

Abstract
Background
People with type 1 diabetes mellitus (T1DM) need treatment with insulin for survival. Whether any particular type of (ultra‐)long‐acting insulin provides benefit especially regarding risk of diabetes complications and hypoglycaemia is unknown.
Objectives
To compare the effects of long‐term treatment with (ultra‐)long‐acting insulin analogues to NPH insulin (neutral protamine Hagedorn) or another (ultra‐)long‐acting insulin analogue in people with type 1 diabetes mellitus.
Search methods
We searched the Cochrane Central Register of Controlled Trials, MEDLINE, Scopus, ClinicalTrials.gov, the World Health Organization (WHO) International Clinical Trials Registry Platform and the reference lists of systematic reviews, articles and health technology assessment reports. We explored the US Food and Drug Administration (FDA) and European Medical Agency (EMA) web pages. We asked pharmaceutical companies, EMA and investigators for additional data and clinical study reports (CSRs). The date of the last search of all databases was 24 August 2020.
Selection criteria
We included randomised controlled trials (RCTs) with a duration of 24 weeks or more comparing one (ultra‐)long‐acting insulin to NPH insulin or another (ultra‐)long‐acting insulin in people with T1DM.
Data collection and analysis
Two review authors assessed risk of bias using the new Cochrane 'Risk of bias' 2 (RoB 2) tool and extracted data. Our main outcomes were all‐cause mortality, health‐related quality of life (QoL), severe hypoglycaemia, non‐fatal myocardial infarction/stroke (NFMI/NFS), severe nocturnal hypoglycaemia, serious adverse events (SAEs) and glycosylated haemoglobin A1c (HbA1c). We used a random‐effects model to perform meta‐analyses and calculated risk ratios (RRs) and odds ratios (ORs) for dichotomous outcomes and mean differences (MDs) for continuous outcomes, using 95% confidence intervals (CIs) and 95% prediction intervals for effect estimates. We evaluated the certainty of the evidence applying the GRADE instrument.
Main results
We included 26 RCTs. Two studies were unpublished. We obtained CSRs, clinical study synopses or both as well as medical reviews from regulatory agencies on 23 studies which contributed to better analysis of risk of bias and improved data extraction. A total of 8784 participants were randomised: 2428 participants were allocated to NPH insulin, 2889 participants to insulin detemir, 2095 participants to insulin glargine and 1372 participants to insulin degludec. Eight studies contributing 21% of all participants comprised children. The duration of the intervention varied from 24 weeks to 104 weeks.
Insulin degludec versus NPH insulin: we identified no studies comparing insulin degludec with NPH insulin.
Insulin detemir versus NPH insulin (9 RCTs): five deaths reported in two studies including adults occurred in the insulin detemir group (Peto OR 4.97, 95% CI 0.79 to 31.38; 9 studies, 3334 participants; moderate‐certainty evidence). Three studies with 870 participants reported QoL showing no true beneficial or harmful effect for either intervention (low‐certainty evidence). There was a reduction in severe hypoglycaemia in favour of insulin detemir: 171/2019 participants (8.5%) in the insulin detemir group compared with 138/1200 participants (11.5%) in the NPH insulin group experienced severe hypoglycaemia (RR 0.69, 95% CI 0.52 to 0.92; 8 studies, 3219 participants; moderate‐certainty evidence). The 95% prediction interval ranged between 0.34 and 1.39. Only 1/331 participants in the insulin detemir group compared with 0/164 participants in the NPH insulin group experienced a NFMI (1 study, 495 participants; low‐certainty evidence). No study reported NFS. A total of 165/2094 participants (7.9%) in the insulin detemir group compared with 102/1238 participants (8.2%) in the NPH insulin group experienced SAEs (RR 0.95, 95% CI 0.75 to 1.21; 9 studies, 3332 participants; moderate‐certainty evidence). Severe nocturnal hypoglycaemia was observed in 70/1823 participants (3.8%) in the insulin detemir group compared with 60/1102 participants (5.4%) in the NPH insulin group (RR 0.67, 95% CI 0.39 to 1.17; 7 studies, 2925 participants; moderate‐certainty evidence). The MD in HbA1c comparing insulin detemir with NPH insulin was 0.01%, 95% CI ‐0.1 to 0.1; 8 studies, 3122 participants; moderate‐certainty evidence.
Insulin glargine versus NPH insulin (9 RCTs): one adult died in the NPH insulin group (Peto OR 0.14, 95% CI 0.00 to 6.98; 8 studies, 2175 participants; moderate‐certainty evidence). Four studies with 1013 participants reported QoL showing no true beneficial effect or harmful effect for either intervention (low‐certainty evidence). Severe hypoglycaemia was observed in 122/1191 participants (10.2%) in the insulin glargine group compared with 145/1159 participants (12.5%) in the NPH insulin group (RR 0.84, 95% CI 0.67 to 1.04; 9 studies, 2350 participants; moderate‐certainty evidence). No participant experienced a NFMI and one participant in the NPH insulin group experienced a NFS in the single study reporting this outcome (585 participants; low‐certainty evidence). A total of 109/1131 participants (9.6%) in the insulin glargine group compared with 110/1098 participants (10.0%) in the NPH insulin group experienced SAEs (RR 1.08, 95% CI 0.63 to 1.84; 8 studies, 2229 participants; moderate‐certainty evidence). Severe nocturnal hypoglycaemia was observed in 69/938 participants (7.4%) in the insulin glargine group compared with 83/955 participants (8.7%) in the NPH insulin group (RR 0.83, 95% CI 0.62 to 1.12; 6 studies, 1893 participants; moderate‐certainty evidence). The MD in HbA1c comparing insulin glargine with NPH insulin was 0.02%, 95% CI ‐0.1 to 0.1; 9 studies, 2285 participants; moderate‐certainty evidence.
Insulin detemir versus insulin glargine (2 RCTs),insulin degludec versus insulin detemir (2 RCTs), insulin degludec versus insulin glargine (4 RCTs): there was no evidence of a clinically relevant difference for all main outcomes comparing (ultra‐)long‐acting insulin analogues with each other.
For all outcomes none of the comparisons indicated differences in tests of interaction for children versus adults.
Authors' conclusions
Comparing insulin detemir with NPH insulin for T1DM showed lower risk of severe hypoglycaemia in favour of insulin detemir (moderate‐certainty evidence). However, the 95% prediction interval indicated inconsistency in this finding. Both insulin detemir and insulin glargine compared with NPH insulin did not show benefits or harms for severe nocturnal hypoglycaemia. For all other main outcomes with overall low risk of bias and comparing insulin analogues with each other, there was no true beneficial or harmful effect for any intervention. Data on patient‐important outcomes such as QoL, macrovascular and microvascular diabetic complications were sparse or missing. No clinically relevant differences were found between children and adults.
Plain language summary
Do people with type 1 diabetes mellitus benefit from using a different type of insulin as their basal insulin?
Background
Diabetes is a condition that causes a person's blood sugar (glucose) level to become too high. Insulin is a hormone that is released by the pancreas (a small organ behind the stomach) which controls the blood levels of glucose. In people with type 1 diabetes mellitus (T1DM) the pancreas does not produce any insulin, so the person has to inject insulin to control the glucose levels and keep well. The goal of insulin therapy is to provide insulin that mimics physiologic insulin secretion. Insulin is given by an injection under the skin (subcutaneous) by means of insulin syringes, insulin pens or insulin pumps. In order to control blood glucose levels in periods of fasting, basal or background insulin is needed. Basal insulin can be given by means of daily or twice‐daily injections of an intermediate‐acting or (ultra‐)long‐acting insulin. Basal insulin can be given as intermediate‐acting human neutral protamine Hagedorn (NPH) insulin or as (ultra‐)long‐acting analogue insulin (synthetic insulin). Bolus insulin is taken at mealtime (prandial insulin) to control blood glucose levels following a meal and is given by means of short‐acting or rapid‐acting insulin. The aim for most people with T1DM is to achieve near‐normal blood glucose levels to avoid long‐term complications such as kidney and eye disease and to allow flexibility regarding time, type and amount of food intake. The major unwanted effect of insulin therapy is hypoglycaemia (low blood glucose) which can be severe.
We wanted to find out whether one type of (ultra‐)long‐acting insulin compared with NPH insulin or another type of (ultra‐)long‐acting insulin is better for people with T1DM. The outcomes we were specifically interested in were death, health‐related quality of life, severe (night‐time) hypoglycaemia, serious unwanted events, non‐fatal complications of diabetes (heart attacks, strokes) and levels of glycosylated haemoglobin A1c (HbA1c) which is an indicator of long‐term glucose control.
What did we look for?
We searched medical databases and contacted pharmaceutical manufacturers and drug regulatory agencies for studies that: — were randomised controlled trials (medical studies where participants are put randomly into one of the treatment groups); — included people with T1DM; — compared one (ultra‐)long‐acting insulin with another (ultra‐)long‐acting insulin or NPH insulin; — lasted at least 24 weeks.
What did we find? We found 26 studies including a total of 8780 participants (21% were children). The studies lasted between 24 weeks and two years. They compared: — NPH insulin with insulin detemir (nine studies); — NPH insulin with insulin glargine (nine studies); — Insulin detemir with insulin glargine (two studies); — Insulin degludec with insulin detemir (two studies); — Insulin degludec with insulin glargine (four studies).
No study compared NPH insulin with insulin degludec.
Key results
There were no clear differences for all main outcomes comparing (ultra‐)long‐acting insulin analogues with each other.
Severe hypoglycaemic episodes were reduced with insulin detemir: among 1000 participants using NPH insulin, 115 would experience severe hypoglycaemia; using insulin detemir there would be 36 participants fewer (9 to 55 participants fewer) experiencing severe hypoglycaemia. However, the results were inconsistent, meaning if another study was performed there may not be a clear difference between insulin detemir and NPH insulin. There was no clear difference regarding the risk of severe night‐time hypoglycaemia. There were no clear differences for health‐related quality of life, serious unwanted effects or HbA1c levels. Very few people experienced a heart attack or died, and stroke was not reported.
There were no clear differences comparing insulin glargine with NPH insulin for all main outcomes. Very few people experienced a heart attack, stroke or died.
There were also no clear differences for all comparisons between children and adults.
Certainty of the evidence In the comparison of the insulin analogues detemir and glargine with NPH insulin, we are moderately confident about the results for death, severe (night‐time) hypoglycaemia, serious unwanted effects and HbA1c levels. We are uncertain about the effects on heart attacks, stroke and health‐related quality of life, mainly because there were only a few studies which did not last long enough to reliably investigate these outcomes.
How up to date is this review? This evidence is up‐to‐date as of 24 August 2020.
Summary of findings
Background
Description of the condition
Onset of type 1 diabetes mellitus (T1DM) can occur at any age and accounts for about 5% to 10% of all diabetes mellitus cases (Daneman 2006). It is a metabolic disease caused by an autoimmune destruction of pancreatic β‐cells which results in a deficiency of insulin secretion. What causes the pathological autoimmune response is not yet fully understood but includes genetic susceptibility in combination with an environmental trigger (Field 1997; Maahs 2010; van der Werf 2007). The incidence of T1DM varies geographically, being highest in Northern Europe (Karvonen 1993). Over the years, a worldwide increase in incidence has been observed, the reasons for which are not yet clear (Onkamo 1999; Pitkaniemi 2004).
Description of the intervention
For people with T1DM, the goal of insulin therapy is to provide insulin that mimics physiologic insulin secretion. The most commonly used administration of insulin is by subcutaneous injection (ADA 2019). Insulin is usually applied through insulin syringes, insulin pens or insulin pumps. In order to control blood glucose levels in periods of fasting and to enable cells to incorporate glucose for production of energy, basal or background insulin is needed, which can be given by means of daily or twice‐daily injections of an intermediate‐acting or (ultra‐)long‐acting insulin preparation. Bolus insulin is taken at mealtime (prandial insulin) to control blood glucose levels following a meal and is given by means of short‐acting or rapid‐acting insulin, usually before meals (ADA 2019). With insulin pump‐based treatments, a continuous delivery of rapid‐acting insulin is administered through the pump, with the addition of mealtime insulin bolus (basal‐bolus regimen). The aim for most people with T1DM is to achieve near‐normal glycaemic levels (ADA 2019) and to allow flexibility regarding time, type and amount of food intake which can best be mastered through structured patient‐education programmes (Pillay 2015).
Since the early 1920s, people with diabetes were treated with insulin, which was purified from bovine or porcine pancreas (animal insulin). Recombinant 'human' insulin was first produced in Escherichia coli in 1978 by combining the expressed insulin A‐ and B‐chains (Chance 1993). In 1982, the first insulin utilising recombinant deoxyribonucleic acid (DNA) technology was marketed. At present, insulin is being produced predominantly in Escherichia coli and yeasts (Chance 1993).
The choice of basal insulin depends upon patient and prescriber preferences, 'lifestyle' and economic and health system considerations. Historically, intermediate‐ and long‐acting insulin preparations were obtained by crystallising either protamine (Neutral Protamin Hagedorn (NPH) type, also known as isophane insulin) or zinc (Lente type). Most insulins have a concentration of 100 units per mL (U100) but more concentrated insulin formulations (U200, U300, U500) are currently available (Heinemann 2019). Soluble human insulin consists of different oligomers (monomers, dimers and hexamers). When administered subcutaneously, insulin monomers and dimers are readily absorbed by blood capillaries. Before dissociation of hexamers into dimers and monomers, the crystalline structures need to dissolve, and this process prolongs the absorption phase and contributes to pharmacokinetic variability between injections. Hence, the rate of insulin absorption is fastest for monomers followed by dimers and hexamers, respectively (Gradel 2018). Treatment with intermediate‐acting human insulins has drawbacks: NPH is associated with a pronounced insulin peak following injection, which seems to be associated with variable absorption (Heinemann 2000; Lepore 2000) and an increased risk of hypoglycaemia (Tricco 2014).
In order to achieve the potential benefits of near‐normal glycaemic control with a reduced risk of hypoglycaemia, new insulins have been introduced to the market. In an effort to provide insulin with a more suitable physiological time course to persons with diabetes mellitus, insulin analogues have been developed. Insulin analogues are insulin‐like molecules, engineered on the basis of the molecular structure of human insulin by changing the amino acid sequence and physiochemical properties. Four main (ultra‐)long‐acting insulin analogues are currently available on the market: two long‐acting insulin analogues (insulin detemir and insulin glargine U100), and two ultra‐long‐acting insulin analogues (insulin degludec and insulin glargine U300). The glargine U300 formulation has a more extended time‐action profile than glargine U100 and is thought to achieve a more stable glycaemic control (Yale 2018).
Because the patent of insulin glargine has expired, biosimilar insulins have become available on the market. Biosimilar insulin glargine is a biological copy of the original insulin glargine which is believed to have comparable quality, efficacy and safety. Biosimilar insulin glargine is cheaper than the original insulin glargine (Soldatov 2019).
Adverse effects of the intervention
The risk of developing hypoglycaemic episodes varies among studies depending on the definition of hypoglycaemia and the desired glycaemic target (Kahler 2014). Due to a more sustainable molecule structure of insulin analogues, studies have indicated a reduced risk of severe hypoglycaemia compared with NPH insulin (Tricco 2014). However, data are conflicting (Laranjeira 2018). Targeting lower glycosylated haemoglobin A1c (HbA1c) levels is often difficult to achieve and leads to a higher incidence of hypoglycaemic events (Kahler 2014). However, targeting near‐normal glucose levels in order to avoid detrimental long‐term consequences of hyperglycaemia is currently recommended in most people with type 1 diabetes (ADA 2019).
Compared to human insulin, some insulin analogues have shown higher mitogenic potency and insulin‐growth factor binding affinity in in‐vitro and animal studies (Grant 1993; Jorgensen 1992; King 1985; Kurtzhals 2000). These effects differ depending on the insulin analogue, but results provided in these studies are unable to clarify their relevance for people with diabetes mellitus. The American and European pharmaceutical registration agencies, the Food and Drug Administration (FDA) and the European Medicines Agency (EMA), have commented on the mitogenic and carcinogenic potency of long‐acting insulin analogues and concluded that there appear to be few detrimental effects (EMA 2003; EMA 2004; EMA 2012; FDA 2000; FDA 2005). Observational studies have shown conflicting results regarding cancer risk with insulin analogues compared with human insulin (Hemkens 2009; Ruiter 2012).
The insulin analogues are usually more expensive than NPH insulin (Ewen 2019). While price differences may not be a major problem for health services in high‐income countries, they may be important in low‐ and middle‐income countries.
How the intervention might work
Based on the altered time‐action profiles of (ultra‐)long‐acting insulin analogues, a number of possible advantages in the therapy of people with T1DM have been suggested. For instance, it has been hypothesised that the longer action and the less pronounced insulin peak will enable both improved glycaemic control and reduced risk of hypoglycaemia (Tricco 2014).
Why it is important to do this review
Although their pharmacokinetic profiles appeared to indicate that (ultra‐)long‐acting insulin analogues improve the insulin therapy of people with diabetes mellitus, their superiority in a clinical setting has still to be demonstrated (Hemmingsen 2019). Systematic reviews comparing the benefits and harms of insulin analogues with NPH insulin exist, but they have methodological deficiencies due to lack of identification of all relevant studies, missing analysis of clinical study reports (CSR) and poor 'Risk of bias' assessment (Laranjeira 2018; Tricco 2014).
Objectives
To compare the effects of long‐term treatment with (ultra‐)long‐acting insulin analogues to NPH insulin (neutral protamine Hagedorn) or another (ultra‐)long‐acting insulin analogue in people with type 1 diabetes mellitus.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs).
Types of participants
Non‐pregnant people withT1DM.
Types of interventions
We planned to investigate the following comparisons of intervention versus comparator.
Intervention
Long‐acting insulin analogues (insulin glargine U100 or insulin detemir) and their biosimilar insulins.
Ultra‐long‐acting insulin analogues (insulin glargine U300 or insulin degludec).
Comparisons
Long‐acting insulin analogue or its biosimilar insulin versus human NPH insulin.
Ultra‐long‐acting insulin analogue or its biosimilar insulin versus human NPH insulin.
(Ultra‐)long‐acting insulin analogue versus another (ultra‐)long‐acting insulin analogue.
Concomitant interventions had to be the same in both the intervention and comparator groups to establish fair comparisons.
Only studies reporting on subcutaneously administered insulin were be considered for inclusion in this review.
If a study included multiple arms, we included any arm that met our inclusion criteria.
Minimum duration of intervention
We included studies with a minimum duration of 24 weeks. In the case of a cross‐over RCT, each intervention period had to be at least 24 weeks.
Minimum duration of follow‐up
Minimum duration of follow‐up was 24 weeks. In the case of a cross‐over RCT, duration of follow‐up for each intervention period had to be at least 24 weeks.
We defined any follow‐up period going beyond the original time frame for the primary outcome measure as specified in the power calculation of the study's protocol as an extended follow‐up period (also called 'open‐label extension study') (Buch 2011; Megan 2012).
Types of outcome measures
We did not exclude a study if it failed to report one or several of our primary or secondary outcome measures. If none of our primary or secondary outcomes was reported in the study, we did not include the study but provided some basic information in the 'Characteristics of studies awaiting classification' table.
We investigated the following outcomes using the methods and time points specified below.
Primary outcomes
All‐cause mortality.
Health‐related quality of life.
Severe hypoglycaemia.
Secondary outcomes
Cardiovascular mortality.
Non‐fatal myocardial infarction.
Non‐fatal stroke.
End‐stage renal disease.
Blindness.
Serious adverse events.
Diabetic ketoacidosis.
Non‐serious adverse events.
Nocturnal hypoglycaemia.
Mild/moderate hypoglycaemia.
Socioeconomic effects.
HbA1c levels.
Combined HbA1c levels and severe hypoglycaemia.
Method of outcome measurement
All‐cause mortality: defined as death from any cause.
Health‐related quality of life: defined as mental and physical health‐related quality of life and evaluated by a validated instrument such as Short‐Form‐36 (SF‐36). Scales focusing on treatment satisfaction and not health‐related quality of life as main outcome were not included.
Severe hypoglycaemia: requiring assistance from another person (was planned to be further categorised into 'assistance from other persons', assistance from medical staff, intravenous glucose administration, subcutaneous glucagon administration, hospitalisation, intensive‐care unit stay, coma).
Cardiovascular mortality, non‐fatal myocardial infarction, non‐fatal stroke, blindness: defined as reported in studies.
End‐stage renal disease: defined as need for dialysis and renal transplantation.
Serious adverse events (SAE): defined according to the International Conference on Harmonization (ICH) guidelines as, "any event that leads to death, that is life‐threatening, required in‐patient hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability, and any important medical event which may have had jeopardised the patient or required intervention to prevent it" (ICH 1997) or as reported in studies.
Diabetic ketoacidosis: potentially life‐threatening condition with high levels of ketones in the body which when building up in the blood make the blood more acidic.
Non‐serious adverse events: all adverse events, not classified as SAEs.
Nocturnal hypoglycaemia: hypoglycaemia during night‐time and defined as reported in studies.
Mild/moderate hypoglycaemia: hypoglycaemic episodes not requiring assistance from another person.
Socioeconomic effects: such as direct costs defined as admission or readmission rates; average length of stay; visits to general practitioner; accident or emergency visits; medication consumption; indirect costs defined as resources lost due to illness by the participant or their family member.
HbA1c levels: expressed as percentage or mmol/mol.
Combined HbA1c levels and severe hypoglycaemia: joint examination of the effects of HbA1c reduction and hypoglycaemia risk.
Timing of outcome measurement
For all outcome measures, we defined short‐term follow‐up as 24 weeks to ≤ 52 weeks, medium‐term follow‐up as > 1 year to ≤ 2 years and long‐term follow‐up as > 2 years.
Search methods for identification of studies
Electronic searches
We searched the following sources from the inception of each database to the date of search and did not place restrictions on the language of publication:
Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO) (searched 24 August 2020);
MEDLINE (Ovid MEDLINE ALL 1946 to Daily Update) (searched 24 August 2020);
ClinicalTrials.gov (www.clinicaltrials.gov) (searched 24 August 2020);
World Health Organization International Clinical Trials Registry Platform (ICTRP) (www.who.int/trialsearch) (searched 24 August 2020);
HTA database (https://database.inahta.org/) (searched 24 August 2020).
We did not include Embase in our search, as RCTs indexed in Embase are now prospectively added to CENTRAL via a highly sensitive screening process (Cochrane 2020).
For detailed search strategies, see Appendix 6.
Searching other resources
We identified other potentially eligible studies or ancillary publications by searching the reference lists of included studies, systematic reviews, meta‐analyses, and health technology assessment reports. In addition, we contacted the investigators of included studies to obtain additional information on the retrieved studies and establish whether we may have missed further studies.
We searched the grey literature, which we defined as searching the HTA database, as well as databases from regulatory agencies (European Medicines Agency (EMA) and Food and Drug Administration (FDA) ‐ Hart 2012;Schroll 2015). We searched for CSRs and clinical study synopses as provided on manufacturers' web sites (e.g. Novo Nordisk Trials) and via contact with manufacturers (Appendix 7).
We did not use abstracts or conference proceedings for data extraction unless full data were available from study authors because this information source does not fulfil the CONSORT requirements which consist of "an evidence‐based, minimum set of recommendations for reporting randomized trials" (CONSORT 2018; Scherer 2018). We presented information on abstracts or conference proceedings in the 'Characteristics of studies awaiting classification' table (Characteristics of studies awaiting classification).
Data collection and analysis
Selection of studies
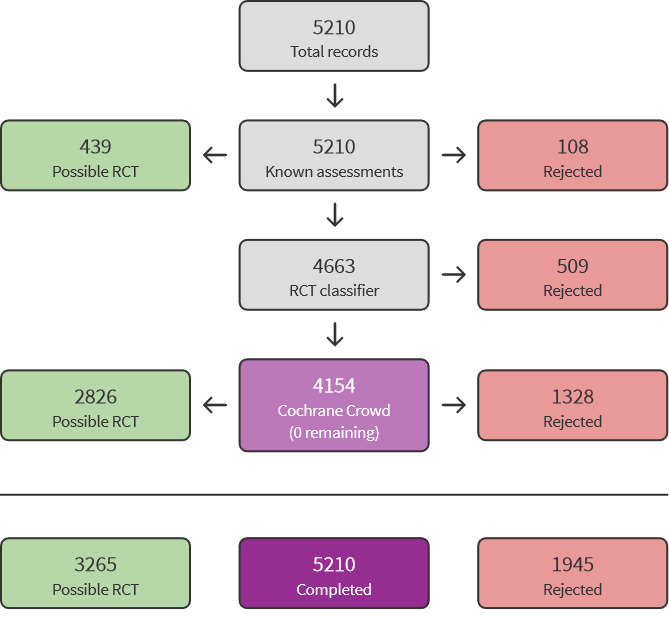
We used Cochrane’s Screen4Me workflow to help assess the search results. Screen4Me comprises three components: known assessments – a service that matches records in the search results to records that have already been screened in Cochrane Crowd and been labelled as an 'RCT' or as 'Not an RCT'; the RCT classifier – a machine learning model that distinguishes RCTs from non‐RCTs, and, if appropriate, Cochrane Crowd – Cochrane’s citizen science platform where the Crowd helped to identify and describe health evidence. Detailed information regarding evaluations of the Screen4Me components can be found in the following publications: Marshall 2018; McDonald 2017; Noel‐Storr 2018; Thomas 2017.
Two review authors (BH, BR) independently screened the abstract, title, or both, of all records remaining after the Screen4Me workflow, to determine which studies we should assess further. We obtained the full text of all potentially relevant records. We would have resolved disagreements through consensus or by recourse to a third review author (MIM), if these had occurred. In case we were unable to resolve a disagreement, we planned to categorise the study as a 'Study awaiting classification' and would have contacted the study authors for clarification. We presented an adapted PRISMA flow diagram to show the process of study selection (Liberati 2009). We listed all articles excluded after full‐text assessment in a 'Characteristics of excluded studies' table and provided the reasons for exclusion (Characteristics of excluded studies).
Data extraction and management
For studies that fulfilled our inclusion criteria, two review authors (BH, BR) independently extracted key participant and intervention characteristics. We described interventions according to an adapted version of the 'template for intervention description and replication' (TIDieR) checklist (Hoffmann 2014; Hoffmann 2017).
We reported data on efficacy outcomes and adverse events using standardised data extraction sheets from the CMED Group. We resolved disagreements by discussion or, if required, by consultation with a third review author (MIM).
We provided information including trial identifier for potentially relevant ongoing trials in the 'Characteristics of ongoing studies' table and in a joint appendix 'Matrix of study endpoints (publications and trial documents)'. We tried to find the protocol and CSR for each included study.
We planned to email all authors of included studies, ongoing trials and studies awaiting classification to enquire whether they would be willing to answer questions regarding their studies. We presented the results of this survey in an appendix. We thereafter sought relevant missing information on the study from the primary study author(s), if required.
Dealing with duplicate and companion publications
In the event of duplicate publications, companion documents, or multiple reports of a primary study, we maximised the information yielded by collating all available data, and we used the most complete data set aggregated across all known publications and records. We listed duplicate publications, companion documents, multiple reports of a primary study, and trial documents of included studies (such as trial registry information and CSRs) as secondary references under the study ID of the included study. Furthermore, we listed duplicate publications, companion documents, multiple reports of a study, and trial documents of excluded studies (such as trial registry information) as secondary references under the study ID of the excluded study.
Data from clinical trials registers and CSR
If data from included studies were available as study results in clinical trials registers, such as ClinicalTrials.gov or as CSR, we made full use of this information and extracted the data. If there also was a full publication of the study, we collated and critically appraised all available data. If an included study was marked as a completed trial in a clinical trials register but no additional information (study results, publication, or both) was available, we added this study to the 'Characteristics of studies awaiting classification' table.
Assessment of risk of bias in included studies
Two review authors (BH, BR) independently assessed the risk of bias for each included study. We would have resolved disagreements by consensus or by consulting a third review author (MIM), if such occurred. If adequate information was unavailable from the publications, trial protocols, CSRs or other sources, we contacted the study authors for more details to request missing data on 'Risk of bias' items.
We undertook ‘Risk of bias’ assessment according to Chapter 7 and Chapter 8 of the CochraneHandbook for Systematic Reviews of Interventions (Boutron 2020; Higgins 2020). We used the Cochrane 'Risk of bias 2' (RoB 2) tool (version 22, August 2019) ‐ (Higgins 2017; Sterne 2019).
We focused on the assessment of the effect of assignment to the interventions at baseline. The effect was analysed as the result of a comparison between interventions on a certain outcome at a specific time point. The RoB 2 tool evaluates the following domains.
Bias arising from the randomisation process.
Bias due to deviations from the intended interventions.
Bias due to missing outcome data.
Bias in measurement of the outcome.
Bias in selection of the reported results.
Within each domain, signalling questions provided information about features of the study that were relevant to risk of bias. Possible answers to the signalling questions were 'Yes', 'Probably yes', 'Probably no', 'No' and 'No information'. After answering the signalling questions, we made a 'Risk of bias' judgement, assigning one of three levels ('low risk of bias', 'some concerns', 'high risk of bias') to each domain. For each specific outcome, we established an overall 'Risk of bias' judgement using the following criteria.
Low risk of bias: the study was judged to be at low risk of bias for all domains for this result.
Some concerns: the study was judged to raise some concern in at least one domain for this result, but not to be at high risk of bias for any domain.
High risk of bias: the study was either judged to be at high risk of bias in at least one domain for this result, or the study was judged to have some concerns for multiple domains in a way that substantially lowers confidence in the result.
We distinguished between participant‐reported outcomes, observer‐reported outcomes not involving judgement, observer‐reported outcomes involving some judgement, outcomes reflecting decisions made by interventions providers and composite outcomes.
Participant‐reported outcomes: health‐related quality of life; mild/moderate and non‐severe nocturnal hypoglycaemia; non‐serious adverse events; socioeconomic effects.
Observer‐reported outcomes not involving judgement: all‐cause mortality, end‐stage renal disease, blindness, HbA1c levels.
Observer‐reported outcomes involving some judgement: cardiovascular mortality, non‐fatal myocardial infarction, non‐fatal stroke, socioeconomic effects.
Outcomes reflecting decisions made by interventions providers: SAEs, severe hypoglycaemia, severe nocturnal hypoglycaemia.
Composite outcomes: combined HbA1c levels and severe hypoglycaemia.
Measures of treatment effect
When at least two included studies were available for a comparison of a given outcome, we expressed dichotomous data as a risk ratio (RR) or an odds ratio (OR) with 95% confidence intervals (CI). For continuous outcomes measured on the same scale (e.g. HbA1c in %), we estimated the intervention effect using the mean difference (MD) with 95% CIs. For continuous outcomes that measured the same underlying concept (e.g. health‐related quality of life) but used different measurement scales, we would have calculated the standardised mean difference (SMD). We would have expressed time‐to‐event data as a hazard ratio (HR) with 95% CIs.
Unit of analysis issues
We took into account the level at which randomisation occurred, such as cross‐over studies, cluster‐randomised studies, and multiple observations for the same outcome. If more than one comparison from the same study had been eligible for inclusion in the same meta‐analysis, we would either have combined groups to create a single pair wise comparison, or we would appropriately reduce the sample size so that the same participants had not contributed data to the meta‐analysis more than once (splitting the 'shared' group into two or more groups). Although the latter approach offers some solution for adjusting the precision of the comparison, it does not account for correlation arising from inclusion of the same set of participants in multiple comparisons (Higgins 2011).
We would have re‐analysed cluster‐RCTs that had not appropriately adjusted for potential clustering of participants within clusters in their analyses. Variance of the intervention effects would have been inflated by a design effect. Calculation of a design effect involves estimation of an intracluster correlation coefficient (ICC). We would have obtained estimates of ICCs by contacting study authors, or by imputing ICC values using either estimates from other included studies that reported ICCs or external estimates from empirical research (e.g. Bell 2013). We would have examined the impact of clustering by performing sensitivity analyses.
Dealing with missing data
If possible, we obtained missing data from the authors of included studies. We carefully evaluated important numerical data such as screened, randomly assigned participants, as well as intention‐to‐treat and as‐treated and per‐protocol populations. We investigated attrition rates (e.g. dropouts, losses to follow‐up, withdrawals), and we critically appraised issues concerning missing data and use of imputation methods (e.g. last‐observation‐carried‐forward).
If studies were identified in which the standard deviation (SD) of the outcome was not available at follow‐up or we could not recreate it, we would have standardised by the mean of the pooled baseline SD from studies that reported this information.
If we had identified included studies not reporting means and SDs for outcomes, and we could not receive the requested information from study authors, we would have imputed these values by estimating the mean and the variance from the median, the range and the size of the sample (Hozo 2005).
We would have investigated the impact of imputation on meta‐analyses by performing sensitivity analyses, and we would have reported for every outcome which studies had imputed SDs.
Assessment of heterogeneity
In the event of clinical or methodological heterogeneity, we planned not to report study results as the pooled effect estimate in a meta‐analysis.
We identified heterogeneity (inconsistency) by visually inspecting the forest plots and by using a standard Chi² test with a significance level of α = 0.1 (Deeks 2017). In view of the low power of this test, we also considered the I² statistic, which quantifies inconsistency across studies, to assess the impact of heterogeneity on the meta‐analysis (Higgins 2002; Higgins 2003).
When we found heterogeneity, we planned to determine possible reasons for this by examining individual study and subgroup characteristics. If possible, we calculated prediction intervals to elucidate the clinical implication of the observed heterogeneity (for details see Data synthesis).
Assessment of reporting biases
If we had included 10 or more studies that investigated a particular outcome, we would have used funnel plots to assess small‐study effects. Several explanations may account for funnel plot asymmetry, including true heterogeneity of effect with respect to study size, poor methodological design (and hence bias of small studies), and publication bias (Sterne 2017). Therefore, we would have interpreted the results carefully (Sterne 2011).
Data synthesis
We undertook (or displayed) a meta‐analysis only if we judged participants, interventions, comparisons, and outcomes to be sufficiently similar to ensure an answer that was clinically meaningful. Unless good evidence showed homogeneous effects across studies of different methodological quality, we would have primarily summarised low risk of bias data using a random‐effects model (Wood 2008). We interpreted random‐effects meta‐analyses with due consideration for the whole distribution of effects and presented a prediction interval (Borenstein 2017a; Borenstein 2017b; Higgins 2009). A prediction interval requires at least three studies to be calculated and specifies a predicted range for the true treatment effect in an individual study (Riley 2011). For rare events such as event rates below 1%, we used the Peto odds ratio method, provided there was no substantial imbalance between intervention and comparator group sizes, and intervention effects were not exceptionally large. In addition, we performed statistical analyses according to the statistical guidelines presented in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2017).
Subgroup analysis and investigation of heterogeneity
We expected the following characteristics to introduce clinical heterogeneity, and we planned to carry out the following subgroup analyses including investigation of interactions (Altman 2003).
Head‐to‐head comparisons of insulin analogues.
Studies designed to blind participants and investigators versus open‐label studies.
NPH once daily versus NPH two‐ or three‐times daily.
Studies of long duration (more than two years) versus studies of short to medium duration (two years or less).
Studies performed in high‐income countries versus middle‐income countries versus low‐income countries.
According to healthcare setting.
Sensitivity analysis
We planned to perform sensitivity analyses to explore the influence of the following factors (when applicable) on effect sizes by restricting analysis to the following.
Published studies.
Effect of risk of bias, as specified in the Assessment of risk of bias in included studies section.
Very long or large studies to establish the extent to which they dominated the results.
Use of the following filters: diagnostic criteria, imputation, language of publication, source of funding (industry versus other), or country.
We tested the robustness of results by repeating analyses using different measures of effect size (i.e. RR, OR, etc.) and different statistical models (fixed‐effect and random‐effects models).
Summary of findings and assessment of the certainty of the evidence
Certainty of the evidence
We presented the overall certainty of the evidence for each outcome specified below, according to the GRADE approach, which takes into account issues related not only to internal validity (risk of bias, inconsistency, imprecision, publication bias), but also to external validity, such as directness of results. Two review authors (BH, BR) independently rated the certainty of the evidence for each outcome. If differences in assessment had occurred, they would have been solved by discussion or by consultation with a third review author (MIM).
We included an appendix entitled 'Checklist to aid consistency and reproducibility of GRADE assessments', to help with standardisation of the 'Summary of findings' tables (Meader 2014). Alternatively, we would have used the GRADEpro Guideline Development Tool (GDT) software and presented evidence profile tables as an appendix (GRADEproGDT 2015). We presented results for outcomes as described in the Types of outcome measures section. If meta‐analysis was not possible, we presented the results in a narrative format in the 'Summary of findings' table. We justified all decisions to downgrade the certainty of the evidence by using footnotes, and we made comments to aid the reader's understanding of the Cochrane Review when necessary.
'Summary of findings' table
We presented a summary of the evidence in a 'Summary of findings' table. This provided key information about the best estimate of the magnitude of effect, in relative terms and as absolute differences for each relevant comparison of alternative management strategies, numbers of participants and studies addressing each important outcome, and a rating of overall confidence in effect estimates for each outcome.
In the 'Summary of findings' table, we reported on the 'intervention' (ultra‐)long‐acting insulin analogue or its biosimilar insulin versus the 'comparator' human NPH insulin or another (ultra‐)long‐acting insulin analogue.
We created the 'Summary of findings' table using the methods described in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2017), along with Review Manager (RevMan 5.3) table editor (RevMan 2014). We reported the following outcomes, listed according to priority.
All‐cause mortality.
Health‐related quality of life.
Severe hypoglycaemia.
Non‐fatal myocardial infarction/stroke.
Severe nocturnal hypoglycaemia.
SAEs.
HbA1c levels.
Results
Description of studies
For a detailed description of studies, see Table 1, Characteristics of included studies, Characteristics of excluded studies and Characteristics of studies awaiting classification tables.
Results of the search
The initial search identified a total of 7747 records. In assessing the studies, we used Cochrane’s Screen4Me workflow to help identify potential reports of randomised studies. The results of the Screen4Me assessment process can be seen in Figure 1. Subsequently, we assessed the remaining 3265 records, as well as the 570 records retrieved by the update search prior to publication. We excluded most of the references on the basis of their titles and abstracts because they clearly did not meet the inclusion criteria. We evaluated a further 47 records identified as CSRs, clinical study synopses, a study protocol and one additional record identified through handsearching of reference lists of included studies (Figure 2).
1.
Screen4Me: Cochrane´s screening service.
2.

Study flow diagram
CSR: clinical study report; EMA: European Medicines Agency; HTA: health technology assessment; Screen4Me: Cochrane's screening service.
Searching the web pages of Novo Nordisk and Sanofi, we identified 23 CSRs, clinical study synopses or both. On request, we received 10 CSRs from Sanofi and six CSRs, sections of two CSRs and one study protocol from Novo Nordisk, respectively. The two studies with sections of CSRs only were Japanese studies (Kobayashi 2007; NCT00605137). For both studies, clinical study synopses were available and we could not get full access to the Japanese versions of the CSRs. For one study, a trial protocol was provided by Novo Nordisk (NCT00605137). One study had a clinical study synopsis only (NCT00595374). The total number of additional references from web pages and contact with manufacturers was 22 CSRs, 23 clinical study synopses and one study protocol.
We identified applications/documents through searching FDA and EMA web sites (EMA 2014; EMA 2015; EMA 2015a; EMA 2015b; FDA 2000; FDA 2002; FDA 2005; FDA 2015). These references did not provide information about additional studies.
In summary, after screening the full texts from the electronic search and additional sources, we identified 26 RCTs published in 202 records that met our inclusion criteria. Two studies were unpublished, but clinical study synopses and parts of the CSRs were obtained and provided data for inclusion (NCT00595374; NCT00605137). The remaining included studies were published. For all studies, except two, it was possible to retrieve additional information from clinical trials registers, documents from regulatory agencies, CSRs, clinical study synopses and investigators (Bolli 2009; Porcellati 2004). The number of records per included studies varied from 1 to 21. Thirteen studies are awaiting assessment.
Included studies
A detailed description of the characteristics of included studies is presented elsewhere (see Characteristics of included studies and Appendix 7; Appendix 8; Appendix 9; Appendix 10; Appendix 11; Appendix 12; Appendix 13; Appendix 14; Appendix 15; Appendix 16; Appendix 17; Appendix 18; Appendix 19; Appendix 20; Appendix 21; Appendix 1; Appendix 2; Appendix 3; Appendix 4; Appendix 5. The following is a succinct overview.
Overview of study populations
Twenty‐five studies reported the total number of participants screened (Bartley 2008; BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; BEGIN Young; Bolli 2009; Chase 2008; Davies 2014; Fulcher 2005; Heller 2009; Home 2005; Kobayashi 2007; Liu 2016; NCT00595374; NCT00605137; Pieber 2007; Porcellati 2004; PRESCHOOL; Ratner 2000; Robertson 2007; Russell‐Jones 2004; Schober 2002; Standl 2004; SWITCH 1; Thalange 2013; Vague 2003).
A total of 8784 participants were randomised: 2428 participants were randomised to NPH insulin, 2889 participants to insulin detemir, 2095 participants to insulin glargine and 1372 participants to insulin degludec (see Table 6). Eight of the studies included children and randomised 1835 participants, i.e. 21% of all participants (BEGIN Young; Chase 2008; Liu 2016; NCT00605137; Robertson 2007; Schober 2002; Thalange 2013; Urakami 2017). The remaining studies included adults.
1. Overview of study populations.
| Study ID (study design) | Intervention(s) and comparator(s) | Description of power and sample size calculation | Screened/eligible (n) | Randomised (n) | Analysed primary outcome) (n) | Finishing study (n) | Randomised finishing study (%) | Follow‐up (extended follow‐up)a |
|
Bartley 2008 (parallel‐group non‐inferiority RCT) |
I: insulin detemir | Quote: "A total of 489 patients were needed to obtain 245 evaluable patients on detemir and 123 on NPH to detect a clinically relevant difference of 0.4% in HbA1c with a power of 85%, assuming a standard deviation (SD) for HbA1c of 1.2 and an expected drop‐out rate of 25%" | 557 | 331 | 320 | 278 | 84.3 | 24 months |
| C: NPH insulin | 166 | 159 | 144 | 86.7 | ||||
| total: | 497 | 479 | 422 | 85.0 | ||||
|
BEGIN Basal‐Bolus Type 1b (parallel‐group non‐inferiority RCT) |
I: insulin degludec | Quote: "Sample size was determined by the primary objective with the assumption of a one sided t test at a significance level of 2·5%, a zero mean treatment difference, and an SD of 1·1% for HbA1c. A total of 624 participants were needed for at least 95% power after adjustment for a 15% dropout rate" | 722 | 472 | 472 | 404 | 85.6 | 52 weeks (104 weeks) |
| C: insulin glargine | 157 | 157 | 137 | 87.0 | ||||
| total: | 629 | 629 | 541 | 86.0 | ||||
|
BEGIN Flex T1c (parallel‐group non‐inferiority RCT) |
I: insulin degludec | Quote: "Sample size was determined on the basis of the primary objective under the assumption of a 1‐sided t test of size 2.5%, a zero mean treatment difference, and standard deviation of 1.1% for HbA1c" | 549 | 165 | 165 | 139 | 84.2 | 26 weeks (52 weeks) |
| C: insulin glargine | 164 | 164 | 152 | 92.7 | ||||
| total: | 329 | 329 | 291 | 88.4 | ||||
|
BEGIN Youngd (parallel‐group non‐inferiority RCT) |
I: insulin degludec | Quote: "The sample size was determined using a t‐statistic under the assumption of a one‐sided test of size 2.5%, a zero mean treatment difference and standard deviation (SD) of 1.25% for HbA1c. A total of 346 participants had to be randomized to achieve at least 80% or greater power in the evaluation of the per protocol (PP) analysis set, after adjustment for a 10% dropout rate" | 363 | 174 | 174 | 170 | 97.7 | 26 weeks (52 weeks) |
| C: insulin detemir | 176 | 176 | 163 | 93.7 | ||||
| total: | 350 | 350 | 333 | 95.1 | ||||
|
Bolli 2009 (parallel‐group superiority RCT) |
I: insulin glargine | Quote: "The expected FBG difference in the two groups at the end of the study treatment was estimated to be 30+/‐60 mg/dL. Using a two‐sided test with ɑ = 0.01 and ß = 0.1 (i.e., power: 1‐ß = 0.9), 240 evaluable patients were to be included. Due to an expected dropout rate of 20% and to the randomization schedule, which was restricted and stratified by centre (26 centres), 312 patients were planned to be enrolled" | 213 | 85 | 85 | 78 | 91.8 | 24 weeks (30 weeks) |
| C: NPH insulin | 90 | 90 | 74 | 82.2 | ||||
| total: | 175 | 175 | 152 | 86.7 | ||||
|
Chase 2008 (parallel‐group non‐inferiority RCT) |
I: insulin glargine | Quote: "The primary clinical outcome (the mean change in A1C from baseline [week 0] to endpoint [week 24 or last post randomization assessment]) was compared in the 2 treatment groups using analysis of covariance (ANCOVA), with treatment group, study centre (pooled), CGMS values, sex, and baseline value as covariates (α = 0.05; 2‐sided test). The 95% confidence intervals (CIs) were computed for the adjusted mean difference between treatment groups from the ANCOVA to test for noninferiority (defined as an upper bound of the 95% CI for the mean difference in A1C of ≤ 0.4%)" | 235 | 85 | 84 | 81 | 95.3 | 24 weeks (25 weeks) |
| C: NPH insulin/Lente | 90 | 84 | 76 | 84.4 | ||||
| total: | 175 | 168 | 157 | 89.7 | ||||
|
Davies 2014e (parallel‐group non‐inferiority RCT) |
I: insulin degludec | Quote: "Assuming a standard deviation (SD) of 1.1% for the primary endpoint, the trial had 90% power with 360 participants randomized 2:1" | 512 | 303 | 302 | 283 | 93.4 | 26 weeks (52 weeks) |
| C: insulin detemir | 153 | 153 | 138 | 90.2 | ||||
| total: | 456 | 455 | 421 | 92.5 | ||||
|
Fulcher 2005 (parallel‐group non‐inferiority RCT) |
I: insulin glargine | Quote: "The sample size was calculated assuming a 20% dropout rate, so that 118 patients (59 in each group) were enrolled in order to have 96 patients (48 in each group) available for evaluation at end‐point. Assuming a SD of 1.2 for HbA1c (based on previous Phase IIIa studies), the study had 80% power to detect a 0.7% difference in HbA1c" | 173f | 62 | 62 | 58 | 94 | 30 weeks |
| C: NPH insulin | 63 | 62 | 49 | 78 | ||||
| total: | 125 | 124f | 107 | 85.6 | ||||
|
Heller 2009 (parallel‐group non‐inferiority RCT) |
I: insulin detemir | Quote: "The sample size was determined for 2:1 (detemir:glargine) randomization and based on a 1‐sided t test at a 2.5% significance level. Assuming an SD of 1.0% for HbA1c and a dropout rate of 15%, a sample size of 435 patients gave 95% power to demonstrate noninferiority" | 515 | 300 | 299 | 263 | 87.7 | 52 weeks |
| C: insulin glargine | 147 | 144 | 122 | 83.0 | ||||
| total: | 447 | 443 | 385 | 86.1 | ||||
|
Home 2005 (parallel‐group superiority RCT) |
I: insulin glargine | Quote from CSR: "It was planned to treat 520 subjects, 260 subjects in each group. Each investigation site was to randomise 10‐20 subjects.The primary efficacy variable for the comparison between HOE 901 and NPH insulin was the change from baseline in GHb at the study endpoint for the individual subject ... The standard deviation for change from baseline in GHb at endpoint was estimated to be 1.6%. Based on 1:1 randomization and using a t‐test, a total number of 440 subjects (220 subjects for each group) was required to detect a mean difference of 0.5% GHb between HOE 901 and NPH with a type I error of α = 5% and a statistical power of 90%. With an expected drop‐out rate of 15% during the course of the study, a total number of 520 subjects (260 subjects in each group) were to be enrolled in order to have 440 subjects (220 subjects in each group) evaluable at week 28" | 655 | 298 | 292 | 276 | 94.5 | 28 weeks |
| C: NPH insulin | 305 | 293 | 272 | 92.8 | ||||
| total: | 603g | 585h | 548 | 93.6 | ||||
|
Kobayashi 2007 (parallel‐group non‐inferiority RCT) |
I: insulin detemir | — | 454i | 197 | 195 | 183 | 93.4 | 48 weeks |
| C: NPH insulin | 99 | 98 | 91 | 92.9 | ||||
| total: | 296 | 293 | 274 | 92.6 | ||||
|
Liu 2016 (parallel‐group non‐inferiority RCT) |
I: insulin glargine | Quote from CSR: "The planned sample size was reduced from 366 to 150 patients in view of extremely difficult recruitment progress over the 2 years since first patient’s enrolment..." | 196 | 107 | 108 | 106 | 99.1 | 24 weeks (25 weeks) |
| C: NPH insulin | 55 | 54 | 50 | 90.9 | ||||
| total: | 162 | 161 | 156 | 96.3 | ||||
|
NCT00595374f (parallel‐group non‐inferiority RCT) |
I: insulin detemir | — | 124 | 75 | — | 70 | 93.3 | 26 weeks |
| C: NPH insulin | 38 | — | 34 | 92.1 | ||||
| total: | 113 | — | 104 | 92.0 | ||||
|
NCT00605137f (parallel‐group non‐inferiority RCT) |
I: insulin detemir | Quote from trial protocol: "This power calculation is based on a two‐sample poisson test at a significance level of 5% for the comparison of the mean rate of nocturnal episodes per four weeks although nocturnal episodes will be analysed as recurrent events using gamma frailty model in the trial analysis" | 88 | 57 | 55 | 55 | 96.5 | 24 weeks |
| C: NPH insulin | 29 | 27 | 27 | 93.1 | ||||
| total: | 86 | 82 | 82 | 95.3 | ||||
|
Pieber 2007 (parallel‐group non‐inferiority RCT) |
I: insulin detemir | Quote: "The sample size was determined in order to test non‐inferiority in a 1:1 randomization. Assuming a standard deviation for HbA1c of 1.2% and a clinically relevant, absolute difference in HbA1c of 0.4%, a total of 286 randomized participants were needed to achieve a power of 80%. Assuming a 10% drop‐out rate, 159 randomized participants were needed in each group" | 415 | 161 | 161 | 147 | 91.3 | 26 weeks |
| C: insulin glargine | 161 | 159 | 146 | 90.7 | ||||
| total: | 322 | 319 | 293 | 91.0 | ||||
|
Porcellati 2004 (parallel‐group superiority RCT) |
I: insulin glargine | Quote: "In this design, a total of 120 participants were required to achieve 90% power to detect a difference of 0.3% among the means with group standard deviations of 0.4 at the significance level (alpha) of 5%" | 130 | 61 | 61 | 61 | 100 | 1 year |
| C: NPH insulin | 60 | 60 | 60 | 100 | ||||
| total: | 121 | 121 | 121 | 100 | ||||
|
PRESCHOOL (parallel‐group non‐inferiority RCT) |
I: insulin glargine | Quote: "Sample size calculation was based on an expected composite hypoglycemia rate of 0.8 events/100 patient‐yr of exposure to insulin glargine or to NPH insulin. The sample size and novel composite outcome was planned to ensure sufficient power so that the upper bound of the two‐sided 95% confidence interval (CI) for the insulin glargine:NPH ratio of the mean composite hypoglycemia rates for the comparison of treatment groups would not exceed 1.15. A sample size of 35 completed patients per treatment group was to provide 96% power to demonstrate noninferiority of insulin glargine vs. NPH" | 165 | 61 | 61 | 57 | 93.4 | 24 weeks (26 weeks) |
| C: NPH insulin | 64 | 64 | 54 | 84.4 | ||||
| total: | 125j | 125 | 111 | 88.8 | ||||
|
Ratner 2000 (parallel‐group superiority RCT) |
I: insulin glargine | Quote: "An estimated 440 participants (220 in each treatment group) were required to detect a mean difference of 0.5% in GHb levels between treatment with a type 1 error of α = 5% and a statistical power of 90%" | 677f | 266 | 256 | 233 | 88.3 | 28 weeks |
| C: NPH insulin | 274 | 262 | 248 | 91.9 | ||||
| total: | 540k | 518 | 481f | 90.1 | ||||
|
Robertson 2007 (parallel‐group non‐inferiority RCT) |
I: insulin detemir | Quote: "The sample size was determined for a 2: 1 randomization based on a two‐sided t ‐test on a 5% significance level. Assuming a SD for HbA 1c of 1.1% and a clinically relevant difference in HbA 1c of 0.4% (absolute), 270 children were needed to achieve a power of 80%. With an expected drop‐out rate of 20%, 338 children were to be allocated to study treatment" | 363f | 232 | 232 | 226 | 97.4 | 26 weeks |
| C: NPH insulin | 115 | 114 | 109 | 94.8 | ||||
| total: | 347 | 347 | 335f | 96.5 | ||||
|
Russell‐Jones 2004 (parallel‐group non‐inferiority RCT) |
I: insulin detemir | Quote: "Sample size was based on an SD for HbA1c of 1.4% and the assumption that a 0.4% absolute difference in HbA1c represents a clinically relevant difference" and " All comparisons were 2‐tailed tests with a 5% level of significance" | 838f | 492 | 491 | 465 | 94.7 | 6 months |
| C: NPH insulin | 257 | 256 | 235 | 91.8 | ||||
| total: | 749f | 747 | 700 | 93.5 | ||||
|
Schober 2002 (parallel‐group superiority RCT) |
I: insulin glargine | Quote: "The sample size was calculated to detect a mean difference in HbA1C from baseline to endpoint of 0.5% with a statistical power of 90%. Assuming a 20% dropout rate, the minimum sample size required was 360 patients" | 385 | 180 | 155 | 169 | 93.9 | 28 weeks |
| C: NPH insulin | 181 | 156 | 168 | 92.8 | ||||
| total: | 361l | 311 | 337f | 93.4 | ||||
|
Standl 2004m (parallel‐group non‐inferiority RCT) |
I: insulin detemir | Quote from CSR: "A total of 440 type 1 participants were planned for randomisation in order to obtain 400 evaluable participants, assuming a dropout rate of approximately 10%" | 505f | 237 | 210 | 212 | 89.5 | 6 months (12 months) |
| C: NPH insulin | 224 | 206 | 209 | 93.3 | ||||
| total: | 461f | 416f | 421 | 91.3 | ||||
|
SWITCH 1n (cross‐over non‐inferiority RCT) |
I: insulin degludec | Quote: "The trial was powered to show noninferiority of the primary end point. Based on the assumption that up to 10% of the randomised patients may not contribute to the analysis, 400 patients needed to contribute to the analysis if 446 patients were randomised to ensure a power of 94%, to demonstrate noninferiority with an expected rate of overall symptomatic hypoglycemia of 5.0 episodes per patient‐years’ exposure (PYE)" | 634 | 249 | 249 | 209 | 83.9 | 32 weeks |
| C: insulin glargine | 252 | 251 | 205 | 81.3 | ||||
| total: | 501 | 414 | 414 | 82.6 | ||||
|
Thalange 2013 (parallel‐group non‐inferiority RCT) |
I: insulin detemir | Quote: "The power calculation was analysed on this basis: using a two‐sided t‐test with a one‐sided significance level of 2.5%, assuming SD of 1.1, a non‐inferiority criterion of 0.4%, a power of 85% and an expected dropout rate of 20%, a total of 344 children were to be randomized" | 381 | 177 | 171 | 164 | 92.7 | 52 weeks (104 weeks) |
| C: NPH insulin | 171 | 168 | 161 | 94.2 | ||||
| total: | 348 | 339 | 325 | 93.4 | ||||
|
Urakami 2017p (cross‐over superiority RCT) |
I: insulin degludec | — | — | 9 | 9 | 9 | 100 | 24 weeks |
| C: insulin glargine | 9 | 9 | 9 | 100 | ||||
| total: | 18 | 18 | 18 | 100 | ||||
|
Vague 2003 (parallel‐group non‐inferiority RCT) |
I: insulin detemir | Quote: "The initial cohort size was calculated to achieve a power of 85% on the basis of non‐inferiority testing at the 5% significance level and a 2:1 randomization" | 471f | 301 | 280 | 284 | 94.4 | 6 months (12 months) |
| C: NPH insulin | 147 | 139 | 141 | 96.6 | ||||
| total: | 448 | 419 | 425 | 95.1 | ||||
| Overall total | All insulin detemir | 2889 | 2648 | |||||
| All insulin degludec | 1372 | 1214 | ||||||
| All insulin glargine | 2095 | 1890 | ||||||
| All NPH insulin | 2428 | 2202 | ||||||
| All interventions and comparators | 8784 | 7954 | ||||||
— denotes not reported
aFollow‐up under randomised conditions until end of study (= duration of intervention + follow‐up post‐intervention or identical to duration of intervention); extended follow‐up refers to follow‐up of participants once the original study was terminated as specified in the power calculation. bData in the table are for the main period.After 52 weeks, the participants of the initial study were invited to an extension study. 74% in the degludec and 75% in the glargine participated. Of the one included in the extension period, 94% (330/351) participants completed in the degludec group and 96% (113/118) participants in the glargine group. cAn additional study arm existed, which was not included in this review. dData in the table are for the main period. In the insulin degludec group, 152 participants entered the extension study and 151 participants completed; in the insulin detemir group, 128 participants entered the extension study and 122 participants completed. eData in the table are for the main period. In the insulin degludec group, 248 participants entered the extension study and 242 participants completed (79.9% of those initially randomised); in the insulin detemir group, 122 participants entered the extension study and 115 participants completed (75.2% of those initially randomised). fData from clinical study report/synopsis. gIn the publication, it was only mentioned that 602 participants were randomised, but not explained how these were divided between the intervention groups. This was reported in the clinical study report. In the publication, there was only information about the allocation of the 585 participants who received the intervention. hIn the main publication, the number of participants analysed was not clearly described; this number was provided by the clinical study report. iBoth people with type 1 diabetes mellitus and type 2 diabetes mellitus were screened. jOne participant randomised to NPH insulin was actually treated with insulin glargine, thus the safety population comprised 62 participants for insulin glargine and 63 participants for NPH insulin. kIn the main publication, it was stated that 534 participants were randomised (264 participants allocated to insulin glargine; 270 participants allocated to NPH insulin). In the clinical study report, it was stated that a total of 540 participants were randomised, but six were never treated (2 participants in the insulin glargine group; 4 participants in the NPH insulin group). lOf the 361 participants randomised, 12 withdrew their consent before being treated, therefore a total 349 participants were treated: 174 participants in the glargine group compared with 175 participants in the NPH group. mData in the table are for the main period. In the insulin detemir group, 154 participants entered the extension study and 118 participants completed (49.8% of those initially randomised); in the NPH insulin group, 135 participants entered the extension study and 134 participants completed (59.8% of those initially randomised). nData from first treatment period before cross‐over (32 weeks). oExtension only performed for the detemir group. pNot reported if any participant dropped out during the study. All randomised participants were included in all analyses.
A1c: glycosylated haemoglobin A1c ANCOVA: analysis of covariance C: comparator CGMS: continuous glucose monitoring system CI: confidence interval CSR: clinical study report FBG: fasting blood glucose GHb: glycated haemoglobin HbA1c: glycosylated haemoglobin A1c HOE 901: insulin glargine I: intervention NPH: neutral protamine Hagedorn PYE: patient‐years’ exposure RCT: randomised controlled trial SD: standard deviation vs: versus
The proportion of participants finishing the studies varied from 78% to 100% (Fulcher 2005; Porcellati 2004).
Study design
Two studies had a cross‐over design (SWITCH 1; Urakami 2017). The remaining studies were parallel‐group RCTs. All studies had an open‐label design, except for one which was double‐blinded (SWITCH 1). The duration of the intervention ranged from 24 weeks to 24 months. Seven studies had an additional extension period (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; BEGIN Young; Davies 2014; Standl 2004; Thalange 2013; Vague 2003).
All studies except two were multicentre studies (Porcellati 2004; Urakami 2017). The number of study centres ranged from 1 to 90. Sixteen studies were multinational (Bartley 2008; BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; BEGIN Young; Davies 2014; Heller 2009; Home 2005; Pieber 2007; PRESCHOOL; Robertson 2007; Russell‐Jones 2004; Schober 2002; Standl 2004; SWITCH 1; Thalange 2013; Vague 2003). None of the studies was performed in low‐ or middle‐income countries. None of the studies was terminated early.
Participants
Twenty‐three studies reported the ethnicity of the participants: 19 studies included mainly white people (Bartley 2008; BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; BEGIN Young; Chase 2008; Fulcher 2005; Heller 2009; Home 2005¸ NCT00595374; Pieber 2007; PRESCHOOL; Ratner 2000; Robertson 2007; Russell‐Jones 2004; Schober 2002; Standl 2004; SWITCH 1; Thalange 2013; Vague 2003), one study mainly Asian people (Davies 2014) and three studies included Asian people only (Kobayashi 2007; Liu 2016; NCT00605137) (Appendix 9).
All studies included both genders. The age of the participants varied from 4.2 to 44 years. The duration of T1DM varied from 2.1 to 23.2 years (Appendix 10).
Interventions
Nine studies compared insulin detemir with NPH insulin (Bartley 2008; Kobayashi 2007; NCT00595374; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003). Nine studies compared insulin glargine with NPH insulin (Bolli 2009; Chase 2008; Fulcher 2005; Home 2005; Liu 2016; Porcellati 2004; PRESCHOOL; Ratner 2000; Schober 2002). Two studies compared insulin detemir with insulin glargine (Heller 2009; Pieber 2007) and two studies compared insulin degludec with insulin detemir (BEGIN Young; Davies 2014). Finally, four studies compared insulin degludec with insulin glargine (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; SWITCH 1; Urakami 2017).
All studies except one applied NPH insulin once or two times daily. Porcellati 2004 applied NPH insulin four times a day.
Studies started insulin administration in different ways: four studies comparing insulin detemir with NPH insulin started with lower doses of insulin detemir compared with NPH insulin (Kobayashi 2007; NCT00605137; Russell‐Jones 2004; Standl 2004). One study comparing insulin degludec with insulin glargine stated that if prior basal insulin was taken more than once daily, then the dose of glargine had to be reduced by 20% to 30% and insulin degludec dose was reduced based on the investigators' decision (BEGIN Flex T1). Another study comparing insulin degludec with insulin glargine stated that if more than one daily dose had been taken prior to the study, then the total daily basal dose was calculated and replaced with insulin degludec in a 1:1 ratio and the insulin glargine dose was recommended to be reduced by 20% to 30% (BEGIN Basal‐Bolus Type 1). One study comparing insulin detemir with insulin glargine stated that the insulin detemir dose was reduced by 30% in both the morning and evening doses from the previous regimen and insulin glargine was started with a dose of 20% to 30% less than the previous regimen (Pieber 2007).
Eleven studies applied insulin aspart as fast‐acting insulin (Bartley 2008; BEGIN Young; Davies 2014; Heller 2009; Kobayashi 2007; Liu 2016; NCT00595374; Pieber 2007; Robertson 2007; Thalange 2013; Vague 2003); five studies applied insulin lispro (Bolli 2009; Chase 2008; Fulcher 2005; Porcellati 2004; PRESCHOOL); five studies applied human insulin (Home 2005; Ratner 2000; Schober 2002; Russell‐Jones 2004; Standl 2004) and one study did not specify the type of fast‐acting insulin applied (NCT00605137).
Outcomes
We could retrieve detailed study information for 23 studies (Bartley 2008; BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; BEGIN Young; Chase 2008; Davies 2014; Fulcher 2005; Heller 2009; Home 2005; Kobayashi 2007; Liu 2016; NCT00595374; NCT00605137; Pieber 2007; PRESCHOOL; Ratner 2000; Robertson 2007; Russell‐Jones 2004; Schober 2002; Standl 2004; SWITCH 1; Thalange 2013; Vague 2003). For six of the studies, trial protocols were available through the CSRs (Fulcher 2005; Home 2005; Ratner 2000; Schober 2002; Standl 2004; Vague 2003). For the remaining studies with a trial registration, information could be retrieved from the clinical trials register (see Appendix 12). Three studies provided data through publications only (Bolli 2009; Porcellati 2004; Urakami 2017) and one study author sent additional data (Urakami 2017).
All studies except three had predefined HbA1c as the primary outcome (NCT00605137; PRESCHOOL; SWITCH 1). All studies reported one or more outcome measures of relevance for this review.
Source of data
We contacted all study authors or investigators through email (see Appendix 14). When important information was lacking on ongoing trials and excluded studies, we contacted investigators for clarification (see Appendix 14).
Excluded studies
We excluded 22 studies after full‐text evaluation: eight studies had a wrong study design (not an RCT), six studies applied the wrong intervention, three studies included the wrong population, four studies had a short study duration and one reference was an irrelevant congress report. We evaluated four systematic reviews for identification of studies (Laranjeira 2018; Monami 2009; Tricco 2014; Tricco 2018). For further details see Characteristics of excluded studies.
Risk of bias in included studies
For the Cochrane RoB 2 assessment, we obtained CSRs, clinical study reports or both for 23 studies (Bartley 2008; BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; BEGIN Young; Chase 2008; Davies 2014; Fulcher 2005; Heller 2009; Home 2005; Kobayashi 2007; Liu 2016; NCT00595374; NCT00605137; Pieber 2007; PRESCHOOL; Ratner 2000; Robertson 2007; Russell‐Jones 2004; Schober 2002; Standl 2004; SWITCH 1; Thalange 2013; Vague 2003). We primarily used data from CSRs to evaluate risk of bias because the CSRs provided detailed information on all risk of bias domains for the RoB 2 tool. For two studies, we could obtain only parts of the original CSRs because the original documentation was written in Japanese and we did not get access to the full CSR (Kobayashi 2007; NCT00605137). For two studies, the clinical study synopses and a study protocol were the only source for data extraction (NCT00595374; NCT00605137).
For each specific outcome, we established an overall 'Risk of bias' judgement, as well as judgements per 'Risk of bias' domain (bias arising from the randomisation process, bias due to deviations from the intended interventions, bias due to missing outcome data, bias in measurement of the outcome, bias in selection of the reported results).
All‐cause mortality
All studies reporting deaths except two had a low overall risk of bias. Porcellati 2004 and Urakami 2017 had 'some concerns' because in these open‐label studies there was scarce information on methodological aspects of the studies.
Health‐related quality of life
All studies reporting health‐related quality of life except one had 'some concerns' for overall risk of bias because in these open‐label studies this outcome measure was primarily participant‐reported. SWITCH 1 had a low overall risk of bias for this outcome measure.
Severe hypoglycaemia
All studies reporting severe hypoglycaemia except three had a low overall risk of bias. Bolli 2009, Porcellati 2004 and Urakami 2017 had 'some concerns' because in these open‐label studies there was scarce information on methodological aspects of the studies.
Cardiovascular mortality
All studies reporting deaths except two had a low overall risk of bias. Porcellati 2004 and Urakami 2017 had 'some concerns' because in these open‐label studies there was scarce information on methodological aspects of the studies.
Non‐fatal myocardial infarction/stroke
All studies reporting non‐fatal myocardial infarction, non‐fatal stroke or both except one had a low overall risk of bias. Urakami 2017 had 'some concerns' because in this open‐label study there was scarce information on methodological aspects of the study.
End‐stage renal disease/blindness
The single study reporting end‐stage renal disease and blindness had a low overall risk of bias.
Serious adverse events
All studies reporting SAEs except two had a low overall risk of bias. Bolli 2009 and Urakami 2017 had 'some concerns' because in these open‐label studies there was scarce information on methodological aspects of the studies.
Diabetic ketoacidosis
All studies reporting diabetic ketoacidosis except one had a low overall risk of bias. Urakami 2017 had 'some concerns' because in this open‐label study there was scarce information on methodological aspects of the study.
Non‐serious adverse events
All studies reporting non‐serious adverse events had 'some concerns' for overall risk of bias because in these open‐label studies this outcome measurement was primarily participant‐reported.
Severe nocturnal hypoglycaemia
All studies reporting severe nocturnal hypoglycaemia except one had a low overall risk of bias. Urakami 2017 had 'some concerns' because in this open‐label study there was scarce information on methodological aspects of the study.
Mild/moderate hypoglycaemia
All studies reporting mild/moderate hypoglycaemia had some concerns for overall risk of bias because in these open‐label studies this outcome measurement was primarily participant‐reported.
Socioeconomic effects
No studies reported the costs of the intervention during the study period.
HbA1c levels
All studies reporting HbA1c except three had a low overall risk of bias. Bolli 2009, Porcellati 2004 and Urakami 2017 had 'some concerns' because in these open‐label studies there was scarce information on methodological aspects of the studies.
Combined HbA1c and severe hypoglycaemia
The studies providing some data on combined HbA1c and severe hypoglycaemia had a low overall risk of bias.
In general, referring to detailed information from the CSRs, the risk of bias evaluation was much more exhaustive compared to details reported in the publications. Most of our outcomes represented hard clinical (semi)objective outcomes with overall low risk of bias. However, for some outcomes, due to their subjective, participant‐reported nature, we attributed 'some concerns' to overall risk of bias for the outcomes health‐related quality of life, non‐serious adverse events, most measures of nocturnal hypoglycaemia and mild/moderate hypoglycaemia.
Risk of bias assessments for each outcome are located in the risk of bias table section after the characteristics of studies awaiting assessment and at the side of forest plots. For further details on the Excel file of risk of bias evaluation stored online in an open repository (Zenodo), please use the following link: https://zenodo.org/record/4549440.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
Summary of findings 1. Summary of findings: insulin detemir versus NPH insulin.
| Insulin detemir compared with NPH insulin for T1DM | ||||||
|
Patients: people with T1DM Settings: outpatients Intervention: insulin detemir Comparison: NPH insulin | ||||||
| Outcomes | NPH insulin | Insulin detemir | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments |
|
All‐cause mortality Follow‐up: 24‐104 weeks |
See comment | Peto OR4.97 (0.79 to 31.38) | 3334 (9) | ⊕⊕⊕⊝ moderatea | All 5 deaths reported in 2 studies including adults occurred in the insulin detemir group | |
|
Health‐related quality of life Description: diabetes health profile; insulin therapy‐related quality of life at night (scale not specified) Follow‐up: 26‐48 weeks |
See comment | 870 (3) | ⊕⊕⊝⊝ lowb | No study reported health‐related quality of life in a format making it suitable for meta‐analysis 1 study including adults reported higher scores in the insulin detemir group vs the NPH insulin group (Kobayashi 2007) 2 studies did not show evidence of a difference between intervention groups (NCT00595374 included children; Standl 2004 included adults) |
||
|
Severe hypoglycaemia (n/N) Definition: hypoglycaemia requiring third party assistance (Bartley 2008; Kobayashi 2007; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003); episodes where the children were semi‐conscious, unconscious or in a coma, with or without convulsions (Thalange 2013) Follow‐up: 24‐104 weeks |
115 per 1000 | 79 per 1000 (60 to 106) | RR 0.69 (0.52 to 0.92) | 3219 (8) | ⊕⊕⊕⊝ moderatec | The 95% prediction interval ranged between 0.34 and 1.39 5 studies included adults, 3 studies included children (the test for subgroup differences did not indicate interaction) |
|
Non‐fatal myocardial infarction/stroke Definition: myocardial infarction Follow‐up: 24 months |
See comment | 495 (1) | ⊕⊕⊝⊝ lowd | 1/331 participants in the insulin detemir group vs 0/164 participants in the NPH insulin group experienced a non‐fatal myocardial infarction (Bartley 2008) Stroke was not reported Study included adults |
||
|
Severe nocturnal hypoglycaemia (n/N) Definition: severe hypoglycaemia occurring 23:00‐06:00 (Bartley 2008; NCT00605137; Russell‐Jones 2004; Standl 2004; Vague 2003); occurring 22:00‐07:00 (Robertson 2007; Thalange 2013) Follow‐up: 24 weeks ‐ 24 months |
54 per 1000 | 36 per 1000 (21 to 64) | RR 0.67 (0.39 to 1.17) | 2925 (7) | ⊕⊕⊕⊝ moderatee | The 95% prediction interval ranged between 0.16 and 2.87 4 studies included adults, 3 studies included children (the test for subgroup differences did not indicate interaction) |
|
Serious adverse events (n/N) Follow‐up: 24‐104 weeks |
82 per 1000 | 78 per 1000 (62 to 100) | RR 0.95 (0.75 to 1.21) | 3332 (9) | ⊕⊕⊕⊝ moderatee | The 95% prediction interval ranged between 0.71 and 1.27 6 studies included adults, 3 studies included children (the test for subgroup differences did not indicate interaction) |
|
HbA1c (%) Follow‐up: 24 weeks ‐ 24 months |
The mean HbA1c ranged across the NPH insulin groups from 7.3% to 8.6% | The mean HbA1c in the insulin detemir groups was 0.01% higher (0.1% lower to 0.1% higher) | — | 3122 (8) | ⊕⊕⊕⊝ moderatee | The 95% prediction interval ranged between ‐0.1% and 0.1% 5 studies included adults, 3 studies included children (the test for subgroup differences did not indicate interaction) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CSR: clinical study report; HbA1c: glycosylated haemoglobin A1c; n/N: number of people experiencing an event; NPH: neutral protamine Hagedorn; OR: odds ratio RR: risk ratio; T1DM: type 1 diabetes mellitus. | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
*Assumed risk was derived from the event rates in the comparator groups.
aDowngraded by one level because of indirectness (insufficient time frame) ‐ see Appendix 1. bDowngraded by two levels because of overall risk of bias ('some concerns') and imprecision (few studies) ‐ see Appendix 1. cDowngraded by one level because of inconsistency (95% prediction interval consistent with benefit and harm) ‐ see Appendix 1. dDowngraded by two levels because of indirectness (insufficient time frame) and imprecision (few studies) ‐ see Appendix 1. eDowngraded by one level because of imprecision (CI consistent with benefit and harm) ‐ see Appendix 1.
Summary of findings 2. Summary of findings: insulin glargine versus NPH insulin.
| Insulin glargine compared with NPH insulin for T1DM | ||||||
|
Patients: people with T1DM Settings: outpatients Intervention: insulin glargine Comparison: NPH insulin | ||||||
| Outcomes | NPH insulin | Insulin glargine | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments |
|
All‐cause mortality Follow‐up: 24‐52 weeks |
See comment | Peto OR0.14 (0.00 to 6.98) | 2175 (8) | ⊕⊕⊕⊝ moderatea | 1 study including adults reported 0/1207 participants died in the insulin glargine group vs 1/1068 participants in the NPH insulin group 4 studies included adults, 4 studies included children (the test for subgroup differences could not be performed) |
|
|
Health‐related quality of life Scales: Well‐Being Enquiry for Diabetics; General Well‐being; Diabetes Quality of Life for Youth and Parents' Diabetes Quality of Life Follow‐up: 24‐28 weeks |
See comment | 1013 (4) | ⊕⊕⊝⊝ lowb | 1 study including adults (Bolli 2009) reported greater improvements in the insulin glargine group compared with NPH insulin in one domain (diabetes‐related worries) There was no evidence of a difference in 3 studies (Chase 2008 included children; Home 2005 and Ratner 2000 included adults) |
||
|
Severe hypoglycaemia (n/N) Definition: symptomatic hypoglycaemia requiring third party assistance, with either a blood glucose level < 2.8 mmol/L or prompt recovery after administration of oral carbohydrate, iv glucose or glucagon (Fulcher 2005; Home 2005; Schober 2002); requiring third party assistance and associated with either blood glucose < 2.0 mmol/L or prompt recovery after oral carbohydrate, iv glucose, or intramuscular or subcutaneous glucagon administration (Chase 2008); hypoglycaemia requiring third party assistance or involving a seizure, coma, unconsciousness or the use of glucagon (Liu 2016); hypoglycaemia requiring third party assistance (Porcellati 2004; PRESCHOOL; Ratner 2000) Follow‐up: 24‐52 weeks |
125 per 1000 | 105 per 1000 (84 to 130) | RR 0.84 (0.67 to 1.04) | 2350 (9) | ⊕⊕⊕⊝ moderatec | The 95% prediction interval ranged between 0.65 and 1.09 5 studies included adults, 4 studies included children (the test for subgroup differences did not indicate interaction) |
|
Non‐fatal myocardial infarction/stroke Definition: myocardial infarction/cerebral ischaemia Follow‐up: 28 weeks |
See comment | 585 (1) | ⊕⊕⊝⊝ lowd | No participant experienced a non‐fatal myocardial infarction 1 study including adults reported 0/292 participants in the insulin glargine group vs 1/293 participants in the NPH insulin group experienced cerebral ischaemia (Home 2005) |
||
|
Severe nocturnal hypoglycaemia (n/N) Definition: severe hypoglycaemia occurring 23:00‐07:00 (PRESCHOOL); severe hypoglycaemia occurring after the evening insulin injection and before the morning insulin dose (Fulcher 2005); severe hypoglycaemia occurring during sleep between bedtime and rising in the morning, or before the morning pre‐breakfast self‐blood glucose measurement and the morning insulin injection (Home 2005); severe hypoglycaemia occurring while asleep after the bedtime insulin dose and before the morning insulin dose and before the morning blood glucose measurement (Ratner 2000); severe hypoglycaemia while the participant was sleeping between bedtime and after the evening injection and before getting up in the morning (Schober 2002); severe hypoglycaemia occurring 00:00‐06:00 (Chase 2008) Follow‐up: 24‐28 weeks |
87 per 1000 | 72 per 1000 (54 to 97) | RR 0.83 (0.62 to 1.12) | 1893 (6) | ⊕⊕⊕⊝ moderatec | The 95% prediction interval ranged between 0.54 and 1.27 3 studies included adults, 3 studies included children (the test for subgroup differences did not indicate interaction) |
|
Serious adverse events (n/N) Follow‐up: 24‐30 weeks |
100 per 1000 | 108 per 1000 (63 to 184) | RR 1.08 (0.63 to 1.84) | 2229 (8) | ⊕⊕⊕⊝ moderatec | The 95% prediction interval ranged between 0.22 and 5.21 4 studies included adults, 4 studies included children (the test for subgroup differences did not indicate interaction) |
|
HbA1c (%) Follow‐up: 24 weeks ‐ 1 year |
The mean HbA1c ranged across the NPH insulin groups from 7.1% to 7.3% | The mean HbA1c in the insulin glargine groups was 0.02% higher (0.1% lower to 0.1% higher) | — | 2285 (9) | ⊕⊕⊕⊝ moderatec | The 95% prediction interval ranged between ‐0.5% and 0.5% 5 studies included adults, 4 studies included children (the test for subgroup differences did not indicate interaction) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). a.m.: ante meridiem; CI: confidence interval; HbA1c: glycosylated haemoglobin A1c; iv: intravenous; n/N: number of people experiencing an event; NPH: neutral protamine Hagedorn; RR: risk ratio; T1DM: type 1 diabetes mellitus. | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
*Assumed risk was derived from the event rates in the comparator groups.
aDowngraded by one level because of indirectness (insufficient time frame) ‐ see Appendix 2. bDowngraded by two levels because of overall risk of bias ('some concerns') and imprecision (few studies) ‐ see Appendix 2. cDowngraded by one level because of imprecision (CI consistent with benefit and harm) ‐ see Appendix 2. dDowngraded by two levels because of indirectness (insufficient time frame) and imprecision (few studies) ‐ see Appendix 2.
Summary of findings 3. Summary of findings: insulin detemir versus insulin glargine.
| Insulin detemir compared with insulin glargine for T1DM | ||||||
|
Patients: people with T1DM Settings: outpatients Intervention: insulin detemir Comparison: insulin glargine | ||||||
| Outcomes | Insulin glargine | Insulin detemir | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments |
|
All‐cause mortality Follow‐up: 26 and 52 weeks |
See comment | 763 (2) | ⊕⊕⊝⊝ lowa | No participant died 2 studies included adults |
||
| Health‐related quality of life | Not reported | |||||
|
Severe hypoglycaemia (n/N) Definition: hypoglycaemia requiring third party assistance Follow‐up: 26 and 52 weeks |
116 per 1000 | 68 per 1000 (15 to 304) | RR 0.59 (0.13 to 2.63) | 763 (2) | ⊕⊝⊝⊝ very lowb | 2 studies included adults |
|
Non‐fatal myocardial infarction/stroke Definition: non‐fatal myocardial infarction/stroke Follow‐up: 52 weeks |
See comment | 443 (1) | ⊕⊕⊝⊝ lowa | 1 study including adults reported 1/299 participants in the insulin detemir group vs 1/144 participants in the insulin glargine group experienced a non‐fatal myocardial infarction One study including adults reported 2/299 participants in the insulin detemir group vs 0/144 participants in the insulin glargine group experienced a non‐fatal stroke |
||
|
Severe nocturnal hypoglycaemia (n/N) Definition: severe hypoglycaemia occurring from 11 p.m. to 6 a.m. Follow‐up: 26 and 52 weeks |
50 per 1000 | 27 per 1000 (3 to 253) | RR 0.55 (0.06 to 5.12) | 763 (2) | ⊕⊝⊝⊝ very lowb | 2 studies included adults |
|
Serious adverse events (n/N) Follow‐up: 26 and 52 weeks |
59 per 1000 | 102 per 1000 (54 to 195) | RR 1.72 (0.91 to 3.28) | 763 (2) | ⊕⊕⊝⊝ lowc | The fixed‐effect statistical model showed an RR of 1.79 (1.04 to 3.08) in favour of insulin glargine 2 studies included adults |
|
HbA1c (%) Follow‐up: 26 and 52 weeks |
The mean HbA1c ranged across the insulin glargine groups from 7.6% to 8.2% | The mean HbA1c in the insulin detemir groups was 0.01% lower (0.1% lower to 0.1% higher) | — | 763 (2) | ⊕⊕⊝⊝ lowc | 2 studies included adults |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). a.m.: ante meridiem; CI: confidence interval; HbA1c: glycosylated haemoglobin A1c; n/N: number of people experiencing an event; p.m.: post meridiem; RR: risk ratio; T1DM: type 1 diabetes mellitus. | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
*Assumed risk was derived from the event rates in the comparator groups.
aDowngraded by two levels because of indirectness (insufficient time frame) and imprecision (few studies) ‐ see Appendix 3. bDowngraded by three levels because of inconsistency (point estimates varied widely, non‐consistent direction of effect) and serious imprecision (CI consistent with benefit and harm, few studies) ‐ see Appendix 3. cDowngraded by two levels because of serious imprecision (CI consistent with benefit and harm, few studies) ‐ see Appendix 3.
Summary of findings 4. Summary of findings: insulin degludec versus insulin detemir.
| Insulin degludec compared with insulin detemir for T1DM | ||||||
|
Patients people with T1DM Settings: outpatients Intervention: insulin degludec Comparison: insulin detemir | ||||||
| Outcomes | Insulin detemir | Insulin degludec | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments |
|
All‐cause mortality Follow‐up: 26 weeks |
See comment | 802 (2) | ⊕⊕⊝⊝ lowa | No participant died 1 study included adults, 1 study included children |
||
|
Health‐related quality of life Scale: Short‐Form 36 version 2 (higher values mean better health‐related quality of life) Follow‐up: 26 weeks |
Physical health score: the mean score in the insulin detemir group was 52.5 Mental health score: the mean score in the insulin detemir group was 52.5 |
Physical health score: the mean score in the insulin degludec group was 0.60 points lower (1.83 points lower to 0.63 points higher) Mental health score: the mean score in the insulin degludec group was 3.00 points lower (4.44 points lower to 1.56 points lower) |
— | 454 (1) | ⊕⊕⊝⊝ lowb | Physical health score: MID is 2‐3 points Mental health score: MID is 3 points Study included adults |
|
Severe hypoglycaemia (n/N) Definition: hypoglycaemia requiring third party assistance (Davies 2014) or altered mental status and cannot assist in their own care, is semiconscious or unconscious, or in a coma ± convulsions and may require parenteral therapy (glucagon or iv glucose) (BEGIN Young) Follow‐up: 26 weeks |
122 per 1000 | 143 per 1000 (99 to 207) | RR 1.17 (0.81 to 1.69) | 802 (2) | ⊕⊕⊝⊝ lowc | 1 study included adults, 1 study included children (the test for subgroup differences did not indicate interaction) |
|
Non‐fatal myocardial infarction/stroke Definition: non‐fatal myocardial infarction/stroke Follow‐up: 26 weeks |
See comment | 453 (1) | ⊕⊕⊝⊝ lowa | No participant experienced a non‐fatal myocardial infarction or stroke Study included adults |
||
|
Severe nocturnal hypoglycaemia (n/N) Definition: severe hypoglycaemia occurring 00:01‐05:59 (Davies 2014) or 23:00‐07:00 (BEGIN Young) Follow‐up: 26 weeks |
31 per 1000 | 34 per 1000 (16 to 75) | RR 1.12 (0.51 to 2.46) | 802 (2) | ⊕⊕⊝⊝ lowc | 1 study included children, 1 study included adults (the test for subgroup differences did not indicate interaction) |
|
Serious adverse events (n/N) Follow‐up: 26 weeks |
73 per 1000 | 92 per 1000 (56 to 150) | RR 1.25 (0.76 to 2.05) | 802 (2) | ⊕⊕⊝⊝ lowc | 1 study included children, 1 study included adults (the test for subgroup differences did not indicate interaction) |
|
HbA1c (%) Follow‐up: 26 weeks |
The mean HbA1c in the insulin glargine groups was 7.3% | The mean HbA1c in the insulin detemir groups was 0.05% lower (0.1% lower to 0.2% higher) | — | 802 (2) | ⊕⊕⊝⊝ lowc | 1 study included children, 1 study included adults (the test for subgroup differences did not indicate interaction) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). a.m.: ante meridiem; CI: confidence interval; HbA1c: glycosylated haemoglobin A1c; iv: intravenous; MID: minimal important difference; n/N: number of people experiencing an event; p.m.: post meridiem; RR: risk ratio; T1DM: type 1 diabetes mellitus. | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
*Assumed risk was derived from the event rates in the comparator groups. aDowngraded by two levels because of indirectness (insufficient time frame) and imprecision (few studies) ‐ see Appendix 4. bDowngraded by two levels because of overall risk of bias ('some concerns') and imprecision (few studies) ‐ see Appendix 4. cDowngraded by two levels because of serious imprecision (CI consistent with benefit and harm, few studies) ‐ see Appendix 4.
Summary of findings 5. Summary of findings: insulin degludec versus insulin glargine.
| Insulin degludec compared with insulin glargine for T1DM | ||||||
|
Patients: people with T1DM Settings: outpatients Intervention: insulin degludec Comparison: insulin glargine | ||||||
| Outcomes | Insulin glargine | Insulin degludec | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments |
|
All‐cause mortality Follow‐up: 26 ‐ 52 weeks |
3 per 1000 | 4 per 1000 (0 to 36) | Peto OR 1.34 (0.15 to 11.93) | 973 (3) | ⊕⊝⊝⊝ very lowa | A total of 3/646 participants in the insulin degludec group vs 1/327 participants in the insulin glargine group died 2 studies included adults 1 study included children |
|
Health‐related quality of life Scale: Short‐Form 36 version 2 (higher values mean better health‐related quality of life) Follow‐up: 32 and 52 weeks |
Physical health score: the mean score ranged across the insulin glargine groups from 50.6 to 51.8 Mental health score: the mean score ranged across the insulin glargine groups from 49.9 to 50.4 |
Physical health score: the mean score in the insulin degludec groups was 0.04 points lower (1.21 points lower to 1.13 points higher) Mental health score: the mean score in the insulin degludec groups was 0.09 points lower (1.03 points lower to 0.85 points higher) |
— | 1042 (2) | ⊕⊝⊝⊝ very lowb | Physical health score: MID is 2‐3 points
Mental health score: MID is 3 points 2 studies included adults |
|
Severe hypoglycaemia (n/N) Definition: hypoglycaemia requiring third party assistance (BEGIN Flex T1; BEGIN Young) or an event associated with impaired consciousness or seizure (Urakami 2017) Follow‐up: 24 and 52 weeks |
102 per 1000 | 124 per 1000 (83 to 185) | RR 1.22 (0.82 to 1.82) | 970 (3) | ⊕⊕⊝⊝ lowc | 2 studies included adults 1 study including children reported no child experienced severe hypoglycaemia (Urakami 2017) |
|
Non‐fatal myocardial infarction/stroke Definition: non‐fatal myocardial infarction/cerebral ischaemia Follow‐up: 24 and 52 weeks |
See comment | 970 (3)/970 (3) | ⊕⊕⊝⊝ lowd | 2 studies including adults reported 1/637 participants in the insulin degludec group vs 0/315 participants in the insulin glargine group experienced a non‐fatal myocardial infarction; there were no events in 1 study including children (Urakami 2017) 2 studies including adults reported 1/637 participants in the insulin degludec group vs 0/315 in the insulin glargine group experienced cerebral ischaemia; there were no events in 1 study including children (Urakami 2017) |
||
|
Severe nocturnal hypoglycaemia (n/N) Definition: severe hypoglycaemia occurring from 22:00 to 06:59 h Follow‐up: 24 ‐ 52 weeks |
25 per 1000 | 35 per 1000 (15 to 83) | RR 1.39 (0.59 to 3.27) | 970 (3) | ⊕⊕⊝⊝ lowc | 2 studies included adults 1 study include children |
|
Serious adverse events (n/N) Follow‐up: 24 and 52 weeks |
77 per 1000 | 71 per 1000 (45 to 113) | RR 0.92 (0.58 to 1.46) | 970 (3) | ⊕⊕⊝⊝ lowc | 2 studies included adults 1 study including children reported no child experienced a serious adverse event (Urakami 2017) |
|
HbA1c (%) Follow‐up: 24 and 52 weeks |
The mean HbA1c ranged across the insulin glargine groups from 6.9% to 7.8% | The mean HbA1c in the insulin degludec groups was 0.1% higher (0% lower to 0.2% higher) | — | 1388 (4) | ⊕⊕⊝⊝ lowc | The 95% prediction interval ranged between ‐0.1% and 0.3% 3 studies included adults, 1 study included children (the test for subgroup differences did not indicate interaction) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CSR: clinical study report; HbA1c: glycosylated haemoglobin A1c; MID: minimal important difference; n/N: number of people experiencing an event; OR: odds ratio; RR: risk ratio; T1DM: type 1 diabetes mellitus: | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
*Assumed risk was derived from the event rates in the comparator groups.
aDowngraded by three levels because of indirectness (insufficient time frame) and serious imprecision (CI consistent with benefit and harm, few studies) ‐ see Appendix 5. bDowngraded by three levels because of overall risk of bias ('some concerns') and serious imprecision (CI consistent with benefit and harm, few studies) ‐ see Appendix 5. cDowngraded by two levels because of serious imprecision (CI consistent with benefit and harm, few studies) ‐ see Appendix 5. dDowngraded by two levels because of indirectness (insufficient time frame) and imprecision (few studies) ‐ see Appendix 5.
Baseline characteristics
For details of baseline characteristics, see Appendix 9; Appendix 10.
Insulin degludec compared with NPH insulin
We identified no studies comparing insulin degludec with NPH insulin.
Insulin detemir compared with NPH insulin
For an overview of main results for this comparison see Table 1.
Nine studies compared insulin detemir with NPH insulin (Bartley 2008; Kobayashi 2007; NCT00595374; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003). A total of 3345 participants were randomised, 2099 participants to insulin detemir and 1246 participants to NPH insulin (see Table 6). Three studies included children and randomised 781 children, 466 children to insulin detemir and 315 children to NPH insulin (NCT00605137; Robertson 2007; Thalange 2013). The mean age of the children varied from 8.4 to 9.9 years. Two of the studies did not have full‐text publications (NCT00595374; NCT00605137). We retrieved unpublished information on baseline variables or outcomes for all studies for this comparison.
Two studies randomised the participants to insulin detemir and NPH insulin once daily (Bartley 2008; Russell‐Jones 2004). However, a second dose of insulin detemir and NPH insulin could be added if necessary. For one of the studies, it was reported that 37% of the participants in the insulin detemir group and 45% of the participants in the NPH insulin group completed the study on a once‐daily regimen (Bartley 2008). Four studies randomised participants to NPH insulin once or twice daily (Kobayashi 2007; NCT00595374; NCT00605137; Robertson 2007). One study applied insulin detemir and NPH insulin once or twice daily according to a pre‐study regimen (Thalange 2013). One study randomised participants to insulin detemir and NPH insulin twice daily (Vague 2003). Six studies applied insulin aspart as fast‐acting insulin at meals (Bartley 2008; Kobayashi 2007; NCT00595374; Robertson 2007; Thalange 2013; Vague 2003). Two studies applied human insulin as fast‐acting insulin (Russell‐Jones 2004; Standl 2004). One study did not specify the type of fast‐acting insulin applied (NCT00605137).
The duration of the intervention varied from 24 weeks to 104 weeks (see Table 6).
Primary outcomes
All‐cause mortality
We could retrieve data on all‐cause mortality from all nine studies. However, only two studies reported mortality in their full‐text publication (Bartley 2008; Thalange 2013). We retrieved the remaining data from CSRs/clinical study synopses and medical reviews from regulatory agencies (Kobayashi 2007; NCT00595374; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Vague 2003).
A total of 5/2095 participants allocated to insulin detemir died compared with 0/1239 participants allocated to NPH insulin (Peto OR 4.97, 95% CI 0.79 to 31.38; P = 0.09; 9 studies, 3334 participants; moderate‐certainty evidence; Analysis 1.1). We judged the overall risk of bias for this outcome as 'low'.
1.1. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 1: All‐cause mortality
Subgroup and sensitivity analyses
Analysing unpublished data only, 1/1587 participants in the insulin detemir group died compared with 0/905 participants in the NPH insulin group (2492 participants; 7 studies; Analysis 1.2). Analysing published data only 4/508 participants in the insulin detemir group compared with 0/334 in the NPH insulin group died (2 studies, 842 participants; Analysis 1.2). All five deaths occurred in studies including adults. The test for subgroup differences did not indicate interaction (P = 0.84). We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
1.2. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 2: All‐cause mortality (published vs. unpublished data)
Health‐related quality of life
We rated the certainty of the evidence of the three studies with 870 participants providing information on health‐related quality of life as low. We judged the overall risk of bias for this outcome as 'some concerns'.
No study reported health‐related quality of life in a format making the data suitable for meta‐analysis. Kobayashi 2007 applied the Insulin Therapy Related Quality of Life at Night questionnaire (ITR‐QOLN); data were reported in the clinical study synopsis. The evaluation of ITR‐QOLN after 48 weeks showed higher scores in the insulin detemir group compared with the NPH insulin group (Kobayashi 2007). Standl 2004 applied the Diabetes Health Profile scale (only one of the three dimensions of the scale 'Barriers to activity'); data were reported in the CSR. After 26 weeks, the 'Barriers to activity' in the Diabetes Health profile was 0.71 (SD 0.75) in 210 participants in the insulin detemir group compared with 0.20 (SD 0.78) in 208 participants in the NPH insulin group. The P value was 0.52 (Standl 2004). Diabetes treatment satisfaction was also reported in the CSR (Standl 2004). Another unpublished trial reported in the clinical study synopsis that health‐related quality of life did not show any statistically significant differences between the interventions after 26 weeks but did not provide numerical data (NCT00595374).
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Severe hypoglycaemia
Eight studies reported data on severe hypoglycaemia (Bartley 2008; Kobayashi 2007; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003).
Analysing all available data showed that 171/2019 participants (8.5%) in the insulin detemir group compared with 138/1200 participants (11.5%) in the NPH insulin group experienced severe hypoglycaemia. There was a reduction in severe hypoglycaemia in favour of insulin detemir (RR 0.69, 95% CI 0.52 to 0.92; P = 0.01; 8 studies, 3219 participants; moderate‐certainty evidence; Analysis 1.3; Figure 3). The 95% prediction interval ranged between 0.34 and 1.39. We judged the overall risk of bias for this outcome as 'low'.
1.3. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 3: Severe hypoglycaemia
3.

Severe hypoglycaemia
One study had an extension period (Standl 2004). We used data from the core period (six months) in the meta‐analysis. From the publication, only data after the end of the extension period (12 months) were available. However, we could retrieve additional data from the FDA medical review and the CSR (FDA 2002; Standl 2004). One study was unpublished, but data were available from a clinical study synopsis (NCT00605137). Another unpublished study reported no statistically significant differences for severe hypoglycaemia between the intervention groups but did not provide numerical data (NCT00595374). Five studies reported severe hypoglycaemia as requiring third party assistance (Bartley 2008; Kobayashi 2007; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004). One study added to this definition that blood glucose < 2.8 mmol/L should be recorded or symptom reversal with food, glucose or glucagon (Vague 2003). One study defined severe hypoglycaemia as episodes where the children were semi‐conscious, unconscious or in a coma, with or without convulsions (Thalange 2013). Bartley 2008 reported most events: data from the CSR of this study showed that 5/331 participants (1.5%) in the insulin detemir group compared with 6/164 participants (3.7%) in the NPH insulin group experienced a hypoglycaemic coma; 2/331 participants (0.6%) in the insulin detemir group compared with 0/164 participants (0%) in the NPH insulin group experienced hypoglycaemic convulsions; 0/331 participants (0%) in the insulin detemir group compared with 1/164 participants (0.6%) in the NPH insulin group experienced loss of consciousness due to hypoglycaemia. Robertson 2007 reported most events in children: 3/232 children (1.3%) in the insulin detemir group compared with 3/115 children (2.6%) in the NPH insulin group were admitted to hospital due to hypoglycaemia; 4/232 children (1.7%) in the insulin detemir group compared with 4/115 children (3.4%) in the NPH insulin group were unconscious due to hypoglycaemia; 2/332 children (0.6%) in the insulin detemir group compared with 4/115 children (3.4%) in the NPH insulin group experienced hypoglycaemia with convulsions; 4/332 children (1.2%) in the insulin detemir group compared with 2/115 children (1.7%) in the NPH insulin group received glucagon treatment. One study stipulated, that the risk of experiencing hypoglycaemia could have been influenced by lack of blinding: "Investigators and patients in this trial may have been reluctant to aggressively increase the dose of a new basal insulin preparation such as insulin detemir because of the fear of hypoglycemia, especially during the night" (Vague 2003).
Subgroup and sensitivity analyses
Analysing studies including adults only indicated an RR of 0.71, 95% CI 0.49 to 1.03; 5 studies, 2443 participants; Analysis 1.3. Analysing studies including children only indicated an RR of 0.61, 95% CI 0.30 to 1.23; 3 studies, 776 children; Analysis 1.3. The test for subgroup differences did not indicate interaction (P = 0.72).
Restricting the analysis to published data only indicated an RR of 0.62, 95% CI 0.50 to 0.78; 6 studies, 2677 participants; Analysis 1.4; favouring insulin detemir. Restricting the analyses to unpublished data only indicated an RR of 1.42, 95% CI 0.77 to 2.62; 2 studies, 498 participants; Analysis 1.4 . The test for subgroup differences indicated interaction (P = 0.01). This has to be interpreted with caution because the subgroup of studies with unpublished data consisted of two studies only and the CIs slightly overlapped.
1.4. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 4: Severe hypoglycaemia (published vs. unpublished data)
A sensitivity analysis excluding the largest study (Russell‐Jones 2004) indicated an RR of 0.68, 95% CI 0.48 to 0.97. A sensitivity analysis excluding the longest study (Bartley 2008) indicated an RR of 0.72, 95% CI 0.51 to 1.04.
A sensitivity analysis with data from studies published in English only (Bartley 2008; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003) indicated an RR of 0.70, 95% CI 0.52 to 0.95.
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Hypoglycaemia reported as a serious adverse event
A total of 30/2019 participants (1.5%) in the insulin detemir group compared with 19/1200 participants (1.6%) in the NPH insulin group had a SAE due to hypoglycaemia. There was no evidence of a difference in hypoglycaemia reported as a SAE (RR 0.93, 95% CI 0.51 to 1.71; P = 0.82; 8 studies, 3219 participants; Analysis 1.5). The 95% prediction interval ranged between 0.44 and 1.99. We judged the overall risk of bias for this outcome as 'low' (data not shown).
1.5. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 5: Hypoglycaemia reported as a serious adverse event
The test for subgroup differences comparing adults with children did not indicate interaction (P = 1.00). We judged the overall risk of bias for this outcome as 'low' (data not shown).
Secondary outcomes
Cardiovascular mortality
We could retrieve data on cardiovascular mortality from all studies. Only two studies reported cardiovascular mortality in their full‐text publication (Bartley 2008; Thalange 2013). We retrieved the remaining data from CSRs/clinical study synopses/medical reviews from regulatory agencies (Kobayashi 2007; NCT00595374; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Vague 2003).
Only one adult participant died due to cardiovascular disease (Analysis 1.6). This participant belonged to the insulin detemir group (1/2069 participants). No participant died in the NPH insulin group (0/1221 participants). We judged the overall risk of bias for this outcome as 'low'.
1.6. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 6: Cardiovascular mortality
Subgroup analysis and sensitivity analysis
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐fatal myocardial infarction
None of the included studies reported non‐fatal myocardial infarction in the publications. One study had data on non‐fatal myocardial infarction from the CSR (Bartley 2008). In this study, 1/331 participants in the insulin detemir group compared with 0/164 participants in the NPH insulin group experienced a non‐fatal myocardial infarction (low‐certainty evidence; Analysis 1.7). One study reported data at the end of the extension period (duration of intervention was six months with an additional six months extension period) with 1/154 participants in the insulin detemir group and 0/135 participants in the NPH insulin group experiencing a myocardial infarction (Standl 2004). We judged the overall risk of bias for this outcome as 'low'.
1.7. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 7: Non‐fatal myocardial infarction
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐fatal stroke
No study reported on non‐fatal stroke.
End‐stage renal disease
No study reported on end‐stage renal disease.
Blindness
No study reported on blindness.
Serious adverse events
We could retrieve data on SAEs from all studies.
In the insulin detemir group, 165/2094 participants (7.9%) reported a SAE compared with 102/1238 participants (8.2%) in the NPH insulin group. There was no evidence of a difference in SAEs (RR 0.95, 95% CI 0.75 to 1.21; P = 0.67; 9 studies, 3332 participants; moderate‐certainty evidence; Analysis 1.8). The 95% prediction interval ranged between 0.71 and 1.27. We judged the overall risk of bias for this outcome as 'low'.
1.8. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 8: Serious adverse events
Three studies reported SAEs in the main publications in a format making data unsuitable for meta‐analysis: one study reported that the frequency and type of adverse events observed during the study were similar with insulin detemir and NPH insulin (Russell‐Jones 2004); one study reported that fewer than 5% in each intervention group reported SAEs (Vague 2003) and one study reported that about 10% of participants in both intervention groups experienced SAEs (Standl 2004). However, in the CSRs of these studies, data were reported in a way making them suitable for meta‐analysis.
Subgroup and sensitivity analyses
Six studies had data on SAEs for adults: 124/1630 participants (7.6%) in the insulin detemir group compared with 71/926 participants (7.7%) in the NPH insulin group experienced SAEs. The RR was 0.97, 95% CI 0.73 to 1.28; 6 studies, 2556 participants; Analysis 1.8. Three studies had data on SAEs for children: 41/464 children (8.8%) in the insulin detemir group compared with 31/312 children (9.9%) in the NPH insulin group experienced SAEs. The RR was 0.89, 95% CI 0.69 to 1.27; 3 studies, 776 children; Analysis 1.8. The test for subgroup differences did not indicate interaction (P = 0.77).
Restricting the analyses to published data only for SAEs indicated an RR of 0.66, 95% CI 0.40 to 1.09; 2 studies, 641 participants; Analysis 1.9. Restricting analysis to unpublished data only indicated an RR of 1.06, 95% CI 0.80 to 1.39; 6 studies, 2691 participants; Analysis 1.9. The test for subgroup differences did not indicate interaction (P = 0.11).
1.9. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 9: Serious adverse events (published vs. unpublished data)
Sensitivity analysis excluding the largest study (Vague 2003) indicated an RR of 0.93, 95% CI 0.70 to 1.25. Sensitivity analysis excluding the longest study (Bartley 2008) indicated an RR of 0.96, 95% CI 0.72 to 1.29.
A sensitivity analysis with data from studies published in English only indicated an RR of 0.89, 95% CI 0.56 to 1.43 (Bartley 2008; NCT00595374; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003).
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Diabetic ketoacidosis
We could retrieve data on diabetic ketoacidosis from six studies (Bartley 2008; Kobayashi 2007; NCT00605137; Robertson 2007; Thalange 2013; Vague 2003). Two studies reported ketoacidosis in their full‐text publications (Robertson 2007; Thalange 2013). One study was unpublished, but we retrieved data from the clinical study synopsis (NCT00605137). Three studies reported diabetic ketoacidosis in CSRs (Bartley 2008; Kobayashi 2007; Vague 2003). It appeared likely that all studies had evaluated this outcome but some did not report this outcome measure (NCT00595374; Russell‐Jones 2004; Standl 2004).
A total of 14/1292 participants (1.1%) experienced diabetic ketoacidosis in the insulin detemir group compared with 10/720 participants (1.4%) in the NPH insulin group. There was no evidence of a difference in diabetic ketoacidosis (RR 0.80, 95% CI 0.36 to 1.76; P = 0.58; 6 studies, 2012 participants; Analysis 1.10). We judged the overall risk of bias for this outcome as 'low'.
1.10. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 10: Diabetic ketoacidosis
Subgroup and sensitivity analyses
Three studies reported diabetic ketoacidosis in adults; the RR was 0.84, 95% CI 0.24 to 2.92; 3 studies, 1236 participants; Analysis 1.10. Three studies reported diabetic ketoacidosis in children; the RR was 0.77, 95% CI 0.27 to 2.15; 3 studies, 776 children; Analysis 1.10. The test for subgroup differences did not indicate interaction (P = 0.91).
Restricting the analyses to only published data for diabetic ketoacidosis indicated an RR of 0.83, 95% CI 0.27 to 2.52; 2 studies, 694 participants; Analysis 1.11. Restricting the analyses to only unpublished data for diabetic ketoacidosis indicated an RR of 0.77, 95% CI 0.25 to 2.38; 4 studies, 1318 participants; Analysis 1.11.
1.11. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 11: Diabetic ketoacidosis (published vs. unpublished data)
Sensitivity analysis excluding the largest study and the longest study indicated an RR of 0.86, 95% CI 0.34 to 2.20 (Bartley 2008).
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐serious adverse events
We could retrieve data on non‐serious adverse events from all studies. Only four studies reported non‐serious adverse events in a format suitable for meta‐analysis in their full‐text publications (Robertson 2007; Standl 2004; Thalange 2013; Vague 2003). For the remaining studies, we retrieved data from CSRs/clinical study synopses (Bartley 2008; Kobayashi 2007; NCT00595374; NCT00605137; Russell‐Jones 2004).
A total of 1622/2094 participants (77.5%) in the insulin detemir group compared with 968/1238 participants (78.2%) in the NPH insulin group experienced a non‐serious adverse event. There was no evidence of a difference in non‐serious adverse events (RR 0.98, 95% CI 0.94 to 1.01; P = 0.22; 9 studies, 3332 participants; Analysis 1.12). The 95% prediction interval ranged between 0.95 and 1.02. We judged the overall risk of bias for this outcome as 'some concerns'.
1.12. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 12: Non‐serious adverse events
Subgroup analysis and sensitivity analysis
Five studies reported non‐serious adverse events in adults. A total of 1242/1630 participants (76.2%) in the insulin detemir group compared with 706/926 participants (76.2%) in the NPH insulin group experienced a non‐serious adverse event. The RR was 0.99, 95% CI 0.95 to 1.03; Analysis 1.12. Three studies including children reported 380/464 children (81.9%) in the insulin detemir group compared with 262/312 (84.0%) children in the NPH insulin group experienced a non‐serious adverse event. The RR was 0.96, 95% CI 0.90 to 1.02; Analysis 1.12. The test for subgroup differences did not indicate interaction (P = 0.40).
Restricting the analyses to only published data indicated 553/710 participants (77.9%) in the insulin detemir group compared with 351/431 participants (81.4%) in the NPH insulin group experienced a non‐serious adverse event. The RR was 0.95, 95% CI 0.90 to 1.01; Analysis 1.13. Restricting the analyses to only unpublished data indicated 1069/1384 participants (77.2%) in the insulin detemir group compared with 617/807 participants (76.5%) in the NPH insulin group experienced a non‐serious adverse event. The RR was 1.00, 95% CI 0.95 to 1.04; Analysis 1.13. The test for subgroup differences did not indicate interaction (P = 0.25).
1.13. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 13: Non‐serious adverse events (published vs unpublished data)
Sensitivity analysis excluding the largest study (Russell‐Jones 2004) indicated an RR of 0.97, 95% CI 0.93 to 1.01 and excluding the longest study (Bartley 2008) indicated an RR of 0.98, 95% CI 0.94 to 1.02.
A sensitivity analysis with data from studies published in English only indicated an RR of 0.98, 95% CI 0.94 to 1.01 (Bartley 2008; NCT00595374; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003).
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Withdrawals due to adverse events
A total of 30/2020 participants (1.5%) in the insulin detemir group compared with 6/1202 participants (0.5%) in the NPH insulin group withdrew because of adverse events. There was no evidence of a difference in withdrawals due to adverse events (RR 2.23, 95% CI 0.98 to 5.05; P = 0.05; 8 studies, 3222 participants; Analysis 1.14). The 95% prediction interval ranged between 0.80 and 6.19. We judged the overall risk of bias for this outcome as 'low' (data not shown).
1.14. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 14: Withdrawals due to adverse events
Nocturnal hypoglycaemia
We could retrieve data on nocturnal hypoglycaemia from eight studies (Bartley 2008; Kobayashi 2007; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003).
Seven studies reported severe nocturnal hypoglycaemia. A total of 70/1823 participants (3.8%) in the insulin detemir group compared with 60/1102 participants (5.4%) in the NPH insulin group experienced a severe nocturnal hypoglycaemic event. There was no evidence of a difference in severe nocturnal hypoglycaemia (RR 0.67, 95% CI 0.39 to 1.17, P = 0.16; 7 studies, 2925 participants; moderate‐certainty evidence; Analysis 1.18). We judged the overall risk of bias for this outcome as 'low'.
1.18. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 18: Severe nocturnal hypoglycaemia
The studies applied different ways of reporting nocturnal hypoglycaemia. In the trial synopsis of one study, authors wanted to investigate major nocturnal hypoglycaemia, minor nocturnal hypoglycaemia, nocturnal hypoglycaemia with symptoms only and biochemical nocturnal hypoglycaemia (defined as asymptomatic plasma glucose value). However, only the outcome of any nocturnal hypoglycaemic events was reported (Kobayashi 2007). In the CSR of this study, data for subtypes of hypoglycaemia were provided in a format making them unsuitable for meta‐analysis: minor nocturnal hypoglycaemia had an RR of 0.67, 95% CI 0.42 to 1.06; symptoms only nocturnal hypoglycaemia had an RR of 0.58, 95% CI 0.31 to 1.09 and biochemical nocturnal hypoglycaemia had an RR of 0.77, 95% CI 0.45 to 1.33 (Kobayashi 2007). One unpublished study reported nocturnal hypoglycaemia in a format suitable for meta‐analysis in the CSR (NCT00605137). Data for another unpublished study (NCT00595374) were reported in a format making them unsuitable for meta‐analysis ('no significant differences between the intervention groups').
The data for the analysis of any type of nocturnal hypoglycaemia were available in the full‐text articles of six studies (Bartley 2008; Kobayashi 2007; Robertson 2007; Russell‐Jones 2004; Thalange 2013; Vague 2003). Two studies provided data in the CSRs (NCT00605137; Standl 2004). Data for mild nocturnal hypoglycaemia and symptomatic nocturnal hypoglycaemia (without confirmed blood glucose values) could be retrieved from seven studies: four studies reported the outcome in the publication (Bartley 2008; Robertson 2007; Russell‐Jones 2004; Thalange 2013) and three studies provided the data from unpublished sources (NCT00605137; Standl 2004; Vague 2003). One study reported data on asymptomatic hypoglycaemia (Thalange 2013).
A total of 1041/1555 participants (66.9%) in the insulin detemir group compared with 877/1200 participants (73.1%) in the NPH insulin group experienced any type of nocturnal hypoglycaemic event. There was a reduction in any type of nocturnal hypoglycaemia in favour of insulin detemir (RR 0.91, 95% CI 0.87 to 0.95; P < 0.001; 8 studies, 3219 participants; Analysis 1.15). The 95% prediction interval ranged between 0.86 and 0.96. There was a reduction in mild nocturnal hypoglycaemia in favour of insulin detemir (RR of 0.90, 95% CI 0.85 to 0.96; P = 0.002; 7 studies, 3073 participants; Analysis 1.16). There was a reduction in nocturnal hypoglycaemia with symptoms in favour of insulin detemir (RR 0.88, 95% CI 0.79 to 0.98; P = 0.02; 6 studies, 2578 participants; Analysis 1.17). One study reported asymptomatic nocturnal hypoglycaemia in 83/177 participants (46.9%) in the insulin detemir group compared with 85/170 participants (50%) in the NPH insulin group (Thalange 2013). We judged the overall risk of bias for all these outcomes except for severe nocturnal hypoglycaemia as 'some concerns' (data not shown).
1.15. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 15: Any nocturnal hypoglycaemia
1.16. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 16: Mild nocturnal hypoglycaemia
1.17. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 17: Nocturnal hypoglycaemia (symptoms)
Subgroup and sensitivity analyses
Five studies reported any type of nocturnal hypoglycaemia in adults. A total of 1041/1555 participants (66.9%) in the insulin detemir group compared with 629/888 participants (70.8%) in the NPH insulin group experienced any type of nocturnal hypoglycaemia. The RR was 0.93, 95% CI 0.88 to 0.98; Analysis 1.15; favouring insulin detemir. Three studies including children reported that 337/464 children (72.6%) in the insulin detemir group compared with 258/312 children (82.7%) in the NPH insulin group experienced any type of nocturnal hypoglycaemia. The RR was 0.87, 95% CI 0.81 to 0.94; Analysis 1.15; favouring insulin detemir. The test for subgroup differences did not indicate interaction (P = 0.23).
Four studies reported mild nocturnal hypoglycaemia in adults. The RR was 0.91, 95% CI 0.83 to 1.00; Analysis 1.16; favouring insulin detemir. Three studies reported mild nocturnal hypoglycaemia in children. The RR was 0.88, 95% CI 0.78 to 1.00; Analysis 1.16; favouring insulin detemir. The test for subgroup differences did not indicate interaction (P = 0.66).
Four studies reported nocturnal hypoglycaemia with symptoms in adults. The RR was 0.91, 95% CI 0.82 to 1.01; Analysis 1.17. Two studies reported nocturnal hypoglycaemia with symptoms in children. The RR was 0.55, 95% CI 0.19 to 1.61; Analysis 1.17. The test for subgroup differences did not indicate interaction (P = 0.36).
Four studies reported severe nocturnal hypoglycaemia in adults. The RR was 0.57, 95% CI 0.35 to 0.93; Analysis 1.8; favouring insulin detemir. Three studies including children reported severe nocturnal hypoglycaemia. The RR was 0.64, 95% CI 0.13 to 3.17; Analysis 1.18. The test for subgroup differences did not indicate interaction (P = 0.88).
Six studies had published information on any type of nocturnal hypoglycaemia. The RR was 0.90, 95% CI 0.86 to 0.95; Analysis 1.19; favouring insulin detemir. Two studies had unpublished data on any type of nocturnal hypoglycaemia. The RR was 0.91, 95% CI 0.80 to 1.04; Analysis 1.19. The test for subgroup differences did not indicate interaction (P = 0.90).
1.19. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 19: Any nocturnal hypoglycaemia (published vs. unpublished data)
Four studies had published information on mild nocturnal hypoglycaemia. The RR was 0.91, 95% CI 0.85 to 0.98; Analysis 1.20; favouring insulin detemir. Three studies had unpublished information on mild nocturnal hypoglycaemia. The RR was 0.89, 95% CI 0.75 to 1.07; Analysis 1.20. The test for subgroup differences did not indicate interaction (P = 0.83).
1.20. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 20: Mild nocturnal hypoglycaemia (published vs. unpublished data)
Three studies had published information on nocturnal hypoglycaemia with symptoms. The RR was 0.90, 95% CI 0.81 to 0.99; Analysis 1.21; favouring insulin detemir. Three studies had unpublished information on nocturnal hypoglycaemia with symptoms. The RR was 0.79, 95% CI 0.57 to 1.08; Analysis 1.21. The test for subgroup differences did not indicate interaction (P = 0.44).
1.21. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 21: Nocturnal hypoglycaemia, symptoms only (published vs. unpublished data)
Five studies had published information on severe nocturnal hypoglycaemia. The RR was 0.63, 95% CI 0.32 to 1.25; Analysis 1.22. Two studies had unpublished information on severe nocturnal hypoglycaemia. The RR was 0.90, 95% CI 0.33 to 2.45; Analysis 1.22. The test for subgroup differences did not indicate interaction (P = 0.56).
1.22. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 22: Severe nocturnal hypoglycaemia (published vs. unpublished data)
Sensitivity analysis excluding the longest study (Bartley 2008) for any type of nocturnal hypoglycaemia indicated an RR of 0.90, 95% CI 0.86 to 0.94 favouring insulin detemir.
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Mild/moderate hypoglycaemia
We could retrieve data on mild/moderate hypoglycaemia from eight studies (Bartley 2008; Kobayashi 2007; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003). One unpublished study reported data for mild/moderate hypoglycaemia in a format making the data unsuitable for meta‐analysis (NCT00595374). One study did not specify mild hypoglycaemia; for this study we used data for any type of hypoglycaemia (NCT00605137). For the remaining studies, data for mild hypoglycaemia were available (Bartley 2008; Kobayashi 2007; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003).
A total of 1726/2019 participants (85.5%) in the insulin detemir compared with 1028/1200 participants (85.7%) in the NPH insulin group experienced mild/moderate hypoglycaemia. There was a reduction in mild/moderate hypoglycaemia in favour of insulin detemir (RR 0.97, 95% CI 0.94 to 0.99; P = 0.01, 8 studies, 3219 participants; Analysis 1.24). The 95% prediction interval ranged between 0.95 and 1.00. We judged the overall risk of bias for this outcome as 'some concerns'.
1.24. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 24: Mild/moderate hypoglycaemia
Subgroup and sensitivity analyses
Five studies reported mild/moderate hypoglycaemia in adults. A total of 1313/1555 participants (84.4%) in the insulin detemir group compared with 742/888 participants (83.4%) in the NPH insulin group experienced mild/moderate hypoglycaemia. The RR was 0.97, 95% CI 0.93 to 1.02; Analysis 1.24. Three studies including children reported 413/464 children (89.0%) in the insulin detemir group compared with 286/312 children (91.7%) in the NPH insulin group experienced mild/moderate hypoglycaemia. The RR was 0.97, 95% CI 0.93 to 1.01; Analysis 1.24. The test for subgroup differences did not indicate interaction (P = 0.82).
Six studies had published information on mild/moderate hypoglycaemia. The RR was 0.97, 95% CI 0.93 to 1.00; Analysis 1.25; favouring insulin detemir. Two studies had unpublished information on mild/moderate hypoglycaemia. The RR was 0.98, 95% CI 0.92 to 1.05; Analysis 1.25. The test for subgroup differences did not indicate interaction (P = 0.69).
1.25. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 25: Mild/moderate hypoglycaemia (published vs. unpublished data)
Sensitivity analysis excluding the largest study indicated an RR of 0.96, 95% CI 0.93 to 0.98 (Russell‐Jones 2004) favouring insulin detemir. Sensitivity analysis excluding the longest study indicated an RR of 0.97, 95% CI 0.94 to 1.00 (Bartley 2008) favouring insulin detemir.
A sensitivity analysis with data from studies published in English only indicated an RR of 0.97, 95% CI 0.94 to 1.00 favouring insulin detemir.
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Socioeconomic effects
No studies reported direct or indirect costs of the intervention during the study period. One study reported economic predictions of the interventions based on simulation cohorts in Belgian, Canadian, French, German, Italian and Spanish, Swedish settings (Bartley 2008).
HbA1c
We could retrieve data on HbA1c levels from eight studies (Bartley 2008; Kobayashi 2007; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003). Six studies reported HbA1c levels in publications (Bartley 2008; Kobayashi 2007; Robertson 2007; Russell‐Jones 2004; Thalange 2013; Vague 2003). Standl 2004 only reported HbA1c after the end of the extension period in publications, but through FDA review and CSR, we could retrieve data at the end of the regular intervention period. One unpublished study reported HbA1c in the clinical study synopsis (NCT00605137).
There was no evidence of a difference in HbA1c (MD 0.01%, 95% CI ‐0.1 to 0.1; P = 0.11; 8 studies, 3122 participants; moderate‐certainty evidence; Analysis 1.26). The 95% prediction interval ranged between ‐0.1% and 0.1%. We judged the overall risk of bias for this outcome as 'low'.
1.26. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 26: HbA1c
Subgroup and sensitivity analyses
Five studies reported HbA1c levels in adults. The MD of HbA1c was ‐0.03%, 95% CI ‐0.1 to 0.1; Analysis 1.26. Three studies including children reported HbA1c levels. The MD of HbA1c was 0.1%, 95% CI ‐0.04 to 0.3; Analysis 1.26. The test for subgroup differences did not indicate interaction (P = 0.11).
Analysing only published data indicated a MD of HbA1c of ‐0.02%, 95% ‐0.1 to 0.1; Analysis 1.27. Analysing only unpublished data indicated a MD of HbA1c of 0.1%, 95% CI ‐0.1 to 0.3; Analysis 1.27. The test for subgroup differences did not indicate interaction (P = 0.28). One unpublished study reported data for HbA1c in a format making the data unsuitable for meta‐analysis (NCT00595374).
1.27. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 27: HbA1c (published vs. unpublished data)
Sensitivity analysis excluding the largest study indicated a MD of HbA1c of 0.02%, 95% CI ‐0.1 to 0.1 (Russell‐Jones 2004). Sensitivity analysis excluding the longest study indicated a MD of HbA1c of 0.04%, 95% CI ‐0.1 to 0.1 (Bartley 2008).
Sensitivity analysis exclusively analysing data from studies published in English indicated a MD of HbA1c of ‐0.01%, 95% CI ‐0.1 to 0.1 (Bartley 2008; NCT00605137; Robertson 2007; Russell‐Jones 2004; Standl 2004; Thalange 2013; Vague 2003).
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Combined HbA1c and severe hypoglycaemia
No study reported on combined HbA1c and severe hypoglycaemia.
One study provided data on the combined outcome HbA1c and hypoglycaemia (Bartley 2008). We extracted these data from the CSR. This specified the percentage of participants who reached HbA1c ≤ 7.0% at the end of the study without symptomatic hypoglycaemia with a plasma glucose < 4.0 mmol/L or any single plasma glucose value < 3.1 mmol/L during the last month of treatment. This number was 71/321 participants (22.2%) in the insulin detemir group compared with 21/159 participants (13.2%) in the NPH insulin group.
Two studies stated that similar results were seen for hypoglycaemia when adjusted for HbA1c (Robertson 2007; Vague 2003). One study reported in the CSR that the observed risk of hypoglycaemia was not explained by differences in HbA1c (Russell‐Jones 2004).
Insulin glargine compared with NPH insulin
For an overview of main results for this comparison see Table 2.
Nine studies compared insulin glargine with NPH insulin (Bolli 2009; Chase 2008; Fulcher 2005; Home 2005; Liu 2016; Porcellati 2004; PRESCHOOL; Ratner 2000; Schober 2002). A total of 2387 participants were randomised, 1205 participants to insulin glargine and 1182 participants to NPH insulin (see Table 6). Four studies included children and randomised 823 children, 433 children to insulin glargine and 390 children to NPH insulin (Chase 2008; Liu 2016; PRESCHOOL; Schober 2002). The mean age of the children varied from 4.2 to 13.2 years.
All studies were published as full‐text articles in English. However, we retrieved unpublished information from most studies for this comparison (Chase 2008; Fulcher 2005; Home 2005; Liu 2016; PRESCHOOL; Ratner 2000; Schober 2002). Two studies had information solely based on full‐text publications (Bolli 2009; Porcellati 2004). We contacted investigators in order to achieve additional information, but did not receive a reply (see Appendix 19).
One study randomised participants to insulin glargine once daily and NPH insulin once daily (Fulcher 2005). One study randomised participants to insulin glargine once daily and NPH insulin or Lente insulin twice daily according to a pre‐study regimen. However, only three participants received Lente insulin (Chase 2008). Six studies randomised participants to insulin glargine once daily and NPH insulin (Bolli 2009; Home 2005; PRESCHOOL; Liu 2016; Ratner 2000; Schober 2002). One study randomised participants to insulin glargine once daily and NPH insulin four times a day (Porcellati 2004).
Five studies applied insulin lispro as fast‐acting insulin at meals (Bolli 2009; Chase 2008; Fulcher 2005; Porcellati 2004; PRESCHOOL). One study applied insulin aspart as fast‐acting insulin (Liu 2016). Three studies applied human insulin as fast‐acting insulin (Home 2005; Ratner 2000; Schober 2002).
The duration of the intervention varied from 24 weeks to 30 weeks.
Primary outcomes
All‐cause mortality
We could retrieve data on all‐cause mortality from eight studies (Chase 2008; Fulcher 2005; Home 2005; Liu 2016; Porcellati 2004; PRESCHOOL; Ratner 2000; Schober 2002). Only one of these studies reported all‐cause mortality in the full‐text publication (Porcellati 2004). We obtained the remaining data from unpublished sources.
A total of 0/1207 participants allocated to insulin glargine died compared with 1/1068 participants allocated to NPH insulin (Peto OR 0.14, 95% CI 0.00 to 6.98; P = 0.32; 8 studies, 2175 participants; moderate‐certainty evidence; Analysis 2.1). We judged the overall risk of bias for this outcome as 'low'.
2.1. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 1: All‐cause mortality
Subgroup and sensitivity analyses
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Health‐related quality of life
Four studies reported health‐related quality of life (Bolli 2009; Chase 2008; Home 2005; Ratner 2000). We judged the certainty of the evidence for these studies with 1013 participants as low. We judged the overall risk of bias for this outcome as 'some concerns'.
One study applied the Well‐Being Enquiry for Diabetics (Bolli 2009), two studies applied the General Well‐being scale (Home 2005; Ratner 2000) and one study applied the Diabetes Quality of Life for Youth and Parents' Diabetes Quality of Life (Chase 2008). Bolli 2009 randomised 175 participants. After six months, data from 133 participants were evaluated for impact domain, 114 participants for level of satisfaction, 108 participants for general worries and 111 participants for diabetes‐related worries. It was not reported how many participants in each intervention arm were included in the analysis. The only domain showing a statistically significant difference after six months was diabetes‐related worries, which showed greater improvements in the insulin glargine group (P = 0.05). At six months, the impact domain score was 77 (quartiles 73 to 82) in the insulin glargine group and 80 (quartiles 73 to 85) in the NPH insulin group. Changes in percentage from baseline were ‐1.4 (quartiles ‐10 to 8) in the insulin glargine group and ‐4.4 (quartiles ‐14 to 7) in the NPH insulin group. At six months, the level of satisfaction score was 31 (quartiles 27 to 35) in the insulin glargine group and 32 (quartiles 27 to 38) in the NPH insulin group. Changes in percentage from baseline were 0.0 (quartiles ‐10 to 8) in the insulin glargine group and ‐3.0 (quartiles ‐7 to 3) in the NPH insulin group. At six months, the general worries score was 32 (quartiles 27 to 34) in the insulin glargine group and 32 (quartiles 26 to 35) in the NPH insulin group. Changes in percentage from baseline were ‐1.4 (quartiles ‐7 to 3) in the insulin glargine group and 0.0 (quartiles ‐11 to 4) in the NPH insulin group. At six months, the diabetes‐related worries score was 32 (quartiles 27 to 34) in the insulin glargine group and 31 (quartiles 25 to 34) in the NPH insulin group. Changes in percentage from baseline were ‐5.7 (quartiles ‐12 to 4) in the insulin glargine group and 0.0 (quartiles ‐8 to 8) in the NPH insulin group (P = 0.05) (Bolli 2009). Two studies applied the General Well‐being scale (Home 2005; Ratner 2000). One study reported health‐related quality of life through a CSR (Ratner 2000). Home 2005 reported in a co‐publication that the mean score for the General Well‐being scale showed an increase (i.e. better well‐being) of 1.44 points at week 28 in the insulin glargine group compared with 1.57 points in the NPH insulin group with all four subscales contributing to these improvements (Home 2005). In the CSR, health‐related quality of life with SDs at the end of intervention were reported (Home 2005). Combining data from the two studies applying the General Well‐being scale did not show evidence of a difference (MD 0.62 points, 95% CI ‐0.71 to 1.96; P = 0.36; 2 studies, 880 participants; Analysis 2.2). For both studies, the difference between the treatments was not statistically significant at the end of follow‐up for each separate item of the General Well‐being scale (depression, anxiety, energy, positive well‐being). One study evaluated health‐related quality of life in children (Chase 2008). Data were available from the clinical study synopsis. This study applied the Diabetes Quality of Life for Youth and Parents' Diabetes Quality of Life (Chase 2008). This study did not find evidence of a difference between the interventions. No information about scores or number of participants included in the analysis was reported.
2.2. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 2: Health‐realted quality of life
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Severe hypoglycaemia
Nine studies reported data on severe hypoglycaemia (Bolli 2009; Chase 2008; Fulcher 2005; Home 2005; Liu 2016; Porcellati 2004; PRESCHOOL; Ratner 2000; Schober 2002). All studies defined severe hypoglycaemia as hypoglycaemia requiring third party assistance. For two studies, we retrieved unpublished data from the CSRs (Fulcher 2005; Ratner 2000).
A total of 122/1191 participants (10.2%) in the insulin glargine group compared with 145/1159 participants (12.5%) in the NPH insulin group experienced severe hypoglycaemia. There was no evidence of a difference in severe hypoglycaemia (RR 0.84, 95% CI 0.67 to 1.04; P = 0.11; 9 studies, 2350 participants; moderate‐certainty evidence; Analysis 2.3; Figure 4). The 95% prediction interval ranged between 0.65 and 1.09. We judged the overall risk of bias for this outcome as 'low'.
2.3. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 3: Severe hypoglycaemia
4.

Severe hypoglycaemia
One study in the main publication defined severe hypoglycaemia as requiring third party assistance in the methods section of the main publication (Ratner 2000). However, the definition of severe hypoglycaemia reported in the results section in the main publication was severe hypoglycaemic event with blood glucose levels < 2.0 mmol/L. In the CSR, severe hypoglycaemia with and without confirmed blood glucose < 2.0 mmol/L was reported. With the definition of severe hypoglycaemia according to the methods section, 23/264 participants (8.7%) in the insulin glargine group compared with 28/270 participants (10.4%) in the NPH insulin group experienced severe hypoglycaemia. This number was used for the meta‐analysis. Using severe hypoglycaemia applying the definition of blood glucose < 2.0 mmol/L showed that 7/264 participants (2.5%) in the insulin glargine group compared with 16/270 participants (5.9%) in the NPH insulin group experienced severe hypoglycaemia (Ratner 2000). From the CSR, it was also apparent, that during the screening phase no participants receiving insulin glargine during the study had an episode of severe hypoglycaemia compared with 6/270 participants (2.2%) receiving NPH insulin (Ratner 2000). One study stated in the FDA report that the participants receiving NPH insulin twice daily tended to have less hypoglycaemia than the participants receiving insulin glargine (FDA 2000; Home 2005). Schober 2002 reported the greatest number of events in children: in the CSR, 1/174 children (0.6%) in the insulin glargine group compared with 1/175 children (0.6%) in the NPH insulin group experienced coma due to hypoglycaemia; 4/174 children (2.3%) in the insulin glargine group compared with 3/175 children (1.7%) in the NPH insulin group experienced convulsions due to hypoglycaemia; 6/174 children (3.4%) in the insulin glargine group compared with 1/175 children (0.6%) in the NPH insulin group experienced syncope due to hypoglycaemia. Home 2005 reported the greatest number of events in adults: in the CSR, 7/292 participants (2.4%) in the insulin glargine group compared with 12/293 participants (4.1%) in the NPH insulin group experienced coma, convulsions or syncope reported as associated symptoms from severe hypoglycaemia (Home 2005).
Subgroup analysis and sensitivity analysis
Analysing studies including only adults indicated an RR of 0.78, 95% CI 0.58 to 1.05; Analysis 2.3. Analysing studies including only children indicated an RR of 1.14, CI 95% CI 0.59 to 2.21; Analysis 2.3. The test for subgroup differences did not indicate interaction (P = 0.31).
Restricting the analysis to only published data indicated an RR of 0.87, 95% CI 0.63 to 1.22; Analysis 2.4. Restricting the analysis to only unpublished data indicated an RR of 0.83. 95% CI 0.56 to 1.25; Analysis 2.4. The test for subgroup differences did not indicate interaction (P = 0.87).
2.4. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 4: Severe hypoglycaemia (published vs. unpublished data)
Sensitivity analysis excluding the largest study and the longest study indicated an RR of 0.88, 95% CI 0.68 to 1.14 (Home 2005).
All studies except one had received funding from the pharmaceutical industry (Porcellati 2004). Porcellati 2004 applied NPH insulin four times a day. Excluding this study from the analysis indicated an RR of 0.83, 95% CI 0.67 to 1.04.
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Hypoglycaemia reported as a serious adverse event
A total of 52/1131 participants (4.6%) in the insulin glargine group compared with 56/1098 participants (5.1%) in the NPH insulin group had a SAE due to hypoglycaemia. There was no evidence of a difference in hypoglycaemia reported as a SAE (RR 0.94, 95% CI 0.64 to 1.39; P = 0.76; 8 studies, 2229 participants; Analysis 2.5). The 95% prediction interval ranged between 0.52 and 1.71. We judged the overall risk of bias for this outcome as 'low' (data not shown).
2.5. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 5: Hypoglycaemia reported as a serious adverse event
The test for subgroup differences comparing adults with children did not indicate interaction (P = 0.90).
Secondary outcomes
Cardiovascular mortality
We could retrieve data on cardiovascular mortality from eight studies (Chase 2008; Fulcher 2005; Home 2005; Liu 2016; Porcellati 2004; PRESCHOOL; Ratner 2000; Schober 2002). Only one of these studies reported cardiovascular mortality in the full‐text publication (Porcellati 2004). We retrieved the remaining data from unpublished sources.
Analysing all available data showed 0/1106 participants allocated to insulin glargine died compared with 1/1068 participants allocated to NPH insulin (Analysis 2.6). We judged the overall risk of bias for this outcome as 'low'.
2.6. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 6: Cardiovascular mortality
Subgroup and sensitivity analyses
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐fatal myocardial infarction
None of the included studies reported non‐fatal myocardial infarction in the publications. One study in adults had data on non‐fatal myocardial infarction from the CSR (Home 2005). In this study, 0/292 participants in the insulin glargine group compared with 0/293 participants in the NPH insulin group experienced a non‐fatal myocardial infarction (low‐certainty evidence; Analysis 2.7). We judged the overall risk of bias for this outcome as 'low'.
2.7. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 7: Non‐fatal myocardial infarction
Non‐fatal stroke
None of the included studies reported non‐fatal stroke in the publications. One study in adults had data on cerebral ischaemia from the CSR (Home 2005). In this study, 0/292 participants in the insulin glargine group compared with 1/293 participants in the NPH insulin group experienced cerebral ischaemia (low‐certainty evidence; Analysis 2.8). We judged the overall risk of bias for this outcome as 'low'.
2.8. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 8: Non‐fatal stroke
End‐stage renal disease
None of the studies reported on end‐stage renal disease.
Blindness
None of the studies reported on blindness.
Serious adverse events
Eight studies reported data on SAEs (Bolli 2009; Chase 2008; Fulcher 2005; Home 2005; Liu 2016; PRESCHOOL; Ratner 2000; Schober 2002).
A total of 109/1131 participants (9.6%) in the insulin glargine group compared with 110/1098 participants (10.0%) in the NPH insulin group experienced SAEs. There was no evidence of a difference in SAEs (RR 1.08, 95% CI 0.63 to 1.84; P = 0.79; 8 studies, 2229 participants; moderate‐certainty evidence; Analysis 2.9). The 95% prediction interval ranged between 0.22 and 5.21. We judged the overall risk of bias for this outcome as 'low'.
2.9. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 9: Serious adverse events
One study did not mention SAEs in the publication (Porcellati 2004). One study reported SAEs other than hypoglycaemia in the main publication (Fulcher 2005): 5/62 participants (8.0%) in the insulin glargine group compared with 3/63 participants (4.7%) in the NPH insulin group experienced a SAE. From the CSR, the number of participants experiencing any SAE was reported and used in the meta‐analysis (Fulcher 2005). Three other studies contributed with data from additional sources (Liu 2016; PRESCHOOL; Ratner 2000). Three studies reported SAEs in the main publication (Bolli 2009; Home 2005; Schober 2002).
Subgroup analysis and sensitivity analysis
Analysing studies including only adults indicated an RR of 0.99, 95% CI 0.72 to 1.35; Analysis 2.9. Analysing studies including only children indicated an RR of 1.02, CI 95% CI 0.28 to 3.64; Analysis 2.9. The test for subgroup differences did not indicate interaction (P = 0.96).
Restricting the analysis to only published data indicated an RR of 1.11, 95% CI 0.11 to 2.70; Analysis 2.10. Restricting the analysis to only unpublished data indicated an RR of 1.10, 95% CI 0.46 to 2.60; Analysis 2.10. The test for subgroup differences did not indicate interaction (P = 0.99).
2.10. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 10: Serious adverse events (published vs. unpublished data)
Sensitivity analysis excluding the largest study and the longest study indicated an RR of 1.15, 95% CI 0.58 to 2.30 (Home 2005).
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Diabetic ketoacidosis
We could retrieve data on diabetic ketoacidosis from seven studies (Chase 2008; Fulcher 2005; Home 2005; Liu 2016; PRESCHOOL; Ratner 2000; Schober 2002). Three studies reported ketoacidosis in their full‐text publications (Chase 2008; Liu 2016; Schober 2002).
A total of 6/1046 participants (0.6%) had ketoacidosis in the insulin glargine group compared with 8/1008 participants (0.1%) in the NPH insulin group. There was no evidence of a difference in diabetic ketoacidosis (RR 0.53, 95% CI 0.19 to 1.44; P = 0.21; 7 studies, 2054 participants; Analysis 2.11). We judged the overall risk of bias for this outcome as 'low'.
2.11. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 11: Diabetic ketoacidosis
Subgroup and sensitivity analyses
Analysing diabetic ketoacidosis in only adults indicated an RR of 1.00, 95% CI 0.11 to 9.58; Analysis 2.11. Analysing diabetic ketoacidosis in only children indicated an RR of 0.45, 95% CI 0.15 to 1.39, Analysis 2.11. The test for subgroup differences did not indicate interaction (P = 0.53).
Analysing only published data indicated that 4/366 participants (1.1%) in the insulin glargine group compared with 8/319 participants (2.5%) in the NPH insulin group experienced diabetic ketoacidosis. The RR was 0.39, 95% CI 0.11 to 1.31; Analysis 2.12. Analysing only unpublished data indicated that 2/680 participants (0.3%) in the insulin glargine group compared with 3/689 participants (0.4%) in the NPH insulin group experienced diabetic ketoacidosis. The RR was 1.01, 95% CI 0.18 to 5.77; Analysis 2.12. The test for subgroup differences did not indicate interaction (P = 0.38).
2.12. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 12: Diabetic ketoacidosis (published vs. unpublished data)
Sensitivity analysis excluding the largest study and the longest study indicated an RR of 0.43, 95% CI 0.16 to 1.17 (Home 2005).
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐serious adverse events
Eight studies reported data on non‐serious adverse events (Bolli 2009; Chase 2008; Fulcher 2005; Home 2005; Liu 2016; PRESCHOOL; Ratner 2000; Schober 2002).
A total of 792/1131 participants (70.0%) in the insulin glargine group compared with 747/1098 (68.0%) participants in the NPH insulin group experienced a non‐serious adverse event. There was no evidence of a difference in non‐serious adverse events (RR 1.01, 95% CI 0.96 to 1.06; P = 0.72; 8 studies, 2229 participants; Analysis 2.13). The 95% prediction interval ranged between 0.95 and 1.07. We judged the overall risk of bias for this outcome as 'some concerns'.
2.13. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 13: Non‐serious adverse events
One study did not mention adverse events in the publication (Porcellati 2004).
Subgroup analysis and sensitivity analysis
Analysing studies including only adults indicated an RR of 1.01, 95% CI 0.95 to 1.07; Analysis 2.13. Analysing studies including only children indicated an RR of 1.02, CI 95% CI 0.93 to 1.12; Analysis 2.13. The test for subgroup differences did not indicate interaction (P = 0.81).
Restricting the analysis to only published data indicated an RR of 1.00, 95% CI 0.94 to 1.05, Analysis 2.14. Restricting the analysis to only unpublished data indicated an RR of 1.03, 95% CI 0.94 to 1.14, Analysis 2.14. The test for subgroup differences did not indicate interaction (P = 0.53).
2.14. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 14: Non‐serious adverse events (published vs. unpublished data)
Sensitivity analysis excluding the largest study and the longest study indicated an RR of 1.00, 95% CI 0.95 to 1.06 (Home 2005).
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Withdrawals due to adverse events
A total of 11/1130 participants (1%) in the insulin glargine group compared with 9/1100 participants (0.8%) in the NPH insulin group withdrew because of adverse events. There was no evidence of a difference in withdrawals due to adverse events (RR 0.80, 95% CI 0.24 to 2.81; P = 0.76; 8 studies, 2130 participants; Analysis 2.15). The 95% prediction interval ranged between 0.07 and 10.27. We judged the overall risk of bias for this outcome as 'low' (data not shown).
2.15. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 15: Withdrawals due to adverse events
Nocturnal hypoglycaemia
We could retrieve data on nocturnal hypoglycaemia from seven studies (Chase 2008; Fulcher 2005; Home 2005; Liu 2016; PRESCHOOL; Ratner 2000; Schober 2002).
Four studies reported severe nocturnal hypoglycaemia in the CSRs (Chase 2008; Fulcher 2005; Home 2005; Ratner 2000) and two studies reported severe nocturnal hypoglycaemia in the publications (PRESCHOOL; Schober 2002). A total of 69/938 participants (7.4%) in the insulin glargine group compared with 83/955 participants (8.7%) in the NPH insulin group experienced severe nocturnal hypoglycaemia. There was no evidence of a difference in severe nocturnal hypoglycaemia (RR 0.83, 95% CI 0.62 to 1.12; P = 0.23; 6 studies, 1893 participants; moderate‐certainty evidence; Analysis 2.19). We judged the overall risk of bias for this outcome as 'low'.
2.19. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 19: Severe nocturnal hypoglycaemia
One study only reported frequency of nocturnal hypoglycaemia for the last month of treatment and not for the whole intervention period (12 months): there were 1.2 (SD 0.2) episodes/patient‐month in the insulin glargine group compared with 3.2 (SD 0.3) episodes/patient‐month in the NPH insulin group (Porcellati 2004). One study reported that there was no statistically significant change in nocturnal hypoglycaemia between the intervention groups (Bolli 2009). Five of the studies reported the number of participants with nocturnal hypoglycaemia in the publications (Fulcher 2005; Home 2005; Liu 2016; PRESCHOOL; Schober 2002). For two studies, we retrieved data from other sources (Chase 2008; Ratner 2000). Chase 2008 reported that no statistically significant change between the intervention groups was identified. Ratner 2000 reported nocturnal hypoglycaemia with confirmed blood glucose < 2 mmol/L and not just hypoglycaemia occurring at night as defined in the method section of the publication. In the CSR of this study, two different definitions of nocturnal hypoglycaemia were stated: hypoglycaemia at night and hypoglycaemia at night with blood glucose < 2 mmol/L. Nocturnal hypoglycaemia was reported for three different time periods in the CSR (after one month, from two months to the end of study, for the entire study period). From the tables in the CSR, it was apparent that the only analysis showing a statistically significant benefit of insulin glargine was nocturnal hypoglycaemia with confirmed blood glucose < 2 mmol/L from two months until the end of the study. This definition and time period were the ones reported in the full‐text publication.
A total of 713/1045 participants (68.2%) in the insulin glargine group compared with 693/1009 participants (68.7%) in the NPH insulin group experienced any nocturnal hypoglycaemia. There was no evidence of a difference in any nocturnal hypoglycaemia (RR 1.00, 95% CI 0.96 to 1.05; P = 0.96; 7 studies, 1054 participants; Analysis 2.16). One study investigated mild nocturnal hypoglycaemia as reported in the CSR (Fulcher 2005): 39/62 participants (62.9%) in the insulin glargine group compared with 47/63 participants (74.6%) in the NPH insulin group experienced mild nocturnal hypoglycaemia (RR 0.84, 95% CI 0.66 to 1.07; Analysis 2.17). Symptomatic nocturnal hypoglycaemia with or without blood glucose validation was reported in four studies (Fulcher 2005; Home 2005; Liu 2016; PRESCHOOL). There was no evidence of a difference in symptomatic nocturnal hypoglycaemia (RR 0.93, 95% CI 0.82 to 1.05; P = 0.26; 4 studies, 996 participants; Analysis 2.18). No study reported on asymptomatic nocturnal hypoglycaemia. Home 2005 reported that the proportion of participants who experienced nocturnal hypoglycaemia confirmed by a blood glucose level < 2.8 mmol/L and < 2.0 mmol/L did not differ significantly between interventions. We judged the overall risk of bias for all these outcomes except for severe nocturnal hypoglycaemia as 'some concerns' (data not shown).
2.16. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 16: Nocturnal hypoglycaemia
2.17. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 17: Mild nocturnal hypoglycaemia
2.18. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 18: Nocturnal hypoglycaemia (symptoms)
Subgroup analysis and sensitivity analysis
Analysing studies for any nocturnal hypoglycaemia including only adults indicated an RR of 0.99, 95% CI 0.92 to 1.06; Analysis 2.16. Analysing studies for any nocturnal hypoglycaemia including only children indicated an RR of 1.01, CI 95% CI 0.95 to 1.08; Analysis 2.16. The test for subgroup differences did not indicate interaction (P = 0.65).
Analysing studies for symptomatic nocturnal hypoglycaemia including only adults indicated an RR of 0.97, 95% CI 0.88 to 1.08; Analysis 2.18. Analysing studies for symptomatic nocturnal hypoglycaemia including only children indicated an RR of 0.74, 95% CI 0.55 to 1.00; Analysis 2.18. The test for subgroup differences did not indicate interaction (P = 0.09).
Analysing studies for severe nocturnal hypoglycaemia including only adults indicated an RR of 0.87, 95% CI 0.60 to 1.27; Analysis 2.19. Analysing studies for severe nocturnal hypoglycaemia including only children indicated an RR of 0.77, 95% CI 0.47 to 1.25; Analysis 2.19. The test for subgroup differences did not indicate interaction (P = 0.68).
Restricting the analysis to only published data for any nocturnal hypoglycaemia indicated an RR of 1.00, 95% CI 0.95 to 1.06; Analysis 2.20. Restricting the analysis to only unpublished data for any nocturnal hypoglycaemia indicated an RR of 1.00, 95% CI 0.91 to 1.08; Analysis 2.20. The test for subgroup differences did not indicate interaction (P = 0.86).
2.20. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 20: Nocturnal hypoglycaemia (published vs. unpublished data)
Sensitivity analysis of any nocturnal hypoglycaemia excluding the largest study and the longest study indicated an RR of 1.00, 95% CI 0.95 to 1.05 (Home 2005).
Restricting the analysis to only published data for symptomatic nocturnal hypoglycaemia indicated an RR of 0.87, 95% CI 0.67 to 1.12; Analysis 2.21. Analysing only unpublished data for symptomatic nocturnal hypoglycaemia indicated an RR of 0.94, 95% CI 0.80 to 1.10; Analysis 2.21.
2.21. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 21: Symptomatic nocturnal hypoglycaemia (published vs. unpublished data)
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Mild/moderate hypoglycaemia
We could retrieve data on mild/moderate hypoglycaemia from seven studies (Chase 2008; Fulcher 2005; Home 2005; Liu 2016; PRESCHOOL; Ratner 2000; Schober 2002).
A total of 951/1045 participants (91.0%) in the insulin glargine group compared with 898/1009 participants (89.0%) in the NPH insulin group experienced mild/moderate hypoglycaemia. There was no evidence of a difference in mild/moderate hypoglycaemia (RR 1.02, 95% CI 1.00 to 1.04; P = 0.09; 7 studies, 2054 participants; Analysis 2.22). We judged the overall risk of bias for this outcome as 'some concerns'.
2.22. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 22: Mild/moderate hypoglycaemia
One study only reported frequency of mild hypoglycaemia for the last month of treatment and not for the whole intervention period (12 months): there were 7.2 (SD 0.5) episodes/patient‐month in the insulin glargine group compared with 13.2 (SD 0.5) episodes/patient‐month in the NPH insulin group (Porcellati 2004). One study reported that there was no statistically significant change in hypoglycaemia between the intervention groups (Bolli 2009). Five studies reported mild/moderate hypoglycaemia in a format making the data suitable for meta‐analysis (Chase 2008; Home 2005; Liu 2016; PRESCHOOL; Schober 2002). For two studies, we retrieved the data from additional sources (Fulcher 2005; Ratner 2000).
Subgroup analysis and sensitivity analysis
Analysing studies including only adults indicated an RR of 1.02, 95% CI 0.99 to 1.06; Analysis 2.22. Analysing studies including only children indicated an RR of 1.01, CI 95% CI 0.99 to 1.04; Analysis 2.22. The test for subgroup differences did not indicate interaction (P = 0.68).
Restricting the analysis to only published data indicated an RR of 1.02, 95% CI 1.00 to 1.05; Analysis 2.23. Restricting the analysis to only unpublished data indicated an RR of 1.01, 95% CI 0.98 to 1.04; Analysis 2.23. The test for subgroup differences did not indicate interaction (P = 0.78).
2.23. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 23: Mild/moderate hypoglycaemia (published vs. unpublished data)
Sensitivity analysis excluding the largest study and the longest study indicated an RR of 1.01, 95% CI 0.99 to 1.04 (Home 2005).
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Socioeconomic effects
We retrieved data for socioeconomic effects from CSRs of three studies (Fulcher 2005; Home 2005; Schober 2002). No studies reported an estimate of the costs of the intervention during the study period. One study had evaluated economic effects, but the supplemental CSR with these data could unfortunately not be retrieved (Ratner 2000). In the CSR for Fulcher 2005, it was reported that very few participants (three in each group) reported a loss of income because of diabetes during the treatment period. Approximately 30 participants in each intervention group reported seeking medical advice (ambulatory care) once or more during the treatment period (Fulcher 2005). Home 2005 could not evaluate all participants for economic data: 6/275 participants (2.1%) changed from employment status to non‐employment status during the study in the insulin glargine group compared with 7/265 participants (2.6%) in the NPH insulin group. Of the participants employed at baseline, 16/287 participants (7.5%) in the insulin glargine group compared with 23/283 participants (10.8%) in the NPH insulin group lost time for work during the study. Reasons for these changes during the study were not reported. Schober 2002 reported that nine of the caregivers (7.5%) employed at baseline had lost time for work during the study in the insulin glargine group compared with 12 of the caregivers (10.3%) in the NPH insulin group.
HbA1c
We retrieved data on HbA1c levels from all studies (Bolli 2009; Chase 2008; Fulcher 2005; Home 2005; Liu 2016; Porcellati 2004; PRESCHOOL; Ratner 2000; Schober 2002).
There was no evidence of a difference in HbA1c (MD 0.02%, 95% CI ‐0.1 to 0.1; P = 0.59; 9 studies, 2285 participants; moderate‐certainty evidence; Analysis 2.24). The 95% prediction interval ranged between ‐0.5% and 0.5%. We judged the overall risk of bias for this outcome as 'low'.
2.24. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 24: HbA1c
One study reported HbA1c at the end of follow‐up as adjusted least square means in the publication (Fulcher 2005). However, in this study, HbA1c at baseline was higher in the participants randomised to NPH insulin compared with insulin glargine (9.2% (SD 1.1) in the insulin glargine group compared with 9.7% (SD 1.3) in the NPH insulin group). In the CSR of this study, data with change from baseline were provided which we included in the meta‐analysis. Chase 2008 reported HbA1c for completers of the study only. However, in the CSR, HbA1c was reported for completers and for the intention‐to‐treat population.
Subgroup and sensitivity analysis
Five studies reported HbA1c in adults with a MD of ‐0.01%, 95% CI ‐0.2 to 0.1; Analysis 2.24. Four studies including only children reported HbA1c with a MD of 0.03%, 95% CI ‐0.1 to 0.2; Analysis 2.24. The test for subgroup differences did not indicate interaction (P = 0.67).
Analysing only published data indicated HbA1c with a MD of 0.02%, 95% CI ‐0.1 to 0.1, Analysis 2.25. Analysing only unpublished data indicated HbA1c with a MD of ‐0.04%, 95% CI ‐0.3 to 0.2; Analysis 2.25. The test for subgroup differences did not indicate interaction (P = 0.60).
2.25. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 25: HbA1c (published vs unpublished data)
Sensitivity analysis excluding the largest study and the longest study indicated a MD in HbA1c of 0.0%, 95% CI ‐0.1 to 0.1 (Home 2005).
All studies, except one had received funding from the pharmaceutical industry (Porcellati 2004). Porcellati 2004 applied NPH four times a day. Excluding this study from the analysis indicated a MD in HbA1c of 0.02%, 95% CI ‐0.1 to 0.1.
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Combined HbA1c and severe hypoglycaemia
None of the studies reported on combined HbA1c and severe hypoglycaemia.
Insulin detemir compared with insulin glargine
For an overview of main results for this comparison, see Table 3.
Two studies compared insulin detemir with insulin glargine (Heller 2009; Pieber 2007). A total of 769 participants were randomised, 461 participants to insulin detemir and 308 participants to insulin glargine (Table 6). Both studies were published as full‐text articles in English. However, we retrieved unpublished information on outcomes for both studies from additional sources. One study administered insulin detemir once daily (evening dose). If necessary, a second dose could be administered in the morning (Heller 2009). One study applied insulin detemir twice daily (Pieber 2007). Insulin glargine was given once daily (evening dose) in both studies. Fast‐acting insulin was insulin aspart in both studies. Both studies included adults with T1DM. The duration of the intervention varied from 24 weeks to 52 weeks (see Table 6). Both studies were sponsored by Novo Nordisk.
Primary outcomes
All‐cause mortality
We retrieved data on all‐cause mortality from the clinical study synopsis of both studies. Heller 2009 reported that 0/299 participants died in the insulin detemir group compared with 1/144 participants (0.7%) in the insulin glargine group and Pieber 2007 reported that 0/161 participants died in the insulin detemir group compared with 0/159 participants in the insulin glargine group (low‐certainty evidence; Analysis 3.1). We judged the overall risk of bias for this outcome as 'low'.
3.1. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 1: All‐cause mortality
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Health‐related quality of life
No study reported scales evaluating health‐related quality of life. One study used the Diabetes Treatment Satisfaction Questionnaire and pain perception (Pieber 2007). One study used the Insulin Treatment Satisfaction Questionnaire (Heller 2009). Both treatment satisfaction questionnaires were reported in CSRs.
Severe hypoglycaemia
Heller 2009 reported the mean number of hypoglycaemic episodes in the insulin detemir group to be 146 in 299 participants in the insulin detemir group compared with 53 in 144 participants in the insulin glargine group. However, we could retrieve the number of participants experiencing one or more severe hypoglycaemic episodes from the associated CSR. Pieber 2007 reported severe hypoglycaemia in the publication.
A total of 57/460 participants (12.4%) in the insulin detemir group compared with 35/303 participants (11.6%) in the insulin glargine group experienced severe hypoglycaemia. There was no evidence of a difference in severe hypoglycaemia (RR 0.59, 95% CI 0.13 to 2.63; P = 0.49; 2 studies, 763 participants; very low‐certainty evidence; Analysis 3.2; Figure 5). We judged the overall risk of bias for this outcome as 'low'.
3.2. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 2: Severe hypoglycaemia
5.

Severe hypoglycaemia
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Hypoglycaemia reported as a serious adverse event
A total of 13/460 participants (2.8%) in the insulin detemir group compared with 5/303 participants (1.7%) in the insulin glargine group experienced hypoglycaemia as a SAE. There was no evidence of a difference in hypoglycaemia reported as a SAE (RR 1.16, 95% CI 0.14 to 9.48; P = 0.89; 2 studies, 763 participants; Analysis 3.4). We judged the overall risk of bias for this outcome as 'low' (data not shown).
3.4. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 4: Hypoglycaemia reported as a serious adverse event
Subgroup and sensitivity analysis
Analysis according to publication status indicated interaction (P = 0.02; Analysis 3.3). However, this has to be interpreted with caution because the 95% CIs slightly overlapped. The remaining subgroup and sensitivity analyses could not be performed due to lack of data (Appendix 20).
3.3. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 3: Severe hypoglycaemia (published vs. unpublished data)
Secondary outcomes
Cardiovascular mortality
We could retrieve data on cardiovascular mortality from additional sources for both studies. Heller 2009 reported that 0/299 participants died due to cardiovascular disease in the insulin detemir group compared with 1/144 participants (0.7%) in the insulin glargine group and Pieber 2007 reported that 0/161 participants in the insulin detemir group compared with 0/159 participants in the insulin glargine group died (Analysis 3.5). We judged the overall risk of bias for this outcome as 'low'.
3.5. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 5: Cardiovascular mortality
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐fatal myocardial infarction
Heller 2009 reported in the CSR that 1/299 participants (0.3%) in the insulin detemir group compared with 1/144 participants (0.7%) in the insulin glargine group experienced a non‐fatal myocardial infarction (low‐certainty evidence; Analysis 3.6). We judged the overall risk of bias for this outcome as 'low'.
3.6. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 6: Non‐fatal myocardial infarction
Non‐fatal stroke
Heller 2009 reported in the CSR that 2/299 participants (0.6%) in the insulin detemir group compared with 0/144 participants in the insulin glargine group experienced a non‐fatal stroke (low‐certainty evidence; Analysis 3.7). We judged the overall risk of bias for this outcome as 'low'.
3.7. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 7: Non‐fatal stroke
End‐stage renal disease
None of the studies for reported on end‐stage renal disease.
Blindness
None of the studies reported on blindness.
Serious adverse events
Both studies reported SAEs in the publications. A total of 49/460 participants (10.7%) in the insulin detemir group compared with 18/303 participants (5.9%) in the insulin glargine group experienced a SAE. There was no evidence of a difference in SAEs (RR 1.72, 95% CI 0.91 to 3.23; P = 0.24; 2 studies, 763 participants; low‐certainty evidence; Analysis 3.8). Analysing data in a fixed‐effect model showed beneficial effects of insulin glargine (RR 1.79, 95% CI 1.04 to 3.08; P = 0.04). We judged the overall risk of bias for this outcome as 'low'.
3.8. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 8: Serious adverse events
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Diabetic ketoacidosis
Heller 2009 reported in the CSR that 1/299 participants (0.3%) in the insulin detemir group compared with 0/144 participants in the insulin glargine group experienced ketoacidosis (Analysis 3.9). We judged the overall risk of bias for this outcome as 'low'.
3.9. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 9: Diabetic ketoacidosis
Non‐serious adverse events
We could retrieve data on non‐serious adverse events from both studies. Heller 2009 reported adverse events in the publication; we retrieved data for Pieber 2007 data from additional sources.
A total of 394/460 participants (85.7%) in the insulin detemir group compared with 250/303 participants (82.5%) in the insulin glargine group reported a non‐serious adverse event. There was no evidence of a difference in non‐serious adverse events (RR 1.01, 95% CI 0.93 to 1.09; 2 studies, 763 participants; Analysis 3.10). We judged the overall risk of bias for this outcome as 'some concerns'.
3.10. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 10: Non‐serious adverse events
Subgroup and sensitivity analysis
Subgroup analysis according to published data compared with unpublished data did not indicate interaction (P = 0.28; Analysis 3.11). We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
3.11. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 11: Non‐serious adverse events (published vs. unpublished data)
Withdrawals due to adverse events
A total of 9/460 participants (2.0%) in the insulin detemir group compared with 5/303 participants (1.7%) in the insulin glargine group withdrew because of adverse events. There was no evidence of a difference in withdrawals due to adverse events (RR 1.06, 95% CI 0.31 to 3.67; P = 0.92; 2 studies, 763 participants; Analysis 3.12).
3.12. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 12: Withdrawals due to adverse events
Nocturnal hypoglycaemia
We could retrieve data on nocturnal hypoglycaemia from both studies. Pieber 2007 reported nocturnal hypoglycaemia in the publication. We retrieved data for Heller 2009 from additional sources. Both studies defined nocturnal hypoglycaemia as an episode occurring between 23.00 and 06.00.
A total of 27/460 participants (5.9%) in the insulin detemir group compared with 15/303 participants (5.0%) in the insulin glargine group experienced severe nocturnal hypoglycaemia. There was no evidence of a difference in severe nocturnal hypoglycaemia (RR 0.55, 95% CI 0.06 to 5.12; P = 0.60; 2 studies, 763 participants; very low‐certainty evidence; Analysis 3.16). We judged the overall risk of bias for this outcome as 'low'.
3.16. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 16: Severe nocturnal hypoglycaemia
Pieber 2007 reported data on nocturnal hypoglycaemia according to different definitions in the publication. Heller 2009 reported there were no significant differences between the interventions in the risk of having a nocturnal hypoglycaemic episode, but the number of participants with an event in each intervention group was not provided in the publication. However, we could obtain these data from the CSR. Both studies had analysed nocturnal hypoglycaemia according to the same subclassifications: there was no evidence of a difference in any nocturnal hypoglycaemia (RR 1.01, 95% CI 0.93 to 1.09; P = 0.84; 2 studies, 763 participants; Analysis 3.13), in confirmed nocturnal hypoglycaemia (plasma glucose < 3.1 mmol/L and no assistance; RR 1.01, 95% CI 0.92 to 1.10; P = 0.90; 2 studies, 763 participants; Analysis 3.14); and in symptomatic nocturnal hypoglycaemia (plasma glucose ≤ 3.1 mmol/L or no plasma glucose, no assistance required; RR 1.02, 95% CI 0.81 to 1.29; P = 0.85; 2 studies, 763 participants; Analysis 3.15). We judged the overall risk of bias for all these outcomes except for severe nocturnal hypoglycaemia as 'some concerns' (data not shown).
3.13. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 13: Any nocturnal hypoglycaemia
3.14. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 14: Confirmed nocturnal hypoglycaemia (PG < 3.1 mmol/L and no assistance)
3.15. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 15: Symptomatic nocturnal hypoglycaemia (PG ≥ 3.1 or no PG and no assistance required)
Subgroup and sensitivity analysis
Analysis could only be performed according to published data compared with unpublished data: none of the definitions of nocturnal hypoglycaemia indicated interactions. We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Mild/moderate hypoglycaemia
We could retrieve data on mild/moderate hypoglycaemia from both studies. Pieber 2007 reported mild/moderate hypoglycaemia in the publication. Heller 2009 reported that the overall risk of having a hypoglycaemic episode during the treatment period was similar between the insulin detemir and the insulin glargine group with a relative risk (insulin detemir/insulin glargine) of 0.94; P = 0.57. The number of participants with mild/moderate hypoglycaemia was not reported in this publication. However, we could retrieve data from the CSR.
A total of 404/460 participants (87.8%) in the insulin detemir group compared with 243/303 participants (80.2%) in the insulin glargine group experienced mild/moderate hypoglycaemia. There was no evidence of a difference in mild/moderate hypoglycaemia (RR 1.04, 95% CI 0.94 to 1.14; P = 0.44; 2 studies, 763 participants; Analysis 3.17). We judged the overall risk of bias for this outcome as 'some concerns'.
3.17. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 17: Mild/moderate hypoglycaemia
Subgroup and sensitivity analysis
Analysis according to published data compared with unpublished data did not indicate interaction. We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Socioeconomic effects
No studies reported the costs of the intervention during the study period. One study published economic data based on simulation cohorts from a US healthcare system perspective (Pieber 2007).
HbA1c
We could retrieve data on HbA1c levels from both studies. There was no evidence of a difference in HbA1c (MD ‐0.01%, 95% CI ‐0.1 to 0.1; P = 0.89; 2 studies, 763 participants; low‐certainty evidence; Analysis 3.18). We judged the overall risk of bias for this outcome as 'low'.
3.18. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 18: HbA1c
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Combined HbA1c and severe hypoglycaemia
Heller 2009 reported that HbA1c ≤ 7% was achieved without major hypoglycaemia during the last month of treatment for 91/285 participants (31.9%) in the insulin detemir group compared with 39/135 participants (28.9%) in the insulin glargine group (RR 1.11, 95% CI 0.81 to 1.51; P = 0.53; Analysis 3.19). Pieber 2007 did not report numerical data, but stated that the adjustment for HbA1c showed that the reduced risk of hypoglycaemia with insulin detemir was not due to differences in glycaemic control. We judged the overall risk of bias for this outcome as 'low' (data not shown).
3.19. Analysis.

Comparison 3: Insulin detemir versus insulin glargine, Outcome 19: Individuals with HbA1c < 7% without severe hypoglycaemia
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Insulin degludec compared with insulin detemir
For an overview of main results for this comparison, see Table 4.
Two studies compared insulin degludec with insulin detemir (BEGIN Young; Davies 2014). A total of 806 participants were randomised, 477 participants to insulin degludec and 329 participants to insulin detemir (see 'Overview of study populations' Table 6). One study included children (BEGIN Young). The mean age of the children was 10 years. Both studies were published as full‐text articles in English. However, for both studies we could retrieve additional information on outcomes from additional sources. Both studies applied insulin degludec once daily and insulin detemir once or twice daily. Both studies applied insulin aspart as fast‐acting insulin. The duration of the intervention was 26 weeks in both studies and both studies had an extension period of 26 weeks. Both studies were sponsored by the same pharmaceutical company (Novo Nordisk).
Primary outcomes
All‐cause mortality
Both studies reported data on all‐cause mortality (BEGIN Young; Davies 2014). No participant died (0/475 participants in the insulin degludec group compared with 0/327 participants in the insulin detemir group; low‐certainty evidence; Analysis 4.1). We judged the overall risk of bias for this outcome as 'low'.
4.1. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 1: All‐cause mortality
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Health‐related quality of life
We retrieved data on health‐related quality of life from additional sources (Davies 2014). The applied questionnaire was the SF‐36. There was no evidence of a difference in health‐related quality of life for the physical health score (MD ‐0.60, 95% CI ‐1.83 to 0.63; P = 0.34; 1 study, 454 participants; low‐certainty evidence; Analysis 4.2) The health‐related quality of life for the mental health score favoured insulin detemir (MD ‐3.00, 95% CI ‐4.44 to ‐1.56; P < 0.001; 1 study, 454 participants; low‐certainty evidence; Analysis 4.2). The minimal important difference for the physical component score is two to three points and for the mental component score three points. We judged the overall risk of bias for this outcome as 'some concerns'.
4.2. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 2: Health‐related quality of life
Severe hypoglycaemia
Both studies reported data on severe hypoglycaemia in the publications. In the insulin degludec group, 63/475 participants (13.3%) experienced severe hypoglycaemia compared with 40/327 participants (12.2%) in the insulin detemir group. There was no evidence of a difference in severe hypoglycaemia (RR 1.17, 95% CI 0.81 to 1.69; P = 0.42; 2 studies, 802 participants; low‐certainty evidence; Analysis 4.3; Figure 6). We judged the overall risk of bias for this outcome as 'low'.
4.3. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 3: Severe hypoglycaemia
6.

Severe hypoglycaemia
Subgroup and sensitivity analysis
Subgroup analysis including only adults compared with studies including only children did not indicate interaction (P = 0.51; Analysis 4.3). We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Hypoglycaemia reported as a serious adverse event
A total of 15/475 participants (3.2%) in the insulin degludec group compared with 10/327 participants (3.1%) in the insulin detemir group had a SAE due to hypoglycaemia. There was no evidence of a difference in SAEs (RR 0.92, 95% CI 0.37 to 2.32; P = 0.86; 2 studies, 802 participants; Analysis 4.4). We judged the overall risk of bias for this outcome as 'low' (data not shown).
4.4. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 4: Hypoglycaemia reported as a serious adverse event
The test for subgroup differences comparing adults with children did not indicate interaction (P = 0.27).
BEGIN Young in the SAE list of the CSR stated that 2/174 participants (1.1%) in the insulin degludec group compared with 4/175 participants (2.3%) in the insulin detemir experienced a hypoglycaemic seizure and 1/174 participants (0.6%) in the insulin degludec group compared with 1/175 participants (0.6%) in the insulin detemir group experienced hypoglycaemic unconsciousness. Davies 2014 in the SAE list of the CSR reported that 3/301 participants (1.0%) in the insulin degludec group compared with 1/152 participants (0.7%) in the insulin detemir group experienced a hypoglycaemic coma and 3/301 participants (1.0%) compared with 1/152 participants (0.7%) experienced hypoglycaemic unconsciousness.
Secondary outcomes
Cardiovascular mortality
Both studies reported data on cardiovascular mortality. No participant died (0/475 participants in the insulin degludec group compared with 0/327 participants in the insulin detemir group; Analysis 4.5). We judged the overall risk of bias for this outcome as 'low'.
4.5. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 5: Cardiovascular mortality
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐fatal myocardial infarction
Davies 2014 reported that no participant experienced a non‐fatal myocardial infarction (0/301 participants in the insulin degludec group compared with 0/152 participants in the insulin detemir group; ; low‐certainty evidence; Analysis 4.6). We retrieved these data from additional sources. We judged the overall risk of bias for this outcome as 'low'.
4.6. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 6: Non‐fatal myocardial infarction
Non‐fatal stroke
Davies 2014 reported that no participant experienced a non‐fatal stroke (0/301 participants in the insulin degludec group compared with 0/152 participants in the insulin detemir group; Analysis 4.7; low‐certainty evidence). We retrieved these data from additional sources. We judged the overall risk of bias for this outcome as 'low'.
4.7. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 7: Non‐fatal stroke
End‐stage renal disease
Davies 2014 reported that 0/301 participants in the insulin degludec group compared with 0/152 participants in the insulin detemir group experienced end‐stage renal disease (Analysis 4.8). We retrieved these data from additional sources. We judged the overall risk of bias for this outcome as 'low'.
4.8. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 8: End stage renal disease
Blindness
Davies 2014 reported that no participant experienced blindness (0/301 participants in the insulin degludec group compared with 0/152 participants in the insulin detemir group; Analysis 4.9). These data were retrieved from additional sources. We judged the overall risk of bias for this outcome as 'low'.
4.9. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 9: Blindness
Serious adverse events
Both studies reported SAEs. In the insulin degludec group, 41/475 participants (8.6%) compared with 24/327 participants (7.3%) in the insulin detemir group experienced a SAE. There was no evidence of a difference in SAEs (RR 1.25, 95% CI 0.76 to 2.05; P = 0.38; 2 studies, 802 participants; low‐certainty evidence; Analysis 4.10). We judged the overall risk of bias for this outcome as 'low'.
4.10. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 10: Serious adverse events
Subgroup and sensitivity analysis
Subgroup analysis including only adults compared with studies including only children did not indicate interaction (P = 0.63; Analysis 4.10). We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Diabetic ketoacidosis
None of the studies reported on ketoacidosis in the publications. However, we retrieved data on diabetic ketoacidosis from additional sources. A total of 2/475 participants (0.4%) in the insulin degludec group compared with 0/327 participants in the insulin detemir group experienced diabetic ketoacidosis (Analysis 4.11). Both participants experiencing diabetic ketoacidosis were children (BEGIN Young). We judged the overall risk of bias for this outcome as 'low'.
4.11. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 11: Diabetic ketoacidosis
We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐serious adverse events
BEGIN Young reported the number of children with non‐serious adverse events in the publication. For Davies 2014, we retrieved this information from additional sources. A total of 380/475 participants (80%) in the insulin degludec group compared with 269/327 participants (82.3%) in the insulin detemir group experienced a non‐serious adverse event. There was no evidence of a difference in non‐serious adverse events (RR 1.02, 95% CI 0.96 to 1.08; P = 0.48; 2 studies, 802 participants; Analysis 4.12). We judged the overall risk of bias for this outcome as 'some concerns'.
4.12. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 12: Non‐serious adverse events
Subgroup and sensitivity analysis
Analyses including only adults compared with studies including only children and analyses comparing only published data with only unpublished data did not indicate interaction (P = 0.53; Analysis 4.12). We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Withdrawals due to adverse events
A total of 5/475 participants (1.1%) in the insulin degludec group compared with 1/327 participants (0.3%) in the insulin detemir group withdrew because of adverse events. There was no evidence of a difference in withdrawals due to adverse events (RR 2.32, 95% CI 0.38 to 14.18; P = 0.36; 2 studies, 802 participants; Analysis 4.13). We judged the overall risk of bias for this outcome as 'low' (data not shown).
4.13. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 13: Withdrawals due to adverse events
Nocturnal hypoglycaemia
Both studies reported data on nocturnal hypoglycaemia. None of the studies reported on any nocturnal hypoglycaemia.
Severe nocturnal hypoglycaemia was reported in the CSRs of both studies. A total of 17/475 participants in the insulin degludec group (3.6%) compared with 10/327 participants (3.1%) in the insulin detemir group experienced severe nocturnal hypoglycaemia. There was no evidence of a difference in severe nocturnal hypoglycaemia (RR 1.12, 95% CI 0.51 to 2.46; P = 0.77; 2 studies, 802 participants; low‐certainty evidence; Analysis 4.18). We judged the overall risk of bias for this outcome as 'low'.
4.18. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 18: Severe nocturnal hypoglycaemia
We retrieved data on nocturnal hypoglycaemia confirmed with blood glucose measurements. There was no evidence of a difference in confirmed nocturnal hypoglycaemia (RR 1.04, 95% CI 0.94 to 1.15; P = 0.40; 2 studies, 802 participants; Analysis 4.14). From the CSRs of both studies, data for mild documented nocturnal hypoglycaemia (plasma glucose ≤ 3.9 mmol/L, able to self‐treat) were available. There was no evidence of a difference in mild documented nocturnal hypoglycaemia (RR 0.97, 95% CI 0.86 to 1.10; P = 0.67; 2 studies, 802 participants; Analysis 4.15). There was no evidence of a difference in symptomatic nocturnal hypoglycaemia without blood glucose measurements (RR of 0.72, 95% CI 0.15 to 3.59; P = 0.69; 2 studies, 802 participants; Analysis 4.16). There was no evidence of a difference in asymptomatic nocturnal hypoglycaemia (RR 0.91, 95% CI 0.80 to 1.03; P = 0.13; 2 studies, 802 participants; Analysis 4.17). We judged the overall risk of bias for all these outcomes except for severe nocturnal hypoglycaemia as 'some concerns' (data not shown).
4.14. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 14: Nocturnal hypoglycaemia
4.15. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 15: Mild nocturnal hypoglycaemia
4.16. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 16: Nocturnal hypoglycaemia (symptomatic)
4.17. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 17: Nocturnal hypoglycaemia (asymptomatic)
Subgroup and sensitivity analysis
Subgroup analysis including only adults only compared with studies including only children did not indicate subgroup interaction (P = 0.82; Analysis 4.18). We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Mild/moderate hypoglycaemia
Both studies reported data on mild/moderate hypoglycaemia in the publications. There was no evidence of a difference in mild/moderate hypoglycaemia (RR 1.02, 95% CI 0.99 to 1.05; P = 0.17; 2 studies, 802 participants; Analysis 4.19). We judged the overall risk of bias for this outcome as 'some concerns'.
4.19. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 19: Mild/moderate hypoglycaemia
Subgroup and sensitivity analysis
Subgroup analysis including only adults compared with studies including only children did not indicate interaction (P = 0.85; Analysis 4.19). We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Socioeconomic effects
No studies reported the costs of the intervention during the study period. One study reported economic predictions of the interventions based on simulation cohorts in an UK setting of children and adolescents (BEGIN Young).
HbA1c
Both studies had data for HbA1c. BEGIN Young reported data until the end of the extension period in the publication and not until the end of the intervention period. However, we could retrieve these data from ClinicalTrials.gov. There was no evidence of a difference in HbA1c (MD 0.05%, 95% CI ‐0.1 to 0.2; P = 0.44; 2 studies, 804 participants; low‐certainty evidence; Analysis 4.20). We judged the overall risk of bias for this outcome as 'low'.
4.20. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 20: HbA1c
Subgroup and sensitivity analysis
Subgroup analyses including only adults only compared with studies including only children and only published data compared with only unpublished data did not indicate interactions (P = 0.42; Analysis 4.20). We did not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Combined HbA1c and severe hypoglycaemia
Davies 2014 reported the combined outcome HbA1c and severe hypoglycaemia in the CSR. At the end of the intervention period, a total of 116/292 participants (39.7%) in the insulin degludec group compared with 53/145 participants (36.6%) in the insulin detemir group achieved an HbA1c < 7% without severe hypoglycaemia during the last 12 weeks of treatment (RR 1.09, 95% CI 0.84 to 1.41; P = 0.53; Analysis 4.21). We judged the overall risk of bias for this outcome as 'low' (data not shown).
4.21. Analysis.

Comparison 4: Insulin degludec versus insulin detemir, Outcome 21: Individuals with HbA1c < 7% without severe hypoglycaemia
Insulin degludec compared with insulin glargine
For an overview of main results for this comparison, see Table 5.
Four studies compared insulin degludec with insulin glargine (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; SWITCH 1; Urakami 2017). A total of 1477 participants were randomised, 895 participants to insulin degludec and 582 participants to insulin glargine (see Table 6). One study included children (Urakami 2017). The mean age of the children was 10.5 years. All studies were published in full text in English. However, for all studies, we could retrieve additional information on outcomes from additional sources. All studies applied insulin degludec once daily and insulin glargine once daily. Urakami 2017 applied insulin aspart or insulin lispro before meals. The remaining studies applied insulin aspart before meals. The duration of the intervention ranged from 26 weeks to 52 weeks. SWITCH 1 and Urakami 2017 had a cross‐over design; the remaining studies were parallel‐group RCTs. Because of carryover effects, we evaluated outcomes before cross‐over. In SWITCH 1, each of the two treatment periods consisted of a 16‐week titration period and a 16‐week maintenance period; only data for health‐related quality of life and HbA1c were available before cross‐over. Three of the studies were sponsored by Novo Nordisk (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; SWITCH 1); one study did not report the funding source (Urakami 2017).
All‐cause mortality
Two studies reported on all‐cause mortality (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1). For one study, we retrieved this information from additional sources (BEGIN Flex T1). SWITCH 1 reported that four deaths occurred. However, these data could not be included in the meta‐analysis because it was not reported if the deaths occurred before or after cross‐over.
All studies reporting all‐cause mortality were performed in adults. A total of 3/646 participants (0.5%) in the insulin degludec group compared with 1/327 participants (0.3%) in the insulin glargine group died. There was no evidence of a difference in all‐cause mortality (Peto OR 1.34, 95% CI 0.15 to 11.93; P = 0.79; 2 studies, 955 participants; very low‐certainty evidence; Analysis 5.1). We judged the overall risk of bias for this outcome as 'low'.
5.1. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 1: All‐cause mortality
Subgroup analysis and sensitivity analysis
Analysis according to only published data compared with only unpublished data did not indicate interaction (P = 0.46; Analysis 5.2). The remaining subgroup and sensitivity analyses could not be performed due to lack of data (Appendix 20).
5.2. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 2: All‐cause mortality (published vs. unpublished data)
Health‐related quality of life
SWITCH 1 reported health‐related quality of life before cross‐over in the CSR. BEGIN Basal‐Bolus Type 1 reported health‐related quality of life in an appendix to the publication. Both studies applied the SF‐36 questionnaire. There was no evidence of a difference in health‐related quality of life (MD for physical health score ‐0.04 points, 95% CI ‐1.21 to 1.13; P = 0.94; 2 studies, 1042 participants; very low‐certainty evidence; Analysis 5.3; and MD of mental health score ‐0.09 points, 95% CI ‐1.03 to 0.85; P = 0.85; 2 studies, 1042 participants; very low‐certainty evidence; Analysis 5.4). The minimal important difference for the physical component score is two to three points and for the mental component score three points. We judged the overall risk of bias for this outcome as 'some concerns'.
5.3. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 3: Health‐related quality of life (physical health)
5.4. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 4: Health‐related quality of life (mental health)
Subgroup analysis and sensitivity analysis
Analysis according to only published data compared with only unpublished data did not indicate subgroup interaction. The remaining subgroup and sensitivity analyses could not be performed due to lack of data (Appendix 20).
Severe hypoglycaemia
We could evaluate severe hypoglycaemia for three studies (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; Urakami 2017). Two studies reported severe hypoglycaemia in the main publication (BEGIN Basal‐Bolus Type 1; Urakami 2017) and for one study we retrieved data from an appendix to the publication (BEGIN Flex T1).
A total of 79/646 participants (12.3%) in the insulin degludec group compared with 32/324 participants (9.9%) in the insulin glargine group reported severe hypoglycaemia. There was no evidence of a difference in severe hypoglycaemia (RR 1.22, 95% CI 0.82 to 1.82; P = 0.32; 3 studies, 970 participants; low‐certainty evidence; Analysis 5.5; Figure 7). We judged the overall risk of bias for this outcome as 'low'.
5.5. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 5: Severe hypoglycaemia
7.

Severe hypoglycaemia
Subgroup analysis and sensitivity analysis
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Hypoglycaemia reported as a serious adverse event
A total of 49/1100 participants (4.5%) in the insulin degludec group compared with 44/784 participants (5.6%) in the insulin glargine group had a SAE due to hypoglycaemia. There was no evidence of a difference in hypoglycaemia reported as a SAE (RR 0.81, 95% CI 0.40 to 1.66; P = 0.57; 4 studies, 1884 participants; Analysis 5.6). We judged the overall risk of bias for this outcome as 'low' (data not shown).
5.6. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 6: Hypoglycaemia reported as a serious adverse event
Cardiovascular mortality
We could retrieve data on cardiovascular mortality from two studies through additional sources (BEGIN Flex T1; Urakami 2017). One study reported the cause of death in the main publication (BEGIN Basal‐Bolus Type 1). Only BEGIN Basal‐Bolus Type 1 reported any deaths due to cardiovascular disease. In this study, 2/472 participants (0.4%) in the insulin degludec group compared with 1/154 participants (0.6%) in the insulin glargine group died due to cardiovascular disease (Analysis 5.7). We judged the overall risk of bias for this outcome as 'low'.
5.7. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 7: Cardiovascular mortality
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐fatal myocardial infarction
We could retrieve data on non‐fatal myocardial infarction for three studies from additional sources (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; Urakami 2017). However, only one study reported any participant experiencing a non‐fatal myocardial infarction. In this study, 1/472 participants (0.2%) in the insulin degludec group compared with 0/154 participants in the insulin glargine group experienced a non‐fatal myocardial infarction ( low‐certainty evidence; Analysis 5.8). We judged the overall risk of bias for this outcome as 'low'.
5.8. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 8: Non‐fatal myocardial infarction
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐fatal stroke
We could retrieve data on non‐fatal stroke for two studies from CSRs (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1). For one study, data were provided by the study author (Urakami 2017). BEGIN Flex T1 reported no event (0/165 participants in the insulin degludec group compared with 0/161 participants in the insulin glargine group). Urakami 2017 also reported no event (0/9 participants in both intervention groups). BEGIN Basal‐Bolus Type 1 reported that 1/472 participants (0.2%) in the insulin degludec group compared with 0/154 participants in the insulin glargine group experienced cerebral ischaemia (low‐certainty evidence; Analysis 5.9). We judged the overall risk of bias for this outcome as 'low'.
5.9. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 9: Non‐fatal stroke
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
End‐stage renal disease
For one study, the study author provided information that no participant developed end‐stage renal disease (Urakami 2017). None of the other studies reported on end‐stage renal disease. We judged the overall risk of bias for this outcome as 'low' (data not shown).
Blindness
For one study, the study author provided information that no participant developed blindness (Urakami 2017). None of the other studies reported on blindness. We judged the overall risk of bias for this outcome as 'low' (data not shown).
Serious adverse events
Three studies reported SAEs (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; Urakami 2017). Two studies reported data in the publications (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1). For one study, the investigator reported that no participant experienced a SAE (Urakami 2017).
A total of 56/646 participants (8.7%) in the insulin degludec group compared with 25/324 participants (7.7%) in the insulin glargine group experienced serious adverse events. There was no evidence of a difference in SAEs (RR 0.92, 95% CI 0.58 to 1.46; P = 0.73; 3 studies, 970 participants; low‐certainty evidence; Analysis 5.10). We judged the overall risk of bias for this outcome as 'low'.
5.10. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 10: Serious adverse events
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Diabetic ketoacidosis
Three studies reported diabetic ketoacidosis (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; Urakami 2017). One study reported data in the publication (BEGIN Basal‐Bolus Type 1). For two studies, we retrieved data from additional sources (BEGIN Flex T1; Urakami 2017).
A total of 3/646 participants (0.5%) in the insulin degludec group compared with 3/324 participants (0.9%) in the insulin glargine group experienced diabetic ketoacidosis. There was no evidence of a difference in diabetic ketoacidosis (RR 0.57, 95% CI 0.05 to 6.89; P = 0.66; 3 studies, 970 participants; Analysis 5.11). We judged the overall risk of bias for this outcome as 'low'.
5.11. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 11: Diabetic ketoacidosis
Subgroup analysis and sensitivity analysis
Analysis according to only published data compared with only unpublished data did not indicate subgroup interaction.
We could not perform the remaining subgroup and sensitivity analyses due to lack of data (Appendix 20).
Non‐serious adverse events
Three studies reported non‐serious adverse events (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; Urakami 2017). Two studies reported data in the publications (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1). For one study, the investigator reported that no participant experienced a non‐serious adverse event (Urakami 2017).
A total of 522/646 participants (80.8%) in the insulin degludec group compared with 244/324 participants (75.3%) in the insulin glargine group experienced a non‐serious adverse event. There was no evidence of a difference in non‐serious adverse events (RR 1.02, 95% CI 0.95 to 1.10; P = 0.52; 3 studies, 970 participants; Analysis 5.13). We judged the overall risk of bias for this outcome as 'some concerns'.
5.13. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 13: Non‐serious adverse events
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Nocturnal hypoglycaemia
Three studies reported nocturnal hypoglycaemia (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; Urakami 2017). Two studies reported data for one or more nocturnal hypoglycaemic outcomes in the publications (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1). For one study, the investigator reported that no participant experienced nocturnal hypoglycaemia (Urakami 2017).
A total of 23/646 participants (3.6%) in the insulin degludec group compared with 8/324 participants (2.5%) in the insulin glargine group experienced severe nocturnal hypoglycaemia. There was no evidence of a difference in severe hypoglycaemia (RR 1.39, 95% CI 0.59 to 3.27; P = 0.46; 3 studies, 970 participants; low‐certainty evidence; Analysis 5.19). We judged the overall risk of bias for this outcome as 'low'.
5.19. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 19: Severe nocturnal hypoglycaemia
A total of 464/646 participants (71.8%) in the insulin degludec group compared with 235/324 participants (72.5%) in the insulin glargine group experienced nocturnal hypoglycaemia. There was no evidence of a difference in nocturnal hypoglycaemia (RR 0.99, 95% CI 0.91 to 1.07; P = 0.76; 3 studies, 970 participants; Analysis 5.15). We retrieved data on mild nocturnal hypoglycaemia from additional sources. There was no evidence of a difference in mild nocturnal hypoglycaemia (RR 0.98, 95% CI 0.90 to 1.07; P = 0.63; 2 studies, 952 participants; Analysis 5.16). Asymptomatic nocturnal hypoglycaemia was reported in the CSRs of two studies. There was no evidence of a difference in asymptomatic nocturnal hypoglycaemia (RR 0.84, 95% CI 0.71 to 1.00; P = 0.05; 2 studies, 952 participants; Analysis 5.17). There was no evidence of a difference in symptomatic nocturnal hypoglycaemia (RR 1.22, 95% CI 0.72 to 2.07; P = 0.46; 2 studies, 952 participants; Analysis 5.18). We judged the overall risk of bias for all these outcomes except for severe nocturnal hypoglycaemia as 'some concerns' (data not shown).
5.15. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 15: Nocturnal hypoglycaemia
5.16. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 16: Mild nocturnal hypoglycaemia
5.17. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 17: Nocturnal hypoglycaemia (asymptomatic)
5.18. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 18: Nocturnal hypoglycaemia (symptomatic)
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Mild/moderate hypoglycaemia
Three studies reported on mild/moderate hypoglycaemia (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; Urakami 2017). Two studies reported data in the publications (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1). For one study, the investigator reported that no participant experienced mild/moderate hypoglycaemia (Urakami 2017).
A total of 624/646 participants (96.6%) in the insulin degludec group compared with 312/324 participants (96.3%) in the insulin glargine group experienced mild/moderate hypoglycaemia. There was no evidence of a difference in mild/moderate hypoglycaemia (RR 1.02, 95% CI 0.99 to 1.04; P = 0.18; 3 studies, 970 participants; Analysis 5.20). We judged the overall risk of bias for this outcome as 'some concerns'.
5.20. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 20: Mild/moderate hypoglycaemia
We did not perform subgroup and sensitivity analyses due to lack of data (Appendix 20).
Socioeconomic effects
No studies reported the costs of the intervention during the study period. One co‐publication analysed the cost‐effectiveness based on applying assumptions from two studies to a UK National Health Service perspective (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1).
HbA1c
Four studies reported HbA1c levels (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; SWITCH 1; Urakami 2017). Three studies reported data in the publications (BEGIN Flex T1; SWITCH 1; Urakami 2017). BEGIN Basal‐Bolus Type 1 only reported HbA1c after the extension period and not after the end of the regular intervention in the publication. However, we could retrieve these data from the CSR.
There was a reduction in HbA1c in favour of insulin glargine (MD 0.1%, 95% CI 0.0 to 0.2; P = 0.05; 1388 participants; 4 studies; Analysis 5.21; low‐certainty evidence). We judged the overall risk of bias for this outcome as 'low'.
5.21. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 21: HbA1c
Subgroup and sensitivity analyses
Three studies reported HbA1c in adults with a MD of 0.1%, 95% CI 0.0 to 0.2; Analysis 5.21. One study reported HbA1c in children with a MD of 0%, 95% CI ‐0.6 to 0.6; Analysis 5.21. The test for subgroup differences did not indicate interaction (P = 0.71).
Analysing only published data indicated a MD in HbA1c of 0.1%, 95% 0.02 to 0.3; Analysis 5.22. Analysing only unpublished data indicated a MD in HbA1c of 0.0%, 95% CI ‐0.2 to 0.2; Analysis 5.22. The test for subgroup differences did not indicate interaction (P = 0.26).
5.22. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 22: HbA1c (published vs. unpublished data)
Sensitivity analysis excluding the largest study and the longest study indicated a MD in HbA1c of 0.1%, 95% CI 0.02 to 0.3 (BEGIN Basal‐Bolus Type 1).
The remaining subgroup analyses could not be performed due to lack of data (Appendix 20).
Combined HbA1c and severe hypoglycaemia
A combined measure of HbA1c and severe hypoglycaemia was available from two studies through the CSRs (BEGIN Basal‐Bolus Type 1; BEGIN Flex T1). BEGIN Basal‐Bolus Type 1 reported that 174/453 participants (38.4%) in the insulin degludec group compared with 63/149 participants (42.3%) in the insulin glargine group achieved the HbA1c target < 7% without severe hypoglycaemia during the last 12 weeks of treatment. BEGIN Flex T1 reported that 56/153 participants (36.6%) in the insulin degludec group compared with 60/156 participants (38.5%) in the insulin glargine group achieved the HbA1c target < 7% without severe hypoglycaemia during the last 12 weeks of treatment.
There was no evidence of a difference in people achieving HbA1c < 7% without severe hypoglycaemia (RR 0.92, 95% CI 0.78 to 1.10; 2 studies, 911 participants; Analysis 5.23). We judged the overall risk of bias for this outcome as 'low' (data not shown).
5.23. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 23: Individuals with HbA1c < 7% without severe hypoglycaemia
Assessment of reporting bias
We did not draw funnel plots due to limited number of studies per outcome included in the analyses.
Ongoing studies
We did not identify ongoing trials of interest for this review.
Studies awaiting assessment
We identified 13 studies with 20 records which we classified as awaiting classification (Agesen 2019; Basal Analog Study; ChiCTR2000032703; EudraCT 2007‐004144‐74; EudraCT 2009‐012317‐22; INEOX; IRCT201203079224N1; J‐Collection; Mianowska 2007; NCT00564018; Sherif 2014; UMIN000020521; UMIN000021046); for details please see 'Studies awaiting classification'.
Three studies randomising 474 participants compared insulin degludec with insulin glargine (Agesen 2019; ChiCTR2000032703; INEOX). Four studies randomising 253 participants compared insulin detemir with insulin glargine (Basal Analog Study; EudraCT 2007‐004144‐74; EudraCT 2009‐012317‐22; J‐Collection). Three studies randomising 154 participants compared insulin glargine with NPH insulin (IRCT201203079224N1; Mianowska 2007; Sherif 2014).
Two studies had more than two intervention groups: one study randomising 33 participants had three intervention groups comparing insulin glargine with insulin detemir with NPH insulin (NCT00564018), one study randomising 100 participants compared insulin degludec with insulin glargine and with continuing existing basal insulin treatment (UMIN000020521).
One study compared insulin degludec with another unspecified long‐acting insulin analogue (UMIN000021046). This study randomising 200 participants included people with T1DM and T2DM.
Seven studies were marked as awaiting classification, as they were listed as completed, but no publications were yet available (Agesen 2019; EudraCT 2007‐004144‐74; EudraCT 2009‐012317‐22; INEOX; IRCT201203079224N1; UMIN000020521; UMIN000021046).
Two studies were published as abstracts (Basal Analog Study; Sherif 2014). One study had results available in the trials register – however, it was stated in the trials register that the trial was ended prematurely. It was not possible through correspondence with authors to clarify how long the trial continued (EudraCT 2007‐004144‐74). One study was listed as completed and prematurely ended with no study data (NCT00564018).
One cross‐over study had a full‐text publication available. No data could be retrieved before cross‐over from the publication (Mianowska 2007).
Investigators were contacted, if this was possible, in order to get the status of the studies clarified (See Appendix 19).
Discussion
Summary of main results
This Cochrane Review is the first systematic review investigating the effects of (ultra‐)long acting insulin analogues in people with T1DM with substantial amounts of information from CSRs and clinical study synopses. We included 26 studies with 8784 participants: 2428 participants were randomised to NPH insulin, 2889 participants to insulin detemir, 2095 participants to insulin glargine and 1372 participants to insulin degludec. Eight studies contributing 21% of all participants included children.
The amount of evidence on patient‐important outcomes was limited from full‐text publications. However, we could retrieve substantial data on patient‐important outcomes from the CSRs. There was moderate‐certainty evidence comparing insulin detemir with NPH insulin for T1DM showing a lower risk of severe hypoglycaemia in favour of insulin detemir. However, the 95% prediction interval indicated inconsistency of this result. Insulin detemir or insulin glargine compared with NPH insulin did not show benefits or harms for severe nocturnal hypoglycaemia. For all other main outcomes, with overall low risk of bias and comparing insulin analogues with each other, there were no clear differences. Data on patient‐important outcomes such as health‐related quality of life, macrovascular and microvascular diabetic complications were sparse or missing.
Comparing the insulin analogues detemir and glargine with NPH insulin, we are moderately confident about the results for all‐cause mortality, severe (nocturnal) hypoglycaemia, SAEs and HbA1c. We are uncertain about the effects on non‐fatal myocardial infarction, non‐fatal stroke and health‐related quality of life, mainly because data were sparse or there were only a few studies which did not last long enough to investigate these outcomes.
There was no evidence of a difference in any outcome between children and adults.
Overall completeness and applicability of evidence
We conducted an extensive search for studies, included publications in all languages, and tried to obtain additional data on all studies. We identified two unpublished studies (NCT00595374; NCT00605137). We managed to retrieve additional unpublished information on all studies, except for three studies which were only available as full‐text publications (Bolli 2009; Porcellati 2004; Urakami 2017). Two study authors provided personal information on their studies (Home 2005; Urakami 2017), One unpublished study did not have a CSR but some data could be retrieved from a clinical study synopsis (NCT00595374). Two Japanese studies had CSRs, but we were unable to obtain the complete version of these (Kobayashi 2007; NCT00605137). Two studies had a cross‐over design and not all data could be analysed or were reported before cross‐over which we needed because of potential carryover effects (SWITCH 1; Urakami 2017). We looked for additional studies and cross‐checked our data with the data from other meta‐analyses of relevance (Laranjeira 2018; Tricco 2014; Tricco 2018). The information obtained from CSRs was clearly the best to establish an adequate 'Risk of bias assessment' and to maximise the yield of information for our prespecified outcomes (Appendix 22; Appendix 23; Appendix 24; Appendix 25; Appendix 26; Appendix 27; Appendix 28; Appendix 29; Appendix 30; Appendix 31; Appendix 32; Appendix 33; Appendix 34; Appendix 35; Appendix 36; Appendix 37; Appendix 38; Appendix 39; Appendix 40; Appendix 41; Appendix 42). We noticed major differences between reported outcomes in publications and CSRs, e.g. all‐cause mortality was documented in 25% of publications compared to 91% in CSRs (Appendix 41). SAEs and non‐serious adverse events were documented in 54% of publications compared to 91% in CSRs (Appendix 41). However, the amount of information within the CSRs varied substantially and we probably did not have access to a single full CSR (Appendix 7).
We investigated a broad spectrum of people with T1DM as both children and adults were included. However, we did not include pregnant women with T1DM, as we anticipated these women would have pronounced fluctuating insulin requirements and a specific hypoglycaemia risk profile. All studies were performed in white or Asian people. Data on people of African origin were lacking. None of the studies was performed in low‐ or middle‐income settings.
Quality of the evidence
Depending on the outcome measures, we judged the certainty of the evidence as moderate for all‐cause mortality, severe hypoglycaemia, severe nocturnal hypoglycaemia, SAEs and HbA1c. For most comparisons, we judged the certainty of the evidence as low for non‐fatal myocardial infarction, non‐fatal stroke and health‐related quality of life. No information or only few data were available for blindness, end‐stage renal disease, combined HbA1c with severe hypoglycaemia and socioeconomic effects.
For all studies, we contacted one or more study authors to obtain supplemental information on baseline data, 'Risk of bias' domains and outcomes (see Appendix 19). However, several investigators advised us to contact the pharmaceutical company of the study, as they did not have access to the full dataset.
All studies but six had a non‐inferiority RCT design which is often required for regulatory approval (Bolli 2009; Home 2005; Porcellati 2004; Ratner 2000; Schober 2002; Urakami 2017). The usual primary endpoint was change in HbA1c which does not minimise the reliability of analysing other outcomes such as hypoglycaemia by means of meta‐analysis, because with a potential benefit of newer compounds in reducing HbA1c, a benefit of the number of hypoglycaemic episodes could be expected. Adjustments of hypoglycaemic events for HbA1c levels or achievement of certain HbA1c thresholds without hypoglycaemia would provide important information. Unfortunately, only a few of our included studies reported on this combined endpoint, and, if done, no clear differences were recorded.
All studies except one had an open‐label design (SWITCH 1). This could have influenced some of the subjective outcome measures, especially health‐related quality of life, non‐serious adverse events, mild/moderate hypoglycaemia and some measures of nocturnal hypoglycaemia. Another factor influencing findings could have been investigators being more careful when adjusting the newer insulin analogues due to less clinical experience with these compounds. Also, some participants might have been more prone to measure blood glucose as they might have anticipated experiencing more hypoglycaemic episodes with human insulin preparations, thereby even affecting hypoglycaemia confirmed with blood glucose measurements.
Improving and maintaining glycaemic control in T1DM is a key objective. However, hypoglycaemia is a serious problem affecting health‐related quality of life and treatment satisfaction of people with diabetes, making it difficult to achieve near‐normal glucose levels in T1DM. Therefore, for any proclaimed benefit of an intervention on hypoglycaemia, it is vital to evaluate the risk of bias in order to establish reliable results. 'Risk of bias' assessment depends considerably on the definition of hypoglycaemia. It appears low if severe hypoglycaemia is also reported as a serious adverse event (SAE) because there is a standard definition of SAEs, or if the combined endpoint of HbA1c levels with severe hypoglycaemia is reported. Unfortunately, no data were available for the combined endpoint HbA1c with severe hypoglycaemia for the comparisons insulin detemir versus NPH insulin and insulin glargine versus NPH insulin. Of note, only about one third of participants being treated with insulin glargine, insulin detemir or insulin degludec achieved an HbA1c < 7% without severe hypoglycaemia. Other definitions of severe hypoglycaemia like hypoglycaemia‐induced coma or convulsions, necessity for intubation or intensive‐care unit stay also reflect hard clinical endpoints. However, the included studies most often defined severe hypoglycaemia as a hypoglycaemic event which needed "third party assistance". This is prone to bias because third party assistance might encompass a broad range of interventions, e.g. giving food or a drink by a relative or friend, subcutaneous glucagon injection or intravenous glucose administration. Only Thalange 2013 made an effort to define third party assistance in a way that minimised risk of bias (the child had to be semiconscious or unconscious or in coma with or without convulsions and may have required parenteral treatment with glucagon or intravenous glucose). A Cochrane Review associated to this systematic review will establish an in‐depth analysis of the definitions and reporting of hypoglycaemia in trials of long‐acting insulin analogues in people with type 1 diabetes mellitus (Ørskov Ipsen 2020).
An overview of the reported definitions of hypoglycaemic episodes in our included studies found no evidence of differences between the various interventions on these outcomes with the exception of insulin detemir compared with NPH insulin, demonstrating a benefit for severe hypoglycaemia, any/mild/symptomatic nocturnal hypoglycaemia and mild/moderate hypoglycaemia (Appendix 42). With the exception of severe hypoglycaemia, we judged the risk of bias as 'some concerns' for measurement of these outcomes. There was no benefit or risk of insulin detemir for hypoglycaemia reported as a SAE or severe nocturnal hypoglycaemia event (Appendix 42).
Long‐term complications of diabetes were sparsely reported. Long‐term complications of diabetes develop over years, and therefore the duration of the included studies might have been too short to identify if an intervention had beneficial or harmful effects. Data on all‐cause mortality were most often retrieved from CSRs and few deaths were observed in the studies. However, to our knowledge, no data from long‐term observational studies indicate that the type of intermediate or (ultra‐)long‐acting insulin influences the risk of death or macrovascular and microvascular complications of diabetes. However, long‐term follow‐up from interventional studies has shown that good glycaemic control in people with T1DM is an important factor for preventing complications (DCCT/EDIC 2016).
No studies reported the direct costs of insulin treatment during the study period. Several studies had co‐publications with economic analyses in different country settings based on assumptions derived from the clinical study (Bartley 2008; BEGIN Basal‐Bolus Type 1; BEGIN Flex T1; BEGIN Young; Pieber 2007). However, these assumptions do not seem to be supported by our meta‐analyses of the clinical trial data. Furthermore, other studies have shown that the direct costs of the long‐acting insulin analogues often are substantially higher than the costs of NPH insulin (Ewen 2019).
Only one study had not received free drugs or financial funding from the pharmaceutical industry (Porcellati 2004). It is known that studies receiving funding or provision of free drugs or devices from a pharmaceutical company lead to more favourable results and conclusions compared to studies sponsored by other sources (Lundh 2017).
Potential biases in the review process
We were unable to draw funnel plots to assess small‐study bias due to lack of data. We tried to explore inconsistency of results and the reasons for it through subgroup and sensitivity analyses. The only factor, comparing insulin detemir with NPH insulin, that indicated an influence on the effect estimate for the sensitivity analysis of one outcome (severe hypoglycaemia) was publication status. This has to be interpreted with caution because the subgroup of studies with unpublished data consisted of two studies only.
We identified 13 studies as 'awaiting classification'. Data from these studies would have added information on an additional 1194 participants. Most of the studies were listed as completed in trials registers, but data, publications or both were not available. For some of the studies, these data might not yet have been analysed, but other studies were completed years ago and are still not published (Basal Analog Study; EudraCT 2007‐004144‐74; J‐Collection; NCT00564018; UMIN000021046). For most studies awaiting classification, we contacted the investigators for clarification.
We were dealing with a heterogeneous group of studies. Our meta‐analyses, when performed, were limited by the inability to use individual participant data to assess whether distinct clinical characteristics may have influenced the effect estimates of the interventions. Many of the included studies were designed and powered to detect changes in HbA1c but, for all studies, we were able to extract most of our predefined outcomes.
Several studies were published in more than one publication which, for some studies, made it difficult to separate the primary publication from companion papers (for details, see Included studies).
Two review authors carried out data extraction. However, the review authors extracting the data were not blinded as to from which study they were extracting data.
We only included studies with a duration of 24 weeks or more to get some information on patient‐relevant outcomes; by not including studies with a shorter duration, we might have underestimated the short‐term risks of the interventions.
A potential selection bias exists as more healthy and motivated people may participate in a clinical study. However, a Cochrane Review observed that clinical outcomes in people participating in RCTs are not substantially different to outcomes in comparable individuals outside the RCT context (Vist 2008).
We requested CSRs and other information from EMA. EMA replied that it "is currently operating within the fourth phase of its business continuity plan to ensure operational continuity during its relocation to Amsterdam. Whilst every effort is being made to process all requests as soon as possible, you should be aware that due to these exceptional circumstances from October 2019 requests cannot be processed immediately and will be dealt in a chronological order from the time they were received". At the moment of publication of this Cochrane Review, the first pieces of information from EMA are arriving. Because we do not know when the last information package of EMA will be available, we plan to make full use of EMA data in a future update of our review. In case of very important EMA data, we will publish an interim updated version of our review as soon as possible.
Agreements and disagreements with other studies or reviews
Other reviews of insulin analogues in people with T1DM have been published. The most recent systematic review was performed for refinement of the WHO Essential Medicine List (EML), which was an update of a systematic review published in 2014 (Tricco 2018). The review for WHO EML included adults with T1DM, but also included pregnant women with T1DM. We did not chose to include the latter cohort as pregnancy causes considerable changes in insulin sensitivity. Tricco 2018 included studies irrespective of study duration. We required a minimum duration of 24 weeks to get more reliable information on patient‐relevant outcome measures. Short‐term studies usually evaluate surrogate markers and often have shorter intervention periods than the titration periods of the longer‐term studies. Tricco 2018 analysed insulin glargine and insulin detemir together; we chose to perform separate analyses. Tricco 2018 included 62 RCTs according to the abstract. However, they missed identifying co‐publications of primary publications. Therefore, several studies were included more than once and handled as independent studies. Another difference to our review is the lack of identification of unpublished data, especially CSRs which provided substantial information to all our analyses including 'Risk of bias' assessment. Tricco 2018 reported a statistically significant decrease in HbA1c with insulin analogues compared with NPH insulin and a statistically significant lower risk of severe hypoglycaemia, which we could not verify in our analyses. One umbrella review of reviews compared long‐acting insulin analogues with NPH insulin (Laranjeira 2018). Eleven systematic reviews were identified and a total of 25 RCTs were included irrespective of age of participants or duration of the intervention. The conclusion of this overview, based on data for all systematic reviews, was that long‐acting insulin analogues were more effective than NPH insulin concerning lowering HbA1c. No statistically significant differences were found for severe hypoglycaemia (Laranjeira 2018).
Authors' conclusions
Implications for practice.
We analysed randomised controlled trials (RCTs) with a duration of 24 weeks or more comparing (ultra‐)long‐acting insulin with neutral protamine Hagedorn (NPH) insulin or another (ultra‐)long‐acting insulin in people with type 1 diabetes mellitus. Nine RCTs compared NPH insulin with insulin detemir or insulin glargine, respectively. Two RCTs each compared insulin detemir with insulin glargine or insulin degludec, respectively. Four RCTs compared insulin degludec with insulin glargine. No studies compared insulin degludec with NPH insulin. There was moderate‐certainty evidence that insulin detemir reduces severe hypoglycaemia compared with NPH insulin. However, the 95% prediction interval indicated inconsistency which means that if we performed an additional study comparing insulin detemir with NPH insulin there may not be a clear difference in the risk of severe hypoglycaemia for this comparison.
There were no clear differences for severe nocturnal hypoglycaemia comparing insulin detemir or insulin glargine with NPH insulin. For all other main outcomes, with overall low risk of bias and comparing (ultra‐)long‐acting insulin analogues with each other, there were also no clear differences.
Definitions of hypoglycaemia varied substantially among the studies. Health‐related quality of life was inconsistently reported and did not show clear benefits or harms for any insulin analogue or NPH insulin. Data on macrovascular and microvascular diabetic complications were sparse or missing.
It remains unclear whether the risk of hypoglycaemia, especially severe and severe nocturnal hypoglycaemia, is associated with clinically relevant differences regarding the type of (ultra‐)long‐acting or intermediate‐acting insulin.
Implications for research.
All studies investigating insulin use in diabetes should report hypoglycaemic episodes in a standard way. 'Risk of bias' assessment depends considerably on the definition of hypoglycaemia. It appears low if severe hypoglycaemia is also reported as a serious adverse event (SAE) because there is a standard definition of SAEs or if the combined endpoint of HbA1c levels with associated hypoglycaemia is reported. Other definitions of severe hypoglycaemia like hypoglycaemia‐induced coma or convulsions, necessity for intubation or intensive‐care unit stay also reflect hard clinical endpoints. However, the included studies most often defined severe hypoglycaemia as a hypoglycaemic event which needed "third party assistance". This is prone to bias because third party assistance encompasses a broad range of interventions, e.g. giving food or a drink by a relative or friend, subcutaneous glucagon injection or intravenous glucose administration. Therefore, any proclaimed benefit of (ultra‐)long‐acting insulin analogues compared with NPH insulin especially for (nocturnal) hypoglycaemia has to demonstrate clinically relevant differences for these outcomes which should be measured in an identical manner to achieve fair comparisons within and between studies.
There is a gap in research on patient‐important outcomes such as health‐related quality of life, macrovascular and microvascular diabetic complications which were rarely reported or missing. Furthermore, studies including people from a wide range of ethnicities and studies in low‐and middle‐income countries are needed.
The availability of clinical study reports (CSRs) provided a substantially improved body of evidence, for both data extraction and 'Risk of bias' analysis. Pharmaceutical companies, the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) should facilitate full access to CSRs to better enable systematic reviewers to establish high‐quality systematic reviews.
What's new
| Date | Event | Description |
|---|---|---|
| 27 April 2021 | Amended | Analysis 2.15 corrected |
| 27 April 2021 | Amended | Analysis 2.15 corrected |
History
Protocol first published: Issue 12, 2019 Review first published: Issue 3, 2021
Notes
We have based parts of the Methods, as well as Appendix 6 of this Cochrane Review, on a standard template established by the CMED Group.
Risk of bias
Risk of bias for analysis 1.1 All‐cause mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.1.1 Adults | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Kobayashi 2007 | Low risk of bias | Randomised, multi‐centre, open‐labelled, parallel‐group trial. No details were provided in the clinical study synopsis. Because this study was performed in cooperation with Novo Nordisk it is likely that randomisation and concealment of allocation were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). The FAS consisted of all randomised participants who had any available efficacy data after receiving the trial product. The last observation carried forward (LOCF) approach was used for all endpoints at week 48 for participants who had at least one valid post‐baseline measurement. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from clinical study synopsis and some translated pages from the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| NCT00595374 | Low risk of bias | Randomisation method was not specified. Because this trial was conducted in cooperation with Novo Nordisk we assumed that randomisation and allocation concealment were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All analyses were conducted for the full analysis set (FAS). | Low risk of bias | Almost all randomised participants completed the study. All analyses were conducted for the full analysis set (FAS). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from clinical study synopsis and ClinicalTrials.gov. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Russell‐Jones 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). Block randomisation within individual trial sites was handled by the system. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. All primary and secondary efficacy analyses were based on the ITT analysis set. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Standl 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 1.1.2 Children | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 1.3 Severe hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.3.1 Adults | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Kobayashi 2007 | Low risk of bias | Randomised, multi‐centre, open‐labelled, parallel‐group trial. No details were provided in the clinical study synopsis. Because this study was performed in cooperation with Novo Nordisk it is likely that randomisation and concealment of allocation were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). The FAS consisted of all randomised participants who had any available efficacy data after receiving the trial product. The last observation carried forward (LOCF) approach was used for all endpoints at week 48 for participants who had at least one valid post‐baseline measurement. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from clinical study synopsis and some translated pages from the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Russell‐Jones 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). Block randomisation within individual trial sites was handled by the system. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. All primary and secondary efficacy analyses were based on the ITT analysis set. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Standl 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 1.3.2 Children | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 1.6 Cardiovascular mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.6.1 Adults | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Kobayashi 2007 | Low risk of bias | Randomised, multi‐centre, open‐labelled, parallel‐group trial. No details were provided in the clinical study synopsis. Because this study was performed in cooperation with Novo Nordisk it is likely that randomisation and concealment of allocation were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). The FAS consisted of all randomised participants who had any available efficacy data after receiving the trial product. The last observation carried forward (LOCF) approach was used for all endpoints at week 48 for participants who had at least one valid post‐baseline measurement. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from clinical study synopsis and some translated pages from the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| NCT00595374 | Low risk of bias | Randomisation method was not specified. Because this trial was conducted in cooperation with Novo Nordisk we assumed that randomisation and allocation concealment were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All analyses were conducted for the full analysis set (FAS). | Low risk of bias | Almost all randomised participants completed the study. All analyses were conducted for the full analysis set (FAS). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from clinical study synopsis and ClinicalTrials.gov. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Russell‐Jones 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). Block randomisation within individual trial sites was handled by the system. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. All primary and secondary efficacy analyses were based on the ITT analysis set. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Standl 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Protocol deviations were explained in the clinical study report. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 1.6.2 Children | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. . A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 1.7 Non‐fatal myocardial infarction.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 1.8 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.8.1 Adults | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Kobayashi 2007 | Low risk of bias | Randomised, multi‐centre, open‐labelled, parallel‐group trial. No details were provided in the clinical study synopsis. Because this study was performed in cooperation with Novo Nordisk it is likely that randomisation and concealment of allocation were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). The FAS consisted of all randomised participants who had any available efficacy data after receiving the trial product. The last observation carried forward (LOCF) approach was used for all endpoints at week 48 for participants who had at least one valid post‐baseline measurement. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from clinical study synopsis and some translated pages from the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| NCT00595374 | Low risk of bias | Randomisation method was not specified. Because this trial was conducted in cooperation with Novo Nordisk we assumed that randomisation and allocation concealment were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All analyses were conducted for the full analysis set (FAS). | Low risk of bias | Almost all randomised participants completed the study. All analyses were conducted for the full analysis set (FAS). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from clinical study synopsis and ClinicalTrials.gov. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Russell‐Jones 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). Block randomisation within individual trial sites was handled by the system. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. All primary and secondary efficacy analyses were based on the ITT analysis set. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Standl 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 1.8.2 Children | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 1.10 Diabetic ketoacidosis.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.10.1 Adults | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Kobayashi 2007 | Low risk of bias | Randomised, multi‐centre, open‐labelled, parallel‐group trial. No details were provided in the clinical study synopsis. Because this study was performed in cooperation with Novo Nordisk it is likely that randomisation and concealment of allocation were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). The FAS consisted of all randomised participants who had any available efficacy data after receiving the trial product. The last observation carried forward (LOCF) approach was used for all endpoints at week 48 for participants who had at least one valid post‐baseline measurement. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from clinical study synopsis and some translated pages from the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 1.10.2 Children | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Protocol deviations were explained in the clinical study report. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Protocol deviations were explained in the clinical study report. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 1.12 Non‐serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.12.1 Adults | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Kobayashi 2007 | Low risk of bias | Randomised, multi‐centre, open‐labelled, parallel‐group trial. No details were provided in the clinical study synopsis. Because this study was performed in cooperation with Novo Nordisk it is likely that randomisation and concealment of allocation were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). The FAS consisted of all randomised participants who had any available efficacy data after receiving the trial product. The last observation carried forward (LOCF) approach was used for all endpoints at week 48 for participants who had at least one valid post‐baseline measurement. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from clinical study synopsis and some translated pages from the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| NCT00595374 | Low risk of bias | Randomisation method was not specified. Because this trial was conducted in cooperation with Novo Nordisk we assumed that randomisation and allocation concealment were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All analyses were conducted for the full analysis set (FAS). | Low risk of bias | Almost all randomised participants completed the study. All analyses were conducted for the full analysis set (FAS). | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from clinical study synopsis and ClinicalTrials.gov. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Russell‐Jones 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). Block randomisation within individual trial sites was handled by the system. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. All primary and secondary efficacy analyses were based on the ITT analysis set. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Standl 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 1.12.2 Children | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 1.18 Severe nocturnal hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.18.1 Adults | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Russell‐Jones 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). Block randomisation within individual trial sites was handled by the system. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. All primary and secondary efficacy analyses were based on the ITT analysis set. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Standl 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 1.18.2 Children | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 1.22 Severe nocturnal hypoglycaemia (published vs. unpublished data).
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.22.1 Published | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Russell‐Jones 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). Block randomisation within individual trial sites was handled by the system. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. All primary and secondary efficacy analyses were based on the ITT analysis set. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 1.22.2 Unpublished | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Standl 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 1.24 Mild/moderate hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.24.1 Adults | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Kobayashi 2007 | Low risk of bias | Randomised, multi‐centre, open‐labelled, parallel‐group trial. No details were provided in the clinical study synopsis. Because this study was performed in cooperation with Novo Nordisk it is likely that randomisation and concealment of allocation were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). The FAS consisted of all randomised participants who had any available efficacy data after receiving the trial product. The last observation carried forward (LOCF) approach was used for all endpoints at week 48 for participants who had at least one valid post‐baseline measurement. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from clinical study synopsis and some translated pages from the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Russell‐Jones 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). Block randomisation within individual trial sites was handled by the system. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. All primary and secondary efficacy analyses were based on the ITT analysis set. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Standl 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 1.24.2 Children | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 1.26 HbA1c.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.26.1 Adults | ||||||||||||
| Bartley 2008 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) was used as the primary analysis for both efficacy and safety. Missing observations were considered missing at random in all analyses. Last observation carried forward (LOCF) was used for missing values on HbA1c, if participants had been treated for a minimum of 12 weeks and had a measurement after this period of time. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Kobayashi 2007 | Low risk of bias | Randomised, multi‐centre, open‐labelled, parallel‐group trial. No details were provided in the clinical study synopsis. Because this study was performed in cooperation with Novo Nordisk it is likely that randomisation and concealment of allocation were performed adequately. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All participants who received at least one dose of trial product were included in the safety analysis. For all efficacy endpoints the analysis was performed on the full analysis set (FAS). The FAS consisted of all randomised participants who had any available efficacy data after receiving the trial product. The last observation carried forward (LOCF) approach was used for all endpoints at week 48 for participants who had at least one valid post‐baseline measurement. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from clinical study synopsis and some translated pages from the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Russell‐Jones 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). Block randomisation within individual trial sites was handled by the system. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis (ITT) set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. All primary and secondary efficacy analyses were based on the ITT analysis set. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Standl 2004 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Vague 2003 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 1.26.2 Children | ||||||||||||
| NCT00605137 | Low risk of bias | Randomisation codes were prepared by a responsible person for randomisation and carried out by the registration centre. Randomisation was stratified by type of bolus insulin. Minor baseline differences did not indicate a problem with randomisation. | Low risk of bias | Open‐label trial design. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. For all efficacy endpoints the analyses were performed on a full analysis set (FAS), i.e. all randomised participants who had any available efficacy data after receiving the trial product. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from study protocol, clinical study synopsis and some translated pages of the clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Robertson 2007 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. In the primary analysis, the missing HbA1c values at the end of trial were substituted by last observation carried forward (LOCF) if the measurement after 18 weeks was available. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Thalange 2013 | Low risk of bias | Randomisation was carried out using a telephone/web based randomisation system called Interactive Voice Response System (IVRS)/Interactive Web Response System (IWRS). Stratification was applied by age group (2‐5 and 6‐16 years old). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. P study report. A modified intention‐to‐treat analysis was applied (all participants exposed to at least one dose of trial product with a post‐baseline observation). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. Last‐observation carried forward (LOCF) was used for HbA1c. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 2.1 All‐cause mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.1.1 Adults | ||||||||||||
| Fulcher 2005 | Low risk of bias | The randomisation schedule for treatment assignments was prepared by centre on a 1:1 basis. A randomisation list was provided to each centre (pharmacy department or appropriate study personal but not to investigator blinded to the study). There were no relevant baseline imbalances. | Low risk of bias | Single‐blind trial design: the investigator responsible for insulin dosage adjustment was blinded to the treatment allocated to the participant and was not involved in any aspect relating to study supplies. The study coordinator and the participant were aware of the treatment allocation. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. The safety population consisted of all randomised participants who received at least one dose of study medication. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Porcellati 2004 | Low risk of bias | A simple randomisation was used based on computer‐generated random numbers by a person who was not involved in establishing eligibility and entry of patients. Concealment of the randomisation was insured by having the allocation codes in a locked unreadable computer file handled by a person not involved in the recruitment of patients. The randomisation schedule was restricted and stratified by centre. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants randomised finished the study. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce information on analysis plan. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label trial design. Scarce information on analysis plan. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pretreatment and a during‐treatment value. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pre‐treatment and a during‐treatment value. An ITT analysis was performed for all variables. Participants with missing baseline values or no value during treatment were excluded from the statistical analysis of the variable in question. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 2.1.2 Children | ||||||||||||
| Chase 2008 | Low risk of bias | Two randomisation schedules with a 1:1 randomisation ratio, one for males another for females, were generated by the sponsor. At the point of randomisation both the investigator/study coordinator and the participant were blinded to the treatment allocation (to randomisation). The randomisation schedule was stored with the randomisation code administrator at Aventis Pharmaceuticals, Bridgewater, NJ. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Liu 2016 | Low risk of bias | For efficacy analyses, patients were analysed in the treatment group allocated by the Interactive Voice Response System/Interactive Web Response System (IVRS/IWRS) at randomisation (as randomised). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy analyses were based on the modified intention‐to‐treat (mITT) population, corresponding to all randomised patients who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable. The safety population was defined as all randomised patients who took at least one dose or partial dose. In the event of patients having received treatments that differed from those assigned according to the randomisation schedule, then the safety analyses were to be conducted according to the treatment received rather than according to the randomisation groups. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| PRESCHOOL | Low risk of bias | Participants were centrally randomised (utilising permuted block randomisation schedule) via Interactive Voice Response System (IVRS) at a 1:1 ratio to one of the two treatment groups. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy population consisted of all randomised patients who received at least one dose of the study medication (modified intent‐to‐treat [mITT] population). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Schober 2002 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. The safety population compromised all participants who were randomised and received study medication. Efficacy was analysed using those ITT participants for whom both a pre‐treatment and an on‐treatment value were available. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 2.2 Health‐realted quality of life.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pretreatment and a during‐treatment value. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pre‐treatment and a during‐treatment value. An ITT analysis was performed for all variables. Participants with missing baseline values or no value during treatment were excluded from the statistical analysis of the variable in question. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 2.3 Severe hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.3.1 Adults | ||||||||||||
| Bolli 2009 | Some concerns | This was a randomised, parallel group, open‐label, multicentre, single country study. The randomisation schedule was restricted and stratified by centre. No more details available. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All efficacy analyses were determined using a modified intention‐to‐treat (ITT) population (taking at least one dose of study drug and providing data at baseline and at least one on‐treatment visit). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All efficacy analyses were determined using a modified intention‐to‐treat (ITT) population (taking at least one dose of study drug and providing data at baseline and at least one on‐treatment visit). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label trial. No details of the randomisation process. |
| Fulcher 2005 | Low risk of bias | The randomisation schedule for treatment assignments was prepared by centre on a 1:1 basis. A randomisation list was provided to each centre (pharmacy department or appropriate study personal but not to investigator blinded to the study). There were no relevant baseline imbalances. | Low risk of bias | Single‐blind trial design: the investigator responsible for insulin dosage adjustment was blinded to the treatment allocated to the participant and was not involved in any aspect relating to study supplies. The study coordinator and the participant were aware of the treatment allocation. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. The safety population consisted of all randomised participants who received at least one dose of study medication. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Porcellati 2004 | Low risk of bias | A simple randomisation was used based on computer‐generated random numbers by a person who was not involved in establishing eligibility and entry of patients. Concealment of the randomisation was insured by having the allocation codes in a locked unreadable computer file handled by a person not involved in the recruitment of patients. The randomisation schedule was restricted and stratified by centre. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants randomised finished the study. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce information on analysis plan. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label trial design. Scarce information on analysis plan. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pretreatment and a during‐treatment value. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pre‐treatment and a during‐treatment value. An ITT analysis was performed for all variables. Participants with missing baseline values or no value during treatment were excluded from the statistical analysis of the variable in question. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 2.3.2 Children | ||||||||||||
| Chase 2008 | Low risk of bias | Two randomisation schedules with a 1:1 randomisation ratio, one for males another for females, were generated by the sponsor. At the point of randomisation both the investigator/study coordinator and the participant were blinded to the treatment allocation (to randomisation). The randomisation schedule was stored with the randomisation code administrator at Aventis Pharmaceuticals, Bridgewater, NJ. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Liu 2016 | Low risk of bias | For efficacy analyses, patients were analysed in the treatment group allocated by the Interactive Voice Response System/Interactive Web Response System (IVRS/IWRS) at randomisation (as randomised). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy analyses were based on the modified intention‐to‐treat (mITT) population, corresponding to all randomised patients who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable. The safety population was defined as all randomised patients who took at least one dose or partial dose. In the event of patients having received treatments that differed from those assigned according to the randomisation schedule, then the safety analyses were to be conducted according to the treatment received rather than according to the randomisation groups. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| PRESCHOOL | Low risk of bias | Participants were centrally randomised (utilising permuted block randomisation schedule) via Interactive Voice Response System (IVRS) at a 1:1 ratio to one of the two treatment groups. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy population consisted of all randomised patients who received at least one dose of the study medication (modified intent‐to‐treat [mITT] population). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Schober 2002 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Protocol deviations were explained in the clinical study report. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. The safety population compromised all participants who were randomised and received study medication. Efficacy was analysed using those ITT participants for whom both a pre‐treatment and an on‐treatment value were available. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 2.6 Cardiovascular mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.6.1 Adults | ||||||||||||
| Fulcher 2005 | Low risk of bias | The randomisation schedule for treatment assignments was prepared by centre on a 1:1 basis. A randomisation list was provided to each centre (pharmacy department or appropriate study personal but not to investigator blinded to the study). There were no relevant baseline imbalances. | Low risk of bias | Single‐blind trial design: the investigator responsible for insulin dosage adjustment was blinded to the treatment allocated to the participant and was not involved in any aspect relating to study supplies. The study coordinator and the participant were aware of the treatment allocation. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. The safety population consisted of all randomised participants who received at least one dose of study medication. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Porcellati 2004 | Low risk of bias | A simple randomisation was used based on computer‐generated random numbers by a person who was not involved in establishing eligibility and entry of patients. Concealment of the randomisation was insured by having the allocation codes in a locked unreadable computer file handled by a person not involved in the recruitment of patients. The randomisation schedule was restricted and stratified by centre. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants randomised finished the study. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce information on analysis plan. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label trial design. Scarce information on analysis plan. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 2.6.2 Children | ||||||||||||
| Chase 2008 | Low risk of bias | Two randomisation schedules with a 1:1 randomisation ratio, one for males another for females, were generated by the sponsor. At the point of randomisation both the investigator/study coordinator and the participant were blinded to the treatment allocation (to randomisation). The randomisation schedule was stored with the randomisation code administrator at Aventis Pharmaceuticals, Bridgewater, NJ. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Liu 2016 | Low risk of bias | For efficacy analyses, patients were analysed in the treatment group allocated by the Interactive Voice Response System/Interactive Web Response System (IVRS/IWRS) at randomisation (as randomised). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy analyses were based on the modified intention‐to‐treat (mITT) population, corresponding to all randomised patients who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable. The safety population was defined as all randomised patients who took at least one dose or partial dose. In the event of patients having received treatments that differed from those assigned according to the randomisation schedule, then the safety analyses were to be conducted according to the treatment received rather than according to the randomisation groups. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| PRESCHOOL | Low risk of bias | Participants were centrally randomised (utilising permuted block randomisation schedule) via Interactive Voice Response System (IVRS) at a 1:1 ratio to one of the two treatment groups. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy population consisted of all randomised patients who received at least one dose of the study medication (modified intent‐to‐treat [mITT] population). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Schober 2002 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. The safety population compromised all participants who were randomised and received study medication. Efficacy was analysed using those ITT participants for whom both a pre‐treatment and an on‐treatment value were available. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 2.7 Non‐fatal myocardial infarction.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 2.8 Non‐fatal stroke.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 2.9 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.9.1 Adults | ||||||||||||
| Bolli 2009 | Some concerns | This was a randomised, parallel group, open‐label, multicentre, single country study. The randomisation schedule was restricted and stratified by centre. No more details available. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All efficacy analyses were determined using a modified intention‐to‐treat (ITT) population (taking at least one dose of study drug and providing data at baseline and at least one on‐treatment visit). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All efficacy analyses were determined using a modified intention‐to‐treat (ITT) population (taking at least one dose of study drug and providing data at baseline and at least one on‐treatment visit). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label trial. No details of the randomisation process. |
| Fulcher 2005 | Low risk of bias | The randomisation schedule for treatment assignments was prepared by centre on a 1:1 basis. A randomisation list was provided to each centre (pharmacy department or appropriate study personal but not to investigator blinded to the study). There were no relevant baseline imbalances. | Low risk of bias | Single‐blind trial design: the investigator responsible for insulin dosage adjustment was blinded to the treatment allocated to the participant and was not involved in any aspect relating to study supplies. The study coordinator and the participant were aware of the treatment allocation. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. The safety population consisted of all randomised participants who received at least one dose of study medication. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Only participants with at least one measurement after baseline were included in the intent‐to‐treat (ITT) analysis. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pretreatment and a during‐treatment value. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pre‐treatment and a during‐treatment value. An ITT analysis was performed for all variables. Participants with missing baseline values or no value during treatment were excluded from the statistical analysis of the variable in question. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 2.9.2 Children | ||||||||||||
| Chase 2008 | Low risk of bias | Two randomisation schedules with a 1:1 randomisation ratio, one for males another for females, were generated by the sponsor. At the point of randomisation both the investigator/study coordinator and the participant were blinded to the treatment allocation (to randomisation). The randomisation schedule was stored with the randomisation code administrator at Aventis Pharmaceuticals, Bridgewater, NJ. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Liu 2016 | Low risk of bias | For efficacy analyses, patients were analysed in the treatment group allocated by the Interactive Voice Response System/Interactive Web Response System (IVRS/IWRS) at randomisation (as randomised). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy analyses were based on the modified intention‐to‐treat (mITT) population, corresponding to all randomised patients who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable. The safety population was defined as all randomised patients who took at least one dose or partial dose. In the event of patients having received treatments that differed from those assigned according to the randomisation schedule, then the safety analyses were to be conducted according to the treatment received rather than according to the randomisation groups. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| PRESCHOOL | Low risk of bias | Participants were centrally randomised (utilising permuted block randomisation schedule) via Interactive Voice Response System (IVRS) at a 1:1 ratio to one of the two treatment groups. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. The safety population compromised all participants who were randomised and received study medication. Efficacy was analysed using those ITT participants for whom both a pre‐treatment and an on‐treatment value were available. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Schober 2002 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 2.11 Diabetic ketoacidosis.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.11.1 Adults | ||||||||||||
| Fulcher 2005 | Low risk of bias | The randomisation schedule for treatment assignments was prepared by centre on a 1:1 basis. A randomisation list was provided to each centre (pharmacy department or appropriate study personal but not to investigator blinded to the study). There were no relevant baseline imbalances. | Low risk of bias | Single‐blind trial design: the investigator responsible for insulin dosage adjustment was blinded to the treatment allocated to the participant and was not involved in any aspect relating to study supplies. The study coordinator and the participant were aware of the treatment allocation. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. The safety population consisted of all randomised participants who received at least one dose of study medication. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pretreatment and a during‐treatment value. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pre‐treatment and a during‐treatment value. An ITT analysis was performed for all variables. Participants with missing baseline values or no value during treatment were excluded from the statistical analysis of the variable in question. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 2.11.2 Children | ||||||||||||
| Chase 2008 | Low risk of bias | Two randomisation schedules with a 1:1 randomisation ratio, one for males another for females, were generated by the sponsor. At the point of randomisation both the investigator/study coordinator and the participant were blinded to the treatment allocation (to randomisation). The randomisation schedule was stored with the randomisation code administrator at Aventis Pharmaceuticals, Bridgewater, NJ. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Liu 2016 | Low risk of bias | For efficacy analyses, patients were analysed in the treatment group allocated by the Interactive Voice Response System/Interactive Web Response System (IVRS/IWRS) at randomisation (as randomised). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy analyses were based on the modified intention‐to‐treat (mITT) population, corresponding to all randomised patients who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable. The safety population was defined as all randomised patients who took at least one dose or partial dose. In the event of patients having received treatments that differed from those assigned according to the randomisation schedule, then the safety analyses were to be conducted according to the treatment received rather than according to the randomisation groups. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| PRESCHOOL | Low risk of bias | Participants were centrally randomised (utilising permuted block randomisation schedule) via Interactive Voice Response System (IVRS) at a 1:1 ratio to one of the two treatment groups. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy population consisted of all randomised patients who received at least one dose of the study medication (modified intent‐to‐treat [mITT] population). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Schober 2002 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. The safety population compromised all participants who were randomised and received study medication. Efficacy was analysed using those ITT participants for whom both a pre‐treatment and an on‐treatment value were available. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 2.13 Non‐serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.13.1 Adults | ||||||||||||
| Bolli 2009 | Some concerns | This was a randomised, parallel group, open‐label, multicentre, single country study. The randomisation schedule was restricted and stratified by centre. No more details available. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All efficacy analyses were determined using a modified intention‐to‐treat (ITT) population (taking at least one dose of study drug and providing data at baseline and at least one on‐treatment visit). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All efficacy analyses were determined using a modified intention‐to‐treat (ITT) population (taking at least one dose of study drug and providing data at baseline and at least one on‐treatment visit). | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. No details of the randomisation process. |
| Fulcher 2005 | Low risk of bias | The randomisation schedule for treatment assignments was prepared by centre on a 1:1 basis. A randomisation list was provided to each centre (pharmacy department or appropriate study personal but not to investigator blinded to the study). There were no relevant baseline imbalances. | Low risk of bias | Single‐blind trial design: the investigator responsible for insulin dosage adjustment was blinded to the treatment allocated to the participant and was not involved in any aspect relating to study supplies. The study coordinator and the participant were aware of the treatment allocation. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. The safety population consisted of all randomised participants who received at least one dose of study medication. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pretreatment and a during‐treatment value. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pre‐treatment and a during‐treatment value. An ITT analysis was performed for all variables. Participants with missing baseline values or no value during treatment were excluded from the statistical analysis of the variable in question. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 2.13.2 Children | ||||||||||||
| Chase 2008 | Low risk of bias | Two randomisation schedules with a 1:1 randomisation ratio, one for males another for females, were generated by the sponsor. At the point of randomisation both the investigator/study coordinator and the participant were blinded to the treatment allocation (to randomisation). The randomisation schedule was stored with the randomisation code administrator at Aventis Pharmaceuticals, Bridgewater, NJ. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Liu 2016 | Low risk of bias | For efficacy analyses, patients were analysed in the treatment group allocated by the Interactive Voice Response System/Interactive Web Response System (IVRS/IWRS) at randomisation (as randomised). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy analyses were based on the modified intention‐to‐treat (mITT) population, corresponding to all randomised patients who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable. The safety population was defined as all randomised patients who took at least one dose or partial dose. In the event of patients having received treatments that differed from those assigned according to the randomisation schedule, then the safety analyses were to be conducted according to the treatment received rather than according to the randomisation groups. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| PRESCHOOL | Low risk of bias | Participants were centrally randomised (utilising permuted block randomisation schedule) via Interactive Voice Response System (IVRS) at a 1:1 ratio to one of the two treatment groups. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy population consisted of all randomised patients who received at least one dose of the study medication (modified intent‐to‐treat [mITT] population). | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Schober 2002 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. The safety population compromised all participants who were randomised and received study medication. Efficacy was analysed using those ITT participants for whom both a pre‐treatment and an on‐treatment value were available. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 2.19 Severe nocturnal hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.19.1 Adults | ||||||||||||
| Fulcher 2005 | Low risk of bias | The randomisation schedule for treatment assignments was prepared by centre on a 1:1 basis. A randomisation list was provided to each centre (pharmacy department or appropriate study personal but not to investigator blinded to the study). There were no relevant baseline imbalances. | Low risk of bias | Single‐blind trial design: the investigator responsible for insulin dosage adjustment was blinded to the treatment allocated to the participant and was not involved in any aspect relating to study supplies. The study coordinator and the participant were aware of the treatment allocation. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. The safety population consisted of all randomised participants who received at least one dose of study medication. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pretreatment and a during‐treatment value. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pre‐treatment and a during‐treatment value. An ITT analysis was performed for all variables. Participants with missing baseline values or no value during treatment were excluded from the statistical analysis of the variable in question. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 2.19.2 Children | ||||||||||||
| Chase 2008 | Low risk of bias | Two randomisation schedules with a 1:1 randomisation ratio, one for males another for females, were generated by the sponsor. At the point of randomisation both the investigator/study coordinator and the participant were blinded to the treatment allocation (to randomisation). The randomisation schedule was stored with the randomisation code administrator at Aventis Pharmaceuticals, Bridgewater, NJ. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| PRESCHOOL | Low risk of bias | Participants were centrally randomised (utilising permuted block randomisation schedule) via Interactive Voice Response System (IVRS) at a 1:1 ratio to one of the two treatment groups. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy population consisted of all randomised patients who received at least one dose of the study medication (modified intent‐to‐treat [mITT] population). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Schober 2002 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. The safety population compromised all participants who were randomised and received study medication. Efficacy was analysed using those ITT participants for whom both a pre‐treatment and an on‐treatment value were available. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 2.22 Mild/moderate hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.22.1 Adults | ||||||||||||
| Fulcher 2005 | Low risk of bias | The randomisation schedule for treatment assignments was prepared by centre on a 1:1 basis. A randomisation list was provided to each centre (pharmacy department or appropriate study personal but not to investigator blinded to the study). There were no relevant baseline imbalances. | Low risk of bias | Single‐blind trial design: the investigator responsible for insulin dosage adjustment was blinded to the treatment allocated to the participant and was not involved in any aspect relating to study supplies. The study coordinator and the participant were aware of the treatment allocation. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. The safety population consisted of all randomised participants who received at least one dose of study medication. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pretreatment and a during‐treatment value. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pre‐treatment and a during‐treatment value. An ITT analysis was performed for all variables. Participants with missing baseline values or no value during treatment were excluded from the statistical analysis of the variable in question. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 2.22.2 Children | ||||||||||||
| Chase 2008 | Low risk of bias | Two randomisation schedules with a 1:1 randomisation ratio, one for males another for females, were generated by the sponsor. At the point of randomisation both the investigator/study coordinator and the participant were blinded to the treatment allocation (to randomisation). The randomisation schedule was stored with the randomisation code administrator at Aventis Pharmaceuticals, Bridgewater, NJ. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Liu 2016 | Low risk of bias | For efficacy analyses, patients were analysed in the treatment group allocated by the Interactive Voice Response System/Interactive Web Response System (IVRS/IWRS) at randomisation (as randomised). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy analyses were based on the modified intention‐to‐treat (mITT) population, corresponding to all randomised patients who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable. The safety population was defined as all randomised patients who took at least one dose or partial dose. In the event of patients having received treatments that differed from those assigned according to the randomisation schedule, then the safety analyses were to be conducted according to the treatment received rather than according to the randomisation groups. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| PRESCHOOL | Low risk of bias | Participants were centrally randomised (utilising permuted block randomisation schedule) via Interactive Voice Response System (IVRS) at a 1:1 ratio to one of the two treatment groups. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy population consisted of all randomised patients who received at least one dose of the study medication (modified intent‐to‐treat [mITT] population). | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Schober 2002 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. The safety population compromised all participants who were randomised and received study medication. Efficacy was analysed using those ITT participants for whom both a pre‐treatment and an on‐treatment value were available. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 2.24 HbA1c.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.24.1 Adults | ||||||||||||
| Bolli 2009 | Some concerns | This was a randomised, parallel group, open‐label, multicentre, single country study. The randomisation schedule was restricted and stratified by centre. No more details available. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All efficacy analyses were determined using a modified intention‐to‐treat (ITT) population (taking at least one dose of study drug and providing data at baseline and at least one on‐treatment visit). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. All efficacy analyses were determined using a modified intention‐to‐treat (ITT) population (taking at least one dose of study drug and providing data at baseline and at least one on‐treatment visit). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label trial. No details of the randomisation process. |
| Fulcher 2005 | Low risk of bias | The randomisation schedule for treatment assignments was prepared by centre on a 1:1 basis. A randomisation list was provided to each centre (pharmacy department or appropriate study personal but not to investigator blinded to the study). There were no relevant baseline imbalances. | Low risk of bias | Single‐blind trial design: the investigator responsible for insulin dosage adjustment was blinded to the treatment allocated to the participant and was not involved in any aspect relating to study supplies. The study coordinator and the participant were aware of the treatment allocation. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The primary efficacy analysis was carried out with a modified intention‐to‐treat population (ITT), which consisted of all randomised patients with at least one dose of study medication. All secondary efficacy variables were also analysed on an ITT basis. The safety population consisted of all randomised participants who received at least one dose of study medication. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Home 2005 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (participants with at least one measurement after baseline). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (efficacy was analysed using those intention‐to‐treat (ITT) participants for whom both a pre‐treatment and an on‐treatment value were available, with the exception of hypoglycaemia, for which the entire ITT population was analysed). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Porcellati 2004 | Low risk of bias | A simple randomisation was used based on computer‐generated random numbers by a person who was not involved in establishing eligibility and entry of patients. Concealment of the randomisation was insured by having the allocation codes in a locked unreadable computer file handled by a person not involved in the recruitment of patients. The randomisation schedule was restricted and stratified by centre. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. All participants randomised finished the study. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce information on analysis plan. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label trial design. Scarce information on analysis plan. |
| Ratner 2000 | Low risk of bias | The randomisation schedule was stratified according to centre and pre‐randomisation basal insulin regimen of once versus twice daily. Forty randomisation numbers were allocated to each basal insulin regimen for each centre. To ensure a balanced number of participants for each treatment group the randomisation schedule was generated using block sizes of 4. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pretreatment and a during‐treatment value. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated and having both a pre‐treatment and a during‐treatment value. An ITT analysis was performed for all variables. Participants with missing baseline values or no value during treatment were excluded from the statistical analysis of the variable in question. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 2.24.2 Children | ||||||||||||
| Chase 2008 | Low risk of bias | Two randomisation schedules with a 1:1 randomisation ratio, one for males another for females, were generated by the sponsor. At the point of randomisation both the investigator/study coordinator and the participant were blinded to the treatment allocation (to randomisation). The randomisation schedule was stored with the randomisation code administrator at Aventis Pharmaceuticals, Bridgewater, NJ. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (participants taking at least one dose of study medication with a baseline measurement and at least one follow‐up measure; the safety population consisted of all randomised participants who received at least one dose of study medication). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Liu 2016 | Low risk of bias | For efficacy analyses, patients were analysed in the treatment group allocated by the Interactive Voice Response System/Interactive Web Response System (IVRS/IWRS) at randomisation (as randomised). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy analyses were based on the modified intention‐to‐treat (mITT) population, corresponding to all randomised patients who received at least one dose, and had both a baseline assessment and at least one post‐baseline assessment for at least one efficacy variable. The safety population was defined as all randomised patients who took at least one dose or partial dose. In the event of patients having received treatments that differed from those assigned according to the randomisation schedule, then the safety analyses were to be conducted according to the treatment received rather than according to the randomisation groups. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| PRESCHOOL | Low risk of bias | Participants were centrally randomised (utilising permuted block randomisation schedule) via Interactive Voice Response System (IVRS) at a 1:1 ratio to one of the two treatment groups. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The efficacy population consisted of all randomised patients who received at least one dose of the study medication (modified intent‐to‐treat [mITT] population). | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Schober 2002 | Low risk of bias | A 1:1 randomisation schedule was generated by the sponsor. This schedule paired sequential numbers with treatment codes allocated at random. The schedule was prepared by centre on a 1:1 basis. An independent agency was used for central telephone randomisation. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The modified intention‐to‐treat (ITT) population was defined as all participants randomised and treated. The safety population compromised all participants who were randomised and received study medication. Efficacy was analysed using those ITT participants for whom both a pre‐treatment and an on‐treatment value were available. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 3.1 All‐cause mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Pieber 2007 | Low risk of bias | Participants were symmetrically randomised (1:1) to either insulin detemir or insulin glargine. Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to trial products). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 3.2 Severe hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Pieber 2007 | Low risk of bias | Participants were symmetrically randomised (1:1) to either insulin detemir or insulin glargine. Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to trial products). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 3.5 Cardiovascular mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Pieber 2007 | Low risk of bias | Participants were symmetrically randomised (1:1) to either insulin detemir or insulin glargine. Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to trial products). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 3.6 Non‐fatal myocardial infarction.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 3.7 Non‐fatal stroke.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 3.8 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Pieber 2007 | Low risk of bias | Participants were symmetrically randomised (1:1) to either insulin detemir or insulin glargine. Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to trial products). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 3.9 Diabetic ketoacidosis.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 3.10 Non‐serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Pieber 2007 | Low risk of bias | Participants were symmetrically randomised (1:1) to either insulin detemir or insulin glargine. Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to trial products). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 3.16 Severe nocturnal hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 3.16.1 Published | ||||||||||||
| Pieber 2007 | Low risk of bias | Participants were symmetrically randomised (1:1) to either insulin detemir or insulin glargine. Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to trial products). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 3.16.2 Unpublished | ||||||||||||
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 3.17 Mild/moderate hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 3.17.1 Published | ||||||||||||
| Pieber 2007 | Low risk of bias | Participants were symmetrically randomised (1:1) to either insulin detemir or insulin glargine. Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to trial products). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 3.17.2 Unpublished | ||||||||||||
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 3.18 HbA1c.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Heller 2009 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to at least one dose of trial product, bolus or basal insulin). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Participants needed to have at least one post‐treatment measurement obtained after 3 month of treatment and a valid baseline measurement in order to qualify for the analysis of HbA1c. Last‐observation‐carried‐forward (LOCF) method was generally used for missing values on the endpoints that were measured after initiation of treatment and on more than one occasion. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Pieber 2007 | Low risk of bias | Participants were symmetrically randomised (1:1) to either insulin detemir or insulin glargine. Randomisation was carried out using a telephone randomisation system, the Interactive Voice Response System (IVRS). | Low risk of bias | Open‐label trial design. A modified intention‐to‐treat analysis was applied (all randomised participants exposed to trial products). | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Modified intention‐to‐treat analysis set was used (all exposed participants were analysed). Missing observations were considered missing at random in all analyses. Missing HbA1c measurement at the end of the trial (after 26 weeks) was extrapolated by last‐observation‐carried‐forward (LOCF) if a measurement after 20 weeks was available. Otherwise, the measurement was considered as missing. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.1 All‐cause mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.1.1 Adults | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 4.1.2 Children | ||||||||||||
| BEGIN Young | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin detemir in a 1:1 ratio. Randomisation was carried out using a central interactive voice/web response system (IV/WRS). Randomisation was stratified according to 3 age groups: 1 to less than 6 years; 6 to less than 12 years; 12 to less than 18 years of age. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The internal Novo Nordisk safety committee and the external data monitoring committee (DMC) reviewed safety data on an ongoing basis. The internal safety committee was blinded and the DMC was unblinded. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The full analysis set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. The safety analysis set included all participants receiving at least one dose of the investigational product. Participants in the safety set were to contribute to the evaluation “as treated”. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Unless otherwise specified, missing values (including intermittent missing values) were imputed using the last‐observation‐carried‐forward (LOCF) method. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.2 Health‐related quality of life.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.2.1 Physical health score | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 4.2.2 Mental health score | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 4.3 Severe hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.3.1 Adults | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 4.3.2 Children | ||||||||||||
| BEGIN Young | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin detemir in a 1:1 ratio. Randomisation was carried out using a central interactive voice/web response system (IV/WRS). Randomisation was stratified according to 3 age groups: 1 to less than 6 years; 6 to less than 12 years; 12 to less than 18 years of age. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The internal Novo Nordisk safety committee and the external data monitoring committee (DMC) reviewed safety data on an ongoing basis. The internal safety committee was blinded and the DMC was unblinded. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The full analysis set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. The safety analysis set included all participants receiving at least one dose of the investigational product. Participants in the safety set were to contribute to the evaluation “as treated”. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Unless otherwise specified, missing values (including intermittent missing values) were imputed using the last‐observation‐carried‐forward (LOCF) method. External classification of severe hypoglycaemia was performed blinded. | Low risk of bias | External classification of severe hypoglycaemia was performed blinded and outcome measure unlikely influenced by potential lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.5 Cardiovascular mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.5.1 Adults | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 4.5.2 Children | ||||||||||||
| BEGIN Young | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin detemir in a 1:1 ratio. Randomisation was carried out using a central interactive voice/web response system (IV/WRS). Randomisation was stratified according to 3 age groups: 1 to less than 6 years; 6 to less than 12 years; 12 to less than 18 years of age. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The internal Novo Nordisk safety committee and the external data monitoring committee (DMC) reviewed safety data on an ongoing basis. The internal safety committee was blinded and the DMC was unblinded. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The full analysis set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. The safety analysis set included all participants receiving at least one dose of the investigational product. Participants in the safety set were to contribute to the evaluation “as treated”. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Unless otherwise specified, missing values (including intermittent missing values) were imputed using the last‐observation‐carried‐forward (LOCF) method. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.6 Non‐fatal myocardial infarction.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.7 Non‐fatal stroke.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.8 End stage renal disease.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.9 Blindness.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.10 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.10.1 Adults | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 4.10.2 Children | ||||||||||||
| BEGIN Young | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin detemir in a 1:1 ratio. Randomisation was carried out using a central interactive voice/web response system (IV/WRS). Randomisation was stratified according to 3 age groups: 1 to less than 6 years; 6 to less than 12 years; 12 to less than 18 years of age. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The internal Novo Nordisk safety committee and the external data monitoring committee (DMC) reviewed safety data on an ongoing basis. The internal safety committee was blinded and the DMC was unblinded. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The full analysis set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. The safety analysis set included all participants receiving at least one dose of the investigational product. Participants in the safety set were to contribute to the evaluation “as treated”. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Unless otherwise specified, missing values (including intermittent missing values) were imputed using the last‐observation‐carried‐forward (LOCF) method. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.11 Diabetic ketoacidosis.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.11.1 Adults | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 4.11.2 Children | ||||||||||||
| BEGIN Young | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin detemir in a 1:1 ratio. Randomisation was carried out using a central interactive voice/web response system (IV/WRS). Randomisation was stratified according to 3 age groups: 1 to less than 6 years; 6 to less than 12 years; 12 to less than 18 years of age. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The internal Novo Nordisk safety committee and the external data monitoring committee (DMC) reviewed safety data on an ongoing basis. The internal safety committee was blinded and the DMC was unblinded. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The full analysis set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. The safety analysis set included all participants receiving at least one dose of the investigational product. Participants in the safety set were to contribute to the evaluation “as treated”. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Unless otherwise specified, missing values (including intermittent missing values) were imputed using the last‐observation‐carried‐forward (LOCF) method. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.12 Non‐serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.12.1 Adults | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 4.12.2 Children | ||||||||||||
| BEGIN Young | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin detemir in a 1:1 ratio. Randomisation was carried out using a central interactive voice/web response system (IV/WRS). Randomisation was stratified according to 3 age groups: 1 to less than 6 years; 6 to less than 12 years; 12 to less than 18 years of age. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The internal Novo Nordisk safety committee and the external data monitoring committee (DMC) reviewed safety data on an ongoing basis. The internal safety committee was blinded and the DMC was unblinded. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The full analysis set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. The safety analysis set included all participants receiving at least one dose of the investigational product. Participants in the safety set were to contribute to the evaluation “as treated”. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Unless otherwise specified, missing values (including intermittent missing values) were imputed using the last‐observation‐carried‐forward (LOCF) method. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 4.18 Severe nocturnal hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.18.1 Adults | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 4.18.2 Children | ||||||||||||
| BEGIN Young | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin detemir in a 1:1 ratio. Randomisation was carried out using a central interactive voice/web response system (IV/WRS). Randomisation was stratified according to 3 age groups: 1 to less than 6 years; 6 to less than 12 years; 12 to less than 18 years of age. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The internal Novo Nordisk safety committee and the external data monitoring committee (DMC) reviewed safety data on an ongoing basis. The internal safety committee was blinded and the DMC was unblinded. External classification of severe hypoglycaemia was performed blinded. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The full analysis set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. The safety analysis set included all participants receiving at least one dose of the investigational product. Participants in the safety set were to contribute to the evaluation “as treated”. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Unless otherwise specified, missing values (including intermittent missing values) were imputed using the last‐observation‐carried‐forward (LOCF) method. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 4.19 Mild/moderate hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.19.1 Adults | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 4.19.2 Children | ||||||||||||
| BEGIN Young | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin detemir in a 1:1 ratio. Randomisation was carried out using a central interactive voice/web response system (IV/WRS). Randomisation was stratified according to 3 age groups: 1 to less than 6 years; 6 to less than 12 years; 12 to less than 18 years of age. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The internal Novo Nordisk safety committee and the external data monitoring committee (DMC) reviewed safety data on an ongoing basis. The internal safety committee was blinded and the DMC was unblinded. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The full analysis set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. The safety analysis set included all participants receiving at least one dose of the investigational product. Participants in the safety set were to contribute to the evaluation “as treated”. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Unless otherwise specified, missing values (including intermittent missing values) were imputed using the last‐observation‐carried‐forward (LOCF) method. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
Risk of bias for analysis 4.20 HbA1c.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.20.1 Adults | ||||||||||||
| Davies 2014 | Low risk of bias | At the randomisation visit (visit 2) the participants were to be randomised to either insulin degludec or insulin detemir, both in combination with insulin aspart. The randomisation was to be carried out in a 2:1 manner using the Interactive Voice/Web Response System IV/WRS. The trial was stratified according to region with 4 levels: Europe (Italy, UK, Macedonia and Finland), Japan, India and South America (Brazil). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. Full Analysis Set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. Safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Participants in the safety set were to contribute to the evaluation “as treated”. Unless otherwise specified missing values (including intermittent missing values) were imputed using the last observation carried forward (LOCF) method. Missing observations were considered missing at random in all analyses. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 4.20.2 Children | ||||||||||||
| BEGIN Young | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin detemir in a 1:1 ratio. Randomisation was carried out using a central interactive voice/web response system (IV/WRS). Randomisation was stratified according to 3 age groups: 1 to less than 6 years; 6 to less than 12 years; 12 to less than 18 years of age. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The internal Novo Nordisk safety committee and the external data monitoring committee (DMC) reviewed safety data on an ongoing basis. The internal safety committee was blinded and the DMC was unblinded. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The full analysis set (FAS) included all randomised participants. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants contributed to the evaluation “as randomised”. The safety analysis set included all participants receiving at least one dose of the investigational product. Participants in the safety set were to contribute to the evaluation “as treated”. All analyses and summary of efficacy endpoints, and formal statistical analyses related to safety endpoints were based on the full analysis set (FAS). Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Unless otherwise specified, missing values (including intermittent missing values) were imputed using the last‐observation‐carried‐forward (LOCF) method. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 5.1 All‐cause mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants finished the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label trial design with outcome measure unlikely influenced by lack of blinding. Scarce information. |
Risk of bias for analysis 5.3 Health‐related quality of life (physical health).
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.3.1 Published | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 5.3.2 Unpublished | ||||||||||||
| SWITCH 1 | Low risk of bias | Participants were randomised in a 1:1 manner to one of the two treatment sequences, using the Interactive Voice/Web Response System IV/WRS. Within each treatment sequence participants were randomised 1:1 to morning or evening dosing. There were no relevant baseline imbalances. | Low risk of bias | Double‐blind crossover study (participants, the clinical study group and the investigator remained blinded throughout the trial). Analyses of all endpoints were based on the full analysis set (FAS). Efficacy endpoints and patient‐reported outcome endpoints were summarised using the FAS. Safety endpoints were summarised using the safety analysis set. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Analyses of all endpoints were based on the FAS. Efficacy endpoints and patient‐reported outcome endpoints were summarised using the FAS. Safety endpoints were summarised using the SAS. | Low risk of bias | Double‐blind design (participants, the clinical study group and the investigator remained blinded throughout the trial). | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 5.4 Health‐related quality of life (mental health).
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.4.1 Published | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 5.4.2 Unpublished | ||||||||||||
| SWITCH 1 | Low risk of bias | Participants were randomised in a 1:1 manner to one of the two treatment sequences, using the Interactive Voice/Web Response System IV/WRS. Within each treatment sequence participants were randomised 1:1 to morning or evening dosing. There were no relevant baseline imbalances. | Low risk of bias | Double‐blind crossover study (participants, the clinical study group and the investigator remained blinded throughout the trial). Analyses of all endpoints were based on the full analysis set (FAS). Efficacy endpoints and patient‐reported outcome endpoints were summarised using the FAS. Safety endpoints were summarised using the safety analysis set. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Analyses of all endpoints were based on the FAS. Efficacy endpoints and patient‐reported outcome endpoints were summarised using the FAS. Safety endpoints were summarised using the SAS. | Low risk of bias | Double‐blind design (participants, the clinical study group and the investigator remained blinded throughout the trial). | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
Risk of bias for analysis 5.5 Severe hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.5.1 Adults | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 5.5.2 Children | ||||||||||||
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label trial design with outcome measure unlikely influenced by lack of blinding. Scarce information. |
Risk of bias for analysis 5.7 Cardiovascular mortality.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.7.1 Adults | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 5.7.2 Children | ||||||||||||
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants finished the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label trial design with outcome measure unlikely influenced by lack of blinding. Scarce information. |
Risk of bias for analysis 5.8 Non‐fatal myocardial infarction.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.8.1 Adults | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 5.8.2 Children | ||||||||||||
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label trial design with outcome measure unlikely influenced by lack of blinding. Scarce information. |
Risk of bias for analysis 5.9 Non‐fatal stroke.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label trial design with outcome measure unlikely influenced by lack of blinding. Scarce information. |
Risk of bias for analysis 5.10 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.10.1 Adults | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 5.10.2 Children | ||||||||||||
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label trial design with outcome measure unlikely influenced by lack of blinding. Scarce information. |
Risk of bias for analysis 5.11 Diabetic ketoacidosis.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.11.1 Adults | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 5.11.2 Children | ||||||||||||
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label trial design with outcome measure unlikely influenced by lack of blinding. Scarce information. |
Risk of bias for analysis 5.13 Non‐serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.13.1 Adults | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 5.13.2 Children | ||||||||||||
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants completed the study. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. Scarce information. |
Risk of bias for analysis 5.19 Severe nocturnal hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label trial design with outcome measure unlikely influenced by lack of blinding. Scarce information. |
Risk of bias for analysis 5.20 Mild/moderate hypoglycaemia.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.20.1 Adults | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. |
| Subgroup 5.20.2 Children | ||||||||||||
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants completed the study. | Some concerns | Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label design. Outcome measure could have been influenced by knowledge of the intervention received. It it unlikely that there were strong beliefs in beneficial or harmful effects of the interventions. Scarce information. |
Risk of bias for analysis 5.21 HbA1c.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.21.1 Adults | ||||||||||||
| BEGIN Basal‐Bolus Type 1 | Low risk of bias | Participants were allocated to treatment with insulin degludec or insulin glargine in a 3:1 ratio. The Interactive Voice/Web Response System IV/WRS allocated the trial product to the participant at each dispensing and randomisation visit. There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the FAS was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 52 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| BEGIN Flex T1 | Low risk of bias | Randomisation was carried out using a telephone randomisation system (Interactive Voice Response System (IVRS)). There were no relevant baseline imbalances. | Low risk of bias | Open‐label trial design. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Only endpoints derived after 26 weeks of treatment were to be analysed statistically. Missing values were imputed by last‐observation‐carried‐forward (LOCF) for all endpoints. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| SWITCH 1 | Low risk of bias | Participants were randomised in a 1:1 manner to one of the two treatment sequences, using the Interactive Voice/Web Response System IV/WRS. Within each treatment sequence participants were randomised 1:1 to morning or evening dosing. There were no relevant baseline imbalances. | Low risk of bias | Double‐blind crossover study (participants, the clinical study group and the investigator remained blinded throughout the trial). Analyses of all endpoints were based on the full analysis set (FAS). Efficacy endpoints and patient‐reported outcome endpoints were summarised using the FAS. Safety endpoints were summarised using the safety analysis set. | Low risk of bias | Withdrawals and reasons for withdrawal were documented, did not differ substantially between intervention groups and did not appear to be related to health status. The statistical evaluation of the full analysis set (FAS) was to follow the intention‐to‐treat (ITT) principle and participants were to contribute to the evaluation “as randomised”. The safety analysis set included all participants who received at least one dose of the investigational product or its comparator. Analyses of all endpoints were based on the FAS. Efficacy endpoints and patient‐reported outcome endpoints were summarised using the FAS. Safety endpoints were summarised using the SAS. | Low risk of bias | Double‐blind design (participants, the clinical study group and the investigator remained blinded throughout the trial). | Low risk of bias | Data from full clinical study report. Only one measurement and result provided for the time point selected by review authors. | Low risk of bias | No risk of bias identified. |
| Subgroup 5.21.2 Children | ||||||||||||
| Urakami 2017 | Some concerns | No details on the randomisation process. There were no relevant baseline imbalances. | Some concerns | Open‐label trial design. Scarce information. | Low risk of bias | All randomised participants completed the study. | Low risk of bias | Open‐label design with outcome measure unlikely influenced by lack of blinding. | Some concerns | Scarce data from publication and study author. Probably only one measurement and result provided for the time point selected by review authors. | Some concerns | No details on the randomisation process. Open‐label trial design with outcome measure. and no protocol available. |
Acknowledgements
The review authors and the CMED editorial base are grateful to the following peer reviewers for their time and comments: Prof. Hans V. Hogerzeil, Global Health Unit, Department of Health Sciences, University Medical Centre Groningen, University of Groningen, Groningen, The Netherlands; Prof. Richard Laing, School of Public Health, Boston University, Boston, Massachusetts, USA & School of Public Health, University of the Western Cape, Cape Town, Western Cape, South Africa; Dr. Gojka Roglic, Department of Noncommunicable Diseases, World Health Organization, Geneva, Switzerland; Dr. Sylvia Kehlenbrink, Division of Endocrinology, Diabetes and Hypertension, Department of Medicine, Brigham and Women's Hospital, Boston, Massachusetts, USA; Prof. John S. Yudkin, Division of Medicine, University College London, London, UK; Dr. David Beran, Division of Tropical and Humanitarian Medicine, University of Geneva Faculty of Medicine, and Geneva University Hospitals, Geneva, Switzerland; Molly Lepeska, Health Action International, Amsterdam, The Netherlands; Dr. Margaret Ewen, Health Action International, Amsterdam, The Netherlands.
The review authors would like to thank Theresa Moore, Methodological Editor in the Cochrane Methods Support Unit, for her valuable comments to improve risk of bias evaluation with the new Cochrane risk of bias 2 (RoB 2) tool.
Thanks to Dr. Philip Home for replying to our request (Home 2005), We thank Dr. Parvaresh Rizi (employee in Novo Nordisk A/S) for replying to our request on Davies 2014. Thanks to Sanofi and Novo Nordisk for providing additional data. Thanks to Dr. Orchard for replying to our request (Orchard 2014). Thanks to Dr. Peter BANG for clarifying the status of the Basal Analog Study.
We would also like to acknowledge and thank the following people for their help in assessing the search results via Cochrane’s Screen4Me workflow: Nicole Edworthy, Susanna Wisniewski, Emmet Farragher, Ruth Suhami, Nicole Askin, Katarina Paunovic, Lenny Vasanthan, Sarah Bruch, Karen Ma, Therese Dalsbø, Louise Murphy, Stella Maria O'Brien, Danial Sayyad, Nikolaos Sideris, Fatai Momodu Akemokwe, Anna Noel‐Storr, Richard Tran, Hebatullah Abdulazeem, Esteban González, Gesiane Pajarinen, Leire Leache, Brian Duncan, Jessica Antretter, Sarah Jane Moll, Daniel Beales, Abhijit Dutta, Mohammed Deeb Zakkor, Tarig Fadalla, Abhijna Vithal Yergolkar, Chet Chaulagai, Charlotte Flahou, Ronak Paul, Niwanda Yogiswara, Ferdy Cayami, Georgina Johnstone.
Appendices
Appendix 1. Checklist to aid consistency and reproducibility of GRADE assessments: insulin detemir compared with NPH insulin
| Items | (1) All‐cause mortality | (2) Health‐related quality of life | (3) Severe hypoglycaemia | (4) Non‐fatal myocardial infarction/stroke | (5) Severe nocturnal hypoglycaemia | (6) Serious adverse events | (7) HbA1c | |
| Study limitations (risk of bias)a | Overall risk of bias | Low risk | Some concerns | Low risk | Low risk/not reported | Low risk | Low risk | Low risk |
| Inconsistencyb | Point estimates did not vary widely? | Yes | NA | No (↓) | NA | Yes | Yes | Yes |
| To what extent did confidence intervals overlap (substantial: all confidence intervals overlap at least one of the included studies point estimate; some: confidence intervals overlap but not all overlap at least one point estimate; no: at least one outlier: where the confidence interval of some of the studies do not overlap with those of most included studies)? | Substantial | Some | Substantial | Substantial | Some | |||
| Was the direction of effect consistent? | Yes | No (↓) | Yes | No (↓) | Yes | |||
| What was the magnitude of statistical heterogeneity (as measured by I²) ‐ low (I² < 40%), moderate (I² 40%‐60%), high I² > 60%)? | Low | Low | Moderate | Low | Low | |||
| Was the test for heterogeneity statistically significant (P < 0.1)? | Not statistically significant | Not statistically significant | Not statistically significant | Not statistically significant | Not statistically significant | |||
| Indirectness | Were the populations in included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable |
| Were the interventions in the included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | |
| Was the included outcome not a surrogate outcome? | Yes | Yes | Yes | Yes | Yes | Yes | No (↓) | |
| Was the outcome timeframe sufficient? | No (↓) | Yes | Yes | No (↓) | Yes | Yes | Yes | |
| Were the conclusions based on direct comparisons? | Yes | NA | Yes | Yes | Yes | Yes | Yes | |
| Imprecisionc | Was the confidence interval for the pooled estimate not consistent with benefit and harm? | Yes | NA | Yes | NA | No (↓) | Yes | No (↓) |
| What is the magnitude of the median sample size (high: 300 participants, intermediate: 100‐300 participants, low: < 100 participants)?e | High | High | High | High | High | High | High | |
| What was the magnitude of the number of included studies (large: > 10 studies, moderate: 5‐10 studies, small: < 5 studies)?e | Moderate | Small (↓) | Moderate | Small (↓) | Moderate | Moderate | Moderate | |
| Was the outcome a common event (e.g. occurs more than 1/100)? | No (↓) | NA | Yes | No (↓) | Yes | Yes | NA | |
| Publication biasd | Was a comprehensive search conducted? | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Was grey literature searched? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Were no restrictions applied to study selection on the basis of language? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| There was no industry influence on studies included in the review? | No | No | No | No | No | No | No | |
| There was no evidence of funnel plot asymmetry? | NA | NA | NA | NA | NA | NA | NA | |
| There was no discrepancy in findings between published and unpublished studies? | Yes | Unclear | No (↓) | Unclear | Yes | Yes | Yes | |
|
aRisk of bias was addressed by the Cochrane 'Risk of bias' 2 tool (RoB 2).
bQuestions on inconsistency are primarily based on visual assessment of forest plots and prediction intervals. cWhen judging the width of the confidence interval, it is recommended to use a clinical decision threshold to assess whether the imprecision is clinically meaningful. dQuestions address comprehensiveness of the search strategy, industry influence, funnel plot asymmetry and discrepancies between published and unpublished studies. eDepends on the context of the systematic review area. (↓): key item for potential downgrading the certainty of the evidence (GRADE) as shown in the footnotes of the 'Summary of finding' table(s). HbA1c: glycosylated haemoglobin A1c;NA: not applicable. | ||||||||
Appendix 2. Checklist to aid consistency and reproducibility of GRADE assessments: insulin glargine compared with NPH insulin
| Items | (1) All‐cause mortality | (2) Health‐related quality of life | (3) Severe hypoglycaemia | (4) Non‐fatal myocardial infarction/stroke | (5) Severe nocturnal hypoglycaemia | (6) Serious adverse events | (7) HbA1c | |
| Study limitations (risk of bias)a | Overall risk of bias | Low risk | Some concerns | Low risk | Low risk / low risk | Low risk | Low risk | Low risk |
| Inconsistencyb | Point estimates did not vary widely? | NA | Unclear | Yes | NA | Yes | No (↓) | Yes |
| To what extent did confidence intervals overlap (substantial: all confidence intervals overlap at least one of the included studies point estimate; some: confidence intervals overlap but not all overlap at least one point estimate; no: at least one outlier: where the confidence interval of some of the studies do not overlap with those of most included studies)? | Substantial | Substantial | Substantial | Some | Some | |||
| Was the direction of effect consistent? | Unclear | Yes | Yes | No (↓) | Yes | |||
| What was the magnitude of statistical heterogeneity (as measured by I²) ‐ low (I² < 40%), moderate (I² 40%‐60%), high I² > 60%)? | Low | Low | Low | High | Low | |||
| Was the test for heterogeneity statistically significant (P < 0.1)? | Not statistically significant | Not statistically significant | Not statistically significant | Statistically significant | Not statistically significant | |||
| Indirectness | Were the populations in included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable |
| Were the interventions in the included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | |
| Was the included outcome not a surrogate outcome? | Yes | Yes | Yes | Yes | Yes | Yes | No (↓) | |
| Was the outcome timeframe sufficient? | No (↓) | Yes | Yes | No (↓) | Yes | Yes | Yes | |
| Were the conclusions based on direct comparisons? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Imprecisionc | Was the confidence interval for the pooled estimate not consistent with benefit and harm? | NA | NA | No (↓) | NA | No (↓) | Yes | No (↓) |
| What is the magnitude of the median sample size (high: 300 participants, intermediate: 100‐300 participants, low: < 100 participants)?e | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | |
| What was the magnitude of the number of included studies (large: > 10 studies, moderate: 5‐10 studies, small: < 5 studies)?e | Small (↓) | Small (↓) | Moderate | Small (↓) | Moderate | Moderate | Moderate | |
| Was the outcome a common event (e.g. occurs more than 1/100)? | No (↓) | NA | Yes | No (↓) | Yes | Yes | NA | |
| Publication biasd | Was a comprehensive search conducted? | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Was grey literature searched? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Were no restrictions applied to study selection on the basis of language? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| There was no industry influence on studies included in the review? | No | No | No | No | No | No | No | |
| There was no evidence of funnel plot asymmetry? | NA | NA | NA | NA | NA | NA | NA | |
| There was no discrepancy in findings between published and unpublished studies? | Yes | Unclear | Yes | Unclear | Yes | Yes | Yes | |
|
aRisk of bias was addressed by the Cochrane 'Risk of bias' 2 tool (RoB 2).
bQuestions on inconsistency are primarily based on visual assessment of forest plots and prediction intervals. cWhen judging the width of the confidence interval, it is recommended to use a clinical decision threshold to assess whether the imprecision is clinically meaningful. dQuestions address comprehensiveness of the search strategy, industry influence, funnel plot asymmetry and discrepancies between published and unpublished studies. eDepends on the context of the systematic review area. (↓): key item for potential downgrading the certainty of the evidence (GRADE) as shown in the footnotes of the 'Summary of finding' table(s). HbA1c: glycosylated haemoglobin A1c;NA: not applicable. | ||||||||
Appendix 3. Checklist to aid consistency and reproducibility of GRADE assessments: insulin detemir compared with insulin glargine
| Items | (1) All‐cause mortality | (2) Health‐related quality of life | (3) Severe hypoglycaemia | (4) Non‐fatal myocardial infarction/stroke | (5) Severe nocturnal hypoglycaemia | (6) Serious adverse events | (7) HbA1c | |
| Study limitations (risk of bias)a | Overall risk of bias | Low risk | Not reported | Low risk | Low risk / low risk | Low risk | Low risk | Low risk |
| Inconsistencyb | Point estimates did not vary widely? | NA | No (↓) | NA | No (↓) | No (↓) | Yes | |
| To what extent did confidence intervals overlap (substantial: all confidence intervals overlap at least one of the included studies point estimate; some: confidence intervals overlap but not all overlap at least one point estimate; no: at least one outlier: where the confidence interval of some of the studies do not overlap with those of most included studies)? | Some | Substantial | Substantial | Substantial | ||||
| Was the direction of effect consistent? | No (↓) | No (↓) | Yes | Yes | ||||
| What was the magnitude of statistical heterogeneity (as measured by I²) ‐ low (I² < 40%), moderate (I² 40%‐60%), high I² > 60%)? | High | High | Low | Low | ||||
| Was the test for heterogeneity statistically significant (P < 0.1)? | Statistically significant | Not statistically significant | Not statistically significant | Not statistically significant | ||||
| Indirectness | Were the populations in included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | |
| Were the interventions in the included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | ||
| Was the included outcome not a surrogate outcome? | Yes | Yes | Yes | Yes | Yes | No (↓) | ||
| Was the outcome timeframe sufficient? | No (↓) | Yes | No (↓) | Yes | Yes | Yes | ||
| Were the conclusions based on direct comparisons? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Imprecisionc | Was the confidence interval for the pooled estimate not consistent with benefit and harm? | NA | No (↓) | NA | No (↓) | No (↓) | No (↓) | |
| What is the magnitude of the median sample size (high: 300 participants, intermediate: 100‐300 participants, low: < 100 participants)?e | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | ||
| What was the magnitude of the number of included studies (large: > 10 studies, moderate: 5‐10 studies, small: < 5 studies)?e | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | ||
| Was the outcome a common event (e.g. occurs more than 1/100)? | Yes (↓) | Yes | Yes (↓) | Yes | Yes | NA | ||
| Publication biasd | Was a comprehensive search conducted? | Yes | Yes | Yes | Yes | Yes | Yes | |
| Was grey literature searched? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Were no restrictions applied to study selection on the basis of language? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| There was no industry influence on studies included in the review? | No | No | No | No | No | No | ||
| There was no evidence of funnel plot asymmetry? | NA | NA | NA | NA | NA | NA | ||
| There was no discrepancy in findings between published and unpublished studies? | Unclear | No (↓) | NA | Yes | NA | Yes | ||
|
aRisk of bias was addressed by the Cochrane 'Risk of bias' 2 tool (RoB 2).
bQuestions on inconsistency are primarily based on visual assessment of forest plots and prediction intervals. cWhen judging the width of the confidence interval, it is recommended to use a clinical decision threshold to assess whether the imprecision is clinically meaningful. dQuestions address comprehensiveness of the search strategy, industry influence, funnel plot asymmetry and discrepancies between published and unpublished studies. eDepends on the context of the systematic review area. (↓): key item for potential downgrading the certainty of the evidence (GRADE) as shown in the footnotes of the 'Summary of finding' table(s). HbA1c: glycosylated haemoglobin A1c;NA: not applicable. | ||||||||
Appendix 4. Checklist to aid consistency and reproducibility of GRADE assessments: insulin degludec compared with insulin detemir
| Items | (1) All‐cause mortality | (2) Health‐related quality of life | (3) Severe hypoglycaemia | (4) Non‐fatal myocardial infarction/stroke | (5) Severe nocturnal hypoglycaemia | (6) Serious adverse events | (7) HbA1c | |
| Study limitations (risk of bias)a | Overall risk of bias | Low risk | Some concerns | Low risk | Low risk / low risk | Low risk | Low risk | Low risk |
| Inconsistencyb | Point estimates did not vary widely? | NA | NA | Yes | NA | Yes | Yes | Yes |
| To what extent did confidence intervals overlap (substantial: all confidence intervals overlap at least one of the included studies point estimate; some: confidence intervals overlap but not all overlap at least one point estimate; no: at least one outlier: where the confidence interval of some of the studies do not overlap with those of most included studies)? | Substantial | Substantial | Substantial | Some | ||||
| Was the direction of effect consistent? | Yes | Yes | Yes | Yes | ||||
| What was the magnitude of statistical heterogeneity (as measured by I²) ‐ low (I² < 40%), moderate (I² 40%‐60%), high I² > 60%)? | Low | Low | Low | Low | ||||
| Was the test for heterogeneity statistically significant (P < 0.1)? | Not statistically significant | Not statistically significant | Not statistically significant | Not statistically significant | ||||
| Indirectness | Were the populations in included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable |
| Were the interventions in the included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | |
| Was the included outcome not a surrogate outcome? | Yes | Yes | Yes | Yes | Yes | Yes | No (↓) | |
| Was the outcome timeframe sufficient? | No (↓) | Yes | Yes | No (↓) | Yes | Yes | Yes | |
| Were the conclusions based on direct comparisons? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Imprecisionc | Was the confidence interval for the pooled estimate not consistent with benefit and harm? | NA | NA | No (↓) | NA | No (↓) | No (↓) | No (↓) |
| What is the magnitude of the median sample size (high: 300 participants, intermediate: 100‐300 participants, low: < 100 participants)?e | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | |
| What was the magnitude of the number of included studies (large: > 10 studies, moderate: 5‐10 studies, small: < 5 studies)?e | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | |
| Was the outcome a common event (e.g. occurs more than 1/100)? | No (↓) | NA | Yes | NA | Yes | Yes | NA | |
| Publication biasd | Was a comprehensive search conducted? | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Was grey literature searched? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Were no restrictions applied to study selection on the basis of language? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| There was no industry influence on studies included in the review? | No | No | No | No | No | No | No | |
| There was no evidence of funnel plot asymmetry? | NA | NA | NA | NA | NA | NA | NA | |
| There was no discrepancy in findings between published and unpublished studies? | NA | NA | NA | NA | Yes | NA | Yes | |
|
aRisk of bias was addressed by the Cochrane 'Risk of bias' 2 tool (RoB 2).
bQuestions on inconsistency are primarily based on visual assessment of forest plots and prediction intervals. cWhen judging the width of the confidence interval, it is recommended to use a clinical decision threshold to assess whether the imprecision is clinically meaningful. dQuestions address comprehensiveness of the search strategy, industry influence, funnel plot asymmetry and discrepancies between published and unpublished studies. eDepends on the context of the systematic review area. (↓): key item for potential downgrading the certainty of the evidence (GRADE) as shown in the footnotes of the 'Summary of finding' table(s). HbA1c: glycosylated haemoglobin A1c;NA: not applicable. | ||||||||
Appendix 5. Checklist to aid consistency and reproducibility of GRADE assessments: insulin degludec compared with insulin glargine
| Items | (1) All‐cause mortality | (2) Health‐related quality of life | (3) Severe hypoglycaemia | (4) Non‐fatal myocardial infarction/stroke | (5) Severe nocturnal hypoglycaemia | (6) Serious adverse events | (7) HbA1c | |
| Study limitations (risk of bias)a | Overall risk of bias | Low risk | Some concerns | Low risk | Low risk / low risk | Low risk | Low risk | Low risk |
| Inconsistencyb | Point estimates did not vary widely? | Yes | Yes | Yes | NA | Yes | Yes | Yes |
| To what extent did confidence intervals overlap (substantial: all confidence intervals overlap at least one of the included studies point estimate; some: confidence intervals overlap but not all overlap at least one point estimate; no: at least one outlier: where the confidence interval of some of the studies do not overlap with those of most included studies)? | Substantial | Some | Substantial | Substantial | Substantial | Some | ||
| Was the direction of effect consistent? | No (↓) | No (↓) | Yes | Yes | Yes | Yes | ||
| What was the magnitude of statistical heterogeneity (as measured by I²) ‐ low (I² < 40%), moderate (I² 40%‐60%), high I² > 60%)? | Low | Low | Low | Low | Low | Low | ||
| Was the test for heterogeneity statistically significant (P < 0.1)? | Not statistically significant | Not statistically significant | Not statistically significant | Not statistically significant | Not statistically significant | Not statistically significant | ||
| Indirectness | Were the populations in included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable |
| Were the interventions in the included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | |
| Was the included outcome not a surrogate outcome? | Yes | Yes | Yes | Yes | Yes | Yes | No (↓) | |
| Was the outcome timeframe sufficient? | No (↓) | Yes | Yes | No (↓) | Yes | Yes | Yes | |
| Were the conclusions based on direct comparisons? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Imprecisionc | Was the confidence interval for the pooled estimate not consistent with benefit and harm? | Yes | Yes | No (↓) | NA | No (↓) | Yes | No (↓) |
| What is the magnitude of the median sample size (high: 300 participants, intermediate: 100‐300 participants, low: < 100 participants)?e | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | Intermediate | |
| What was the magnitude of the number of included studies (large: > 10 studies, moderate: 5‐10 studies, small: < 5 studies)?e | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | |
| Was the outcome a common event (e.g. occurs more than 1/100)? | No (↓) | NA | Yes | No (↓) | Yes | Yes | NA | |
| Publication biasd | Was a comprehensive search conducted? | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Was grey literature searched? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Were no restrictions applied to study selection on the basis of language? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| There was no industry influence on studies included in the review? | No | No | No | No | No | No | No | |
| There was no evidence of funnel plot asymmetry? | NA | NA | NA | NA | NA | NA | NA | |
| There was no discrepancy in findings between published and unpublished studies? | Unclear | Unclear | NA | NA | Yes | NA | Yes | |
|
aRisk of bias was addressed by the Cochrane 'Risk of bias' 2 tool (RoB 2).
bQuestions on inconsistency are primarily based on visual assessment of forest plots and prediction intervals. cWhen judging the width of the confidence interval, it is recommended to use a clinical decision threshold to assess whether the imprecision is clinically meaningful. dQuestions address comprehensiveness of the search strategy, industry influence, funnel plot asymmetry and discrepancies between published and unpublished studies. eDepends on the context of the systematic review area. (↓): key item for potential downgrading the certainty of the evidence (GRADE) as shown in the footnotes of the 'Summary of finding' table(s). HbA1c: glycosylated haemoglobin A1c;NA: not applicable. | ||||||||
Appendix 6. Search strategies
| Cochrane Central Register of Controlled Trials (Cochrane Register of Studies Online) |
| 1. MESH DESCRIPTOR Insulin Glargine 2. glargin*:TI,AB,KY 3. ("2ZM8CX04RZ" OR "160337‐95‐1"):TI,AB,KY 4. (lantus* or basaglar* or abasaglar* or abasria* or t?ujeo* or optisulin* or suliqua* or soliqua* or solostar* or lusduna* or nexvue* or basalin* or bonglixan* or basalog* or vibrenta* or glaritus* or basagin* or glarine* or semglee*):TI,AB,KY 5. ("HOE 901" or HOE901 or "HOE 71GT" or "HOE71GT" or "LY 2963016"):TI,AB,KY 6. (gly?A21 OR A21gly* OR (gly* ADJ1 A21)):TI,AB,KY 7. (arg?B31 OR B31arg* OR (arg* ADJ1 B31)):TI,AB,KY 8. (arg?B32 OR B32?arg* OR (arg* ADJ1 B32)):TI,AB,KY 9. ("MK‐1293" or "MK1293"):TI,AB,KY 10. #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 11. MESH DESCRIPTOR Insulin Detemir 12. detemir*:TI,AB,KY 13. ("169148‐63‐4" or "4FT78T86XV"):TI,AB,KY 14. levemir*:TI,AB,KY 15. (lys?B29 OR B29lys* OR (lys* ADJ1 B29)):TI,AB,KY 16. (ala?B30 OR B30ala* OR (ala* ADJ1 B30)):TI,AB,KY 17. ("NN 304" OR NN304):TI,AB,KY 18. #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 19. degludec:TI,AB,KY 20. ("844439‐96‐9" or "54Q18076QB"):TI,AB,KY 21. (tresiba OR ryzodeg OR xultrophy):TI,AB,KY 22. (B29N* OR (29B ADJ1 N6)):TI,AB,KY 23. ("NN 1250" OR NN1250):TI,AB,KY 24. #19 OR #20 OR #21 OR #22 OR #23 25. #10 OR #18 OR #24 26. MESH DESCRIPTOR Diabetes Mellitus EXPLODE ALL TREES 27. diabet*:TI,AB,KY 28. (IDDM OR MODY OR NIDDM OR T1D* OR T2D*):TI,AB,KY 29. #26 OR #27 OR #28 30. #25 AND #29 |
| MEDLINE (Ovid) |
|
[Glargine insulin and biosimilars] 1. Insulin Glargine/ 2. glargin*.mp. 3. ("2ZM8CX04RZ" or "160337‐95‐1").mp. 4. (lantus* or basaglar* or abasaglar* or abasria* or t?ujeo* or optisulin* or suliqua* or soliqua* or solostar* or lusduna* or nexvue* or basalin* or bonglixan* or basalog* or vibrenta* or glaritus* or basagin* or glarine* or semglee*).mp. 5. ("HOE 901" or HOE901 or "HOE 71GT" or "HOE71GT" or "LY 2963016").mp. 6. (gly?A21 or A21gly* or (gly* adj1 A21)).mp. 7. (arg?B31 or B31arg* or (arg* adj1 B31)).mp. 8. (arg?B32 or B32?arg* or (arg* adj1 B32)).mp. 9. ("MK‐1293" or "MK1293").mp. 10. or/1‐9 [Detemir insulin] 11. Insulin Detemir/ 12. detemir*.mp. 13. ("169148‐63‐4" or "4FT78T86XV").mp. 14. levemir*.mp. 15. (lys?B29 or B29lys* or (lys* adj1 B29)).mp. 16. (ala?B30 or B30ala* or (ala* adj1 B30)).mp. 17. (NN 304 or NN304).mp. 18. or/11‐17 [Degludec insulin] 19. degludec*.mp. 20. ("844439‐96‐9" or "54Q18076QB").mp. 21. (tresiba* or ryzodeg or xultrophy).mp. 22. (B29N* or (29B adj1 N6)).mp. 23. (NN 1250 or NN1250).mp. 24. or/19‐23 25. 10 or 18 or 24 [ Condition: diabetes ] 26. exp Diabetes Mellitus/ 27. diabet*.mp. 28. (IDDM or T1D* or NIDDM or T2D* or MODY).tw. 29. or/26‐28 [ Combination of intervention and population ] 30. 25 and 29 [Cochrane Handbook 2019 RCT filter, sensitivity max version (Lefebvre 2019)] 31. randomized controlled trial.pt. 32. controlled clinical trial.pt. 33. randomi?ed.ab. 34. placebo.ab. 35. drug therapy.fs. 36. randomly.ab. 37. trial.ab. 38. groups.ab. 39. or/31‐38 40. exp animals/ not humans/ 41. 39 not 40 [ “Phase 3” filter (Cooper 2019)] 42. Clinical Trial, Phase III/ 43. ("phase 3" or "phase3" or p3 or "pIII").ti,ab,kw. 44. 42 or 43 [ RCT or "phase 3" filter ] 45. 41 or 44 [ Combination of intervention, population and filters ] 46. 30 and 45 |
| WHO ICTRP Search Portal (Standard search) |
| glargine AND diabet* OR levemir AND diabet* OR detemir AND diabet* OR degludec AND diabet* |
| ClinicalTrials.gov (Expert search) |
| (glargine OR lantus OR basaglar OR abasaglar OR abasria OR toujeo OR tujeo OR optisulin OR soliqua OR suliqua OR solostar OR lusduna OR nexvue OR basalin OR bonglixan OR basalog OR vibrenta OR glaritus OR basagin OR glarine OR semglee OR "HOE 901" OR HOE901 OR "HOE 71GT" OR HOE71GT OR "LY 2963016" OR MK‐1293 OR MK1293 OR detemir OR levemir OR "NN 304" OR NN304 OR degludec OR tresiba OR ryzodeg OR xultrophy OR "NN 1250" OR NN1250) [TREATMENT] AND EXACT "Interventional" [STUDY‐TYPES] AND ( diabetes OR diabetic OR IDDM OR MODY OR NIDDM OR T1DM OR T2DM OR T1D OR T2D ) [DISEASE] |
| HTA database |
| (glargine) OR (levemir) OR (detemir) OR (degludec) |
Appendix 7. Overview of sources of unpublished additional data
| Study ID (Trial ID) | Accessible pages from clinical study report | Accessible pages from clinical study synopsis | Accessible pages from EMA | Accessible pages from FDA |
|
Bartley 2008 (NN304‐1595) |
731 No appendices |
5 | — | — |
|
BEGIN Basal‐Bolus Type 1 (NN1250‐3583) |
2581 (+3564 CSR pages of extension trial NN1250‐3644) No appendices |
17 | 134 | 419 |
|
BEGIN Flex T1 (NN1250‐3770) |
1675 (+ 2212 CSR pages of extension trial NN1250‐3770‐ext) No appendices |
9 (12 synopsis of main trial period + extension trial period) | 134 | 559 |
|
BEGIN Young (NN1250‐3561) |
1914 (+ 3350 CSR pages of extension trial NN1250‐3561) No appendices |
16 | 81 | 559 |
| Bolli 2009 | — | — | — | — |
|
Chase 2008 (HOE901/4030) |
150 No end‐of‐text tables, no appendices (additional 4182+ pages) |
7 | — | — |
|
Davies 2014 (NN1250‐3585) |
1645 (+ 2086 CSR pages of extension trial NN1250‐3725) No appendices |
16 | 134 | 419 |
|
Fulcher 2005 (HOE901/4010) |
127 No summary tables, no appendices |
8 | — | — |
|
Heller 2009 (NN304‐1430) |
386 No appendices |
8 | — | — |
|
Home 2005 (HOE901/3001) |
317 (+ 40 CSR pages on health‐related quality of life; + 342 CSR pages on health economics) No appendices |
3 | 34 | |
|
Kobayashi 2007 (NN304‐1476) |
3 Translated pages |
8 | — | — |
|
Liu 2016 (HOE901; EFC11681) |
154 No appendices |
9 | 21 | — |
|
NCT00595374 (NN304‐1582) |
— | 4 | — | — |
|
NCT00605137 (NN304‐1604) |
4 (+ 80 CSR protocol pages) Translated pages |
6 | — | — |
|
Pieber 2007 (NN304‐1372) |
97 No appendices |
4 | — | 145 |
| Porcellati 2004 | — | — | — | — |
|
PRESCHOOL (HOE901; EFC11202) |
188 No appendices |
7 | 36 | — |
|
Ratner 2000 (HOE301/3004) |
331 No appendices, some tables ("participant listing") missing (additional 11.990+ pages) |
5 | — | 34 |
|
Robertson 2007 (NN304‐1379) |
647 (+ 653 CSR pages on extension trial NN304‐1690) No appendices |
5 | — | 11 |
|
Russell‐Jones 2004 (NN304‐1335) |
314 No appendices |
5 | 29 | 145 |
|
Schober 2002 (HOE901/3003) |
330 (+ 196 CSR pages on health economics) No appendices (additional 7087+ pages) |
3 | — | 34 |
|
Standl 2004 (NN304‐1181) |
108 No end‐of‐text tables, no end‐of‐text figures, no selected listings, no appendices |
5 | 29 | 145 |
|
SWITCH 1 (NN1250‐3995) |
3042 No appendices |
9 | — | 559 |
|
Thalange 2013 (NN304‐1689) |
1055 (+653 CSR pages of extension trial NN304‐1690) No appendices |
7 | 38 | — |
| Urakami 2017 | — | — | — | — |
|
Vague 2003 (NN304‐1205) |
256 No end‐of‐text tables, no end‐of‐text figures, no selected listings, no appendices |
5 | 29 | 145 |
| —: indicates source not available CSR: clinical study report; EMA: European Medicines Agency; FDA: Food and Drug Administration. | ||||
Appendix 8. Description of interventions
| Bartley 2008 | Intervention | Description |
| Interventiona | I: detemir | Once daily at any time during the evening (Levemir®, Novo Nordisk A/S, Bagsvaerd, Denmark 100 U/mL), administered in the thigh, sc, a second basal insulin dose could be added in the morning |
| C: NPH | Once daily NPH at any time during the evening (Insulatard®, Novo Nordisk A/S, 100 U/mL), administered in the thigh, sc, a second basal insulin dose could be added in the morning | |
| Titration period | Assuming 12 weeks ("During the first 12 weeks, patients were in weekly contact with the investigator or research team") | |
| Strength of insulin | Based in titration regimen, then 1 U detemir = 1 unit NPH | |
| Rapid‐acting insulin | Aspart (NovoRapid®, Novo Nordisk A/S, 100 U/mL) was injected immediately before each main meal, administered in the abdomen. Aspart was titrated according to local practice to achieve a post‐prandial PG level ≤ 9.0 mmol/L | |
| Glycaemic targets | Basal insulin was titrated aiming for a PG target ≤ 6.0 mmol/L before breakfast and dinner; post‐prandial glucose < 10 mmol/L; BG 2:00‐4:00 4‐7 mmol/L | |
| Interval of blood glucose measurement | Participants were asked to measure PG pre‐breakfast and pre‐dinner on three consecutive days prior to each contact | |
| Calibration of blood glucose measurement device | Participants were instructed in the use and calibration of blood glucose meters | |
| Adjusting insulin doses | Patients transferred from a once daily basal insulin regimen started treatment with detemir or NPH at an identical number of units, while those transferred from a twice‐daily regimen initiated treatment at 70% of the previous total daily basal insulin dose. If it was necessary to add more than once daily insulin dose, then the additional basal morning dose was initiated at 4 U and titrated according to the same algorithm as used for the evening dose. Algorithm: FPG or pre‐evening dinner meal: Insulin adjustment > 15 mmol/L +6 U 10.1–15.0 mmol/L +4 U 6.1–10.0 mmol/L +2 U ≤ 6.0 mmol/L no adjustment If one SMPG measurement: 3.1–4.0 mmol/L −2 U < 3.1 mmol/L −4 U If the FPG target was achieved while pre‐dinner PG values remained above target, the basal evening dose could be increased as long as nocturnal hypoglycaemia did not occur. A second basal insulin dose could be added in the morning if the pre‐dinner PG target was not achieved with use of the algorithm and after optimisation of bolus insulin. |
|
| Interval for insulin adjustments | After the first 12 weeks, weekly contact between the investigators and the participants. A central surveillance committee reviewed the PG concentrations and the prescribed basal insulin doses throughout the study | |
| Other concomitant intervention | None | |
| BEGIN Basal‐Bolus Type 1 | Intervention | Description |
| Intervention | I: degludec | Once daily with main evening meal, 100 U/mL, sc, 3 mL FlexPen®, insulin and insulin pen manufactured by Novo Nordisk, Bagsværd, Denmark, sc, abdomen or deltoid or thigh |
| C: glargine | Lantus ®, Once daily at any time, 100 U/mL, sc, 3 mL SoloStar®, Sanofi, Paris, France, sc, abdomen or deltoid or thigh | |
| Titration period | None | |
| Strength of insulin | If previous basal insulin was used once daily, initial doses were replaced with insulin degludec or insulin glargine in a 1:1 ratio. If more than one daily dose had been taken, the total daily basal dose was calculated and replaced with insulin degludec in a 1:1 ratio, with the recommendation that the dose be reduced by 20% to 30% for participants in the insulin glargine group, and administered once daily | |
| Rapid‐acting insulin | Insulin aspart before each meal (NovoRapid/NovoLog®, 100 U/mL, subcutaneously, 3 mL FlexPen®, Novo Nordisk, Bagsvaerd, Denmark). Additional doses were allowed with a fourth meal and snacks | |
| Glycaemic targets | Pre‐breakfast plasma glucose values of 3.9–4.9 mmol/L | |
| Interval of blood glucose measurement | Measurements before breakfast, lunch, main evening meal and bedtime. Measurements were preferably performed on 3 consecutive days just before each scheduled visit or telephone contact using the glucose meter provided. a 9‐point profile with an additional 4‐point profile on the 3 days immediately before some predefined visits | |
| Calibration of blood glucose measurement device | Glucose meter and instructions for use and calibration for measurement | |
| Adjusting insulin doses | Changes to basal insulin were recommended before changes to the bolus insulin were considered | |
| Interval for insulin adjustments | Basal insulin: Pre‐breakfast plasma glucose (mmol/L) (footnote: mean of 3 measures before visit) and adjustment of insulin dose < 3.1 Insulin dose: –4 (If dose > 45U, reduce by 10%) 3.1‐3.8 Insulin dose: –2 (If dose > 45U, reduce by 5%) 3.9 ‐< 5.0 Insulin dose: 0 5‐9.9 Insulin dose: +2 10‐14.9 Insulin dose: +4 ≥ 15.0 Insulin dose: +6 Titration basal bolus: Pre‐prandial/bedtime PG and adjustment of insulin aspart 3.9 −< 5.0 Insulin dose: 0 5.0–7.9 Insulin dose: +2 8.0–9.9 Insulin dose: +3 ≥ 10.0 Insulin dose: +4 |
|
| Other concomitant intervention | None | |
| BEGIN Flex T1 | Intervention | Description |
| Intervention | I: degludec | Once daily with evening meal, 100 U/mL, 3 mL FlexPen®; Novo Nordisk, Bagsvaerd, Denmark, sc (abdomen or deltoid or thigh) |
| C: glargine | Once daily, Lantus®, 100 U/mL, 3 mL SoloStar®, Sanofi, Paris, France, sc (abdomen or deltoid or thigh) | |
| Titration period | None | |
| Strength of insulin | If once daily regimen, then prescribe same number of units. If prior basal insulin was taken more than once daily, then dose of glargine was reduced by 20% to 30% and degludec reduction based on the investigators decision | |
| Rapid‐acting insulin | Insulin aspart, three‐times daily or more | |
| Glycaemic targets | Basal: pre‐breakfast SMPG target of 4.0–5.0 mmol/L; mean premeal SMPG: a mean premeal SMPG target of less than 5.0 mmol/L | |
| Interval of blood glucose measurement | Daily | |
| Calibration of blood glucose measurement device | Glucose measurements were performed with drawn capillary blood automatically calibrated to plasma‐equivalent glucose values | |
| Adjusting insulin doses | Titration of basal insulin Previous days’ mean pre‐breakfast SMPG (mmol/L) and insulin adjustment (U) < 4.0 Insulin dose: ‐2 4.0–5.0 Insulin dose: 0 > 5.0 Insulin dose: +2 Titration of bolus insulin Pre‐prandial (mmol/L) and titration of insulin aspart < 5.0 Insulin dose: 0 5.0‐8.0 Insulin dose: +2 8.0‐10.0 Insulin dose: +3 ≥ 10 Insulin dose: +4 |
|
| Interval for insulin adjustments | Self‐adjustment of basal insulin dose was to be performed three‐times weekly (Monday, Wednesday, Friday) based on daily pre‐breakfast SMPG | |
| Other concomitant intervention | None | |
| BEGIN Young | Intervention | Description |
| Intervention | I: degludec | Once daily (approximately same time of the day), 100U/mL, Penfill® 3‐mL cartridge, Novo Nordisk, Bagsværd, Denmark, sc |
| C: detemir | Once or twice daily (approximately same tome of the day), 100U/mL,Penfill®3‐mLcartridge; Novo Nordisk, sc | |
| Titration period | — | |
| Strength of insulin | Participants were to continue on the previous dose of basal insulin if randomised to detemir. Detemir doses were consistently higher than degludec doses | |
| Rapid‐acting insulin | Insulin aspart at meals, 100 U/ml 3 ml Penfill® cartridge. It was aiming for a basal:bolus ratio of between 50:50 and 30:70. The choice of basal:bolus split for each participant was made at the discretion of the investigator | |
| Glycaemic targets | Pre‐breakfast SMPG target of 5–8 mmol/L | |
| Interval of blood glucose measurement | Daily (morning, premeal and evening). Four‐point profiles were performed weekly and 8‐point profiles were performed at randomisation, 12, 26, 38 and 52) | |
| Calibration of blood glucose measurement device | Glucose meters calibrated to plasma values | |
| Adjusting insulin doses | Basal insulin titration was based on the lowest pre‐breakfast SMPG value, on the 3 days prior to each weekly visit/phone contact Current basal dose < 5U 5–15U > 15U Pre‐breakfast or pre‐dinner PG (mmol/L) Adjustment (U) < 5 −1/2 −1 −2 5.0–8.0 0 0 0 8.1–10.0 +0.5 +1 +2 10.1–15.0 +1 +2 +4 > 15.0 +1.5 +3 +6 Current bolus dose ≤ 5U > 5U Lowest pre‐meal or bedtime PG (mmol/L) Adjustment (U) < 5.0 −1 −2 5.0–8.0 0 0 8.1–10.0 +0.5 +1 10.1–15.0 +1 +2 > 15.0 +1.5 +3 |
|
| Interval for insulin adjustments | Weekly | |
| Other concomitant intervention | None | |
| Bolli 2009b | Intervention | Description |
| Intervention | I: glargine | Glargine (Lantus, SanofieAventis) once daily at dinner time by means of pen device (OptiPen pro 1®) |
| C: NPH | NPH (Humulin I, Eli Lilly and Co.) twice (or more) daily (bedtime and lunchtime) by pen (Humapen Lilly®) | |
| Titration period | — | |
| Strength of insulin | — | |
| Rapid‐acting insulin | Lispro | |
| Glycaemic targets | FBG target value 5.0‐6.7 mmol/L; NPH pre‐dinner BG 5.0‐6.7 mmol/L | |
| Interval of blood glucose measurement | During the last 2 weeks before the scheduled visits, participants measured BG 2 hours after meals and at 3 a.m., in addition to FBG and pre‐prandial BG to provide 7‐point BG profile | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses |
Long‐acting insulin Dinnertime glargine and bedtime NPH were titrated to achieve the FBG target value 5.0‐6.7 mmol/L, but avoiding nocturnal hypoglycaemia. The lunchtime dose of NPH was adjusted to a target pre‐dinner 5.0‐6.7 mmol/L Bolus insulin The dose of lispro was adjusted to a target post‐prandial BG of < 7.8 mmol/L. Additional doses (1 or 2 U) of lispro were also used to correct unexpected hyperglycaemia |
|
| Interval for insulin adjustments | — | |
| Other concomitant intervention | None | |
| Chase 2008 | Intervention | Description |
| Intervention | I: glargine | Once daily, sc, before breakfast, 10 mL vial (1 mL contains 100 U) |
| C: NPH | Twice daily, sc, before breakfast and in the evening, 10 mL vial (1 mL contains 100 U)c | |
| Titration period | — | |
| Strength of insulin | Anticipated to be 1 U glargine = 1 U NPH. The starting doses of basal insulin were determined by the investigator | |
| Rapid‐acting insulin | Insulin lispro, sc, before each meal based on insulin:carbohydrate ratio and correction factor (proactive sliding scale), 10 mL vial (1 mL contains 100 IU) and 3 mL pen cartridges | |
| Glycaemic targets | FPG between 3.9 ‐ 5.6 mmol/L | |
| Interval of blood glucose measurement | CGMS applied to most participantsd. Everyday (FBG, pre‐prandial and bedtime SMBG) | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | 40%‐50% of the total daily dose of insulin was basal insulin and 50%‐60% of the total daily dose was bolus insulin. The total daily dose of insulin glargine and the evening dose of NPH/Lente were titrated weekly by the investigator to achieve FPG between 3.9 ‐ 5.6 mmol/L. The pre‐breakfast dose of NPH was titrated based on the investigator's clinical judgement. The weekly increase in the insulin dose could be divided across 2 or more incremental doses over the course of the week at the investigator's discretion | |
| Interval for insulin adjustments | Basal dose changes were made at scheduled study visits, titration contacts (weekly) or in the event of unexplained hypoglycaemia | |
| Other concomitant intervention | None | |
| Davies 2014 | Intervention | Description |
| Intervention | I: degludec | Once daily (between evening meal and bedtime), FlexPen®, sc (abdomen or deltoid or thigh), 100 U/mL, 3 mL |
| C: detemir | Once daily (between evening meal and bedtime, an additional morning dose could be added) FlexPen®. sc (abdomen or deltoid or thigh), 100 U/mL, 3 mL | |
| Titration period | Not reported, but optimisation of basal insulin dose was to be prioritised the first 8 weeks of the study | |
| Strength of insulin | 1 U of degludec was estimated to have the same BG lowering activity as 1 U detemir. If basal insulin was taken in a once daily regimen prior to the study, the same number of units once daily was prescribed. If basal insulin was taken more than once daily prior to the study, the total daily basal dose was calculated and transferred 1:1 as the once daily starting dose for both degludec and detemir | |
| Rapid‐acting insulin | Insulin aspart was administered immediately prior to breakfast, lunch and dinner, and an additional dose was permitted to cover an additional meal/snack. The dose of insulin aspart was adjusted weekly based on the mean of three self measured pre‐prandial PG values | |
| Glycaemic targets | On the basis of pre‐breakfast SMBG (mean value from 3 consecutive days), insulins were titrated individually once a week to a glucose of 3.9–4.9 mmol/L Criteria according for splitting detemir doses in two also pre‐dinner: plasma glucose > 6.0mmol/L |
|
| Interval of blood glucose measurement | — | |
| Calibration of blood glucose measurement device | All capillary blood measurements were calibrated to plasma‐equivalent glucose values (SMPG), using the plasma glucose meter and documented by the participant | |
| Adjusting insulin doses |
Titration algorithm for basal insulin < 3.1 mmol/L Insulin dose: decrease by 4 U 3.1–3.8 mmol/L Insulin dose: decrease by 2 U 3.9–4.9 mmol/L Insulin dose: no adjustment 5.0–9.9 mmol/L Insulin dose: increase by 2 U 10.0–14.9 mmol/L Insulin dose: increase by 4 U ≥ 15.0 mmol/L Insulin dose: increase by 6 U In the insulin detemir group, a second detemir dose could be added if there was inadequate glycaemic control after ≥ 8 weeks of treatment (defined as < 0.5%‐point improvement in HbA1c (participants with baseline HbA1c ≥ 8.0% or any deterioration of HbA1c; participants with baseline HbA1c < 8.0% in conjunction with a mean pre‐dinner PG > 6.0 mmol/L and no diagnosis of a treatable concurrent disease causing hyperglycaemia) Titration algorithm for bolus insulin ‐ pre‐prandial plasma glucose < 5.0 mmol/L Insulin dose: no adjustment 5.0–7.9 mmol/L Insulin dose: increase by 2 U 8.0–9.9 mmol/L Insulin dose: increase by 3 U ≥ 10.0 mmol/L Insulin dose: increase by 4 U |
|
| Interval for insulin adjustments | Once a week | |
| Other concomitant intervention | None | |
| Fulcher 2005 | Intervention | Description |
| Intervention | I: glargine | Once daily at bedtime (10 p.m.), sc, delivered by OptiPen Pro® device, cartridge containing 3 mL (1mL contains 100 IU), Aventis Pharma |
| C: NPH | Once daily at bedtime (10 p.m.), sc, delivered by HumaPen® device, cartridge containing 3 mL (1mL contains 100 IU), Eli Lilly | |
| Titration period | 6 weeks | |
| Strength of insulin | Based in titration regimen, then 1 U glargine = 1 unit NPH | |
| Rapid‐acting insulin | Lispro (before meals) | |
| Glycaemic targets | Targets were as follows: FBG = 5.5 mmol/L, pre‐prandial BG 3.9–6.7 mmol/L, 2‐h post‐prandial BG <8 mmol/L and 3 a.m. BG >3.6 mmol/L | |
| Interval of blood glucose measurement | Not explicit stated, but mentioned that targets were as follows: FBG = 5.5 mmol/L, pre‐prandial BG 3.9–6.7 mmol/L, 2‐h post‐prandial BG < 8 mmol/L and 3 a.m. BG > 3.6 mmol/L, then 7 times a day | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | Basal insulin dose adjustments were made twice weekly during the titration phase and fortnightly in the treatment follow‐up phase based on FBG measurements. Initiation dose: decided by the investigator > 7.7 mmol/L Insulin dose: increased by 4–6 IU 6.6–7.7 mmol/L Insulin dose: increased by 2–4 IU 5.5–6.6 mmol/L Insulin dose: increased by 2 IU All glycaemic measures should be for at least one of the two consecutive days before the visit, no episodes of severe hypoglycaemia or an FBG or overnight BG of = 3.6 mmol/L |
|
| Interval for insulin adjustments | Twice weekly (during titration phase), thereafter every second week | |
| Other concomitant intervention | None | |
| Heller 2009 | Intervention | Description |
| Intervention | I: detemir | Detemir, 100 U/mL (2400 nmol/mL) FlexPen®, initially administered once daily (in the evening). If patients in the detemir arm were achieving the PG target before breakfast but not before dinner, a second daily dose (initially 4 U) administered in the morning was added to the usual evening dose |
| C: glargine | Glargine, 100 U/mL (600 nmol/mL) in 3 mL cartridges in Europe and in 10 mL vials in the United States, initially administered once daily (in the evening). In the glargine arm, the dose was administered once daily regardless of the pre‐dinner PG measurement | |
| Titration period | — | |
| Strength of insulin | Based in titration regimen, then 1 U detemir = 1 U glargine. If pre‐study basal insulin was administered more than once daily, the total daily basal insulin dose was reduced by 30% | |
| Rapid‐acting insulin | NovoRapid® (insulin aspart), 100 U/mL FlexPen® 3 mL solution for injection in a pre‐filled pen (Novo Nordisk, Denmark). The dose was individually titrated and administered as subcutaneous injections | |
| Glycaemic targets | PG target of ≤ 6.0 mmol/L before breakfast and dinner, with no episodes of significant hypoglycaemia. Post‐prandial PG target ≤ 9.0 mmol/L |
|
| Interval of blood glucose measurement | Patients measured their FPG before breakfast and dinner on the 3 days before each study visit using standard glucose meters and test strips calibrated to PG levels. All patients were asked to record a 10‐point SMPG profile on a typical day during the weeks before the randomisation visit, the 24‐week visit, and the 52‐week visit | |
| Calibration of blood glucose measurement device | Yes | |
| Adjusting insulin doses | If the pre‐trial basal insulin had been administered more frequently, the total daily basal insulin dose was reduced by 30% and given once daily, followed by dose titration Mean pre‐breakfast PG values were used for titration of the evening dose; mean pre‐dinner PG values were used for titration of the morning dose Mean PG change in basal insulin dose (without significant hypoglycaemia) Target: ≤ 6.0 mmol/L (≤ 108 mg/dL) Insulin dose: no adjustment 6.1–10.0 mmol/L (109–180 mg/dL) Insulin dose: + 2 U 10.1–15.0 mmol/L (181–270 mg/dL) Insulin dose: + 4 U > 15.0 mmol/L (> 270 mg/dL) Insulin dose: + 6 U |
|
| Interval for insulin adjustments | The increase of the basal insulin was not to be more frequent than every 2 days | |
| Other concomitant intervention | None | |
| Home 2005 | Intervention | Description |
| Intervention | I: glargine | Once daily at bedtime. The dose was determined on the first treatment day by the total basal insulin dose the day before |
| C: NPH | NPH according to previous regimen (people who were treated previously with NPH insulin and continued to receive NPH insulin in the study remained on a regimen similar to their previous basal insulin regimen: those on once‐daily injections continued on once‐daily (bedtime) and those on more than once daily injections were put on a twice‐daily injection regimen (morning and at bedtime). Starting evening doses were the same as those on the immediate pre‐treatment day | |
| Titration period | — | |
| Strength of insulin | Not reported, but based on initiation regimen then 1 U glargine = 1 U NPH | |
| Rapid‐acting insulin | Unmodified human insulin was injected before meals according to the participant's habit | |
| Glycaemic targets |
Titration of basal insulin: the protocol suggested dose titration by 10% or greater increments, according to self‐monitored FBG levels, with a target of 4.4–6.7 mmol/L averaged over at least 2–4 days and an absence of nocturnal hypoglycaemia. All dose adjustments were at the discretion of the investigator/person with diabetes Titration of bolus insulin: 4.4–6.7 mmol/L, in the absence of hypoglycaemia |
|
| Interval of blood glucose measurement | Self‐measurement of FBG on the 7 consecutive days immediately preceding baseline and the 8‐, 20‐ and 28‐week visits. On the day immediately preceding each of these visits, the participants were asked to perform a 24‐hour blood glucose profile at 03:00 hours, just prior to and 2 h after breakfast, lunch and dinner, and at bedtime | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | Given the large number of centres and, therefore, the small number of people per centre, it was recognised that it was premature to enforce any algorithm for insulin dose adjustment | |
| Interval for insulin adjustments | Insulin dose adjustment was made throughout the study based on advice from the investigators during the scheduled visits (week 1, 4, 8, 12, 20 and 28) and informal contacts, and SMBG results between visits. Basal insulin regulated with at least two days in between | |
| Other concomitant intervention | None | |
| Kobayashi 2017 | Intervention | Description |
| Intervention | I: detemir | Detemir, sc once (bedtime) or twice (morning and bedtime) daily, 2400 nmol/mL (100 U/mL), 3 mL Penfill®. |
| C: NPH | NPH, sc once (bedtime) or twice (morning and bedtime) daily, 600 nmol/mL (100 U/mL), 3 mL Penfill® | |
| Titration period | 4 weeks | |
| Strength of insulin | All participants in the detemir group started treatment on approximately 70% of basal insulin dose (insulin detemir units) as their pre‐study intermediate/long‐acting human insulin dose. All participants in NPH group started the treatment on the same basal insulin dose as their pre‐study intermediate/long‐acting human insulin dose | |
| Rapid‐acting insulin | Insulin aspart as bolus insulin 3 times daily before each main meal | |
| Glycaemic targets | During the entire study, insulin dose was adjusted in accordance with treatment targets: FPG < 5.6 mmol/L and HbA1c < 6.2% | |
| Interval of blood glucose measurement | Assumed daily | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | — | |
| Interval for insulin adjustments | — | |
| Other concomitant intervention | — | |
| Liu 2016 | Intervention | Desciption |
| Intervention | I: glargine | Lantus®, 100 U/mL, sc, once daily at bedtime (22:00 ‐ 22:00), Solostar® device |
| C: NPH | Novolin N®, 100 U/mL, sc, once (at bedtime 20:00 to 22:00) or twice daily in the morning (before breakfast) and at bedtime (20:00 to 22:00). Decided by the investigator if it should be given once or twice daily | |
| Titration period | — | |
| Strength of insulin | The initial glargine dose for participants whose prestudy regimen was based on NPH insulin was recommended to take entire daily dose of basal insulin as on the pre‐treatment day (reduced by 20% if NPH insulin given more than once daily), then adjusted at the discretion of the Investigator to achieve glycaemic targets without an increase of hypoglycaemia | |
| Rapid‐acting insulin | Insulin aspart, 100 U/mL, sc, before each meal. The doses of insulin aspart were adjusted to optimise glycaemic control after basal insulin doses had been optimised and could be reduced as basal insulin doses are increased | |
| Glycaemic targets | Metabolic control without hypoglycaemia, defined by: FBG 5.0–8.0 mmol/L, bedtime BG 6.7–10.0 mmol/L, nocturnal BG 4.4–9.0 mmol/L and HbA1c < 7.5% | |
| Interval of blood glucose measurement | Not reported, but probably daily | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | On the investigators discretion | |
| Interval for insulin adjustments | Week 1, 2, 4, 6, 8, 10, 12, 16, 20 and 24 | |
| Other concomitant intervention | Diet and lifestyle counselling every 3rd months | |
| NCT00595374 | Intervention | Description |
| Intervention | I: detemir | sc, once or twice daily |
| C: NPH | sc, once or twice daily | |
| Titration period | 6 weeks | |
| Strength of insulin | The starting dose of basal insulin was equal to previous basal insulin dose | |
| Rapid‐acting insulin | Insulin aspart | |
| Glycaemic targets | — | |
| Interval of blood glucose measurement | — | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | — | |
| Interval for insulin adjustments | — | |
| Other concomitant intervention | — | |
| NCT00605137 | Intervention | Description |
| Intervention | I: detemir | 2400 nmol/mL (100 U/mL), 3 mL cartridge in FlexPen®, sc once daily at bedtime or twice daily before breakfast and at bedtime, according to the same treatment regimen as pre‐study basal insulin |
| C: NPH | 600 nmol/mL (100 IU/mL), 3 mL cartridge, FlexPen®, sconce daily at bedtime or twice daily before breakfast and at bedtime, according to the same treatment regimen as pre‐study basal insulin | |
| Titration period | 6 weeks | |
| Strength of insulin | Start of detemir was 70% basal insulin dose (insulin detemir unit) as their pre‐study intermediate/long‐acting human insulin dose. The start dose of NPH was the same as the pre‐study dose | |
| Rapid‐acting insulin | Not reported, probably the same type of rapid‐acting insulin as pre‐study (insulin aspart and/or soluble human insulin) | |
| Glycaemic targetsb | 7‐12 years; pre‐breakfast 4.4 to 8.3 mmol/L; post‐prandial (2 hours after meal) < 11.1. mmol/L: HbA1c: 6.5% to 7.4%; 13 years or older; pre‐breakfast 4.4 to 7.8 mmol/L; post‐prandial (2 hours after meal) < 10.0 mmol/L: HbA1c: 6.5% to 7.4% | |
| Interval of blood glucose measurement | — | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin dosesb | Algorithm for adjustment of the bedtime dose (guidance only) FBG Change in basal insulin dose > 4.4 mmol/L Should be reduced 4.4 to 8.3 mmol/L (7‐12 years) Investigators' judgement 4.4 to 7.8 mmol/L (13 years and older) Investigators' judgement > 8.3 to 10 mmol/L (7‐12 years) +10% > 7.8 to 10 mmolL (13 years and older) +10% > 10 mmol/L +20% Algorithm for adjustment of the morning dose in participants in twice daily regimen (guidance only) FBG Change in basal insulin dose > 4.4 mmol/L Should be reduced 4.4 to 8.3 mmol/L (7‐12 years) Investigators' judgement 4.4 to 7.8 mmol/L (13 years and older) Investigators' judgement > 8.3 to 10 mmol/L (7‐12 years) +10% > 7.8 to 10 mmolL (13 years and older) +10% > 10 mmol/L +20% |
|
| Interval for insulin adjustments | — | |
| Other concomitant intervention | Throughout the study period, instructions for diet and exercise (if any) therapy to participants was continued | |
| Pieber 2007 | Intervention | Description |
| Intervention | I: detemir | Detemir (Levemir®), 100 U/mL, morning and bedtime, NovoPen 3® |
| C: glargine | Glargine (Lantus®), 100 U/mL, bedtime, OptiPen® | |
| Titration period | 6 weeks with contact at least twice a week (during the first 6 weeks, the detemir doses were titrated aiming for a pre‐breakfast and pre‐evening meal PG of ≤ 7.3 mmol/L, whereas the glargine doses were titrated only to a pre‐breakfast PG). The insulin aspart dose was kept constant during the titration period. Based on two 5‐point PG profiles and the data recorded on insulin therapy, all participants were instructed about their starting dose of study medication | |
| Strength of insulin | Detemir with a 30% reduction in both the morning and evening doses from previous regimen. Glargine was initiated at a dose of 20–30% less than the participants previous total basal insulin dose | |
| Rapid‐acting insulin | Insulin aspart before meals | |
| Glycaemic targets | Doses were optimised according to the following algorithm: PG ≤ 7.3 mmol/L resulted in no change in dose; PG > 7.3–11.2 mmol/L resulted in a 10% increase in dose; PG > 11.2–16.8 mmol/L resulted in a 20% increase in dose; PG > 16.8 mmol/L resulted in a 25% increase in dose Post‐prandial PG target (90 min after a meal) of ≤ 10.1 mmol/L |
|
| Interval of blood glucose measurement | Detemir: recommended to measure FPG before breakfast (prior to insulin injection) and before dinner on a normal weekday before the next contact Glargine: recommended to measure FPG before breakfast on a normal weekday before the next contact |
|
| Calibration of blood glucose measurement device | Test strips for glucose meters were plasma‐calibrated | |
| Adjusting insulin doses | See 'Glycaemic targets' | |
| Interval for insulin adjustments | After titration period, intervals for insulin adjustments were decided by the investigator | |
| Other concomitant intervention | None | |
| Porcellati 2004 | Intervention | Description |
| Intervention | I: glargine | Glargine was given once daily at dinner time (20:00 h), injected in anterior part of one thigh, either pens or syringes. sc |
| C: NPH | NPH was administered 4 times daily (NPH insulin at each meal, and NPH at bedtime), injected in anterior part of one thigh, either pens or syringese, sc | |
| Titration period | — | |
| Strength of insulin | 1 U glargine = 1 U NPH | |
| Rapid‐acting insulin | Lispro | |
| Glycaemic targets | FBG and BG before meals and at bedtime 6.4–7.2 mmol/L, 2 hours after meal 8.0–9.2 mmol/L | |
| Interval of blood glucose measurement | Every day: capillary BG before meals and bedtime Every other day: BG 2 hours after meals Twice a week: BG at 03.00 o'clock |
|
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | For the first 2 days of treatment, the daily glargine dose was assumed to be identical to the total daily NPH units of the run‐in period. Afterwards, the dose of glargine was varied by 1–2 units every 2–3 days, if necessary, to meet the target FBG. Similar adjustments were made with the NPH treatment Basal insulin: participants were advised to decrease or increase the dose of basal insulin if FBG was repeatedly below 6.0 mmol/L or above 7.8 mmol/L, and to decrease or increase the dose of rapid‐acting insulin at meals if the 2 hour post‐prandial BG was repeatedly below 7.0 mmol/L or above 9.5 mmol/L NPH doses at each meal were adjusted based on BG values observed the previous days prior to meals Adjusting bolus insulin: adjustments of lispro dose was made according to carbohydrate content of meal Mealtime doses of lispro were 0.04–0.08 U/kg at breakfast, and 0.10–0.17 U/kg at lunch and dinner. The lispro doses were adjusted daily on the basis of pre‐prandial BG, as well as 2 hours after meal BG of previous days, as well as composition and size of meal and physical activity |
|
| Interval for insulin adjustments | Not reported, but probably continuously based on information from publication (all participants were in daily telephone contact with the investigators, and were seen weekly in the outpatient unit). | |
| Other concomitant intervention | None | |
| PRESCHOOL | Intervention | Description |
| Intervention | I: glargine | Once daily, 100 U/mL, Solostar® each containing 300 U and as 10 mL vials each containing 1000 U, sc |
| C: NPH | Once or twice daily, 100 U/mL Huminsulin Basal®, Huminsulin Basal Pen® each containing 300 U and as 10 mL vials each containing 1000 U, sc | |
| Titration period | Best efforts were made to complete the up‐titration of both basal insulins by week 12 | |
| Strength of insulin | Estimated to be 1 U glargine = 1 U NPH | |
| Rapid‐acting insulin | Insulin lispro used as the principal bolus insulin; regular human insulin permitted. Administration: multiple injection before meals and/or at bedtime at the discretion of the investigator | |
| Glycaemic targets | FBG between 5.0 to 8.0 mmol/L; bedtime BG between 6.7 to 10.0 mmol/L; nocturnal BG between 4.4 to 9.0 mmol/L; HbA1c < 7.5% | |
| Interval of blood glucose measurement | Assuming daily, participants had CGM during the study | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | Titration schedule not provided | |
| Interval for insulin adjustments | Doses of insulin glargine and NPH insulin were increased no more often than once a week, but doses could be reduced due to hypoglycaemia at any time | |
| Other concomitant intervention | None | |
| Ratner 2000 | Intervention | Description |
| Intervention | I: glargine | Once daily at bedtime, vial containing 5 mL solution (1 mL contains 100 U), sc |
| C: NPH | Once daily at bedtime or twice (at bedtime and before breakfast) depending on pre‐trial insulin regimens, vial containing 10 mL suspension (1 mL contains 100 U), sc | |
| Titration period | 4 weeks | |
| Strength of insulin | Switching from insulin glargine to once daily NPH was done 1:1. Slight dose decrease was done when switching from twice‐daily NPH to glargine. From clinical study report: investigators were advised at the study initiation meeting to reduce glargine dose with 10% — however, this was not specified in protocol | |
| Rapid‐acting insulin | Recombinant human insulin about 30 min before meals, vial containing 10 mL solution (1 mL contains 100 U) | |
| Glycaemic targets | Based on capillary FBG; goal was 4.4 to 6.7 mmol/L and a bedtime BG value of 6.7 to 8.0 mmol/L | |
| Interval of blood glucose measurement | Daily. Glucose measurements were evaluated on 7 consecutive days preceding baseline and visit at week 8, 20 and 28 | |
| Calibration of blood glucose measurement device | — | |
| Adjusting of insulin doses | Dose increases were made if morning capillary FBG levels were constantly > 6.7 mmol/L with no symptomatic hypoglycaemia. Dose decreases were done if fasting capillary BG were < 4.4 mmol/L or if symptomatic nocturnal hypoglycaemia was present. Glargine: the dose increase was to be at least 10% of the total dose of glargine while not exceeding 4 units. Dose increases were not to be made any more frequently than every 2 to 4 days. Dose decreases were to be made if any pre‐breakfast BG was less than 4.4 mmol/L or there had been any symptomatic hypoglycaemia during sleep or BG values less than 5.0 mmol/L during sleep in the last 2 to 4 days. The dose was decreased for the next evening dose following the occurrence of the hypoglycaemia or low pre‐breakfast BG. The dose of glargine was generally not lowered because of daytime hypoglycaemia unless repeated episodes of daytime hypoglycaemia had occurred after total elimination of the previous dose of regular insulin. NPH: evening dose adjustments as for glargine The pre‐breakfast dose of NPH human insulin, if part of the pre‐treatment basal insulin regimen, was administered at a standard time in conjunction with the pre‐breakfast dose of regular insulin. If a participants had BG values less than 4.4 mmol/L or symptomatic hypoglycaemia occurred between lunch and dinner, either the morning NPH was lowered, the prelunch regular insulin dose was lowered or the afternoon snack was increased. If the majority of pre‐supper BG values were greater than 6.7 mmol/L over a 2‐ to 4‐day period, either the morning NPH was increased, the prelunch regular insulin was increased, or the afternoon snack was decreased |
|
| Interval for insulin adjustments | Baseline, week 8, 20 and 28 | |
| Other concomitant intervention | None | |
| Robertson 2007 | Intervention | Description |
| Intervention | I: detemir | Detemir (Levemir®; Novo Nordisk A/S, Bagsvaerd, Denmark; 100 U/mL), once (at bedtime) or twice (morning and bedtime) daily, sc, thigh or abdomen, Penfill |
| C: NPH | NPH (NPH, human isophane insulin®; Novo Nordisk A/S; 100 IU/mlL, once (at bedtime) or twice (morning and bedtime) daily, sc, thigh or abdomen, Penfill | |
| Titration period | 6 weeks | |
| Strength of insulin | Equivalence. The initial basal insulin dose was 70% of the prestudy basal insulin dose | |
| Rapid‐acting insulin | Insulin aspart (NovoRapid®/NovoLog®; Novo Nordisk A/S; 100 U/mL) before meals, thigh or abdomen | |
| Glycaemic targets | FPG was 4.5–7.8 mmol/L and evening basal insulin doses were adjusted by the investigator FPG: < 4.5 mmol/L: adjustment according to local practice FPG 4.5‐7.8 mmol/L: no adjustment FPG > 7.8–11.2 mmol/L: bedtime dose increased by 10% FPG > 11.2–16.8 mmol/L: bedtime dose increased by 20% FPG > 16.8 mmol/L: bedtime dose increased by 25% A similar guidance algorithm was used for pre‐evening meal plasma glucose for children on a twice‐daily regimen to adjust the morning dose of basal insulin During the 20‐week maintenance period, the insulin aspart dose was optimised by aiming for a post‐prandial (90 min after each meal) plasma glucose guidance level of 6.7–10.1 mmol/L. Further adjustment of basal insulin doses in this period was also allowed |
|
| Interval of blood glucose measurement | The number and regularity of self‐measured plasma glucose testing was individualised depending on acceptance by the child and the plasma glucose level, but was at least twice weekly during the 6‐week titration period | |
| Calibration of blood glucose measurement device | Regular calibration | |
| Adjusting of insulin doses | See glycaemic targets. A change between once‐daily and twice‐daily regimens during the study was allowed | |
| Interval for insulin adjustments | In titration period, basal insulins were adjusted twice weekly | |
| Other concomitant intervention | None | |
| Russell‐Jones 2004 | Intervention | Description |
| Intervention | I: detemir | Detemir (100 U/mL) at bedtime, 2400 nmol/mL, supplied in 3.0 mL cartridges |
| C: NPH | NPH (100 U/mL) at bedtime, supplied in 3.0 mL cartridges | |
| Titration period | During the first two weeks, mealtime bolus insulin doses should (preferably) be kept unchanged and only basal insulin dose was titrated according to treatment goals. The following weeks were used to optimise the dose ratio between mealtime bolus insulin and basal insulin | |
| Strength of insulin | The starting dose for participants switching to insulin detemir was 50% of the usual pre‐trial basal insulin dose. Patients assigned to NPH started on their pre‐trial basal insulin dose. Participants randomised to NPH insulin were to continue on the same dose as their pre‐trial NPH insulin dose | |
| Rapid‐acting insulin | Regular human insulin (100 U/mL), supplied in 3.0 mL cartridges | |
| Glycaemic targets | FBG, pre‐breakfast/night 4.0–7.0 mmol/L; 90 minutes post‐prandial <10.0 mmol/L | |
| Interval of blood glucose measurement | Daily; SMBG was performed regularly throughout the study | |
| Calibration of blood glucose measurement device | Patients were instructed in the calibration and use of blood glucose meters (OneTouch Profile, LifeScan, Inc., Milpitas, California), and were asked to perform SMBG regularly throughout the study to allow continuous adjustment of insulin doses | |
| Adjusting of insulin doses | — | |
| Interval for insulin adjustments | — | |
| Other concomitant intervention | None | |
| Schober 2002 | Intervention | Description |
| Intervention | I: glargine | Once daily at bedtime (19:00 – 22:00), cartridge containing 3 mL solution (1 mL contains 100 U) |
| C: NPH | Once (at bedtime) or twice daily (before breakfast and bedtime) depending in pre‐treatment insulin regimen, cartridge containing 3 mL solution (1 mL contains 100 U) | |
| Titration period | — | |
| Strength of insulin | 1:1 | |
| Rapid‐acting insulin | Regular human insulin before meals according to individual habits, premeal goal was 4.4 ‐ 8.8 mmol/L | |
| Glycaemic targets | Titration of bedtime insulin was FBG 4.4‐8.8 mmol/L. Morning dose for NPH adjusted as required (not further specified) | |
| Interval of blood glucose measurement | Daily | |
| Calibration of blood glucose measurement device | — | |
| Adjusting of insulin doses | — | |
| Interval for insulin adjustments | Increase of basal insulin was not to be more frequent than every 4‐5 days; dose decrease was decided by the investigator | |
| Other concomitant intervention | None | |
| Standl 2004 | Intervention | Description |
| Intervention | I: detemir | 100 U/mL (100 U = 1200 nmol), Penfill, twice daily |
| C: NPH | 100 U/mL, twice daily, only the basal insulins were titrated during the initial 2 weeks | |
| Titration period | First month of study | |
| Strength of insulin | 1 U of detemir was estimated to have the same BG lowering activity as 1 U NPH. At study start, the initial detemir dose was half the unit dose of the patients’ previous basal insulin, with the expectation of upward titration | |
| Rapid‐acting insulin | Human soluble insulin (Actrapid) before meals | |
| Glycaemic targets | FBG < 4–7 mmol/L; 90 minutes post‐prandial < 10 mmol/L; at 02:00 and 04:00 a.m. < 4–7 mmol/L | |
| Interval of blood glucose measurement | Not reported, but based on "aiming for the following targets: fasting, 4–7 mmol/L; 90‐min post‐prandial < 10 mmol/L; at 0200 and 0400 a.m., 4–7 mmol/L" then 5 times a day | |
| Calibration of blood glucose measurement device | — | |
| Adjusting of insulin doses | Doses were adjusted continuously at investigators’ discretion based on patients’ SMBG measurements | |
| Interval for insulin adjustments | In titration period every second day, thereafter at week 2, 4, 9, 13, 19 and 26 | |
| Other concomitant intervention | None | |
| SWITCH 1 | Intervention | Description |
| Intervention | I: degludec | Degludec® 100 U/mL (Novo Nordisk) (about 50% of the participants were randomised to morning dose (from waking up to breakfast) and 50% to evening dose (from main evening meal to bedtime)), 10 mL vial, sc |
| C: glargine | Lantus® 100 U/mL (Sanofi) (about 50% of the participants were randomised to morning dose (from waking up to breakfast) and 50% to evening dose (from main evening meal to bedtime)), 10 mL vial, sc | |
| Titration period | Not clearly stated, but participants had a 16 week wash‐out period at initiation of study and after cross‐over in order to stabilise HbA1c | |
| Strength of insulin | The starting dose of basal insulin and total bolus insulin (algorithm users only) was reduced by 20% at randomisation and at cross‐over (i.e. after 32 weeks) | |
| Rapid‐acting insulin | Insulin aspart 100 U/mL was administered using a prefilled pen (FlexPen®; Novo Nordisk), 2‐4 times/daily, sc | |
| Glycaemic targets | For basal insulin adjustment: FBG between 4.0–5.0 mmol/L Pre‐prandial BG between 3.9 and 6.0 mmol/Lb |
|
| Interval of blood glucose measurement | Participants were supplied with a blood glucose meter and instructed to measure their BG before breakfast, lunch, main evening meal, and bedtime on all days throughout the study. Their BG levels were also measured whenever a hypoglycaemic episode was suspected | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | Titration of basal insulin was performed once weekly according to the study algorithm, based on the lowest of 3 previous pre‐breakfast SMBGs.
Basal insulin titration regimen: lowest pre‐breakfast BG measurement (mmol/L) and adjustment (U)
< 3.1 Insulin dose: –4
3.1–3.9 Insulin dose: –2
4.0–5.0 Insulin dose: 0
5.1–10.0 Insulin dose: +2
10.1–15.0 Insulin dose: +4
> 15.0 Insulin dose: +6 Titration of bolus insulin was either performed twice weekly based on the previous 3 or 4 days’ readings according to the provided algorithm, or several times daily based on the insulin:carbohydrate ratio and insulin sensitivity factor Insulin aspart was titrated individually based either on carbohydrate counting or sliding scale |
|
| Interval for insulin adjustments | 4‐point profiles were evaluated with weekly telephone contacts | |
| Other concomitant intervention | None | |
| Thalange 2013 | Desription | Intervention |
| Intervention | I: detemir | Levemir®; Novo Nordisk A/S, Bagsvaerd, Denmark; 100 U/mL, sc, once or twice daily, according to pre‐trial insulin regimen and dose |
| C: NPH | Human isophane insulin®; Novo Nordisk A/S; 100 IU/mL, sc, once or twice daily, according to pre‐trial insulin regimen and dose | |
| Titration period | — | |
| Strength of insulin | Anticipated to be 1 U detemir = 1 U NPH | |
| Rapid‐acting insulin | Insulin aspart (NovoRapid®/NovoLog®; Novo Nordisk A/S; 100 U/ml) 2–4 times daily with main meals, was to be taken 0–15 min prior to or immediately after the meal | |
| Glycaemic targets | Pre‐prandial PG 4.0–7.0 mmol/L; post‐prandial PG 5.0–11.0 mmol/L | |
| Interval of blood glucose measurement | Participants were asked to measure their PG before breakfast and dinner on the last 3 days prior to each contact; nine‐point SMPG profiles, including nocturnal plasma glucose at 03.00 o'clock, were assessed by the children on a normal weekday 4–7 days prior to randomisation, and after 26 and 52 weeks of treatment | |
| Calibration of blood glucose measurement device | Use of test strips calibrated to plasma glucose values ensured that capillary blood concentrations were displayed as plasma glucose values | |
| Adjusting insulin doses (foot note ‐ only adjustment for dose intervals 5‐15 written in table) | Pre‐breakfast or pre‐dinner plasma glucose Insulin adjustment (varies with insulin dose in intervals < 5 U, 5‐15 U, > 15 U) < 4.0 mmol/L Reduce according to local practice 4.0‐7.0 mmol/L 0 7.1‐10.0 mmol/L +1 10.1‐15.0 mmol/L +2 > 15 mmol/L +3 Rapid‐acting insulin: adjusted according to local practice |
|
| Interval for insulin adjustments | Long‐acting insulin: each contact Rapid‐acting insulin: adjusted according to local practice |
|
| Other concomitant intervention | None | |
| Urakami 2017 | Description | Intervention |
| Intervention | I: degludec | Once daily at bedtime |
| C: glargine | Once daily at bedtime | |
| Titration period | One week stabilisation period was reported | |
| Strength of insulin | — | |
| Rapid‐acting insulin | Insulin aspart or insulin lispro before meals | |
| Glycaemic targets | — | |
| Interval of blood glucose measurement | Daily before each meal, at bedtime and if symptoms on hypoglycaemia | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | — | |
| Interval for insulin adjustments | — | |
| Other concomitant intervention | — | |
| Vague 2003 | Description | Intervention |
| Intervention | I: detemir | Before breakfast and bedtime, 1200 nmol/mL (1 U = 24 nmol) |
| C: NPH | Before breakfast and bedtime, 600 nmol/mL, 100 U/mL | |
| Titration period | 1 months | |
| Strength of insulin | Anticipated to be 1:1f | |
| Rapid‐acting insulin | Insulin aspart at main meals | |
| Glycaemic targets | Fasting/pre‐prandial, 4–7 mmol/L; post‐prandial < 10 mmol/L; from 02:00 to 04:00, 4–7 mmol/L | |
| Interval of blood glucose measurement | Daily | |
| Calibration of blood glucose measurement device | — | |
| Adjusting insulin doses | In titration phase, basal insulin was titrated every second day. Thereafter, basal and bolus doses were adjusted according to investigator recommendations, based on BG measurements | |
| Interval for insulin adjustments | Continuously during study | |
| Other concomitant intervention | None | |
| —: denotes not reported a37% of participants treated with insulin detemir versus 45% treated with NPH insulin completed the study on a once‐daily basal insulin regimen. bValues converted from mg/dL to mmol/L using: https://www.diabetes.co.uk/blood-sugar-converter.html. cOnly 3 participants stayed on Lente insulin, remaining on NPH insulin, administered twice daily, before breakfast and in the evening. d75 participants in each intervention group received CGMS at baseline. Data were available for 33participants at baseline and after 24 weeks in the insulin glargine group and 36 participants in the NPH insulin group. eSyringes with Lispro insulin and NPH insulin were mixed and administered together. fApproximately three‐ to fourfold higher molar dose of insulin detemir was required (resulting in an approximately twofold ratio by volume using the formulation in the study). This result may have further discouraged upward titration of dose, a factor that would not be an issue with the more concentrated and bioequivalent preparation of insulin detemir to be marketed (which has a four times higher molar concentration than that of NPH insulin in order to establish unit‐to‐unit conversion). a.m.: ante meridiem; BG: blood glucose; CGM: continuous glucose measurement;CGMS: continuous glucose measurement system;FBG: fasting blood glucose; FPG: fasting plasma glucose; HbA1c: glycosylated haemoglobin A1c; HSI: human soluble insulin; IU: international unit;NPH: neutral protamine Hagedorn insulin; PG: plasma glucose; p.m.: post meridiem; sc: subcutaneous; SMBG: self‐measured blood glucose; SMPG: self‐measured plasma glucose; T1DM: type 1 diabetes mellitus;TRIM‐HYPO: treatment‐related impact measure ‐ hypoglycaemic events; U: units. | ||
Appendix 9. Baseline characteristics (I)
| Study ID | Intervention(s) and comparator(s) | Duration of intervention (duration of follow‐up)a | Description of participants | Study period | Country | Setting | Ethnic groups (%) |
Duration of diabetes (mean/range years (SD)) |
| Bartley 2008 | I: detemir | 24 months (24 months) | T1DM, adults | June 2004 ‐ September 2006 | Argentina, Australia, Bulgaria, Croatia, India, Macedonia, The Former Yugoslav Republic, Malaysia, Romania, South Africa, Turkey | Outpatients | White: 73.7 Black: 0.9 Asian/Pacific Islander: 19.9 Other: 5.4 |
12.7 (9.4) |
| C: NPH | White: 78.7 Black: 0.6 Asian/Pacific Islander: 19.5 Other: 1.2 |
13.5 (9.9) | ||||||
| BEGIN Basal‐Bolus Type 1 | I: degludec | 52 weeks (104 weeks) | T1DM, adults | September 2009 ‐ November 2010 | France, Germany, Russia, South Africa, UK, USA | Outpatients | White: 93 Black: 2 Asian: 1 Other: 4 |
19.2 (12.2) |
| C: glargine | White: 94 Black: 2 Asian: 2 Other: 2 |
18.2 (11.4) | ||||||
| BEGIN Flex T1 | I: degludec | 26 weeks (52 weeks) | T1DM, adults | March 2010‐ November 2010b | Belgium, Germany, Norway, Poland, UK, USA | Outpatients | White: 97.6 Black: 1.8 Asian: 0.0 Other: 0.6 |
20.0 (12.5) |
| C: glargine | White: 98.8 Black: 0.6 Asian: 0.6 Other: 0.0 |
18.2 (11.9) | ||||||
| BEGIN Young | I: degludec | 26 weeks (52 weeks) | T1DM, children (1–17 years) | January 2012 ‐ February 2013b | Bulgaria, Finland, France, Germany, Italy, Japan, the Netherlands, Republic of Macedonia, Russian Federation, South Africa, UK and USA | Outpatients |
White: 78.2 Black: 2.9 Asian: 13.2 Other: 5.7 |
3.9 (3.6) |
| C: detemir | White: 86.0 Black: 2.3 Asian: 2.3 Other: 9.3 |
4.0 (3.4) | ||||||
| Bolli 2009 | I: glargine | 24 weeks (30 weeks) | T1DM, adults | — | Italy | Outpatients | — | 12.9 (8.3) |
| C: NPH | — | 14.8 (9.6) | ||||||
| Chase 2008 | I: glargine | 24 weeks (25 weeks) | T1DM, children (9‐17 years) | December 2002 ‐ February 2005 | USA, Canada | Outpatients | White: 84.5 Black: 0 Asian: 2.4 Hispanic: 8.3 Multiracial: 2.3 Other: 2.4 |
5.1 (3.4) |
| C: NPH/Lente | White: 81.0 Black: 8.3 Asian: 2.4 Hispanic: 4.8 Multiracial: 1.2 Other: 2.4 |
5.4 (3.7) | ||||||
| Davies 2014 | I: degludec | 26 weeks (52 weeks) | T1DM, adults | February 2010‐ December 2010 | Brazil, Finland, India, Italy, Japan, Macedonia and UK | Outpatient | White: 44.0 Black:0.7 Asian:54.6 Other: 0.7 |
13.7 (10.6) |
| C: detemir | White: 45.8 Black:0.0 Asian: 53.6 Other: 0.7 |
14.4 (9.7) | ||||||
| Fulcher 2005 | I: glargine | 30 weeks (30 weeks) | T1DM, adults | November 2000 ‐ November 2001 | Australia | Outpatient | White: 98.4 | 17.9 (10.5) |
| C: NPH | 17.1 (9.7) | |||||||
| Heller 2009 | I: detemir | 52 weeks (52 weeks) | T1DM, adults | September 2004 ‐ December 2005 | USA, UK, Germany, France, the Netherlands, Finland and Sweden | Outpatient | Black: 2.0b Hispanic: 2.3 White: 95.7 |
17.2 (11.7) |
| C: glargine | Black: 1.4 Hispanic: 2.8 White: 95.8 |
17.3 (10.7) | ||||||
| Home 2005c | I: glargine | 28 weeks (28 weeks) | T1DM, adults | August 1997 ‐ August 1998b | 12 European countries (Austria, Czech Republic, Denmark, Finland, France, Germany, Greece, Netherlands, Norway, Sweden, Switzerland, UK) | Outpatient | White: 99.7b Other: 0.3 |
16 (12) |
| C: NPH | White: 99.0 Other: 1.0 |
15 (9) | ||||||
| Kobayashi 2007 | I: detemir | 48 weeks (48 weeks) | T1DM, adults | May 2003 ‐ March 2005b | Japan | Outpatient | Asian (Japanese): 100 | 13.4 (8.18) |
| C: NPH | Asian (Japanese): 100 | 13.01 (8.5) | ||||||
| Liu 2016 | I: glargine | 24 weeks (25 weeks) | T1DM, children (≥ 6 to < 18 years) | February 2011 ‐ August 2013 | China | Outpatient | Asian (Chinese): 100 | 3.8 (2.9) |
| C: NPH | Asian (Chinese): 100 | 3.6 (2.3) | ||||||
| NCT00595374 | I: detemir | 26 weeks (26 weeks) | T1DM, adults | December 2003 ‐ October 2004 | Netherlands | Outpatients | White: 98.7 Asian/Pacific islander: 1.3 |
— |
| C: NPH | White: 97.4 Asian/Pacific islander: 2.6 |
— | ||||||
| NCT00605137 | I: detemir | 24 weeks (24 weeks) | T1DM, children (7 to 18 years) | May 2004 ‐ April 2005 | Japan | Outpatients | Asian (Japanese): 100 | 4.7 (3.2) |
| C: NPH | Asian (Japanese): 100 | 6.5 (4.0) | ||||||
| Pieber 2007 | I: detemir | 26 weeks (26 weeks) | T1DM, adults | April 2002 ‐ March 2003 | Germany, Austria, South Africa | Outpatient | From co‐publication:
White: 95.3 Other: 4.7 |
17 (range 1‐57) |
| C: glargine | 16 (range 1‐48) | |||||||
| Porcellati 2004 | I: glargine | 1 year (1 year) | T1DM, adults | — | Italy | Outpatient | — | 13 (2.4)d |
| C: NPH | 15 (2.3) | |||||||
| PRESCHOOL | I: glargine | 24 weeks (26 weeks) | T1DM, children (1‐6 years) | October 2009 ‐ March 2011 | Argentina, Austria, Brazik, Chile, Czech Republic, Germany, Hungary, India, Mexico, Peru, Poland, Romania, Russia, South Africa, Spain, USA | Outpatient | White: 86.9 Black: 3.3 Asian: 6.6 Other: 3.3 |
2.1 (1.2) |
| C: NPH | White: 75.0 Black: 3.1 Asian: 17.2 Other: 4.7 |
2.1 (1.0) | ||||||
| Ratner 2000 | I: glargine | 28 weeks (28 weeks) | T1DM, adults | June 1997 ‐ June 1998b | USA | Outpatient | White: 95.1b Black:4.2 Asian: — Hispanic: 3.0 Other: 0.8 |
17.9 (11.7) |
| C: NPH | White: 95.6 Black: 3.0 Asian: — Hispanic: 3.3 Other: — |
16.9 (10.0) | ||||||
| Robertson 2007 | I: detemir | 26 weeks (26 weeks) | T1DM, children (6‐17 years) | August 2002 ‐ August 2003 | Europe (Belgium, Croatia, Denmark, Finland, Germany, Ireland, Macedonia, Netherlands, Norway, Slovenia, Spain, Sweden, Switzerland, UK) and Israel | Outpatient | White: 99.6 Acian/Pacific islander: 0.4 |
5.1 (3.1) |
| C: NPH | White: 100 | 4.8 (2.8) | ||||||
| Russell‐Jones 2004 | I1; detemir | 6 months (6 months) | T1DM, adults | February 2001 ‐ November 2001 | United Kingdom, France, Sweden, Norway, Australia, Netherlands, Denmark, Finland, Belgium, Ireland and Luxembourg | Outpatient | White: 98.7 Other: 1.3 |
17.1 (11.3) |
| C: NPH | 16.4 (9.5) | |||||||
| Schober 2002 | I: glargine | 28 weeks (28 weeks) | T1DM, Children (5‐16 years) | June 1997 ‐ March 1999 | Austria, Belgium, Croatia, Czech Republic, Finland, Germany, Switzerland, Netherlands, UK and South Africa | Outpatient | White: 96.6 Black: 0.0 Asian/Oriental: 1.7 Multiracial: 1.7 |
5.8 (3.02) |
| C: NPH | White: 97.1 Black: 0.0 Asian/Oriental: 2.9 Multiracial: 0.0 |
4.7 (3.08) | ||||||
| Standl 2004e | I: detemir | 6 months (12 months) | T1DM, adults | October 1999 ‐ September 2000b | Germany, Switzerland, Austriaf, Australia, New Zealand | Outpatient | White 99b | 14.9 (9.4) |
| C: NPH | 15.5 (10.8) | |||||||
| SWITCH 1 | I: degludec | 32 weeks (32 weeks) | T1DM, adults | January 2014 ‐ January 2016 | USA, Poland | Outpatient | White: 93.6 Black: 5.2 Asian: 0.4 Hispanic or Latino: 9.2 Other: 0.8 |
23.2 (13.5) |
| C: glargine | White: 90.9 Black: 7.5 Asian: 0.4 Hispanic or Latino: 11.1 Other: 1.2 |
23.6 (13.4) | ||||||
| Thalange 2013 | I: detemir | 52 weeks (104 weeks) | T1DM, children (2‐16 years) | February 2007 ‐ September 2008 | Bulgaria, Czech Republic, Denmark, Finland, France, Hungary, Macedonia, Poland, Russia, Turkey and UK | Outpatient | White: 98b Other: 1 | 3.7 (2.7) |
| C: NPH | 3.7 (2.5) | |||||||
| Urakami 2017 | I: degludec | 24 weeks (24 weeks) | T1DM, children | — | Japan | Outpatient | — | — |
| C: glargine | ||||||||
| Vague 2003 | I: detemir | 6 months (12 months) | T1DM, adults | November 1999 ‐ October 2000b | France, Belgium. Luxembourg, Netherlands, Norway | Outpatient | White: 99.5b | 17.1 (9.9) |
| C: NPH | 17.4 (11.0) | |||||||
| —: denotes not reported aFollow‐up under randomised conditions until end of study (= duration of intervention + follow‐up post‐intervention or identical to duration of intervention). bData from clinical study report/synopsis. cBaseline characteristics only available for the 585 participants who were treated with study medication (292 participants with insulin glargine and 293 participants with NPH). dNot reported if SD or standard error was provided. Assumed to be standard error, so SD was calculated from anticipated standard error. eIn publication, baseline characteristics were only available for the participants completing the 6 months treatment period and participating in the 6 months extension period (NPH; N = 135; Detemir; N = 154). Data on randomised participants available from clinical study report. fIn publication, just stated that Europe ‐ countries in Europa provided by clinical study report. C: comparator; I: intervention; NPH: neutral protamine Hagedorn; SD: standard deviation; T1DM: type 1 diabetes mellitus; UK: United Kingdom; USA: United States of America. | ||||||||
Appendix 10. Baseline characteristics (II)
| Study ID | Intervention(s) and comparator(s) | Sex (% women) | Age (mean/range years (SD)) | HbA1c (mean % (SD)) | BMI (mean kg/m² (SD)) | Comedications/cointerventions (% of participants) | Comorbidities (% of participants) |
| Bartley 2008 | I: detemir | 44.4 | 35 (12) | 8.3 (1.2) | 24.7 (3.7) | Enalapril 5.4a Acetylsalicyl acid 6.9 Paracetamol 2.4 |
Concomitant illness 57.1a Hypertension 16.0 Metabolism and nutrition disorder 14.2 Eye disorder 7.6 Cardiac disorder 7.9 |
| C: NPH | 47.0 | 35 (11) | 8.4 (1.3) | 24.7 (3.7) | Enalapril 8.5 Acetylsalicyl acid 4.3 Paracetamol 6.1 |
Concomitant illness 55.5 Hypertension 14.6 Metabolism and nutrition disorder 14.0 Eye disorder 11.0 Cardiac disorder 7.3 |
|
| BEGIN Basal‐Bolus Type 1 | I: degludec | 41.1 | 42.8 (13.7) | 7.7 (0.9) | 26.3 (3.7) | Simvastatin 14.4a Lisonipril 13.8 Acetylsalicyl acid 28.0 |
Ophthalmic complications 18.6a Neurological complications 14.0 Renal complications 7.6 Cardiovascular complications 0.8 Hypothyroidism 13.6 |
| C: glargine | 42.7 | 43.7 (13.3) | 7.7 (1.0) | 26.4 (4.2) | Simvastatin 12.1 Lisonipril 12.7 Acetylsalicyl acid 25.5 |
Ophthalmic complications 17.2 Neurological complications 12.1 Renal complications 6.4 Cardiovascular complications 1.3 Hypothyroidism 19.1 |
|
| BEGIN Flex T1 | I: degludec | 43.0 | 44.5 (13.1) | 7.7 (0.9) | — | Acetylsalicyl acid 21.8a Simvastatin 21.2 Lisonopril 11.5 |
Ophthalmic complications 9.1a Neurological complications 7.3 Renal complications 4.8 |
| C: glargine | 46.3 | 44.1 (12.6) | 7.7 (0.9) | — | Acetylsalicyl acid 24.4 Simvastatin 20.7 Lisonopril 11.0 |
Ophthalmic complications 6.7 Neurological complications 6.7 Renal complications 3.7 |
|
| BEGIN Young | I: degludec | 44.8 | 10.0 (4.4) | 8.2 (1.1) | 18.7 (3.6) | Ibuprofen 9.3a Paracetamol 5.2 Salbutamol 3.4 Loratadine 3.4 |
Diabetes complications 0.6a Seasonal allergy 8.0 Asthma 2.9 |
| C: detemir | 44.3 | 10.0 (4.4) | 8.0 (1.1) | 18.5 (3.6) | Ibuprofen 2.3 Paracetamol 4.0 Salbutamol 2.3 Loratadine 1.7 |
Diabetes complications 0.3 Seasonal allergy 6.3 Asthma 3.4 |
|
| Bolli 2009 | I: glargine | 43.5 | 35.5 (10.6) | 7.8 (0.7) | 23.3 (2.0) | — | — |
| C: NPH | 45.6 | 37.0 (9.4) | 7.8 (0.6) | 23.6 (1.9) | — | — | |
| Chase 2008 | I: glargine | 53.6 | 13.1 (2.4) | 7.8 (0.8) | 22.6 (3.8) | — | Neuropathy 0a Nephropathy 2.4 Retinopathy 0 Hypertension 1.2 Hyperlipidaemia 1.2 |
| C: NPH/Lente | 52.4 | 13.4 (2.4) | 8.0 (0.8) | 22.9 (5.0) | — | Neuropathy 0 Nephropathy 3.3 Retinopathy 0 Hypertension: 3.3 Hyperlipidaemia 5.6 |
|
| Davies 2014 | I: degludec | 50.3 | 41.1. (14.9) | 8.0 (1.0) | 24.0 (3.5) | Acetylsalicyl acid 7.3a Ramipril 4.0 Simvastatin 6.3 |
Diabetic complications 26.2a Hypertension 28.2 |
| C: detemir | 43.8 | 41.7 (14.4) | 8.0 (0.9) | 23.7 (3.4) | Acetylsalicyl acid 7.8 Ramipril 3.9 Simvastatin 7.2 |
Diabetic complications 28.8 Hypertension 21.6 |
|
| Fulcher 2005 | I: glargine | 61.3 | 41.6 (12.9) | 9.2 (1.1) | 27.0 (3.6) | — | — |
| C: NPH | 60.3 | 39.3 (13.9) | 9.7 (1.3) | 26.0 (3.9) | — | — | |
| Heller 2009 | I: detemir | 44.1 | 42 (13) | 8.1 (1.1) | 26.5 (4.0) | Acetylsalicyl acid 11.7a Levothyroxine 11.4 Simvastatin 10.0 |
Retinopathy 26.8a Neuropathy 17.7 Nephropathy 10.0 Macroangiopathy 3.3 |
| C: glargine | 43.8 | 41 (12) | 8.1 (1.2) | 26.3 (3.9) | Acetylsalicyl acid 11.1 Levothyroxine 9.7 Simvastatin 11.1 |
Retinopathy 27.1 Neuropathy 10.4 Nephropathy 6.9 Macroangiopathy 0.7 |
|
| Home 2005 | I: glargine | 45.2 | 39 (12) | 7.9 (1.2) | 24.6 (3.1) | Retinopathy 31.2a Neuropathy 17.5 Nephropathy 6.2 Macroangiopathy 3.1 |
|
| C: NPH | 43.3 | 39 (12) | 8.0 (1.2) | 25.1 (3.3) | Retinopathy 29.4 Neuropathy 16.7 Nephropathy 7.2 Macroangiopathy 3.4 |
||
| Kobayashi 2007 | I: detemir | 58.7 | 42.4 (14.2) | 7.4 (1.0) | 22.4 (1.7) | — | — |
| C: NPH | 50 | 41.8 (13.5) | 7.4 (1.2) | 22.4 (2.7) | — | — | |
| Liu 2016 | I: glargine | 58.6 | 12.2 (3.2) | 8.9 (1.2) | 18.7 (2.9) | Unspecified herbal and traditional medicine 32.7a Anti‐infective for systemic use 25.2 |
Retinopathy 0 Nephropathy 0 Neuropathy 0 |
| C: NPH | 64.8 | 12.2 (3.5) | 9.1 (1.3) | 18.2 (2.6) | Unspecified herbal and traditional medicine 47.3 Anti‐infective for systemic use 23.6 |
Retinopathy 0 Nephropathy 1.9 Neuropathy 0 |
|
| NCT00595374 | I: detemir | 49.3 | 39 (13.3) | 8.5 (0.9) | — | — | Diabetic complications 25.3 |
| C: NPH | 36.8 | 42.8 (12.7) | 8.3 (1.0) | — | — | Diabetic complications 34.2 | |
| NCT00605137 | I: detemir | 63.6 | 13.2 (2.5) | 7.2 (0.9) | 20.5 (3.5) | — | — |
| C: NPH | 40.7 | 14.1 (2.5) | 7.5 (1.3) | 20.8 (3.7) | — | — | |
| Pieber 2007 | I: detemir | 45.3 | 40 (range 18‐79) | 8.9 (range 7.6 ‐ 11.9) | 25.6 (range 18.2 ‐ 35.1) | Acetylsalicylic acid 17a Paracetamol 13 Ibuprofen 7 |
From co‐publication: Angina pectoris 2.4 Myocardial infarction 0.3 Heart failure 0.3 Stroke 0.4 Atrial fibrillation 0.5 Microalbuminuria 27.2 |
| C: glargine | 52.2 | 41 (range 18‐70) | 8.8 (range 7.6‐11.9) | 15.5 (range 16.8 ‐ 34.4) | |||
| Porcellati 2004b | I: glargine | 44.3 | 36 (7.8) | 7.1 (0.8) | 22.9 (0.4) | — | Smoker: 17.4 Cardiovascular disease: 11.0 |
| C: NPH | 45.0 | 34 (7.8) | 7.1 (1.6) | 23.2 (1.2) | — | ||
| PRESCHOOL | I: glargine | 47.5 | 4.3 (0.9) | 8.0 (1.0) | — | Dermatologicals 23a Cardiovascular system 23 Repiratory system 13.1 |
Diabetic retinopathy 0a Motor neuropathy 0 Autonomic neuropathy 1.6 Nephropathy 0 Albuminuria 3.3 |
| C: NPH | 53.1 | 4.1 (1.0) | 8.2 (1.4) | — | Dermatologicals 15.6 Cardiovascular system 12.5 Repiratory system 15.6 |
Diabetic retinopathy 0 Motor neuropathy 0 Autonomic neuropathy 1.6 Nephropathy 0 Albuminuria 1.6 |
|
| Ratner 2000 | I: glargine | 46.6 | 38.2 (12.2) | 7.7 (1.2) | 25.6 (4.0) | — | Smoker 14.1 Cardiovascular disease 10.4 |
| C: NPH | 52.2 | 38.9 (11.9) | 7.7 (1.1) | 25.9 (4.6) | — | ||
| Robertson 2007 | I: detemir | 48.7 | 11.9 (2.8) | 8.8 (1.2) | 19.2c | Paracetamol 49.6a Ibuprofen 15.1 Acetylsalicyl acid 5.6 |
Retinopathy 0a Neuropathy 0 Nephropathy 0.4 |
| C: NPH | 52.2 | 11.7 (2.7) | 8.7 (1.1) | 19.1 | Paracetamol 46.1 Ibuprofen 11.3 Acetylsalicyl acid 7.0 |
Retinopathy 1.7 Neuropathy 0 Nephropathy 0.9 |
|
| Russell‐Jones 2004 | I: detemir | 34.4 | 40.9 (12.4) | 8.4 (1.2) | 25.1 (3.4) | Paracetamol 35.6a Ibuprofen 12.6 Acetylsalicylic acid 7.4 | Essential hypertension 13 Retinal disorders 11 Disorder of lipid metabolism 10 |
| C: NPH | 38.7 | 39.8 (12.3) | 8.4 (1.2) | 25.4 (3.4) | |||
| Schober 2002 | I: glargine | 44.3 | 11.8 (2.5) | 8.5 (1.4) | 18.8 (2.8) | Concomitant medication other than glucose‐lowering drugs 52.6a | One patient presented with macroalbuminuria and three presented with microalbuminuria at study entry |
| C: NPH | 52.0 | 11.5 (2.4) | 8.8 (1.4) | 18.9 (2.9) | Concomitant medication other than glucose‐lowering drugs 56.6 | ||
| Standl 2004d | I: detemir | 38.6 | 38.6 (13.4) | 7.6 (1.2) | 25.3 (3.2) | Paracetamol 20a Acetylsalicylic acid 17 |
Neuropathy 9 Both groups: Essential hypertension 21 Retinal disorders 5 Neuropathy 13 Disorders of the lipoid metabolism 10 |
| C: NPH | 36.6 | 39.8 (12.2) | 7.7 (1.2) | 25.2 (3.3) | Neuropathy 16 | ||
| SWITCH 1 | I: degludec | 49.4 | 45.4 (13.7) | 7.7 (1.0) | 27.9 (5.1) | — | — |
| C: glargine | 43.3 | 46.4 (14.6) | 7.5 (1.0) | 27.0 (4.5) | — | — | |
| Thalange 2013 | I: detemir | 53.1 | 10.0 (4.1) | 8.4 (1.1) | 18 (2.7)a | Paracetamol 37.3a Ibuprofen 19.2 |
Diabetic nephropathy 1.7a Diabetic neuropathy 2.3 Diabetic retinopathy 1.7 Macroangiopathy 0 |
| C: NPH | 42.9 | 9.8 (3.9) | 8.4 (1.1) | 18 (2.7) | Paracetamol 34.7 Ibuprofen 19.4 |
Diabetic nephropathy 1.8 Diabetic neuropathy 2.4 Diabetic retinopathy 0 Macroangiopathy 0 |
|
| Urakami 2017 | I: degludec | 30.0 | 10 (1.5) | 7.7 (0.9) | 16.0 (4.5) | — | None had microvascular complications |
| C: glargine | 44.4 | 11 (1.5) | 7.8 (0.9) | ||||
| Vague 2003 | I: detemir | 46.2 | 38.9 (13.3) | 8.18 (1.14) | 24.5 (3.2) | — | — |
| C: NPH | 49.3 | 41.8 (14.2) | 8.11 (1.12) | 24.6 (3.4) | — | — | |
| —: denotes not reported aData from clinical study report/synopsis. Additional information available in clinical study report/synopsis. bNot reported if SD or standard error was provided. Assumed to be standard error, so SD was calculated from anticipated standard error. cData from Food and Drug Administration medical review. dIn publication, baseline characteristics were only available for the participants completing the 6 months treatment period and participating in the 6 months extension period (NPH; N = 135; Detemir; N = 154). Data on randomised participants available from clinical study report. BMI: body mass index; C: comparator; HbA1c: glycosylated haemoglobin A1c; I: intervention; NPH: neutral protamine Hagedorn; SD: standard deviation. | |||||||
Appendix 11. Study endpoints and timing of outcome measurement
| Study ID | Review's primary and secondary outcomes | Timing of outcome measurement in study |
| Bartley 2008 | Hypoglycaemia and safety data | At each visit (baseline, 4 weeks, 8 weeks, 12 weeks, 24 weeks, 36 weeks, 52 weeks, 64 weeks, 76 weeks, 88 weeks, 104 weeks)a |
| HbA1c | Baseline, 12 weeks, 24 weeks, 36 weeks, 52 weeks, 64 weeks, 76 weeks, 88 weeks, 104 weeksa | |
| BEGIN Basal‐Bolus Type 1 | Hypoglycaemia and safety data | Baseline, every second week during study (main study) (for extension study every 4th week)a |
| HbA1c | Baseline, 12 weeks, 16 weeks, 26 weeks | |
| BEGIN Flex T1 | Hypoglycaemia and safety | Baseline, every second week during study |
| HbA1c | Baseline, 12 weeks, 16 weeks, 26 weeks | |
| BEGIN Young | Hypoglycaemia and safety | Every visit (i.e. every third week during study) |
| HbA1c | Baseline, 12 weeks, 16 weeks, 38 weeks, 52 weeks | |
| Bolli 2009 | Quality of life | Baseline, 12 weeks, 24 weeks |
| HbA1c | Baseline, 8 weeks, 16 weeks, 24 weeks | |
| Safety | At each visit (number of visits not described) | |
| Chase 2008 | Quality of life | Baseline, 2 weeks, 6 weeks, 12 weeks, 18 weeks, 24 weeksa |
| HbA1c and hypoglycaemia | During clinical visits at 6 weeks, 12 weeks, 18 weeks, 24 weeks | |
| Adverse events | Every 1 week of follow‐up | |
| Davies 2014 | Quality of life | Baseline, 12 weeks, 26 weeks |
| Adverse events and hypoglycaemia | Baseline, 1 week, 2 weeks and thereafter every second week | |
| HbA1c | Baseline, 12 weeks, 16 weeks, 26 weeks | |
|
Fulcher 2005a |
Quality of life | Baseline, 14 weeks, 30 weeks |
| Adverse events | 6 weeks, 12 weeks, 18 weeks, 14 weeks, 30 weeks | |
| HbA1c | 6 weeks, 14 weeks, 22 weeks, 30 weeks | |
| Hypoglycaemia | 6 weeks, 12 weeks, 18 weeks, 14 weeks, 30 weeks | |
| Economic data | 14 weeks, 30 weeks | |
| Heller 2009 | Hypoglycaemic and adverse events | Baseline, 2 weeks, 4 weeks, 6 weeks, 8 weeks, 12 weeks, 18 weeks, 24 weeks, 50 weeks, 36 weeks, 44 weeks, 52 weeks |
| HbA1c | Baseline, 12 weeks, 24 weeks, 36 weeks, 52 weeks | |
| Home 2005 | Quality of life | Baseline, 8 weeks, 20 weeks, 28 weeks |
| Hypoglycaemia, adverse events, HbA1c | Baseline, 1 week, 4 weeks, 8 weeks, 12 weeks, 20 weeks, 28 weeks | |
| Kobayashi 2007 | Hypoglycaemia, adverse events, HbA1c | Baseline, 48 weeks |
| Liu 2016 | HbA1c | Baseline, 12 weeks, 24 weeks |
| Hypoglycaemia, safety | Baseline, 1 week, 2 weeks, 4 weeks, 6 weeks, 8 weeks, 10 weeks, 12 weeks, 16 weeks, 20 weeks, 24 weeks, 25 weeksa | |
| NCT00595374 | Mortality, adverse events | — |
| NCT00605137 | Hypoglycaemia, adverse events | Baseline, 1 week, 2 weeks, 3 weeks, 4 weeks, 6 weeks, 8 weeks, 10 weeks, 12 weeks, 14 weeks, 16 weeks, 18 weeks, 20 weeks, 22 weeks, 24 weeks |
| HbA1c | Baseline, 4 weeks, 8 weeks, 12 weeks, 16 weeks, 20 weeks, 24 weeks | |
| Pieber 2007 | Hypoglycaemia and adverse events | Baseline, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 8 weeks, 14 weeks, 20 weeks, 26 weeks |
| HbA1c | Baseline, 20 weeks, 26 weeks | |
| Porcellati 2004 | Hypoglycaemia | — |
| HbA1c | Not reported, but based on figure 2 in main publication, then every second month (0, 2, 4, 6, 8, 10, 12 months) | |
| PRESCHOOL | Adverse events, hypoglycaemia | Baseline, 1 week, 2 weeks, 4 weeks, 6 weeks, 8 weeks, 10 weeks, 12 weeks, 16 weeks, 20 weeks, 24 weeks, 26 weeks |
| HbA1c | Baseline, 12 weeks, 24 weeks | |
| Ratner 2000 | Hypoglycaemia and adverse events | Baseline, 1 week, 4 weeks, 8 weeks, 12 weeks, 20 weeks, 28 weeksa |
| Quality of life | Baseline, 8 weeks, 20 weeks, 28 weeksa | |
| Pharmacoeconomic assessment | Baseline, 1 week, 4 weeks, 8 weeks, 12 weeks, 20 weeks, 28 weeksa | |
| HbA1c | Baseline, 8 weeks, 20 weeks, 28 weeks | |
| Robertson 2007a | Hypoglycaemia and safety | Baseline, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 10 weeks, 18 weeks, 26 weeks |
| HbA1c | Baseline, 18 weeks, 24 weeks | |
| Russell‐Jones 2004 | Hypoglycaemia and adverse events | Baseline, 2 weeks, 4 weeks, 9 weeks, 13 weeks, 19 weeks, 26 weeks |
| HbA1c | Baseline, 3 months, 6 months | |
| Schober 2002 | Hypoglycaemia and adverse events | Baseline, 4 weeks, 16 weeks, 28 weeks |
| HbA1c | Baseline, 4 weeks, 16 weeks, 28 weeks | |
| Standl 2004 | Quality of life | Baseline, 13 weeks, 26 weeks |
| Safety and hypoglycaemia | Baseline, 3 months, 6 months (extension: 9 months, 12 months) | |
| HbA1c | Baseline, 3 months, 6 months (extension: 9 months, 12 months) | |
| SWITCH 1 | Quality of life | Baseline, 32 weeks |
| Hypoglycaemia/adverse events | Weekly during study period | |
| HbA1c | Baseline, 8 weeks, 12 weeks, 16 weeks, 20 weeks, 24 weeks, 28 weeks, 32 weeks | |
| Thalange 2013 | Adverse events, hypoglycaemia | Collected during the study |
| HbA1c | Baseline, 12 weeks, 24 weeks, 38 weeks, 52 weeks | |
| Urakami 2017 | HbA1c and hypoglycaemia | Baseline, 4 weeks, 12 weeks, 24 weeks |
| Vague 2003a | Hypoglycaemia, safety | Baseline, 2 weeks, 4 weeks, 9 weeks, 13 weeks, 19 weeks, 26 weeks, 27 weeks |
| HbA1c | Baseline, 13 weeks, 26 weeks | |
| —: denotes not reported
aInformation retrieved from clinical study report. HbA1c: glycosylated haemoglobin A1c. | ||
Appendix 12. Matrix of study endpoints (publications and trial documents)
| Study ID | |
| Bartley 2008 | Endpoints quoted in trial registersa |
|
Source:NCT00184665 Primary outcome measure(s): HbA1c Secondary outcome measure(s): adverse events, body weight, antibodies, body composition, blood glucose, hypoglycaemia Other outcome measure(s): — Trial results available in trials register: yes | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): FPG, nocturnal hypoglycaemia, weight, safety, insulin antibodies Other outcome measure(s): — | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): FPG, nocturnal hypoglycaemia, weight, safety Other outcome measure(s): — | |
| BEGIN Basal‐Bolus Type 1 | Endpoints quoted in trial registersa |
|
Source:NCT00982228 (main study); NCT00982228 (extension) Primary outcome measure(s): Main study: change in HbA1c after 52 weeks Extension: adverse events from week 0 to 104 + 7 days, confirmed hypoglycaemic episodes from week 0 to 104 + 7 days, cross‐reacting antibodies to human insulin (extension study) Secondary outcome measure(s): Main study: confirmed hypoglycaemic episodes, nocturnal confirmed hypoglycaemic episodes, mean of 9‐point SMBG profile at week 52 Extension: nocturnal confirmed hypoglycaemic episodes, change in HbA1c after 104 weeks, mean of 9‐point SMPG profile at week 104 Other outcome measure(s): — Trial results available in trials register: no | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): Main study: HbA1c Extension: hypoglycaemia, AEs Secondary outcome measure(s): Main study: all predefined outcomes Extension: all predefined outcomes Other outcome measure(s): Main study: adverse events, QoL Extension: insulin dose | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): main study: HbA1c, extension study: hypoglycaemia Secondary outcome measure(s): main study: hypoglycaemia, extension study: glycaemic measures Other outcome measure(s): main study: adverse events, extension study: insulin dose | |
| BEGIN Flex T1 | Endpoints quoted in trial registersa |
|
Source:NCT01079234 Primary outcome measure(s): Main study: HbA1c Extension: confirmed hypoglycaemic episodes, nocturnal confirmed hypoglycaemic episodes Secondary outcome measure(s): Main study: FPG Extension: HbA1c, FPG Other outcome measure(s): — Trial results available in trials register: yes | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): main study: HbA1c, extension study: rate of confirmed hypoglycaemic episodes, rate of nocturnal confirmed hypoglycaemic episodes Secondary outcome measure(s): main study: FPG; extension study: HbA1c, FPG Other outcome measure(s): safety, insulin dose, weight | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): main study: HbA1c, extension study: rate of confirmed hypoglycaemic episodes, rate of nocturnal confirmed hypoglycaemic episodes Secondary outcome measure(s): — Other outcome measure(s): — | |
| BEGIN Young | Endpoints quoted in trial registersa |
|
Source:NCT01513473 Primary outcome measure(s): change in HbA1c at 26 weeks Secondary outcome measure(s): change in HbA1c at 52 weeks, change in FPG at 26 weeks and 52 weeks, adverse events at 26 weeks and 52 weeks, hypoglycaemia at 26 weeks and 52 weeks, self‐measured hyperglycaemia at 26 and 52 weeks, episodes with blood ketones above 1.5 mmol/L at 26 weeks and 52 weeks, steady‐state plasma concentrations of insulin during study, insulin antibodies Other outcome measure(s): — Trial results available in trials register: yes | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): main publication: HbA1c Secondary outcome measure(s): main publication: FPG, hypoglycaemia, adverse events, hyperglycaemia with ketosis Other outcome measure(s): insulin dose | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): FPG, hypoglycaemia, adverse events, hyperglycaemia with ketosis Other outcome measure(s): insulin dose | |
| Bolli 2009 | Endpoints quoted in trial registersa |
| Source: NT | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): glycaemic measures Secondary outcome measure(s): QoL, safety Other outcome measure(s): — | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): glycaemic measures Secondary outcome measure(s): safety Other outcome measure(s): — | |
| Chase 2008 | Endpoints quoted in trial registersa |
|
Source:NCT00046501 Primary outcome measure(s): HbA1c from baseline to end of follow‐up Secondary outcome measure(s): HbA1c at different time points, percentage achieving HbA1c target, change in SMBG, albumin/creatinine ratio, insulin dose, lipids, hypoglycaemia, adverse events, weight Other outcome measure(s): — Trial results available in trials register: no | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia, blood glucose, insulin dose, SAEs, percentage achieving HbA1c target Other outcome measure(s): — | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia Other outcome measure(s): — | |
| Davies 2014 | Endpoints quoted in trial registersa |
|
Source:NCT01074268 Primary outcome measure(s): Main study: change from baseline in HbA1c after 26 weeks of treatment Extension: adverse events Secondary outcome measure(s): Main study: mean of 9‐point SMPG profile, change in FPG, confirmed hypoglycaemic episodes, nocturnal confirmed hypoglycaemic episodes Extension: change in HbA1c, mean of 9‐point SMPG profile, change in FPG, confirmed hypoglycaemic episodes, nocturnal confirmed hypoglycaemic episodes Other outcome measure(s): none Trial results available in trials register: yes | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): Main publication: change in HbA1c Extension: adverse events Secondary outcome measure(s): Main study: FPG, 9‐point SMPG profiles and doses of basal and mealtime insulin Extension: hypoglycaemia, immunogenicity, insulin dose and body weight Other outcome measure(s): safety variables included number of hypoglycaemic episodes, adverse events, body weight, standard clinical and laboratory assessments (including insulin antibodies), electrocardiogram, fundoscopy/fundus photography and injection‐site reactions | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): FPG Other outcome measure(s): hypoglycaemia, adverse events | |
| Fulcher 2005 | Endpoints quoted in trial registersa |
| Source: NT | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): change in HbA1c Secondary outcome measure(s): FBG, FBG variability, HbA1c response rates, hypoglycaemia, body weight, lipid profiles, adverse events Other outcome measure(s): insulin dose, titration index | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): FBG, hypoglycaemia Other outcome measure(s): — | |
| Heller 2009 | Endpoints quoted in trial registersa |
|
Source:NCT00095082; Eudra-CT 2004-000086-35 Primary outcome measure(s): HbA1c Secondary outcome measure(s): adverse events, body weight, hypoglycaemia, blood glucose, insulin treatment satisfaction (clinical trials register EU: proportion of participants with HbA1c ≤ 7.0 % without any episodes of major hypoglycaemia during the last month of treatment) Other outcome measure(s): — Trial results available in trials register: no | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): FPG; within‐patient variation in SMPG before breakfast and dinner; and 10‐point SMPG profiles, hypoglycaemia, adverse events, weight, proportion of participants with HbA1c ≤ 7.0 % without any episodes of major hypoglycaemia during the last month of treatment Other outcome measure(s): — | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): withdrawal due to adverse events, nocturnal hypoglycaemia, proportion of participants with HbA1c ≤ 7.0 % without any episodes of major hypoglycaemia during the last month of treatment Other outcome measure(s): insulin dose | |
| Home 2005 | Endpoints quoted in trial registersa |
| Source: NT | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): plasma glucose, SMBG and hypoglycaemia Other outcome measure(s): retinopathy, insulin antibodies, adverse events | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): FBG, hypoglycaemia Other outcome measure(s): adverse events, clamp investigations | |
| Kobayashi 2007 | Endpoints quoted in trial registersa |
|
Source:NCT00604344 Primary outcome measure(s): HbA1c Secondary outcome measure(s): blood glucose, hypoglycaemia, adverse events, body weight, insulin antibodies Other outcome measure(s): — (from synopsis: insulin treatment questionnaire (questions concerning glycaemic control, insulin therapy related QoL at night [ITR‐QOLN] and insulin treatment satisfaction questionnaire Japan [ITSQ‐J])) Trial results available in trials register: no | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): blood glucose, hypoglycaemia, adverse events, body weight, insulin antibodies Other outcome measure(s): — | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): blood glucose, hypoglycaemia, adverse events, body weight Other outcome measure(s): — | |
| Liu 2016 | Endpoints quoted in trial registersa |
|
Source:NCT01223131; Eudra-CT 2014-004640-35 Primary outcome measure(s): HbA1c over 24 weeks Secondary outcome measure(s): percentage achieving HbA1c < 7.5%, blood glucose, SMBG, insulin dose, hypoglycaemia, safety, antibodies, pharmacokinetics Other outcome measure(s): — Trial results available in trials register: yes (EudraCT) | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): percentage achieving HbA1c < 7.5%, blood glucose, SMBG, insulin dose, hypoglycaemia, safety, antibodies, pharmacokinetics Other outcome measure(s): — | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia Other outcome measure(s): — | |
| NCT00595374 | Endpoints quoted in trial registersa |
|
Source:NCT00595374 Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia, adverse events, blood glucose, body weight, QoL Other outcome measure(s): — Trial results available in trials register: no | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): — Secondary outcome measure(s): — Other outcome measure(s): — | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): — Secondary outcome measure(s): — Other outcome measure(s): — | |
| NCT00605137 | Endpoints quoted in trial registersa |
|
Source:NCT00605137 Primary outcome measure(s): safety profile (incidence of hypoglycaemia, adverse events, laboratory assessments, BMI, blood pressure, fundoscopy) Secondary outcome measure(s): laboratory assessments and other safety endpoints, HbA1c, blood glucose Other outcome measure(s): height, insulin dose Trial results available in trials register: no | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): — Secondary outcome measure(s): — Other outcome measure(s): — | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): — Secondary outcome measure(s): — Other outcome measure(s): — | |
| Pieber 2007 | Endpoints quoted in trial registersa |
|
Source:NCT00312104 Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia, adverse events, blood glucose (from CSR: treatment satisfaction and pain perception) Other outcome measure(s): Trial results available in trials register: no | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia, adverse events, blood glucose Other outcome measure(s): insulin dose, weight | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia, adverse events, blood glucose Other outcome measure(s): insulin dose, weight | |
| Porcellati 2004 | Endpoints quoted in trial registersa |
| Source: NT | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): — Other outcome measure(s): glycaemic control, hypoglycaemia, clamp data, weight (co‐publication: well‐being and treatment satisfaction) | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): — Other outcome measure(s): glycaemic control, hypoglycaemia (co‐publication: well‐being and treatment satisfaction) | |
| PRESCHOOL | Endpoints quoted in trial registersa |
|
Source:NCT00993473; Eudra CT 2009-011231-12 Primary outcome measure(s): all hypoglycaemia Secondary outcome measure(s): symptomatic hypoglycaemia, severe hypoglycaemia, nocturnal hypoglycaemia, severe nocturnal hypoglycaemia, HbA1c, percentage with HbA1c < 7.5%, CGM Other outcome measure(s): — Trial results available in trials register: yes | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): all hypoglycaemia Secondary outcome measure(s): symptomatic hypoglycaemia, severe hypoglycaemia, nocturnal hypoglycaemia, severe nocturnal hypoglycaemia, HbA1c, percentage with HbA1c < 7.5%, CGM Other outcome measure(s): treatment emergent adverse events | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): all hypoglycaemia Secondary outcome measure(s): symptomatic hypoglycaemia, severe hypoglycaemia, nocturnal hypoglycaemia, severe nocturnal hypoglycaemia, HbA1c, CGM Other outcome measure(s): — | |
| Ratner 2000 | Endpoints quoted in trial registersa |
| Source: NT | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): glycaemic control and hypoglycaemia Secondary outcome measure(s): — Other outcome measure(s): safety, insulin dose | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): glycaemic control, hypoglycaemia Secondary outcome measure(s): — Other outcome measure(s): — | |
| Robertson 2007 | Endpoints quoted in trial registersa |
|
Source:NCT00312156 Primary outcome measure(s): HbA1c Secondary outcome measure(s): adverse events, body weight, antibodies, blood glucose, hypoglycaemia (in CSR also: incidence of diabetic ketoacidosis requiring hospitalisation) Other outcome measure(s): — Trial results available in trials register: no | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): adverse events, body weight, antibodies, blood glucose, hypoglycaemia, (ketoacidosis) Other outcome measure(s): — | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): adverse events, blood glucose, hypoglycaemia Other outcome measure(s): — | |
| Russell‐Jones 2004 | Endpoints quoted in trial registersa |
|
Source:NCT03220425 Primary outcome measure(s): HbA1c Secondary outcome measure(s): (from CSR: blood glucose, hypoglycaemia, safety profile, antibodies) Other outcome measure(s): — Trial results available in trials register: no | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): — Other outcome measure(s): FPG, nocturnal hypoglycaemia, weight, adverse events | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): — Other outcome measure(s): FPG, nocturnal hypoglycaemia, weight, adverse events | |
| Schober 2002 | Endpoints quoted in trial registersa |
| Source: NT | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): no study protocol available, but HbA1c described as primary outcome in main publication Secondary outcome measure(s): no study protocol available, but FBG and hypoglycaemia described as secondary outcomes in main publication Other outcome measure(s): antibodies, adverse events | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): FBG and hypoglycaemia Other outcome measure(s): adverse events | |
| Standl 2004 | Endpoints quoted in trial registersa |
| Source: NT | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): 9‐point blood glucose profiles, hypoglycaemia, FPG, adverse events Other outcome measure(s): weight | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): 9‐point blood glucose profiles, hypoglycaemia, FPG, adverse events Other outcome measure(s): weight | |
| SWITCH 1 | Endpoints quoted in trial registersa |
|
Source:NCT02034513 Primary outcome measure(s): severe or blood glucose confirmed symptomatic hypoglycaemic episodes (maintenance period) Secondary outcome measure(s): severe or blood glucose confirmed symptomatic nocturnal hypoglycaemic episodes (maintenance period), proportion of participants with one or more severe hypoglycaemic episodes (maintenance period), incidence of adverse events (32 weeks for each treatment period), change from baseline in HbA1c, FPG Other outcome measure(s): (according to appendix to main publication: Treatment Related Impact Measure for minor HYPOglycaemic events (TRIM‐HYPO) and SF‐36 v2) Trial results available in trials register: yes | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): hypoglycaemia Secondary outcome measure(s): glycaemic variables Other outcome measure(s): insulin dose, (co‐publication: cost, HbA1c/severe hypoglycaemia) | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): hypoglycaemia
Secondary outcome measure(s): Other outcome measure(s): (co‐publication: cost, HbA1c/severe hypoglycaemia) | |
| Thalange 2013 | Endpoints quoted in trial registersa |
|
Source:NCT00435019 (main study); NCT00623194 (extension study) Primary outcome measure(s): Main study: HbA1c (after 52 weeks of treatment) Extension: insulin antibodies Secondary outcome measure(s): Main study: adverse events, insulin antibodies; extension study: insulin antibodies, HbA1c, FPG, hypoglycaemia, BMI, body weight, ketoacidosis, insulin dose, laboratory values, fundoscopy, blood pressure, pulse Other outcome measure(s): — Trial results available in trials register: yes | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): adverse events Other outcome measure(s): weight, insulin dose, hypoglycaemia | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia Other outcome measure(s): weight, insulin dose, glucose | |
| Urakami 2017 | Endpoints quoted in trial registersa |
| Source: NT | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): — Secondary outcome measure(s): — Other outcome measure(s): | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): — Secondary outcome measure(s): — Other outcome measure(s): | |
| Vague 2003 | Endpoints quoted in trial registersa |
| Source: NT | |
| Endpoints quoted in publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia, safety, glucose Other outcome measure(s): weight, insulin dose | |
| Endpoints quoted in abstract of publication(s)a,b | |
|
Primary outcome measure(s): HbA1c Secondary outcome measure(s): hypoglycaemia, safety, glucose Other outcome measure(s): weight, insulin dose | |
| — denotes not reported
aPrimary and secondary outcomes refer to verbatim specifications in publication/records. Unspecified outcome measures refer to all outcomes not described as primary or secondary outcome measures. bPublication(s) refers to study information published in scientific journals (primary reference, duplicate publications, companion documents or multiple reports of a primary study). AE: adverse event; BMI: body mass index; CGM: continuous glucose monitoring; CSR: clinical study report;EMA: European Medicines Agency; FBG: fasting blood glucose; FDA: Food and Drug Administration (US); FPG: fasting plasma glucose; HbA1c: glycosylated haemoglobin A1c; ITR‐QOLN: insulin therapy related quality of life at night; ITSQ‐J: insulin treatment satisfaction questionnaire ‐ Japan; NT: no trial register document available; QoL: quality of life; SAE: serious adverse event; SF‐36: short‐form 36; SMBG: self‐measured blood glucose; SMPG: self‐measured plasma glucose. | |
Appendix 13. High risk of outcome reporting bias according to Outcome Reporting Bias In Trials (ORBIT) classification
| Study ID | Outcome | High risk of bias (category A)a | High risk of bias (category D)b | High risk of bias (category E)c | High risk of bias (category G)d |
| Bartley 2008 | ND | ||||
| BEGIN Basal‐Bolus Type 1 | ND | ||||
| BEGIN Flex T1 | ND | ||||
| BEGIN Young | ND | ||||
| Bolli 2009 | Severe hypoglycaemia | Yes | |||
| Moderate/mild/nocturnal hypoglycaemia | Yes | ||||
| Chase 2008 | ND | ||||
| Davies 2014 | ND | ||||
| Fulcher 2005 | ND | ||||
| Heller 2009 | ND | ||||
| Home 2005 | ND | ||||
| Kobayashi 2007 | Health‐related quality of life | Yes | |||
| Liu 2016 | ND | ||||
| NCT00595374 | Health‐related quality of life | Yes | |||
| Diabetic ketoacidosis | Yes | ||||
| Mild/moderate hypoglycaemia | Yes | ||||
| Nocturnal hypoglycaemia | Yes | ||||
| HbA1c | Yes | ||||
| NCT00605137 | ND | ||||
| Pieber 2007 | Severe hypoglycaemia combined with HbA1c | Yes | |||
| Porcellati 2004 | Adverse events (severe and non‐severe) | Yes | |||
| PRESCHOOL | ND | ||||
| Ratner 2000 | ND | ||||
| Robertson 2007 | HbA1c combined with hypoglycaemia | Yes | |||
| Russell‐Jones 2004 | Diabetic ketoacidosis | Yes | |||
| HbA1c combined with hypoglycaemia | |||||
| Schober 2002 | All‐cause mortality | Yes | |||
| Standl 2004 | Diabetic ketoacidosis | Yes | |||
| Non‐fatal myocardial infarction | Yes | ||||
| Non‐fatal stroke | Yes | ||||
| SWITCH 1 | Mortality, hypoglycaemia, safety | Yes (not reported at cross‐over) | |||
| Thalange 2013 | ND | ||||
| Urakami 2017 | ND | ||||
| Vague 2003 | HbA1c combined with hypoglycaemia | Yes | |||
|
aClear that outcome was measured and analysed; study report stated that outcome was analysed but reported only that the result was not significant
(Classification 'A', table 2, Kirkham 2010)
bClear that outcome was measured and analysed;study report stated that outcome was analysed but reported no results
(Classification 'D', table 2, Kirkham 2010)
cClear that outcome was measured but was not necessarily analysed; judgement suggests that likely to have been analysed but not reported due to non‐significant results
(Classification 'E', table 2, Kirkham 2010)
dUnclear whether outcome was measured; not mentioned, but clinical judgement suggests likely to have been measured and analysed but not reported on the basis of non‐significant results
(Classification 'G', table 2, Kirkham 2010) CSR: clinical study report;HbA1c: glycosylated haemoglobin A1c; ND: none detected. | |||||
Appendix 14. Definition of endpoint measurementa
| Study ID | Endpoints | Definition |
| Bartley 2008 | All‐cause mortality | ND (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Hypoglycaemia requiring third party assistance (IO) | |
| Cardiovascular mortality | One participant died due to cardiovascular disease (IO) | |
| Non‐fatal myocardial infarction | Acute myocardial infarctionb (IO) | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosisb (IO) | |
| Non‐serious adverse events | An adverse event is any undesirable medical event occurring to a participant in a clinical study, whether or not related to the study product(s). A non‐serious adverse event is any adverse event which does not fulfil the definition of a serious adverse eventb (SO) | |
| Noctural hypoglycaemia | Hypoglycaemia between 23:00 to 06:00 h (SO) | |
| Mild/moderate hypoglycaemia | All SMPG values < 3.1 mmol/L as well as signs and symptoms of hypoglycaemia minor if plasma glucose < 3.1 mmol/L and the individual dealt with the episode him/herself, and as symptoms only if episodes were not confirmed by a plasma glucose measurement and no assistance was required (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | Percentage of participants reaching HbA1c ≤ 7.0% at the end of the study without symptomatic hypoglycaemia with a plasma glucose < 4.0 mmol/L or any single plasma glucose value < 3.1 mmol/L during the last month of treatment" (IO) | |
| BEGIN Basal‐Bolus Type 1 | All‐cause mortality | All‐cause mortality (IO, AO) |
| Health‐related quality of life | Short Form‐36 v2 (SO) | |
| Severe hypoglycaemia | Hypoglycaemia requiring third party assistance (IO) | |
| Cardiovascular mortality | Cause of each death described separately (myocardial infarction event; sudden death; ventricular tachycardia event) (IO, AO) | |
| Non‐fatal myocardial infarction | Myocardial infarction (IO, AO) | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Ketoacidosis (IO) | |
| Non‐serious adverse events | A non‐serious adverse event is any adverse event which does not fulfil the definition of a serious adverse eventb (SO) | |
| Noctural hypoglycaemia | Hypoglycaemic episodes occurring from 00:01 to 05:59 h (SO) | |
| Mild/moderate hypoglycaemia | Confirmed hypoglycaemic episodes included those with a plasma glucose value of < 3.1 mmol/L (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND | |
| Combined HbA1c and severe hypoglycaemia | HbA1c < 7% without severe hypoglycaemiab (IO) | |
| BEGIN Flex T1 | All‐cause mortality | Fatal serious adverse events (one committed suicide) (IO, AO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Hypoglycaemia requiring third party assistance (IO, AO) | |
| Cardiovascular mortality | ND | |
| Non‐fatal myocardial infarction | Acute coronary syndrome (IO, AO) | |
| Non‐fatal stroke | Stroke (IO, AO) | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect; • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | A non‐serious adverse event is any adverse event that does not fulfil the definition of a serious adverse eventb (SO) | |
| Nocturnal hypoglycaemia | Episodes occurring between 00:01 and 05:59 hours (inclusive) (SO) | |
| Mild/moderate hypoglycaemia | Minor hypoglycaemic episodes are defined as participants able to treat her/himself and plasma glucose below 3.1 mmol/L (OBS page 68 + 69 in CSR ‐ different definitions) (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | Treatment targets at the end of study achieved without hypoglycaemic episodes in the last 12 weeks of treatment considering severe episodes only, and severe and minor episodes togetherb (IO) | |
| BEGIN Young | All‐cause mortality | ND (IO, AO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | The child has altered mental status and cannot assist in their own care, is semiconscious or unconscious, or in a coma ± convulsions and may require parenteral therapy (glucagon or iv glucose) (IO) | |
| Cardiovascular mortality | ND (IO, AO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect; • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | A non‐serious AE is any AE which does not fulfil the definition of an SAEb (IO) | |
| Nocturnal hypoglycaemia | Hypoglycaemic episodes occurring between 11 p.m. and 7 a.m. inclusive were classified as nocturnal (SO) | |
| Mild/moderate hypoglycaemia | Confirmed hypoglycaemia was defined as SMPG < 3.1 mmol/Lc (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Bolli 2009 | All‐cause mortality | — |
| Health‐related quality of life | Well‐Being Enquiry for Diabetics questionnaire (SO) | |
| Severe hypoglycaemia | Serious hypoglycaemia was defined as an event with blood glucose < 2.3 mmol/L, severe hypoglycaemia an event with symptoms consistent with hypoglycaemia, during which the participant required the assistance of another person, or with prompt recovery after oral carbohydrate, iv glucose or glucagon administration (IO) | |
| Cardiovascular mortality | — | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | Serious adverse events (IO) | |
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | Adverse events (SO) | |
| Nocturnal hypoglycaemia | Serious nocturnal hypoglycaemia (blood glucose < 2.3 mmol/Lc); hypoglycaemia which occurred between bedtime and before getting up in the morning (IO) | |
| Mild/moderate hypoglycaemia | Blood glucose ≤ 4.0 mmol/Lc (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Chase 2008 | All‐cause mortality | — |
| Health‐related quality of life | The Diabetes Quality of Life for Youth questionnaire and Parents' Diabetes Quality of Lifeb (SO) | |
| Severe hypoglycaemia | Severe hypoglycaemia was defined as an event requiring assistance from another person and associated with either BG < 2.0 mmol/L or prompt recovery after oral carbohydrate, iv glucose, or intramuscular or subcutaneous glucagon administration (IO) | |
| Cardiovascular mortality | — | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect; • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | The term adverse event covered any unfavourable and unintended sign, symptom, syndrome, or illness that developed or worsened during the period of observation in the clinical studyb (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia from midnight and 6 a.m. (SO) | |
| Mild/moderate hypoglycaemia | The rates of biochemical hypoglycaemia were ascertained by analysis of SMBG data and divided into 3 categories: < 3.9 mmol/L, < 2.8 mmol/L and < 2.0 mmol/Lc (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Davies 2014 | All‐cause mortality | All‐cause mortality (IO, AO) |
| Health‐related quality of life | Short Form‐36 v2b (SO) | |
| Severe hypoglycaemia | Hypoglycaemia requiring third party assistance (IO) | |
| Cardiovascular mortality | Cardiovascular mortality (IO, AO) | |
| Non‐fatal myocardial infarction | Non‐fatal myocardial infarction (IO, AO) | |
| Non‐fatal stroke | Non‐fatal stroke (IO, AO) | |
| End‐stage renal disease | End‐stage renal disease (IO) | |
| Blindness | Blindness (IO) | |
| Serious adverse events | Serious adverse events: adverse event that at any dose results in any of the following death, a life‐threatening experience, in‐participant hospitalisations/prolongation of existing hospitalisation, persistent/significant disability/incapacity/congenital anomaly/birth defect or important medical issues (IO) | |
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | Mild: no/transient symptoms, no interference with participant's daily activities. Moderate: marked symptoms, moderate interference with participant's daily activities (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia between 00:01 and 05:59 hours (SO) | |
| Mild/moderate hypoglycaemia | Confirmed hypoglycaemia was defined as plasma glucose < 3.1 mmol/L regardless of symptoms (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | HbA1c < 7% without confirmed severe hypoglycaemia during the last 12 weeks of treatmentb (IO) | |
| Fulcher 2005 | All‐cause mortality | ND (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Symptoms consistent with hypoglycaemia required the assistance of another person and was associated with a blood glucose level < 2.8 mmol/L or prompt recovery after oral carbohydrate, iv glucose or sc glucagon administration (IO) | |
| Cardiovascular mortality | ND (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect; • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | Adverse event covers any sign, symptom, syndrome, or illness that appears or worsens in a patient during the period of observation in the clinical study and that may impair the well‐being of the patient. The term also covers laboratory findings or results of other diagnostic procedures that are considered to be clinically relevantb. A non‐serious adverse event is any adverse event not meeting the serious adverse event criteriab (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia occurring after the evening insulin injection and before the morning insulin dose | |
| Mild/moderate hypoglycaemia | Symptomatic hypoglycaemia was defined as an event with symptoms consistent with hypoglycaemia that was mild (2.8–3.6 mmol/L) or moderate (< 2.8 mmol/L) | |
| Socioeconomic effects | Information in relation to whether participants had suffered any income loss because of diabetes during the study (SO, IO) | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Heller 2009 | All‐cause mortality | ND (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | The patient could not treat the episode by himself/herself (IO) | |
| Cardiovascular mortality | One patient died from acute myocardial infarction (IO) | |
| Non‐fatal myocardial infarction | Myocardial ischaemia (IO) | |
| Non‐fatal stroke | Cerebrovascular accident (IO) | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect; • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | A non‐serious adverse event is any adverse event which does not fulfil the definition of a serious adverse eventb (SO) | |
| Nocturnal hypoglycaemia | Episodes of hypoglycaemia occurring from 11 p.m. up to but not including 6 a.m. (SO) | |
| Mild/moderate hypoglycaemia | Minor: the patient could treat himself/herself and the measured plasma glucose value was < 3.1 mmol/L; symptoms only: the patient could treat himself/herself and no plasma glucose measurement was taken or the measured plasma glucose value was ≥ 3.1 mmol/L (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | HbA1c ≤ 7% without major hypoglycaemia during the last month of treatment (IO) | |
| Home 2005 | All‐cause mortality | ND (IO) |
| Health‐related quality of life | Well‐being Questionnaire (W‐BQ) (SO) | |
| Severe hypoglycaemia | Severe symptomatic hypoglycaemia was defined as an event consistent with symptomatic hypoglycaemia requiring the assistance of another person, with either a blood glucose level < 2.8 mmol/L or prompt recovery after administration of oral carbohydrate, iv glucose or glucagon (IO) | |
| Cardiovascular mortality | ND (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect; • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosis | |
| Non‐serious adverse events | A non‐serious adverse event is any adverse event not meeting the serious adverse event criteria (SO) | |
| Nocturnal hypoglycaemia | Symptomatic hypoglycaemia occurring during sleep between bedtime and rising in the morning, or before the morning pre‐breakfast self‐blood glucose measurement and the morning insulin injection. Only participants with confirmed blood glucose < 2.0 mmol/L were considered clinically relevant (SO) | |
| Mild/moderate hypoglycaemia | Hypoglycaemia was categorised as symptomatic (clinical symptoms confirmed by blood glucose < 2.8 mmol/L) or asymptomatic (confirmed by blood glucose < 2.8 mmol/L without symptoms) (SO) | |
| Socioeconomic effects | Information about loss of income during the study (SO, IO) | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Kobayashi 2007 | All‐cause mortality | "No participants died" (IO) |
| Health‐related quality of life | Insulin Therapy Related Quality of Life at Night | |
| Severe hypoglycaemia | Any event requiring assistance of another person to recover from hypoglycaemic symptoms with or without measurement of blood glucose levels (IO) | |
| Cardiovascular mortality | "No participants died" (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | Serious adverse events (IO) | |
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | Adverse events (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia occurring between 23:00 to 06:00 (SO) | |
| Mild/moderate hypoglycaemia | Any symptoms consistent with hypoglycaemia (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Liu 2016 | All‐cause mortality | ND (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Hypoglycaemia requiring the assistance of a third party or involving a seizure, coma, unconsciousness or the use of glucagon (IO) | |
| Cardiovascular mortality | ND (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | An adverse event is any untoward medical occurrence in a patient or clinical investigation where a patient administered a pharmaceutical product and which does not necessarily have to have a causal relationship with the treatmentb (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia occurring between 23:00–07:00 (SO) | |
| Mild/moderate hypoglycaemia | Hypoglycaemia was defined as asymptomatic (blood glucose values < 3.9 mmol/L without clinical symptoms), symptomatic (blood glucose < 3.9 mmol/L with associated clinical symptoms) (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| NCT00595374 | All‐cause mortality | Reported no one died (IO) |
| Health‐related quality of life | Quality of life (SO) | |
| Severe hypoglycaemia | — | |
| Cardiovascular mortality | Reported no one died (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | Serious adverse event (IO) | |
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | Any adverse event that started one day or more after the start of active medication (SO) | |
| Nocturnal hypoglycaemia | — | |
| Mild/moderate hypoglycaemia | — | |
| Socioeconomic effects | — | |
| HbA1c | — | |
| Combined HbA1c and severe hypoglycaemia | — | |
| NCT00605137 | All‐cause mortality | No patients died (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Hypoglycaemia requiring third party assistance (IO) | |
| Cardiovascular mortality | No patients died (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definition(IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | An adverse event is any untoward medical occurrence in a patient or clinical investigation where a patient administered a pharmaceutical product and which does not necessarily have to have a causal relationship with the treatment. A non‐serious adverse event is any adverse event which does not fulfil the definition of a serious adverse event (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia from 23:00 ‐ 06:00, inclusive (SO) | |
| Mild/moderate hypoglycaemia | Minor hypoglycaemic episodes blood glucose < 3.1 mmol/L and able treat the period themselves), symptoms only (no blood glucose measurement or blood glucose > 3.1 mmol/L) and biochemical hypoglycaemia (defined as asymptomatic hypoglycaemic with blood glucose value < 3.1 mmol/L) (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Pieber 2007 | All‐cause mortality | No patients died (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Hypoglycaemia requiring third party assistance (IO) | |
| Cardiovascular mortality | No patients died (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | From CSR: An adverse event (AE) is any undesirable medical event occurring to a participant in a clinical study, whether or not related to the study product(s). A non‐serious adverse event is any AE that does not fulfil the definition of an SAE (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia between 23:00 and 06:00 (SO) | |
| Mild/moderate hypoglycaemia | Confirmed hypoglycaemia if plasma glucose was < 3.1 mmol/L and the individuals dealt with the episode themselves (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | Risk of severe hypoglycaemia adjusted for HbA1c (IO) | |
| Porcellati 2004 | All‐cause mortality | ND (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Hypoglycaemia requiring external help (IO) | |
| Cardiovascular mortality | ND (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | — | |
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | — | |
| Nocturnal hypoglycaemia | Nocturnal episodes of hypoglycaemia were calculated from values measured at 03.00 h or any time between 01.00 and 07.30 h when participants awoke with symptoms suggestive of hypoglycaemia (SO) | |
| Mild/moderate hypoglycaemia | Hypoglycaemia was defined as any episode associated with measurement of blood glucose ≤ 4.0 mmol/L irrespective of symptoms. Hypoglycaemia was considered mild when the episodes were self‐treated by the patients (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| PRESCHOOL | All‐cause mortality | All‐cause mortality (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Severe hypoglycaemia was defined as an event requiring assistance from another person, as a result of altered consciousness, to administer carbohydrate, glucagon or to take other actions (IO) | |
| Cardiovascular mortality | ND (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect; • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | An adverse event is any untoward medical occurrence in a patient or clinical investigation patient administered a pharmaceutical product and which does not necessarily have to have a causal relationship with this treatmentb (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia between 23:00 hours and 07:00 hours (SO) | |
| Mild/moderate hypoglycaemia | Composite hypoglycaemia rate consisting of (i) symptomatic hypoglycaemia episodes, which were recorded in patient diaries, then validated by study investigators; (ii) low CGM glucose excursions (< 3.9 mmol/L), which were confirmed by finger stick blood glucose < 3.9mmol/L 10 min before to 10 min after the low CGM excursion (i.e. confirmed low CGM); (iii) FSBG < 3.9 mmol/L, which was recorded ≥ 1 h from the end of a confirmed low CGM excursion (SO) |
|
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Ratner 2000 | All‐cause mortality | ND (IO) |
| Health‐related quality of life | Well‐being Questionnaireb (SO) | |
| Severe hypoglycaemia | Symptomatic hypoglycaemia requiring third party assistance (IO) | |
| Cardiovascular mortality | One died secondary to cardiopulmonary arrest (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | Events causing death, life‐threatening, hospitalisations, medical intervention to prevent impairment (IO) | |
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | The term adverse event covers any sign, symptom, syndrome, or illness that appears or worsens in a participant during the period of observation in the clinical study and that may impair the well‐being of the participant, but do not meet the criteria of severeness (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia occurring while asleep after the bedtime insulin dose and before the morning insulin dose and before the morning blood glucose measurement (SO) | |
| Mild/moderate hypoglycaemia | Hypoglycaemia was divided into 3 subsets; all events, severe hypoglycaemia and nocturnal hypoglycaemia (SO) | |
| Socioeconomic effects | Pharmacoeconomics was assessed throughout the treatment phase in terms of direct costs (volumes of health care resource utilisation) and indirect costs (time lost from work and other usual activities, and time lost by informal caregivers)b (SO, IO) | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Robertson 2007 | All‐cause mortality | No patients died (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Episodes requiring assistance from another person due to severe central nervous system dysfunction (IO) | |
| Cardiovascular mortality | No patients died (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | An adverse event is any undesirable medical event occurring to a participant in a clinical study, whether or not related to the study product(s). A non‐serious adverse event is any adverse event which does not fulfil the definition of a serious adverse eventb (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemic between 22.00 (included) − 07.00 h (excluded) (SO) | |
| Mild/moderate hypoglycaemia | Self‐treated episodes of hypoglycaemia with plasma glucose measurements < 3.1 mmol/L whether symptomatic or notb (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | Quote: "HbA 1c as a covariate, since there is an association between HbA1c and hypoglycaemia" (IO) | |
| Russell‐Jones 2004 | All‐cause mortality | No participants died (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Requiring third party assistance (from CSR: an episode with severe central nervous system symptoms consistent with hypoglycaemia in which the participant is unable to treat himself/herself and which has one of the following characteristics: Blood glucose < 2.8 mmol/L or reversal of symptoms after either food intake or glucagon/iv glucose administration) (IO) | |
| Cardiovascular mortality | No participants died (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | Serious adverse events A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect; • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | An adverse event is any undesirable medical event occurring to a participant in a clinical study, whether or not considered related to the study product(s). A non‐serious adverse event is any adverse event that does not fulfil the definition of a serious adverse eventb (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia between 11 p.m. to 6 a.m. (SO) | |
| Mild/moderate hypoglycaemia | Minor if the blood glucose value was < 2.8 mmol/L and the patient dealt with the episode alone; and as symptoms only if no assistance was required and the event was not confirmed by a blood glucose measurement (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Schober 2002 | All‐cause mortality | No patients died (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | An event with symptoms consistent with hypoglycaemia in which the participant required assistance from another person, and which was associated with a blood glucose level below 2.8 mmol/L or prompt recovery after oral carbohydrate or iv glucose or glucagon administrationb(IO) | |
| Cardiovascular mortality | No patients died (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | Adverse events were considered 'serious' because they either required hospitalisations, were life‐threatening or medically important (quote: "If a symptomatic hypoglycaemic event led to hospitalisation or was considered life‐threatening or medically important, it had to be reported as a serious adverse event") (IO) | |
| Diabetic ketoacidosis | Ketoacidosis (IO) | |
| Non‐serious adverse events | Quote from CSR: "The term adverse event covers any sign, symptom, syndrome, or illness that appears or worsens in a participant during the period of observation in the clinical study and that may impair the well‐being of the participant." (SO) | |
| Nocturnal hypoglycaemia | Nocturnal hypoglycaemia was defined as hypoglycaemia while the participant was sleeping between bedtime and after the evening injection and before getting up in the morning (SO) | |
| Mild/moderate hypoglycaemia | Hypoglycaemia was categorised as either symptomatic, i.e. with clinical symptoms that could be confirmed by blood glucose levels below 2.8 mmol/L, or asymptomatic, i.e. any event with a confirmed blood glucose level below 2.8 mmol/L but without any symptoms (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Standl 2004 | All‐cause mortality | One participant died (IO) |
| Health‐related quality of life | Diabetes Health Profileb(SO) | |
| Severe hypoglycaemia | Requiring third party assistance (IO) | |
| Cardiovascular mortality | — | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | ND | |
| Non‐serious adverse events | Adverse events were considered treatment‐emergent if reported during treatment and not present beforehand, or if they increased in severity during treatment (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia between 23:00 to 06:00 h (SO) | |
| Mild/moderate hypoglycaemia | If blood glucose was below 2.8 mmol/L and the patient handled the episode him‐ or herself (footnote: the study had an additional definition 'symptoms only' if not confirmed by BG measurement) (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | ND | |
| SWITCH 1 | All‐cause mortality | All‐cause death (IO, AO) |
| Health‐related quality of life | SF‐36 v2 (SO) | |
| Severe hypoglycaemia | Episode requiring assistance of another person to actively administer carbohydrate, glucagon, or take other corrective actions, neurological recovery following the return of plasma glucose to normal, or both (IO, AO) | |
| Cardiovascular mortality | ND | |
| Non‐fatal myocardial infarction | All types of myocardial infarction:
|
|
| Non‐fatal stroke | Cerebrovascular event is defined: Any acute episode of focal or global neurological dysfunction caused by brain, spinal cord or retinal vascular injury as a result of haemorrhage or infarction (IO, AO) | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | A serious adverse event is an experience that at any dose results in any of the following: • death • a life‐threatening experience • in‐participant hospitalisation or prolongation of existing hospitalisation • a persistent or significant disability/incapacity • a congenital anomaly/birth defect • important medical events that may not result in death, be life‐threatening, or require hospitalisation may be considered a serious adverse event when, based upon appropriate medical judgement, they may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definitionb (IO) |
|
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | Any untoward medical occurrence in a participant administered a product, and which does not necessarily have a causal relationship with this treatment. A non‐serious adverse event is any adverse event which does not fulfil the definition of a serious adverse event (SO) | |
| Nocturnal hypoglycaemia | Episodes between 12:01 a.m. and 5:59 a.m. (SO) | |
| Mild/moderate hypoglycaemia | Blood glucose ≤ 3.9 mmol/L or > 3.9 mmol/L when they occur in conjunction with hypoglycaemic symptoms, able to treat themselves (SO) | |
| Socioeconomic effects | Cost‐effectiveness analysis/quality‐adjusted life years (IO) | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | Association between the individual patient‐level risk of hypoglycaemia and HbA1c was investigated (IO) | |
| Thalange 2013 | All‐cause mortality | All‐cause mortality (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Severe hypoglycaemia was defined as episodes where the persons were semi‐conscious, unconscious or in a coma, with or without convulsions (IO) | |
| Cardiovascular mortality | ND (IO) | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | Serious adverse events were defined as, amongst others, a life‐threatening experience, inpatient hospitalisations or prolongation of existing hospitalisations, a persistent or significant disability/incapacity or death (IO) | |
| Diabetic ketoacidosis | Diabetic ketoacidosis (IO) | |
| Non‐serious adverse events | Any undesirable medical event occurring to a participant in a clinical study, whether or not related to the study product(s) (SO) | |
| Nocturnal hypoglycaemia | Nocturnal if they occurred between 22:00 and 07:00 h (SO) | |
| Mild/moderate hypoglycaemia | Mild hypoglycaemia was defined as episodes where the participants were able to treat themselves. Moderate hypoglycaemia was categorised as episodes where participants required assistance, but responded to oral treatment (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Urakami 2017 | All‐cause mortality | — |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Severe hypoglycaemia is defined as an event associated with impaired consciousness or seizure (IO) | |
| Cardiovascular mortality | Cardiovascular mortality (IO) | |
| Non‐fatal myocardial infarction | Non‐fatal myocardial infarction (IO) | |
| Non‐fatal stroke | Non‐fatal stroke (IO) | |
| End‐stage renal disease | End‐stage renal disease (IO) | |
| Blindness | Blindness (IO) | |
| Serious adverse events | Serious adverse events (IO) | |
| Non‐serious adverse events | Non‐serious adverse events (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia occurring between 22:00 h – 06:59 h. Nocturnal hypoglycaemia was defined as when the person noted symptoms of hypoglycaemia with self‐monitored plasma glucose levels < 70 mg/dL (SO) | |
| Mild/moderate hypoglycaemia | Hypoglycaemia was defined as a self‐monitored plasma glucose level < 3.9 mmol/L (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | — | |
| Vague 2003 | All‐cause mortality | No patients died (IO) |
| Health‐related quality of life | — | |
| Severe hypoglycaemia | Hypoglycaemic episode with severe central nervous system symptoms consistent with hypoglycaemia, in which the participant was unable to treat himself/herself and which had one of the following characteristics: blood glucose recorded as < 2.8 mmol/L or symptom reversal achieved with food, glucose or glucagon (IO) | |
| Cardiovascular mortality | — | |
| Non‐fatal myocardial infarction | — | |
| Non‐fatal stroke | — | |
| End‐stage renal disease | — | |
| Blindness | — | |
| Serious adverse events | Serious adverse events if resulting in a fatal or life‐threatening illness, prolonged significant disability, hospitalisations or prolongation of hospitalisations (IO) | |
| Diabetic ketoacidosis | — | |
| Non‐serious adverse events | An adverse event was defined as an undesirable medical incident occurring during the study, irrespective of its relation to study products (SO) | |
| Nocturnal hypoglycaemia | Hypoglycaemia between 23:00 to 06:00 (SO) | |
| Mild/moderate hypoglycaemia | Minor if blood glucose was < 2.8 mmol/L and the patients dealt with the episode themselves (in addition according to CSR: any asymptomatic blood glucose measurement) (SO) | |
| Socioeconomic effects | — | |
| HbA1c | ND (IO) | |
| Combined HbA1c and severe hypoglycaemia | HbA1c adjustment and risk of severe hypoglycaemia (IO) | |
| —: denotes not reported
aIn addition to definition of endpoint measurement, description of who measured the outcome (AO: adjudicated outcome measurement; IO: investigator‐assessed outcome measurement; SO: self‐reported outcome measurement).
bDefinition of outcome from clinical study report.
cConverted from mg/dL to mmol/L from https://www.diabetes.co.uk/blood-sugar-converter.html). AE: adverse events; a.m.: ante meridiem; BG: blood glucose;CGM: continuous glucose monitoring;CSR: clinical study report; FSBG: finger stick blood glucose; HbA1c: glycosylated haemoglobin A1c; iv: intravenous; ND: not defined; p.m.: post meridiem; SAE: serious adverse events; sc: subcutaneous; SF‐36: short‐form 36; SMPG: self‐monitored plasma glucose; W‐BQ: well‐being questionnaire. | ||
Appendix 15. Adverse events (I)
| Study ID | Intervention(s) and comparator(s) | Participants included in analysis (n) | Deaths (n) | Deaths (% of participants) | Participants with at least one adverse event (n) | Participants with at least one adverse event (%) | Participants with at least one severe/serious adverse event (n) | Participants with at least one severe/serious adverse event (%) |
| Bartley 2008 | I: detemir | 331 | 4 | 1.2 | 265a | 80.1 | 50a | 15.1 |
| C: NPH | 164 | 0 | 0 | 135 | 82.3 | 27 | 16.5 | |
| BEGIN Basal‐Bolus Type 1 | I: degludec | 472 | 2 | 0.2 | 397 | 84.1 | 49 | 10 |
| C: glargine | 155 | 1 | 0.6 | 128 | 83.1 | 17 | 11 | |
| BEGIN Flex T1 | I: degludec | 165 | 1b | 0.6 | 125 | 75.8 | 7 | 4.2 |
| C: glargine | 161 | 0 | 0 | 161 | 72.0 | 8 | 5.0 | |
| BEGIN Young | I: degludec | 174 | 0 | 0 | 161 | 92.5 | 18 | 10.3 |
| C: detemir | 175 | 0 | 0 | 157 | 89.7 | 16 | 9.1 | |
| Bolli 2009 | I: glargine | 90 | — | — | 19 | 22.3 | 2 | 2.2 |
| C: NPH | 85 | — | — | 13 | 15.1 | 0 | 0 | |
| Chase 2008 | I: glargine | 85 | 0 | 0 | 71b | 83.5 | 18 | 21.2 |
| C: NPH/Lente | 90 | 0 | 0 | 67 | 74.4 | 7 | 7.8 | |
| Davies 2014 | I: degludec | 301 | 0 | 0 | 216 | 71.2 | 23 | 7 |
| C: detemir | 152 | 0 | 0 | 112 | 73.7 | 8 | 5 | |
| Fulcher 2005 | I: glargine | 62 | 0 | 0 | 57 | 91.9 | 5b | 8.1 |
| C: NPH | 63 | 0 | 0 | 56 | 88.9 | 3 | 4.8 | |
| Heller 2009 | I: detemir | 299 | 0b | 0 | 277 | 92.6 | 35 | 11.7 |
| C: glargine | 144 | 1 | 0.7 | 129 | 89.6 | 7 | 4.9 | |
| Home 2005 | I: glargine | 292 | 0b | 0 | 192b | 65.8 | 26c | 9.0 |
| C: NPH | 293 | 0 | 0 | 185 | 63.1 | 29 | 10.0 | |
| Kobayashi 2007 | I: detemir | 196 | 0b | 0 | 173b | 88.3 | 13 | 6.6 |
| C: NPH | 98 | 0 | 0 | 87b | 88.8 | 10b | 10.2 | |
| Liu 2016 | I: glargine | 107 | 0 | 0 | 81 | 75.7 | 3 | 2.8 |
| C: NPH | 54 | 0 | 0 | 44 | 81.5 | 6 | 11.1 | |
| NCT00595374b | I: detemir | 75 | 0 | 0 | 60 | 80.0 | 4 | 5.3 |
| C: NPH | 38 | 0 | 0 | 29 | 76.3 | 1 | 2.6 | |
| NCT00605137b | I: detemir | 55 | 0 | 0 | 36 | 83.6 | 3 | 5.5 |
| C: NPH | 27 | 0 | 0 | 23 | 85.2 | 1 | 3.7 | |
| Pieber 2007 | I: detemir | 161 | 0b | 0 | 117b | 72.7 | 14 | 8.7 |
| C: glargine | 159 | 0 | 0 | 121 | 76.1 | 11 | 6.9 | |
| Porcellati 2004 | I: glargine | 61 | 0 | 0 | — | — | — | — |
| C: NPH | 60 | 0 | 0 | — | — | — | — | |
| PRESCHOOL | I: glargine | 62 | 0 | 0 | 30 | 48.4 | 8b | 12.9 |
| C: NPH | 63 | 0 | 0 | 33 | 52.4 | 2 | 3.2 | |
| Ratner 2000 | I: glargine | 264 | 0b | 0 | 223 | 84.5 | 33b | 12.5 |
| C: NPH | 270 | 1 | 0.4 | 234 | 86.7 | 37 | 13.7 | |
| Robertson 2007 | I: detemir | 232 | 0b | 0 | 202 | 87.0 | 24b | 10 |
| C: NPH | 115 | 0 | 0 | 104 | 90.0 | 10 | 9 | |
| Russell‐Jones 2004 | I: detemir | 491 | 0b | 0 | 361b | 73.5 | 26b | 5.3 |
| C: NPH | 256 | 0 | 0 | 183 | 71.5 | 11 | 4.3 | |
| Schober 2002 | I: glargine | 174 | 0b | 0 | 109 | 62.6 | 10 | 5.7 |
| C: NPH | 175 | 0 | 0 | 105 | 60.0 | 24 | 13.7 | |
| Standl 2004 | I: detemir | 236 | 1b | 0.4 | 164b | 69.5 | 17b | 7.2 |
| C: NPH | 224 | 0 | 0 | 156 | 69.6 | 18 | 8.0 | |
| SWITCH 1d | I: degludec | 249 | — | — | — | — | — | — |
| C: glargine | 251 | — | — | — | — | — | — | |
| Thalange 2013 | I: detemir | 177 | 0 | 0 | 132 | 74.6 | 14 | 7.9 |
| C: NPH | 170 | 0 | 0 | 135 | 79.4 | 20 | 11.7 | |
| Urakami 2017 | I: degludec | 9 | — | — | 0 | 0 | 0 | 0 |
| C: glargine | 9 | — | — | 0 | 0 | 0 | 0 | |
| Vague 2003 | I: detemir | 301 | 0b | 0 | 219 | 72.7 | 14b | 4.7 |
| C: NPH | 146 | 0 | 0 | 112 | 76.8 | 4 | 2.7 | |
| —: denotes not reported aData from CSR. In publication, exact number was no stated. For adverse events, it was stated that adverse events were about 80% in both groups; in publication, it was reported that serious adverse events were reported for about 15%–17%. bData from CSR/synopsis. cData from CSR. The publication stated that 53 participants in total experienced serious adverse events ‐ this number does not completely apply when calculating the percentage. dNo data for this adverse events table was reported before cross‐over. C: comparator; CSR; clinical study report; I: intervention; N: number of participants; NPH: neutral protamine Hagedorn. | ||||||||
Appendix 16. Adverse events (II)
| Study ID | Intervention(s) and comparator(s) | Participants included in analysis (n) | Participants discontinuing study due to an adverse event (n) | Participants discontinuing study due to an adverse event (%) | Participants with at least one hospitalisation (n) | Participants with at least one hospitalisation (%) | Participants with at least one outpatient treatment (n) | Participants with at least one outpatient treatment (%) |
| Bartley 2008 | I: detemir | 331 | 13a | 3.9 | — | — | — | — |
| C: NPH | 164 | 1 | 0.6 | — | — | — | — | |
| BEGIN Basal‐Bolus Type 1 | I: degludec | 472 | 12 | 2.5 | — | — | — | — |
| C: glargine | 157 | 2 | 1.3 | — | — | — | — | |
| BEGIN Flex T1 | I: degludec | 165 | 4 | 2.4 | — | — | — | — |
| C: glargine | 161 | 1 | 0.6 | — | — | — | — | |
| BEGIN Young | I: degludec | 174 | 2 | 1.1 | — | — | — | — |
| C: detemir | 175 | 0 | 0 | — | — | — | — | |
| Bolli 2009 | I: glargine | 90 | 0 | 0 | — | — | — | — |
| C: NPH | 85 | 0 | 0 | — | — | — | — | |
| Chase 2008 | I: glargine | 85 | 1 | 1.2 | — | — | — | — |
| C: NPH/Lente | 90 | 2 | 2.2 | — | — | — | — | |
| Davies 2014 | I: degludec | 301 | 3 | 1.0 | — | — | — | — |
| C: detemir | 152 | 1 | 0.7 | — | — | — | — | |
| Fulcher 2005 | I: glargine | 62 | 0b | 0 | 6b | 9.7 | 37b | 59.7 |
| C: NPH | 63 | 1 | 1.6 | 4 | 6.3 | 31 | 49.2 | |
| Heller 2009 | I: detemir | 299 | 6 | 2.0 | — | — | — | — |
| C: glargine | 144 | 4 | 2.8 | — | — | — | — | |
| Home 2005 | I: glargine | 292 | 2 | 0.7 | 3b | 1.0 | 131b | 45.6 |
| C: NPH | 293 | 2 | 0.7 | 3 | 1.0 | 118 | 41.7 | |
| Kobayashi 2007 | I: detemir | 197 | 3 | 1.5 | — | — | — | — |
| C: NPH | 99 | 1 | 1.0 | — | — | — | — | |
| Liu 2016 | I: glargine | 107 | 0 | 0 | — | — | — | — |
| C: NPH | 55 | 1 | 1.8 | — | — | — | — | |
| NCT00595374 | I: detemir | 75 | — | — | — | — | — | — |
| C: NPH | 38 | — | — | — | — | — | — | |
| NCT00605137 | I: detemir | 55 | 0 | 0 | — | — | — | — |
| C: NPH | 27 | 0 | 0 | — | — | — | — | |
| Pieber 2007 | I: detemir | 161 | 3 | 1.9 | — | — | — | — |
| C: glargine | 159 | 1 | 0.6 | — | — | — | — | |
| Porcellati 2004 | I: glargine | 61 | 0 | 0 | — | — | — | — |
| C: NPH | 60 | 0 | 0 | — | — | — | — | |
| PRESCHOOL | I: glargine | 61 | 0 | 0 | — | — | — | — |
| C: NPH | 64 | 2 | 3.1 | — | — | — | — | |
| Ratner 2000 | I: glargine | 264 | 8 | 3.0 | 7b | 2.7 | 28c | 10.6 |
| C: NPH | 270 | 1 | 0.4 | 11 | 4.1 | 28 | 10.4 | |
| Robertson 2007 | I: detemir | 232 | 1 | 0.4 | — | — | — | — |
| C: NPH | 115 | 0 | 0 | — | — | — | — | |
| Russell‐Jones 2004 | I: detemir | 491 | 5 | 1.0 | — | — | — | — |
| C: NPH | 256 | 2 | 0.8 | — | — | — | — | |
| Schober 2002 | I: glargine | 174 | 0b | 0 | 12d | 7.2 | 75d | 44.9 |
| C: NPH | 175 | 0 | 0 | 25 | 14.7 | 81 | 47.7 | |
| Standl 2004 | I: detemir | 236 | 5b | 2.1 | — | — | — | — |
| C: NPH | 224 | 2 | 0.9 | — | — | — | — | |
| SWITCH 1 | I: degludec | 249 | 5e | 2.0 | — | |||
| C: glargine | 251 | 5 | 2.0 | |||||
| Thalange 2013 | I: detemir | 177 | 1 | 0.6 | — | — | — | — |
| C: NPH | 171 | 0 | 0 | — | — | — | — | |
| Urakami 2017 | I: degludec | 9 | — | — | — | — | — | — |
| C: glargine | 9 | — | — | — | — | — | — | |
| Vague 2003 | I: detemir | 301 | 2 | 0.7 | — | — | — | — |
| C: NPH | 146 | 0 | 0 | — | — | — | — | |
| —: denotes not reported aReasons for withdrawals described in CSR. bData available from CSR/synopsis. cFrom CSR: reported as medically important/required medical intervention. dData from CSR: in the glargine group, 167 participants were included, in the NPH group, 170 participants. eReported before cross‐over. C: comparator; CSR: clinical study report; I: intervention; N: number of participants; NPH: neutral protamine Hagedorn. | ||||||||
Appendix 17. Adverse events (III)
| Study ID | Intervention(s) and comparator(s) | Participants included in analysis (n) | Participants with a specific adverse event (description) | Participants with at least one specific adverse event (n) | Participants with at least one specific adverse event (%) |
| Bartley 2008 | I: detemir | 331 | (1) Upper respiratory tract infectiona (2) Nasopharyngitis (3) Influenza (4) Urinary tract infection (5) Pharyngitis (6) Gastroenteritis (7) Diabetic retinopathy (8) Diarrhoea (9) Headache (10) Pharyngolaryngeal pain (11) Application site disorder |
(1) 69 (2) 59 (3) 46 (4) 19 (5) 19 (6) 17 (7) 26 (8) 20 (9) 19 (10) 17 (11) 19 |
(1) 20.8 (2) 17.8 (3) 13.9 (4) 5.7 (5) 5.7 (6) 5.1 (7) 7.9 (8) 6.0 (9) 5.7 (10) 5.1 (11) 5.7 |
| C: NPH | 164 | (1) Upper respiratory tract infection (2) Nasopharyngitis (3) Influenza (4) Urinary tract infection (5) Pharyngitis (6) Gastroenteritis (7) Diabetic retinopathy (8) Diarrhoea (9) Headache (10) Pharyngolaryngeal pain (11) Application site disorder |
(1) 28 (2) 37 (3) 21 (4) 9 (5) 10 (6) 13 (7) 16 (8) 9 (9) 13 (10) 8 (11) 10 |
(1) 17.1 (2) 22.6 (3) 12.8 (4) 5.5 (5) 6.1 (6) 7.9 (7) 9.8 (8) 5.5 (9) 7.9 (10) 4.9 (11) 6.1 |
|
| BEGIN Basal‐Bolus Type 1 | I: degludec | 472 | (1) Infections and infestationsb (2) Gastrointestinal disorders (3) Nervous system disorders (4) Injury, poisoning and procedural complications (5) Musculoskeletal and connective tissue disorders (6) Respiratory, thoracic and mediastinal disorders (7) Metabolism and nutrition disorders (8) General disorders and administration site conditions (9) Skin and subcutaneous tissue disorders (10) Eye disorders (11) Cardiovascular disorders (12) Psychiatric disorders (13) Investigations (14) Immune system disorders (15) Renal and urinary disorders (16) Ear and labyrinth disorders (17) Reproductive system and breast disorders (18) Endocrine disorders (19) Blood and lymphatic system disorders (20) Neoplasms benign, malignant, and unspecified (including cysts and polyps) (21) Surgical and medical procedures (22) Hepatobiliary disorders (23) Congenital, familial and genetic disorders |
(1) 292 (2) 105 (3) 94 (4) 99 (5) 90 (6) 80 (7) 83 (8) 57 (9) 43 (10) 38 (11) 24 (12) 25 (13) 20 (14) 11 (15) 12 (16) 10 (17) 10 (18) 6 (19) 5 (20) 3 (21) 2 (22) 1 (23) 0 |
(1) 61.9 (2) 22.2 (3) 19.9 (4) 21.0 (5) 19.1 (6) 16.9 (7) 17.6 (8) 12.1 (9) 9.1 (10) 6.1 (11) 5.1 (12) 5.3 (13) 4.2 (14) 2.3 (15) 2.5 (16) 2.1 (17) 2.1 (18) 1.3 (19) 1.1 (20) 0.6 (21) 0.4 (22) 0.4 (23) 0 |
| C: glargine | 154 | (1) Infections and infestations (2) Gastrointesinal disorders (3) Nervous system disorders (4) Injury, poisoning and procedural complications (5) Musculoskeletal and connective tissue disorders (6) Respiratory, thoracic and mediastinal disorders (7) Metabolism and nutrition disorders (8) General disorders and administration site conditions (9) Skin and subcutaneous tissue disorders (10) Eye disorders (11) Cardiovascular disorders (12) Psychiatric disorders (13) Investigations (14) Immune system disorders (15) Renal and urinary disorders (16) Ear and labyrinth disorders (17) Reproductive system and breast disorders (18) Endocrine disorders (19) Blood and lymphatic system disorders (20) Neoplasms benign, malignant, and unspecified (inclusive cysts and polyps) (21) Surgical and medical procedures (22) Hepatobiliary disorders (23) Congenital, familial and genetic disorders |
(1) 97 (2) 33 (3) 39 (4) 31 (5) 31 (6) 29 (7) 20 (8) 23 (9) 16 (10) 10 (11) 7 (12) 6 (13) 6 (14) 11 (15) 4 (16) 3 (17) 2 (18) 1 (19) 1 (20) 3 (21) 1 (22) 1 (23) 1 |
(1) 63.0 (2) 21.4 (3) 25.3 (4) 20.1 (5) 20.1 (6) 18.8 (7) 13.0 (8) 14.9 (9) 10.4 (10) 6.5 (11) 4.5 (12) 3.9 (13) 3.9 (14) 7.1 (15) 2.6 (16) 1.9 (17) 1.3 (18) 0.6 (19) 0.6 (20) 1.9 (21) 0.6 (22) 0.6 (23) 0.6 |
|
| BEGIN Flex T1 | I: degludec | 165 | (1) Gastroenteritisa (2) Nasopharyngitis (3) Sinusitis (4) Upper respiratory tract infections (5) Headache (6) Diarrhoea (7) Nausea (8) Vomiting (9) Cough (10) Oropharyngeal pain (11) Wrong drug administered (12) Injection‐site reactions |
(1) 9 (2) 43 (3) 10 (4) 9 (5) 16 (6) 1 (7) 7 (8) 9 (9) 4 (10) 11 (11) 9 (12) 3 |
(1) 5.5 (2) 26.1 (3) 6.1 (4) 5.5 (5) 9.7 (6) 0.6 (7) 4.2 (8) 5.5 (9) 2.4 (10) 6.7 (11) 5.5 (12) 1.8 |
| C: glargine | 161 | (1) Gastroenteritis (2) Nasopharyngitis (3) Sinusitis (4) Upper respiratory tract infections (5) Headache (6) Diarrhoea (7) Nausea (8) Vomiting (9) Cough (10) Oropharyngeal pain (11) Wrong drug administered (12) Injection‐site reactions |
(1) 5 (2) 29 (3) 7 (4) 13 (5) 18 (6) 9 (7) 8 (8) 5 (9) 10 (10) 11 (11) 7 (12) 4 |
(1) 3.1 (2) 18 (3) 4.3 (4) 8.1 (5) 11.2 (6) 5.6 (7) 5.0 (8) 3.1 (9) 6.2 (10) 6.8 (11) 4.3 (12) 2.5 |
|
| BEGIN Young | I: degludec | 174 | (1) Ear paina,b (2) Abdominal pain (3) Abdominal upper pain (4) Diarrhoea (5) Nausea (6) Vomiting (7) Pyrexia (8) Bronchitis (9) Ear infection (10) Viral gastroenteritis |
(1) 10 (2) 12 (3) 28 (4) 22 (5) 13 (6) 26 (7) 30 (8) 9 (9) 9 (10) 15 |
(1) 5.8 (2) 6.9 (3) 16.1 (4) 12.6 (5) 7.5 (6) 14.9 (7) 17.2 (8) 5.2 (9) 5.2 (10) 8.6 |
| C: detemir | 175 | (1) Ear pain (2) Abdominal pain (3) Abdominal upper pain (4) Diarrhoea (5) Nausea (6) Vomiting (7) Pyrexia (8) Bronchitis (9) Ear infection (10) Viral gastroenteritis |
(1) 5 (2) 8 (3) 17 (4) 17 (5) 9 (6) 22 (7) 28 (8) 8 (9) 11 (10) 22 |
(1) 2.9 (2) 4.6 (3) 9.7 (4) 9.7 (5) 5.1 (6) 12.6 (7) 16.0 (8) 4.6 (9) 6.3 (10) 12.6 |
|
| Bolli 2009 | I: glargine | 90 | — | — | — |
| C: NPH | 85 | — | — | — | |
| Chase 2008 | I: glargine | 85 | — | — | — |
| C: NPH/Lente | 90 | — | — | — | |
| Davies 2014 | I: degludec | 301 | (1) Eye disordera,b (2) Gastrointestinal disorders (3) Nasopharyngitis (4) Metabolism and nutrition disorders (5) Nervous system disorders (6) Musculoskeletal disorders (7) Respiratory disorders |
(1) 20 (2) 20 (3) 94 (4) 15 (5) 21 (6) 42 (7) 21 |
(1) 6.6 (2) 6.6 (3) 31.2 (4) 5.0 (5) 7.0 (6) 14.0 (7) 7.0 |
| C: detemir | 152 | (1) Eye disorder (2) Gastrointestinal disorders (3) Infections (4) Metabolism and nutrition disorders (5) Nervous system disorders (6) Musculoskeletal disorders (7) Respiratory disorders |
(1) 7 (2) 9 (3) 49 (4) 11 (5) 5 (6) 12 (7) 8 |
(1) 4.6 (2) 5.9 (3) 32.2 (4) 7.2 (5) 3.3 (6) 7.9 (7) 5.3 |
|
| Fulcher 2005 | I: glargine | 62 | (1) Upper respiratory tract infectionsa, c (2) Infections (3) Rhinitis (4) Headache (5) Diarrhoea (6) Injection site pain/reactions (7) Ecchomysis (8) Sore throat (9) Flu syndrome (10) Nausea |
(1) 4 (2) 4 (3) 7 (4) 6 (5) 3 (6) 5 (7) 5 (8) 4 (9) 7 (10) 6 |
(1) 7.2 (2) 7.2 (3) 7.2 (4) 9.8 (5) 4.3 (6) 8.1 (7) 8.1 (8) 6.5 (9) 11.3 (10) 9.7 |
| C: NPH | 63 | (1) Upper respiratory tract infections (2) Infections (3) Rhinitis (4) Headache (5) Diarrhoea (6) Injection site pain/reactions (7) Ecchomysis (8) Sore throat (9) Flu syndrome (10) Nausea |
(1) 7 (2) 4 (3) 3 (4) 3 (5) 1 (6) 7 (7) 8 (8) 7 (9) 7 (10) 6 |
(1) 11.2 (2) 6.2 (3) 5.4 (4) 4.2 (5) 0.8 (6) 11.1 (7) 12.7 (8) 11.1 (9) 11.1 (10) 9.5 |
|
| Heller 2009 | I: detemir | 299 | (1) Injection site reactiona (2) Headache (3) Pharyngolaryngeal pain (4) Arthralgia |
(1) 24 (2) 66 (3) 159 (4) 16 |
(1) 8 (2) 22.1 (3) 53.2 (4) 5.4 |
| C: glargine | 144 | (1) Injection site reaction (2) Headache (3) Pharyngolaryngeal pain (4) Arthralgia |
(1) 2 (2) 27 (3) 70 (4) 0 |
(1) 1.4 (2) 18.8 (3) 48.6 (4) 0 |
|
| Home 2005 | I: glargine | 292 | (1) Injection site massa (2) Injection site reaction (3) Respiratory system |
(1) 8 (2) 3 (3) 77 |
(1) 3 (2) 1 (3) 35.2 |
| C: NPH | 293 | (1) Injection site mass (2) Injection site reaction (3) Respiratory system |
(1) 9 (2) 6 (3) 79 |
(1) 3 (2) 2 (3) 27.0 |
|
| Kobayashi 2007 | I: detemir | 197 | (1) Metabolism and nutrition disordera (2) Infections and infestations (3) Respiratory, thoracic and mediastinal disorders (4) Injury, poisoning, procedural disorders (5) Nervous system disorder |
(1) 8 (2) 4 (3) 2 (4) 1 (5) 1 |
(1) 4.1 (2) 2.0 (3) 1.0 (4) 0.5 (5) 0.5 |
| C: NPH | 99 | (1) Metabolism and nutrition disorder (2) Infections and infestations (3) Respiratory, thoracic and mediastinal disorders (4) Injury, poisoning, procedural disorders (5) Nervous system disorder |
(1) 1 (2) 1 (3) 0 (4) 4 (5) 2 |
(1) 1.0 (2) 1.0 (3) 0.0 (4) 4.1 (5) 2.0 |
|
| Liu 2016 | I: glargine | 107 | (1) Respiratory, thoracic and mediastinal disordersa (2) Hypoglycaemia (3) Nasopharyngitis (4) Upper respiratory tract infection |
(1) 3 (2) 74 (3) 28 (4) 18 |
(1) 2.8 (2) 69.2 (3) 26.2 (4) 16.8 |
| C: NPH | 54 | (1) Respiratory, thoracic and mediastinal disorders (2) Hypoglycaemia (3) Nasopharyngitis (4) Upper respiratory tract infection |
(1) 3 (2) 41 (3) 17 (4) 11 |
(1) 5.6 (2) 75.9 (3) 31.5 (4) 20.4 |
|
| NCT00595374 | I: detemir | 75 | — | — | — |
| C: NPH | 38 | — | — | — | |
| NCT00605137 | I: detemir | 55 | (1) Infections and infestations (2) Increased albumin/creatinine ratio |
(1) 29 (2) 3 |
(1) 52.7 (2) 5.5 |
| C: NPH | 27 | (1) Infections and infestations (2) Increased albumin/creatinine ratio |
(1) 14 (2) 1 |
(1) 51.9 (2) 3.7 |
|
| Pieber 2007 | I: detemir | 161 | (1) Respiratory system disordera (2) Gastrointestinal system disorder (3) Headache (4) Skin and appendages disorder |
(1) 53 (2) 33 (3) 23 (4) 6 |
(1) 36 (2) 20.5 (3) 14.3 (4) 3.7 |
| C: glargine | 159 | (1) Respiratory system disorder (2) Gastrointestinal system disorder (3) Headache (4) Skin and appendages disorder |
(1) 67 (2) 30 (3) 31 (4) 9 |
(1) 42.1 (2) 18.9 (3) 19.5 (4) 5.7 |
|
| Porcellati 2004 | I: glargine | 61 | — | — | — |
| C: NPH | 60 | — | — | — | |
| PRESCHOOL | I: glargine | 62 | (1) Vomitingb,c (2) Device lead damage (3) Pyrexia (4) Gastroenteritis (5) Nasopharyngitis (6) Pharyngitis (7) Upper respiratory tract infection (8) Bronchitis (9) Otitis media (10) Tonsilitis (11) Cough |
(1) 5 (2) 5 (3) 3 (4) 6 (5) 6 (6) 6 (7) 4 (8) 3 (9) 1 (10) 1 (11) 2 |
(1) 8.1 (2) 8.1 (3) 4.8 (4) 9.7 (5) 9.7 (6) 9.7 (7) 6.5 (8) 4.8 (9) 1.6 (10) 2.6 (11) 3.2 |
| C: NPH | 63 | (1) Vomiting (2) Device lead damage (3) Pyrexia (4) Gastroenteritis (5) Nasopharyngitis (6) Pharyngitis (7) Upper respiratory tract infection (8) Bronchitis (9) Otitis media (10) Tonsilitis (11) Cough |
(1) 4 (2) 2 (3) 7 (4) 6 (5) 5 (6) 2 (7) 6 (8) 5 (9) 4 (10) 4 (11) 4 |
(1) 6.4 (2) 3.2 (3) 11.1 (4) 9.5 (5) 7.9 (6) 3.2 (7) 9.5 (8) 7.9 (9) 6.4 (10) 6.4 (11) 6.4 |
|
| Ratner 2000 | I: glargine | 264 | (1) Injection site reactiona (2) Respiratory system (3) Body as whole (4) Digestive system (5) Nervous system (6) Metabolic and nutritional disorder (7) Cardiovascular system (8) Muscoskeletal system (9) Special senses (10) Urogenital systems (11) Lymphatic systems (12) Endocrine system |
(1) 40 (2) 123 (3) 90 (4) 60 (5) 43 (6) 33 (7) 32 (8) 28 (9) 27 (10) 19 (11) 6 (12) 4 |
(1) 15.2 (2) 46.6 (3) 34.1 (4) 22.7 (5) 16.3 (6) 12.5 (7) 12.1 (8) 10.6 (9) 10.2 (10) 7.2 (11) 2.3 (12) 1.5 |
| C: NPH | 270 | (1) Injection site reaction (2) Respiratory system (3) Body as whole (4) Digestive system (5) Nervous system (6) Metabolic and nutritional disorder (7) Cardiovascular system (8) Muscoskeletal system (9) Special senses (10) Urogenital systems (11) Lymphatic systems (12) Endocrine system |
(1) 28 (2) 139 (3) 112 (4) 71 (5) 50 (6) 41 (7) 41 (8) 45 (9) 26 (10) 32 (11) 8 (12) 4 |
(1) 10.4 (2) 51.5 (3) 41.5 (4) 26.3 (5) 18.5 (6) 15.2 (7) 15.2 (8) 16.7 (9) 9.6 (10) 11.9 (11) 3.0 (12) 1.5 |
|
| Robertson 2007 | I: detemir | 232 | (1) Injection site reactiona (2) Respiratory system disorder (3) Gastrointestinal system disorder (4) Headache (5) Influenza‐like symptoms |
(1) 8 (2) 134 (3) 91 (4) 72 (5) 32 |
(1) 2.4 (2) 57.8 (3) 39.2 (4) 31.2 (5) 13.8 |
| C: NPH | 115 | (1) Injection site reaction (2) Respiratory system disorder (3) Gastrointestinal system disorder (4) Headache (5) Influenza‐like symptoms |
(1) 2 (2) 64 (3) 43 (4) 37 (5) 24 |
(1) 1.7 (2) 55.7 (3) 37.4 (4) 32.2 (5) 20.9 |
|
| Russell‐Jones 2004 | I: detemir | 491 | (1) Respiratory system disordera (2) Headache (3) Gastrointestinal system disorder (4) Influenza‐like symptoms |
(1) 179 (2) 108 (3) 107 (4) 37 |
(1) 36.5 (2) 22 (3) 21.8 (4) 7.5 |
| C: NPH | 256 | (1) Respiratory system disorder (2) Headache (3) Gastrointestinal system disorder (4) Influenza‐like symptoms |
(1) 77 (2) 58 (3) 56 (4) 15 |
(1) 30.1 (2) 22.7 (3) 21.9 (4) 5.9 |
|
| Schober 2002 | I: glargine | 174 | (1) Injection site reactiona | 16 | 9.2 |
| C: NPH | 175 | (1) Injection site reaction | 15 | 8.6 | |
| Standl 2004 | I: detemir | 236 | (1) Respiratory tract disordera (2) Headache (3) Diarrhoea (4) Accidental injury (5) Skin and appendages disorder |
(1) 89 (2) 60 (3) 16 (4) 6 (5) 5 |
(1) 37.7 (2) 25.4 (3) 6.9 (4) 2.5 (5) 2.1 |
| C: NPH | 224 | (1) Respiratory tract disorder (2) Headache (3) Diarrhoea (4) Accidental injury (5) Skin and appendages disorder |
(1) 73 (2) 48 (3) 15 (4) 12 (5) 17 |
(1) 32.6 (2) 21.4 (3) 6.7 (4) 5.4 (5) 7.6 |
|
| SWITCH 1d | I: degludec | — | — | — | — |
| C: glargine | — | — | — | — | |
| Thalange 2013 | I: detemir | 177 | (1) Nasopharyngitis (2) Pharyngitis (3) Upper respiratory tract infection (4) Headache (5) Gastroenteritis (6) Influenza | (1) 75 (2) 19 (3) 18 (4) 26 (5) 18 (6) 10 |
(1) 42.4 (2) 10.7 (3) 10.2 (4) 14.7 (5) 10.2 (6) 5.6 |
| C: NPH | 170 | (1) Nasopharyngitis (2) Pharyngitis (3) Upper respiratory tract infection (4) Headache (5) Gastroenteritis (6) Influenza | (1) 81 (2) 15 (3) 16 (4) 23 (5) 14 (6) 18 |
(1) 47.6 (2) 8.8 (3) 9.4 (4) 13.5 (5) 8.2 (6) 10.6 |
|
| Urakami 2017 | I: degludec | 9 | — | — | — |
| C: glargine | 9 | — | — | — | |
| Vague 2003 | I: detemir | 301 | (1) Respiratory system disordersa (2) Central and peripheral nervous system disorder (3) Gastrointestinal system disorder (4) Back pain (5) Skin and appendages disorder |
(1) 97 (2) 69 (3) 62 (4) 17 (5) 21 |
(1) 32.2 (2) 22.9 (3) 20.6 (4) 5.6 (5) 7.0 |
| C: NPH | 146 | (1) Respiratory system disorders (2) Central and peripheral nervous system disorder (3) Gastrointestinal system disorder (4) Back pain (5) Skin and appendages disorder |
(1) 51 (2) 36 (3) 31 (4) 6 (5) 2 |
(1) 34.9 (2) 24.7 (3) 21.2 (4) 4.1 (5) 1.4 |
|
| —: denotes not reported aFrom CSR (a very detailed description available from CSR). bDetailed description available at ClinicalTrials.gov. cNumber varies from publication and CSR. Quote: "The most frequently reported AEs were upper respiratory tract infections (glargine: 7.2%; NPH: 11.2%), infections (glargine: 7.2%; NPH: 6.2%) and rhinitis (glargine: 7.2%; NPH: 5.4%)." Quote from CSR: " Most patients in both treatment groups suffered from upper respiratory tract infections (glargine: 24%, NPH: 32%)". dNone of the information for this adverse events table was reported before cross‐over. C: comparator; CSR: clinical study report; I: intervention; N: number of participants; NPH: neutral protamine Hagedorn. |
|||||
Appendix 18. Adverse events (IV)
| Study ID | Intervention(s) and comparator(s) | Participants included in analysis (n) | Participants with at least one hypoglycaemic episode (n) | Participants with at least one hypoglycaemic episode (%) | Participants with at least one nocturnal hypoglycaemic episode (n) | Participants with at least one nocturnal hypoglycaemic episode (% participants) | Participants with at least one severe/serious hypoglycaemic episode (n) | Participants with at least one severe/serious hypoglycaemic episode (%) |
| Bartley 2008 | I: detemir | 331 | 309 | 93.4 | 237 | 71.6 | 49 | 14.8 |
| C: NPH | 164 | 159 | 97.0 | 124 | 75.6 | 42 | 25.6 | |
| BEGIN Basal‐Bolus Type 1 | I: degludec | 472 | 451 | 96 | 341 | 72 | 58a | 12 |
| C: glargine | 154 | 147 | 95 | 114 | 74 | 16 | 10 | |
| BEGIN Flex T1 | I: degludec | 165 | 164 | 99.4 | 121 | 73.3 | 21 | 12.7 |
| C: glargine | 161 | 156 | 96.9 | 117 | 72.6 | 16 | 9.9 | |
| BEGIN Young | I: degludec | 174 | 171 | 98.3 | 133 | 76.4 | 31 | 17.8 |
| C: detemir | 175 | 168 | 96.0 | 125 | 71.4 | 24 | 13.8 | |
| Bolli 2009 | I: glargine | 90 | — | — | — | — | — | — |
| C: NPH | 85 | — | — | — | — | — | — | |
| Chase 2008 | I: glargine | 85 | 85 | 100 | 55a | 64.7 | 9 | 10.6 |
| C: NPH/Lente | 90 | 88 | 97.8 | 61 | 67.8 | 4 | 4.4 | |
| Davies 2014 | I: degludec | 301 | 280 | 93.0 | 176 | 58.5 | 32 | 10.6 |
| C: detemir | 152 | 139 | 91.4 | 89 | 58.6 | 16 | 10.5 | |
| Fulcher 2005 | I: glargine | 62 | 62a | 100 | 50 | 81 | 13a | 21 |
| C: NPH | 63 | 59 | 93.7 | 54 | 86 | 16 | 25.4 | |
| Heller 2009 | I: detemir | 299 | 291a | 97.3 | 256a | 97.3 | 54a | 18.1 |
| C: glargine | 144 | 140 | 97.2 | 121 | 84.0 | 23 | 16.0 | |
| Home 2005 | I: glargine | 292 | 260 | 89.0 | 178 | 61.0 | 31 | 10.6 |
| C: NPH | 293 | 248 | 84.6 | 179 | 61.1 | 44 | 15.0 | |
| Kobayashi 2007 | I: detemir | 196 | 178 | 92.7 | 133 | 69.3 | 2 | 1.0 |
| C: NPH | 98 | 95 | 95.6 | 78 | 79.6 | 3 | 3.0 | |
| Liu 2016 | I: glargine | 107 | 99 | 92.5 | 83 | 77.6 | 1 | 0.9 |
| C: NPH | 54 | 51 | 94.4 | 42 | 77.8 | 1 | 1.9 | |
| NCT00595374 | I: detemir | 75 | — | — | — | — | — | — |
| C: NPH | 38 | — | — | — | — | — | — | |
| NCT00605137 | I: detemir | 55 | 53a | 96.4 | — | — | 5 | 9.1 |
| C: NPH | 27 | 27 | 100 | — | — | 3 | 11.1 | |
| Pieber 2007 | I: detemir | 161 | 120 | 75.9 | 47 | 29.3 | 3 | 1.9 |
| C: glargine | 159 | 108 | 70.1 | 50 | 31.4 | 12 | 7.8 | |
| Porcellati 2004 | I: glargine | 61 | — | — | — | — | 0 | 0 |
| C: NPH | 60 | — | — | — | — | 0 | 0 | |
| PRESCHOOL | I: glargine | 61 | 61 | 100 | 59 | 96.7 | 4 | 6.6 |
| C: NPH | 60 | 63 | 98.4 | 60 | 93.8 | 2 | 3.1 | |
| Ratner 2000 | I: glargine | 264 | 251a | 95.1 | 204a | 77.3 | 23a | 8.7 |
| C: NPH | 270 | 254 | 94.1 | 208 | 77.0 | 28 | 10.4 | |
| Robertson 2007 | I: detemir | 232 | 223 | 96.1 | 174 | 75.0 | 37 | 15.9 |
| C: NPH | 115 | 113 | 98.3 | 101 | 87.8 | 23 | 20.0 | |
| Russell‐Jones 2004 | I: detemir | 491 | 448 | 93.3 | 339 | 70.6 | 31 | 6.5 |
| C: NPH | 256 | 229 | 92.7 | 180 | 72.9 | 22 | 8.9 | |
| Schober 2002 | I: glargine | 174 | 138 | 79.3 | 85 | 48.3 | 40 | 25.0 |
| C: NPH | 175 | 138 | 78.9 | 89 | 50.9 | 50 | 28.8 | |
| Standl 2004 | I: detemir | 236 | 184 | 80.3 | 134a | 58.5 | 20a | 8.7 |
| C: NPH | 224 | 169 | 76.8 | 137 | 62.3 | 12 | 5.5 | |
| SWITCH 1b | I: degludec | — | — | — | — | — | — | — |
| C: glargine | — | — | — | — | — | — | — | |
| Thalange 2013 | I: detemir | 177 | 146 | 82.5 | 100 | 56.5 | 3 | 1.7 |
| C: NPH | 170 | 150 | 88.2 | 111 | 65.3 | 12 | 7.0 | |
| Urakami 2017 | I: degludec | 9 | 9 | 100 | 2 | 22.2 | 0 | 0 |
| C: glargine | 9 | 9 | 100 | 4 | 44.4 | 0 | 0 | |
| Vague 2003 | I: Detemir | 301 | 271 | 90.0 | 198 | 65.8 | 24 | 8.0 |
| C: NPH | 146 | 138 | 94.5 | 110 | 75.3 | 21 | 14.4 | |
| —: denotes not reported aData from CSR. bNone of the information for this table was reported before cross‐over. C: comparator; CSR: clinical study report; I: intervention; N: number of participants; NPH: neutral protamine Hagedorn. | ||||||||
Appendix 19. Survey of study investigators providing information on included studies
| Included studies | Date study author contacted | Date study author replied | Type of additional information | Type of additional data | |
| Bartley 2008 | 11 December 2019 | No reply | |||
| BEGIN Basal‐Bolus Type 1 | 15 January 2020 | 19 January 2020: Dr Heller replied that he would like to try to help with the request | Additional data | No reply with additional data | |
| BEGIN Flex T1 | 21 January 2020 | No reply | |||
| BEGIN Young | 6 February 2020 | No reply | |||
| Bolli 2009 | 11 December 2019 | 11 December 2019: would like to help, but did no longer have access to study data | Study protocol, 'Risk of bias' items, data on safety, additional data | Replied 13 December 2019: no additional data provided | |
| Chase 2008 | 19 February 2020 | No reply | |||
| Davies 2014 | 9 December 2019 | 30 December 2019: Novo Nordisk received the request from Dr. Davies and assured assistance | Study protocol, 'Risk of bias' items, data on safety, additional data | 20 January 2020: Novo Nordisk provided additional data | |
| Fulcher 2005 | 8 December 2019 | Corresponding author was initially contacted. Due to lack of response, Sanofi was contacted on 29 January 2020 (replied same day that they would look further into the request) | Study protocol, 'Risk of bias' items, data on safety, additional data | 31 January 2020: CSR was provided | |
| Heller 2009 | 12 December 2019 | Replied 19 January 2020 that he would try to help. As no further action, Novo Nordisk was contacted | Study protocol, 'Risk of bias' items, data on safety, additional data | Novo Nordisk provided CSR | |
| Home 2005 | 12 December 2019 | 12 December 2019: would try to help, although data were old | Study protocol, 'Risk of bias' items, data on safety, additional data | Comments on data and suggestions | |
| Kobayashi 2007 | No contact information retrieved | 21 March 2020: Novo Nordisk was contacted | Study protocol, 'Risk of bias' items, data on safety, additional data | 26 May 2020: translated pages from CSRs provided | |
| Liu 2016 | 17 February 2020 | No reply from study authors. Sanofi was contacted 23/3‐20. | Asked for study protocol, 'Risk of bias' items and outcomes | 26 March 2020: Sanofi provided CSR | |
| NCT00595374 | 27 January 2020: Novo Nordisk was asked if the study was published | 28 January 2020: Novo Nordisk replied they would look into the request | Asked for additional information | No CSR or additional information could be provided | |
| NCT00605137 | 28 February 2020: Novo Nordisk was asked if the study was published | 2 March 2020: Novo Nordisk replied they would look further into the request. Replied 9 March 2020 that CSR was only available in Japanese, but they were willing to provide some translated pages. | Asked for study protocol, 'Risk of bias' items and outcomes | 24 May 2020: pages from CSR provided, study protocol provided | |
| Pieber 2007 | 25 January 2020 | No reply by the investigators, Novo Nordisk was contacted | Study protocol, 'Risk of bias' items, data on safety, additional data | Novo Nordisk provided CSR | |
| Porcellati 2004 | 12 December 2019 | 11 December 2019: would like to help, but did no longer have access to study data | NA | NA | |
| PRESCHOOL | 19 February 2020 | No reply from investigators. Sanofi was contacted. | 'Risk of bias' items, outcomes | Sanofi provided CSR | |
| Ratner 2000 | 13 January 2020: no reply 12 February 2020: Sanofi contacted |
12 February 2020: Sanofi replied they were willing to assist | Study protocol, 'Risk of bias' items, data on safety, additional data | 14 February 2020: Sanofi provided CSR | |
| Robertson 2007 | 12 February 2020 | 12 February 2020: investigator did not have access to the data (retired). Novo Nordisk was contacted | 'Risk of bias' items, additional data | 12 February 2020: Novo Nordisk provided CSR | |
| Russell‐Jones 2004 | 16 January 2020 | 19 January 2020: authors would like to help, but never replied. Novo Nordisk was contacted. | Study protocol, 'Risk of bias' items, data on safety, additional data | Novo Nordisk provided CSR | |
| Schober 2002 | 17 February 2020 | No reply from authors. Sanofi was contacted | 'Risk of bias' items, additional data | Sanofi provided CSR | |
| Standl 2004 | 28 November 2019: no reply; 29 January 2020: Novo Nordisk was contacted | No reply from investigator, Novo Nordisk was contacted | 'Risk of bias' items, additional data | Novo Nordisk provided CSR | |
| SWITCH 1 | 28 January 2020 | 1 February 2020: corresponding author replied that they would like to help. As investigator did not have access to data, Novo Nordisk was contacted | Data before cross‐over | 24 March 2020: Novo Nordisk replied that the requested analyses for data before cross‐over were not performed | |
| Thalange 2013 | 21 February 2020 | No reply | |||
| Urakami 2017 | 12 February 2020 | 14 February 2020: corresponding author replied they would like to help | Study protocol, 'Risk of bias' items, data before cross‐over | No study protocol provided, but provided data on outcomes | |
| Vague 2003 | 23 January 2020: no valid contact information available for the first author; Novo Nordisk was contacted | 25 January 2020: Novo Nordisk replied that they would assist | Study protocol, 'Risk of bias' items, data on safety, additional data | Novo Nordisk provided CSR | |
| Studies awaiting assessment | Study completion date | Date study author contacted | Date study author replied | Type of additional information | Type of additional data |
| Basal Analog Study | RT/CA | 11 February 2020: asked for full‐text publication of study | 11 February 2020: study never published. A new request if data could be provided. | 11 February 2020: no additional data provided | NA |
| EudraCT 2007‐004144‐74 | RT | 11 February 2020: asked if full‐text publication was available and duration of the intervention | 12/2‐20 ‐ investigator replied that no full‐text article was currently available, but might be in the future. No reply on duration of intervention | NA | NA |
| IRCT201203079224N1 | RT | 11 February 2020: asked for status of study | No reply | ||
| J‐Collection | RT | 11 February 2020: asked for status of study | No reply | ||
| Mianowska 2007 | PM | 17 February 2020: asked for data before cross‐over | No reply | ||
| NCT00564018 | RT | 11 February 2020: asked for status of study | No reply | ||
| NCT01854723 | RT | 12 February 2020: investigator asked for full‐text publication or data on study | No reply | ||
| UMIN000001562 | RT | 12 February 2020: asked for study duration and full‐text | No reply | ||
| UMIN000020521 | RT | 11 February 2020: asked for status of study | No reply | ||
| UMIN000021046 | RT | 11 February 2020: asked for status of study | No reply | ||
| Excluded studies | Study completion date | Date study author contacted | Date study author replied | Date study author was asked for additional information (short summary) | Date study author provided data (short summary) |
| Orchard 2014 | CA | 11 February 2020: asked for full‐text publication of study | 26 February 2020: investigator replied that no full‐text publication was planned. On the same day, the author was asked if additional information could be provided ‐ study protocol and power point presentation provided by the authors | Information about study design and data, publications of study | Based on information from investigator, the study could be excluded |
| —: denotes not reported CA: conference abstract; CSR: clinical study report; NA: not applicable; PM: published manuscript; RT: registered trial. | |||||
Appendix 20. Subgroup and sensitivity analyses
| Comparison/outcome | Outcome | Published versus unpublished | Adults versus children | Blinding | NPH once daily versus multiple doses | Duration of intervention | Income | Setting |
| Insulin detemir versus NPH insulin | All‐cause mortality | P = 0.85 | — | — | — | — | — | — |
| Cardiovascular mortality, non‐fatal myocardial infarction/stroke, blindness, end‐stage renal disease, socioeconomic effects, HbA1c/severe hypoglycaemia combined | — | — | — | — | — | — | — | |
| Severe hypoglycaemia | P = 0.01 | P = 0.72 | — | — | — | — | — | |
| Serious adverse events | P = 0.11 | P = 0.77 | — | — | — | — | — | |
| Diabetic ketoacidosis | P = 0.93 | P = 0.91 | — | — | — | — | — | |
| Adverse events | P = 0.25 | P = 0.40 | — | — | — | — | — | |
| Any nocturnal hypoglycaemia | P = 0.90 | P = 0.36 | — | — | — | — | — | |
| Mild/moderate hypoglycaemia | P = 0.89 | P = 0.82 | — | — | — | — | — | |
| HbA1c | P = 0.30 | P = 0.11 | — | — | — | — | — | |
| Insulin glargine versus NPH insulin | All‐cause mortality, health‐related quality of life, cardiovascular mortality, non‐fatal myocardial infarction/stroke, blindness, end‐stage renal disease, diabetic ketoacidosis, socioeconomic effects, HbA1c/severe hypoglycaemia combined | — | — | — | — | — | — | — |
| Severe hypoglycaemia | P = 0.87 | P = 0.29 | — | — | — | — | — | |
| Serious adverse events | P = 0.99 | P = 0.96 | — | — | — | — | — | |
| Diabetic ketoacidosis | P = 0.48 | P = 0.69 | — | — | — | — | — | |
| Non‐serious adverse events | P = 0.88 | P = 0.64 | — | — | — | — | — | |
| Nocturnal hypoglycaemia | P = 0.99 | P = 0.57 | — | — | — | — | — | |
| Mild/moderate hypoglycaemia | P = 0.80 | P = 0.65 | — | — | — | — | — | |
| HbA1c | P = 0.47 | P = 0.36 | — | — | — | — | — | |
| Insulin detemir versus insulin glargine | All‐cause mortality, health‐related quality of life, cardiovascular mortality, non‐fatal myocardial infarction/stroke, blindness, end‐stage renal disease, serious adverse events, diabetic ketoacidosis, socioeconomic effects, HbA1c, HbA1c/severe hypoglycaemia combined | — | — | — | — | — | — | — |
| Severe hypoglycaemia | P = 0.02 | — | — | — | — | — | — | |
| Adverse events | P = 0.28 | — | — | — | — | — | — | |
| Nocturnal hypoglycaemia | Not significant for multiple comparisons | — | — | — | — | — | — | |
| Mild/moderate hypoglycaemia | P = 0.29 | — | — | — | — | — | — | |
| Insulin degludec versus insulin detemir | All‐cause mortality, health‐related quality of life, cardiovascular mortality, non‐fatal myocardial infarction/stroke, blindness, end‐stage renal disease, HbA1c/severe hypoglycaemia combined | — | — | — | — | — | — | — |
| Severe hypoglycaemia | — | P = 0.51 | — | — | — | — | — | |
| Serious adverse events | — | P = 0.63 | — | — | — | — | — | |
| Non‐serious adverse events | P = 0.53 | P = 0.53 | — | — | — | — | — | |
| Nocturnal hypoglycaemia | — | P = 0.51 | — | — | — | — | — | |
| Mild/moderate hypoglycaemia | — | P = 0.85 | — | — | — | — | — | |
| HbA1c | — | P = 0.42 | — | — | — | — | — | |
| Insulin degludec versus insulin glargine | All‐cause mortality | P = 0.46 | — | — | — | — | — | — |
| Health‐related quality of life | P = 0.27 / 0.51 | — | — | — | — | — | — | |
| Severe hypoglycaemia | ‐ | — | — | — | — | — | — | |
| Non‐fatal myocardial infarction/stroke, blindness, end‐stage renal disease, serious adverse events, non‐serious adverse events, nocturnal hypoglycaemia, socioeconomic effects | — | — | — | — | — | — | — | |
| Diabetic ketoacidosis | P = 0.16 | — | — | — | — | — | — | |
| HbA1c | P = 0.26 | P = 0.71 | — | — | — | — | — | |
| —: denotes not possible to perform subgroup analysis HbA1c: glycosylated haemoglobin A1c; NPH: neutral protamine Hagedorn insulin. | ||||||||
Appendix 21. Health‐related quality of life: instruments
| Instrument | Dimensions (subscales) (no. of items) | Validated instrument | Answer options | Scores |
Minimum score Maximum score |
Weighting of scores | Direction of scales | Minimal important difference |
| Diabetes Health Profile employed in Standl 2004 |
Three dimensions: barriers to activity, psychological distress, and disinhibited eating. Only the dimension barriers to activity was included | Yes | — | — | — | — | — | — |
| ITR‐QOLN (Fujimoto 2018) employed in Kobayashi 2007 |
21 questions divided into 4 domains (1) Anxiety before sleep (2) Disturbances during sleep (3) Glycaemic control before breakfast (4) Overall well‐being |
Yes | Seven‐point scale (0–6) | . | Maximum 126 points | — | The higher score the better well‐being | — |
| W‐BQ employed in Home 2005; Ratner 2000 |
22‐item incorporating four subscales to measure depression (6 items), anxiety (6 items), energy (4 items) and positive well‐being (6 items) | Yes | Of the 22 items, then 8 of which are negatively phrased and 14 positively phrased | Each item is scored from 0 to 3, where 0 = not at all, and 3 = all the time | Depression: 0‐18; Anxiety: 0‐28; Energy: 0‐12; Positive Well‐being 0‐18 General Well‐being (total score): 0‐66 |
— | The higher score the better well‐being (higher scores for negatively‐phrased statements indicate worse well‐being while higher scores for positively‐phrased statements indicate better well‐being. In calculating the subscale scores for the Depression and Anxiety subscales of the W‐BQ, the scores on the positively‐worded items have to be reversed while for the Energy subscale the negatively‐worded item scores have to be reversed. The Positive Wellbeing subscale of the W‐BQ (6 items) is simply added, as all the items are positively worded) | Effect size of 0.20 or more is considered clinically meaningful for psychological outcomes |
| Well‐Being Enquiry for Diabetics (WED) questionnaire employed in Bolli 2009 |
50‐item questionnaire providing an evaluation of four aspects of quality of life: symptoms (10), discomfort (10), serenity (10) and impact (20) | Yes | 4‐point Likert scale (‘always/usually’ to ‘never/very infrequently’) | Total score is the sum of the subscale scores | — | — | Sums of item scores, with higher scores indicating better quality of life | — |
| Diabetes Quality of Life for Youth employed in Chase 2008 |
(1) Life satisfaction (2) Disease impact (3) Diabetes related worries |
Yes | (1) Very satisfied, moderately satisfied, neither satisfied nor dissatisfied, moderately dissatisfied, very dissatisfied (2) Never, seldom, sometimes, often, all the time (3) Does not apply, never, seldom, sometimes, often, all the time |
Total score is the sum of the subscale scores | — | — | Higher score indicates better quality of life, except for one item (in subscale 2 ‐ question B ‐7); here lower scores represent higher quality of life | — |
| Parents' Diabetes Quality of Life employed in Chase 2008 |
(1) Emotional burden of disease (2) Child‐related worries (3) Satisfaction |
Yes | (1) Very satisfied, moderately satisfied, neither satisfied nor dissatisfied, moderately dissatisfied, very dissatisfied (2) Never, seldom, sometimes, often, all the time (3) Does not apply, never, seldom, sometimes, often, all the time |
Total score is the sum of the subscale scores | — | — | Higher score indicates better quality of life | — |
| SF‐36 v2 employed in BEGIN Basal‐Bolus Type 1; Davies 2014; SWITCH 1 |
Physical functioning (10) Role‐physical (4) Bodily pain (2) General health (5) Vitality (4) Social functioning (2) Role‐emotional (3) Mental health (5) Reported health transition (1) | Yes | 3, 5 and 6‐point Likert scale | Scores for dimensions
Physical component summary (PCS) Mental component summary (MCS) |
Minimum scores:
scores for dimensions/PCS/MCS:
norm‐based scale Maximum scores: scores for dimensions/PCS/MCS: norm‐based scale |
No | Higher values mean better assessment | PCS: 2‐3 points
MCS: 3 points Dimensions: physical functioning/bodily pain/vitality: 2 points, if score < 40 3 points, if score ≥ 40 Role physical: 2 points Social functioning/mental health: 3 points Role emotional: 4 points |
| ITR‐QOLN: insulin therapy related quality of life at night; MCS: mental health component summary score; PCS: physical component summary score; SF‐36: short‐form 36; W‐BQ: well‐being questionnaire; WED: well‐being enquiry for diabetics. | ||||||||
Appendix 22. Source of information for outcome data: all‐cause mortality
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Yes | — | No | Yes | Yes | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | No | Yes | Yes | No | No |
| BEGIN Flex T1 | No | — | No | Yes | Yes | No | No |
| BEGIN Young | Yes | — | No | Yes | No | Yes | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | No | — | No | Yes | No | — | — |
| Davies 2014 | Yes | Yes | No | Yes | No | No | No |
| Fulcher 2005 | No | — | — | Yes | — | — | — |
| Heller 2009 | No | — | No | Yes | Yes | — | — |
| Home 2005 | No | Yes | — | Yes | — | No | Yes |
| Kobayashi 2007 | No | — | No | Yes | Yes | — | — |
| Liu 2016 | No | — | No | Yes | Yes | Yes | — |
| NCT00595374 | — | — | No | — | Yes | — | — |
| NCT00605137 | — | — | No | No | Yes | — | — |
| Pieber 2007 | No | — | No | Yes | Yes | — | Yes |
| Porcellati 2004 | Yes | — | — | — | — | — | — |
| PRESCHOOL | No | — | Yes | Yes | — | Yes | — |
| Ratner 2000 | No | — | — | Yes | — | No | Yes |
| Robertson 2007 | No | No | No | Yes | Yes | — | Yes |
| Russell‐Jones 2004 | No | No | No | Yes | Yes | No | Yes |
| Schober 2002 | No | — | — | Yes | — | — | No |
| Standl 2004 | No | — | — | Yes | Yes | No | Yes |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | Yes | — | No | Yes | Yes | Yes | — |
| Urakami 2017 | No | No | — | — | — | — | — |
| Vague 2003 | No | — | — | Yes | Yes | No | Yes |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 23. Source of information for outcome data: health‐related quality of life
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | No | — | No | No | No | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | No | Yes | No | No | No |
| BEGIN Flex T1 | No | — | No | No | No | No | No |
| BEGIN Young | No | — | No | No | No | No | No |
| Bolli 2009 | Yes | — | — | — | — | — | — |
| Chase 2008 | No | — | No | Yes | Yes | — | — |
| Davies 2014 | No | Yes | No | Yes | No | No | No |
| Fulcher 2005 | No | — | — | — | — | — | — |
| Heller 2009 | No | — | No | Yes | No | — | — |
| Home 2005 | No | No | — | Yes | — | No | No |
| Kobayashi 2007 | No | — | No | — | Yes | — | — |
| Liu 2016 | No | — | No | No | No | No | — |
| NCT00595374 | — | — | No | — | Yes | — | — |
| NCT00605137 | — | — | No | No | No | — | — |
| Pieber 2007 | No | — | No | Yes | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | No | — | No | No | — | No | — |
| Ratner 2000 | No | — | — | Yes | — | No | No |
| Robertson 2007 | No | No | No | No | No | — | No |
| Russell‐Jones 2004 | No | No | No | No | No | No | No |
| Schober 2002 | No | — | — | No | — | — | No |
| Standl 2004 | Yes | — | — | Yes | No | No | No |
| SWITCH 1 | No | No | No | Yes | No | — | No |
| Thalange 2013 | Yes | — | No | No | No | — | No |
| Urakami 2017 | No | No | — | — | — | — | — |
| Vague 2003 | No | — | — | No | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 24. Source of information for outcome data: severe hypoglycaemia
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Yes | — | No | Yes | No | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | No | Yes | No | Yes | Yes |
| BEGIN Flex T1 | Yes | — | No | Yes | Yes | Yes | Yes |
| BEGIN Young | Yes | — | No | Yes | No | Yes | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | Yes | — | No | Yes | Yes | — | — |
| Davies 2014 | Yes | Yes | No | Yes | No | Yes | Yes |
| Fulcher 2005 | No | — | — | Yes | — | — | — |
| Heller 2009 | No | — | No | No | Yes | — | — |
| Home 2005 | Yes | No | — | Yes | — | No | No |
| Kobayashi 2007 | Yes | — | No | Yes | No | — | — |
| Liu 2016 | Yes | — | Yes | Yes | No | Yes | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | Yes | — | — |
| Pieber 2007 | Yes | — | No | Yes | Yes | — | Yes |
| Porcellati 2004 | Yes | — | — | — | — | — | — |
| PRESCHOOL | Yes | — | Yes | Yes | — | No | — |
| Ratner 2000 | No | — | — | Yes | — | No | No |
| Robertson 2007 | Yes | No | No | Yes | Yes | — | Yes |
| Russell‐Jones 2004 | Yes | No | No | Yes | Yes | No | Yes |
| Schober 2002 | Yes | — | — | Yes | — | — | Yes |
| Standl 2004 | No | — | — | Yes | No | No | Yes |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | Yes | — | No | Yes | No | Yes | — |
| Urakami 2017 | Yes | Yes | — | — | — | — | — |
| Vague 2003 | Yes | — | — | Yes | No | No | Yes |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 25. Definition/type of outcome data: severe hypoglycaemia
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Hypoglycaemia requiring third party assistance | — | ND | Major hypoglycaemic episode: person not able to treat episode him/herself | Mentioned under serious adverse events | — | — |
| BEGIN Basal‐Bolus Type 1 | Hypoglycaemia requiring third party assistance | — | Severe hypoglycaemic episodes are defined as requiring assistance to administer carbohydrate, glucagon, or other resuscitative actions | Severe hypoglycaemic episodes are defined as requiring assistance | Severe hypoglycaemia | Severe hypoglycaemic episodes, where the patient is not able to treat himself | Defined as an episode requiring assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions |
| BEGIN Flex T1 | Hypoglycaemia requiring third party assistance | — | Severe hypoglycaemic episodes are defined as requiring assistance to administer carbohydrate, glucagon, or other resuscitative actions | Severe hypoglycaemia: an episode requiring assistance of another person to actively administer carbohydrate, glucagon or other resuscitative actions | Severe hypoglycaemia | Severe hypoglycaemic episodes, where the patient is not able to treat himself | An episode requiring assistance of another person to actively administer carbohydrate, glucagon, or resuscitative measures |
| BEGIN Young | The child has altered mental status and cannot assist in his/her own care, is semiconscious or unconscious, or in a coma ± convulsions and may require parenteral therapy (glucagon or iv glucose) | — | "Severe episodes or episodes with plasma glucose (PG) below or equal to 3.9 mmol/L (70 mg/dL) with or without symptoms of hypoglycaemia" | "Severe hypoglycaemia: The child has altered mental status and cannot assist in his own care, is semiconscious or unconscious, or in coma ± convulsions and may require parenteral therapy (glucagon or iv glucose)" | Severe hypoglycaemia | Children and adolescents ‐ severe hypoglycaemia: the child has altered mental status and cannot assist in his own care, is semiconscious or unconscious, or in coma ± convulsions and may require parenteral therapy (glucagon or iv glucose) | "The child has altered mental status and cannot assist in his own care, is semiconscious or unconscious, or in coma ± convulsions and may require parenteral therapy (glucagon or iv glucose)" |
| Bolli 2009 | Serious hypoglycaemia was defined as an event with blood glucose < 2.3 mmol/L, severe hypoglycaemia as an event with symptoms consistent with hypoglycaemia, during which the participant required the assistance of another person, or with prompt recovery after oral carbohydrate, intravenous glucose or glucagon administration | — | — | — | — | — | — |
| Chase 2008 | Severe hypoglycaemia was defined as an event requiring assistance from another person and associated with either BG < 2.0 mmol/L or prompt recovery after oral carbohydrate, intravenous glucose, or intramuscular or subcutaneous glucagon administration | — | ND | "Severe hypoglycemia was defined, as an event with clinical symptoms that was considered to result from hypoglycemia in which the participant required the assistance of another person and one of the following was true:
For further clarification, the definition of severe hypoglycaemia included all episodes in which neurological impairment was severe enough to prevent self‐treatment and because of which the participant was thought to be at risk for injury to themselves or others. Required assistance indicated that the participant could not help her/himself." |
ND | — | — |
| Davies 2014 | Hypoglycaemia requiring third party assistance | Hypoglycaemia requiring third party assistance | Severe hypoglycaemic episodes: episodes requiring active assistance of another person to administer carbohydrate, glucagon, or other resuscitative actions | "An episode requiring assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions" | Severe hypoglycaemia | Severe hypoglycaemic episodes, where the patient is not able to treat himself | Defined as an episode requiring assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions |
| Fulcher 2005 | Symptoms consistent with hypoglycaemia required the assistance of another person and was associated with a blood glucose level < 2.8 mmol/L or prompt recovery after oral carbohydrate, iv glucose or sc glucagon administration | — | — | "Was defined as an event with symptoms consistent with hypoglycaemia in which the participant required the assistance of another person and which was associated with a blood glucose level below 2.8 mmol/L or with prompt recovery after oral carbohydrate, intravenous glucose or glucagon administration" | — | — | — |
| Heller 2009 | The patient could not treat the episode by himself/herself | — | ND | The patient could not treat the episode by himself/herself | Major hypoglycaemia | — | — |
| Home 2005 | Severe symptomatic hypoglycaemia was defined as an event consistent with symptomatic hypoglycaemia requiring the assistance of another person, with either a blood glucose level < 2.8 mmol/L or prompt recovery after administration of oral carbohydrate, iv glucose or glucagon | ND | — | Severe hypoglycaemia was as an event with symptoms consistent with hypoglycaemia in which the participants required the assistance of another person and which was associated with a blood glucose level < 2.8 mmol/L (50 mg/dL) or prompt recovery after oral carbohydrate, intravenous glucose or glucagon administration | — | ND | Severe hypoglycaemia was as an event with symptoms consistent with hypoglycaemia in which the participants required the assistance of another person and with blood glucose level < 2.8 mmol/L (50 mg/dL) or prompt recovery after oral carbohydrate, intravenous glucose or glucagon administration |
| Kobayashi 2007 | Any event requiring assistance of another person to recover from hypoglycaemic symptoms with or without measurement of blood glucose levels | — | Major hypoglycaemia | Major hypoglycaemia | Major hypoglycaemia | — | — |
| Liu 2016 | Hypoglycaemia requiring the assistance of a third party or involving a seizure, coma, unconsciousness or the use of glucagon | — | "Severe symptomatic hypoglycemia: Any event with clinical symptoms considered to result from a hypoglycemic episode for which the participants required the assistance of a third party (other than the participant or a parent/usual caregiver), with acute neurological impairment directly resulting from the hypoglycemic event." | "Any event with clinical symptoms considered to result from a hypoglycemic episode for which the participants required the assistance of a third party (i.e. other than the patient or a parent/usual caregiver, e.g. from emergency personnel) because the participants/parents could not treat the event, with acute neurological impairment directly resulting from the hypoglycemic event. If a patient was assisted when necessary and not due to generosity, it would qualify as “require assistance”. The occurrence of seizure, coma, unconsciousness, or the use of glucagon, would also qualify a hypoglycemic episode as severe." | Severe hypoglycaemia | "Severe symptomatic hypoglycaemia: Any event with clinical symptoms considered to result from a hypoglycaemic episode for which the participants required the assistance of a third party (i.e. other than the patient or a parent/usual caregiver, e.g. from emergency personnel) because the participants/parents could not treat the event, with acute neurological impairment directly resulting from the hypoglycaemic event" | — |
| NCT00595374 | — | — | ND | — | Major hypoglycaemia | — | — |
| NCT00605137 | — | — | ND | Major hypoglycaemia | Hypoglycaemia requiring third party assistance | — | — |
| Pieber 2007 | Hypoglycaemia requiring third party assistance | — | ND | The patient could not treat the episode by himself/herself | Major hypoglycaemia | — | ND |
| Porcellati 2004 | Hypoglycaemia requiring external help | — | — | — | — | — | — |
| PRESCHOOL | Severe hypoglycaemia was defined as an event requiring assistance from another person, as a result of altered consciousness, to administer carbohydrate, glucagon or to take other actions | — | "Severe symptomatic hypoglycemia: any event with clinical symptoms considered to result from a hypoglycemic episode for which the patients required the assistance of a third party (i.e. other than the patient, or a parent/usual caregiver; e.g. from emergency personnel), because the patients/parents could not treat the event with acute neurological impairment directly resulting from the hypoglycemic event. The occurrence of seizure, coma, unconsciousness, or the use of glucagon, were also to qualify a hypoglycemic episode as severe." | "Severe symptomatic hypoglycemia: any event with clinical symptoms considered to result from a hypoglycemic episode for which the patients required the assistance of a third party (i.e. other than the patient, or a parent/usual caregiver; e.g. from emergency personnel), because the patients/parents could not treat the event with acute neurological impairment directly resulting from the hypoglycemic event. The occurrence of seizure, coma, unconsciousness, or the use of glucagon, were also to qualify a hypoglycemic episode as severe" | — | Severe hypoglycaemia | — |
| Ratner 2000 | Symptomatic hypoglycaemia requiring third party assistance | — | — | "Severe hypoglycemia was defined as an event with symptoms consistent with hypoglycemia in which the participant required the assistance of another person and which was associated with a blood glucose level below 2.8 mmol/L (50 mg/dL) or prompt recovery after oral carbohydrate, intravenous glucose or glucagon administration" | — | ND | Severe hypoglycaemia was as an event with symptoms consistent with hypoglycaemia in which the participants required the assistance of another person and with blood glucose level < 2.8 mmol/L (50 mg/dL) or prompt recovery after oral carbohydrate, intravenous glucose or glucagon administration |
| Robertson 2007 | Episodes requiring assistance from another person due to severe central nervous system dysfunction | ND | ND | Hypoglycaemic episode requiring assistance from another person | Major hypoglycaemia | — | Severe CNS symptoms consistent with hypoglycaemia in which the patients required assistance with glucose < 3.1 mmol/L or reversal by food or glucagon |
| Russell‐Jones 2004 | Requiring third party assistance | ND | Major hypoglycaemia | An episode with severe central nervous system symptoms consistent with hypoglycaemia in which the participant is unable to treat himself/herself and which has one of the following characteristics: blood glucose < 2.8 mmol/L or reversal of symptoms after either food intake or glucagon/iv glucose administration | Major hypoglycaemia | Hypoglycaemic episodes were classified in the trials as:
|
|
| Schober 2002 | An event with symptoms consistent with hypoglycaemia in which the participant required assistance from another person, and which was associated with a blood glucose level < 2.8 mmol/L or prompt recovery after oral carbohydrate or intravenous glucose or glucagon administration | — | — | An event with symptoms consistent with hypoglycaemia in which the participant required assistance from another person, and which was associated with a blood glucose level < 2.8 mmol/L or prompt recovery after oral carbohydrate or intravenous glucose or glucagon administration | — | — | Hypoglycaemia in which the participant required assistance from another person and with a blood glucose level below 2.8 mmol/L or prompt recovery administration of glucose or glucagon |
| Standl 2004 | Hypoglycaemia requiring third party assistance | — | — | An episode with severe CNS symptoms consistent with hypoglycaemia in which the participant is unable to treat himself/herself and which has one of the following characteristics:
|
Hypoglycaemic episodes were classified in the trials as:
|
Major hypoglycaemia was defined as severe CNS symptoms consistent with hyperglycaemia in which patients requires assistance, with blood glucose < 2.8 mmol/L or reversal by food or glucagon | |
| SWITCH 1 | Episode requiring assistance of another person to actively administer carbohydrate, glucagon, or take other corrective actions, neurological recovery following the return of plasma glucose to normal, or both | ND | A hypoglycaemic episode requiring assistance of another person to actively administer carbohydrate, glucagon, or take other corrective actions. Plasma glucose values may not be available during an event, but neurological recovery following the return of plasma glucose to normal is considered sufficient evidence that the event was induced by a low plasma glucose concentration | Severe hypoglycaemia was defined as an episode requiring assistance of another person to actively administer carbohydrate, glucagon, or take other corrective actions. Plasma glucose concentrations may not be available during an event, but neurological recovery following the return of plasma glucose to normal is considered sufficient evidence that the event was induced by a low plasma glucose concentration | An episode requiring assistance of another person to actively administer carbohydrate, glucagon, or take other corrective actions | — | ND |
| Thalange 2013 | Severe hypoglycaemia was defined as episodes where the persons were semi‐conscious, unconscious or in a coma, with or without convulsions | — | ND | "participant is semiconscious/ unconscious/in coma ± convulsion and may require parenteral treatment (glucagon or iv glucose)" | Severe hypoglycaemia | Severe hypoglycaemia may lead to unconsciousness and/or convulsions and may result in temporary or permanent impairment of brain function or even death | — |
| Urakami 2017 | Severe hypoglycaemia is defined as an event associated with impaired consciousness or seizure | "Severe hypoglycemia is defined as an event associated with impaired consciousness or seizure" |
— | — | — | — | — |
| Vague 2003 | Hypoglycaemic episode with severe central nervous system symptoms consistent with hypoglycaemia, in which the participant was unable to treat himself/herself and which had one of the following characteristics: blood glucose recorded as < 2.8 mmol/L or symptom reversal achieved with food, glucose or glucagon | — | — | An episode with severe CNS symptoms consistent with hypoglycaemia in which the participant is unable to treat himself/herself and which has one of the following characteristics:
|
Major hypoglycaemia | Hypoglycaemic episodes were classified in the trials as:
|
Major hypoglycaemia was defined as severe CNS symptoms consistent with hypoglycaemia in which patient requires assistance, with blood glucose < 2.8 mmol/L or reversal by food or glucagon |
| —: indicates source not available CNS: central nervous system; EMA: European Medicines Agency; FDA: Food and Drug Administration; iv:intravenous;ND: not defined; NR: not reported; PG: plasma glucose; sc: subcutaneous. | |||||||
Appendix 26. Source of information for outcome data: cardiovascular mortality
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Yes | — | No | Yes | Yes | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | No | Yes | Yes | No | No |
| BEGIN Flex T1 | No | — | No | Yes | Yes | No | No |
| BEGIN Young | Yes | — | No | Yes | Yes | Yes | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | No | — | No | Yes | No | — | — |
| Davies 2014 | Yes | — | No | Yes | No | No | No |
| Fulcher 2005 | No | — | — | Yes | — | — | — |
| Heller 2009 | No | — | No | Yes | Yes | — | — |
| Home 2005 | No | Yes | — | Yes | — | No | No |
| Kobayashi 2007 | No | — | No | Yes | Yes | — | — |
| Liu 2016 | No | — | No | Yes | Yes | Yes | — |
| NCT00595374 | — | — | No | — | Yes | — | — |
| NCT00605137 | — | — | No | No | Yes | — | — |
| Pieber 2007 | No | — | No | Yes | Yes | — | No |
| Porcellati 2004 | Yes | — | — | — | — | — | — |
| PRESCHOOL | No | — | Yes | Yes | — | Yes | — |
| Ratner 2000 | No | — | — | Yes | No | No | No |
| Robertson 2007 | No | No | No | Yes | Yes | — | Yes |
| Russell‐Jones 2004 | No | No | No | Yes | Yes | No | Yes |
| Schober 2002 | No | — | — | Yes | — | — | No |
| Standl 2004 | No | — | — | Yes | Yes | No | Yes |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | Yes | — | No | Yes | Yes | Yes | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | No | — | — | Yes | Yes | No | Yes |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 27. Source of information for outcome data: non‐fatal myocardial infarction
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | No | — | No | Yes | No | — | — |
| BEGIN Basal‐Bolus Type 1 | No | — | Yes | Yes | No | No | No |
| BEGIN Flex T1 | No | — | No | Yes | No | No | No |
| BEGIN Young | No | — | No | No | No | No | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | No | — | No | No | No | — | — |
| Davies 2014 | No | Yes | No | No | No | No | No |
| Fulcher 2005 | No | No | No | No | No | — | — |
| Heller 2009 | No | — | No | Yes | No | — | — |
| Home 2005 | No | No | — | Yes | — | No | No |
| Kobayashi 2007 | No | — | No | No | No | — | — |
| Liu 2016 | No | — | No | No | No | No | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | No | — | — |
| Pieber 2007 | No | No | No | No | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | No | — | No | No | — | No | — |
| Ratner 2000 | No | — | — | No | — | No | No |
| Robertson 2007 | No | No | No | No | No | — | No |
| Russell‐Jones 2004 | No | No | No | No | No | No | No |
| Schober 2002 | No | — | — | No | — | — | No |
| Standl 2004 | No | — | — | No | No | No | No |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | No | — | No | No | No | No | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | No | — | — | No | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 28. Source of information for outcome data: non‐fatal stroke
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | No | — | No | No | No | — | — |
| BEGIN Basal‐Bolus Type 1 | No | — | No | Yes | No | No | No |
| BEGIN Flex T1 | No | — | No | Yes | No | No | No |
| BEGIN Young | No | — | No | No | No | No | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | No | — | No | No | No | — | — |
| Davies 2014 | No | Yes | No | No | No | No | No |
| Fulcher 2005 | No | — | — | No | — | — | — |
| Heller 2009 | No | — | No | Yes | No | — | — |
| Home 2005 | No | No | — | Yes | — | No | No |
| Kobayashi 2007 | No | — | No | No | No | — | — |
| Liu 2016 | No | — | No | No | No | No | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | No | — | — |
| Pieber 2007 | No | No | No | No | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | No | — | No | No | — | No | — |
| Ratner 2000 | No | — | — | No | — | No | No |
| Robertson 2007 | No | No | No | No | No | — | No |
| Russell‐Jones 2004 | No | No | No | No | No | No | No |
| Schober 2002 | No | — | — | No | — | — | No |
| Standl 2004 | No | — | — | No | No | No | No |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | No | — | No | No | No | No | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | No | — | — | No | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 29. Source of information for outcome data: end‐stage renal disease
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | No | — | No | No | No | — | — |
| BEGIN Basal‐Bolus Type 1 | No | — | No | No | No | No | No |
| BEGIN Flex T1 | No | — | No | No | No | No | No |
| BEGIN Young | No | — | No | No | No | No | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | No | — | No | No | No | — | — |
| Davies 2014 | No | Yes | No | No | No | No | No |
| Fulcher 2005 | No | — | — | No | — | — | — |
| Heller 2009 | No | — | No | No | No | — | — |
| Home 2005 | No | No | — | No | — | No | No |
| Kobayashi 2007 | No | — | No | No | No | — | — |
| Liu 2016 | No | — | No | No | No | No | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | No | — | — |
| Pieber 2007 | No | — | No | No | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | No | — | No | No | — | No | — |
| Ratner 2000 | No | — | — | No | — | No | No |
| Robertson 2007 | No | No | No | No | No | — | No |
| Russell‐Jones 2004 | No | No | No | No | No | No | No |
| Schober 2002 | No | — | — | No | — | — | No |
| Standl 2004 | No | — | — | No | No | No | No |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | No | — | No | No | No | No | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | No | — | — | No | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 30. Source of information for outcome data: blindness
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | No | — | No | No | No | — | — |
| BEGIN Basal‐Bolus Type 1 | No | — | No | No | No | No | No |
| BEGIN Flex T1 | No | — | No | No | No | No | No |
| BEGIN Young | No | — | No | No | No | No | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | No | No | No | No | No | — | — |
| Davies 2014 | No | Yes | No | No | No | No | No |
| Fulcher 2005 | No | — | — | No | — | — | — |
| Heller 2009 | No | — | No | No | No | — | — |
| Home 2005 | No | No | — | No | — | No | No |
| Kobayashi 2007 | No | — | No | No | No | — | — |
| Liu 2016 | No | — | No | No | No | No | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | No | — | — |
| Pieber 2007 | No | — | No | No | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | No | — | No | No | — | No | — |
| Ratner 2000 | No | — | — | No | — | No | No |
| Robertson 2007 | No | No | No | No | No | — | No |
| Russell‐Jones 2004 | No | No | No | No | No | No | No |
| Schober 2002 | No | — | — | No | — | — | No |
| Standl 2004 | No | — | — | No | No | No | No |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | No | — | No | No | No | No | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | No | — | — | No | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 31. Source of information for outcome data: serious adverse events
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Yes | — | No | Yes | Yes | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | Yes | Yes | Yes | No | No |
| BEGIN Flex T1 | Yes | — | Yes | Yes | Yes | No | No |
| BEGIN Young | Yes | — | Yes | Yes | Yes | Yes | No |
| Bolli 2009 | Yes | — | — | — | — | — | — |
| Chase 2008 | Yes | — | No | Yes | No | — | — |
| Davies 2014 | Yes | Yes | Yes | Yes | No | No | No |
| Fulcher 2005 | No | — | — | Yes | — | — | — |
| Heller 2009 | Yes | — | No | Yes | Yes | — | — |
| Home 2005 | Yes | No | — | Yes | — | No | No |
| Kobayashi 2007 | Yes | — | No | Yes | Yes | — | — |
| Liu 2016 | No | — | Yes | Yes | Yes | Yes | — |
| NCT00595374 | — | — | No | — | Yes | — | — |
| NCT00605137 | — | — | No | No | Yes | — | — |
| Pieber 2007 | Yes | — | No | Yes | Yes | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | No | — | Yes | Yes | — | Yes | — |
| Ratner 2000 | No | — | — | Yes | — | No | No |
| Robertson 2007 | No | No | No | Yes | Yes | — | Yes |
| Russell‐Jones 2004 | No | No | No | Yes | No | No | No |
| Schober 2002 | Yes | — | — | Yes | — | — | No |
| Standl 2004 | No | — | — | Yes | Yes | No | No |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | Yes | — | Yes | Yes | No | No | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | No | — | — | Yes | Yes | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 32. Source of information for outcome data: diabetic ketoacidosis
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | No | — | No | Yes | Yes | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | Yes | Yes | No | No | No |
| BEGIN Flex T1 | No | — | Yes | Yes | No | No | No |
| BEGIN Young | No | — | No | Yes | No | No | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | Yes | — | No | Yes | No | — | — |
| Davies 2014 | No | Yes | Yes | Yes | No | No | No |
| Fulcher 2005 | No | — | — | Yes | — | — | — |
| Heller 2009 | No | — | No | Yes | No | — | — |
| Home 2005 | No | No | — | Yes | — | No | No |
| Kobayashi 2007 | No | — | No | Yes | No | — | — |
| Liu 2016 | Yes | — | No | Yes | No | Yes | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | Yes | — | — |
| Pieber 2007 | No | — | No | No | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | No | — | Yes | Yes | — | Yes | — |
| Ratner 2000 | No | — | — | Yes | — | No | No |
| Robertson 2007 | Yes | No | No | Yes | Yes | — | Yes |
| Russell‐Jones 2004 | No | No | No | No | No | No | No |
| Schober 2002 | Yes | — | — | Yes | — | — | No |
| Standl 2004 | No | — | — | No | No | No | No |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | Yes | — | Yes | Yes | No | No | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | No | — | — | Yes | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 33. Source of information for outcome data: non‐serious adverse events
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | No | — | No | Yes | Yes | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | Yes | Yes | Yes | No | No |
| BEGIN Flex T1 | Yes | — | Yes | Yes | Yes | No | No |
| BEGIN Young | Yes | — | Yes | Yes | Yes | Yes | No |
| Bolli 2009 | Yes | — | — | — | — | — | — |
| Chase 2008 | Yes | — | No | Yes | No | — | — |
| Davies 2014 | No | Yes | Yes | Yes | No | No | No |
| Fulcher 2005 | Yes | — | — | Yes | — | — | — |
| Heller 2009 | Yes | — | No | Yes | Yes | — | — |
| Home 2005 | No | No | — | Yes | — | No | No |
| Kobayashi 2007 | No | — | No | Yes | Yes | — | — |
| Liu 2016 | No | — | Yes | Yes | Yes | Yes | — |
| NCT00595374 | — | — | No | — | Yes | — | — |
| NCT00605137 | — | — | No | Yes | No | — | — |
| Pieber 2007 | No | — | No | Yes | Yes | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | Yes | — | Yes | Yes | — | No | — |
| Ratner 2000 | Yes | — | — | Yes | — | No | No |
| Robertson 2007 | Yes | No | No | Yes | No | — | No |
| Russell‐Jones 2004 | No | No | No | Yes | No | No | No |
| Schober 2002 | Yes | — | — | Yes | — | — | No |
| Standl 2004 | No | — | — | Yes | No | No | No |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | Yes | — | Yes | Yes | No | No | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | Yes | — | — | Yes | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 34. Source of information for outcome data: nocturnal hypoglycaemia
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Yes | — | No | Yes | No | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | No | Yes | No | Yes | No |
| BEGIN Flex T1 | Yes | — | Yes | Yes | Yes | Yes | No |
| BEGIN Young | Yes | — | No | Yes | No | Yes | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | No | — | No | Yes | No | — | — |
| Davies 2014 | Yes | Yes | No | Yes | No | Yes | No |
| Fulcher 2005 | Yes | — | — | Yes | — | — | — |
| Heller 2009 | Yes | — | No | Yes | Yes | — | — |
| Home 2005 | Yes | No | — | Yes | — | No | No |
| Kobayashi 2007 | Yes | — | No | No | Yes | — | — |
| Liu 2016 | Yes | — | Yes | Yes | No | Yes | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | Yes | — | — |
| Pieber 2007 | Yes | — | No | Yes | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | Yes | — | Yes | Yes | — | No | — |
| Ratner 2000 | No | — | — | Yes | — | No | No |
| Robertson 2007 | Yes | No | No | Yes | No | — | No |
| Russell‐Jones 2004 | Yes | No | No | Yes | No | No | No |
| Schober 2002 | Yes | — | — | Yes | — | — | No |
| Standl 2004 | No | — | — | Yes | No | No | — |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | Yes | — | No | Yes | No | Yes | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | Yes | — | — | Yes | No | No | — |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 35. Definition/type of outcome data: nocturnal hypoglycaemia
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Hypoglycaemia between 23:00 to 06:00 | — | ND | Hypoglycaemia between 23:00 to 06:00 | Hypoglycaemia between 23:00 to 06:00 | — | — |
| BEGIN Basal‐Bolus Type 1 | Hypoglycaemic episodes occurring from 00:01 to 05:59 | — | Nocturnal hypoglycaemic episodes are defined as occurring between 00:01 and 05:59 a.m. | Nocturnal hypoglycaemic episodes are defined as occurring between 00:01 and 05:59 a.m. | Nocturnal hypoglycaemic episodes are defined as occurring between 00:01 and 05:59 a.m. | Hypoglycaemic episodes occurring from 00:01 to 05:59 | Nocturnal hypoglycaemic episodes are defined as occurring between 00:01 and 05:59 a.m. |
| BEGIN Flex T1 | Episodes occurring between 00:01 and 05:59 (inclusive) | — | Nocturnal hypoglycaemic episodes are defined as occurring between 00:01 and 05:59 a.m. | Hypoglycaemia between 00:01 to 05:50 a.m. | "A hypoglycaemic episode with time of onset between 00:01 and 05:59 (both included) was considered nocturnal" | Hypoglycaemic episodes occurring from 00:01 to 05:59 | Hypoglycaemic episodes in the timeframe 00:00 to 06:00 |
| BEGIN Young | Hypoglycaemic episodes occurring between 11 p.m. and 7 a.m. inclusive were classified as nocturnal | — | Hypoglycaemia from 11 p.m. ‐ 7 a.m./23:00 ‐ 07:00 | "Hypoglycaemic episodes were defined as nocturnal if the time of onset was between 11 p.m.‐7 a.m./23:00‐7:00" | "Nocturnal (11 p.m. ‐ 7 a.m.]" | Hypoglycaemia from 11 p.m. ‐ 7 a.m. | Hypoglycaemia from 11 p.m. ‐ 7 a.m. |
| Bolli 2009 | Hypoglycaemia which occurred between bedtime and before getting up in the morning | — | — | — | — | — | — |
| Chase 2008 | Hypoglycaemia from midnight and 6 a.m. | — | ND | Hypoglycaemia from midnight and 6 a.m. | ND | — | — |
| Davies 2014 | Hypoglycaemia between 00:01 and 05:59 hours | Hypoglycaemia between 00:01 and 05:59 hours | Nocturnal hypoglycaemic episodes are defined as occurring between 00:01 and 05:59 a.m. | "The nocturnal period was considered as the period between 00:01 and 05:59 a.m." | Nocturnal hypoglycaemia (00:01‐05:59 a.m.) | Hypoglycaemic episodes occurring from 00:01 to 05:59 h | Nocturnal hypoglycaemic episodes are defined as occurring between 00:01 and 05:59 a.m. |
| Fulcher 2005 | Hypoglycaemia occurring after the evening insulin injection and before the morning insulin dose | — | — | "Nocturnal hypoglycaemia was defined as hypoglycaemia occurring between bedtime after the evening injection and before getting up in the morning (i.e. before the morning determination of fasting blood glucose and before any morning insulin dose)" | — | — | — |
| Heller 2009 | Hypoglycaemia between 23:00 and 06:00 | — | ND | Hypoglycaemia between 23:00 and 06:00 | Hypoglycaemia between 23:00 and 06:00 | — | — |
| Home 2005 | Symptomatic hypoglycaemia occurring during sleep between bedtime and rising in the morning, or before the morning pre‐breakfast self‐blood glucose measurement and the morning insulin injection. Only participants with confirmed blood glucose < 2.0 mmol/L were considered clinically relevant | "Nocturnal hypoglycemia was defined as hypoglycemia occurring while the participant was asleep, between bedtime after the evening injection and before getting up in the morning, i.e. before the morning determination of FBG and before the morning injection" | ND | ND | |||
| Kobayashi 2007 | Hypoglycaemia between 00:01 and 05:59 | — | ND | Nocturnal hypoglycaemia | Hypoglycaemia between 23:00 and 06:00 | — | — |
| Liu 2016 | Hypoglycaemia occurring between 23:00–07:00 | — | "Any asymptomatic and/or symptomatic hypoglycemic event that occurred between 23:00 to 07:00" | "Any asymptomatic and/or symptomatic hypoglycemic event that occurred between 23:00 to 07:00" | Nocturnal hypoglycaemia | Any asymptomatic and/or symptomatic hypoglycaemic event that occurred between 23:00 to 07:00 | — |
| NCT00595374 | — | — | ND | — | Nocturnal hypoglycaemia | — | — |
| NCT00605137 | — | — | ND | Nocturnal hypoglycaemia | Hypoglycaemia from 23:00 ‐ 06:00, inclusive | — | — |
| Pieber 2007 | Hypoglycaemia between 23:00 and 06:00 | — | ND | Hypoglycaemia between 23:00 and 06:00 | Hypoglycaemia between 23:00 and 06:00 | — | ND |
| Porcellati 2004 | Nocturnal episodes of hypoglycaemia were calculated from values measured at 03.00 or any time between 01.00 and 07.30 when participants awoke with symptoms suggestive of hypoglycaemia | — | — | — | — | — | — |
| PRESCHOOL | Hypoglycaemia between 23:00 hours and 07:00 | — | "Nocturnal hypoglycemia: any event from the "all hypoglycemia" total that occurred between 23:00 and 07:00" | "Nocturnal hypoglycemia: any event from the “all hypoglycemia” total that occurred between 23:00 and 07:00" | — | ND | — |
| Ratner 2000 | Hypoglycaemia occurring while asleep after the bedtime insulin dose and before the morning insulin dose and before the morning blood glucose measurement | — | — | "Nocturnal hypoglycemia was defined as hypoglycemia which occurred while the participant was asleep between bedtime after the evening injection and before getting up in the morning (i.e. before the morning determination of fasting blood glucose and before the morning injection)" | — | ND | ND |
| Robertson 2007 | Hypoglycaemic between 22.00 (included) − 07.00(excluded) | ND | ND | Hypoglycaemic between 22.00 (included) − 07.00(excluded) | Hypoglycaemia between (22:00 to 07:00) | — | ND |
| Russell‐Jones 2004 | Hypoglycaemia between 11 p.m. to 6 a.m. | ND | ND | Hypoglycaemia from 23:00 to 06:00 | Hypoglycaemia from 23:00 to 06:00 | Hypoglycaemia episodes occurring between 23:00 and 6:00 | Hypoglycaemia episodes occurring between 23:00 and 06:00 |
| Schober 2002 | Nocturnal hypoglycaemia was defined as hypoglycaemia while the participants was sleeping between bedtime and after the evening injection and before getting up in the morning | "Nocturnal hypoglycemia was defined as hypoglycemia occurring while the participant was asleep, between bedtime after the evening injection and before getting up in the morning, i.e. before the morning determination of FBG and before the morning injection" | — | ND | |||
| Standl 2004 | Hypoglycaemia between 23:00 to 06:00 | — | — | Hypoglycaemia from 23:00 to 06:00 | Hypoglycaemia from 23:00 to 06:00 | Hypoglycaemia episodes occurring between 23:00 and 6:00 | Hypoglycaemia episodes occurring between 23:00 and 6:00 |
| SWITCH 1 | Episodes between 12:01 a.m. and 5:59 a.m. | ND | Hypoglycaemia between 00:01 and 05.59 a.m. | Hypoglycaemia between 00:01 and 05.59 a.m. | Nocturnal hypoglycaemia | — | ND |
| Thalange 2013 | Nocturnal if they occurred between 22:00 and 07:00 | — | ND | "Episodes occurring between 22:00 (included) and 07:00 (excluded) were defined as nocturnal" | ND | ND | — |
| Urakami 2017 | Hypoglycaemia occurring between 22:00 – 06:59 | Hypoglycaemia occurring between 22:00 – 06:59 | — | — | — | — | — |
| Vague 2003 | Hypoglycaemia between 23:00 to 06:00 | — | — | Hypoglycaemia between 23:00 (included) and 06:00 (excluded) | Hypoglycaemia between 23:00 to 06:00 | Hypoglycaemia episodes occurring between 23:00 and 6:00 hours | Hypoglycaemia episodes occurring between 23:00 and 6:00 |
| —: indicates source not available a.m.: ante meridiem; EMA: European Medicines Agency; FBG: fasting blood glucose; FDA: Food and Drug Administration; ND: not defined; p.m.: post meridiem. | |||||||
Appendix 36. Source of information for outcome data: mild/moderate hypoglycaemia
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Yes | — | No | Yes | Yes | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | No | Yes | No | Yes | No |
| BEGIN Flex T1 | Yes | — | No | Yes | No | Yes | No |
| BEGIN Young | Yes | — | No | Yes | No | Yes | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | Yes | — | No | Yes | — | — | — |
| Davies 2014 | Yes | Yes | No | Yes | No | Yes | No |
| Fulcher 2005 | No | — | — | Yes | — | — | — |
| Heller 2009 | No | — | No | Yes | No | — | — |
| Home 2005 | Yes | No | — | Yes | — | No | Yes |
| Kobayashi 2007 | Yes | — | No | No | Yes | — | — |
| Liu 2016 | Yes | — | Yes | Yes | No | Yes | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | Yes | Yes | — | — |
| Pieber 2007 | Yes | — | No | Yes | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | Yes | — | Yes | Yes | — | No | — |
| Ratner 2000 | No | — | — | Yes | — | No | Yes |
| Robertson 2007 | Yes | No | No | Yes | No | — | Yes |
| Russell‐Jones 2004 | Yes | No | No | Yes | No | No | No |
| Schober 2002 | Yes | — | — | Yes | — | — | No |
| Standl 2004 | No | — | — | Yes | No | — | — |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | Yes | — | No | Yes | No | Yes | — |
| Urakami 2017 | No | Yes | — | — | — | — | — |
| Vague 2003 | Yes | — | — | Yes | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 37. Definition/type of outcome data: mild/moderate hypoglycaemia
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | All SMPG values < 3.1 mmol/L as well as signs and symptoms of hypoglycaemia minor if plasma glucose < 3.1 mmol/L and the individual dealt with the episode him/herself, and as symptoms only if episodes were not confirmed by a plasma glucose measurement and no assistance was required | — | ND | Plasma glucose < 3.1 mmol/L as well as signs and symptoms of hypoglycaemia minor if plasma glucose < 3.1 mmol/L and the individual dealt with the episode him/herself, and as symptoms only if episodes were not confirmed by a plasma glucose measurement and no assistance was required | ND | — | — |
| BEGIN Basal‐Bolus Type 1 | Confirmed hypoglycaemic episodes included those with a plasma glucose value of < 3.1 mmol/L | — | No | An episode with symptoms consistent with hypoglycaemia with confirmation by plasma glucose < 3.1 mmol/L or full blood glucose < 2.8 mmol/L and which is handled by the participant himself/herself | Confirmed hypoglycaemic episodes included those with a plasma glucose value of < 3.1 mmol/L | Mild hypoglycaemic episodes can be treated by oral administration of glucose or other products containing sugar | An episode not requiring third party assistance where a plasma glucose < 3.1 mmol/L or whole blood glucose < 2.8 mmol/L was recorded (with or without symptoms of hypoglycaemia) |
| BEGIN Flex T1 | Minor hypoglycaemic episodes are defined as able to treat her/himself and plasma glucose below 3.1 mmol/L | — | Minor hypoglycaemic episodes are defined as able to treat her/himself and plasma glucose below 3.1 mmol/L. | Minor hypoglycaemic episode was defined as: an episode with symptoms consistent with hypoglycaemia with confirmation by PG < 3.1 mmol/L or full blood glucose < 2.8 mmol/L and which was handled by the participants themselves | Minor hypoglycaemia with a confirmed PG < 3.1 mmol/L | Mild hypoglycaemic episodes can be treated by oral administration of glucose or other products containing sugar | An episode not requiring third party assistance where a plasma glucose < 3.1 mmol/L or whole blood glucose < 2.8 mmol/L was recorded (with or without symptoms of hypoglycaemia) |
| BEGIN Young | Confirmed hypoglycaemia was defined as SMPG < 3.1 mmol/L | — | PG below or equal to 3.9 mmol/L (70 mg/dL) with or without symptoms of hypoglycaemia | "An episode with symptoms consistent with hypoglycaemia with confirmation by PG" | PG ≤ to 3.9 mmol/L (70 mg/dL) with or without symptoms of hypoglycaemia | An episode with symptoms consistent with hypoglycaemia with confirmation by plasma glucose < 3.1 mmol/L or full blood glucose < 2.8 mmol/L and which does not fulfil the requirements for being classified as a severe hypoglycaemic episode | Blood glucose < 3.1 mmol/L, self‐treated |
| Bolli 2009 | Blood glucose ≤ 4.0 mmol/L | — | — | — | — | — | — |
| Chase 2008 | The rates of biochemical hypoglycaemia were ascertained by analysis of SMBG data and divided into 3 categories: < 3.9 mmol/L, < 2.8 mmol/L and < 2.0 mmol/L | — | ND | "The study co‐ordinator also reviewed the participant’s diary for any blood glucose values (< 70 mg/dL [3.9 mmol/L]) without symptoms and recorded these events in the CRF if, in the opinion of the investigator/study co‐ordinator, they represented true hypoglycemia " | ND | — | — |
| Davies 2014 | Confirmed hypoglycaemia was defined as plasma glucose < 3.1 mmol/L regardless of symptoms | Confirmed hypoglycaemia was defined as PG < 3.1mmol/L regardless of symptoms | Minor hypoglycaemic episodes: episodes where participant was able to treat her/himself and plasma glucose < 3.1 mmol/L, with or without symptoms | Able to treat him/herself and blood glucose ≤ 3.1 mmol/L | Mild hypoglycaemia with PG < 3.1 mmol/L | Mild hypoglycaemic episodes can be treated by oral administration of glucose or other products containing sugar | An episode not requiring third party assistance where a plasma glucose < 3.1 mmol/L or whole blood glucose < 2.8 mmol/L was recorded (with or without symptoms of hypoglycaemia) |
| Fulcher 2005 | Symptomatic hypoglycaemia was defined as an event with symptoms consistent with hypoglycaemia that was mild (2.8–3.6 mmol/L) or moderate (< 2.8 mmol/L) | — | — | "It could be either mild (between 2.8 and 3.6 mmol/L), moderate (below 2.8 mmol/L but did not require the assistance of another person)" | — | — | — |
| Heller 2009 | Minor: the patient could treat himself/herself and the measured plasma glucose value was < 3.1 mmol/L Symptoms only: the patient could treat himself/herself and no plasma glucose measurement was taken or the measured plasma glucose value was ≥ 3.1 mmol/L |
— | ND | Minor: the patient could treat himself/herself and the measured plasma glucose value was < 3.1 mmol/L Symptoms only: the patient could treat himself/herself and no plasma glucose measurement was taken or the measured plasma glucose value was ≥ 3.1 mmol/L |
Minor and moderate hypoglycaemia | — | — |
| Home 2005 | Hypoglycaemia was categorised as symptomatic (clinical symptoms confirmed by blood glucose < 2.8 mmol/L) or asymptomatic (confirmed by blood glucose < 2.8 mmol/L without symptoms) | ND | — | "Hypoglycemia was either symptomatic, i.e. with clinical symptoms that could be confirmed by blood glucose below 2.8 mmol/L (50 mg/dL), or asymptomatic, i.e. any event with a confirmed blood glucose level below 2.8 mmol/L (50 mg/dL) but without any symptoms" | — | ND | Hypoglycaemia was either symptomatic, i.e. with clinical symptoms that could be confirmed by blood glucose below 2.8 mmol/L (50 mg/dL), or asymptomatic, i.e. any event with a confirmed blood glucose level below 2.8 mmol/L (50 mg/dL) but without any symptoms |
| Kobayashi 2007 | Any symptoms consistent with hypoglycaemia | — | ND | Minor hypoglycaemia | Minor hypoglycaemia | — | — |
| Liu 2016 | Hypoglycaemia was defined as asymptomatic (blood glucose values < 3.9 mmol/L without clinical symptoms), symptomatic (blood glucose < 3.9 mmol/L with associated clinical symptoms) | — | "Asymptomatic hypoglycemia: Blood glucose values < 70 mg/dL (3.9 mmol/L) without clinical symptoms and/or signs. Symptomatic hypoglycemia: Any event with clinical symptoms that were considered to result from a hypoglycemic episode with an accompanying blood glucose < 70 mg/dL (3.9 mmol/L)" | "Symptomatic hypoglycemia: Any event with clinical symptoms that were considered to result from a hypoglycemic episode with an accompanying blood glucose" | Asymptomatic and symptomatic hypoglycaemia | Any event with clinical symptoms that were considered to result from a hypoglycaemic episode with an accompanying blood glucose | — |
| NCT00595374 | — | — | ND | — | Minor hypoglycaemia | — | — |
| NCT00605137 | — | — | ND | Minor hypoglycaemia | Minor hypoglycaemic episodes: blood glucose < 3.1 mmol/L and able treat the period themselves) Symptoms only: no blood glucose measurement or blood glucose > 3.1 mmol/L Biochemical hypoglycaemia: defined as asymptomatic hypoglycaemic with blood glucose value < 3.1 mmol/L |
— | — |
| Pieber 2007 | Confirmed hypoglycaemia if plasma glucose was < 3.1 mmol/L and the individuals dealt with the episode themselves | — | ND | Minor: the patient could treat himself/herself and the measured plasma glucose value was <3.1 mmol/L Symptoms only: the patient could treat himself/herself and no plasma glucose measurement was taken or the measured plasma glucose value was ≥ 3.1 mmol/L |
Minor and moderate hypoglycaemia | — | ND |
| Porcellati 2004 | Hypoglycaemia was defined as any episode associated with measurement of blood glucose ≤ 4.0 mmol/L irrespective of symptoms. Hypoglycaemia was considered mild when the episodes were self‐treated by the patients |
— | — | — | — | — | — |
| PRESCHOOL | Composite hypoglycaemia rate consisting of: (i) Symptomatic hypoglycaemia episodes, which were recorded in patient diaries, then validated by study investigators (ii) Low CGM glucose excursions (< 3.9 mmol/L), which were confirmed by finger stick blood glucose < 3.9 mmol/L 10 min before to 10 min after the low CGM excursion (i.e., confirmed low CGM) (iii) FSBG <3.9 mmol/L, which was recorded ≥1 h from the end of a confirmed low CGM excursion |
— | "Symptomatic hypoglycemia episodes validated by the study investigator based on entries in patients' diaries, ‐ low continuous glucose monitoring system (CGMS) excursions (interstitial glucose < 70 mg/dL [3.9 mmol/L]) confirmed by fingerstick blood glucose (FSBG) < 70 mg/dL, ‐ low FSBG readings (values < 70 mg/dL) performed at other times" | "Symptomatic hypoglycemia: any event with clinical symptoms considered to result from hypoglycemia, validated by site based on data from patient diaries" | — | ND | — |
| Ratner 2000 | Hypoglycaemia was divided into 3 subsets: all events, severe hypoglycaemia and nocturnal hypoglycaemia | — | — | "Hypoglycemia was either symptomatic (physical symptoms of hypoglycemia were present and was to be confirmed by blood glucose below 2.8 mmol/L [50 mg/dL]) or asymptomatic (no physical symptoms of hypoglycemia present but fasting blood glucose level from the SMBG measurements was below 2.8 mmol/L [50 mg/dL])" | — | ND | Hypoglycaemia was either symptomatic, i.e. with clinical symptoms that could be confirmed by blood glucose < 2.8 mmol/L (50 mg/dL), or asymptomatic, i.e. any event with a confirmed blood glucose level < 2.8 mmol/L (50 mg/dL) but without any symptoms |
| Robertson 2007 | Confirmed episodes: all self‐treated episodes of hypoglycaemia with plasma glucose measurements < 3.1 mmol/L whether symptomatic or not | ND | ND | Self‐treated episodes of hypoglycaemia with plasma glucose measurements < 3.1 mmol/L whether symptomatic or not | Minor hypoglycaemia | — | Episode with blood glucose < 3.1 mmol/L handled by the patient or asymptomatic |
| Russell‐Jones 2004 | Minor, if the blood glucose value was < 2.8 mmol/L and the patient dealt with the episode alone Symptoms only, if no assistance was required and the event was not confirmed by a blood glucose measurement |
ND | ND | An episode with symptoms consistent with hypoglycaemia with confirmation by a blood glucose measurement < 2.8 mmol/L and which was handled by the participant himself/herself or any asymptomatic blood glucose measurement | ND | Minor:
|
ND |
| Schober 2002 | Hypoglycaemia was categorised as either symptomatic, i.e. with clinical symptoms that could be confirmed by blood glucose levels < 2.8 mmol/L, or asymptomatic, i.e. any event with a confirmed blood glucose level < 2.8 mmol/L but without any symptoms | — | — | "Hypoglycemia was either symptomatic, i.e. any event with clinical symptoms related to hypoglycemia regardless of whether it could be confirmed by blood glucose below 2.8 mmol/L (50 mg/dL), or asymptomatic, i.e. any event with a confirmed blood glucose level below 2.8 mmol/L (50 mg/dL) but without any symptoms" | — | — | ND |
| Standl 2004 | If blood glucose was < 2.8 mmol/L and the patient handled the episode him‐ or herself | — | — | An episode with symptoms consistent with hypoglycaemia with confirmation by blood glucose measurement < 2.8 mmol/L and which was handled by the participant himself/herself, or any asymptomatic blood glucose measurement < 2.8 mmol/L | ND | Minor:
|
Hypoglycaemia with blood glucose < 2.8 mmol/L handled by the patient or asymptomatic |
| SWITCH 1 | Blood glucose ≤ 3.9 mmol/L or > 3.9 mmol/L when they occur in conjunction with hypoglycaemic symptoms, able to treat themselves | ND | ND | Symptoms of hypoglycaemia and/or episode with low glucose measurement ≤ 3.9 mmol/L, able to self‐treat | Asymptomatic hypoglycaemia: an episode not accompanied by typical symptoms of hypoglycaemia, but with a measured plasma glucose concentration ≤ 3.9 mmol/L Documented symptomatic hypoglycaemia: an episode during which typical symptoms of hypoglycaemia are accompanied by a measured plasma glucose concentration ≤ 3.9 mmol/L Pseudo‐hypoglycaemia: an episode during which the person with diabetes reports any of the typical symptoms of hypoglycaemia with a measured plasma glucose concentration > 3.9 mmol/L but approaching that level Probable symptomatic hypoglycaemia: an episode during which symptoms typical of hypoglycaemia are not accompanied by a plasma glucose determination but that was presumably caused by a plasma glucose concentration ≤ 3.9 mmol/L |
— | ND |
| Thalange 2013 | Mild hypoglycaemia was defined as episodes where the participants were able to treat themselves Moderate hypoglycaemia was categorised as episodes where participants required assistance, but responded to oral treatment |
— | ND | Mild hypoglycaemia was defined as episodes where the participants were able to treat themselves Moderate hypoglycaemia was categorised as episodes where participants required assistance, but responded to oral treatment |
ND | Able to self‐treat and confirmed by capillary blood glucose < 2.8 mmol/L or 3.1 mmol/L if expressed as plasma glucose | — |
| Urakami 2017 | Hypoglycaemia was defined as a self‐monitored PG level < 70 mg/dL |
Hypoglycaemia was defined as a self‐monitored PG level < 70 mg/dL |
— | — | — | — | — |
| Vague 2003 | Minor if blood glucose was < 2.8 mmol/L and the patients dealt with the episode themselves | — | — | Minor if blood glucose was < 2.8 mmol/L and the patients dealt with the episode themselves and any asymptomatic blood glucose measurement < 2.8 mmol/L | ND |
|
Hypoglycaemia with blood glucose < 2.8 mmol/L handled by the patient or asymptomatic |
| —: indicates source not available CGM: continuous glucose monitoring;CGMS: continuous glucose monitoring system; CRF: case record form; EMA: European Medicines Agency; FDA: Food and Drug Administration; FSBG: fingerstick blood glucose; ND: not defined; NR: not reported; PG: plasma glucose; SMBG: self‐measured blood glucose; SMPG: self‐monitored plasma glucose. | |||||||
Appendix 38. Source of information for outcome data: socioeconomic effects
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Yes | — | No | No | No | — | — |
| BEGIN Basal‐Bolus Type 1 | Yes | — | No | No | No | No | No |
| BEGIN Flex T1 | Yes | — | No | No | No | No | No |
| BEGIN Young | Yes | — | No | No | No | No | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | No | — | No | No | No | — | — |
| Davies 2014 | No | No | No | No | No | No | No |
| Fulcher 2005 | No | — | — | Yes | — | — | — |
| Heller 2009 | No | — | No | No | No | — | — |
| Home 2005 | No | No | — | Yes | — | No | No |
| Kobayashi 2007 | No | — | No | No | No | — | — |
| Liu 2016 | No | — | No | No | No | No | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | No | — | — |
| Pieber 2007 | Yes | — | No | No | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | No | — | No | No | — | No | — |
| Ratner 2000 | No | No | No | No | No | No | No |
| Robertson 2007 | No | No | No | No | No | — | No |
| Russell‐Jones 2004 | No | No | No | No | No | No | No |
| Schober 2002 | No | — | — | Yes | — | — | No |
| Standl 2004 | No | — | — | No | No | No | No |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | No | — | No | No | No | No | — |
| Urakami 2017 | No | No | — | — | — | — | — |
| Vague 2003 | No | — | — | No | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration. | |||||||
Appendix 39. Source of information for outcome data: HbA1c
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | Yes | — | No | Yes | No | — | — |
| BEGIN Basal‐Bolus Type 1 | No | — | Yes | Yes | Yes | Yes | No |
| BEGIN Flex T1 | Yes | — | Yes | Yes | Yes | Yes | No |
| BEGIN Young | Yes | — | Yes | Yes | Yes | Yes | No |
| Bolli 2009 | Yes | — | — | — | — | — | — |
| Chase 2008 | No | — | No | Yes | No | — | — |
| Davies 2014 | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Fulcher 2005 | No | — | — | Yes | — | — | — |
| Heller 2009 | Yes | — | No | Yes | No | — | — |
| Home 2005 | Yes | No | — | Yes | — | No | Yes |
| Kobayashi 2007 | Yes | — | No | No | Yes | — | — |
| Liu 2016 | Yes | — | Yes | Yes | Yes | Yes | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | Yes | — | — |
| Pieber 2007 | Yes | — | No | Yes | No | — | Yes |
| Porcellati 2004 | Yes | — | — | — | — | — | — |
| PRESCHOOL | No | — | No | Yes | — | No | — |
| Ratner 2000 | Yes | — | — | Yes | — | No | No |
| Robertson 2007 | Yes | No | No | Yes | Yes | — | Yes |
| Russell‐Jones 2004 | Yes | No | No | Yes | No | Yes | Yes |
| Schober 2002 | Yes | — | — | Yes | — | — | No |
| Standl 2004 | No | — | — | Yes | No | Yes | Yes |
| SWITCH 1 | No | No | No | Yes | No | — | No |
| Thalange 2013 | Yes | — | Yes | Yes | No | Yes | — |
| Urakami 2017 | Yes | Yes | — | — | — | — | — |
| Vague 2003 | Yes | — | — | Yes | No | Yes | Yes |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration; HbA1c: glycosylated haemoglobin A1c. | |||||||
Appendix 40. Source of information for outcome data: combined HbA1c + severe hypoglycaemia
| Study ID | Publication | Study author request | Trials register | Clinical study report | Clinical study synopsis | EMA report | FDA report |
| Bartley 2008 | No | — | No | No | No | — | — |
| BEGIN Basal‐Bolus Type 1 | No | — | No | Yes | Yes | No | No |
| BEGIN Flex T1 | No | — | No | Yes | No | No | No |
| BEGIN Young | No | — | No | No | No | No | No |
| Bolli 2009 | No | — | — | — | — | — | — |
| Chase 2008 | No | — | No | No | No | — | — |
| Davies 2014 | No | No | No | Yes | No | No | No |
| Fulcher 2005 | No | — | — | No | — | — | — |
| Heller 2009 | Yes | — | No | Yes | Yes | — | — |
| Home 2005 | No | No | — | No | — | No | No |
| Kobayashi 2007 | No | — | No | No | No | — | — |
| Liu 2016 | No | — | No | No | No | No | — |
| NCT00595374 | — | — | No | — | No | — | — |
| NCT00605137 | — | — | No | No | No | — | — |
| Pieber 2007 | No | — | No | No | No | — | No |
| Porcellati 2004 | No | — | — | — | — | — | — |
| PRESCHOOL | No | — | No | No | — | No | — |
| Ratner 2000 | No | — | — | — | — | No | No |
| Robertson 2007 | No | No | No | No | No | — | No |
| Russell‐Jones 2004 | No | No | No | No | No | No | No |
| Schober 2002 | No | — | — | No | — | — | No |
| Standl 2004 | No | — | — | No | No | No | No |
| SWITCH 1 | No | No | No | No | No | — | No |
| Thalange 2013 | No | — | No | No | No | No | — |
| Urakami 2017 | No | No | — | — | — | — | — |
| Vague 2003 | No | — | — | No | No | No | No |
| —: indicates source not available EMA: European Medicines Agency; FDA: Food and Drug Administration;´HbA1c: glycosylated haemoglobin A1c. | |||||||
Appendix 41. Overview of source of information for outcome data
| Outcome measure | Publicationa | Study author requesta | Trials register with resultsa | Clinical study reporta | Clinical study synopsisa | EMA report | FDA report |
| All‐cause mortality | 6/24 | 2/24 | 1/8 | 20/22 | 14/23 | 4 | 7 |
| Cardiovascular mortality | 6/24 | 2/24 | 1/8 | 20/22 | 15/23 | 4 | 4 |
| Non‐fatal myocardial infarction | 1/24 | 2/24 | 1/8 | 5/22 | 0 | 0 | 0 |
| Non‐fatal stroke | 1/24 | 2/24 | 0 | 4/22 | 0 | 0 | 0 |
| End‐stage renal disease | 1/24 | 2/24 | 0 | 0 | 0 | 0 | 0 |
| Blindness | 1/24 | 2/24 | 0 | 0 | 0 | 0 | 0 |
| Diabetic ketoacidosis | 6/24 | 2/24 | 5/8 | 17/22 | 3/23 | 2 | 1 |
| Serious adverse events | 13/24 | 2/24 | 7/8 | 20/22 | 13/23 | 3 | 1 |
| Non‐serious adverse events | 13/24 | 2/24 | 7/8 | 21/22 | 9/23 | 2 | 0 |
| Severe hypoglycaemia | 18/24 | 2/24 | 2/8 | 19/22 | 7/23 | 6 | 9 |
| Nocturnal hypoglycaemia | 17/24 | 2/24 | 3/8 | 19/22 | 4/23 | 6 | 0 |
| Mild/moderate hypoglycaemia | 16/24 | 2/24 | 2/8 | 20/22 | 3/23 | 6 | 3 |
| Health‐related quality of life | 4/24 | 2/24 | 1/8 | 9/22 | 3/23 | 4 | 7 |
| HbA1c | 19/24 | 2/24 | 6/8 | 20 /22 | 8/23 | 9 | 6 |
| HbA1c + severe hypoglycaemia | 2/24 | 0 | 0 | 5/22 | 4/23 | 0 | 0 |
| Socioeconomic effects | 5/24 | 0 | 0 | 3/22 | 0 | 0 | 0 |
|
aRecords with information / total number of available records EMA: European Medicines Agency; FDA: Food and Drug Administration; HbA1c: glycosylated haemoglobin A1c. | |||||||
Appendix 42. Overview of comparisons using various definitions of hypoglycaemia
| Outcome measure | Detemir vs NPH | Glargine vs NPH | Detemir vs glargine | Degludec vs detemir | Degludec vs glargine |
| Severe hypoglycaemia | RR 0.69, 95% CI 0.52 to 0.92a | RR 0.84, 95% CI 0.67 to 1.04 | RR 0.59, 95% CI 0.13 to 2.63 | RR 1.17, 95% CI 0.81 to 1.69 | RR 1.22, 95% CI 0.82 to 1.82 |
| Hypoglycaemia reported as a serious adverse event | RR 0.93, 95% CI 0.51 to 1.71 | RR 0.94, 95% CI 0.64 to 1.39 | RR 1.16, 95% CI 0.14 to 9.48 | RR 0.92, 95% CI 0.37 to 2.32 | RR 0.81, 95% CI 0.40 to 1.66 |
| Severe nocturnal hypoglycaemia | RR 0.67, 95% CI 0.39 to 1.17 | RR 0.83, 95% CI 0.62 to 1.12 | RR 0.55, 95% CI 0.06 to 5.12 | RR 1.12, 95% CI 0.51 to 2.46 | RR 1.39, 95% CI 0.59 to 3.27 |
| Any nocturnal hypoglycaemia | RR 0.91, 95% CI 0.87 to 0.95a | RR 1.00, 95% CI 0.96 to 1.05 | RR 1.01, 95% CI 0.93 to 1.09 | No data | RR 0.99, 95% CI 0.91 to 1.07 |
| Confirmed nocturnal hypoglycaemia | No data | No data | RR 1.01, 95% CI 0.92 to 1.10 | RR 1.04, 95% CI 0.94 to 1.15 | No data |
| Mild nocturnal hypoglycaemia | RR 0.90, 95% CI 0.85 to 0.96a | RR 0.84, 95% CI 0.66 to 1.07 | No data | RR 0.97, 95% CI 0.86 to 1.10 (documented) | RR 0.98, 95% CI 0.90 to 1.07 |
| Symptomatic nocturnal hypoglycaemia | RR 0.88, 95% CI 0.79 to 0.98a | RR 0.93, 95% CI 0.82 to 1.05 | RR 1.02, 95% CI 0.81 to 1.29 | RR 0.72, 95% CI 0.15 to 3.59 | RR 1.22, 95% CI 0.72 to 2.07 |
| Asymptomatic nocturnal hypoglycaemia | No evidence of a difference | Not reported | No data | RR 0.91, 95% CI 0.80 to 1.03 | RR 0.84, 95% CI 0.71 to 1.00 |
| Mild/moderate hypoglycaemia | RR 0.97, 95% CI 0.94 to 0.99a | RR 1.02, 95% CI 1.00 to 1.04 | RR 1.04, 95% CI 0.94 to 1.14 | RR 1.02, 95% CI 0.99 to 1.05 | RR 1.02, 95% CI 0.99 to 1.04 |
| HbA1c < 7.0% without major/severe hypoglycaemia | No data | No data | RR 1.11, 95% CI 0.81 to 1.51 | RR 1.09, 95% CI 0.84 to 1.41 | RR 0.92, 95% CI 0.78 to 1.10 |
|
aFavouring insulin detemir CI: confidence interval; HbA1c: glycosylated haemoglobin A1c; NPH: neutral protamine Hagedorn; RR: risk ratio. | |||||
Data and analyses
Comparison 1. Insulin detemir versus NPH insulin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 All‐cause mortality | 9 | 3334 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.97 [0.79, 31.38] |
| 1.1.1 Adults | 6 | 2558 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.97 [0.79, 31.38] |
| 1.1.2 Children | 3 | 776 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Not estimable |
| 1.2 All‐cause mortality (published vs. unpublished data) | 9 | 3334 | Risk Ratio (M‐H, Random, 95% CI) | 3.64 [0.42, 31.40] |
| 1.2.1 Published | 2 | 842 | Risk Ratio (M‐H, Random, 95% CI) | 4.47 [0.24, 82.58] |
| 1.2.2 Unpublished | 7 | 2492 | Risk Ratio (M‐H, Random, 95% CI) | 2.85 [0.12, 69.55] |
| 1.3 Severe hypoglycaemia | 8 | 3219 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.52, 0.92] |
| 1.3.1 Adults | 5 | 2443 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.49, 1.03] |
| 1.3.2 Children | 3 | 776 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.30, 1.23] |
| 1.4 Severe hypoglycaemia (published vs. unpublished data) | 8 | 3175 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.51, 0.93] |
| 1.4.1 Published | 6 | 2677 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.50, 0.78] |
| 1.4.2 Unpublished | 2 | 498 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.77, 2.62] |
| 1.5 Hypoglycaemia reported as a serious adverse event | 8 | 3219 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.51, 1.71] |
| 1.5.1 Adults | 5 | 2443 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.48, 1.86] |
| 1.5.2 Children | 3 | 776 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.16, 5.57] |
| 1.6 Cardiovascular mortality | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.6.1 Adults | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.6.2 Children | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.7 Non‐fatal myocardial infarction | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.8 Serious adverse events | 9 | 3332 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.75, 1.21] |
| 1.8.1 Adults | 6 | 2556 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.73, 1.28] |
| 1.8.2 Children | 3 | 776 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.56, 1.43] |
| 1.9 Serious adverse events (published vs. unpublished data) | 9 | 3332 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.75, 1.21] |
| 1.9.1 Published | 2 | 641 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.40, 1.09] |
| 1.9.2 Unpublished | 7 | 2691 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.80, 1.39] |
| 1.10 Diabetic ketoacidosis | 6 | 2012 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.36, 1.76] |
| 1.10.1 Adults | 3 | 1236 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.24, 2.92] |
| 1.10.2 Children | 3 | 776 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.27, 2.15] |
| 1.11 Diabetic ketoacidosis (published vs. unpublished data) | 6 | 2012 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.36, 1.76] |
| 1.11.1 Published data | 2 | 694 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.27, 2.52] |
| 1.11.2 Unpublished data | 4 | 1318 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.25, 2.38] |
| 1.12 Non‐serious adverse events | 9 | 3332 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.95, 1.01] |
| 1.12.1 Adults | 6 | 2556 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.95, 1.03] |
| 1.12.2 Children | 3 | 776 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.90, 1.02] |
| 1.13 Non‐serious adverse events (published vs unpublished data) | 9 | 3332 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.94, 1.01] |
| 1.13.1 Published data | 3 | 1141 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.90, 1.01] |
| 1.13.2 Unpublished data | 6 | 2191 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.95, 1.04] |
| 1.14 Withdrawals due to adverse events | 8 | 3222 | Risk Ratio (M‐H, Random, 95% CI) | 2.23 [0.98, 5.05] |
| 1.14.1 Adults | 5 | 2445 | Risk Ratio (M‐H, Random, 95% CI) | 2.25 [0.94, 5.41] |
| 1.14.2 Children | 3 | 777 | Risk Ratio (M‐H, Random, 95% CI) | 2.08 [0.22, 19.90] |
| 1.15 Any nocturnal hypoglycaemia | 8 | 3219 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.87, 0.95] |
| 1.15.1 Adults | 5 | 2443 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.88, 0.98] |
| 1.15.2 Children | 3 | 776 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.81, 0.94] |
| 1.16 Mild nocturnal hypoglycaemia | 7 | 3073 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.85, 0.96] |
| 1.16.1 Adults | 4 | 2149 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.83, 1.00] |
| 1.16.2 Children | 3 | 924 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.78, 1.00] |
| 1.17 Nocturnal hypoglycaemia (symptoms) | 6 | 2578 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.79, 0.98] |
| 1.17.1 Adults | 4 | 2149 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.82, 1.01] |
| 1.17.2 Children | 2 | 429 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.19, 1.61] |
| 1.18 Severe nocturnal hypoglycaemia | 7 | 2925 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.39, 1.17] |
| 1.18.1 Adults | 4 | 2149 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.35, 0.93] |
| 1.18.2 Children | 3 | 776 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.13, 3.17] |
| 1.19 Any nocturnal hypoglycaemia (published vs. unpublished data) | 8 | 3219 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.87, 0.95] |
| 1.19.1 Published | 6 | 2677 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.86, 0.95] |
| 1.19.2 Unpublished | 2 | 542 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.80, 1.04] |
| 1.20 Mild nocturnal hypoglycaemia (published vs. unpublished data) | 7 | 3073 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.85, 0.96] |
| 1.20.1 Published | 4 | 2084 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.85, 0.98] |
| 1.20.2 Unpublished | 3 | 989 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.75, 1.07] |
| 1.21 Nocturnal hypoglycaemia, symptoms only (published vs. unpublished data) | 6 | 2578 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.79, 0.98] |
| 1.21.1 Published | 3 | 1589 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.81, 0.99] |
| 1.21.2 Unpublished | 3 | 989 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.57, 1.08] |
| 1.22 Severe nocturnal hypoglycaemia (published vs. unpublished data) | 7 | 2925 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.39, 1.17] |
| 1.22.1 Published | 5 | 2383 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.32, 1.25] |
| 1.22.2 Unpublished | 2 | 542 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.33, 2.45] |
| 1.23 Nocturnal hypoglycaemia, asymptomatic (children vs. adults) | 2 | 429 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.78, 1.18] |
| 1.24 Mild/moderate hypoglycaemia | 8 | 3219 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.94, 0.99] |
| 1.24.1 Adults | 5 | 2443 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.93, 1.02] |
| 1.24.2 Children | 3 | 776 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.93, 1.01] |
| 1.25 Mild/moderate hypoglycaemia (published vs. unpublished data) | 8 | 3219 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.94, 0.99] |
| 1.25.1 Published | 6 | 2677 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.93, 1.00] |
| 1.25.2 Unpublished | 2 | 542 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.92, 1.05] |
| 1.26 HbA1c | 8 | 3122 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.08, 0.10] |
| 1.26.1 Adults | 5 | 2354 | Mean Difference (IV, Random, 95% CI) | ‐0.03 [‐0.14, 0.07] |
| 1.26.2 Children | 3 | 768 | Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.04, 0.31] |
| 1.27 HbA1c (published vs. unpublished data) | 8 | 3122 | Mean Difference (IV, Random, 95% CI) | 0.00 [‐0.09, 0.09] |
| 1.27.1 Published | 6 | 2624 | Mean Difference (IV, Random, 95% CI) | ‐0.02 [‐0.13, 0.09] |
| 1.27.2 Unpublished | 2 | 498 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.08, 0.28] |
1.23. Analysis.

Comparison 1: Insulin detemir versus NPH insulin, Outcome 23: Nocturnal hypoglycaemia, asymptomatic (children vs. adults)
Comparison 2. Insulin glargine versus NPH insulin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 All‐cause mortality | 8 | 2175 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.14 [0.00, 6.98] |
| 2.1.1 Adults | 4 | 1365 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.14 [0.00, 6.98] |
| 2.1.2 Children | 4 | 810 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Not estimable |
| 2.2 Health‐realted quality of life | 2 | 880 | Mean Difference (IV, Random, 95% CI) | 0.62 [‐0.71, 1.96] |
| 2.3 Severe hypoglycaemia | 9 | 2350 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.67, 1.04] |
| 2.3.1 Adults | 5 | 1540 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.58, 1.05] |
| 2.3.2 Children | 4 | 810 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.59, 2.21] |
| 2.4 Severe hypoglycaemia (published vs. unpublished data) | 9 | 2350 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.67, 1.04] |
| 2.4.1 Published | 7 | 1691 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.63, 1.22] |
| 2.4.2 Unpublished | 2 | 659 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.56, 1.25] |
| 2.5 Hypoglycaemia reported as a serious adverse event | 8 | 2229 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.64, 1.39] |
| 2.5.1 Adults | 4 | 1419 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.57, 1.37] |
| 2.5.2 Children | 4 | 810 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.32, 2.87] |
| 2.6 Cardiovascular mortality | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.6.1 Adults | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.6.2 Children | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.7 Non‐fatal myocardial infarction | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.8 Non‐fatal stroke | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.9 Serious adverse events | 8 | 2229 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.63, 1.84] |
| 2.9.1 Adults | 4 | 1419 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.72, 1.35] |
| 2.9.2 Children | 4 | 810 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.28, 3.64] |
| 2.10 Serious adverse events (published vs. unpublished data) | 8 | 2229 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.63, 1.84] |
| 2.10.1 Published | 4 | 1284 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.45, 2.70] |
| 2.10.2 Unpublished | 4 | 945 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.46, 2.60] |
| 2.11 Diabetic ketoacidosis | 7 | 2054 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.19, 1.44] |
| 2.11.1 Adults | 3 | 1244 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.11, 9.58] |
| 2.11.2 Children | 4 | 810 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.15, 1.39] |
| 2.12 Diabetic ketoacidosis (published vs. unpublished data) | 7 | 2054 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.19, 1.44] |
| 2.12.1 Published | 3 | 685 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.11, 1.31] |
| 2.12.2 Unpublished | 4 | 1369 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.18, 5.77] |
| 2.13 Non‐serious adverse events | 8 | 2229 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.96, 1.06] |
| 2.13.1 Adults | 4 | 1419 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.95, 1.07] |
| 2.13.2 Children | 4 | 810 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.93, 1.12] |
| 2.14 Non‐serious adverse events (published vs. unpublished data) | 8 | 2229 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.96, 1.06] |
| 2.14.1 Published | 5 | 1308 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.94, 1.05] |
| 2.14.2 Unpublished | 3 | 921 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.94, 1.14] |
| 2.15 Withdrawals due to adverse events | 8 | 2230 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.24, 2.81] |
| 2.15.1 Adults | 4 | 1419 | Risk Ratio (M‐H, Random, 95% CI) | 1.74 [0.29, 10.39] |
| 2.15.2 Children | 4 | 811 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.06, 1.53] |
| 2.16 Nocturnal hypoglycaemia | 7 | 2054 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.96, 1.05] |
| 2.16.1 Adults | 3 | 1244 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.92, 1.06] |
| 2.16.2 Children | 4 | 810 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.95, 1.08] |
| 2.17 Mild nocturnal hypoglycaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.18 Nocturnal hypoglycaemia (symptoms) | 4 | 996 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.82, 1.05] |
| 2.18.1 Adults | 2 | 710 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.88, 1.08] |
| 2.18.2 Children | 2 | 286 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.55, 1.00] |
| 2.19 Severe nocturnal hypoglycaemia | 6 | 1893 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.62, 1.12] |
| 2.19.1 Adults | 3 | 1244 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.60, 1.27] |
| 2.19.2 Children | 3 | 649 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.47, 1.25] |
| 2.20 Nocturnal hypoglycaemia (published vs. unpublished data) | 7 | 2054 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.96, 1.05] |
| 2.20.1 Published | 5 | 1345 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.95, 1.06] |
| 2.20.2 Unpublished | 2 | 709 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.91, 1.08] |
| 2.21 Symptomatic nocturnal hypoglycaemia (published vs. unpublished data) | 4 | 996 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.82, 1.05] |
| 2.21.1 Published | 3 | 871 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.67, 1.12] |
| 2.21.2 Unpublished | 1 | 125 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.80, 1.10] |
| 2.22 Mild/moderate hypoglycaemia | 7 | 2054 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [1.00, 1.04] |
| 2.22.1 Adults | 3 | 1244 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.99, 1.06] |
| 2.22.2 Children | 4 | 810 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.99, 1.04] |
| 2.23 Mild/moderate hypoglycaemia (published vs. unpublished data) | 7 | 2054 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [1.00, 1.04] |
| 2.23.1 Published | 5 | 1395 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [1.00, 1.05] |
| 2.23.2 Unpublished | 2 | 659 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.98, 1.05] |
| 2.24 HbA1c | 9 | 2285 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.06, 0.11] |
| 2.24.1 Adults | 5 | 1523 | Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.16, 0.13] |
| 2.24.2 Children | 4 | 762 | Mean Difference (IV, Random, 95% CI) | 0.03 [‐0.13, 0.20] |
| 2.25 HbA1c (published vs unpublished data) | 9 | 2285 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.06, 0.11] |
| 2.25.1 Published | 6 | 1868 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.09, 0.14] |
| 2.25.2 Unpublished | 3 | 417 | Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.26, 0.18] |
| 2.26 HbA1c (NPH < 2x/day vs ≥ 2x/day) | 9 | 2285 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.06, 0.11] |
| 2.26.1 NPH up to twice a day | 8 | 2164 | Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.03, 0.13] |
| 2.26.2 NPH more than twice a day | 1 | 121 | Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐0.93, ‐0.07] |
2.26. Analysis.

Comparison 2: Insulin glargine versus NPH insulin, Outcome 26: HbA1c (NPH < 2x/day vs ≥ 2x/day)
Comparison 3. Insulin detemir versus insulin glargine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 All‐cause mortality | 2 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 3.2 Severe hypoglycaemia | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.13, 2.63] |
| 3.3 Severe hypoglycaemia (published vs. unpublished data) | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.13, 2.63] |
| 3.3.1 Published | 1 | 320 | Risk Ratio (M‐H, Random, 95% CI) | 0.25 [0.07, 0.86] |
| 3.3.2 Unpublished | 1 | 443 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.72, 1.77] |
| 3.4 Hypoglycaemia reported as a serious adverse event | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.14, 9.48] |
| 3.5 Cardiovascular mortality | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.6 Non‐fatal myocardial infarction | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.7 Non‐fatal stroke | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.8 Serious adverse events | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 1.72 [0.91, 3.28] |
| 3.9 Diabetic ketoacidosis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.10 Non‐serious adverse events | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.93, 1.09] |
| 3.11 Non‐serious adverse events (published vs. unpublished data) | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.93, 1.09] |
| 3.11.1 Published | 1 | 443 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.97, 1.10] |
| 3.11.2 Unpublished | 1 | 320 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.84, 1.09] |
| 3.12 Withdrawals due to adverse events | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.31, 3.67] |
| 3.13 Any nocturnal hypoglycaemia | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.93, 1.09] |
| 3.13.1 Published | 1 | 320 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.75, 1.17] |
| 3.13.2 Unpublished | 1 | 443 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.94, 1.11] |
| 3.14 Confirmed nocturnal hypoglycaemia (PG < 3.1 mmol/L and no assistance) | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.92, 1.10] |
| 3.14.1 Published | 1 | 320 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.71, 1.16] |
| 3.14.2 Unpublished | 1 | 443 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.93, 1.12] |
| 3.15 Symptomatic nocturnal hypoglycaemia (PG ≥ 3.1 or no PG and no assistance required) | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.81, 1.29] |
| 3.15.1 Published | 1 | 320 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.78, 2.12] |
| 3.15.2 Unpublished | 1 | 443 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.77, 1.21] |
| 3.16 Severe nocturnal hypoglycaemia | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.06, 5.12] |
| 3.16.1 Published | 1 | 320 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.01, 2.02] |
| 3.16.2 Unpublished | 1 | 443 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.60, 2.32] |
| 3.17 Mild/moderate hypoglycaemia | 2 | 763 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.94, 1.14] |
| 3.17.1 Published | 1 | 320 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.95, 1.26] |
| 3.17.2 Unpublished | 1 | 443 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.96, 1.06] |
| 3.18 HbA1c | 2 | 717 | Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.13, 0.12] |
| 3.19 Individuals with HbA1c < 7% without severe hypoglycaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 4. Insulin degludec versus insulin detemir.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 All‐cause mortality | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.1 Adults | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.2 Children | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2 Health‐related quality of life | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.2.1 Physical health score | 1 | 454 | Mean Difference (IV, Fixed, 95% CI) | ‐0.60 [‐1.83, 0.63] |
| 4.2.2 Mental health score | 1 | 454 | Mean Difference (IV, Fixed, 95% CI) | ‐3.00 [‐4.44, ‐1.56] |
| 4.3 Severe hypoglycaemia | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.81, 1.69] |
| 4.3.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.57, 1.78] |
| 4.3.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.80, 2.12] |
| 4.4 Hypoglycaemia reported as a serious adverse event | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.37, 2.32] |
| 4.4.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.29, 1.69] |
| 4.4.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 2.01 [0.37, 10.84] |
| 4.5 Cardiovascular mortality | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.5.1 Adults | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.5.2 Children | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.6 Non‐fatal myocardial infarction | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.7 Non‐fatal stroke | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.8 End stage renal disease | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.9 Blindness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.10 Serious adverse events | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.76, 2.05] |
| 4.10.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [0.67, 3.17] |
| 4.10.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.60, 2.15] |
| 4.11 Diabetic ketoacidosis | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.11.1 Adults | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.11.2 Children | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.12 Non‐serious adverse events | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.96, 1.08] |
| 4.12.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.88, 1.11] |
| 4.12.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.97, 1.10] |
| 4.13 Withdrawals due to adverse events | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 2.32 [0.38, 14.18] |
| 4.13.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.16, 14.44] |
| 4.13.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 5.03 [0.24, 103.99] |
| 4.14 Nocturnal hypoglycaemia | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.94, 1.15] |
| 4.14.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.85, 1.18] |
| 4.14.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.94, 1.21] |
| 4.15 Mild nocturnal hypoglycaemia | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.86, 1.10] |
| 4.15.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.85, 1.16] |
| 4.15.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.79, 1.15] |
| 4.16 Nocturnal hypoglycaemia (symptomatic) | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.15, 3.59] |
| 4.16.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.20, 0.72] |
| 4.16.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 2.01 [0.37, 10.84] |
| 4.17 Nocturnal hypoglycaemia (asymptomatic) | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.80, 1.03] |
| 4.17.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.70, 1.23] |
| 4.17.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.79, 1.04] |
| 4.18 Severe nocturnal hypoglycaemia | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.51, 2.46] |
| 4.18.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.43, 3.38] |
| 4.18.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.30, 3.41] |
| 4.19 Mild/moderate hypoglycaemia | 2 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.99, 1.05] |
| 4.19.1 Adults | 1 | 453 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.96, 1.08] |
| 4.19.2 Children | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.99, 1.06] |
| 4.20 HbA1c | 2 | 805 | Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.08, 0.18] |
| 4.20.1 Adults | 1 | 455 | Mean Difference (IV, Random, 95% CI) | 0.00 [‐0.18, 0.18] |
| 4.20.2 Children | 1 | 350 | Mean Difference (IV, Random, 95% CI) | 0.11 [‐0.08, 0.30] |
| 4.21 Individuals with HbA1c < 7% without severe hypoglycaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 5. Insulin degludec versus insulin glargine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 All‐cause mortality | 3 | 973 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.34 [0.15, 11.93] |
| 5.2 All‐cause mortality (published vs. unpublished data) | 3 | 973 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.17, 7.65] |
| 5.2.1 Published | 1 | 626 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.06, 7.15] |
| 5.2.2 Unpublished | 2 | 347 | Risk Ratio (M‐H, Random, 95% CI) | 2.98 [0.12, 72.67] |
| 5.3 Health‐related quality of life (physical health) | 2 | 1043 | Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐1.21, 1.13] |
| 5.3.1 Published | 1 | 629 | Mean Difference (IV, Random, 95% CI) | 0.50 [‐0.93, 1.93] |
| 5.3.2 Unpublished | 1 | 414 | Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐2.30, 0.90] |
| 5.4 Health‐related quality of life (mental health) | 2 | 1539 | Mean Difference (IV, Random, 95% CI) | ‐0.09 [‐1.03, 0.85] |
| 5.4.1 Published | 1 | 629 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐1.33, 2.13] |
| 5.4.2 Unpublished | 1 | 910 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐1.42, 0.82] |
| 5.5 Severe hypoglycaemia | 3 | 970 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.82, 1.82] |
| 5.5.1 Adults | 2 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.82, 1.82] |
| 5.5.2 Children | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 5.6 Hypoglycaemia reported as a serious adverse event | 4 | 1884 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.40, 1.66] |
| 5.6.1 Adults | 3 | 1866 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.40, 1.66] |
| 5.6.2 Children | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 5.7 Cardiovascular mortality | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.7.1 Adults | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.7.2 Children | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.8 Non‐fatal myocardial infarction | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.8.1 Adults | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.8.2 Children | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.9 Non‐fatal stroke | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.10 Serious adverse events | 3 | 970 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.58, 1.46] |
| 5.10.1 Adults | 2 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.58, 1.46] |
| 5.10.2 Children | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 5.11 Diabetic ketoacidosis | 3 | 970 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.05, 6.89] |
| 5.11.1 Adults | 2 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.05, 6.89] |
| 5.11.2 Children | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 5.12 Diabetic ketoacidosis (published vs. unpublished data) | 3 | 970 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.05, 6.89] |
| 5.12.1 Published | 1 | 626 | Risk Ratio (M‐H, Random, 95% CI) | 0.22 [0.04, 1.29] |
| 5.12.2 Unpublished | 2 | 344 | Risk Ratio (M‐H, Random, 95% CI) | 2.93 [0.12, 71.34] |
| 5.13 Non‐serious adverse events | 3 | 970 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.95, 1.10] |
| 5.13.1 Adults | 2 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.95, 1.10] |
| 5.13.2 Children | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 5.14 Withdrawals due to adverse events | 2 | 955 | Risk Ratio (M‐H, Random, 95% CI) | 2.47 [0.72, 8.43] |
| 5.15 Nocturnal hypoglycaemia | 3 | 970 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.91, 1.07] |
| 5.15.1 Adults | 2 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.91, 1.08] |
| 5.15.2 Chlidren | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.12, 2.08] |
| 5.16 Mild nocturnal hypoglycaemia | 2 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.90, 1.07] |
| 5.17 Nocturnal hypoglycaemia (asymptomatic) | 2 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.71, 1.00] |
| 5.18 Nocturnal hypoglycaemia (symptomatic) | 2 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.72, 2.07] |
| 5.19 Severe nocturnal hypoglycaemia | 3 | 970 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [0.59, 3.27] |
| 5.20 Mild/moderate hypoglycaemia | 3 | 970 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.99, 1.04] |
| 5.20.1 Adults | 2 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.99, 1.04] |
| 5.20.2 Children | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.82, 1.22] |
| 5.21 HbA1c | 4 | 1388 | Mean Difference (IV, Random, 95% CI) | 0.10 [0.00, 0.21] |
| 5.21.1 Adults | 3 | 1370 | Mean Difference (IV, Random, 95% CI) | 0.11 [0.00, 0.21] |
| 5.21.2 Children | 1 | 18 | Mean Difference (IV, Random, 95% CI) | 0.00 [‐0.55, 0.55] |
| 5.22 HbA1c (published vs. unpublished data) | 4 | 1388 | Mean Difference (IV, Random, 95% CI) | 0.10 [0.00, 0.21] |
| 5.22.1 Published | 3 | 847 | Mean Difference (IV, Random, 95% CI) | 0.14 [0.02, 0.25] |
| 5.22.2 Unpublished | 1 | 541 | Mean Difference (IV, Random, 95% CI) | 0.00 [‐0.21, 0.21] |
| 5.23 Individuals with HbA1c < 7% without severe hypoglycaemia | 2 | 911 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.78, 1.10] |
5.12. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 12: Diabetic ketoacidosis (published vs. unpublished data)
5.14. Analysis.

Comparison 5: Insulin degludec versus insulin glargine, Outcome 14: Withdrawals due to adverse events
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bartley 2008.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 2:1 | |
| Participants |
Inclusion criteria: ≥ 18 years; with an HbA1c ≤ 11.0% and BMI ≤ 35.0 kg/m2 with a history of T1DM ≥ 1 year treated on a basal–bolus insulin regimen for ≥ 3 months and able and willing to SMPG Exclusion criteria: proliferative retinopathy or maculopathy, other significant medical disorders, recurrent major hypoglycaemia, allergy to insulin and pregnant or breast feeding Diagnostic criteria: — Number of study centres: 33 |
|
| Interventions |
Intervention(s): detemir Comparator(s): NPH Duration of intervention: 24 months Duration of follow‐up: 24 months (plus 4 to 8 days) Run‐in period: none |
|
| Outcomes | Reported outcome(s) in full text of publication: glycaemic control, hypoglycaemia, safety | |
| Study registration |
Trial identifier: NCT00184665; NN304‐1595 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal and conference abstract |
|
| Stated aim of study | Quote: "This 24‐month, multi‐national, open‐label, parallel group trial investigated the long‐term efficacy and safety of insulin detemir and Neutral Protamine Hagedorn insulin in combination with mealtime insulin aspart in patients with Type 1 diabetes using a treat‐to‐target concept" | |
| Notes | Quote: "Six months into the trial, blinded review of the pre‐breakfast and pre‐evening meal PG concentrations revealed that PG targets were not achieved in a substantial proportion of patients and a protocol amendment was implemented to ensure more frequent contact between patients and investigators during the last year of the trial".CSR identified: from CSR data from hypoglycaemia combined with HbA1c, adverse events, serious adverse events, ketoacidosis and myocardial infarction | |
BEGIN Basal‐Bolus Type 1.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 3:1 | |
| Participants |
Inclusion criteria: T1DM for at least 12 months; current treatment with any basal bolus insulin for at least 12 months; HbA1c below or equal to 10.0%, BMI below or equal to 35.0 kg/m2; for the extension study only: completion of the 52‐week treatment period in study Exclusion criteria: use within the last 3 months of any other antidiabetic glucose‐lowering drug than insulin; anticipated change in concomitant medication known to interfere significantly with glucose metabolism, such as systemic corticosteroids, beta‐blockers, monoamine oxidase (MAO) inhibitors; cardiovascular disease, within the last 6 months defined as: stroke, decompensated heart failure NYHA Class III or IV, myocardial infarction, unstable angina pectoris or coronary arterial bypass graft or angioplasty; uncontrolled treated/untreated severe hypertension (systolic BP ≥ 180 mmHg and/or diastolic BP ≥ 100 mmHg); impaired liver function, defined as ALAT ≥ 2.5 times upper limit of normal (one re‐test analysed at the central laboratory within a week from receipt of the result was permitted with the result of the last sample being conclusive); impaired renal function defined as serum creatinine ≥ 180 µmol/L (≥ 2.0 mg/dL); recurrent severe hypoglycaemia (more than 1 severe hypoglycaemic event during the last 12 months) or hypoglycaemic unawareness or hospitalisation for diabetic ketoacidosis during the previous 6 months; proliferative retinopathy or maculopathy requiring treatment as determined by the investigator; pregnancy, breastfeeding, the intention of becoming pregnant or not using adequate contraceptive measures according to local requirements (for Germany: implants, injectables, combined oral contraceptives, hormonal intrauterine device, sexual abstinence or vasectomised partner) (for United Kingdom: adequate contraceptive measures were defined as established use of oral, injected or implanted hormonal methods of contraception, sterilisation, intrauterine device or intrauterine system, or consistent use of barrier methods); cancer and medical history of cancer (except basal cell skin cancer or squamous cell skin cancer); any clinically significant disease or disorder, except for conditions associated with T1DM, which in the investigator’s opinion could interfere with the results of the study; mental incapacity, psychiatric disorder, unwillingness or language barriers precluding adequate understanding or co‐operation, including not able to read or write; previous participation in this study; receipt of any investigational drug within 1 month prior to screening visit; donation of blood or participation in other trials within 1 month prior to screening visit; known or suspected abuse of alcohol, narcotics or illicit drugs Diagnostic criteria: — Number of study centres: 79 |
|
| Interventions |
Intervention(s): degludec Comparator(s): glargine Duration of intervention: 52 weeks Duration of follow‐up: 52 weeks (104 weeks) Run‐in period: none |
|
| Outcomes | Reported outcome(s) in full text of publication: mortality, cardiovascular outcomes, safety, glycaemic measures | |
| Study registration |
Trial identifier: main study: NCT00982228, obsolete identifier: NCT0119804, NN1250‐3583, EudraCT number 2008‐005774‐13; WHO identifier U1111‐1116‐1578; extension study: NN1250‐3644, EudraCT Number 2009‐015755‐24; NCT01198041; WHO identifier U1111‐1111‐8789 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal and conference abstract |
|
| Stated aim of study | Quote: "We therefore compared the efficacy and safety of insulin degludec and insulin glargine, both administered once daily with mealtime insulin aspart, in basal‐bolus therapy for type 1 diabetes". | |
| Notes | BEGIN Basal–Bolus Type 1 refers to the first 52 weeks, thereafter there was the extension study BEGIN Conference abstract did not reveal any additional data At selected study sites (25), participants underwent assessment of their 24‐hour interstitial glucose profile with a CGM device for 3 consecutive days at baseline (72 hours before visit 2), and at visits 28 and 41 (weeks 26 and 52, respectively) CSR and trial synopsis available. Provided outcome data on severe hypoglycaemia/HbA1c combined Study also reported in FDA 2015 (FDA 2015) ‐ 2 deaths in each intervention arm ‐ but unknown whether this was before or after extension period. No additional data from EMA 2012 (EMA 2012) |
|
BEGIN Flex T1.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: informed consent; males or females 18 years or more; T1DM for ≥ 12 months, the last 3 months with injection‐based therapies; current treatment with any basal insulin using one or two daily injections and no fewer than three injections with bolus insulin as mealtime bolus insulin therapy; HbA1c ≤ 10.0% by central laboratory analysis; BMI ≤ 35.0 kg/m2; ability to self‐manage insulin therapy as assessed by confirmation (verbal confirmation at screening visit) of a changed bolus insulin dose the preceding 2 months prior to screening; ability and willingness to adhere to the protocol, including performance of SMPG readings and self‐adjustment of insulin doses according to protocol Exclusion criteria: use within the last 3 months of any glucose‐lowering drug other than insulin; initiation or significant change of any systemic treatment which, in the investigator’s opinion, could interfere with glucose metabolism, such as systemic corticosteroids, beta‐blockers or monoamine oxidase inhibitors (inhaled corticosteroids were allowed); cardiovascular disease, within the last 6 months (defined as: stroke; decompensated heart failure NYHA class III or IV; myocardial infarction; unstable angina pectoris; or coronary arterial bypass graft or angioplasty); uncontrolled treated/untreated severe hypertension (systolic BP ≥ 180 mmHg and/or diastolic BP ≥ 100 mmHg); impaired liver function, defined as ALAT ≥ 2.5 times upper limit of normal; impaired renal function defined as serum‐creatinine ≥ 180 µmol/L or 2.0 mg/dL; recurrent severe hypoglycaemia (more than 1 severe hypoglycaemic event during the last 12 months) or hypoglycaemic unawareness as judged by the investigator or hospitalisations for diabetic ketoacidosis during the previous 6 months; proliferative retinopathy or maculopathy requiring treatment, according to the investigator; pregnancy, breastfeeding, the intention of becoming pregnant or not using adequate contraceptive measures according to local requirements; cancer and medical history of cancer (except basal cell skin cancer or squamous cell skin cancer); any clinically significant disease or disorder, except for conditions associated with T1DM, which in the investigator’s opinion could interfere with the results of the study; mental incapacity, psychiatric disorder, unwillingness or language barriers precluding adequate understanding or co‐operation, including participants not able to read or write; previous participation in this study; known or suspected allergy to any of the study products or related products; receipt of any investigational drug within 1 month; donation of blood or participation in other trials within 1 month prior; known or suspected abuse of alcohol, narcotics, or illicit drugs Diagnostic criteria: clinically diagnosed (from CSR) Number of study centres: 71 |
|
| Interventions |
Intervention(s): degludec Comparator(s): glargine Duration of intervention: 26 weeks (52 weeks) Duration of follow‐up: 26 weeks (26 weeks) Run‐in period: none |
|
| Outcomes | Reported outcome(s) in full text of publication: adverse events, hypoglycaemia, glycaemic variables | |
| Study registration |
Trial identifier: NCT01079234, NN1250‐3770, WHO U1111‐1112‐8813, EudraCT Number 2009‐012923‐27 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "The aim of this trial is to investigate the efficacy and safety of NN1250 (insulin degludec) in participants with type 1 diabetes". | |
| Notes | The participants were randomised to three intervention arms ‐ insulin degludec forced‐Flex, insulin degludec and insulin glargine. We have only included data from the insulin degludec and insulin glargine groups as they had identical titration regimens. The study consisted of a 26‐week main period and 26‐week extension period. Only data from the main period were included, as the two degludec groups were combined into one group in the extension period. Abstract revealed no additional data CSR and synopsis available. Data provided for all‐cause mortality, cardiovascular mortality, myocardial infarction, stroke, ketoacidosis, severe hypo/HbA1c combined Study also reported in FDA 2015 (FDA 2015) ‐ no additional data. No additional data from EMA 2012 (EMA 2012) |
|
BEGIN Young.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: informed consent, 1–17 years of age, T1DM, ongoing daily treatment with insulin (any regimen) for at least 3 months prior to screening. No oral anti‐diabetic drugs, HbA1c maximum 11% Exclusion criteria: known or suspected hypersensitivity to study product(s) or related products, previous participation in this study, pregnancy, breastfeeding or intend to become pregnant, menarche and are not using adequate contraceptive, known hypoglycaemic unawareness or recurrent severe hypoglycaemic events, more than 1 diabetic ketoacidosis requiring hospitalisation within the last 3 months prior to screening, significant concomitant disease (except for conditions associated with T1DM) which in the investigator's opinion could interfere with the study, receipt of any investigational drug within 1 month prior to screening Diagnostic criteria: based on clinical judgement and supported by laboratory analysis as per local guidelines Number of study centres: 72 |
|
| Interventions |
Intervention(s): degludec Comparator(s): detemir Duration of intervention: 26 weeks (plus 26 weeks of extension) Duration of follow‐up: 26 weeks (plus 26 weeks of extension) Run‐in period: — |
|
| Outcomes | Reported outcome(s) in full text of publication: mortality, adverse events, hypoglycaemia, HbA1c | |
| Study registration |
Trial identifier: NCT01513473, NN1250‐3561, EudraCT 2011‐003148‐39; EMA (ODCO) P/44/2010; WHO U1111‐1122‐4758; JapicCTI‐121824 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal and abstract |
|
| Stated aim of study | Quote: "The objective of this trial was to investigate the efficacy and safety of IDeg vs. IDet, both in combination with bolus insulin aspart (IAsp), in children and adolescents with T1D" | |
| Notes | All participants who completed 26 weeks of treatment (main period) were encouraged to continue in an extension of the study under similar conditions, for an additional 6 months (extension period). The South African sites did not participate in the 26 weeks of extension. Socioeconomic effects were reported in the abstract (Thalange 2017). Selected countries/sites participants underwent assessment of their 24‐hour interstitial glucose levels with a continuous glucose monitoring (CGM) device. CSR available ‐ in there data on diabetic ketoacidosis were available Study also reported in FDA 2015 and EMA 2014 and EMA 2015 reports ‐ no additional data (FDA 2015; EMA 2014; EMA 2015). In FDA, medical review data for adverse events (including ketoacidosis) in table 36 (page 77) |
|
Bolli 2009.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: 18 to 60 years with T1DM (> 3 years duration), with fasting plasma C‐peptide < 0.1 nmol/L and HbA1c 7‐9%, and who were on intensive insulin therapy (NPH twice or more daily and lispro or regular human insulin at mealtimes), no micro‐ or macro‐angiopathic complications and BMI 18‐26 kg/m2 Exclusion criteria: — Diagnostic criteria: fasting plasma C‐peptide < 0.1 nmol/L (not directly described, but is an inclusion criterion) Number of study centres: 21 |
|
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of intervention: 28 weeks (4‐week run‐in phase, 24‐week treatment period) Duration of follow‐up: 30 weeks (4‐week run‐in phase, 24‐week treatment period and 2‐week safety assessment) Run‐in period: 4 weeks |
|
| Outcomes | Reported outcome(s) in full text of publication: glycaemic control, safety, quality of life | |
| Study registration |
Trial identifier: — Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Sanofi‐Aventis) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "To compare switching from NPH insulin (NPH) to insulin glargine (glargine) with continuing NPH for changes in fasting blood glucose (FBG) in patients with Type 1 diabetes on basal bolus therapy with insulin lispro as bolus insulin." | |
| Notes | ||
Chase 2008.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: ≥ 9 to ≤ 17 years; Tanner stage ≥ 2; HbA1c ≥ 7.0% to ≤ 9.5%) who had a diagnosis of T1DM for at least 1 year and were receiving any daily insulin regimen consisting of 2 or more injections or a continuous subcutaneous insulin infusion, ability and willingness to count carbohydrates and perform SMBG testing at least 4 times per day Exclusion criteria: clinically relevant cardiovascular, hepatic, renal, neurologic, endocrine, or other major systemic diseases; psychiatric problems; laboratory test abnormalities; a history of 2 or more episodes of severe hypoglycaemia within the past 12 months or diabetic ketoacidosis in the past 3 months; or hypersensitivity to the investigational product or treatment; lipohypertrophy, a history of drug or alcohol abuse, current use of systemic corticosteroids or large doses of inhaled corticosteroids, and pregnancy Diagnostic criteria: fasting C‐peptide concentration of ≤ 0.5 nmol/L Number of study centres: 40 |
|
| Interventions |
Intervention(s): glargine Comparator(s): NPH/Lente Duration of intervention: 24 weeks Duration of follow‐up: 25 weeks (the treatment period was followed by a 1‐week follow‐up) Run‐in period: 4 weeks (during the educational run‐in period, patients received instruction from a certified diabetes educator on carbohydrate counting and basal/bolus insulin regimens) |
|
| Outcomes | Reported outcome(s) in full text of publication: serious adverse events, hypoglycaemia, HbA1c | |
| Study registration |
Trial identifier: HOE901/4030; NCT00046501 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Sanofi) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "To compare long‐acting insulin glargine (Lantus) with intermediate‐acting insulin (neutral protamine Hagedorn [NPH]/Lente) when used as the basal component of a multiple daily injection (MDI) regimen with prandial insulin lispro (Humalog) in adolescents with type 1 diabetes mellitus (T1DM)" | |
| Notes | Only three participants in the NPH/Lente group received Lente Subset of participants had CGM Clinical study summary available from Sanofis web page. This stated that The Diabetes Quality of Life for Youth questionnaire was applied. In the study summary, it was mentioned that more reported treatment emergent adverse events were observed in the glargine group compared with the NPH group From CSR, data for mortality and adverse events were retrieved |
|
Davies 2014.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: adults (≥ 18 years or ≥ 20 years for Japan) diagnosed with T1DM for ≥ 12 months, currently treated with any basal–bolus insulin regimen for ≥ 12 months prior to screening and with HbA1c ≤ 10.0% (85.8 mmol/mol) and BMI ≤ 35.0 kg/m2, For Japan only: minimum age was 20 years For the extension study only: completed the six‐month treatment period in study NN1250‐3585 (NCT01074268) Exclusion criteria: clinically significant concomitant diseases, including impaired renal and hepatic function; recurrent severe hypoglycaemia or hypoglycaemic unawareness or hospitalisation for diabetic ketoacidosis during the previous 6 months; and cardiovascular disease within the previous 6 months prior to the study, use of any other antidiabetic drug than insulin within the last 3 months, uncontrolled treated/untreated severe hypertension, pregnancy, breast‐feeding, the intention of becoming pregnant or not using adequate contraceptive measures, cancer and medical history of cancer Diagnostic criteria: — Number of study centres: 55 sites (in 7 countries) |
|
| Interventions |
Intervention(s): degludec Comparator(s): detemir Duration of intervention: 26 weeks Duration of follow‐up: 26 weeks (52 weeks) Run‐in period: none Number of study centres: 55 sites (in 7 countries) |
|
| Outcomes | Reported outcome(s) in full text of publication: glycaemic control, hypoglycaemia, safety | |
| Study registration |
Trial identifier: NCT01074268; NN1250‐3585 (26 weeks); NCT01190956; obsolete identifiers: NCT01190956; EudraCT number: 2009‐011672‐29 and 2009‐015721‐36; WHO identifier: U1111‐1111‐7249 and U1111‐1114‐9479; JAPIC Identifier: JapicCTI‐10106 and JapicCTI‐22‐0677; extension study: NN1250‐3725; main study: CTRI/2010/091/000145; extension study: CTRI/2010/091/001097 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "The primary outcome was non‐inferiority of IDeg to IDet in glycated haemoglobin (HbA1c) reduction after 26 weeks" | |
| Notes | Participants who completed the core study were invited to participate in a 26‐week extension study Data were entered after 26 weeks of intervention DiabMedSat (Diabetes Medication Satisfaction), DPM (Diabetes Productivity Measure), TRIM‐D (Treatment Related Impact Measure for Diabetes) and SF‐36 v2 were reported by the investigators and CSR CSR and synopsis available ‐ added information in combined HbA1c and severe hypoglycaemia Study also reported in FDA 2015 document (FDA 2015)‐ no additional data. No additional data from EMA 2012 (EMA 2012) |
|
Fulcher 2005.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: T1DM, 18‐80 years, treated with insulin for 1 year or more, HbA1c 8% or more, Additional from CSR: BMI < 35 kg/m2, Ability and willingness to perform frequent SMBG using a blood glucose meter and to perform continuous blood glucose measurements on numerous occasions Exclusion criteria: nightshift workers, patients with known sensitivity to the study drug or related drugs, and patients with impaired hepatic function or any other clinically relevant physiological or psychological medical conditions were excluded, Additional from CSR: treatment with any blood glucose altering drugs other than insulin in the last 4 weeks before study entry e.g. corticosteroids; pregnancy, breastfeeding; treatment with any investigational drug in the last 2 months before study entry Diagnostic criteria: post‐prandial C‐peptide level ≤ 0.5 nmol/L (≤ 1.5 ng/mL) in the presence of a blood glucose level ≥ 5.5 mmol/L Number of study centres: 9 |
|
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of intervention: 30 weeks (6‐week forced titration phase + 24‐week phase) Duration of follow‐up: 30 weeks Run‐in period: — (but had a one‐ to two‐week screening phase before the treatment phase) |
|
| Outcomes | Reported outcome(s) in full text of publication: glycaemic control, hypoglycaemia, weight, lipid status, safety | |
| Study registration |
Trial identifier: HOE901/4010 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Aventis) Publication status: peer‐reviewed journal and conference abstract |
|
| Stated aim of study | Quote: "To compare glycaemic control and symptomatic hypoglycaemia rates with glargine versus neutral protamine Hagedorn (NPH) in poorly controlled type 1 diabetes patients." | |
| Notes | Conference abstract added no additional information CSR was provided by Sanofi. CSR provided protocol, diagnostic criteria for T1DM, additional outcome data (e.g. mortality, ketoacidosis, hypoglycaemia) and information on bias. |
|
Heller 2009.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: ≥ 18 years who had T1DM for at least 12 months, had been taking a basal–bolus insulin regimen for at least 3 months, and had a HbA1c value ≤ 11.0% Exclusion criteria: proliferative retinopathy or maculopathy requiring acute treatment within 6 months before the study; any recurrent major hypoglycaemia; an anticipated change in any medication known to interfere with glucose metabolism; impaired hepatic or renal function; cardiac problems or uncontrolled hypertension believed to affect study participation Diagnostic criteria: — Number of study centres: 38 (number from synopsis/CSR) |
|
| Interventions |
Intervention(s): detemir Comparator(s): glargine Duration of intervention: 52 weeks Duration of follow‐up: 52 weeks Run‐in period: — (but there might have been one based on the following sentence: "All patients were asked to record a 10‐point self‐monitored PG (SMPG) profile on a typical day during the weeks before the randomization visit") |
|
| Outcomes | Reported outcome(s) in full text of publication: glycaemic measures, safety, hypoglycaemia | |
| Study registration |
Trial identifier: NN304‐1430; EUDRACT 2004‐000086‐35; NCT00095082 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "The primary study objective was to determine whether insulin detemir (detemir) was noninferior to insulin glargine (glargine) as the basal insulin in a basal–bolus regimen, with insulin aspart as the mealtime insulin, in terms of glycemic control at the end of 52 weeks in patients with type 1 diabetes mellitus (T1DM)." | |
| Notes | Each participant attended 13 study visits and received 16 scheduled telephone calls from the study site
From the clinical study synopsis: "The risk of having a nocturnal hypoglycaemic episode during the treatment period was similar in the two groups with a relative risk of 1.12 (P = 0.375)." and "The overall risk of having a hypoglycaemic episode during the treatment period was similar between the insulin detemir and the insulin glargine groups with a relative risk (insulin detemir/insulin glargine) of 0.94 (P = 0.571)." Data for mortality extracted from synopsis. From CSR, data on mortality, severe hypoglycaemia, nocturnal hypoglycaemia, mild hypoglycaemia, acute myocardial infarction, stroke and diabetic ketoacidosis could be retrieved |
|
Home 2005.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: T1DM and post‐prandial serum C‐peptide levels of < 0.50 nmol/L or < 1.50 µg/L when the capillary blood glucose level was ≥ 5.5 mmol/L (≥ 100 mg/dL) at the first visit. All had been treated with insulin for at least 1 year, aged 17–77 years Exclusion criteria: from FDA document (FDA 2000): pregnancy, surgical treatment for diabetic retinopathy, other glucose‐lowering drugs within 4 weeks, impaired renal function, abnormal liver tests Diagnostic criteria: C‐peptide < 0.05 nmol/L Number of study centres: 63 |
|
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of intervention: 28 weeks Duration of follow‐up: 28 weeks Run‐in period: 4 weeks |
|
| Outcomes | Reported outcome(s) in full text of publication: glycaemic variables, adverse events, safety | |
| Study registration |
Trial identifier: HOE 901/3001 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Aventis Pharma) Publication status: peer‐reviewed journal and conference abstract |
|
| Stated aim of study | Quote: "To compare insulin glargine with NPH human insulin for basal insulin supply in adults with type 1 diabetes" | |
| Notes | Of the 655 people entering the screening phase, 602 were randomised and 585 were treated with study medication ‐ 292 with insulin glargine and 293 with NPH insulin (147 people received once‐daily NPH insulin and 146 received twice‐daily NPH insulin) ‐ not reported how the 602 were randomised The corresponding author, Dr. Home, assumed that no participants died, as otherwise it would have been stated in the published paper. Dr. Home made us aware that the publication Witthaus et al. 2001 included the same population. No additional data from conference abstract Study included in FDA 2000 document (FDA 2000)‐ no additional outcome data |
|
Kobayashi 2007.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 2:1 | |
| Participants |
Inclusion criteria: duration of diabetes mellitus for at least 2 years; current treatment of basal‐bolus regimen for at least 12 weeks using an intermediate/long‐acting human insulin and insulin aspart; HbA1c < 11.0%; BMI < 30 kg/m2 Exclusion criteria: impaired renal function; impaired hepatic function; serious heart diseases; known hypoglycaemia unawareness or recurrent major hypoglycaemia; proliferative retinopathy or maculopathy requiring acute treatment; uncontrolled treated/untreated hypertension; current treatment with total insulin dose of more than 100 IU/day; current treatment or expected at the screening to start treatment with systemic corticosteroids Diagnostic criteria: — Number of study centres: 52 |
|
| Interventions |
Intervention(s): detemir Comparator(s): NPH Duration of intervention: 48 weeks Duration of follow‐up: 48 weeks (plus 2 to 9 days) Run‐in period: none |
|
| Outcomes | Reported outcome(s) in full text of publication: all‐cause mortality, hypoglycaemia, adverse events, HbA1c | |
| Study registration |
Trial identifier: NN304‐1476; JapicCTI‐R070008; NCT00604344 Study terminated early: no |
|
| Publication details |
Language of publication: Japanese Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal, conference abstracts and clinical study synopsis, 3 pages from CSR |
|
| Stated aim of study | Quote: "A 48‐week, randomised, multi‐centre, open‐labelled, parallel‐group trial to compare the efficacy and the safety of NN304 (insulin detemir) and NPH human insulin in participants with insulin requiring diabetes mellitus on a basal‐bolus regimen" | |
| Notes | Included both people with T1DM and T2DM, but separate data provided CSR provided data on diabetic ketoacidosis |
|
Liu 2016.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 2:1 | |
| Participants |
Inclusion criteria: T1DM, aged at least 6 years to less than 18 years Exclusion criteria: treatment with other glucose‐lowering medications other than insulin, HbA1c < 7% or > 12 %; Added from CSR: treated with insulin pump during the two months prior to screening; had undergone pancreas or islet cell transplantation; pancreatectomised; anticipated duration of life < 1 year for parents; history of primary seizure disorder; history of severe hypoglycaemic episode accompanied by seizure and/or coma, or diabetic ketoacidosis leading to hospitalisations or to care in the emergency ward, in the 2 months prior to the screening visit; known history of eating disorder such as anorexia or bulimia; known history of drug or alcohol abuse within 6 months prior to screening; treatment with systemic glucocorticoids within the month prior to screening; history of treatment for diabetic retinopathy (laser photocoagulation or vitrectomy) in the 6 months prior to screening, or diabetic retinopathy that may require treatment (e.g. laser photocoagulation) during the year following screening; treatment with any non‐insulin anti‐hyperglycaemic medication during the 3 months prior to screening; serum creatinine > 177 µmol/L; ALAT/ASAT greater than 3 times upper limit of normal); pregnancy, lactation Diagnostic criteria: — Number of study centres: 10 |
|
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of intervention: 24 weeks Duration of follow‐up: 28 weeks (up to 2 weeks screening + 1‐week run‐in + 24 week‐treatment + 1‐week follow‐up) Run‐in period: 1 week |
|
| Outcomes | Reported outcome(s) in full text of publication: hypoglycaemia, ketoacidosis, HbA1c | |
| Study registration |
Trial identifier: NCT01223131; EFC11681; U1111‐1116‐3661; EudraCT 2014‐004640‐35 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Sanofi) Publication status: peer‐reviewed journal and abstract |
|
| Stated aim of study | Quote: "Therefore, the purpose of the present study was to describe the safety and efficacy of once‐daily insulin glargine over a period of 24 weeks in Chinese paediatric patients with T1DM" | |
| Notes | Clinic consultations occurred at screening/run‐in (week –3 to – 2 and week –1), randomisation (week 0), weeks 1, 2, 4, 6, 8, 10, 12, 16, 20, 24 (end of treatment) and week 25 (follow‐up) Two years after enrolment of the first patient, a total of 108 patients were screened and 93 randomised, which constituted only 25% of the original enrolment target. Therefore, the study protocol was amended to reduce the planned number of enrolled patients to 150, with 100 patients randomised to insulin glargine and 50 to NPH insulin CSR synopsis did not report new outcome compared with clinical trials registers. EMA documents did not add additional outcomes (EMA 2015a; EMA 2015b) |
|
NCT00595374.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 2:1 | |
| Participants |
Inclusion criteria: duration of T1DM > 12 months, > 18 years; BMI below 35 kg/m2, HbA1c between 7.0‐12.0%; current treatment with pre‐prandial short‐acting insulin and insulin NPH once or twice daily for at least 6 months Exclusion criteria: known or suspected allergy to study product or related products, receipt of any investigational products within the last 2 months prior to this study; drug or alcohol dependence, pregnancy, breastfeeding or intention of becoming pregnant Diagnostic criteria: — Number of study centres: 17 |
|
| Interventions |
Intervention(s): detemir Comparator(s): NPH Duration of intervention: 26 weeks Duration of follow‐up: 26 weeks (+ 7 days) Run‐in period: 2 weeks |
|
| Outcomes | Reported outcome(s) in full text of publication: no full text available | |
| Study registration |
Trial identifier: NCT00595374; NN304‐1582 Study terminated early: no |
|
| Publication details |
Language of publication: not published Funding: commercial funding (Novo Nordisk) Publication status: unpublished study. Data extraction based on ClinicalTrials.gov and clinical study synopsis |
|
| Stated aim of study | Quote: "The aim of this trial is to compare the efficacy and safety of insulin detemir and insulin NPH in adults with type 1 diabetes on blood glucose control" | |
| Notes | "The primary efficacy variable, the HbA1c showed no statistically significant difference between NPH insulin and insulin detemir for both the Full Analysis Set (FAS) and the Per‐Protocol‐Set (PPS)" and "Both overall and nocturnal analyses show no statistically significant difference in incidence of hypoglycaemic episodes." and "The results indicate that the mean class level of nocturnal hypoglycaemic episode shows no statistically significant difference between NPH insulin and insulin detemir for the FAS (P = 0.2119)" and "Seven patients experienced a total of 10 serious adverse events" Novo Nordisk replied that no CSR was available for this study |
|
NCT00605137.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 2:1 | |
| Participants |
Inclusion criteria: T1DM for at least one year; current treatment of basal‐bolus regimen for at least 12 weeks using an intermediate/long‐acting human insulin and insulin aspart and/or soluble human insulin; HbA1c below 11.0%; able and willing to perform self‐monitoring of capillary blood glucose and to take measures in case of hypoglycaemia Exclusion criteria: impaired renal function; impaired hepatic function; known hypoglycaemia unawareness or recurrent major hypoglycaemia; proliferative retinopathy or maculopathy requiring acute treatment; uncontrolled treated/untreated hypertension; current treatment with total daily insulin dose of more than 2.00 IU/kg; current treatment or expected at the screening to start treatment with systemic corticosteroids; history of serious allergy or serious anaphylactic reaction Diagnostic criteria: — Number of study centres: 17 |
|
| Interventions |
Intervention(s): detemir Comparator(s): NPH Duration of intervention: 24 weeks Duration of follow‐up: 24 weeks Run‐in period: the participants were randomised 6 weeks after the screening visit |
|
| Outcomes | Reported outcome(s) in full text of publication: no full text available (outcomes reported in synopsis: mortality, adverse events, hypoglycaemia, HbA1c) | |
| Study registration |
Trial identifier: NCT00605137; NN304‐1604; JapicCTI‐R070014 Study terminated early: no |
|
| Publication details |
Language of publication: not published Funding: commercial funding (Novo Nordisk) Publication status: unpublished study. Data extraction based on ClinicalTrials.gov, clinical study synopsis, CSR (Novo Nordisk provided 4 pages of the CSR) and the trial protocol |
|
| Stated aim of study | Quote: "To investigate the safety profile of NN304 compared to NPH human insulin during a 24‐week treatment period in children with type 1 diabetes on a basal‐bolus regimen" | |
| Notes | The maintenance period was defined as the interval from 6 weeks after the first day on the study product to the last day on study product (including the last day) "The same trend was seen in nocturnal hypoglycaemic episodes." |
|
Pieber 2007.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: 18 years or more, T1DM ≥ 1 year, BMI ≤ 35 kg/m2, HbA1c between 7.5 and 12.0%, Prior to the study, treated with either intermediate‐/long‐acting insulin twice daily and three to four pre‐meal human soluble insulin injections for ≥ 6 months, or biphasic insulin morning and evening and pre‐lunch human soluble insulin injection for ≥ 6 months; total daily insulin dose was < 1.4 units/kg Exclusion criteria: significant medical problems, including proliferative retinopathy or maculopathy requiring acute treatment; recurrent severe hypoglycaemia; hypoglycaemic unawareness; impaired hepatic or renal function, or uncontrolled cardiovascular problems; pregnant or breastfeeding women Diagnostic criteria: — Number of study centres: 39 (from synopsis) |
|
| Interventions |
Intervention(s): detemir Comparator(s): glargine Duration of intervention: 26 weeks Duration of follow‐up: 26 weeks Run‐in period: — |
|
| Outcomes | Reported outcome(s) in full text of publication: severe adverse events, hypoglycaemia, HbA1c | |
| Study registration |
Trial identifier: NCT00312104; NN304‐1372 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "To compare glycaemic control and risk of hypoglycaemia of twice‐daily insulin detemir with once‐daily insulin glargine in participants with Type 1 diabetes" | |
| Notes | At end of study, higher dose of insulin in the detemir group vs. the glargine group. From synopsis: "The mean daily dose of basal insulin was 34% higher for insulin detemir than for insulin glargine". Data on mortality and adverse events were extracted from synopsis/CSR. FDA medical review (FDA 2002) did not provide additional data |
|
Porcellati 2004.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; superiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: T1DM; C‐peptide ≤ 0.15 nmol/L; on intensified treatment with multiple daily combinations of lispro and NPH insulin at each meal, and NPH at bedtime for at least 2 years Exclusion criteria: microangiopathy; autonomic neuropathy Diagnostic criteria: based on inclusion criteria, it is anticipated to be C‐peptide ≤ 0.15 nmol/L Number of study centres: 1 |
|
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of intervention: 1 year Duration of follow‐up: 1 year Run‐in period: 1 month |
|
| Outcomes | Reported outcome(s) in full text of publication: glycaemic control, hypoglycaemia | |
| Study registration |
Trial identifier: — Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: non‐commercial funding (National Ministery of Scientific Research and University of Perugia) Publication status: peer‐reviewed journal and conference abstract |
|
| Stated aim of study | Quote: "The aim of this study was to test superiority of glargine on long‐term blood glucose (BG) as well as on responses to hypoglycaemia vs. NPH." | |
| Notes | Conference abstract did not report any additional data | |
PRESCHOOL.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: paediatric patients with T1DM aged at least one year to less than 6 years at screening Exclusion criteria: T1DM for less than one year; HbA1c at screening > 12% or < 6%; diabetes other than T1DM; parents and patients not willing to undergo all study assessments and treatments; treated with insulin pump therapy during the two months prior to screening; history of primary seizure disorder; history of severe hypoglycaemic episode accompanied by seizure and/or coma, or diabetic ketoacidosis leading to hospitalisation or to care in the emergency ward in the 2 months prior to the screening; need for chronic treatment with acetaminophen (paracetamol)‐containing medications; serum creatinine > 2.0 mg/dL at screening; serum ALAT or ASAT greater than 3x upper limit of normal for the patient's age and gender; haemoglobin < 10 g/dL, or platelet count less than 100,000/cu mm; treatment with any pharmacologic anti‐hyperglycaemic oral agent for more than 3 months at any time; treatment with any non‐insulin antihyperglycaemic medication for the 3 months prior to screening; treatment with systemic glucocorticoids within the month prior to screening Diagnostic criteria: — Number of study centres: 61 |
|
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of intervention: 24 weeks Duration of follow‐up: 28 to 30 weeks (screening period 2 to 4 weeks, treatment period 24 weeks, and post‐treatment observation period 2 weeks) Run‐in period: 2 weeks |
|
| Outcomes | Reported outcome(s) in full text of publication: HbA1c, hypoglycaemia | |
| Study registration |
Trial identifier: NCT00993473; Eudra CT: 2009‐011231‐12; EFC11202; CTRI/2009/091/000912 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Sanofi) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "To evaluate hypoglycemia with insulin glargine vs. neutral protamine Hagedorn (NPH) insulin in young children, using continuous glucose monitoring (CGM)" | |
| Notes | Additional data from trials registers on serious adverse events, adverse events, ketoacidosis and mortality. CSR did not report any new data but reported HbA1c in more analyses ‐ most appropriate was HbA1c change from baseline, which was used for the analysis and retrieved from the CSR | |
Ratner 2000.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: 18‐80 years; T1DM for at least 1 year; HbA1c ≤ 12%; ability and willingness to perform SMBG using a blood glucose meter at home, as evidenced by 7 consecutive daily FBG values during the screening phase Exclusion criteria: treatment with other glucose‐lowering drugs than insulin within 1 month of study entry, pregnancy, impaired hepatic function, impaired renal function, night shift, glucocorticoids Diagnostic criteria: post‐prandial C‐peptide levels ≤ 0.5 nmol/L Number of study centres: 49 |
|
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of intervention: 28 weeks Duration of follow‐up: 28 weeks Run‐in period: none |
|
| Outcomes | Reported outcome(s) in full text of publication: HbA1c, hypoglycaemia, safety | |
| Study registration |
Trial identifier: HOE 901/3004 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Hoechst Marion Roussel, Aventis) Publication status: peer‐reviewed journal and conference abstract |
|
| Stated aim of study | Quote: "This study compared insulin glargine with NPH human insulin in participants with type 1 diabetes who had been previously treated with multiple daily injections of NPH insulin and regular insulin" | |
| Notes | Herschon 2004 reported on a subgroup of participants (394 out of 534). Part of the study was published in an abstract ‐ this abstract was not retrieved Sanofi provided a CSR. From CSR: amendment 1 (21 May 1997) shortened the treatment period from 52 to 28 weeks; this was achieved by omitting 3 visits, but the interval between visits was not affected. The decision to shorten the treatment period from 52 weeks to 28 weeks was based on the outcome of a meeting with representatives of the US Food and Drug Administration (FDA). The conclusion of this meeting was that a 6‐month treatment period would be sufficient to demonstrate the efficacy and safety of HOE 901 in a Phase III study for regulatory purposes From CSR: additional mortality, hypoglycaemia data, serious adverse events, cost, quality of life Study included in FDA 2000 (FDA 2000) ‐ data on hypoglycaemia and serious adverse events could be retrieved (but these data were also available from CSR) |
|
Robertson 2007.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 2:1 | |
| Participants |
Inclusion criteria: T1DM for at least 12 months; age 6‐17 years; 6‐7 years: BMI less than or equal to 19 kg/m2, 8‐9 years: BMI less than or equal to 20 kg/m2, 10‐11 years: BMI less than or equal to 22 kg/m2, 12‐13 years: BMI less than or equal to 24 kg/m2 and 14‐17 years: BMI less than or equal to 27 kg/m2; HbA1c equal to or less than 12.0% Exclusion criteria: proliferate retinopathy or maculopathy; total daily insulin dose greater than 2.00 IU/kg; any condition or disease that ruled out study participation according to the judgement of the investigator; mental incapacity, unwillingness or language barriers precluding understanding or co‐operation; life‐style incompatible with study participation Diagnostic criteria: — Number of study centres: 44 |
|
| Interventions |
Intervention(s): detemir Comparator(s): NPH Duration of intervention: 26 weeks Duration of follow‐up: 26 weeks Run‐in period: none |
|
| Outcomes | Reported outcome(s) in full text of publication: adverse events, hypoglycaemia, HbA1c | |
| Study registration |
Trial identifier: NCT00312156; NN304‐1379 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "This study compared the effect of insulin detemir on glycaemic control (HbA1c, fasting plasma glucose and variability thereof) with that of Neutral Protamine Hagedorn human isophane (NPH) insulin, both combined with insulin aspart, in children with Type 1 diabetes mellitus, and compared the safety of these treatments." | |
| Notes | Trial synopsis ‐ this provided additional information on serious adverse events A post‐treatment follow‐up visit was performed 2‐4 days after the last visit FDA Medical review 2005 provided information on mortality (FDA 2005) |
|
Russell‐Jones 2004.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 2:1 | |
| Participants |
Inclusion criteria: ≥ 18 years with T1DM for ≥ 1 year who were already using basal or premixed insulin once daily in the evening (between 5 PM and 11 PM) and human insulin before meals for ≥ 2 months Exclusion criteria: very poorly controlled diabetes using the current once daily therapy (as determined by HbA1c > 12% and/or a total basal insulin dose > 100 IU/d); pregnant or breastfeeding; significant medical problems including proliferative retinopathy, impaired hepatic or renal function, recurrent major hypoglycaemia, uncontrolled hypertension, or severe cardiac problems; concomitant use of medications known to interfere with glucose metabolism was not permitted Diagnostic criteria: — Number of study centres: 92 |
|
| Interventions |
Intervention(s): detemir Comparator(s): NPH Duration of intervention: 6 months Duration of follow‐up: 6 months Run‐in period: — (3 weeks screening period, not further specified) |
|
| Outcomes | Reported outcome(s) in full text of publication: HbA1c, safety, hypoglycaemia | |
| Study registration |
Trial identifier: NCT03220425; NN304‐1335 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal/conference abstract |
|
| Stated aim of study | Quote: "The purpose of this trial was to compare the effects of QD basal insulin replacement using insulin detemir versus neutral protamine Hagedorn (NPH) insulin in basal‐bolus therapy in combination with regular human insulin (HI) in patients with type 1 diabetes mellitus (DM)." | |
| Notes | The study consisted of an initial 1‐month titration period (2 visits and telephone contact), during which dosing was optimised to meet individual requirements, and a 5‐month maintenance period (4 visits) Twenty‐four–hour continuous blood glucose profiles were measured in a subgroup of patients from both treatment groups during the last month of treatment. Patients from 18 selected investigational sites were asked (but not required) to wear the Continuous Glucose Monitoring System (CGMS; Medtronic MiniMed, Northridge, California) for 72 hours. For logistic reasons, as well as for optimising compliance, investigational sites were selected based on previous experience with the device and willingness to participate The relative risk of hypoglycaemia was estimated from the incidence of all hypoglycaemic episodes occurring during the maintenance period (i.e. 5 months) (the interval from 30 days after first dose to last day on study product). Conference abstract did not provide any new information CSR provided data on mortality, serious adverse events and adverse events. Study described in FDA medical review (FDA 2002) which provided data on mortality. EMA provided no additional data (EMA 2004) |
|
Schober 2002.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: T1DM; age 5‐16 years; treated with insulin for at least one year; using at least three daily injections; HbA1c < 12% Exclusion criteria: other glucose‐lowering treatment than insulin within the last month; postmenarchal, sexually active girls not using adequate contraception; treatment with hyperglycaemic drugs; impaired liver function; impaired renal function Diagnostic criteria: — Number of study centres: 30 |
|
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of intervention: 28 weeks Duration of follow‐up: 28 weeks (run‐in period included) Run‐in period: 4 weeks |
|
| Outcomes | Reported outcome(s) in full text of publication: hypoglycaemia, adverse events, HbA1c | |
| Study registration |
Trial identifier: HOE901/3003 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: not reported in main publication, but co‐publication reported funding from Sanofi Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "The objective of this 28‐week, multicenter, centrally randomized and controlled study was to compare the effects of insulin glargine and NPH insulin on glycosylated hemoglobin (HbA1c) in children and adolescents with T1DM." | |
| Notes | From Herwig 2007: "This study included those patients from the previous study who continued with insulin glargine treatment." Study in reference is Schober 2002. Herwig and colleagues reported funding from Sanofi CSR provided by Sanofi: added mortality data and a trial protocol. FDA 2000 did not provide additional data (FDA 2000) CSR provided data on economics |
|
Standl 2004.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: adults (aged 18–74 years); T1DM of 12 months or more; treated with twice‐daily basal insulin in combination with meal‐related bolus insulin for at least 2 months; BMI ≤ 35.0 kg/m2; HbA1c ≤ 12%; total basal insulin dosage ≤ 100 IU/day
Exclusion criteria: proliferative retinopathy; impaired hepatic or renal function; severe cardiac disease; uncontrolled hypertension; recurrent major hypoglycaemia; insulin allergy; pregnant or breastfeeding women
Diagnostic criteria: — Number of study centres: 47 |
|
| Interventions |
Intervention(s): detemir Comparator(s): NPH Duration of intervention: 6 months Duration of follow‐up: 6 months (12 months) Run‐in period: none |
|
| Outcomes | Reported outcomes in full text of publication: glycaemic control, hypoglycaemia, weight, safety | |
| Study registration |
Trial identifier: NN304‐1181 (extension NN304‐1243) Study terminated early: no |
|
| Publication details | Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal | |
| Stated aim of study | Quote: "This trial compared the long‐term safety and efficacy of the basal insulin preparations, insulin detemir and NPH insulin, in basal‐bolus therapy for patients with type 1 diabetes". | |
| Notes | After an initial 6‐month treatment period, patients were invited to participate in a 6‐month extension period Data were entered after the 6‐month main period Additional data on this study were available from the FDA Medical Review of Levemir (FDA 2002) for severe hypoglycaemia after 6 months and mortality. EMA document provided no additional data (EMA 2004). CSR provided by Novo Nordisk. From this, it was apparent that quality of life had been evaluated. A trial protocol was provided as well. Data for adverse events could be added as well as exact values for people included in the analysis of the study |
|
SWITCH 1.
| Study characteristics | ||
| Methods | Design: cross‐over RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: fulfilling at least one of the below criteria: experienced at least one severe hypoglycaemic episode within the last year (according to the ADA definition, April 2013); moderate chronic renal failure, defined as glomerular filtration rate 30 ‐ 59 mL/min/1.73 m2 per chronic kidney disease epidemiology collaboration; hypoglycaemic symptom unawareness; diabetes mellitus duration for more than 15 years; recent episode of hypoglycaemia within the last 12 weeks: male or female; age at least 18 years at the time of signing informed consent; T1DM (diagnosed clinically) for at least 52 weeks; current treatment with a basal‐bolus regimen consisting of NPH insulin once daily/twice daily or insulin detemir once daily/twice daily plus 2‐4 daily injections of any rapid‐acting meal time insulin or continuous subcutaneous insulin infusion (with rapid‐acting insulin) for at least 26 weeks; HbA1c below or equal to 10%; BMI below or equal to 45 kg/m2 Exclusion criteria: known or suspected hypersensitivity to study product(s) or related products; previous participation in this study; female who is pregnant, breastfeeding or intends to become pregnant or is of child‐bearing potential and not using adequate contraceptive methods; treatment with glargine or degludec within the last 26 weeks; use of any other glucose‐lowering drug than those stated in the inclusion criteria within the last 26 weeks; receipt of any investigational medicinal product within 4 weeks prior to screening; any chronic disorder or severe disease which, in the opinion of the investigator, might jeopardise the safety or compliance with the protocol: current or past (within the last 5 years) malignant neoplasms (except basal cell and squamous cell carcinoma); stroke, decompensated NYHA class III or IV, myocardial infarction, unstable angina pectoris, or coronary arterial bypass graft or angioplasty, all within the last 26 weeks; uncontrolled or untreated severe hypertension defined as systolic BP ≥ 180 mmHg and/or diastolic BP ≥ 100 mmHg; impaired liver function defined as ALAT or ASAT ≥ 2.5 times upper limit of normal; severe renal impairment defined as glomerular filtration rate < 30 mL/min/1.73 m2; proliferative retinopathy or maculopathy requiring acute treatment according to the investigator verification by fundoscopy or fundus photography performed within 12 weeks Diagnostic criteria: clinically diagnosed Number of study centres: 90 |
|
| Interventions |
Intervention(s): degludec Comparator(s): glargine Duration of intervention: 32 weeks Duration of follow‐up: 32 weeks Run‐in period: none |
|
| Outcomes | Reported outcome(s) in full text of publication: hypoglycaemia, safety, at cross‐over: HbA1c and quality of life | |
| Study registration |
Trial identifier: NCT02034513; NN1250‐3995; WHO ID: U1111‐1129‐9668; EudraCT number: 2012‐001930‐32 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "To determine whether insulin degludec is noninferior or superior to insulin glargine U100 in reducing the rate of symptomatic hypoglycemic episodes" | |
| Notes | The study had a cross‐over design ‐ each intervention period was 32 weeks before cross‐over. Main analyses of the study were performed in the last 16 weeks of each cross‐over period: weeks during the maintenance period (weeks 16‐32 and 48‐64) Study also reported in FDA 2015 report; this trial is described as ongoing and no additional data could be retrieved (FDA 2015) |
|
Thalange 2013.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: T1DM for at least 12 months; age 2‐16 years; total daily insulin dose ≤ 2.0 U/kg; insulin detemir naive; HbA1c less or equal to 11%; BMI ≤ 27 kg/m2 Exclusion criteria: significant concomitant disease Diagnostic criteria: — Number of study centres: 35 |
|
| Interventions |
Intervention(s): detemir Comparator(s): NPH Duration of intervention: 52 weeks Duration of follow‐up: 104 weeks (only for the detemir group) Run‐in period: none |
|
| Outcomes | Reported outcome(s) in full text of publication: adverse events, ketoacidosis, HbA1c, hypoglycaemia | |
| Study registration |
Trial identifier: NN304‐1689; EudraCT 2006‐000051‐18; NCT00435019 (main study); NCT00623194 (extension study); NN304‐1690 (extension study) Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "This 52‐week, randomized, multinational, open‐label, parallel‐group, non‐inferiority trial investigated the efficacy and safety of basal–bolus treatment with insulin detemir vs. NPH (neutral protamine Hagedorn) insulin, in combination with insulin aspart, in participants aged 2–16 years with Type 1 diabetes mellitus" | |
| Notes | A total of 10 scheduled visits to the clinical study sites and 8 telephone contacts. Only participants in the detemir group were invited to extended follow‐up Quote: "Children in the IDet arm who completed this study were offered the option to continue treatment with IDet (once or twice daily) together with IAsp (2–4 times daily with meals) for a further 52 weeks (extension study), for a total of 104 weeks of treatment (total treatment period)" The CSR did not add any additional information on outcomes. Additional information on baseline variables were identified Described in EMA 2011 report, but no additional outcomes provided (EMA 2011) |
|
Urakami 2017.
| Study characteristics | ||
| Methods | Design: cross‐over RCT; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: T1DM; children; Tanner stage 1‐3; previously received a once‐daily injection of glargine at bedtime as a basal insulin regimen Exclusion criteria: — Diagnostic criteria: — Number of study centres: 1 |
|
| Interventions |
Intervention(s): degludec Comparator(s): glargine Duration of intervention: 24 weeks Duration of follow‐up: 24 weeks Run‐in period: — |
|
| Outcomes | Reported outcome(s) in full text of publication: hypoglycaemia, HbA1c | |
| Study registration |
Trial identifier: — Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: not reported Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "In the present study, we have compared the efficacy and safety of IGlar vs. IDeg as a basal‐bolus therapy during sequential 24‐week periods in a randomized crossover study of Japanese children with type 1 diabetes". | |
| Notes | Study authors provided outcomes on request. No study protocol provided | |
Vague 2003.
| Study characteristics | ||
| Methods | Design: parallel‐group RCT; non‐inferiority design; randomisation ratio: 1:1 | |
| Participants |
Inclusion criteria: T1DM for at least 1 year; received basal (once or multiple times daily) bolus insulin treatment for at least 2 months; HbA1c level ≤ 12%, BMI ≤ 35kg/m2; total basal insulin dosage of ≤ 100 IU/day Exclusion criteria: proliferative retinopathy; impaired hepatic or renal function; severe cardiac problems; uncontrolled hypertension; recurrent major hypoglycaemia; allergy to insulin; pregnancy and breastfeeding Diagnostic criteria: — Number of study centres: 46 |
|
| Interventions |
Intervention(s): detemir Comparator(s): NPH Duration of intervention: 6 months Duration of follow‐up: 6 months (12 months) Run‐in period: none |
|
| Outcomes | Reported outcome(s) in full text of publication: HbA1c, safety, hypoglycaemia | |
| Study registration |
Trial identifier: NN304‐1205; extension trial: NN304‐1316 Study terminated early: no |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novo Nordisk) Publication status: peer‐reviewed journal |
|
| Stated aim of study | Quote: "The aim of this trial was to evaluate the metabolic control, risk of hypoglycemia, and other potential effects of treatment with insulin detemir in patients with type 1 diabetes on such a basal‐bolus regimen" | |
| Notes | Patients completing the initial 6‐month trial were invited to participate in the extension phase, with 316 of 425 accepting CSR reported mortality, serious adverse events and ketoacidosis Additional information available from FDA review (mortality) (FDA 2002). EMA document provided no additional data (EMA 2004) |
|
—: denotes not reported
ADA: American Diabetes Association ALAT: alanine aminotransferase ASAT: aspartate‐aminotransferase BG: blood glucose BMI: body mass index BP: blood pressure FBG: fasting blood glucose CGM: continuous glucose monitoring CGMS: continuous glucose monitoring system CSR: clinical study report DM: diabetes mellitus DiabMedSat: diabetes medication satisfaction DPM: diabetes productivity measure EMA: European Medicine Agency EudraCT: European Union Drug Regulating Authorities Clinical Trials Database FAS: full analysis set FDA: Food and Drug Administration HbA1c: glycosylated haemoglobin A1c HI: human insulin IAsp: insulin aspart IDeg: insulin degludec IDet: insulin detemir IGlar: insulin glargine IU: international units MAO: monoamine oxidase MDI: multiple daily injection NPH: neutral protamine Hagedorn NYHA: New York Heart Association PG: plasma glucose PM: post meridiem PPS: per‐protocol set QD: quaque die (daily) SF‐36: short‐form 36 SMBG: self‐monitoring of blood glucose SMPG: self‐measured plasma glucose RCT: randomised controlled trial T1DM: type 1 diabetes mellitus T2DM: type 2 diabetes mellitus Trim‐D: treatment related impact measure for diabetes
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| 21st Brazilian Diabetes Society Congressa | Congress report containing no studies of relevance |
| Bin‐Abbas 2006 | Wrong study design: not a randomised clinical trial |
| Bolli 2016 | Wrong study design: pooled data from four randomised clinical trials |
| Chacra 2010 | Wrong intervention (applied basal insulin no longer available) |
| Hirsch 2012 | Wrong study drug: compared insulin degludec/aspart combined with detemir + aspart (NCT00978627; NN5401‐3645; NN5401‐3645; Eudra: 2008‐005769‐71; U1111‐1111‐8943; 2009‐013412‐13; U1111‐1113‐2475) |
| HypoANA | Wrong study drug: applied different type of rapid‐acting insulin analogue in the intervention arms |
| Iga 2017 | Short duration of the intervention |
| Kiess 2004 | Wrong study design: letter |
| Manini 2007 | Wrong study design: not a randomised clinical trial |
| NCT00788840 | Wrong population: people with T2DM |
| NCT01854723 | Wrong population: people with insulin resistance |
| Orchard 2014 | Wrong intervention: different co‐intervention |
| Ota 2017 | Trial combined outcomes of people with T1DM and T2DM. No separate data available for the 12 people with T1DM included in the trial |
| Perez‐Maraver 2013 | Applied different type of rapid‐acting insulin analogue in the intervention arms |
| Polonsky 2014 | Wrong study design: not a randomised clinical trial |
| Prikhodina 2007 | Wrong study design: not a randomised clinical trial |
| Tentolouris 2018 | Wrong study design: not a randomised clinical trial |
| UMIN000001562 | Wrong study design: one intervention arm |
| UMIN000009965 | Study protocol for a study with short duration |
| UMIN000013817 | Study protocol for a study with short duration |
| Yamada 2014 | Short duration of the intervention |
| Ziemen 2015 | Wrong intervention: comparing insulin glargine in different concentrations |
T1DM: type 1 diabetes mellitus T2DM: type 2 diabetes mellitus
Characteristics of studies awaiting classification [ordered by study ID]
Agesen 2019.
| Methods |
Allocation: randomised Intervention model: cross‐over Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 154 Inclusion criteria: T1DM for more than five years, one or more episodes of nocturnal severe hypoglycaemia in the previous two years (defined as need for third party assistance to restore blood glucose level), age > 18 years, treatment with multiple dose insulin injection (more than 2) or insulin pump allowing for both human insulin and insulin analogues, a negative pregnancy test, willingness to a once‐daily regimen concerning basal insulin, willingness to do self‐monitoring of blood glucose and keep a diary Exclusion criteria: history of primary or secondary adrenal or growth hormone insufficiency, untreated hypothyroidism, history of unstable angina or major cardiovascular events, heart failure (NYHA class IV), history of malignancy unless a disease‐free period exceeding five years, history of alcohol or drug abuse, pregnancy or lactation, and women of childbearing potential who are not using chemical or mechanical contraception, HbA1c > 86 mmol/mol (10%), and shifting working hours |
| Interventions |
Intervention(s): degludec Comparator(s): glargine Duration of the intervention: 12 months before cross‐over (24 months in total) |
| Outcomes |
Primary outcome(s): symptomatic nocturnal hypoglycaemia Secondary outcome(s): hypoglycaemia (severe, any nocturnal, CGM recorded, any in hospital), HbA1c, insulin dose, quality of life, change in glycaemic variability Other outcome(s): — Relevant proposed outcome measures for SoF table: health‐related quality of life, hypoglycaemia |
| Reason for awaiting classification | Marked as 'completed' in Clinicaltrials.gov but no publication was available |
| Study details |
Study identifier: NCT02192450; 2014‐001942‐24 Study start date: July 2014 Study completion date: June 2019 Responsible party/principal investigator: Ulrik Pedersen‐Bjergaard, Nordsjaellands Hospital, Denmark |
| Official title and purpose of study | Insulin Degludec and Symptomatic Nocturnal Hypoglycaemia (HypoDeg) Quote: "The purpose of this study is to determine whether insulin degludec compared to insulin glargine can reduce the risk of symptomatic nocturnal hypoglycaemia in participants with the greatest potential benefit from optimised insulin treatment, which are patients with type 1 diabetes and high risk of nocturnal severe hypoglycaemia" |
| Notes |
Basal Analog Study.
| Methods |
Allocation: randomised Intervention model: parallel‐group assignment Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: newly diagnosed T1DM Estimated number of participants: 120 Inclusion criteria: diagnosis of T1DM and novel to insulin therapy, age 7 to 17 years Exclusion criteria: moderate to severe ketoacidosis (pH < 7.2 and/or standard bicarbonate < 10 mmol/L); suspected non‐type 1 IA2 and GAD65: all antibody negative; celiac disease or other chronic disease; hypothyroidism, if not well controlled syndromes; previous anorexia nervosa; neuro‐psychiatric disease; malignancy |
| Interventions |
Intervention(1): glargine Comparator(1): detemir Comparator (2): NPH Duration of the intervention: 12 months |
| Outcomes |
Primary outcome(s): HbA1c Secondary outcome(s): stimulated C‐peptide, IGF‐1 (from EudraCT: quality of life, hypoglycaemia) Other outcome(s): — Relevant proposed outcome measures for SoF table: health‐related quality of life, hypoglycaemia |
| Reason for awaiting classification | Marked as 'completed' in Clinicaltrials.gov and conference abstract available. No publication available. HbA1c reported in a format making it unsuitable for meta‐analysis |
| Study details |
Study identifier: EudraCT‐number 2005‐001726‐80; NCT01271517 Study start date: September 2005 Study completion date: March 2005 Responsible party/principal investigator: Peter Bang, Karolinska Institutet, Sweden |
| Official title and purpose of study | Basal Analog Study ‐ Comparison of lantus or levemir with NPH insulin from T1DM diagnosis (BAS) Quote: "To study if the use of long acting insulin analog treatment from diagnosis of pediatric type 1 diabetes mellitus (T1DM) improves metabolic control and IGF‐I levels" |
| Notes | Corresponding author contacted. No full‐text publication was available. Published as conference abstract |
ChiCTR2000032703.
| Methods |
Allocation: randomised Intervention model: cross‐over study Masking: not stated Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 20 Inclusion criteria: 1. At the time of screening, the age of patients >= 18 years old; both male and female considered; 2. Participants who were all type 1 diabetic patients, with the serum C‐peptide concentration confirmed as low level (< 0.07 nmol/L) at least twice; 3. Participants who had been continuously using the basic plus meal insulin treatment scheme in the past 3 months; 4. Patients with HbA1c meeting the standard: 6.9% <= HbAlc <= 10.0%; 5. Body mass index (BMI) of 18.0‐35.0kg/m2; 6. Patients who could understand and abide by the test process, voluntarily participate in the test and provide informed consent. Exclusion criteria: 1. Patients with diabetic ketoacidosis or diabetic hyperosmotic nonketotic coma in the past 6 months; 2. Patients with severe infection, surgery or severe trauma in the past month; 3. Patients with any of the following history and conditions of heart disease in the past 6 months: (1) Decompensated cardiac insufficiency (NYHA grade III or IV) (2) Unstable angina, myocardial infarction, coronary artery bypass grafting or coronary stent implantation (3) Uncontrolled or serious arrhythmias (such as long QT interval syndrome) according to the evaluation of researchers; 4. Patients with haemorrhagic stroke or ischaemic stroke in the past 6 months as assessed by the researchers; 5. At present, patients with any disease that may cause haemolysis or red blood cell instability and affect the detection of glycosylated haemoglobin; 6. Patients with a history of acute or chronic pancreatitis; 7. Liver function damaged, AST/ALT > 3 times of the upper limit of reference range, total bilirubin > 1.5 times of the upper limit of reference range; 8. Renal insufficiency, glomerular filtration rate (EGFR) < 60 mL/min/1.73m2 9. Patients with diseases that may cause tissue hypoxia (especially the deterioration of acute disease or chronic respiratory disease); 10. Patients with severe chronic gastrointestinal diseases with malnutrition, hunger or weakness; 11. Patients with adrenal dysfunction; 12. Patients that were habitual heavy drinkers; 13. Patients with dehydration or gastrointestinal symptoms, such as diarrhoea or vomiting related to dehydration risk; 14. Patients with malignant tumours requiring treatment in the past 5 years; 15. Patients who had received or were receiving any other investigational drug in the past 3 months; 16. Patients with serious mental illness or language disorder who were unwilling or unable to fully understand co‐operation; 17. Patients who were or might be allergic to insulin or similar drugs; 18. Pregnant or lactating women; 19. Patients who had used CGMS system in the past 6 months; 20. Patients who were receiving systemic glucocorticoid treatment (oral and intravenous) due to any disease; 21. Patients taking vitamin C and aspirin with daily dose greater than 60 mg; 22. Honeymoon patients with type 1 diabetes; 23. Patients known to be allergic to medical grade glue; 24. Where the researchers believed that the participants had other important diseases that were not suitable for the study. |
| Interventions |
Intervention: glargine Comparator: degludec Duration of the intervention: unclear |
| Outcomes |
Primary outcome(s): 24‐h mean glucose levels (SD, coefficient of variation), mean (largest) amplitude of glycaemic excursions, mean of daily difference, time in hypoglycaemia (< 2.8/3.9 mmol/L) during a 24‐h period, time in hyperglycaemia (> 7.8/10.0/13.9 mmol/L) during a 24‐h period; Secondary outcome(s): HbA1c, insulin dose, nocturnal hypoglycaemia, self‐perceived satisfaction rating scale Other outcome(s): — Relevant proposed outcome measures for SoF table: HbA1c, nocturnal hypoglycaemia |
| Reason for awaiting classification | No publication available. Unclear duration of intervention/follow‐up |
| Study details |
Study identifier: ChiCTR2000032703 Study start date: May 2020 Study completion date: unclear Responsible party/principal investigator: Kuang Hongyu, The First Affiliated Hospital of Harbin Medical University, 23 Post Street, Nangang District, Harbin, Heilongjiang, China |
| Official title and purpose of study | Comparision of insulin degludec and insulin glargine on blood glucose variability in northern Chinese patients with type 1 diabetes Quote: "To compare blood glucose variability in northern Chinese patients with type 1 diabetes treated with insulin glargine (IGla) versus insulin degludec (IDeg) using flash glucose monitoring (FGM)" |
| Notes |
EudraCT 2007‐004144‐74.
| Methods |
Allocation: randomised Intervention model: parallel‐group assignment Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 97 Inclusion criteria: T1DM, female, 13‐20 years, diagnosed over 1 year or C‐peptide negative, postmenarchal or in late puberty, HbA1c < 12%, BMI less than or equal to +2.5 for age, no active or untreated concurrent disease Exclusion criteria: non‐T1DM including those secondary to an existing pathology, any other physical or psychological disease likely to interfere with the normal conduct of the study and interpretation of the results, pregnant or breastfeeding women, females of reproductive age who are unwilling to take appropriate measures of contraception, taking medication likely to affect glucose metabolism |
| Interventions |
Intervention: glargine Comparator: detemir Duration of the intervention: 1 year according to protocol, but study ended prematurely, therefore, unknown how long the study duration was |
| Outcomes |
Primary outcome(s): BMI Secondary outcome(s): (results available for adverse events at EudraCT) Other outcome(s): — Relevant proposed outcome measures for SoF table: serious adverse events |
| Reason for awaiting classification | The trial is listed as prematurely ended, but duration of trial unknown |
| Study details |
Study identifier: EudraCT 2007‐004144‐74; ISRCTN49492872 Study start date: October 2007 Study completion date: December 2016 Responsible party/principal investigator: David Dunger, University of Cambridge, United Kingdom |
| Official title and purpose of study | A comparison of the effects of insulin detemir with insulin glargine on weight gain in female adolescents and young adults with Type 1 Diabetes (T1D) on a basal bolus regimen Quote: "To explore the hypothesis that use of insulin Detemir vs. insulin Glargine will lead to reduced weight gain in young women with Type 1 Diabetes" |
| Notes | ISRCTN49492872; EudraCT 2007‐004144‐74 Results are available on https://www.clinicaltrialsregister.eu/ctr-search/trial/2007-004144-74/results |
EudraCT 2009‐012317‐22.
| Methods |
Allocation: randomised Intervention model: parallel‐group assignment Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 16 Inclusion criteria: aged >= 6 and < 11 years; HbA1c < 7.5%; basal C‐peptide < 0.1 nmol/L Exclusion criteria: clinical signs of puberty illness associated with T1DM, using any drug except insulin, clinically relevant microalbuminuria, non‐availability of blood samples |
| Interventions |
Intervention(s): glargine Comparator(s): detemir Duration of the intervention: 1 year (not explicitly stated ‐ could also be 4 months) |
| Outcomes |
Primary outcome(s): GH and IGF‐1 levels Secondary outcome(s): — Other outcome(s): — Relevant proposed outcome measures for SoF table: — |
| Reason for awaiting classification | Marked as 'completed' in EU Clinical Trial Register but no publication available |
| Study details |
Study identifier: EudraCT 2009‐012317‐22 Study start date: June 2009 Study completion date: — (listed as completed) Responsible party/principal investigator: GM Lancise, Azienda Ospedaliero Universitaria Ospedali Ruinti Umberte, Italy |
| Official title and purpose of study | Pediatric basal bolus therapy ‐ Basal‐bolus regimen in the treatment of children with type 1 diabetes Quote: "...to study the difference of GH/IGF1 axis in children treated with glargine or detemir" |
| Notes | Primary investigator contacted. No reply |
INEOX.
| Methods |
Allocation: randomised Intervention model: parallel‐group assignment Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 300 Inclusion criteria: 18 to 65 years; T1DM of more than two years; HbA1c ≤ 10%; intensive treatment with basal multiple doses of insulin Exclusion criteria: chronic kidney disease, liver disease, thyroid dysfunction (except hypothyroidism correctly treated and controlled); pregnancy or pregnancy planning; T2DM; hyperuricaemia |
| Interventions |
Intervention(s): degludec Comparator(s): glargine Duration of the intervention: 6 months |
| Outcomes |
Primary outcome(s): oxidative stress markers Secondary outcome(s): glycaemic measures, hypoglycaemia, ketosis, quality of life, treatment satisfaction Other outcome(s): — Relevant proposed outcome measures for SoF table: hypoglycaemia, health‐related quality of life |
| Reason for awaiting classification | Marked as expected to be completed December 2019 in ClinicalTrials.gov. No data available |
| Study details |
Study identifier: FIM‐EOX‐2016‐01; EudraCT 2016‐002915‐17; NCT03328845 Study start date: January 2017 Study completion date: December 2019 Responsible party/principal investigator: Maria Soledad Ruiz de Adana, Regional University Hospital of Málaga, Spain |
| Official title and purpose of study | Impact on the oxidative stress of the different analogues of insulin in people with type 1 diabetes (Ineox Study) (INEOX) Quote: "This study evaluates in a group of people with DM 1 the influence in parameters of oxidative stress of the treatments with the different current analogs of insulin" |
| Notes |
IRCT201203079224N1.
| Methods |
Allocation: randomised Intervention model: parallel‐group assignment Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 40 Inclusion criteria: age 6 to 11 years; T1DM under treatment of insulin at least 6 months; BMI below the 90th percentile at baseline and having the desire and ability to measure blood glucose self‐monitoring using glucometer devices Exclusion criteria: mental and physical disorders; patients who did not complete the study period and patients with diabetes who were not suitable for regular tracking and checking |
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of the intervention: 6 months |
| Outcomes |
Primary outcome(s): fasting blood glucose, HbA1c, lipid profile Secondary outcome(s): — Other outcome(s): — Relevant proposed outcome measures for SoF table: none |
| Reason for awaiting classification | Marked as 'completed' in Clinicaltrials.gov but no publication available |
| Study details |
Study identifier: IRCT201203079224N1 Study start date: May 2012 Study completion date: not reported, but marked as complete Responsible party/principal investigator: Dr. Aria Setoodeh, Tehran University of Medical Sciences |
| Official title and purpose of study | Insulin glargine + insulin aspart vs NPH insulin + regular insulin for people with type 1 diabetes |
| Notes | Primary investigator contacted. No reply |
J‐Collection.
| Methods |
Allocation: randomised Intervention model: cross‐over Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 20 Inclusion criteria: 18 years or more, T1DM, receiving basal‐bolus insulin therapy Exclusion criteria: T2DM |
| Interventions |
Intervention(s): glargine Comparator(s): detemir Duration of the intervention: unknown |
| Outcomes |
Primary outcome(s): continuous glucose value of 24 hours by CGM Secondary outcome(s): — Other outcome(s): — Relevant proposed outcome measures for SoF table: none |
| Reason for awaiting classification | Marked as 'completed' in UMIN‐CTR Clinical Trial but no publication available |
| Study details |
Study identifier: UMIN000001402 Study start date: May 2008 Study completion date: December 2012 Responsible party/principal investigator: Daisuke Tsujino, The Jikei University School of Medicine, Division of Diabetes, Metabolism and Endocrinology, Department of Internal Medicine, Japan |
| Official title and purpose of study | Quote: "We compare glucose control of Detemir to Glargine in Japanese patient with type 1 diabetes". |
| Notes |
Mianowska 2007.
| Methods |
Allocation: randomised Intervention model: cross‐over assignment Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 14 Inclusion criteria: T1DM, 6 to 12 years, unsatisfactory glycaemic control defined as the presence of at least one of the following: (i) mean HbA1c from the preceding 6 months > 7.5% or (ii) large daily blood glucose excursions (from < 3.1 mmol/L to > 13.9 mmol/L) or (iii) strong dawn phenomenon (without an extra insulin injection at 3.00‐4.00 a.m. and most blood glucose measurements before breakfast > 8.9 mmol/L) Exclusion criteria: inadequate results of baseline laboratory tests and clinical remission (total daily insulin dose < 0.3 U/kg/day with HbA1c < 6.5%) |
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of the intervention: 6 months |
| Outcomes |
Primary outcome(s): — Secondary outcome(s): — Other outcome(s): HbA1c, hypoglycaemia, ketoacidosis, glucose, weight, insulin dose Relevant proposed outcome measures for SoF table: hypoglycaemia |
| Reason for awaiting classification | Study was published ‐ no data before cross‐over reported |
| Study details |
Study identifier: — Study start date: — Study completion date: — (but publication from 2007) Responsible party/principal investigator: Dr. Mianowska, Klinika Chorób Dzieci, Katedry Pediatrii UM, Poland |
| Official title and purpose of study | Quote: "The aim of this prospective cross‐over study was to compare glycemic control on NPH insulin (NPH) and on glargine in unsatisfactorily controlled type 1 diabetic prepubertal children." |
| Notes | No severe hypoglycaemia or ketoacidosis occurred during the trial. |
NCT00564018.
| Methods |
Allocation: randomised Intervention model: parallel‐group assignment Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 33 Inclusion criteria: newly diagnosed T1DM within 1 week of diagnosis; age 6 to 18 years Exclusion criteria: actual treatment with oral drugs influencing beta cell function or blood glucose levels (e.g. oral hypoglycaemic agents); actual treatment with drugs influencing insulin sensitivity (e.g. metformin or systemic steroids); significant concomitant disease likely to interfere with glucose metabolism (children with active bacterial infections at the time of diagnosis must be cured prior to entry); expected poor compliance; pregnancy; any other condition that by the judgement of the investigator may be potentially harmful to the patients |
| Interventions |
Intervention(s): detemir Comparator (1): glargine Comparator (2): NPH Duration of the intervention: planned to 1 year (but terminated early ‐ unknown when) |
| Outcomes |
Primary outcome(s): C‐peptide Secondary outcome(s): HbA1c Other outcome(s): adverse events Relevant proposed outcome measures for SoF table: serious adverse events |
| Reason for awaiting classification | Marked as terminated early ‐ the duration of the trial was not reported prior to termination |
| Study details |
Study identifier: NCT00564018; UTSW‐052006‐056 Study start date: September 2006 Study completion date: April 2011 Responsible party/principal investigator: Soumya Adhikari, University of Texas Southwestern Medical Center, USA |
| Official title and purpose of study | Quote: "To determine whether using a long‐acting insulin analog at the time of diagnosis, instead of intermediate‐acting insulin, affects the rate of loss of the body's ability to make insulin in children with newly diagnosed type 1 diabetes." |
| Notes |
Sherif 2014.
| Methods |
Allocation:randomised Intervention model: parallel‐group assignment Masking:open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM Estimated number of participants: 100 Inclusion criteria: T1DM, age 3 to 8 years Exclusion criteria: — |
| Interventions |
Intervention(s): glargine Comparator(s): NPH Duration of the intervention: 6 months |
| Outcomes |
Primary outcome(s): — Secondary outcome(s): — Other outcome(s): glycaemic control, frequency of hypoglycaemia, quality of life and serum level of C‐reactive protein as an inflammatory marker Relevant proposed outcome measures for SoF table: hypoglycaemia, health‐related quality of life |
| Reason for awaiting classification | Abstract of trial available from ISPAD 2014 conference. No full text identified |
| Study details |
Study identifier: — Study start date: — Study completion date: — Responsible party/principal investigator: — (first author of abstract is EM Sherif, Ain Shams University, Pediatric Department, Cairo, Egypt) |
| Official title and purpose of study | Quote: "To compare the efficacy and safety of insulin glargine with NPH insulin in children with type 1 diabetes mellitus (T1DM) below years old regarding glycemic control, frequency of hypoglycemia, quality of life and serum level of hsC‐reactive protein (C‐RP) as an inflammatory marker" |
| Notes | No contact information could be retrieved. Published as conference abstract. Performed in Egypt. Reported in abstract that quality of life improved in all children receiving insulin glargine but not with NPH insulin (no other data provided). Frequency of severe and nocturnal hypoglycaemia was lower with insulin glargine (no other data provided). HbA1c at the end of the study was 6.6% (SD 0.5) for the insulin glargine group versus 7.4% (SD 0.7) for the NPH insulin group |
UMIN000020521.
| Methods |
Allocation: randomised Intervention model: parallel‐group Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: diabetes (not specified if T1DM or T2DM) Estimated number of participants: 100 Inclusion criteria: T1DM, HbA1c more than 8.0%, already using insulin Exclusion criteria: hypoglycaemic risk, serious heart trouble, severe hepatic dysfunction, severe renal dysfunction, internal secretion disease, steroids |
| Interventions |
Intervention(s): degludec Comparator(1): glargine Comparator (2): continuing basal insulin treatment Duration of the intervention: 24 weeks |
| Outcomes |
Primary outcome(s): change in HbA1c, body weight, the custom‐built Diabetes Treatment Satisfaction questionnaire result and adverse events including hypoglycaemia Secondary outcome(s): achievement rate of HbA1c < 7.0% and < 8.0%; fasting blood glucose; glycaemic variability by SMBG; change in insulin dose Other outcome(s): — Relevant proposed outcome measures for SoF table: hypoglycaemia, adverse events |
| Reason for awaiting classification | Marked as 'completed' in UMIN000020521 but no publication or results available |
| Study details |
Study identifier: UMIN000020521 Study start date: January 2016 Study completion date: July 2019 Responsible party/principal investigator: Koichiro Yasuda, Osaka Saiseikai Noe Hospital, Japan |
| Official title and purpose of study | The efficacy and the safety of the new long‐acting insulin in patient with diabetes Quote: "To compare a new long‐acting insulin with existing diabetes therapeutic drug for efficacy and safety in diabetes" |
| Notes |
UMIN000021046.
| Methods |
Allocation: cluster‐randomised Intervention model: parallel‐group assignment Masking: open‐label Primary purpose: treatment |
| Participants |
Condition: T1DM and T2DM Estimated number of participants: 200 Inclusion criteria: 20 years or more, diabetes receiving basal‐bolus insulin therapy in outpatients for > 4 months prior to screening; if T2DM then a duration of a disease more than 12 months; available for self‐monitoring of blood glucose Exclusion criteria: hypersensitivity to insulin; severe ketosis, diabetic coma or formerly comatose; severe renal dysfunction including patients needing haemodialysis or peritoneal dialysis; pre or proliferative retinopathy, including vitreous haemorrhage risk; serious infection; perioperative period; serious trauma; pregnancy or possible pregnancy |
| Interventions |
Intervention(s): degludec Comparator(s): another long acting insulin analogue Duration of the intervention: 24 weeks |
| Outcomes |
Primary outcome(s): change in HbA1c Secondary outcome(s): hypoglycaemia, glucose levels, diabetes treatment satisfaction Other outcome(s): — Relevant proposed outcome measures for SoF table: hypoglycaemia |
| Reason for awaiting classification | Marked as 'completed' in UMIN‐CTR Clinical Trial but no publication available |
| Study details |
Study identifier: UMIN000021046 Study start date: April 2013 Study completion date: February 2015 Responsible party/principal investigator: Tomoyasu Fukui, Department of Medicine, Division of Diabetes, Metabolism and Endocrinology, Showa University School of Medicine, Japan |
| Official title and purpose of study | Showa University examines the effects of insulin degludec Quote: "To compare glucose lowering effect of insulin degludec to conventional basal insulin analogue in Japanese patients with type 1 and type 2 diabetes in basal‐bolus treatment" |
| Notes | Combines people with T1DM and T2DM ‐ unknown if separate data might be available. Not specified which other long‐acting insulin analogue was applied in the comparator arm |
— denotes not reported
a.m.: ante meridiem BAS: basal analog study BMI: body mass index CGM: continuous glucose monitoring CGMS: continuous glucose monitoring system DM: diabetes mellitus EudraCT: European Union Drug Regulating Authorities Clinical Trials Database FGM: flash glucose monitoring GAD: glutamic acid decarboxylase GH: growth hormone HbA1c: glycosylated haemoglobin A1c HypoDeg: insulin degludec and symptomatic nocturnal hypoglycaemia study IA2: islet tyrosine phosphatase 2 IDeg: insulin degludec IGF‐1: insulin‐Like Growth Factor 1 IGla: insulin glargine INEOX: impact on the oxidative stress of the different analogues of insulin in people with type 1 diabetes study ISPAD: International Society for Pediatric and Adolescent Diabetes NPH: neutral protamine Hagedorn NYHA: New York Heart Association pH: potentia hydrogenii QT: time from the start of the Q wave to the end of the T wave (recorded by electrocardiogram) SD: standard deviation SMBG: self‐monitoring of blood glucose SoF: Summary of Findings T1DM: type 1 diabetes mellitus T2DM: type 2 diabetes mellitus USA: United States of America
Differences between protocol and review
In addition to the databases mentioned in the protocol, we searched the Health Technology Assessment (HTA) database, which became available in the meantime.
Because of scarce data, we changed the following outcome measures in the 'Summary of findings' tables.
Instead of end‐stage renal disease, we used severe nocturnal hypoglycaemia.
Instead of combined glycosylated haemoglobin A1c (HbA1c) with severe hypoglycaemia, we used HbA1c only.
We renamed the outcome 'serious/severe hypoglycaemia' to 'severe hypoglycaemia' because this term was mainly used in the publications and clinical study reports. For the same reason, we renamed the outcome 'HbA1c combined with serious/severe hypoglycaemia' to 'HbA1c combined with severe hypoglycaemia'.
In addition to the outcome measure 'non‐serious adverse events', we analysed 'withdrawals due to adverse events' because this outcome was detailed in the clinical study reports.
In addition to the outcome measure 'severe hypoglycaemia', we analysed 'hypoglycaemia reported as a serious adverse event' because this outcome was detailed in the clinical study reports and is the hardest clinical endpoint with regard to hypoglycaemic episodes.
We additionally evaluated the subgroup adults versus children because appropriate data were available and it appeared to be important to report this information for consumers and decision makers.
Contributions of authors
All review authors read and approved the final review draft.
BH: protocol and review draft, data interpretation and review of drafts, contact with pharmaceutical companies and investigators, study selection, data extraction, data analysis, data interpretation, future review updates
MIM: search strategy development, performed electronic searches, searched regulatory agencies web pages, review of drafts
BR: protocol and review draft, study selection, data analysis, data interpretation and review of drafts, future review updates
Sources of support
Internal sources
No sources of support supplied
External sources
-
Health Action International's ACCISS Study, Netherlands
Health Action International’s ACCISS Study was started in 2015 to identify and address the inequities and inefficiencies in the global insulin market. Health Action International is a not‐for‐profit foundation, based in Amsterdam The Netherlands, committed to advancing access to medicines globally.
Declarations of interest
BH: this review was funded by The Leona M. and Harry B. Helmsley Charitable Trust as part of the Addressing the Challenge and Constraints of Insulin Sources and Supply (ACCISS) Study. Statements and conclusions presented in this report are those of the authors alone and do not necessarily reflect the views of the Helmsley Charitable Trust. All references and conclusions are intended for educational and informative purposes and do not constitute an endorsement or recommendation from the Helmsley Charitable Trust.
BR: none known.
MIM: none known.
Edited (no change to conclusions)
References
References to studies included in this review
Bartley 2008 {published and unpublished data}
- Bartley PC, Bogoev M, Larsen J, Philotheou A. Long-term efficacy and safety of insulin detemir compared to neutral protamine Hagedorn insulin in patients with type 1 diabetes using a treat-to-target basal-bolus regimen with insulin aspart at meals: a 2-year, randomized, controlled trial. Diabetic Medicine 2008;25(4):442-9. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwend MH, Aagren M, Valentine WJ. Cost-effectiveness of insulin detemir compared with neutral protamine Hagedorn insulin in patients with type 1 diabetes using a basal-bolus regimen in five European countries. Journal of Medical Economics 2009;12(2):114-23. [PMID: ] [DOI] [PubMed] [Google Scholar]
- NCT00184665. Comparison of insulin detemir with NPH insulin in type 1 diabetes. clinicaltrials.gov/ct2/show/NCT00184665 (first received 6 February 2020).
- Novo Nordisk. Clinical trial report trial ID: NN304-1595. www.novonordisk-trials.com/studie/522 (accessed 24 February 2020).
- Novo Nordisk. Trial synopsis NN304-1595. www.novonordisk-trials.com/studie/522 (accessed 3 March 2020).
- Tunis SL, Minshall ME, Conner C, McCormick JI, Kapor J, Yale JF, et al. Cost-effectiveness of insulin detemir compared to NPH insulin for type 1 and type 2 diabetes mellitus in the Canadian payer setting: modeling analysis. Current Medical Research and Opinion 2009;25(5):1273-84. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Valentine WJ, Aagren M, Haglund M, Ericsson A, Gschwend MH. Evaluation of the long-term cost-effectiveness of insulin detemir compared with neutral protamine Hagedorn insulin in patients with type 1 diabetes using a basal-bolus regimen in Sweden. Scandinavian Journal of Public Health 2011;39(1):79-87. [PMID: ] [DOI] [PubMed] [Google Scholar]
BEGIN Basal‐Bolus Type 1 {published and unpublished data}
- 2008-005774-13/GB. A 52 week randomised, controlled, open label, multicentre, multinational, parallel, treat-to-target trial comparing efficacy and safety of SIBA and insulin glargine both administered once daily in a basal-bolus regimen with insulin aspart as mealtime insulin in subjects with type 1 diabetes. clinicaltrialsregister.eu/ctr-search/trial/2008-005774-13/GB (first received 10 February 2020).
- Bode BW, Buse JB, Fisher M, Garg SK, Marre M, Merker L, et al. Insulin degludec improves glycaemic control with lower nocturnal hypoglycaemia risk than insulin glargine in basal–bolus treatment with mealtime insulin aspart in Type 1 diabetes (BEGIN(®) Basal–Bolus Type 1): 2-year results of a randomized clinical trial. Diabetic Medicine 2013;30(11):1293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode BW, Heller S, Hansen CT, Rana A, Russell-Jones DL. Insulin degludec improves glycemic control with lower nocturnal hypoglycemia risk than insulin glargine: a 2-year randomized trial in type 1 diabetes. Canadian Journal of Diabetes 2012;36(5 Suppl 1):S57-8. [Google Scholar]
- Evans M, Wolden M, Gundgaard J, Chubb B, Christensen T. Cost-effectiveness of insulin degludec compared with insulin glargine in a basal-bolus regimen in patients with type 1 diabetes mellitus in the UK. Journal of Medical Economics 2015;18(1):56-68. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Heller S, Buse J, Fisher M, Garg S, Marre M, Merker L, et al. Insulin degludec, an ultra-long acting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Bolus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet 2012;379(9825):1489-97. [DOI] [PubMed] [Google Scholar]
- Heller S, Francisco AMO, Pei H, Russell-Jones D. Basal–bolus therapy with insulin degludec improves long-term glycaemic control with less nocturnal hypoglycaemia compared with insulin glargine in type 1 diabetes: results of a one year trial. Diabetic Medicine 2012;29:23-5. [Google Scholar]
- Heller S, Francisco AMO, Pei HL, Russell-Jones D. Insulin degludec improves long-term glycemic control with less nocturnal hypoglycemia compared with insulin glargine: 1-year results from a randomized basal-bolus trial in type 1 diabetes. Diabetes 2011;60:A19. [Google Scholar]
- Kerlan V, Gouet D, Marre M, Renard E. Use of insulin degludec, a new basal insulin with an ultra-long duration of action, in basal-bolus therapy in type 1 and type 2 diabetes. Annales d'Endocrinologie 2013;74(5-6):487-90. [DOI] [PubMed] [Google Scholar]
- NCT00982228. Comparison of NN1250 plus insulin aspart with insulin glargine plus insulin aspart in type 1 diabetes (BEGIN™). clinicaltrials.gov/ct2/show/NCT00982228 (first received 15 January 2020).
- Novo Nordisk. Clinical trial report. www.novonordisk-trials.com/studie/309 (accessed 3 March 2020).
- Novo Nordisk. Synopsis NN1250-3583. www.novonordisk-trials.com/studie/309 (accessed 3 March 2020).
- Russell-Jones DL, Francisco AO, Pei H, Heller SR. Basal-bolus therapy with insulin degludec improves long-term glycaemic control with less nocturnal hypoglycaemia compared with insulin glargine in type 1 diabetes: results of a 1-year trial. Diabetologia 2011;54 Suppl 1:S1-543. [Google Scholar]
- Russell-Jones DL, Heller S, Hansen CT, Chang D, Bode B. A two year randomised trial: improved glycaemic control and lower risk of nocturnal hypoglycaemia with insulin degludec compared with insulin glargine in type 1 diabetes. Diabetic Medicine 2013;30:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestakova MV, Antciferov MB, Mayorov AY, Ruyatkina LA, Suplotova LA, Dogadin SA, et al. Insulin degludec: a new basal insulin analogue with an ultra-long duration of action. Safety and efficacy in Russian patients with diabetes. Diabetes Mellitus 2015;18(4):130-41. [Google Scholar]
- Siegmund T, Westrup D. Insulin degludec vs. insulin glargine 100 U/ml in diabetes: 2-year results. Diabetes, Stoffwechsel und Herz 2017;26(1):21-8. [Google Scholar]
BEGIN Flex T1 {published and unpublished data}
- 2009-012923-27. Begin™ Flex T1. A 26-week trial investigating the dosing flexibility, efficacy and safety of NN1250 in subjects with type 1 diabetes with a 26-week extension. www.clinicaltrialsregister.eu/ctr-search/search?query=2009-012923-27 (first received).
- Danne T, Bolinder J. New insulins and insulin therapy. Diabetes Technology & Therapeutics 2014;16(Suppl 1):S34-43. [DOI] [PubMed] [Google Scholar]
- EUCTR2009-012923-27-NO. Begin™ Flex T1. A 26-week trial investigating the dosing flexibility, efficacy and safety of NN1250 in subjects with type 1 diabetes with a 26-week extension. apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2009-012923-27-NO (first received).
- Evans M, Wolden M, Gundgaard J, Chubb B, Christensen T. Cost-effectiveness of insulin degludec compared with insulin glargine in a basal-bolus regimen in patients with type 1 diabetes mellitus in the UK. Journal of Medical Economics 2015;18(1):56-68. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Mathieu C, Hollander P, Miranda-Palma B, Cooper J, Franek E, Russell-Jones D. Efficacy and safety of insulin degludec in a flexible dosing regimen vs insulin glargine in patients with type 1 diabetes (BEGIN: Flex T1): a 26-week randomized, treat-to-target trial with a 26-week extension. Journal of Clinical Endocrinology and Metabolism 2013;98(3):1154-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01079234. Comparison of NN1250 with insulin glargine in type 1 diabetes (BEGIN™). clinicaltrials.gov/ct2/show/NCT01079234 (first received 21January 2020).
- Novo Nordisk. Clinical trial report. Trial ID: NN1250-3770. www.novonordisk-trials.com/studie/310 (accessed 3 March 2020).
- Novo Nordisk. Synopsis trial NN1250-3770. www.novonordisk-trials.com/studie/310 (accessed 3 March 2020).
BEGIN Young {published and unpublished data}
- 2011-003148-39. A 26-week, multinational, multi-centre, open-labelled, randomised, parallel, efficacy and safety comparison of insulin degludec and insulin detemir in children and adolescents 1 to less than 18 years with type 1 diabetes mellitus on a basal-bolus regimen with insulin aspart as bolus insulin, followed by a 26-week extension investigating long term safety. clinicaltrialsregister.eu/ctr-search/trial/2011-003148-39/results (first received 28 July 2015 ).
- NCT01513473. A trial investigating the efficacy and safety of insulin degludec in children and adolescents with type 1 diabetes mellitus (BEGIN™). clinicaltrials.gov/ct2/show/NCT01513473 (first received 20 January 2012).
- Novo Nordisk. Clinical trial report trial ID NN1250-3561. www.novonordisk-trials.com/studie/440 (accessed 29 February 2020).
- Novo Nordisk. Synopsis trial ID NN1250-3561. www.novonordisk-trials.com/studie/440 (accessed 29 February 2020).
- Thalange N, Deeb L, Iotova V, Kawamura T, Klingensmith G, Philotheou A, et al. Insulin degludec in combination with bolus insulin aspart is safe and effective in children and adolescents with type 1 diabetes. Pediatric Diabetes 2015;16(3):164-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalange N, Deeb L, Iotova V, Kawamura T, Silverstein J, Francisco AM, et al. Safety and efficacy of insulin degludec in children and adolescents with type 1 diabetes. Diabetic Medicine 2015;32:67-8. [Google Scholar]
- Thalange N, Deeb L, Klingensmith G, Franco D, Hanas R, Bardtrum L, et al. The incidence of hyperglycemia and ketosis with insulin degludec-based treatment compared with insulin detemir in pediatric patients with type 1 diabetes: an analysis of data from two randomized trials. Pediatric Diabetes 2016;17:36-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalange N, Deeb L, Klingensmith G, Franco DR, Bardtrum L, Tutkunkardas D, et al. The rate of hyperglycemia and ketosis with insulin degludec-based treatment compared with insulin detemir in pediatric patients with type 1 diabetes: an analysis of data from two randomized trials. Pediatric Diabetes 2019;20(3):314-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalange N, Deeb LC, Iotova V, Kawamura T, Klingensmith G, Philotheou A, et al. Long-term efficacy and safety of insulin degludec (IDeg) in combination with bolus insulin aspart (IAsp) in children and adolescents with type 1 diabetes (T1D). Pediatric Diabetes 2014;15:16-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalange N, Gundgaard J, Parekh W, Tutkunkardas D. Cost analysis of insulin degludec in comparison with insulin detemir in treatment of children and adolescents with type 1 diabetes in the UK. BMJ Open Diabetes Research & Care 2019;7(1):e000664. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalange N, Hakan-Bloch J, Parekh W. Cost analysis of insulin degludec (IDeg) in comparison with insulin detemir (IDet) in treatment of children and adolescents with type 1 diabetes (T1D) in the UK. Pediatric Diabetes 2017;18 Suppl 25:47-137. [Google Scholar]
Bolli 2009 {published data only}
- Bolli GB, Songini M, Trovati M, Del Prato S, Ghirlanda G, Cordera R, et al. Lower fasting blood glucose, glucose variability and nocturnal hypoglycaemia with glargine vs NPH basal insulin in subjects with type 1 diabetes. Nutrition, Metabolism, and Cardiovascular Diseases 2009;19(8):571-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Chase 2008 {published and unpublished data}
- Chase HP, Arslanian S, White NH, Tamborlane WV. Insulin glargine versus intermediate-acting insulin as the basal component of multiple daily injection regimens for adolescents with type 1 diabetes mellitus. Journal of Pediatrics 2008;153(4):547-53. [DOI] [PubMed] [Google Scholar]
- NCT00046501. Compare blood sugar level between Lantus in the morning and other insulins in type 1 diabetes adolescents. clinicaltrials.gov/ct2/show/NCT00046501 (first received 1 February 2020).
- Sanofi Aventis. Clinical study report - HOE901/4030 [personal communication]. Conversation with Sanofi Aventis 21 February 2020.
- Sanofi Aventis. Study number HOE901/4030. www.sanofi.com/en/science-and-innovation/clinical-trials-and-results/our-disclosure-commitments/pharma/-/media/Project/One-Sanofi-Web/Websites/Global/Sanofi-COM/Home/common/docs/clinical-study-results/HOE901_4030_summary.pdf (accessed 21 February 2020).
- White NH, Chase HP, Arslanian S, Tamborlane WV, Study Group. Comparison of glycemic variability associated with insulin glargine and intermediate-acting insulin when used as the basal component of multiple daily injections for adolescents with type 1 diabetes. Diabetes Care 2009;32(3):387-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Davies 2014 {published and unpublished data}
- 1358&EncHid=&modid=&compid=%27, %271358det%27. Comparison of NN1250 plus insulin aspart with insulin detemir plus insulin aspart in type 1 diabetes. ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=1358&EncHid=&modid=&compid=%27,%271358det%27 (first received 10 February 2020).
- 1906&EncHid=&modid=&compid=%27, %271906det%27. Comparison of NN1250 plus insulin aspart with insulin detemir plus insulin aspart in type 1 diabetes: an extension trial to NN1250-3585. ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=1906&EncHid=&modid=&compid=%27,%271906det%27 (first received 10 February 2020).
- 2009-011672-29. A trial investigating the efficacy and safety of NN1250 compared to insulin detemir in subjects with type 1 diabetes mellitus in a basal/bolus treatment regimen. www.clinicaltrialsregister.eu/ctr-search/search?query=2009-011672-29 (first received 20 February 2020).
- Davies M, Sasaki T, Gross JL, Bantwal G, Ono Y, Nishida T, et al. Comparison of insulin degludec with insulin detemir in type 1 diabetes: a 1-year treat-to-target trial. Diabetes, Obesity & Metabolism 2016;18(1):96-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Davies MJ, Gross JL, Ono Y, Sasaki T, Bantwal G, Gall MA, et al. Efficacy and safety of insulin degludec given as part of basal-bolus treatment with mealtime insulin aspart in type 1 diabetes: a 26-week randomized, open-label, treat-to-target non-inferiority trial. Diabetes, Obesity & Metabolism 2014;16(10):922-30. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01074268. Comparison of NN1250 plus insulin aspart with insulin detemir plus insulin aspart in type 1 diabetes (BEGIN™). clinicaltrials.gov/ct2/show/NCT01074268 (first received 21 January 2020).
- Novo Nordisk. Clinical trial report trial ID NN1250-3585. www.novonordisk-trials.com/studie/446 (accessed 3 March 2020).
- Novo Nordisk. Synopsis trial NN1250-3585. www.novonordisk-trials.com/studie/446 (accessed 3 March 2020).
- Ono Y, Nishida T, Hyllested-Winge J, Seino H, Sasaki T. A comparison of IDeg + IAsp versus IDet + IAsp in subjects with type 1 diabetes: subgroup analysis of Japanese subjects. Diabetology International 2016;7(4):404-12. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fulcher 2005 {published and unpublished data}
- Aventis Pharma. Clinical study report - HOE901/4010 [personal communication]. Provided by Sanofi Aventis 31 January 2020.
- Fulcher GR, Gilbert RE, Yue DK. Glargine is superior to neutral protamine Hagedorn for improving glycated haemoglobin and fasting blood glucose levels during intensive insulin therapy. Internal Medicine Journal 2005;35(9):536-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Fulcher GR, Yue DK, Gilbert RE. Insulin glargine vs. NPH insulin in patients with type 1 diabetes: the effects of intensive insulin therapy on glycaemic control, hypoglycaemia and quality of life. Diabetologia 2002;45 (Suppl 2):A258. [Google Scholar]
Heller 2009 {published and unpublished data}
- 2004-000086-35/FI. A one-year, multi-national, open-labelled, parallel-group, 2:1 randomised treat-to-target trial comparing efficacy and safety of insulin detemir with insulin glargine using a basal-bolus regimen with insulin aspart as mealtime insulin in subjects with type 1 diabetes. www.clinicaltrialsregister.eu/ctr-search/trial/ (first received 10 February 2020).
- Heller S, Koenen C, Bode B. Comparison of insulin detemir and insulin glargine in a basal-bolus regimen, with insulin aspart as the mealtime insulin, in patients with type 1 diabetes: a 52-week, multinational, randomized, open-label, parallel-group, treat-to-target noninferiority trial. Clinical Therapeutics 2009;31(10):2086-97. [PMID: ] [DOI] [PubMed] [Google Scholar]
- NCT00095082. Efficacy and safety comparison of insulin detemir plus insulin aspart versus insulin glargine plus insulin aspart in type 1 diabetes. clinicaltrials.gov/ct2/show/NCT00095082 (first received 10 February 2020).
- Novo Nordisk. Clinical trial report NN 304-1430. s3.amazonaws.com/ctr-nvo-7271/NN304-1430/105a0bac-2d8a-4868-8963-5911cf45f823/5a60a40f-33f1-41ed-96b6-5f1a0107cdb6/1430-ctr-redacted-v1.pdf (accessed 3 April 2020).
- Novo Nordisk. Synopsis trial NN304-1430. www.novonordisk-trials.com/studie/512 (accessed 28 February 2020).
Home 2005 {published and unpublished data}
- Bradley C, Plowright R, Stewart J, Valentine J, Witthaus E. The diabetes treatment satisfaction questionnaire change version (DTSQc) evaluated in insulin glargine trials shows greater responsiveness to improvements than the original DTSQ. Health and Quality of Life Outcomes 2007;5:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoechst Marion Roussel. 28-week multicentre, controlled, randomized, open clinical trial comparing HOE 901 insulin with NPH human insulin in subjects with type I diabetes [personal communication]. Clinical study report HOE 901/3001 31 January 2020.
- Hoechst Marion Roussel. International report no. K1999CLN0073. Pharmacoeconomic data from a 28-week multicentre, controlled, randomized, open clinical trial comparing HOE 901 insulin with NPH human insulin in subjects with type 1 diabetes [personal communication]. Clinical Study Report 131 January 2020.
- Hoechst Marion Roussel. QoL study report No. F1998CLN0002 [personal communication]. Clinical Study Report 31 January 2020.
- Home P. A randomized, muIticentre trial of insulin glargine versus NPH insulin in people with type 1 diabetes. Diabetologia 2002;35 (Suppl 2):A258. [Google Scholar]
- Home PD, Rosskamp R, Forjanic-Klapproth J, Dressler A. A randomized multicentre trial of insulin glargine compared with NPH insulin in people with type 1 diabetes. Diabetes/Metabolism Research and Reviews 2005;21(6):545-53. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Witthaus E, Stewart J, Bradley C. Treatment satisfaction and psychological well-being with insulin glargine compared with NPH in patients with type 1 diabetes. Diabetic Medicine 2001;18(8):619-25. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kobayashi 2007 {published and unpublished data}
- Ishii H, Iwamoto Y, Kaku K, Kawamori R, Tajima N, Kobayashi M. Assessment of insulin detemir and NPH human insulin in Japanese subjects with diabetes on basal-bolus regimen. Diabetes 2007;56(Suppl 1):A170 (2805-PO). [Google Scholar]
- Kobayashi M, Iwamoto Y, Kaku K, Kawamori R, Tajima N. 48-week randomized multicenter open-label parallel group phase 3 trial to compare insulin detemir and NPH insulin efficacy and safety in subjects with insulin requiring diabetes mellitus in a basal-bolus regimen. T̄ōnyōbyō (Journal of the Japan Diabetes Society) 2007;50(9):649-63. [Google Scholar]
- Kobayashi M, Iwamoto Y, Kawamori R, Tajima N, Nishida T, Kaku K. Insulin detemir achieves lower, less variable fasting glucose and a reduced risk of nocturnal hypoglycaemia and weight gain compared to NPH insulin in basal bolus therapy of Japanese patients with type 1 diabetes. Diabetologia 2006;49(Suppl 1):A608-A609 [Abstract 995]. [Google Scholar]
- NCT00604344. Efficacy and the safety of Insulin detemir in subjects with insulin requiring diabetes. clinicaltrials.gov/ct2/show/NCT00604344 (first received 11 February 2020).
- Novo Nordisk. Clinical trial report NN304-1476 [personal communication]. Provided by Novo Nordisk (pages 232, 235, 236) 26 May 2020.
- Novo Nordisk. Synopsis. A 48-week, randomised, multi-centre, open-labelled, parallel-group trial to compare the efficacy and the safety of NN304 (insulin detemir) and NPH human insulin in subjects with insulin requiring diabetes mellitus on a basal-bolus regimen. www.novonordisk-trials.com/studie/517 (accessed 26 February 2020).
Liu 2016 {published and unpublished data}
- 2014-004640-35. A 24-week, randomized, open-label, parallel group, multicenter comparison of Lantus® (insulin glargine) given once daily versus neutral protamine Hagedorn (NPH) insulin in children with type 1 diabetes mellitus aged at least 6 years to less than 18 year. www.clinicaltrialsregister.eu/ctr-search/trial/2014-004640-35/results (first received 11 February 2020).
- Gong C, Liu M, Zhou Z, Yan J, Li P, Song W, et al. Efficacy and safety of once-daily insulin glargine in Chinese T1DM children aged between 6 to 17 years. Pediatric Diabetes 2015;16:50-150. [Google Scholar]
- Liu M, Zhou Z, Yan J, Li P, Song W, Fu J, et al. A randomised, open-label study of insulin glargine or neutral protamine Hagedorn insulin in Chinese paediatric patients with type 1 diabetes mellitus. BMC Endocrine Disorders 2016;16(1):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01223131. Efficacy and safety of insulin glargine versus. neutral protamine Hagedorn (NPH) insulin in children with type 1 diabetes above 6 years old. apps.who.int/trialsearch/Trial2.aspx?TrialID=NCT01223131 (first received 11 February 2020).
- NCT01223131. Efficacy and safety of insulin glargine versus neutral protamine Hagedorn (NPH) insulin in children with type 1 diabetes above 6 years old. clinicaltrials.gov/ct2/show/NCT01223131 (first received 11 February 2020).
- Sanofi Aventis. A 24-week, randomized, open-label, parallel group, multicenter comparison of Lantus® (insulin glargine) given once daily versus neutral protamine Hagedorn (NPH) insulin in children with type 1 diabetes mellitus aged at least 6 years to less than 18 years. www.sanofi.com/en/science-and-innovation/clinical-trials-and-results/our-disclosure-commitments/pharma/-/media/Project/One-Sanofi-Web/Websites/Global/Sanofi-COM/Home/common/docs/clinical-study-results/EFC11681_summary.pdf (accessed 11 February 2020).
- Sanofi Aventis. Clinical study report - Study EFC11681 [personal communication]. Provided by Sanofi Aventis 26 March 2020.
NCT00595374 {unpublished data only}
- NCT00595374. Efficacy and safety of Insulin detemir in type 1 diabetes. clinicaltrials.gov/ct2/show/NCT00595374 (first received 13 February 2020).
- Novo Nordisk. Synopsis trial ID NN304-1582. www.novonordisk-trials.com/studie/621 (accessed 13 February 2020).
NCT00605137 {unpublished data only}
- NCT00605137. Safety of insulin detemir in children with type 1 diabetes. www.clinicaltrials.gov/ct2/show/NCT00605137 (first received 11 February 2020).
- Novo Nordisk. Clinical trial report NN304-1604 [personal communication]. Provided by Novo Nordisk (pages 121, 122, 199, 200) 24 May 2020.
- Novo Nordisk. Protocol trial ID NN304-1604 [personal communication]. Provided by Novo Nordisk 24 May 2020.
- Novo Nordisk. Synopsis trial ID NN304-1604. www.novonordisk-trials.com/studie/624 (accessed 21 February 2020).
Pieber 2007 {published and unpublished data}
- Alcolado J, Poole CD, Peters JR, Currie CJ. Potential flaws and biases in a randomized controlled trial (RCT) of insulin detemir vs. insulin glargine by Pieber and colleagues. Diabetic Medicine 2008;25(1):115-6. [DOI] [PubMed] [Google Scholar]
- NCT00312104. Comparison of efficacy and safety of insulin detemir and insulin glargine in patients with type 1 diabetes. clinicaltrials.gov/ct2/show/NCT00312104 (first received 11 February 2020).
- Novo Nordisk. Clinical trial synopsis trial NN304-1372. s3.amazonaws.com/ctr-nvo-7271/NN304-1372/7ebd4cb1-935c-4a00-b514-215862cfe66e/3700973c-aa26-4ac0-8857-0e24d1355fe4/1372-ctr-redacted-v1.pdf (accessed 3 April 2020).
- Novo Nordisk. Synopsis trial NN 304-1372. www.novonordisk-trials.com/studie/508 (accessed 28 January 2020).
- Pieber TR, Treichel HC, Hompesch B, Philotheou A, Mordhorst L, Gall MA, et al. Comparison of insulin detemir and insulin glargine in subjects with type 1 diabetes using intensive insulin therapy. Diabetic Medicine 2007;24(6):635-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Valentine WJ, Palmer AJ, Erny-Albrecht KM, Ray JA, Cobden D, Foos V, et al. Cost-effectiveness of basal insulin from a US health system perspective: comparative analyses of detemir, glargine, and NPH. Advances in Therapy 2006;23(2):191-207. [PMID: ] [DOI] [PubMed] [Google Scholar]
Porcellati 2004 {published data only}
- Porcellati F, Rossetti P, Fanelli CG, Scionti L, Brunetti P, Bolli GB. Glargine vs NPH as basal insulin in intensive treatment of TIDM given lispro at meals: one year comparison. Diabetologia 2002;45 (Suppl. 2):A51. [Google Scholar]
- Porcellati F, Rossetti P, Pampanelli S, Fanelli CG, Torlone E, Scionti L, et al. Better long-term glycaemic control with the basal insulin glargine as compared with NPH in patients with type 1 diabetes mellitus given meal-time lispro insulin. Diabetic Medicine 2004;21(11):1213-20. [PMID: ] [DOI] [PubMed] [Google Scholar]
PRESCHOOL {published and unpublished data}
- %27, %271067det%27. A phase 3b trial, in type 1 diabetes mellitus, on children who are at least 1 year old to less than 6 years and are given either Lantus (insulin glargine) or neutral protamine Hagedorn (NPH) insulin. ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=1067&EncHid=&modid=&compid=%27,%271067det%27 (first received 10 February 2020).
- 114-09. A 24-week, randomized, open-label, parallel group multinational comparison of Lantus® (insulin glargine) given in the morning as once-a-day basal insulin versus neutral protamine Hagedorn (NPH) insulin, in children with type 1 diabetes mellitus aged at least 1 year to less than 6 years - PRESCHOOL (preschool children with type 1 diabetes on morning Lantus). www.ins.gob.pe/ensayosclinicos/rpec/recuperarECPBNuevoEN.asp?numec=114-09 (first received 11 February 2020).
- 2009-011231-12/DE. A 24-week, randomized, open-label, parallel group multinational comparison of Lantus® (insulin glargine) given in the morning as once-a-day basal insulin versus neutral protamine Hagedorn (NPH) insulin, in children with type 1 diabetes mellitus aged at least 1 year to less than 6 years. www.clinicaltrialsregister.eu/ctr-search/trial/2009-011231-12/DE (first received 10 February 2020).
- Danne T, Philotheou A, Goldman D, Guo X, Ping L, Cali A, et al. A randomized trial comparing the rate of hypoglycemia-assessed using continuous glucose monitoring in 125 preschool children with type 1 diabetes treated with insulin glargine or NPH insulin (the PRESCHOOL study). Pediatric Diabetes 2013;14(8):593-601 (corrigendum: Pediatric Diabetes 2015; 16(6):462). [DOI] [PubMed] [Google Scholar]
- NCT00993473. 6-month comparison of morning Lantus versus neutral protamine Hagedorn insulin in young children with type 1 diabetes (PRESCHOOL). clinicaltrials.gov/ct2/show/NCT00993473 (first received 10 February 2020).
- Sanofi Aventis. A 24-week, randomized, open-label, parallel group, multinational comparison of Lantus® (insulin glargine) given in the morning as once-a-day basal insulin versus neutral protamine Hagedorn (NPH) insulin, in children with type 1 diabetes mellitus aged at least 1 year to less than 6 years [personal communication]. Clinical study report - HOE901 / EFC11202 PRESCHOOL (provided by Sanofi Aventis) 14 February 2020.
Ratner 2000 {published and unpublished data}
- Hershon KS, Blevins TC, Mayo CA, Rosskamp R. Once-daily insulin glargine compared with twice-daily NPH insulin in patients with type 1 diabetes. Endocrine Practice 2004;10(1):10-7. [DOI] [PubMed] [Google Scholar]
- Hoechst Marion Roussel. Clinical Study Report No. K1998CLN0001 (HOE 901/3004) [personal communication]. Provided by Hoechst Marion Roussel 14 February 2020.
- Ratner RE, Hirsch IB, Neifing JL, Garg SK, Mecca TE, Wilson CA. Less hypoglycemia with insulin glargine in intensive insulin therapy for type 1 diabetes. U.S. study group of Insulin glargine in type 1 diabetes. Diabetes Care 2000;23(5):639-43. [PMID: ] [DOI] [PubMed] [Google Scholar]
Robertson 2007 {published and unpublished data}
- NCT00312156. Comparison of efficacy and safety of insulin detemir and NPH insulin in children and adolescents with type 1 diabetes. clinicaltrials.gov/ct2/show/NCT00312156 (first received 11 February 2020).
- Novo Nordisk. A 26-week, multinational, multi-centre, open-labelled, randomised, parallel efficacy and safety comparison of insulin detemir and NPH insulin in children and adolescents with type 1 diabetes on a basal-bolus regimen. www.novonordisk-trials.com/studie/291 (accessed 13 January 2020).
- Novo Nordisk. Clinical trial report NN304-1379. s3.amazonaws.com/ctr-nvo-7271/NN304-1379/9c6f472d-1d67-4309-b5cf-6b7412b007ae/e7d585e9-95db-487c-a7ef-4916166661ac/1379-ctr-nn-trials-redacted-v1.pdf (accessed 3 March 2020).
- Robertson KJ, Schoenle E, Gucev Z, Mordhorst L, Gall MA, Ludvigsson J. Insulin detemir compared with NPH insulin in children and adolescents with type 1 diabetes. Diabetic Medicine 2007;24(1):27-34. [DOI] [PubMed] [Google Scholar]
Russell‐Jones 2004 {published and unpublished data}
- NCT03220425. Evaluation of the efficacy and safety of insulin detemir compared with that of NPH insulin in subjects with type 1 diabetes. apps.who.int/trialsearch/Trial2.aspx?TrialID=NCT03220425 (first received 3 March 2020).
- NCT03220425. Evaluation of the efficacy and safety of insulin detemir compared with that of NPH insulin in subjects with type 1 diabetes. clinicaltrials.gov/ct2/show/NCT03220425 (first received 11 February 2020).
- Novo Nordisk. Clinical trial report NN304-1335. s3.amazonaws.com/ctr-nvo-7271/NN304-1335/318871ce-8b98-4af4-9d87-7b592268dcc6/5a8066b6-a561-422e-b741-39ddd62894b0/1335-ctr-redacted-v1.pdf (accessed 2 April 2020).
- Novo Nordisk. Synopsis trial NN304-1335. s3.amazonaws.com/ctr-nvo-7271/NN304-1335/d676d57d-c1d9-4914-887c-9df26687b9d5/672c2a20-f769-4c16-9dff-29d13a4d06bd/1335-ctr-synopsis-redacted-v1.pdf (accessed 2 April 2020).
- Russell-Jones D, Bolinder J, Simpson R. Lower and more predictable fasting blood glucose and reduced risk of nocturnal hypoglycaemia with once daily insulin detemir versus NPH in subjects with type 1 diabetes. Diabetologia 2002;45 (Suppl 2):A51. [Google Scholar]
- Russell-Jones D, Simpson R, Hylleberg B, Draeger E, Bolinder J. Effects of QD insulin detemir or neutral protamine Hagedorn on blood glucose control in patients with type I diabetes mellitus using a basal-bolus regimen. Clinical Therapeutics 2004;26(5):724-36. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schober 2002 {published and unpublished data}
- Herwig J, Scholl-Schilling G, Bohles H. Glycaemic control and hypoglycaemia in children, adolescents and young adults with unstable type 1 diabetes mellitus treated with insulin glargine or intermediate-acting insulin. Journal of Pediatric Endocrinology & Metabolism 2007;20(4):517-25. [DOI] [PubMed] [Google Scholar]
- Hoechst Marion Roussel. Clinical study report - HOE901/3003. Pharmaeconomic data [personal communication]. Provided by Sanofi Aventis 20 February 2020.
- Hoechst Marion Roussel. Clinical Study Report - HOE901/3003 [personal communication]. Provided by Sanofi Aventis 20 February 2020.
- Mohn A, Strang S, Wernicke-Panten K, Lang AM, Edge JA, Dunger DB. Nocturnal glucose control and free insulin levels in children with type 1 diabetes by use of the long-acting insulin HOE 901 as part of a three-injection regimen. Diabetes Care 2000;23(4):557-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Schober E, Schoenle E, Van Dyk J, Wernicke-Panten K, Pediatric Study Group of Insulin Glargine. Comparative trial between insulin glargine and NPH insulin in children and adolescents with type 1 diabetes. Diabetes Care 2001;24(11):2005-6. [DOI] [PubMed] [Google Scholar]
- Schober E, Schoenle E, Van Dyk J, Wernicke-Panten K, Pediatric Study Group of Insulin Glargine. Comparative trial between insulin glargine and NPH insulin in children and adolescents with type 1 diabetes mellitus. Journal of Pediatric Endocrinology & Metabolism 2002;15(4):369-76. [DOI] [PubMed] [Google Scholar]
Standl 2004 {published and unpublished data}
- Novo Nordisk. Clinical trial report NN304-1181. s3.amazonaws.com/ctr-nvo-7271/NN304-1181/9590fb43-a575-4272-baf9-c9abe645c63b/90f140bd-90f8-40bf-80f2-010945dc66a6/1181-ctr-redacted-v1.pdf (accessed 31 March 2020).
- Novo Nordisk. Synopsis NN304-1181. s3.amazonaws.com/ctr-nvo-7271/NN304-1181/dda30fa0-6147-4528-809c-9d814d85cf32/29ecec7a-c124-432b-b5fd-face38df5cb3/1181-ctr-synopsis-redacted-v1.pdf (accessed 1 April 2020).
- Roberts A, Bayer T, Munksgaard E, Lang H, Standl E. Efficacy and safety of 6-month treatment with insulin detemir in type 1 diabetic patients on a basal/bolus regimen. Diabetes 2001;50:A129. [Google Scholar]
- Standl E, Lang H, Roberts A. The 12-month efficacy and safety of insulin detemir and NPH insulin in basal-bolus therapy for the treatment of type 1 diabetes. Diabetes Technology & Therapeutics 2004;6(5):579-88. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Standl E, Roberts A, Lang H. One-year safety and efficacy of insulin detemir in subjects with type 1 diabetes. Favourable weight development and reduced nocturnal hypoglycaemia compared to NPH. Diabetologia 2002;45 (Suppl 2):A51. [Google Scholar]
SWITCH 1 {published and unpublished data}
- Bantwal G, Bailey TS, Bhargava A, De Vries JH, Gerety G, Gumprecht J, et al. Day-to-day variability of fasting self-measured plasma glucose correlates with risk of hypoglycemia in adults with type 1 and type 2 diabetes. Indian Journal of Endocrinology and Metabolism 2017;21(8):S25-6. [Google Scholar]
- Bossi CA, Bailey TS, Bhargave A, De Vries JH. Day-to-day variability of fasting self-measured plasma glucose correlates with risk of hypoglycaemia in adults with type 1 and type 2 diabetes. Italian Journal of Medicine 2018;12:4. [Google Scholar]
- Danne T, Thalange N, Tutkunkardas D, Troelsen LN, Lane W. Randomised, double-blind, crossover trial comparing the safety and efficacy of insulin degludec (IDeg) and insulin glargine U100 (IGlarU100) in young adults with type 1 diabetes (T1D): SWITCH 1 subgroup analysis. Pediatric Diabetes 2017;18:18-46. [Google Scholar]
- De Vries JH, Bailey TS, Bhargava A, Gerety G, Gumprecht J, Heller S, et al. Day-to-day fasting self-monitored blood glucose variability is associated with risk of hypoglycaemia in insulin-treated patients with type 1 and type 2 diabetes: a post hoc analysis of the SWITCH Trials. Diabetes, Obesity & Metabolism 2019;21(3):622-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EUCTR2012-001930-32-PL. A trial comparing the safety and efficacy of insulin degludec and insulin glargine, both with insulin aspart as mealtime insulin in subjects with type 1 diabetes. apps.who.int/trialsearch/Trial3.aspx?trialid=EUCTR2012-001930-32-PL (first received 20 February 2020).
- Evans M, Mehta R, Gundgaard J, Chubb B. Cost-effectiveness of insulin degludec vs. insulin glargine u100 in type 1 and type 2 diabetes mellitus in a UK setting. Diabetes Therapy: Research, Treatment and Education of Diabetes and Related Disorders 2018;9(5):1919-30. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G, Wysham C, Gumprecht J, Lane W, Troelsen LN, Tutkunkardas D, et al. Insulin degludec shows consistent risk reductions across hypoglycaemia definitions vs insulin glargine U100 in the SWITCH 1 and 2 trials. Italian Journal of Medicine 2018;12(2):66. [Google Scholar]
- Heller S, Gumprecht J, Lane W, Troelsen LN, Tutkunkardas D, Wysham CH. Insulin degludec (IDeg) shows consistent risk reductions across hypoglycaemia definitions vs insulin glargine U100 (IGlar U100) in the SWITCH 1 and SWITCH 2 trials. Diabetic Medicine 2018;35(Suppl 1):145. [Google Scholar]
- Heller SR, Buse JB, Ratner R, Seaquist E, Bardtrum L, Hansen CT, et al. Redefining hypoglycemia in clinical trials: validation of definitions recently adopted by the American Diabetes Association/European Association for the Study of Diabetes. Diabetes Care 2019;28:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane W, Bailey TS, Gerety G, Gumprecht J, Philis-Tsimikas A, Hansen CT, et al. Effect of insulin degludec vs insulin glargine u100 on hypoglycemia in patients with type 1 diabetes: the SWITCH 1 randomized clinical trial. JAMA 2017;318(1):33-44. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane W, Bailey TS, Gerety G, Gumprecht J, Philis-Tsimikas A, Hansen CT, et al. SWITCH 1: reduced risk of hypoglycaemia with insulin degludec vs insulin glargine U100 in patients with type 1 diabetes - a randomised, double-blind, crossover trial. Clinical Endocrinology 2018;89:9-15. [Google Scholar]
- NCT02034513. A trial comparing the safety and efficacy of insulin degludec and insulin glargine, both with insulin aspart as mealtime insulin in subjects with type 1 diabetes (SWITCH 1). clinicaltrials.gov/ct2/show/NCT02034513 (first received 15 January 2020).
- Novo Nordisk. Clinical trial report trial ID: NN1250-3995. www.novonordisk-trials.com/studie/128 (accessed 24 February 2020).
- Novo Nordisk. Synopsis trial ID: NN1250-3995. s3.amazonaws.com/ctr-nvo-7271/NN1250-3995/1fee3a46-263c-45b1-855c-716f83202cdf/5cd3a1dc-2202-42d8-acdc-9813925af59c/3995-ctr-synopsis-redacted-v1.pdf (accessed 24 February 2020).
- Philis-Tsimikas A, Lane W, Pedersen-Bjergaard U, Wysham C, Bardtrum L, Harring S, et al. The relationship between HbA1c and hypoglycaemia in patients with diabetes treated with insulin degludec versus insulin glargine 100 units/mL. Diabetes, Obesity & Metabolism 2020;22(5):779-87. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philis-Tsimikas A, Lane W, Pedersen-Bjergaard U, Wysham CH, Bardtrum L, Ostoft SH, et al. Relationship between a1c and hypoglycemia risk in individual patients comparing insulin degludec with insulin glargine u100. Diabetes 2018;67:A80. [Google Scholar]
- Ramirez de Arellano, Serna A, Darba J, Tikkanen C, Conde V. Cost-effectiveness analysis of insulin degludec versus insulin glargine u100 in type 1 and type 2 diabetes patients from the Portuguese national healthcare system perspective: evidence from the SWITCH 1&2 trials. Value in Health 2017;20(9):A481. [Google Scholar]
- Wysham CH, Lane W, Ladelund S, Tutkunkardas D, Heller S. Target fasting plasma glucose (FPG) without nocturnal hypoglycemia in patients with type 1 diabetes or type 2 diabetes - results from SWITCH trials. Diabetes 2018;67:A104-5. [Google Scholar]
Thalange 2013 {published and unpublished data}
- 2006-000051-18/DK. A 52-week, multinational, multi-centre, open-labelled, randomised, parallel, efficacy and safety comparison of insulin detemir and NPH insulin in children and adolescents 2-16 years with type 1 diabetes on a basal-bolus regimen with insulin aspart as bolus insulin. www.clinicaltrialsregister.eu/ctr-search/trial/2006-000051-18/DK (first received 10 February 2020).
- 2006-002478-23. A 52-week, multinational, multi-centre, open-labelled, randomised, parallel, efficacy and safety comparison of insulin detemir and NPH insulin in children and adolescents 2-16 years with type 1 diabetes on a basal-bolus regimen with insulin aspart as bolus insulin. www.clinicaltrialsregister.eu/ctr-search/search?query=2006-002478-23 (first received 28 February 2020).
- NCT00435019. Comparison of NPH insulin and insulin detemir in children and adolescents with type 1 diabetes. clinicaltrials.gov/ct2/show/NCT00435019 (first received 11 February).
- NCT00623194. Safety follow-up on children and adolescents with type 1 diabetes treated with insulin detemir. An extension to trial NN304-1689. clinicaltrials.gov/ct2/show/NCT00623194 (first received 21 February 2020).
- Novo Nordisk. Revised clinical trial report trial ID NN304-1689. www.novonordisk-trials.com/studie/527 (accessed 28 February 2020).
- Novo Nordisk. Synopsis trial NN304-1689. www.novonordisk-trials.com/studie/527 (accessed 28 February 2020).
- Thalange N, Bereket A, Jensen LB, Hiort LC, Peterkova V. Development of insulin detemir/insulin aspart cross-reacting antibodies following treatment with insulin detemir: 104-week study in children and adolescents with type 1 diabetes aged 2-16 years. Diabetes Technology & Therapeutics 2016;7(4):713-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalange N, Bereket A, Larsen J, Hiort LC, Peterkova V. Insulin analogues in children with type 1 diabetes: a 52-week randomized clinical trial. Diabetic Medicine 2013;30(2):216-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalange N, Bereket A, Larsen J, Hiort LC, Peterkova V. Treatment with insulin detemir or NPH insulin in children aged 2-5 yr with type 1 diabetes mellitus. Pediatric Diabetes 2011;12(7):632-41. [DOI] [PubMed] [Google Scholar]
Urakami 2017 {published and unpublished data}
- Urakami T, Mine Y, Aoki M, Okuno M, Suzuki J. A randomized crossover study of the efficacy and safety of switching from insulin glargine to insulin degludec in children with type 1 diabetes. Endocrine Journal 2017;64(2):133-40. [DOI] [PubMed] [Google Scholar]
Vague 2003 {published and unpublished data}
- De Leeuw I, Vague P, Selam JL, Skeie S, Elte JWF, Lang H, et al. Lower risk of nocturnal hypoglycaemia and favourable weight development in type 1 diabetic subjects after 12 months treatment with insulin detemir vs. NPH insulin. Diabetologia 2002;45 (Suppl 2):A257. [Google Scholar]
- De Leeuw I, Vague P, Selam JL, Skeie S, Lang H, Draeger E, et al. Insulin detemir used in basal-bolus therapy in people with type 1 diabetes is associated with a lower risk of nocturnal hypoglycaemia and less weight gain over 12 months in comparison to NPH insulin. Diabetes, Obesity & Metabolism 2005;7(1):73-82. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Novo Nordisk. Clinical trial report NN304-1205. s3.amazonaws.com/ctr-nvo-7271/NN304-1205/52ea98e6-dcf4-42f2-8e30-d285b734646e/ec830729-e19e-46bf-a5fa-3a280cc70cfc/1205-ctr-redacted-v1.pdf (accessed 1 April 2020).
- Novo Nordisk. Synopsis NN304-1205. www.novonordisk-trials.com/en/studie/?id=NN304-1205&arrayList=NN304-1205,NN304-1761,NN304-1690,NN304-1927,NN304-1951,NN304-3701,NN304-3699,NN304-1449,NN304-1595,NN304-1613,NN304-3023,NN304-1859,NN304-1604,NN304-1438,NN304-1813&Conditions=&AgeRanges=&Phases=&SearchTerm=%E2%80%A2%09NN304-1205&Status=&Treatment=&AttachmentTypes=&country=&zip= (accessed 1 April 2020).
- Vague P, Selam JL, Skeie S, De Leeuw I, Elte JW, Haahr H, et al. Insulin detemir is associated with more predictable glycemic control and reduced risk of hypoglycemia than NPH insulin in patients with type 1 diabetes on a basal-bolus regimen with premeal insulin aspart. Diabetes Care 2003;26(3):590-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
21st Brazilian Diabetes Society Congressa {published data only}
- 21st Brazilian Diabetes Society Congress. In: Diabetology & Metabolic Syndrome. Vol. 10 (S1). 2018. [DOI] [PMC free article] [PubMed]
Bin‐Abbas 2006 {published data only}
- Bin-Abbas BS, Al-Agha AE, Sakati NA, Al-Ashwal AA. Multiple daily insulin regimen using insulin glargine in type 1 diabetic Saudi children. Saudi Medical Journal 2006;27(2):262-4. [PubMed] [Google Scholar]
Bolli 2016 {published data only}
- Bolli G, Owens DR, Fulcher GR, Home PD, Frier BM, Gao L, et al. Poster Tours. Pediatric Diabetes 2016;17:90-91. [Google Scholar]
Chacra 2010 {published data only}
- 2006-006375-21/HU. The COMPLETE T1D trial: comparison of insulin lispro protamine suspension and detemir in type 1 diabetes comparison of two basal insulin analogs (insulin lispro protamine suspension and insulin detemir) in basal-bolus therapy for patients with type 1 diabetes. www.clinicaltrialsregister.eu/ctr-search/trial/2006-006375-21/HU (first received 10 February 2020).
- Chacra AR, Kipnes M, Ilag LL, Sarwat S, Giaconia J, Chan J, et al. Comparison of insulin lispro protamine suspension and insulin detemir in basal-bolus therapy in patients with type 1 diabetes. Diabetic Medicine 2010;27(5):563-9. [DOI] [PubMed] [Google Scholar]
- NCT00487240. Comparison of two basal insulin therapies for patients with type 1 diabetes. clinicaltrials.gov/ct2/show/NCT00487240 (first received 12 February 2020).
Hirsch 2012 {published data only}
- 2009-013412-13/GB. An extension trial comparing safety and efficacy of NN5401 plus meal-time insulin aspart for the remaining meals with insulin detemir plus meal-time insulin aspart in type 1 diabetes. www.clinicaltrialsregister.eu/ctr-search/trial/2009-013412-13/GB (first received 10 February 2020).
- Hirsch IB, Bode B, Courreges JP, Dykiel P, Franek E, Hermansen K, et al. Insulin degludec/insulin aspart administered once daily at any meal, with insulin aspart at other meals versus a standard basal-bolus regimen in patients with type 1 diabetes: a 26-week, phase 3, randomized, open-label, treat-to-target trial. Diabetes Care 2012;35(11):2174-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch IB, Franek E, Mersebach H, Bardtrum L, Hermansen K. Safety and efficacy of insulin degludec/insulin aspart with bolus mealtime insulin aspart compared with standard basal-bolus treatment in people with type 1 diabetes: 1-year results from a randomized clinical trial (BOOST™). Diabetic Medicine 2017;34(2):167-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00978627. Comparison of NN5401 plus insulin aspart with insulin detemir plus insulin aspart in type 1 diabetes (BOOST™). clinicaltrials.gov/ct2/show/NCT00978627 (first received 11 February 2020).
HypoANA {published data only}
- 2006-003630-15. The effect of insulin analogues and human insulin on the incidence of severe hypoglycaemia in hypoglycaemia prone type 1 diabetic patients. www.clinicaltrialsregister.eu/ctr-search/search?query=2006-003630-15 (first received 10 February 2020).
- Agesen RM, Kristensen PL, Beck-Nielsen H, Norgaard K, Perrild H, Christiansen JS, et al. Effect of insulin analogues on frequency of non-severe hypoglycaemia in patients with type 1 diabetes prone to severe hypoglycaemia: the HypoAna trial. Diabetes/Metabolism Reviews 2016;42(4):249-55. [DOI] [PubMed] [Google Scholar]
- Agesen RM, Kristensen PL, Beck-Nielsen H, Norgaard K, Perrild H, Jensen T, et al. Effect of insulin analogs on frequency of non-severe hypoglycemia in patients with type 1 diabetes prone to severe hypoglycemia: much higher rates detected by continuous glucose monitoring than by self-monitoring of blood glucose - the HypoAna trial. Diabetes Technology & Therapeutics 2018;20(3):247-56. [DOI] [PubMed] [Google Scholar]
- Kristensen PL, Pedersen-Bjergaard U, Beck-Nielsen H, Norgaard K, Perrild H, Christiansen JS, et al. A prospective randomised cross-over study of the effect of insulin analogues and human insulin on the frequency of severe hypoglycaemia in patients with type 1 diabetes and recurrent hypoglycaemia (the HypoAna trial): study rationale and design. BMC Endocrine Disorders 2012;12:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen PL, Tarnow L, Bay C, Norgaard K, Jensen T, Parving HH, et al. Comparing effects of insulin analogues and human insulin on nocturnal glycaemia in hypoglycaemia-prone people with type 1 diabetes. Diabetic Medicine 2017;34(5):625-31. [DOI] [PubMed] [Google Scholar]
- NCT00346996. Insulin analogues and severe hypoglycaemia. clinicaltrials.gov/ct2/show/NCT00346996 (first received 11 February 2020).
- Pedersen-Bjergaard U, Kristensen PL, Beck-Nielsen H, Norgaard K, Perrild H, Christiansen JS, et al. Effect of insulin analogues on risk of severe hypoglycaemia in patients with type 1 diabetes prone to recurrent severe hypoglycaemia (HypoAna trial): a prospective, randomised, open-label, blinded-endpoint crossover trial. Lancet Diabetes Endocrinology 2014;2(7):553-61. [DOI] [PubMed] [Google Scholar]
- Pedersen-Bjergaard U, Kristensen PL, Beck-Nielsen H, Norgaard K, Perrild H, Christiansen JS, et al. The effect on insulin analogues on the risk of severe hypoglycaemia in patients with type 1 diabetes and recurrent severe hypoglycaemia: the HypoAna trial. Diabetologia 2013;56 Suppl 1:S84. [Google Scholar]
- Pedersen-Bjergaard U, Kristensen PL, Beck-Nielsen H, Norgaard K, Perrild H, Christiansen JS, et al. The potential for improvement of outcomes by personalized insulin treatment of type 1 diabetes as assessed by analysis of single-patient data from a randomized controlled cross-over insulin trial. Diabetes Research and Clinical Practice 2017;123:143-8. [DOI] [PubMed] [Google Scholar]
- Pedersen-Bjergaard U, Kristensen PL, Beck-Nielsen H, Norgaard K, Perrild H, Christiansen JS, et al. The significance of obtaining single-patient evidence for the best insulin treatment in patients with type 1 diabetes with recurrent severe hypoglycemia-lessons from the Hypoana trial. Diabetes 2015;64:A249. [Google Scholar]
Iga 2017 {published data only}
- Iga R, Uchino H, Kanazawa K, Usui S, Miyagi M, Kumashiro N, et al. Glycemic variability in type 1 diabetes compared with degludec and glargine on the morning injection: an open-label randomized controlled trial. Diabetes Therapy: Research, Treatment and Education of Diabetes and Related Disorders 2017;8(4):783-92. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- R000014228. Study of the efficacy and safety of insulin degludec who treating with basal insulin glargine or insulin detemir in type 1 diabetes with the basal-bolus therapy. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000014228 (first received 10 February 2020).
Kiess 2004 {published data only}
- Kiess W, Raile K, Galler A, Kapellen T. Insulin detemir offers improved glycemic control compared with NPH insulin in people with type 1 diabetes. Diabetes Care 2004;27(10):2567-8. [DOI] [PubMed] [Google Scholar]
Manini 2007 {published data only}
- Manini R, Forlani G, Moscatiello S, Zannoni C, Marzocchi R, Marchesini G. Insulin glargine improves glycemic control and health-related quality of life in type 1 diabetes. Nutrition, Metabolism, and Cardiovascular Diseases 2007;17(7):493-8. [DOI] [PubMed] [Google Scholar]
NCT00788840 {published data only}
- NCT00788840. Detemir energy expenditure study (DEES). clinicaltrials.gov/ct2/show/NCT00788840 (first received 11 February 2020).
NCT01854723 {published data only}
- NCT01854723. Comparison study of insulin glargine and NPH insulin. clinicaltrials.gov/ct2/show/NCT01854723 (first received 11 February 2020).
Orchard 2014 {published data only}
- Orchard TJ, Sibomana L, Miller R. Evaluation of differing type 1 diabetes treatment regimens in youth in Rwanda. Pediatric Diabetes 2014;15:16-48. [Google Scholar]
- Sibomana L, Rwabufigiri B, Kaberuka V, Gishoma C, Rubanzana W, Miller RG, et al. Type 1 diabetes-related quality of life in Rwanda. In: Diabetes. Vol. 64. American Diabetes Association (Clinical Therapeutics/New Technology), 2015:A368.
Ota 2017 {published data only}
- Ota M, Morita S, Kitamoto Y, Santi T, Fukushima M, Yasuda K. The efficacy of the treatment with insulin degludec in Japanese diabetes mellitus and the QOL of the treated patients. Journal of the Japanese Diabetic Society 2017;60(12):791-9. [Google Scholar]
- R000013521. The efficacy of a new long-acting insulin degludec in patient with diabetes. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000013521 (first received 10 February 2020).
Perez‐Maraver 2013 {published data only}
- 2006-005817-36/ES. Ensayo clínico en fase IV para la comparación entre dos pautas de tratamiento (insulina humana vs análogos de insulina) respecto al riesgo de hipoglicemia y la variabilidad del control glicémico en pacientes con diabetes mellitus tipo 1: impacto sobre el HYPO score y el LI (lability index). www.clinicaltrialsregister.eu/ctr-search/trial/2006-005817-36/ES (first received 11 February 2020).
- Perez-Maraver M, Caballero-Corchuelo J, Boltana A, Insa R, Soler J, Montanya E. Comparison of human insulin and insulin analogues on hypoglycaemia and metabolic variability in type 1 diabetes using standardized measurements (HYPO score and lability index). Acta Diabetologica 2013;50(4):529-35. [DOI] [PubMed] [Google Scholar]
Polonsky 2014 {published data only}
- Polonsky W, Traylor L, Gao L, Wei W, Ameer B, Stuhr A, et al. Improved treatment satisfaction in patients with type 1 diabetes mellitus treated with insulin glargine vs neutral protamine Hagedorn insulin. Endocrine Reviews 2014;35. [DOI] [PubMed] [Google Scholar]
Prikhodina 2007 {published data only}
- Prikhodina OA, Surikova SV, Girsh YV. Basal insulin analogue versus traditional NPH insulin in basal bolus therapy of children and adolescents with type 1 diabetes. Problemy Endokrinologii 2007;53(6):11-5. [DOI] [PubMed] [Google Scholar]
Tentolouris 2018 {published data only}
- Tentolouris A, Eleftheriadou I, Tentolouris N. Insulin degludec U100 is associated with lower risk for severe and symptomatic hypoglycemia as compared with insulin glargine U100 in subjects with type 1 diabetes. Annals of Translational Medicine 2018;6(3):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
UMIN000001562 {published data only}
- R000001889. Comparison of insulin detemir and insulin glargine on glucose variability in type 1 and type 2 diabetes. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000001889 (first received 10 February 2020).
UMIN000009965 {published data only}
- R000011669. Investigation of the difference among long acting insulin products in type 1 diabetes. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000011669 (first received 10 February 2020).
UMIN000013817 {published data only}
- R000016126. Jikei-evaluation of basal insulin analogue on (nocturnal) glycemic variability with continuous glucose monitoring - existing basal insulin analogue versus Tresiba - insulin degludec - a new basal insulin analogue, in basal-bolus treatment in type 1 diabetes. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000016126 (first received 10 February 2020).
Yamada 2014 {published data only}
- Yamada K, Nakayama H, Sato S, Tajiri Y, Kaku H, Tokubuchi I, et al. A randomized crossover study of the efficacy and safety of switching from insulin glargine to insulin degludec among patients with type 1 diabetes. Diabetology International 2014;5(1):74-7. [Google Scholar]
Ziemen 2015 {published data only}
- Ziemen M, Bergenstal RM, Riddle MC, Rojeski M, Espinasse M, Bolli GB, et al. Glycaemic control and hypoglycaemia with new insulin glargine 300 U/mL in people with type 1 diabetes (EDITION 4). Diabetologie und Stoffwechsel 2015;10:P227. [DOI: 10.1055/s-0035-1549733] [DOI] [Google Scholar]
References to studies awaiting assessment
Agesen 2019 {published data only}
- 2014-001942-24. The effect of insulin degludec on risk of symptomatic nocturnal hypoglycaemia in subjects with type 1 diabetes and high risk of nocturnal severe hypoglycaemia. www.clinicaltrialsregister.eu/ctr-search/search?query=2014-001942-24 (first received 10 February 2020).
- Agesen RM, Alibegovic AC, Andersen HU, Beck-Nielsen H, Gustenhoff P, Hansen K, et al. The effect of insulin degludec on risk of symptomatic nocturnal hypoglycaemia in adults with type 1 diabetes and high risk of nocturnal severe hypoglycaemia (the HypoDeg trial): study rationale and design. BMC Endocrine Disorders 2019;19(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT02192450. Insulin degludec and symptomatic nocturnal hypoglycaemia (HypoDeg). clinicaltrials.gov/ct2/show/NCT02192450 (first received 11 February 2020).
Basal Analog Study {published data only}
- 2005-001726-80. Effects of new long acting insulin analogs on metabolic control, endogenous insulin production, GH/IGF-I axis and quality of life. www.clinicaltrialsregister.eu/ctr-search/search?query=2005-001726-80 (first received 10 February 2020).
- EUCTR2005-001726-80-SE. Effects of new long acting insulin analogs on metabolic control, endogenous insulin production, GH/IGF-I axis and quality of life – comparison of NPH, glargine and detemir insulin from the debut of T1DM in adolescents - basal analog study. apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2005-001726-80-SE (first received 10 February 2020).
- NCT01271517. Basal analog study - comparison of Lantus or Levemir with NPH insulin from t1dm diagnosis (BAS). clinicaltrials.gov/ct2/show/NCT01271517 (first received 10 February 2020).
- Salemyr J, Ekström K, Örtqvist E, Pulkkinen M, Brorsson AL, Carlsson-Skwirut C, et al. Better first year HbA1c in type 1 diabetes mellitus adolescents on long acting insulin analogs vs. NPH insulin is not associated with reversal of their IGF-I deficiency. Growth Hormone and IGF Research 2012;22:S51-2. [Google Scholar]
ChiCTR2000032703 {published and unpublished data}
- ChiCTR2000032703. Comparision of insulin degludec and insulin glargine on blood glucose variability in northern Chinese patients with type 1 diabetes. www.chictr.org.cn/showproj.aspx?proj=53208 (first received 25 September 2020).
EudraCT 2007‐004144‐74 {published data only}
- 2007-004144-74/GB. A comparison of the effects of insulin detemir with insulin glargine on weight gain in female adolescents and young adults with type 1 diabetes (T1D) on a basal bolus regime. www.clinicaltrialsregister.eu/ctr-search/trial/2007-004144-74/GB (first received 10 February 2020).
- ISRCTN49492872. Determir versus glargine for weight gain in adolescents with type 1 diabetes. www.isrctn.com/ISRCTN49492872 (first received 10 February 2020).
EudraCT 2009‐012317‐22 {published data only}
- 2009-012317-22/IT. Pediatric basal bolus therapy - basal-bolus regimen in the treatment of children with type 1 diabetes. www.clinicaltrialsregister.eu/ctr-search/trial/2009-012317-22/IT (first received 10 February 2020).
INEOX {published data only}
- 2016-002915-17/ES. Impact on oxidative stress of novel analogues of insulin in people with type 1 diabetes. Low- intervention clinical trial. Ineox study. www.clinicaltrialsregister.eu/ctr-search/trial/2016-002915-17/ES (first received 10 February 2020).
- NCT03328845. Impact on the oxidative stress of the different analogues of insulin in people with type 1 diabetes (INEOX study). clinicaltrials.gov/ct2/show/NCT03328845 (first received 20 February 2020).
IRCT201203079224N1 {published data only}
- IRCT201203079224N1. Therapeutic effect of glargine combined aspart comparing regiment NPH combined regular for treatment patients with type 1 diabetes. apps.who.int/trialsearch/Trial3.aspx?trialid=IRCT201203079224N1 (first received 10 February 2020).
J‐Collection {published data only}
- R000001705. Jikei-comparison of Lantus and Levemir with CGM for thinking insulin optimization. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000001705 (first received 10 February 2020).
Mianowska 2007 {published data only}
- Mianowska B, Szadkowska A, Czerniawska E, Pietrzak I, Bodalski J. Insulin glargine improves fasting blood glucose levels in prepubertal children with unsatisfactorily controlled type 1 diabetes. Pediatric Endocrinology, Diabetes, and Metabolism 2007;13(4):189-93. [PubMed] [Google Scholar]
NCT00564018 {published data only}
- NCT00564018. Duration of the honeymoon phase of type 1 diabetes: a comparison of insulins detemir, glargine and NPH. clinicaltrials.gov/ct2/show/NCT00564018 (first received 11 February 2020).
Sherif 2014 {published data only}
- Sherif EM, El Tonbary KY, Abd Aziz MM. Comparative study between the use of insulin glargine and intermediate acting insulin (NPH) in type 1 diabetic children less than eight years old. Pediatric Diabetes 2014;15:16-48. [Google Scholar]
UMIN000020521 {published data only}
- R000023691. The efficacy and the safety of the new long-acting insulin in patient with diabetes. upload.umin.ac.jp/cgi-open-bin/ctr/ctr.cgi?function=brows&action=brows&type=summary&recptno=R000023691&language=E (first received 10 February 2020).
UMIN000021046 {published data only}
- UMIN000021046. Insulin degludec compared with conventional basal insulin in basal-bolus therapy with type 1 and type 2 diabetes in outpatient: a 24-week, randomized, open-label, treat-to-target trial. rctportal.niph.go.jp/en/detail?trial_id=UMIN000021046 (first received 2 April 2020).
Additional references
Boutron 2020
- Boutron I, Page MJ, Higgins JP, Altman DG, Lundh A, Hróbjartsson A. Chapter 7: Considering bias and conflicts of interest among the included studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Cochrane 2020
- Cochrane. How CENTRAL is created. www.cochranelibrary.com/central/central-creation (accessed 6 July 2020).
Cooper 2019
- Cooper C, Varley-Campbell J, Carter P. Established search filters may miss studies when identifying randomized controlled trials. Journal of Clinical Epidemiology 2019;112:12-9. [DOI: 10.1016/j.jclinepi.2019.04.002] [PMID: ] [DOI] [PubMed] [Google Scholar]
DCCT/EDIC 2016
- Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) study research group. Intensive diabetes treatment and cardiovascular outcomes in type 1 diabetes: the DCCT/EDIC study 30-year follow-up. Diabetes Care 2016;39(5):686-93. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
EMA 2004
- European Medicines Agency. Levemir: EPAR - scientific discussion. www.ema.europa.eu/en/documents/scientific-discussion/levemir-epar-scientific-discussion_en.pdf (accessed 3 March 2003).
EMA 2011
- European Medicines Agency. Assessment report - procedure No: EMEA/H/C/000528/II/0051. www.ema.europa.eu/en/documents/variation-report/levemir-h-c-528-ii-0051-epar-assessment-report-variation_en.pdf (accessed 3 March 2020).
EMA 2012
- European Medicines Agency. Tresiba: EPAR - Public assessment report. www.ema.europa.eu/en/documents/assessment-report/tresiba-epar-public-assessment-report_en.pdf (accessed 3 March 2020).
EMA 2014
- European Medicines Agency. Tresiba-H-C-2498-II-0011: EPAR - Assessment report - variation. www.ema.europa.eu/en/documents/variation-report/tresiba-h-c-2498-ii-0011-epar-assessment-report-variation_en.pdf (accessed 3 March 2020).
EMA 2015
- European Medicines Agency. Levemir-H-C-528-II-0070: EPAR - Assessment report - variation. www.ema.europa.eu/en/documents/variation-report/levemir-h-c-528-ii-0070-epar-assessment-report-variation_en.pdf (accessed 3 March 2020).
EMA 2015a
- European Medicines Agency. Assessment report for paediatric studies submitted according to Article 46 of the Regulation (EC) No 1901/2006. www.ema.europa.eu/en/documents/variation-report/lantus-h-c-284-p46-0521-epar-assessment-report_en.pdf (accessed 1 March 2020).
EMA 2015b
- European Medicines Agency. Assessment report for paediatric studies submitted according to Article 46 of the Regulation (EC) No 1901/2006. www.ema.europa.eu/en/documents/variation-report/toujeo-h-c-309-p46-0511-epar-assessment-report_en.pdf (accessed 1 March 2020).
Ewen 2019
- Ewen M, Joosse HJ, Beran D, Laing R. Insulin prices, availability and affordability in 13 low-income and middle-income countries. BMJ Global Health 2019;4(3):e001410. [DOI] [PMC free article] [PubMed] [Google Scholar]
FDA 2000
- Center for Drug Evaluation and Research. Medical Review NDA #021081. www.accessdata.fda.gov/drugsatfda_docs/nda/2000/21081_Lantus_medr.pdf (accessed 3 March 2020).
FDA 2002
- Center for Drug Evaluation and Research. Medical review (insulin detemir). www.accessdata.fda.gov/drugsatfda_docs/nda/2005/021-536_Levemir_medr.PDF (accessed 3 March 2020).
FDA 2005
- Center for Drug Evaluation and Research. Medical review (insulin detemir). www.accessdata.fda.gov/drugsatfda_docs/nda/2005/021878s000_Levemir_MedR.pdf (accessed 3 March 2020).
FDA 2015
- Center for Drug Evaluation and Research. Medical review Levemir. www.accessdata.fda.gov/drugsatfda_docs/nda/2015/203313Orig1s000_203314Orig1s000MedR.pdf (accessed 3 March 2020).
Fujimoto 2018
- Fujimoto K, Iwakura T, Aburaya M, Matsuoka N. Twice-daily insulin degludec/insulin aspart effectively improved morning and evening glucose levels and quality of life in patients previously treated with premixed insulin: an observational study. Diabetology & Metabolic Syndrome 2018;10:64. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2020
- Higgins JP, Savović J, Page MJ, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [DOI: 10.1136/bmj.c365] [DOI] [PubMed] [Google Scholar]
Laranjeira 2018
- Laranjeira FO, Andrade KR, Figueiredo AC, Silva EN, Pereira MG. Long‐acting insulin analogues for type 1 diabetes: an overview of systematic reviews and meta‐analysis of randomized controlled trials. PLOS One 2018;13(4):e0194801. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lefebvre 2019
- Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf MI, et al. Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Marshall 2018
- Marshall IJ, Noel-Storr AH, Kuiper J, Thomas J, Wallace BC. Machine learning for identifying randomized controlled trials: an evaluation and practitioner’s guide. Research Synthesis Methods 2018;9(4):602-14. [DOI: 10.1002/jrsm.1287] [DOI] [PMC free article] [PubMed] [Google Scholar]
McDonald 2017
- McDonald S, Noel-Storr AH, Thomas J. Harnessing the efficiencies of machine learning and Cochrane Crowd to identify randomised trials for individual Cochrane reviews. In: Global Evidence Summit. 2017 Sept 13-16; Cape Town, South Africa. 2017.
Monami 2009
- Monami M, Marchionni N, Mannucci E. Long-acting insulin analogues vs. NPH human insulin in type 1 diabetes. A meta-analysis. Diabetes, Obesity & Metabolism 2009;11(4):372-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Noel‐Storr 2018
- Noel-Storr AH, Project Transform team. Cochrane Crowd: new ways of working together to produce health evidence. In: Evidence Live. 2018 June 18-20; Oxford, UK. 2018.
Sterne 2019
- Sterne JA, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366(l4898). [DOI: 10.1136/bmj.l4898] [DOI] [PubMed] [Google Scholar]
Thomas 2017
- Thomas J, Noel-Storr AH, Marshall I, Wallace B, McDonald S, Mavergames C, et al. Living systematic reviews: 2. combining human and machine effort. Journal of Clinical Epidemiology 2017;91:31-7. [DOI: 10.1016/j.jclinepi.2017.08.011] [DOI] [PubMed] [Google Scholar]
Tricco 2014
- Tricco AC, Ashoor HM, Antony J, Beyene J, Veroniki AA, Isaranuwatchai W, et al. Safety, effectiveness, and cost effectiveness of long acting versus intermediate acting insulin for patients with type 1 diabetes: systematic review and network meta-analysis. BMJ 2014;349:g5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tricco 2018
- Tricco AC, Huda HM, Atnony J. Comparative efficacy and safety of Intermediate-acting, long-acting and biosimilar insulins for type 1 diabetes mellitus. www.who.int/selection_medicines/committees/expert/22/applications/s18.5_insulin-analogues.pdf?ua=1 (accessed 1 March 2020).
Ørskov Ipsen 2020
- Ørskov Ipsen E, Hemmingsen B, Østrup Petersen L, Metzendorf Maria-Inti, Richter B. Definitions and reporting of hypoglycaemia in trials of long‐acting insulin analogues in people with type 1 diabetes mellitus. Cochrane Database of Systematic Reviews 2020, Issue Issue 12. Art. No: CD013824. [DOI: 10.1002/14651858.CD013824] [DOI] [Google Scholar]
References to other published versions of this review
Hemmingsen 2019
- Hemmingsen B, Richter B, Metzendorf MM. (Ultra‐)long‐acting insulin analogues for people with type 1 diabetes mellitus. Cochrane Database of Systematic Reviews 2019, Issue 12. Art. No: CD013498. [DOI: 10.1002/14651858.CD013498] [DOI] [PMC free article] [PubMed] [Google Scholar]