory | 1.916 | 1.960 | 1.960 | |
| CuII | 2.027(2) | 2.058(4) | 2.020(1) | |
| CuIII−CuII | −0.127 | −0.108 | −0.072 | |
| Cu-O1 | CuIII | 1.799(3) | 1.812(12) | 1.849(5) |
| theory | 1.783 | 1.818 | 1.817 | |
| CuII | 1.867(2) | 1.857(4) | 1.838(2) | |
| CuIII−CuII | −0.068 | −0.045 | 0.011 | |
| mean | CuIII−CuII | −0.102 | −0.081 | −0.051 |
Averaged values are presented for carboxamide Cu–N1 and Cu–N3 bonds. N2 is pyridyl donor. Estimated standard deviations are in parentheses.
The expected electronic perturbations of the LOMe and −OCH2CF3 moieties are reflected in cyclic voltammograms (THF, 0.2 M Bu4NPF6), which contain quasi-reversible waves associated with [CuOR]2+/[CuOR]+ couples (Table 2). Good reversibility of the waves for the −OCH2CF3 complexes is apparent at scan rates as slow as 10 mV/s (Figure S20), consistent with low reactivity for the oxidized species (vide infra). A more modest enhancement of reversibility is seen for [NBu4][LOMe CuOH] relative to the analog supported by LH (Figure S18).5 The replacement of −OH with −OCH2CF3 shifts the E1/2 by +0.191 V on average, whereas the alteration of LY had a lesser effect, with an average difference of only −0.049 V induced by the introduction of the p-OMe substituent.
Table 2.
Reduction Potentials for the [CuOR]2+/+ Couple and Rate Constants for Reactions with ttbPhOH
| compound | E1/2 (mV)a | k2 (M−1 s−1)b |
|---|---|---|
| LHCuOH | −167c | 15 900 |
| LOMeCuOH | −202 | 11 800 |
| LCuOCH2CF3 | +37 | 3.0 |
| LOMeCuOCH2CF3 | −25 | 1.5 |
Conditions: THF, 0.2 M Bu4NPF6, values vs Fc+/0.
Conditions: −25 °C in DFB using either 1 equiv (LYCuOH) or 50 equiv (LYCuOCH2CF3).
The chemical oxidation of [NBu4][LOMe CuOH] or [NBu4]-[LYCuOCH2CF3] (Y = H or OMe) was performed by adding 1 equiv of FcBArF4 or AcFcBArF4, respectively, in THF (−80 °C) or in 1,2-difluorobenzene (DFB, −25 °C). Immediate color changes (deep violet or blue) and intense electronic absorption features (Figure 2) diagnostic of the formation of [CuOH]2+ and [CuOCH2CF3]2+ cores were observed, and reversible one-electron processes were confirmed through titrations and the sequential cyclic additions of oxidant and decamethylferrocene (Figures S22–S27).13 TDDFT UV–vis transitions exhibit λmax values in agreement with experiment that are predominantly HOMO to LUMO (Figure 2) and shift from 546.1 and 537.1 nm in LHCuOH and LOMeCuOH to 578. 7 and 573. 6 nm in LHCuOCH2CF3 and LOMeCuOCH2CF3, respectively, as a result of the stabilization of the LUMO (Table S4, Figures S50–S58). Also in agreement with experiment (and precedent17a), additional transitions with partial HOMO-to-LUMO character contribute at longer wavelengths to the spectra for the −OCH2CF3 complexes. Resonance Raman spectra of frozen solutions (λex = 561 nm) of all LYCuOR complexes contain a signal at ~635 cm−1 that we assign as νCu‑OR by analogy to data acquired for LHCuOH and theory (Figures S45–S49 and S59, Table S15).13
Figure 2.
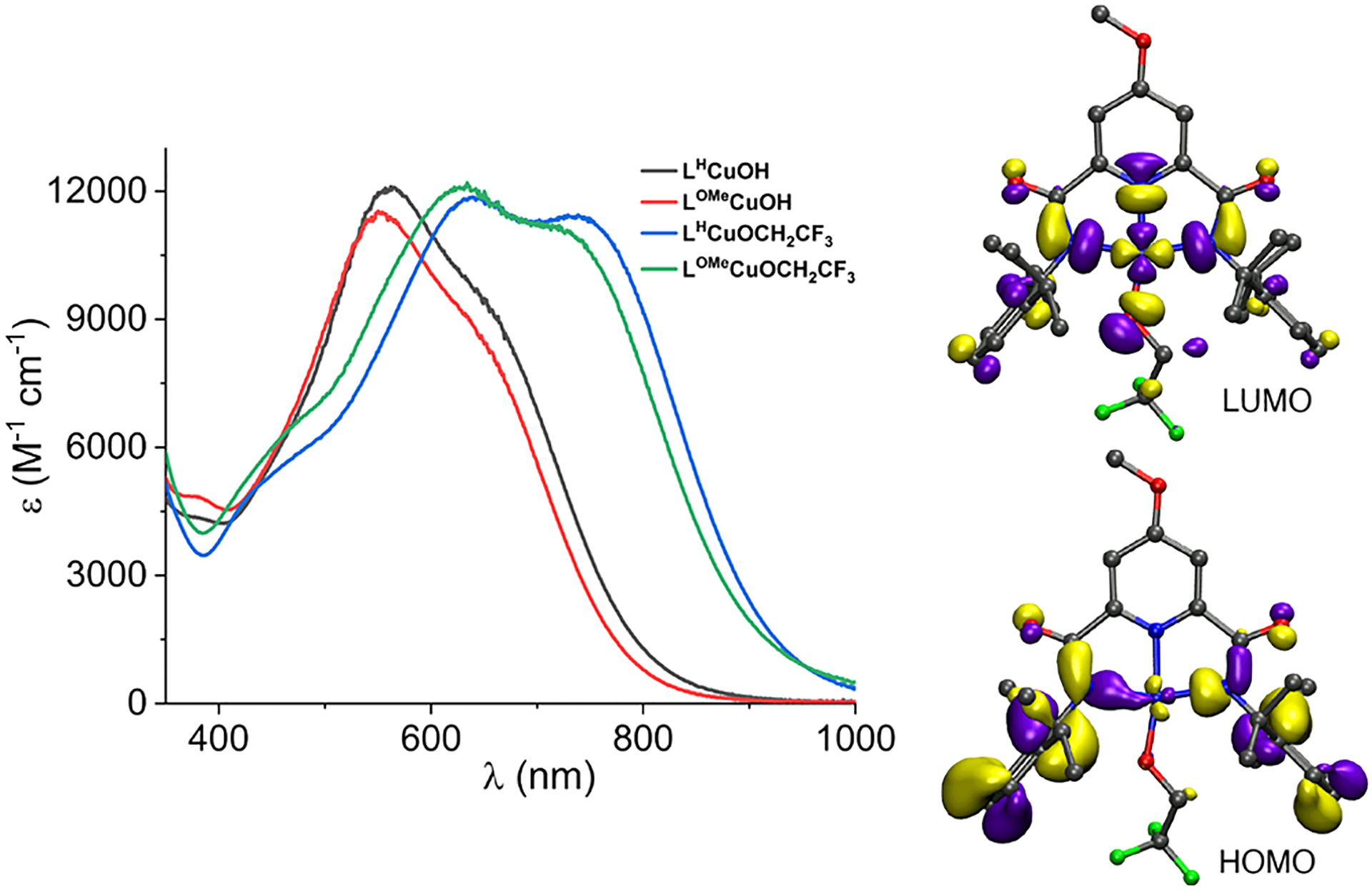
(Left) UV–visible absorption spectra of the indicated compounds in DFB at −25 °C. (Right) Orbitals for LOMeCuOCH2CF3 plotted with an isovalue of 0.04 au from the B98 functional.
To evaluate how ligand variation influences stability and reactivity, we compared the reactions of LYCuOH and LYCuOCH2CF3 (Y = H or OMe) with 2,4,6-tri-tert-butylphenol (ttbPhOH) to yield the stable phenoxyl radical.11,19,20 Second-order rate constants were measured using either 1 equiv (LYCuOH) or 50 equiv (LYCuOCH2CF3) of ttbPhOH at −25 °C in DFB (Table 2). The data show significantly higher reactivity for the hydroxide complexes (≳5000-fold), a difference that may be attributed to the higher basicity of the −OH vs −OCH2CF3 moieties and/or steric inhibition for the latter. Modest decreases in the rate constants for the cases where Y = OMe at parity for −OR may be rationalized by stabilization of the [CuOR]2+ core by the electron-donating methoxide substituent. Monitoring the room-temperature decays of the four [CuOR]2+ species in the absence of substrate in THF and DFB revealed complicated kinetic traces, but trends in the overall lifetimes paralleled the ttbPhOH reactivity trends (SI; cf. t1/2 ≈ 4 h vs <1 h for LOMeCuOCH2CF3 vs LCuOH in DFB).
The enhanced stability of the new complexes led us to attempt characterization by X-ray crystallography. We discovered that the complex prepared by the treatment of [NBu4][LOMeCuOH] with FcBArF4 in DFB could be isolated as suitable deep-purple crystals via the layered diffusion of pentane at −30 °C. Similar attempts with [NBu4][LYCuOCH2CF3] (Y = H or OMe) failed to give suitable crystals, likely in part due to the formation of highly intractable viscous residues containing [NBu4][BArF4]. To circumvent this issue, we employed reactants that would yield insoluble inorganic salts as byproducts. Thus, we reacted LCu-(CH3CN)4 and LOMeCu(CH3CN) with NaOCH2CF3 and then oxidized the resulting crude materials with AcFcSbF6 in CH2Cl2 or DFB. After the removal of a light-colored precipitate (presumably NaSbF6), suitable crystals of the oxidized products were obtained at −30 °C.
Representations of the X-ray structures of LOMeCuOH and LOMeCuOCH2CF3 (Figure 3) as well as LHCuOCH2CF3 (Figure S42) show similar square-planar geometries compared to their [CuOR]+ progenitors (τ4 = 0.11–0.15), but they are neutral species as expected for one-electron oxidation products. Comparison of metal–ligand bond distances between the oxidized and reduced forms (Table 1) indicates in all but one case shortening upon oxidation, by as much as 0.127 Å. The average Cu–N/O bond contraction in LOMeCuOH, 0.102 Å, is in excellent agreement with previously reported EXAFS analyses of LHCuOH (0.1 Å).8 The trifluoroethoxides show somewhat less contraction and in LOMeCuOCH2CF3 the Cu–O bond even lengthens slightly, by 0.011 Å, but disorder in the trifluoroethoxide ligand imparts an inherent inaccuracy to the O atom’s position. Gas-phase geometry optimizations (SI) for the S = 0 ground states of the oxidized species are in excellent agreement with the experimentally determined values (theory in Table 1). Overall, the bond length differences between the precursor and oxidation products are consistent with the loss of an electron from orbitals spanning the Cu center and/or its immediate environment, as reported for less-reactive complexes LCuZ (Z = F, Cl, Br).16 Furthermore, the lack of significant structural changes associated with this redox event agrees with the low reorganization energy of 0.95 eV previously measured for the [LHCuOH]−/LHCuOH couple.14
Figure 3.
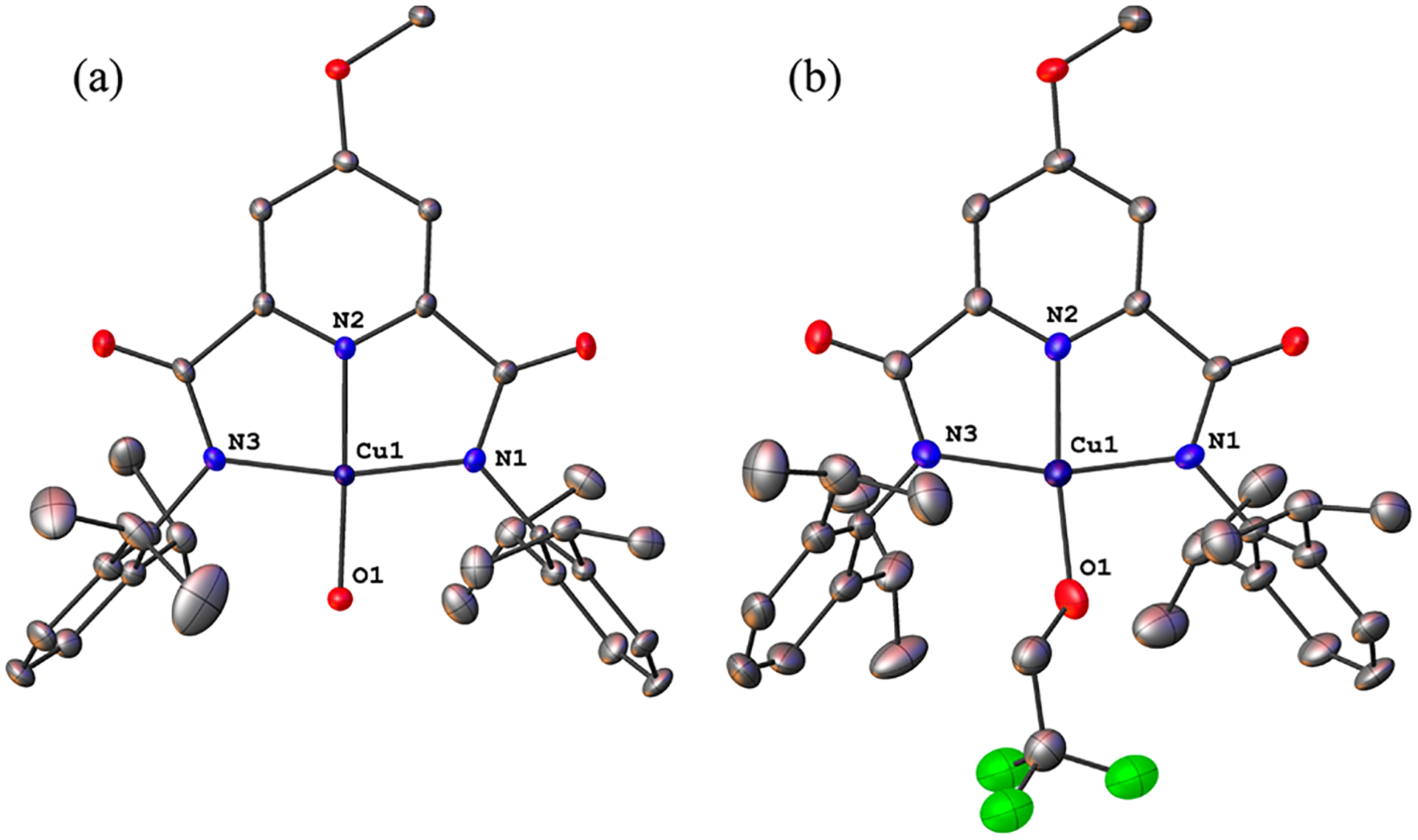
Representations of the X-ray structures of (a) LOMeCuOH and (b) LOMeCuOCH2CF3, showing all nonhydrogen atoms as 50% thermal ellipsoids.
While theoretical calculations support a closed-shell S = 0 ground state for the [CuOH]2+ core (Table S5),8 the only experimental corroboration has come from a dearth of signal in the X-band EPR spectrum, an observation consistent with either the S = 0 or S = 1 ground state. Acquiring NMR spectra, which would distinguish the two spin states, presents challenges due to the formation of paramagnetic Cu(II) decay species. We were nonetheless able to observe sharp peaks in the diamagnetic region of 1H NMR spectra for both LYCuOH species in 1,2-dichlorobenzene-d4 at −15 °C (Figure S28–S29), although broadening due to decomposition was evident for Y = H. The 1H NMR spectrum of the more robust complex LHCuOCH2CF3 in THF-d8 at −80 °C (Figure S30) displayed negligible broadening, but some resonances were obscured by solvent/byproduct signals. Since LOMeCuOCH2CF3 could be isolated in neat form as a crystalline solid, NMR spectra of isolated material were collected (CD2Cl2, −15 °C), and all expected 1H NMR resonances and J couplings (Figure 4) as well as 13C{1H} NMR peaks (Figure S32) were observed. The sharpness of the observed 1H and 13C{1H} NMR features in the diamagnetic chemical shift region confirms an S = 0 ground state, in agreement with predictions.8
Figure 4.
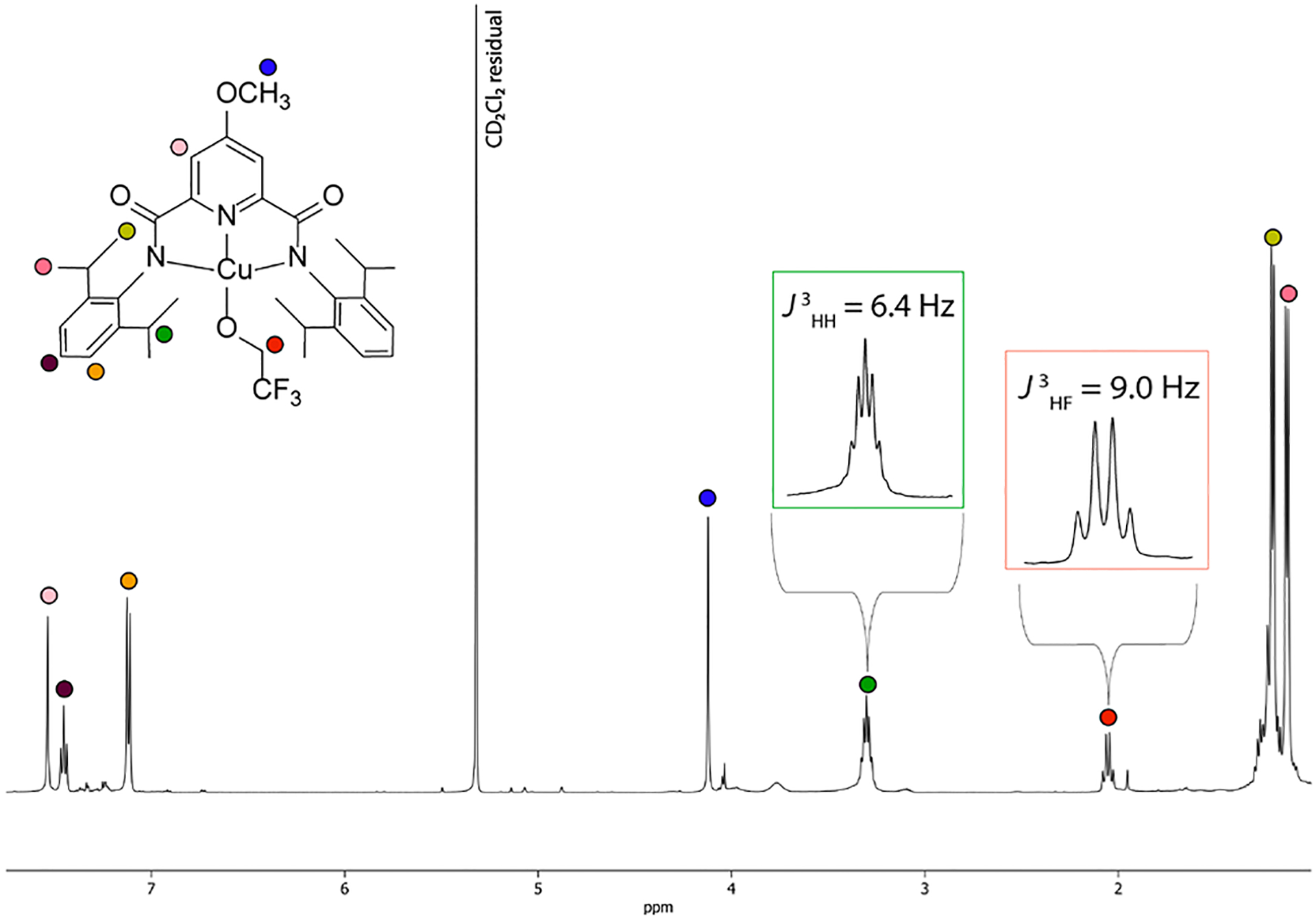
1H NMR spectrum of LOMeCuOCH2CF3 (CD2Cl2, −15 °C).
In conclusion, we prepared and characterized a new set of complexes with [CuOR]+/2+ cores using a modified supporting ligand and/or core (R = CH2CF3). Both changes attenuate the PCET reactivity of the oxidized state, the former by lowering its oxidizing potential and the latter by lowering the basicity of the proton-accepting site. While each modification has the opposite effect on the opposite property (e.g., the less-basic proton acceptor also leads to a more oxidizing species), the dominant impact is on electronics for the supporting ligand and basicity for the core, in line with previously observed reactivity trends and demonstrating how PCET reactivity can be tuned. These stabilization effects were sufficient to permit, for the first time, the successful characterization of complexes with [CuOR]2+ cores by X-ray crystallography and NMR spectroscopy. These data are consistent with several predictions made about LHCuOH, in particular, the conservation of geometry with minimal reorganization upon oxidation,14 the EXAFS-derived contraction of Cu–L bonds by ~0.1 Å on oxidation,8 the calculated geometries, and the diamagnetic S = 0 ground state.8 To the best of our knowledge, the complexes with R = CH2CF3 are the first alkoxo analogs bearing the formal Cu(III) oxidation state, and with the discovery that they share many similarities with their hydroxide counterparts, including PCET reactivity with phenols, the potential for steric and electronic tuning that is unavailable with hydroxide makes this class of compounds promising for further research into the bond-activation properties of high-valent copper species.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institutes of Health (GM 47365) for financial support. X-ray diffraction data were collected using diffractometers acquired through NSF-MRI award no. CHE-1827756. Computations were performed on high performance computing systems at the University of South Dakota, funded by NSF award no. OAC-1626516.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c13470
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c13470.
DFT structural coordinate file (XYZ)
Experimental details and figures (PDF)
Accession Codes
CCDC 2053118–2053123 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).Mirica LM; Ottenwaelder X; Stack TDP Sructure and Spectroscopy of Copper-Dioxygen Complexes. Chem. Rev 2004, 104, 1013–1045. [DOI] [PubMed] [Google Scholar]
- (2).Lewis EA; Tolman WB Reactivity of Copper-Dioxygen Systems. Chem. Rev 2004, 104, 1047–1076. [DOI] [PubMed] [Google Scholar]
- (3).Elwell CE; Gagnon NL; Neisen BD; Dhar D; Spaeth AD; Yee GM; Tolman WB Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev 2017, 117, 2059–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of dioxygen by copper metalloproteins and insights from model complexes. JBIC, J. Biol. Inorg. Chem 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Liu JJ; Diaz DE; Quist DA; Karlin KD Copper(I)-Dioxygen Adducts and Copper Enzyme Mechanisms. Isr. J. Chem 2016, 56, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ciano L; Davies GJ; Tolman WB; Walton PH Bracing copper for the catalytic oxidation of C–H bonds. Nature Catal. 2018, 1, 571–577. [Google Scholar]
- (7).Trammell R; Rajabimoghadam K; Garcia-Bosch I Copper-Promoted Functionalization of Organic Molecules: from Biologically Relevant Cu/O2 Model Systems to Organometallic Transformations. Chem. Rev 2019, 119, 2954–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Donoghue PJ; Tehranchi J; Cramer CJ; Sarangi R; Solomon EI; Tolman WB Rapid C–H Bond Activation by a Monocopper(III)–Hydroxide Complex. J. Am. Chem. Soc 2011, 133, 17602–17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dhar D; Tolman WB Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex. J. Am. Chem. Soc 2015, 137, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gagnon N; Tolman WB [CuO]+ and [CuOH]2+ complexes: intermediates in oxidation catalysis? Acc. Chem. Res 2015, 48, 2126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dhar D; Yee GM; Spaeth AD; Boyce DW; Zhang H; Dereli B; Cramer CJ; Tolman WB Perturbing the Copper(III)–Hydroxide Unit through Ligand Structural Variation. J. Am. Chem. Soc 2016, 138, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dhar D; Yee GM; Markle TF; Mayer JM; Tolman WB Reactivity of the copper(III)-hydroxide unit with phenols. Chem. Sci 2017, 8, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Spaeth AD; Gagnon NL; Dhar D; Yee GM; Tolman WB Determination of the Cu(III)–OH Bond Distance by Resonance Raman Spectroscopy Using a Normalized Version of Badger’s Rule. J. Am. Chem. Soc 2017, 139, 4477–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zerk TJ; Saouma CT; Mayer JM; Tolman WB Low Reorganization Energy for Electron Self-Exchange by a Formally Copper(III,II) Redox Couple. Inorg. Chem 2019, 58, 14151–14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mandal M; Elwell CE; Bouchey CJ; Zerk TJ; Tolman WB; Cramer CJ Mechanisms for Hydrogen-Atom Abstraction by Mononuclear Copper(III) Cores: Hydrogen-Atom Transfer or Concerted Proton-Coupled Electron Transfer? J. Am. Chem. Soc 2019, 141, 17236–17244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bower JK; Cypcar AD; Henriquez B; Stieber SCE; Zhang S C(sp3)-H Fluorination with a Copper(II)/(III) Redox Couple. J. Am. Chem. Soc 2020, 142, 8514–8521. [DOI] [PubMed] [Google Scholar]
- (17).(a) Elwell CE; Mandal M; Bouchey CJ; Que L Jr.; Cramer CJ; Tolman WB Carboxylate Structural Effects on the Properties and Proton-Coupled Electron Transfer Reactivity of [CuO2CR]2+ Cores. Inorg. Chem 2019, 58, 15872–15879. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Unjaroen D; Gericke R; Lovisari M; Nelis D; Mondal P; Pirovano P; Twamley B; Farquhar ER; McDonald AR High-Valent d(7) Ni(III) versus d(8) Cu(III) Oxidants in PCET. Inorg. Chem 2019, 58, 16838–16848. [DOI] [PubMed] [Google Scholar]
- (18).Yang L; Powell D; Houser R Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, t4. Dalton Trans. 2007, 955–964. [DOI] [PubMed] [Google Scholar]
- (19).Porter TR; Capitao D; Kaminsky W; Qian Z; Mayer JM Synthesis, Radical Reactivity, and Thermochemistry of Monomeric Cu(II) Alkoxide Complexes Relevant to Cu/Radical Alcohol Oxidation Catalysis. Inorg. Chem 2016, 55, 5467–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Manner VW; Markle TF; Freudenthal JH; Roth JP; Mayer JM The First Crystal Structure of a Monomeric Phenoxyl Radical: 2,4,6-Tri-tert-butylphenoxyl Radical. Chem. Commun 2008, 256–258. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.