

Abstract
The Fragile X Mental Retardation 1 protein FMRP is the product of FMR1, a gene whose epigenetic inactivation by a triplet nucleotide repeat expansion causes Fragile X Syndrome (FXS), a neurodevelopmental disorder. FMRP is a widely expressed RNA binding protein whose activity is essential for proper synaptic plasticity and architecture, aspects of neural function that are known to go awry in FXS. For over two decades, FMRP has been known as a translational repressor protein; in its absence, protein synthesis in the brain is excessive, which in turn impairs neuronal circuit formation and higher cognitive function. However, although the neurophysiology of Fragile X Syndrome has been described in remarkable detail, research focusing on the molecular biology of FMRP has only scratched the surface. Recent advances in whole transcriptome and translatome analysis from mouse and human models of the disorder show that FMRP is involved in nearly all aspects of gene expression. Emerging mechanistic details of FMRP regulation offer ways to consider new therapies to treat FXS.
Keywords: FMRP, Fragile X Syndrome, Neurodevelopmental disorder, RNA binding protein
Introduction
Fragile X Syndrome (FXS) is a neurodevelopmental disorder characterized by intellectual disability and behavioral symptoms such as hyperactivity and anxiety. Nearly all FXS cases result from expansion of what are normally ~30 CGG triplet repeats in the 5’ untranslated region of the X-linked Fragile X Mental Retardation 1 (FMR1) gene to over 200, leading to DNA methylation, transcriptional inactivation, and loss of the gene product Fragile X Mental Retardation Protein (FMRP)1,2. FXS afflicts about 1 in 4000 male and 1 in 7000 females and is the most common inherited cause of intellectual impairment3,4. There is strong association between FXS and autism; about half of all individuals with FXS and 2 in 3 males with FXS also meet the criteria for this disorder. Consequently, FXS contributes to 2–6% of all autism cases, making it the largest single gene cause of this disability3,4. In the brain, FMRP is expressed in neural stem cells and probably all neuronal and glial cell types. Its importance is illustrated by the fact that FMR1 inactivation impairs brain development and cell function at multiple levels including dendrite morphogenesis, circuit integration, and axon targeting (Figure 1).
Figure 1. Brain cell types affected in FXS.

FXS is a neurodevelopmental disorder resulting from FMRP deficiency, which adversely affects nearly every aspect brain development and functioning of many brain cell types. FMRP-deficient neural stem (progenitor) cells (NSCs) exhibit altered proliferation and fate specification in both developing brains as well as adult neurogenic zones. FMRP-deficient neurons display stunted dendritic morphogenesis, altered axonal targeting, reduced synaptogenesis, and abnormal circuit integration. FMRP is also expressed in astrocytes where it regulates astrocyte-neuron interactions. FMRP deficiency may lead to transient impairment in white matter development.
The discovery of FMR1 silencing as the cause of FXS has prompted extensive investigations into the functions of FMRP using animal and human stem cell models5,6. Studies of FMRP protein have shown that it is a brain-enriched RNA binding protein7,8,9 and have identified its mRNA targets and molecular activities, particularly as a regulator of translation9,10. Through genetic and pharmacologic “rescue” experiments primarily in mice, both upstream and downstream pathways that coalesce on and emanate from FMRP have been identified and suggested as possible therapeutic targets for the treatment of FXS11,12,13. However, despite advances towards the mitigation of FXS-relative phenotypes in animal models, recent clinical trials of candidate drugs have been unsuccessful at achieving primary endpoints5,12,13.
Although enormously useful for understanding FMRP and some aspects of FXS, Fmr1 mutant mice (one of the most commonly-used animal models of FXS) carry a deletion in the Fmr1 gene as opposed to CGG expansion-induced gene methylation and silencing, which may be at least partly responsible for the subtly of the behavioral abnormalities found in these mice compared to humans with FXS13,14. In addition, it is clear that many aspects of human brain development cannot be modeled in animals6,15. Therefore, FXS individual-derived or gene edited human pluripotent stem cells (hPSCs) have been used to mimic FXS pathogenesis in a dish, which may reveal human-specific FMRP functions that can be targeted for therapeutic intervention6.
Despite being a single gene disorder, individuals with FXS vary widely in intellectual impairment; for example, some males with FXS function nearly normally while others are profoundly disabled16,17. Most individuals with FXS have considerable anxiety and about a third of males with FXS exhibit irritability and aggression18. Although genetic background is certainly responsible for heterogeneity of symptoms, the molecular function(s) of FMRP may also be a contributing factor. For example, FMRP regulates mRNA splicing in a stoichiometric manner which may lead to differential splicing changes among FXS individuals. In addition, FMRP-targeted mRNAs may be regulated by other proteins (e.g. MTL3 or ADAR) that may be present at various levels among FXS individuals.
To move a field forward sometimes it is useful to a step back. This is what we do in this Review by taking a fresh look at what FMRP does and how its loss drives FXS pathophysiology. This survey, which includes recent advances in next generation sequencing technology, multi-omics gene network analysis, hPSCs, and CRISPR/Cas9 human gene editing, allows us to delve into novel molecular mechanisms regulated by FMRP. We will summarize how new studies on cell type-specific FMRP targets in mouse and human neurons inform us as to how FMRP selects its target transcripts. We will discuss innovative methods that unveil how FMRP regulates translation, alternative splicing, and RNA stability through recognition of mRNA codon bias and m6A modifications. Finally, we will discuss how this new knowledge base influences our understanding FXS which may lead to innovative therapeutic development.
Identifying FMRP’s targets
FMRP domain structure
The most prevalent form of FMRP in humans contains 632 amino acids and is a classic RNA binding protein containing at least three canonical RNA binding motifs (Figure 2) : two hnRNP K homology (KH) domains and an arginine-glycine-glycine (RGG) box19. A third putative KH domain, KH0, has been identified through x-ray crystallography20, although its RNA binding capabilities have not been tested. The amino terminal region of FMRP contains two Agenet/Tudor domains that interact with RNA, chromatin and other proteins20,21,22. FMRP also contains nuclear localization and nuclear export sequences (NLS, NES, respectively) that direct its shuttling between nucleus and cytoplasm23. At steady state, however, the protein is predominantly cytoplasmic23. FMRP is post-translationally modified by phosphorylation, that is triggered by metabotropic glutamate receptor (mGluR) signaling. This phosphorylation controls FMRP’s ubiquitin-mediated destruction at synapses24, coordination with miRNAs to regulate translation25, phase separation into liquid droplets by way of its intrinsically disordered domain26 and association with RNA-containing granules that may influence translational regulation27.
Figure 2. Functions of FMRP targets.

FMRP protein contains multiple domains that are important for its functions, including two Agenet/Tudor domains (Agn) for binding DNA and other proteins, nuclear localization (NLS) and exit (NES) sequences, and several RNA binding domains, KH0, KH1, KH2, and RGG box. FMRP binds >1000 mRNAs in the brain that are involved in neural processes such as synaptogenesis (synaptic proteins), cell-cell communication (neurotransmitter receptors), cytoskeleton and microtubule modulators, and metabolic regulators. FMRP also binds to mRNAs encoding transcription factors and epigenetic chromatin modulators, especially in immature cells (e.g. NSCs and immature neurons), although transcription factors and chromatin modulators are not among the top categories identified in neurons or brain tissue.
The modular nature of the FMRP RNA binding domains allows for in vitro identification of RNA recognition sequences. For example, assays with FMRP fragment polypeptides show that, unlike other KH motif-containing RNA binding proteins, all three FMRP KH domains bind very weakly to single stranded RNAs and may require higher order secondary structures to confer specificity28. The RGG motif preferentially interacts with a G quadruplex (G4) pseudoknot structure in mRNA28,29,30. KH domain 2 recognizes a variable sequence within a complex pseudoknot structure that is important for FMRP to associate with polyribosome31.
FMRP target mRNAs
Nearly three decades have passed since FMRP was shown to bind mRNA7,8, which has laser-focused the field on answering these questions: what mRNAs are bound by FMRP? Is there cell type-specificity of its mRNA targets? How does FMRP select its mRNA targets? What does FMRP do to its mRNA targets?
In an important initial study, Brown et al32 co-immunoprecipitated FMRP–RNA complexes (through RNP immunoprecipitation (RIP)) from the mouse brain followed by microarray analysis, which identified 432 FMRP-bound mRNAs. G4 structures were detected in 8 of the 12 top ranked targets. In another assay, FMRP antibody-directed amplification of mRNA sequestered in FMRP-containing mRNPs (APRA) also found G4 structures in many FMRP-bound mRNAs33. These early studies suggested that FMRP associated with unique sets of mRNAs and that the presence of G4 structures was one prominent determinant for the interaction; however, because both RIP and APRA lack specificity and are unable to identify protein binding sites on mRNA, a more rigorous and high-resolution method was required. In a seminal study, Darnell et al9 covalently crosslinked FMRP to RNA by ultraviolet irradiation, which was followed by stringent FMRP immunoprecipitation and high throughput sequencing (HITS-CLIP, also called CLIP-seq) to identify associated RNAs. They identified 842 high confidence FMRP target RNAs in juvenile mouse forebrain, 76% of which were not identified by RIP32. Surprisingly, the RNAs identified by HITS-CLIP did not contain noticeable G4 structures. However, nearly a third encoded postsynaptic proteins such as metabotropic glutamate and N-methyl-D-aspartate (NMDA) receptors, whose signaling is impaired in FXS. The FMRP HITS-CLIP target list has been an enormously useful resource for investigators analyzing the biology of FMRP (Table 1). Two additional observations by Darnell et al9 have catalyzed studies in the molecular biology of RNA-protein interactions. First, there was no clearly identifiable FMRP interacting sequence motif within mRNAs (i.e., cis elements), contradicting the previous studies using RIP9,32. Second, FMRP predominantly bound to the coding regions (CDS) of mRNAs rather than 5’ or 3’-UTRs, unlike most other RNA binding proteins.
Table 1.
High throughput identification of FMRP targeted mRNAs
| Publication | species | tissue/cell types | Method | numbers of target | binding regions | motif of targets | other features | Top categories of targets |
|---|---|---|---|---|---|---|---|---|
| Brown 2001 (32) | mouse | juvenile mouse forebrains | Anti-FMRP antibody (7G1–1 mAb; Brown et al 1998 (89), RIP-microarray | 432 | unknown | 67% (8 out of 12 top ranked targets has G4 | no category specified. | |
| Miyashiro 2003 (33) | mouse | primary hippocampal neurons | Anti-FMRP antibody (1C3 mAb; Devys et al 1993 (90); APRA | 83 | unknown | 23% (18 of 83) has G4 | Cell signaling, cell-cell communications, cell structure/mobility, gene/protein expressions. | |
| Darnell 2011 (9) | mouse | juvenile mouse forebrains | Anti-FMRP antibody (7G1–1 mAb; Brown et al 1998 (89), CLIP-seq | 842 | mostly coding sequence, also 3’UTR | none (no G4 abundance) | 661 are new compared to Brown 2001 | synaptic transmission, neuronal activity, membrane, cell signaling, autism |
| Maurin 2018 (34) | mouse | hippocampus, cortex, cerebellum | Anti-FMRP antibody (Rb#11, pAb; this study), CLIP-seq | 4174 total from all 3 brain regions | coding sequence enriched with GAC codon | CTGKA; GCTGYY; GWRGA; CATCRYC and TAY motifs; G4 is enriched. | 680 shared with Darnell 2011; 192 new | about 1/4 are distantly transported mRNAs; Top pathways include: cell signaling, neuronal activity, synaptic function, microtubule. |
| Van Driesche 2019 (35) | mouse | cerebellar Purkinje cells (Pcp2-Cre) and granule cells (NeuroD1-Cre), | Anti-GFP mAbs (HtzGFP19C8 and HtzGFP19F7, Heiman 2008 (91); from FMRP-cTag mice | 135 high confidence FMRP CLIP targets in Purkinje cells; 259 in granule cells, they are distinct | Long, coding sequences | none | has ranked targets based on TRAP-seq of same cell types; | Synaptic functions; microtubule, Cell signaling, GTPase signaling in Purkinje cells, GABAergic transmission in cerebellar granule cells |
| Sawicka 2019 (36) | mouse | CA1 neurons (Camk2a-Cre) | Anti-GFP mAbs (HtzGFP19C8 and HtzGFP19F7, Heiman 2008 (91); from FMRP-cTag mice | 327; different from cb granule neurons | Long, coding sequences | none | has ranked targets based on TRAP-seq of same cell types; | Signaling, transcription, synaptic development and function, microtubule organization, axon transport, autism, circadian rhythm |
| Ascano 2012 (42) | Human | HEK293 cell lines | Transfected HA-tagged FMRP, anti-HA ab (MMS-101P, Convance), PAR-CLIP | >6000 | coding sequence and 3’UTR | ACUK, WGGA | many | |
| Tran 2019 (43) | Human | postmortem adult human frontal cortex | Anti-FMRP pAb (MBL, RN016P), CLIP-seq | 4895 | binds to A-to-I edited site; predominantly 3’UTR | ACUG | Synaptic functions, (focus was editing in autism, not FMRP targets) | |
| Li 2020 (44) | Human | human iPSC differentiated dorsal and ventral forebrain NPCs and neurons (four cell types) | FLAG tag knocked into human FMR1 locus, FMRP-FLAG, anti-FLAG ab (Sigma Aldrich F1804), CLIP-seq | 1653 total (1232 in dNPCs, 1234 in vNPCs, 629 in dNeurons, 721 in vNeurons | coding sequences, long genes | CUAC; UGGA; GAUG, AGGU, CAGC; unclear about G4 | Neurogenesis, axon and dendrite morphogenesis, cytoskeleton, microtubule, RNA transport, histone modification (especially in NPCs), autism. |
Because FMRP is thought to be expressed in all cells in the brain6,11, a key question is whether it binds mRNAs with cell-type specificity. FMRP HITS-CLIP of juvenile mouse cortex, hippocampus, and cerebellum showed that the target RNAs in each region generally overlapped and that the FMRP binding sequences were mostly in CDS34. In a refinement of HITS-CLIP, Van Driesche et al35 generated a conditional FMRP-GFP mouse line and through crossing with different Cre driver lines, created mice expressing a FMRP-GFP fusion protein in specific cell types. Then using antibodies against GFP for HITS-CLIP, they identified both common and unique FMRP-bound RNAs in cerebellar Purkinje and granule cell neurons as well as in hippocampal area CA1 neurons35, 36. Among the new FMRP target RNAs identified in hippocampal CA1 neurons are several that control circadian rhythms especially those that influence behaviors associated with Fmr1-deficient mice such as contextual fear memory36.. These results support an earlier study showing dysregulation of circadian rhythms in FMRP-deficient mice37. Confirming the findings of the earlier FMRP mouse forebrain CLIP study9, Van Driesche et al35 and Sawicka et al36 found that many FMRP CLIP targets are encoded by autism spectrum disorder (ASD) high risk genes. The CLIP RNAs identified in these two studies are also particularly long, although the biological significance of this observation is unclear. Most importantly, the FMRP CLIP studies generally found that a number of FMRP’s target RNAs are translationally dysregulated in Fmr1-deficient animals9, 35,36.
Studies showing that Fmr1 deficiency in mouse models does not faithfully recapitulate all aspects of brain biology of FXS in humans38 and that mouse embryonic stem cells do not completely mimic their human counterparts39,40,41 suggest that interspecies differences in brain development and FMRP function might be significant. One question is whether the FMRP targets identified in mouse are the same as those in human cells. To assess this, ectopic expression of tagged FMRP in human embryonic kidney (HEK293) cells was followed by a modified CLIP procedure (PAR-CLIP, where cells are metabolically labeled with the photoactivatable nucleoside analog 4-thiouridine (4-TU)), which revealed that half of the expressed mRNAs (>6000) were bound by FMRP42, raising concerns about specificity and biological relevance. More recent work using an antibody against endogenous FMRP and a modified CLIP-seq (eCLIP) method identified FMRP targets in post-mortem adult human frontal cortex43, although the drawbacks here are mixed cell types and questionable applicability to neurodevelopment. In a recent study to interrogate developing neural cells, FLAG-tagged FMRP knock-in hPSC lines differentiated into dorsal forebrain excitatory neuroprogenitors and neurons (dNPC, dNeurons) or ventral forebrain inhibitory linages (vNPCs, vNeurons) were generated44. FLAG antibody immunoprecipitation and an optimized CLIP protocol for small numbers of primary cells identified FMRP target RNAs in four human neural cell types: vNPCs, vNeurons, dNPCs, dNeurons. As in the mouse brain, human FMRP preferentially bound long transcripts and mostly to CDS. Furthermore, many FMRP targets (39%−77%) were shared among the four cell types, suggesting a common FMRP function during neurogenesis. However, some FMRP targets were specific to NPCs, including those involved in cell cycle regulation, which may be relevant to the macrocephaly observed in some individuals with FXS and autism. FMRP targets enriched in the ventral but not dorsal neural cells encoded proteins involved in cell junction, ATP binding, and cytoskeleton pathways, suggesting that FMR1 might play a role in the unique tangential migration of the ventrally developed interneurons. The majority of FMRP targets were not shared with those identified in adult human brains, which might be attributed to differences in both cell-type specificity and stage of development. Interestingly, a previous application of network-based integrative analysis45 for reconciling FMRP targets with transcriptomic changes revealed that FMRP regulates the production of transcription factors, which in turn could coordinate the synthesis of the downstream RNAs observed in RNA-seq datasets (Table 1)
How does FMRP select its target RNAs? Recognition of the G4 structure reported in mouse brain34 is not a consistent preference of human FMRP44. A more detailed analysis of the original mouse and human FMRP CLIP studies9,42 has identified WGGA and UGGA among the top preferred binding motifs of FMRP46,47, which was also observed in new CLIP-seq studies of mouse brain34 and human neuron studies44. In general, FMRP binding motifs are more degenerate than those of many other RNA binding proteins and are thus poor predictors of FMRP target transcripts. Table 1 summarizes the key features of FMRP target mRNAs identified using high throughput methods. The top biological processes that are regulated by these mRNAs include cellular signaling, synaptic development and function, axonal and dendritic development, cytoskeleton and microtubule, RNA transport, as well as transcriptional and epigenetic regulations (Figure 2).
Several consensus observations can be drawn from the CLIP studies discussed above. First, FMRP preferentially binds to mRNAs that are implicated in autism, indicating a molecular convergence between these developmental neural disorders9,43. Second, FMRP preferentially binds to long RNAs35,36,44. Although this in and of itself does not indicate a biological function, it is worth noting that the topoisomerase TOP1 is particularly important for the transcription of long genes associated with autism48, and there is a direct interaction between topoisomerase TOP3beta and FMRP49,50,51. Third, FMRP predominantly binds to coding regions of mRNAs, which suggests that it mediates a novel translational control mechanism9.
FMRP and the regulation of translation
FMRP loss alters translation
The identification of FMRP CLIP targets does not necessarily indicate that the binding of FMRP to a particular target has a molecular function. The observation that hippocampal slices derived from Fmr1 knockout mice incorporate 15–20% more 35S-methionine into protein compared to wild type mice has experimentally supported original suggestions that FMRP is primarily a translational inhibitor52,53,54. However, changes in transcription or in RNA stability can also lead to increased protein levels.
The first of two high-resolution methods that have been used to assess FMRP-regulated translation is TRAP-seq (translating ribosome affinity purification; also call RiboTag-seq) where an epitope-tagged ribosomal protein (rp) introduced into animals (genetically or by viral injection) is incorporated into translating ribosomes followed by immunoprecipitation of ribosome-mRNA complexes and RNA-seq (Figure 3). Because the epitope tagged rp can be expressed in particular cell types, this method is useful for assessing the translatome in, for example, excitatory neurons derived from a whole brain extract. In one TRAP-seq study, GFP-tagged rpL10a expressed in mouse hippocampal CA1 pyramidal neurons exhibited increased co-precipitation with Chrm4 mRNA in Fmr1-KO versus control WT mice. Chrm4 encodes muscarinic acetylcholine receptor 4 (M4), a protein that is translated excessively upon Fmr1 depletion55. Another TRAP-seq study (with Cre-dependent HA-tagged rpL22) was combined with Cre-mediated CLIP in mouse CA1 pyramidal neurons to assess both FMRP binding and translation in the same cells36. These two experiments reported in references36 and55 revealed that in Fmr1-deficient neurons, the levels of many FMRP target mRNAs are reduced (as measured by the amount of RNA co-precipitated with HA-tagged ribosomes) and that the stronger the FMRP binding to a particular mRNA in wild type neurons, the greater the reduction of the RNA level in Fmr1-lacking neurons (for related studies cf35,56).
Figure 3. Methods for the analysis of transcriptome-wide translation.
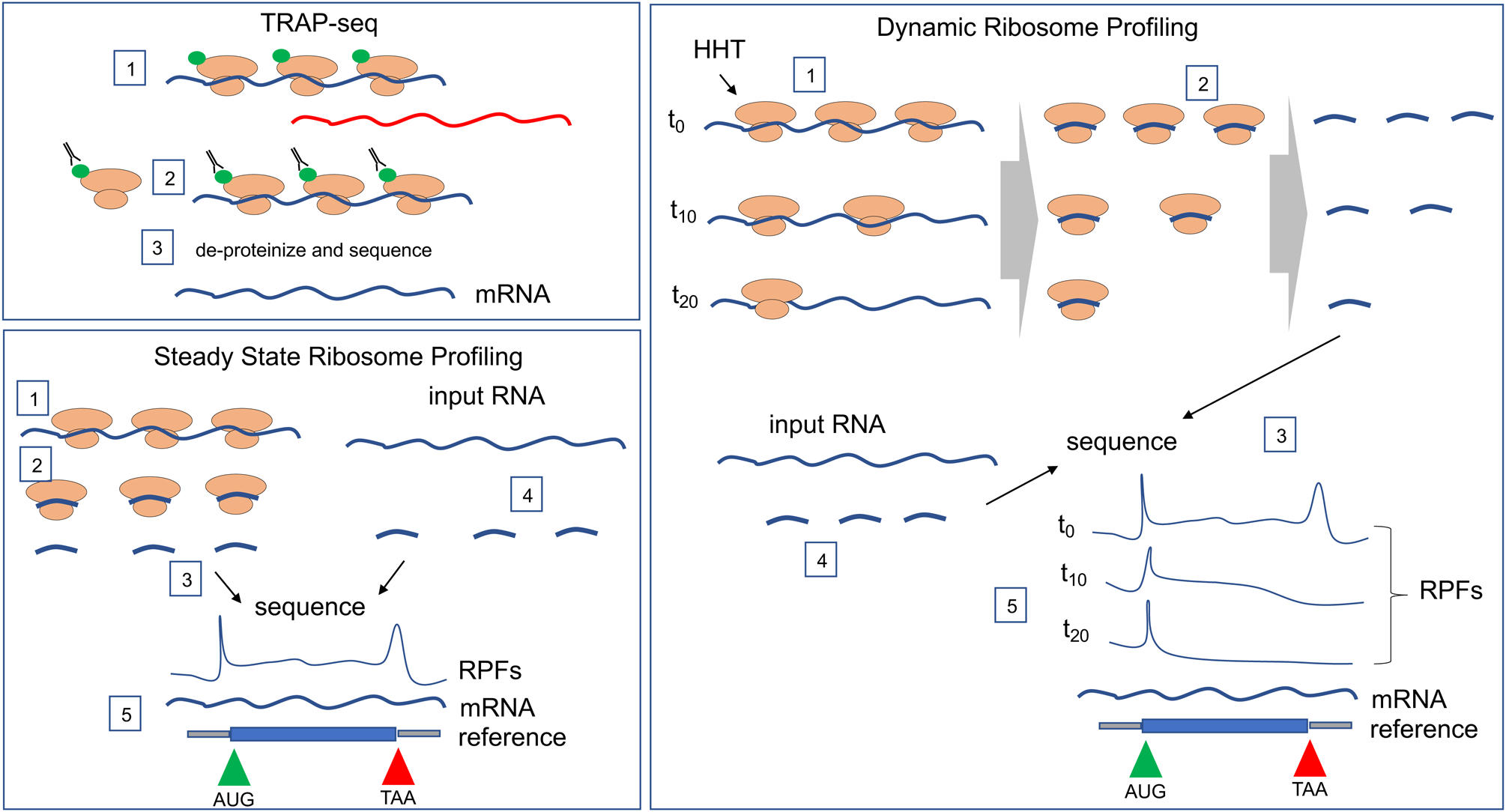
TRAP (translating ribosome affinity purification)-seq. Cells or tissues are transduced with an epitope-tagged ribosomal protein (green), which can be expressed in specific cells or tissues (step 1). The cells/tissues are then subjected to immunoprecipitation with antibody against the epitope (usually HA, FLAG, or GFP) (step 2); the co-precipitating RNA is then de-proteinized and sequenced (blue line) (step 3). Note that mRNAs associated with few or no ribosomes are not immunoprecipitted (red line). TRAP-seq has been performed in WT and Fmr1-deficient mouse brain35, 55 and has identified mRNAs under translational control by FMRP in specific neuron subtypes.
Steady state ribosome profiling. In cells or tissues, ribosomes are “frozen” on RNA by treatment with cycloheximide (step 1). The RNA is digested with RNase (step 2) and the small mRNA fragments protected from hydrolysis by the ribosomes are collected and sequenced (step 3). Input total RNA, which serves as a measure of all mRNA in the cells is also sequenced (step 4). The ribosome protected fragments (RPFs) and input mRNA are then aligned to the reference genome (step 5). RPFs are generally higher of the start (AUG) and stop (TAA) codons and are not present on 5’ or 3’ untranslated regions. Steady state ribosome profiling from WT and Fmr1-deficient cells or brain tissue has identified mRNAs whose translation is up and down regulated by FMRP58,59,60,61. Dynamic ribosome profiling. Cells or tissues are treated with homoherringtonine (HHT), which freezes ribosomes on initiation codons but allows translocating ribosomes to continue to elongate polypeptides (step 1). The ribosomes are evident at t10 and t20 when compared to t0. At various time points after HHT, the translocating ribosomes are frozen by treatment with cycloheximide (step 2), which is followed by ribosome profiling, as described above (step 3). Input RNA is also sequenced (step 4). Because there are fewer ribosomes at, for example, t20 relative to t0, there are fewer ribosome footprints. Ribosome footprints on the AUG start codon (green) are maintained throughout the time course where as footprints on the TAA stop codon (red) are generally reduced over time (step 5). The time it takes for ribosomes runoff following HHT treatment has demonstrated that on most mRNAs, ribosomes translocate quickly whereas on other mRNAs, the ribosomes translocate slowly or nearly not at all62. Dynamic ribosome profiling performed using WT and Fmr1-deficient mouse brain has revealed that FMRP stalls ribosomes on specific mRNAs62. Note that dynamic ribosome profiling, but not steady state ribosome profiling, can distinguish between translocating and stalled ribosomes.
Although TRAP-seq is a valuable method for identifying ribosome-bound mRNAs in specific cell types, it is perhaps less useful for examining detailed mechanisms of translational control. For example, the amount of any particular RNA immunoprecipitated by, say 4 or 8 epitope-tagged ribosomes might be similar (that is, the assay may not be sufficiently sensitive to distinguish 4 versus 8 epitopes, 1 epitope per ribosome), yet these transcripts would be expected to produce different amounts of protein. An alternative high-resolution method is ribosome profiling57 in which tissues or cells are incubated with cycloheximide to ‘freeze’ ribosomes that are translating mRNA, followed by RNase digestion, collection of the small (~29 base) ribosome-protected mRNA fragments, and sequencing. Input mRNA is sequenced as well, thereby yielding the number and positions of ribosome footprints relative to mRNA transcriptome-wide (Figure 3). This ratio, number of ribosome footprints relative to amount of input mRNA is referred to as translational efficiency (TE), a useful proxy for protein output. However, TE does not allow for the assessment of mRNAs associated with stalled ribosomes (that is, ribosomes that have initiated translation but that become stalled during the process of polypeptide elongation) (see below). Several studies have used ribosome profiling to investigate translational control by FMRP in various tissues and cells36,58,59,60,61,62,63. A case in point is adult mouse neural stem cells (aNSCs); they give rise to neurons and glia but when FMRP is absent, the cells over-proliferate and primarily differentiate into more glia at the expense of neurons64,65. Ribosome profiling of these cells demonstrated that Fmr1 deficiency resulted in increased or decreased ribosomes on specific transcripts, indicating that FMRP can be both an activator and repressor of translation58. This study also showed a remarkable degree of translational “buffering” in FMRP deficient aNSCs and mitochondrial ribosome mRNAs are enriched in this category. Buffering is a phenomenon where, for example, a reduction in the amount of an RNA is accompanied by an increased number of ribosomes such that the (predicted) protein output would be unaffected. In a similar vein, an increase in an RNA may be compensated by reduced ribosome number – again the predicted protein output would be unaffected. How translational buffering in the absence of FMRP occurs is unknown, but genetic and dosage compensatory mechanisms in general occur in probably most cell and animal models66. Liu et al58 also revealed remarkable biology through ribosome profiling; their studies demonstrated that the transcription factor Necdin was responsible for aNSC over-proliferation and improper differentiation into glia.
FMRP regulation of ribosome stalling
How does FMRP regulate translation? There are several clues: it binds primarily to CDS9, it represses general translation54 and it co-sediments with polyribosomes in sucrose gradients 502 These connections suggest that FMRP slows or stalls ribosome translocation, thereby reducing the rate of polypeptide elongation. Indirect assessments of ribosome translocation in brain extracts9,10 support this idea but the clearest evidence comes from dynamic ribosome profiling of the mouse hippocampus (Figure 3)62. Hippocampal slices of WT mice were treated with homoherringtonine (HHT), which freezes initiating ribosomes on translation start codons (usually AUG), but allows translocating ribosomes to continue to elongate polypeptides. Stopping ribosome translocation at various times by treatment with cycloheximide followed by ribosome profiling revealed ribosome transit rates on all mRNAs. This showed that although the ribosomes on most mRNAs wild type mice translocate quickly, other RNAs are associated with stalled or slowly moving ribosomes including many of those involved in neural and synaptic function Treatment of slices with HHT for 30 minutes – most of the ribosomes that can run off (that is, translocate on mRNAs until they terminate at a stop codon) have done so by this time – resulted in FMRP co-sedimenting in sucrose gradients with the few ribosomes that remained on mRNA62. RNA-seq from this 6–8 ribosome region of the gradient showed that about 50 mRNAs were reduced in amount in Fmr1-deficient slices relative to wild type slices. Because the input amounts of these RNAs were unaffected by genotype, one may infer that the reduction of RNAs in the polysome gradient was caused by the lack of FMRP to stall ribosomes. These RNAs encode many proteins with neural and synaptic functions as well as several that establish epigenetic chromatin marks (see FMRP and Chromatin).
By what mechanism could FMRP slow or stall ribosome translocation and how could it do so on specific mRNAs? FMRP binds RNA, so it could potentially act as a simple roadblock. Translocating ribosomes generate 13 picoNewtons of force, which is sufficient to unwind most stable secondary structures in RNA67 and perhaps not be impeded by RNA binding proteins such as FMRP (Figure 4). However, if FMRP binding to RNA is adequate to stop or slow a ribosome, then specific sequences identified by CLIP would be expected to strongly overlap with those showing elevated ribosome occupancy, which is not necessarily the case58,59,60,62. These observations suggest that FMRP may not merely act as roadblock to ribosome translocation. Thus, alternative mechanisms of ribosome stalling may underlie the effects of FMRP, including codon bias and inhibition of the association of tRNA and elongation factors with the ribosome.
Figure 4. Mechanisms of FMRP-regulated translation.
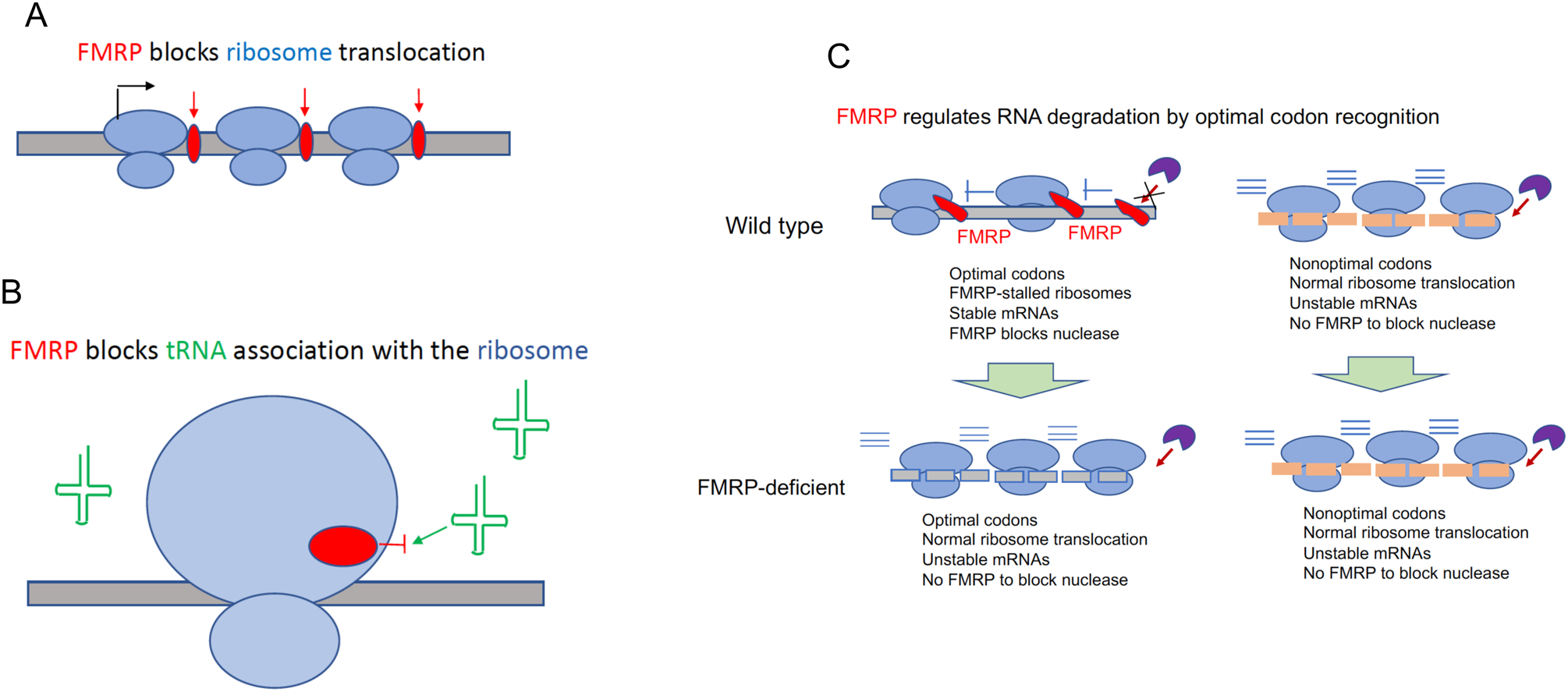
A. FMRP as a roadblock to ribosome translocation. FMRP binding to mRNA might impede ribosome translocation, thereby leading in reduced polypeptide elongation.
B. FMRP as a ribosomal protein that inhibits tRNA association with the ribosome. In vitro reconstitution experiments followed by cryo-EM analysis suggest that FMRP binds the 60S ribosomal subunit in such a manner as to prevent tRNA and/or elongation assembly into the ribosome, thereby causing ribosome stalling70
C. FMRP and codon optimality. According to one current model, FMRP directly or indirectly associates with the translational machinery on wild type mouse cortex mRNAs that have optimal codons and stalls ribosomes (as indicated by the blue |-). In this model, FMRP also blocks a hypothetical nuclease(s) (purple pac-man) to prevent RNA degradation. Because there is no FMRP association with the translational machinery on RNAs with nonoptimal codons, these RNAs have normal ribosome translocation but the mRNAs are unstable because nuclease attack is not hindered by FMRP. Because of this RNA instability, there is reduced protein production. In FMRP-deficient cortex, RNAs with optimal codons are associated with normally translocating ribosomes, but the RNAs are unstable because there is no FMRP to block prevent nuclease hydrolysis. RNAs with nonoptimal codons behave similarly in FMRP-deficient cortex as they do in wild type cortex63. According to this model, FMRP regulates protein synthesis primarily at the level of RNA degradation as opposed to translational control.
Codon bias in mRNAs affects ribosome translocation as well as RNA stability68,69. That is, mRNAs may have optimal codons whose cognate tRNAs are abundant, or nonoptimal codons whose cognate tRNAs are relatively rare. Ribosome stalling has been shown to occur on RNAs with nonoptimal codons68,69. In the brain, FMRP modulates the relationship between ribosome stalling and codon optimality63. A current model is depicted in Figure 4. In the wild type mouse cortex, FMRP directly or indirectly associates with the translational machinery on mRNAs with optimal codons and stalls ribosomes63. FMRP also blocks an as yet unidentified nuclease(s) to prevent RNA degradation. FMRP does not associate with the translational machinery on RNAs with nonoptimal codons63. On these RNAs, ribosome translocation is normal but the mRNAs tend to be unstable because there is no FMRP to block nuclease attack. In FMRP-deficient cortex, RNAs with nonoptimal codons behave similarly in FMRP-deficient cortex as they do in wild type cortex63. However, RNAs with optimal codons are associated with normally translocating ribosomes, but the RNAs are unstable because there is no FMRP to block nucleases. Therefore, FMRP deficiency can lead to either increased or decreased RNA stabilities depending on their codon optimality status, which impacts the gene network controlling cellular functions62.
In an intriguing study, recombinant, but not full-length, Drosophila FMRP was reconstituted with mammalian ribosomes in vitro and its structure analyzed by cryo-electron microscopy (cryo-EM)70. FMRP bound rpL5 with a stereo-configuration that would be predicted to inhibit association of tRNA and elongation factors to the ribosome and thereby induce stalling (Figure 4). Because these reconstitution experiments did not include mRNA or other translation factors, their biological relevance as yet is unclear. However, if similar results using mammalian FMRP and including mRNA and translation factors can be obtained, then this would offer a window into the mechanism of ribosome stalling. Even so, it is unclear how mRNA specificity could be achieved.
FMRP regulation of translation initiation
Studies have shown that FMRP may also influence the initiation of translation. CYFIP1/2 (Cytoplasmic FMRP Interacting Proteins 1 and 2), identified by a yeast two-hybrid screen71, are the most studied FMRP-associated proteins. Although CYFIP1 interacts only with FMRP, CYFIP2 interacts with FMRP and the paralogs FXR1 and 2 as well. The CYFIP proteins are components of the five subunit WAVE regulatory complex (WRC) that remodels the actin cytoskeleton72, which may indirectly influence translation. The authors of this study show that CYFIP bridges FMRP and the translation initiation factor eIF4E; by doing so, CYFIP prevents the scaffold protein eIF4G from binding eIF4E, which inhibits cap-dependent72 translation. It is possible that the conglomeration of FMRP/CYFIP/WRC-regulated translation of actin cytoskeleton dynamics controls dendritic spine morphogenesis, which is impaired in FXS72.
FMRP and RNA Modifications
In addition to its role as a translational repressor, recent studies indicate that FMRP contributes to other aspects of gene expression, including its regulation of RNA modifications.
FMRP Interactions with m6A
N6-methyladenosine, commonly referred to as m6A, is an RNA modification that has received considerable attention in the last several years as a multi-faceted regulator of posttranscriptional gene expression73. FMRP influences RNA stability74, translation75, as well as nucleus-to-cytoplasm export through interactions with m6A76. Mechanistically, it is not clear how FMRP affects these molecular processes that depend on this RNA modification, but it is noteworthy that there appears to be some overlap between FMRP CLIP targets and those harboring m6A77. This suggests that RNA modifications may assist in the selection of FMRP targets. However, how such selection is mediated and how FMRP recognize m6A marked transcripts remain unknown.
FMRP and RNA Editing
Adenosine deaminase acting on RNA (ADAR) is an editing enzyme that converts adenosine to inosine in double stranded RNA, which the cell recognizes as a guanosine. Consequently, translation of edited RNAs can result in amino acid substitutions, thereby increasing protein diversity78. FMRP interacts with ADAR in mice79, Drosophila,80, and zebrafish81, indicating an important function conserved through evolution. In human postmortem FXS brain, RNAs are generally hypo-edited, many of which are also hypo-edited in human postmortem autistic brain43. RNA editing, therefore, is another remarkable point of convergence between FXS and autism. Hints of how FMRP affects RNA editing come from experiments in Drosophila, where expression of an FMRP-ADAR transgene causes editing within RNA CDS82. These data indicate that ADAR catalytic activity is directed to certain sequences through FMRP-mediated RNA binding.
FMRP and Chromatin
It has recently become apparent that FMRP also has profound effects on nuclear events such as DNA damage response, transcription, and splicing. Several studies have shown that FMRP controls RNA synthesis through regulated translation of critical transcriptional factors or chromatin modulators62, 83. In a remarkable series of experiments, re-analysis of the original FMRP CLIP data discussed above (cf ref9) revealed a preponderance of RNAs encoding proteins with nuclear functions (110 out of 842), 44 of which modify chromatin, particularly histone acetylation and methylation83. In line with the identity of these enzymes as chromatin modifiers, large increases in H3K4me3, H4K8ac, and H4K19ac levels were observed in Fmr1 deficient neurons, which were accompanied by commensurate changes in the epigenetic chromatin landscapes. It was shown that Brd4, a bromodomain-containing transcription factor elevated in Fmr1 deficient mice could be targeted by JQ1, a small molecule inhibitor of this class of proteins. JQ1 mitigated aberrant dendritic spine density and several FXS-associated behaviors, suggesting a possible therapy to treat the disorder in humans.
Another chromatin modifier that is regulated by FMRP is SETD2, which catalyzes trimethylation of lysine 36 on histone H3 (H3K36me3). SETD2 mRNA is associated with FMRP-stalled ribosomes, but when FMRP is absent, translation of this transcript is elevated and results in an increase of SETD2 protein by ~2.5 fold62. A chromatin immunoprecipitation experiment followed by DNA sequencing (ChIP-seq) showed that the H3K36me3 landscape was altered in Fmr1 KO hippocampus. H3K36me3 predominantly resides within the body of genes and mediates alternative splicing. Not surprisingly then, there is widespread dysregulation of alternative splicing, particularly exon skipping, in Fmr1 deficient hippocampus. Interestingly, a number of these mis-splicing events also occur in human postmortem autistic brain, indicating a molecular convergence between FXS and other ASDs62.
A complementary study observed elevated levels of the histone acetyltransferase EP300 and ubiquitin-mediated destruction of the histone deacetylase HDAC1 in Fmr1-depleted adult mouse hippocampal neural stem cells84. Treating the cells with nutlin-3, a compound that inhibits the interaction between the ubiquitin ligase MDM2 and HDAC1 restored histone acetylation to normal levels in Fmr1-deficient NPCs, which not only rescued characteristic over-proliferation and differentiation defects of NSCs but also several behavioral deficits of Fmr1 deficient mice.
Not only does FMRP modify chromatin indirectly by regulating the translation of mRNAs encoding epigenetic and transcription factors62,83, in non-neuronal cells, it also acts directly in the nucleus to bind and alter chromatin structure85. By way of its Agenet domain, FMRP regulates the levels and positioning of gammaH2A.x, a histone H2 subtype associated with cell death, in response to replicative stress in mouse embryo fibroblasts and mammalian spermatocytes. Without FMRP, spermatocytes are unable to undergo DNA repair and resolve single stranded chromatin intermediates at the pachytene stage, a necessary event for meiotic progression. Whether FMRP directly regulates chromatin in neurons and if so whether it plays a role in activity and/or stress-induced cellular responses has not been assessed.
Linking FMRP molecular biology to FXS
By highlighting recent advances of the molecular biology of FMRP, we can now ask what new mechanisms need to be explored, what FMRP activity tells us about FXS, and whether this information can form the basis new therapeutic development to treat the disorder. One clear message is that FMRP acts at multiple levels of gene expression, and that an original driving force for investigating this protein – that it is a translational repressor – is certainly true but is far too simplistic. It is also too simplistic to think that FMRP performs the same molecular tasks in all cells. The fact that FMRP interacts with many different proteins and RNAs and in certain combinations in particular cells as noted in this essay makes it self-evident that molecular readouts of the protein will have cell type specificity (Figure 5). Even so, there are several general themes of FMRP molecular biology that are useful to consider, particularly where new avenues of investigation are needed.
Figure 5. Summary of FMRP activities.

FMRP is a multi-functional protein with diverse mechanisms of action; it binds mRNAs and may regulate their translation, stability, editing, or intracellular transport. FMRP also directly interacts with proteins (such as ion channels) and regulates their function. Finally, FMRP can regulate RNA synthesis by either controlling the expression of or modulating the activities of transcription factors and chromatin modifying enzymes
FMRP target selection
The observation that FMRP binds mainly coding sequences of mRNA has now been made several times in several cell types, but what remains unclear is how this occurs. Candidate cis recognition elements in RNA have been proposed, but they are far from absolute requirements46, 47. The G4 structure identified by FMRP in in vitro binding assays appears clear, yet the structure generally is not detected in CLIP studies and has been questioned as to whether it even exists in vivo86. What distinguishes CDS from UTRs are, of course, ribosomes, so perhaps ribosomes are involved in positioning where FMRP binds9. In any event, the mechanism by which FMRP binds CDS remains to be determined. Recent identification of cell type specific targets for FMRP strongly suggests that FMRP has distinct roles in different cell types in different brain regions. Therefore, studies of cell type-specific FMRP targets may point to certain brain circuitry that goes awry in FXS, which in turn could suggest more targeted therapies to mitigate pathophysiology of the syndrome.
FMRP regulates translation
This is certainly true, but it can repress or activate specific RNAs58. Many studies have now shown that FMRP regulates ribosome translocation, but how it does so is unknown. A key extant question is whether FMRP acts directly on the ribosome as suggested by in vitro reconstitution studies70. Even if FMRP does interact with the ribosome, it is not clear how mRNA specificity would simultaneously be achieved. The importance of understanding how FMRP controls translation is evidenced by a number of FXS rescue paradigms in mice that focus on the translational machinery or signaling pathways leading to it11. Specific molecules targeting translation factors or the ribosome could lead to new therapeutic development.
FMRP regulates nuclear activities including transcription and splicing
These are newly emerging functions of FMRP requiring further investigation. Two studies62,83 indicate that transcription and splicing are likely to be downstream activities of FMRP-regulated translation. On the other hand, another study demonstrates that FMRP acts directly on chromatin85. FMRP has an NLS and NES and although it is predominantly cytoplasmic at steady state, shuttling in and out of the nucleus may occur rapidly, which could be why potential nuclear activities of FMRP have been overlooked. Indeed, the etiology of autism is associated with changes of chromatin-modifying factors in early development and synaptic proteins primarily during postnatal periods. Because FMRP directly binds and regulates many mRNAs related to autism genes, one avenue for FXS treatment at early developmental stages could target epigenetic chromatin modulators.
FMRP and RNA modifications
Here, we refer to m6A and RNA editing by ADAR. These fields are rapidly evolving and are likely to expand the FMRP molecular biology universe in substantial ways. Key questions include: to what extent does FMRP recognize m6A and does it influence translation as well as RNA stability? how widespread is RNA editing by the FMRP-ADAR complex? and does RNA editing drive FXS pathophysiology? Although this field is still in its infancy, FMRP-ADAR and RNA editing pathways may provide a rich source of targets for therapeutic development.
FMRP does not act in isolation
FMRP likely forms unique protein complexes in different cell types and cellular compartments, and in response to certain stimuli87. We described some of them in this review (e.g., FMRP-CYFIP1, FMRP-ADAR), but a comprehensive identification of FMRP interacting proteins at a resolution that mimics that of CLIP-seq has not been performed in neural cell types. Even so, it is already evident that such proteins help define FMRP functions including mRNA target selection. A key extant question is whether differing patterns of expression of FMRP-interacting proteins help promote phenotypic diversity in FXS full mutation populations. The identification of FMRP-interacting proteins in specific neural cell types, while technically challenging, may provide a gateway to understanding how FXS is a spectrum of deficits.
Translation to therapy
A number of therapies to treat FXS are based on the neural activities that go awry when FMRP is absent, and for the most part do not take into account, nor do they need to account, just what FMRP does. For example, agonists or antagonists for certain synaptic receptors that modulate the hyper-excitability of neurons in FXS have been used frequently as therapies, albeit with mixed results5. Agents that regulate other signaling pathways to or from FMRP have also been used, for example the diabetes drug metformin, which shows some promise88. One possible new therapy could be based on the observation that ribosome translocation is excessive in FXS. Certain antibiotics slow or stop ribosomes, so perhaps these may useful therapeutically9. Going further, JQ1, the inhibitor of bromodomain-containing transcription factors that are over-synthesized in FXS ameliorates some pathophysiological symptoms of the disorder in mice83, and perhaps it may be extendable to humans.
The vast majority of successful therapeutic studies of FXS are based on highly in-bred mouse models but when limited tests are extended to human FXS populations the results have been mixed. Although general inter-species differences can certainly account for the mouse-human disparity, the genetic and epigenetic diversity among FXS individuals may limit the utility of mouse models. A deep molecular understanding of FMRP, its interactors and modulators, will be key elements in future clinical trials and therapeutic applications.
References
- 1.Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, & Nelson DL Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822 (1991). [DOI] [PubMed] [Google Scholar]
- 2.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, & Zhang FP Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991). [DOI] [PubMed] [Google Scholar]
- 3.Schaefer GB, & Mendelsohn NJ Genetics evaluation for the etiologic diagnosis of autism spectrum disorders. Genet Med 10, 4–12 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Wang LW, Berry-Kravis E, & Hagerman RJ Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics 7, 264–274 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berry-Kravis EM, Lindemann L, Jonch AE, Apostol G, Bear MF, Carpenter RL, Crawley JN, Curie A, Des Portes V, Hossain F, et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat Rev Drug Discov 17, 280–299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao X, & Bhattacharyya A Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am J Hum Genet 103, 829–857 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashley CT Jr., Wilkinson KD, Reines D, & Warren ST FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Siomi H, Siomi MC, Nussbaum RL, & Dreyfuss G The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74, 291–298 (1993). [DOI] [PubMed] [Google Scholar]
- 9.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism. Cell 146, 247–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Udagawa T, Farny NG, Jakovcevski M, Kaphzan H, Alarcon JM, Anilkumar S, Ivshina M, Hurt JA, Nagaoka K, Nalavadi VC, et al. Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat Med 19, 1473–1477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richter JD, Bassell GJ, & Klann E Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 16, 595–605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ligsay A, & Hagerman RJ Review of targeted treatments in fragile X syndrome. Intractable & rare diseases research 5, 158–167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gross C, Hoffmann A, Bassell GJ, & Berry-Kravis EM Therapeutic Strategies in Fragile X Syndrome: From Bench to Bedside and Back. Neurotherapeutics 12, 584–608 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakker CE, & Oostra BA Understanding fragile X syndrome: insights from animal models. Cytogenet Genome Res 100, 111–123 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Bhattacharyya A, & Zhao X Human pluripotent stem cell models of Fragile X syndrome. Molecular and cellular neurosciences 73, 43–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garber KB, Visootsak J, & Warren ST (2008). Fragile X syndrome. Eur J Hum Genet 16, 666–672 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sansone SM, Schneider A, Bickel E, Berry-Kravis E, Prescott C, & Hessl D Improving IQ measurement in intellectual disabilities using true deviation from population norms. J Neurodev Disord 6, 16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaufmann WE, Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics 139(Suppl 3), S194–S206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson DL, Orr HT, & Warren ST The unstable repeats – three evolving faces of neurologic disease. Neuron 77, 825–843 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myrick LK, Hashimoto H, Cheng X, & Warren ST Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain. Hum Mol Genet. 24, 1733–1740 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adinolfi S, Ramos A, Martin SR, Dal Piaz F, Pucci P, Bardoni B, Mandel JL, & Pastore A The N-terminus of the fragile X mental retardation protein contains a novel domain involved in dimerization and RNA binding. Biochemistry 42, 10437–10444 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Ramos A, Hollingworth D, Adinolfi S, Castets M, Kelly G, Frenkiel TA, Bardoni B, & Pastore A The structure of the N-terminal domain of the fragile X mental retardation protein: a platform for protein–protein interaction. Structure 14, 21–31 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Eberhart DE, Malter HE, Feng Y, & Warren ST The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet 5, 1083–1091 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Ikeuchi Y, Malumbres M, & Bonni A A Cdh1-APC/FMRP ubiquitin signaling link drives mGluR-dependent synaptic plasticity in the mammalian rain. Neuron 86, 726–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O, Warren ST, & Bassell GJ Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation and mGluR signaling. Mol Cell. 42, 673–688 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim TH, Tsang B, Vernon RM, Sonenberg N, Kay LE, & Forman-Kay JD Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 365, 825–829 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Tsang B, Arsenault J, Vernon RM, Lin H, Sonenberg N, Wang L, Bah A, & Forman-Kay JD Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc Natl Acad Sci U S A. 116, 4218–4227 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Althar YM, & Joseph S RNA binding specificity of the human fragile X mental retardation protein. J Mol Biol 432, 3851–3868 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, & Darnell RB Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107, 489–499 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Vasilyev N, Polonskaia A, Darnell JC, Darnell RB, Patel DJ, & Serganov A Crystal structure reveals specific recognition of a G-quadruplex RNA by a β-turn in the RGG motif of FMRP. Proc Natl Acad Sci U S A. 112, E5391–E5400 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darnell JC, Fraser CE, Mostovetsky O, Stefani G, Jones TA, Eddy SR, & Darnell RB Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 19, 903–918 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, & Eberwine J RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37, 417–431 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Maurin T, Lebrigand K, Castagnola S, Paquet A, Jarjat M, Popa A, Grossi M, Rage F, & Bardoni B HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic Acids Research, gky267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Driesche SJ, Sawicka K, Zhang C, Hung SKY, Park CY, Fak JJ, Yang C, Darnell RB, & Darnell JC FMRP binding to a ranked subset of long genes is revealed by coupled CLIP and TRAP in specific neuronal cell types. bioRxiv 762500 (2019). [Google Scholar]
- 36.Sawicka K, Hale CR, Park CY, Fak JJ, Gresack JE, Van Driesche SJ, Kang JJ, Darnell JC, & Darnell RB FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. Elife 8, e46919 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Fang Z, Jud C, Vansteensel MJ, Kaasik K, Lee CC, Albrecht U, Tamanini F, Meijer JH, Oostra BA, & Nelson D L Fragile X-related proteins regulate mammalian circadian behavioral rhythms. Am J Hum Genet. 83, 43–52 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwan KY, Lam MM, Johnson MB, Dube U, Shim S, Rasin MR, Sousa AM, Fertuzinhos S, Chen JG, Arellano JI, et al. Species-dependent posttranscriptional regulation of NOS1 by FMRP in the developing cerebral cortex. Cell 149, 899–911 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, Zhang SC, Abbeduto L, & Bhattacharyya A iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev 23, 1777–1787 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khalfallah O, Jarjat M, Davidovic L, Nottet N, Cestèle S, Mantegazza M, & Bardoni B Depletion of the fragile X mental retardation protein in embryonic stem cells alters the kinetics of neurogenesis. Stem Cells 35, 374–385 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Telias M, Mayshar Y, Amit A, & Ben-Yosef D Molecular mechanisms regulating impaired neurogenesis of fragile X syndrome human embryonic stem cells. Stem cells and development 24, 2353–2365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ascano M Jr., Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tran SS, Jun HI, Bahn JH, Azghadi A, Ramaswami G, Van Nostrand EL, Nguyen TB, Hsiao YE, Lee C, Pratt GA, et al. Widespread RNA editing dysregulation in brains from autistic individuals. Nature Neuroscience 22, 25–36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li M, Shin J, Risgaard RD, Parries MJ, Wang J, Chasman D, Liu S, Roy S, Bhattacharyya A, & Zhao X Identification of FMR1-regulated molecular networks in human neurodevelopment. Genome Res. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chasman D, Fotuhi Siahpirani A, & Roy S Network-based approaches for analysis of complex biological systems. Current Opinion in Biotechnology 39, 157–166 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Anderson BR, Chopra P, Suhl JA, Warren ST, & Bassell GJ Identification of consensus binding sites clarifies FMRP binding determinants. Nucleic Acids Res 44, 6649–6659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suhl JA, Chopra P, Anderson BR, Bassell GJ, & Warren ST Analysis of FMRP mRNA target datasets reveals highly associated mRNAs mediated by G-quadruplex structures formed via clustered WGGA sequences. Hum Mol Genet 23, 5479–5491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmad M, Shen W, Li W, Xue Y, Zou S, Xu D, & Wang W Topoisomerase 3beta is the major topoisomerase for mRNAs and linked to neurodevelopment and mental dysfunction. Nucleic Acids Res 45, 2704–2713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SK, Xue Y, Shen W, Zhang Y, Joo Y, Ahmad M, Chinen M, Ding Y, Ku WL, De S, et al. (2018). Topoisomerase 3beta interacts with RNAi machinery to promote heterochromatin formation and transcriptional silencing in Drosophila. Nat Commun 9, 4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu D, Shen W, Guo R, Xue Y, Peng W, Sima J, Yang J, Sharov A, Srikantan S, Yang J, et al. Top3beta is an RNA topoisomerase that works with fragile X syndrome protein to promote synapse formation. Nat Neurosci 16, 1238–1247 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, & Warren ST FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell 1, 109–118 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Khandjian EW, Corbin F, Woerly S, & Rousseau F The fragile X mental retardation protein is associated with ribosomes. Nat Genet. 12, 91–93 (1996). [DOI] [PubMed] [Google Scholar]
- 54.Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, & Bear MF Correction of fragile X syndrome in mice. Neuron 56, 955–962 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomson SR, Seo SS, Barnes SA, Louros SR, Muscas M, Dando O, Kirby C, Wyllie DJA, Hardingham GE, Kind PC, & Osterweil EK Cell-type-specific translation profiling reveals novel strategy for treating fragile X syndrome. Neuron 95, 550–563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ceolin L, Bouquier N, Vitre-Boubaker J, Rialle S, Severac D, Valjent E, Perroy J, & Puighermanal E Cell Type-Specific mRNA dysregulation in hippocampal CA1 pyramidal neurons of the fragile X syndrome mouse model. Front Mol Neurosci.10, 340 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu B, Li Y, Stackpole EE, Novak A, Gao Y, Zhao Y, Zhao X, & Richter JD Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation.. Proc Natl Acad Sci U S A. :E11397–E11405 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu B, Molinaro G, Shu H, Stackpole EE, Huber KM, & Richter JD Optimization of ribosome profiling using low-input brain tissue from fragile X syndrome model mice. Nucleic Acids Res. 47:e25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das Sharma S, Metz JB, Li H, Hobson BD, Hornstein N, Sulzer D, Tang G, & Sims PA Widespread alterations in translation elongation in the brain of juvenile Fmr1 knockout mice. Cell Rep. 26, 3313–3322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greenblatt EJ, & Spradling AC Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science 361, 709–712 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shah S, Molinaro G, Liu B, Wang R, Huber KM, & Richter JD FMRP control of ribosome translocation promotes chromatin modifications and alternative splicing of neuronal genes linked to autism. Cell Rep. 30, 4459–4472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shu S, Donnard E, Liu B, Wang R, & Richter JD FMRP links optimal codons to mRNA stability in neurons. bioRxiv 801449; doi: 10.1101/801449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo Y, et al. Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 6, e1000898 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo W, Murthy AC, Zhang L, Johnson EB, Schaller EG, Allan AM, & Zhao X Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet 21, 681–691 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El-Brolosy MA, & Stainier DYR Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 13, e1006780 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu T, Kaplan A, Alexander L, Yan S, Wen J, Lancaster L, Wickersham CE, Fredrick K, Noller H, Tinoco I Jr, & Bustamante CJ Direct measurement of the mechanical work during translocation by the ribosome. eLife 3, e03406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Richter JD, & Coller J Pausing on polysomes: make way for elongation in translational control. Cell 16, 292–300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanson G, & Coller J Codon optimality, bias and usage in translation and mRNA decay. Nat Rev. Mol. Cell. Biol 19, 20–30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen E, Sharma MR, Shi X, Agrawal RK, & Joseph S Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol Cell. 54, 407–417 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schenck A, Bardoni B, Moro A, Bagni C, & Mandel JL A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci U S A 98, 8844–8849 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.De Rubeis S, et al. CYFIP1 Coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 79, 1169–1182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meyer KD, & Jaffrey SR Rethinking m6A readers, writers, and erasers. Ann. Rev. Cell Dev. Biol 33, 319–342 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, Song H, Wu H, Shu Q, & Jin P Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet. 27, 3936–3950 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edupuganti RR., Geiger S, Lindeboom RGH, Shi H, Hsu PJ, Lu Z, Wang SY, Baltissen MPA, Jansen PW & Rossa M N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol 24, 870–878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Edens BM, Vissers C, Su J, Arumugam S, Xu Z, Shi H, Miller N, Ringeling FR, Ming G, He C, Song H, & Ma YC FMRP modulates neural differentiation through m6A-dependent mRNA nuclear export;. Cell Rep. 28, 845–854 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Engel M et al. The role of m6A/m-RNA methylation in stress response regulation. Neuron 99, 389–403 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reich DP, & Bass BL Mapping the dsRNA world. Cold Spring Harb Perspect Biol. 11, a035352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan MH et al. Dynamic landscape and regulation of RNA editing in mammals Nature 550, 249–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bhogal B, Jepsen JE, Savva YA, Pepper A, Reenan RA, & Jongens TA Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat Neurosci 14, 1517–1524 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shamay-Ramot A et al. Fmrp interacts with Adar and regulates RNA editing, synaptic density and locomotor activity in zebrafish. PLoS Genet. 11, e1005702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McMahon AC, Rahman R, Jin H, Shen JL, Fieldsend A, Luo W, & Rosbash M TRIBE: hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell 165, 742–753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Korb E, Herre M, Zucker-Scharff I, Gresack J, Allis CD, & Darnell RB Excess translation of epigenetic regulators contributes to fragile X syndrome and Is alleviated by Brd4 inhibition. Cell 170, 1209–1223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Y, Stockton ME, Eisinger BE, Zhao Y, Miller JL, Bhuiyan I, Gao Y, Wu Z, Peng J, & Zhao X Reducing histone acetylation rescues cognitive deficits in a mouse model of Fragile X syndrome. Nat Commun 9, 2494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alpatov R, et al. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell 157, 869–881 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guo JU, & Bartel DP RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 353, aaf5371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pasciuto E, & Bagni C SnapShot: FMRP interacting proteins. Cell 159, 218. [DOI] [PubMed] [Google Scholar]
- 88.Protic D, Salcedo-Arellano MJ, Dy J, Potter LA, & Hagerman RJ New targeted treatments for fragile X syndrome. Curr Pediatr Rev. 15, 251–258 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown V, Small K, Lakkis L, Feng Y, Gunter C, Wilkinson KD, & Warren ST Purified recombinant Fmrp exhibits selective RNA binding as an intrinsic property of the fragile X mental retardation protein. J Biol Chem. 273, 15521–15527 (1998). [DOI] [PubMed] [Google Scholar]
- 90.Devys D, Lutz Y, Rouyer N, Bellocq JP, & Mandel JL The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 4, 335–340 (1993). [DOI] [PubMed] [Google Scholar]
- 91.Heiman M, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]