

Abstract
In human spina bifida aperta (SBA), cerebral pathogenesis [hydrocephalus, Sylvius aqueduct (SA) stenosis and heterotopias] is poorly understood. In animal models, loss of ventricular lining (ependymal denudation) causes SA stenosis and hydrocephalus. We aimed to investigate whether ependymal denudation also takes place in human foetal SBA. Considering that ependymal denudation would be related to alterations in junction proteins, sections through SA of five SBA and six control foetuses (gestational ages ranged between 37 and 40 weeks) were immunostained for markers of ependyma (caveolin 1, βIV‐tubulin, S100), junction proteins (N‐cadherin, connexin‐43, neural cell adhesion molecule (NCAM), blood vessels (Glut‐1) and astrocytes [glial fibrillary acidic protein (GFAP)]. In control foetuses, ependymal denudation was absent. In SBA foetuses different stages of ependymal denudation were observed: (i) intact ependyma/neuroepithelium; (ii) imminent ependymal denudation (with abnormal subcellular location of junction proteins); (iii) ependymal denudation (with protrusion of neuropile into SA, formation of rosettes and macrophage invasion); (iv) astroglial reaction. It is suggested that abnormalities in the formation of gap and adherent junctions result in defective ependymal coupling, desynchronized ciliary beating and ependymal denudation, leading to hydrocephalus. The presence of various stages of ependymal denudation within the same full‐term SBA foetuses suggests continuation of the process after birth.
Keywords: adherent junctions, ependymal denudation, gap junctions, hydrocephalus, spina bifida aperta
INTRODUCTION
Spina bifida aperta (SBA) is associated with cerebral and spinal malformations such as meningomyelocele, Chiari II type malformation, hydrocephalus (HC) 19, 28, 41 periventricular gray matter heterotopia and cortical polygyration (20). However, in SBA patients the information about the nature and degree of the cortical brain alterations is still sparse. In many of the SBA patients HC appears to be caused by crowding of the posterior fossa, which interferes with the CSF flow out the fourth ventricle(11). Less frequently, HC would be associated to stenosis of the cerebral aqueduct 5, 30. Furthermore, little is known about the underlying mechanism triggering the early pathological events. In human SBA foetuses, we have previously shown that disruption of the neuroepithelium/ependyma (neuroepithelium/ependymal denudation) starts during the first trimester, which is before the development of HC (13). This process is associated with the formation of rosette‐like structures and migration of neuroblasts into the Sylvius aqueduct (SA) lumen 14, 36. Interestingly, neuroepithelium/ependymal denudation and formation of subependymal rosettes (at the site of denudation) have been observed in chick embryos after intraventricular injection of functional blocking antibodies against N‐cadherin, a major component of adherent junctions between neuroepithelial/ependymal cells (16). In several other animal models, ependymal denudation is ascribed to a function loss of different genes involved in the maintenance of junctions between the cells forming the neuroepithelium/ependyma 6, 8, 21, 27, 29, 38.
The aim of the present investigation was twofold. First, to establish whether in full‐term in SBA ependymal denudation is an ongoing process. The point is relevant as a positive answer would imply that such a process could continue after birth and be associated with aqueduct stenosis and the onset of high pressure HC. Second, to gain more insight into the actual process of ependymal denudation occurring in SBA foetuses, paying special attention to junction proteins.
PATIENTS
We retrospectively investigated histological sections and clinical data of five SBA [21, 22, 37, 39 and 40 weeks gestational age; (GA)] and six control foetuses (20, 21, 22, 33, 37 and 41 weeks GA). Tissue blocks were obtained at the University Hospital Groningen Netherlands and the Regional Hospital, Valdivia. Parents had given verbal informed consent. The medical ethical committee of the University Hospital Groningen Netherlands and of the Hospital Regional de Valdivia, Chile approved the study. In one SBA foetus, abruptio placentae caused delivery. In the other foetuses, delivery was induced [prostaglandine‐E2 medication (n = 4)] and/or assisted [cephalocentesis (n = 2) or vacuum extraction (n = 1)]. Elective termination of the pregnancies was done after ultrasound demonstration [and/or magnetic resonance imaging (MRI) confirmation] of spinal meningomyeolocele and/or HC. After initiation of delivery, all foetuses died during or shortly after delivery (median 12 h). Patient data concerning the level of the meningomyelocele and the development of HC are indicated in Table 1. For comparison with fetal control data, we excluded all cerebral conditions that might (potentially) relate with impaired integrity and/or damage of the foetal ependymal lining. Exclusion criteria for foetal controls involved: congenital cerebral malformations or chromosomal abnormalities, HC, macrocephaly, fetal intracerebral bleedings and infections.
Table 1.
Clinical data of spina bifida cases. Abbreviations: GA = gestational age in weeks; HC = hydrocephalus; L‐S = lumbar‐sacral; MMC = meningomyeolocele; Th‐L = thoracic‐lumbar; V = ventriculomegaly; + = present, − = absent.
| Case | GA | MMC | V | HC |
|---|---|---|---|---|
| 1 | 21 | L‐S | − | − |
| 2 | 22 | L‐S | + | − |
| 3 | 37 | L‐S | + | + |
| 4 | 39 | L‐S | − | − |
| 5 | 40 | Th‐L | + | + |
Control foetuses had died from: premature labor (3), maternal diabetes, umbilical cord strangulation and complicated twin pregnancy. In all five control foetuses, signs of cerebral pathology prior to delivery and/or congenital malformations were not found.
MATERIALS AND METHODS
Immunocytochemistry
Post‐mortem time before fixation ranged between 2 h and 3 days for the SBA patients and between 12 h and 3 days for the control cases. The brain stem containing the SA of all SBA patients and of four controls were fixed by immersion in 4% formalin in phosphate‐buffered saline, pH 7.4, and embedded in paraffin. The SA has three distinct segments in its rostrocaudal axis (see Results). The middle segment (MSA) is the longest. MSA was cut transversally. All tissue blocks containing the aqueduct were serially cut. Several hundreds of 5‐µm‐thick sections were obtained in each series. From each series, every 20 sections were mounted together, thus obtaining 20 series per tissue block. These series were used for haematoxylin–eosin stain and for immunocytochemistry according to the immunoperoxidase method or the streptoavidin‐biotin method (Vectastain kit, VECTOR, SERVA, Heidelberg, Germany), with diaminobenzidine (DAB) as electron donor. Some series were processed for immunofluorescence. For immunocytochemistry, antisera and the PAP complex were diluted in Tris buffer, pH 7.8, containing 0.7% non‐gelling seaweed lambda carrageenan (Sigma, St. Louis, MO, USA) and 0.5% Triton X‐100 (Sigma). The following primary antibodies were used: (1) anti‐glial fibrillary acidic protein (GFAP; an astroglial marker), raised in rabbit (Sigma), 1:500 dilution (2); anti‐S100 (AbCam, Cambridge, UK), dilution 1:100 (3); anti βIV‐tubulin (an ependymal and cilium marker) (AbCam), dilution 1:100 (4); anti‐caveolin 1 (antibodies against the cholesterol‐binding proteins of the invaginations (caveola) of the plasma membrane, serving as an ependymal marker), raised in rabbit, affinity purified (N‐20; Santa Cruz Biotechnology, Inc., San Diego, CA, USA), 1:200 dilution (5); anti‐glucose transporter 1, raised in rabbits (Glut‐1, blood capillaries marker, kindly provided by Coralia I. Ribas, Memorial Sloan‐Kettering Cancer Center, NY, USA), 1:1000 dilution (6); anti‐N‐cadherin, raised in rabbits (Santa Cruz Biotechnology), dilution 1:50 (7); anti‐neural cell adhesion molecule (NCAM), monoclonal, which recognizes polysialylated and nonsialylated forms of neural cell adhesion molecules (5B8 clone; Developmental StudiesHybridoma Bank, Iowa City, IA), supernatant was used undiluted (8); anti‐connexin 43, raised in rabbits (kindly provided by JC Saéz, Universidad Católica de Chile), dilution 1:750. We used omission of incubation in the primary antiserum as control test for immunostaining. This procedure resulted in no staining.
Double immunofluorescence and confocal microscopy
Double immunofluorescence staining was performed by combining anti βIV‐tubulin (ependymal marker) with antibodies against S100, connexin 43 or N‐cadherin. Additional combination was NCAM and N‐cadherin.
Appropriate secondary antibodies conjugated with Alexa Fluor 488 or 594 (1:500 dilution; Invitrogen) were used. Omission of the primary antibody was used as a control test. This procedure resulted in no fluorescence. Sections were mounted in Vectashield (Dako) and inspected with an epifluorescence microscope provided with the multidimensional acquisition software AxioVision Rel (version 4.6) of Zeiss (Jena, Germany). Confocal microscopy by a Fluoview 1000 (Miami, FL, USA) laser‐scanning microscope was performed on the same sections used for immunofluorescence.
RESULTS
Different ependymal cell types line the SA
In all (SBA and control) foetuses, the cerebral aqueduct was open. The central nervous system of the youngest control fetus (20 weeks) was cut sagittaly. A midsagittal section through the SA allowed to distinguish three regions in the rostro‐caudal axis, namely a narrow rostral segment (rostral SA = RSA), about 1 mm in length, corresponding to the region occupied by the subcommissural organ, a wide open middle segment (MSA), about 3.5 mm in length, with a dorsal expansion corresponding to the collicular recess and a narrow and flat caudal segment (CSA), about 1 mm in length, opening to the fourth ventricle (Figure 2A). This shape of SA correlates completely with that of SA of mice and rats (cf. 42). Worth indicating is that measurements of SA mentioned earlier were done in young brain tissue fixed in formalin and embedded in paraffin, under which conditions shrinkage must be expected to occur.
Figure 2.

(A–F) Sylvius aqueduct (SA) of a control foetus [20 weeks gestational age (GA)]. A. Sagittal section through the middle (MSA) [collicular recess (cr)] and caudal (CSA) third of SA. Haematoxylin–eosin stain. An area similar to that framed is shown in B. Cerebellum (Ce); 4°, fourth ventricle. Bar 250 µm. B. Magnification of area framed in A. Double‐immunofluorescence for βIV‐tubulin (red) and caveolin 1 (green). The ventral wall (V) of CSA is multiciliated (red) and expresses caveolin strongly, whereas the dorsal wall (D) is not multiciliated and expresses caveolin 1 poorly. Areas framed in the dorsal and ventral walls are shown in C–F. Bar 70 µm. Insert: Detailed magnification of ventral wall showing N‐cadherin‐based adherent junctions (green). Bar 12 µm. C. Detailed magnification of area framed in B. Double‐immunofluorescence for βIV‐tubulin (red) and connexin 43 (green) showing the multiciliated ventral (V) ependyma of SA. Note the strong expression of connexin 43. Bar 8 µm. D. Detailed magnification of area framed in B showing the monociliated dorsal (D) ependyma of SA. The few multiciliated cells and the monociliated cells (full arrow) all express connexin 43 (broken arrow). Bar 8 µm. Insert: Detailed of area shown in D showing a multiciliated cell and cross and longitudinal views of the single cilium of monociliated cells (arrow). Bar 4 µm. E,F. Detailed views of areas of ventral and dorsal ependyma framed in B. E. The multiciliated ventral ependyma (V) expresses caveolin 1 in the apical (arrow) and subnuclear regions of the cells. Bar 10 µm. F. The Dorsal ependyma (D) expresses caveolin 1 mostly in the apical region of the cells (arrow). Bar 10 µm. G–J. Cross section through the middle third of SA of a spina bifida aperta (SBA) fetus (22 weeks GA). G. Double‐immunofluorescence for βIV‐tubulin (red) and N‐cadherin (green). Bar 30 µm. H. A region of previous section showing the image only for N‐cadherin. Some cells display N‐cadherin‐based adherent junctions (full arrows) whereas others do not (broken arrow). Bar 15 µm. I. Section adjacent to that of Figure G, immunostained for glial fibrillary acidic protein (GFAP). In the dorsal wall there are many immunoreactive elongated cells. The area framed is shown in J. Bar 30 µm. J. Magnification of area framed in Figure I. GFAP+ cells displaying a basal process intermingled with non‐reactive cells. Bar 8 µm. Insert: Detailed view of previous figure showing that GFAP+ are not multiciliated (arrows). C = cilia. Bar 4 µm.
In the youngest control foetus (20 weeks), the neuroepithelium/ependyma lining revealed different features at the ventral and the dorsal CSA walls (Figure 2A,B). The ventral neuroepithelium/ependyma was multiciliated and strongly expressed connexin 43 and caveolin 1 throughout the cytoplasm (Figure 2B–D). In the dorsal neuroepithelium/ependyma, most cells were monociliated and expressed connexin 43 and caveolin 1 at the apical cell pole (Figure 2B,D,F). Cells of the dorsal and ventral walls expressed N‐cadherin (Figure 2B insert). In the dorsal wall of MSA there was a mixed population of multiciliated and monociliated cells. This was also found in cross sections through the MSA of the two youngest SBA foetuses (21 and 22 weeks GA). The dorsal wall of MSA was lined by a pseudo‐stratified neuroepithelium formed by multiciliated βIV‐tubulin+/GFAP– cells (Figure 2G,I). The non‐ciliated (and/or monociliated) cells were GFAP+ and displayed a long basal process and were likely to correspond with radial glia/stem cells (Figure 2I,J). In full term foetuses GFAP+ cells were scarce and circumscribed to small areas (Figure 3B) or scattered (Figure 6F).
Figure 3.

Sylvius aqueduct (transverse section) of a full‐term spina bifida aperta (SBA) foetus [(37 weeks gestational age (GA)]. Immunofluorescence for N‐cadherin, neural cell adhesion molecule (NCAM) and immunocytochemistry for glial fibrillary acidic protein (GFAP). A. Immunocytochemistry for N‐cadherin. This figure is used for orientation. Framed areas 1, 2, 3 and 4 are shown as C,E,D and G, respectively. Bar 360 µm. B. Area similar to that framed by red rectangle in A. GFAP+ cells with and without basal processes intermingle with GFAP– cells. Bar 35 µm. C and D show areas resembling normal ependyma. C. Area framed by rectangle 1 in A. Immunofluorescence for N‐cadherin. Ventral ependymal cells display strong reaction at their lateral plasma membrane (arrows). Bar 10 µm. D. Area framed by rectangle 3 in A. Immunofluorescence of N‐cadherin was located at the lateral plasma membrane (full arrow) or as abnormal masses in the cytoplasm (broken arrow). Bar 10 µm. E. Area framed by rectangle 2 in A. Immunofluorescence for N‐cadherin. Only a minority of cells were immunoreactive for N‐cadherin at the lateral plasma membrane (full arrow), whereas all others have abnormal immunoreactive masses in the cytoplasm (broken arrows). Bar 14 µm. F. Magnification of previous figure showing masses of N‐cadherin in the cytoplasm (broken arrows) and a weak reaction throughout the cytoplasm. Bar 8 µm. G. Same area shown in square 4 of A. Section adjacent to Figure 7O. This is an area undergoing ependymal denudation; the adjacent ependyma (E) and the ependyma of the forming rosettes (R) have little or no N‐cadherin (broken arrow). Bar 32 µm. H. Stretch of ependyma of SA of a full‐term SBA patient (40 weeks GA). N‐cadherin appears located at the lateral plasma membrane of ependymal cells (arrow). Bar 48 µm. I. Detailed view of previous figure showing the normal location of N‐cadherin, namely, as elongated patches along the lateral plasma membrane (broken arrow) and as small patches in the most apical region of the lateral plasma membrane (full arrows). Bar 9 µm. J. Area framed by rectangle 2 in A. Immunofluorescence for the neural cell adhesion molecule (NCAM). Only at few places NCAM appears to be at the lateral membrane of ependymal cells (full arrow), otherwise it displays at abnormal locations in the cytoplasm or at the apical plasma membrane (broken arrows). Bar 10 µm. K. Double immunolabeling for NCAM (red) and N‐cadherin (green). Both molecules display an abnormal location in the ependymal layer (broken and full arrows). Bar 10 µm.
Figure 6.

Small and large ependymal rosettes are a distinct feature of the Sylvius aqueduct (SA) of spina bifida aperta (SBA) foetus. A. SA (transverse section) of a full‐term SBA fetus (40 weeks GA). Haematoxylin–eosin stain. Abnormalities, such as forking (broken arrow), numerous small (arrows) and large rosettes are shown. Bar 160 µm. B. Immunostaining with anti‐caveolin 1. The ependymal lining and the subependymal small rosettes (arrow) are reactive. The dorsal (dw), lateral (lw) and ventral (vw) of SA are lined by different ependymal cells. Bar 120 µm. C. Detailed magnification of area framed by top rectangle in B. Deep enfolding of the ependyma (forking) and subependymal small rosettes (arrows) are observed. The areas framed are shown in top and bottom inserts. Bar 40 µm. Top insert: Dorsal ependymal cells display caveolin 1 at their apical and basal poles. Bar 10 µm. Bottom insert: lateral ependyma displayed caveolin 1 at the apical cell pole. Bar 10 µm. D. Detailed magnification of area framed in A. A large rosette (R) and numerous small rosettes (arrows) are shown. Bar 40 µm. E. Adjacent section to that of previous figure immunostained for caveolin 1. The large rosette (R) and the numerous small rosettes (arrows) are reactive. Bar 40 µm. Insert: Small rosette. Immunofluorescence for βIV‐tubulin. Elongated structures most likely corresponding to cilia are strongly reactive (arrow). Bar 5 µm. F. Anti‐glial fibrillary acidic protein (GFAP) immunostains a mesh of glial processes embedding the rosettes but the large (R) and small rosettes (arrows) are not reactive. Bar 40 µm. Insert: Area similar to that framed by bottom rectangle in B. Scarce and isolated GFAP+ cells in the ependymal lining are seen (arrows). Bar 40 µm. The cell indicated by broken arrow is shown in top insert. G. Large (R) and small rosettes (arrows) do not react with anti‐Glut‐1 but capillaries (c) do. Bar 40 µm. Insert: Double Immunofluorescence for connexin 43 (green) and βIV‐tubulin (red). The ependyma lining the ventricle (broken arrow) and the cells lining the small rosettes (r, full arrow) are multiciliated. Bar 8 µm.
In full‐term control foetuses (33, 37 and 41 weeks), the dorsal wall of the MSA was lined by a mixed population of multiciliated and monociliated cells (Figure 1A,C,D,F), whereas the latero‐ventral walls were lined by multiciliated ependyma (Figure 1F,G). The dorsal neuroepithelium/ependyma was about 15 µm height (Figure 4A) and expressed βIV‐tubulin (Figure 1D,F) and caveolin 1 (Figure 6B,C). At the lateral walls of MSA there were areas lined by cylindrical cells alternating with others lined by flat multiciliated ependyma (Figure 1A,B,E,F). The cytoplasm of these cells was poorly reactive with anti‐βIV‐tubulin and caveolin 1, which was mainly present at the apical cell pole (Figure 6B,C). The ventral MSA wall was endowed with tall multiciliated ependymal cells, about 17‐µm height. This ependyma was strongly reactive with anti‐βIV‐tubulin and anti‐caveolin 1. Most of the cells displayed short basal processes, which projected into the neuropile (4, 5). Double immunostaining with anti‐S100 and anti‐βIV‐tubulin revealed that most cells lining the MSA reacted with both antibodies. There were, however, a few cells that only reacted with anti‐βIV‐tubulin and others that only reacted with anti‐S100 (Figure 5G,H).
Figure 1.
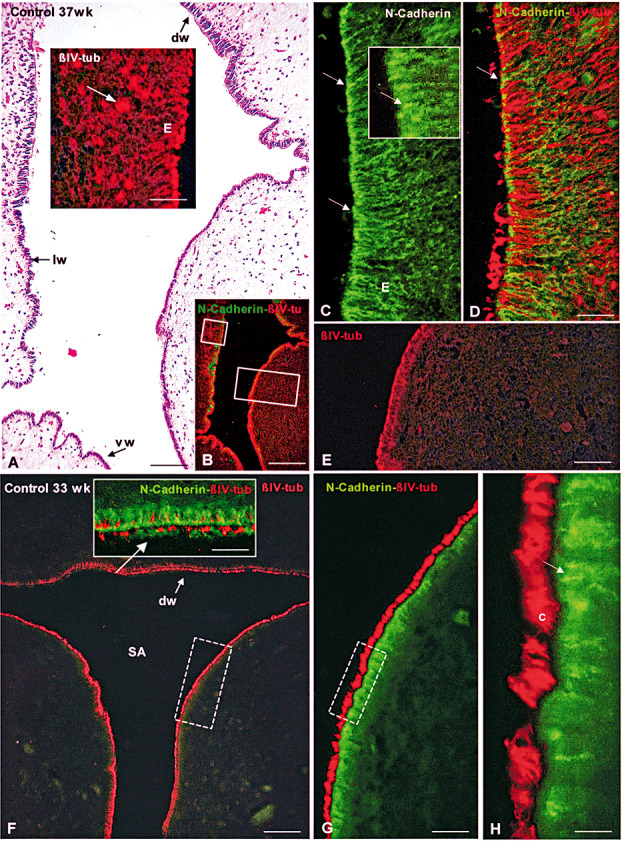
Sylvius aqueduct (SA; transverse section) of control foetuses [33 and 37 weeks gestational age (GA)]. Immunofluorescence for βIV‐tubulin and N‐cadherin. A–E. Control foetus 37 weeks GA. A. Haematoxylin–eosin stain. The dorsal (dw), lateral (lw) and ventral wall (vw) of the SA are lined by different ependymal cells. Bar 100 µm. Insert: Detailed magnification of area framed in small rectangle in B. βIV‐tubulin immunofluorescence. A few scattered small rosettes (arrows) located under the ependyma (E) are shown. Bar 24 µm. B. Adjacent section to that of previous figure. Double immunolabeling with anti‐βIV‐tubulin (red) and anti‐N‐cadherin (green). The areas framed are shown in insert of A and in E. Bar 300 µm. C. Laterodorsal ependyma (E) of SA shown in A. Ependyma with a normal appearance immunostained for N‐cadherin. Immunoreaction is mostly confined to the apical region of lateral plasma membrane (arrows). Bar 24 µm. Insert: High magnification of an area shown in C. Bar 10 µm. D Same section shown in C. An overlay of a double‐immunofluorescence image to visualize βIV‐tubulin (red) and N‐cadherin (green). Arrow points to an adherent junction. Bar 24 µm. E. Detailed magnification of area framed in large rectangle in B. βIV‐tubulin immunofluorescence showing the selective fluorescence of the ependyma (E) and the absence of rosettes in the periaqueductal neuropile. F–H. Control fetus 33 weeks GA. F. Transverse section of SA. βIV‐tubulin immunofluorescence showing the selective fluorescence of the ependyma and the absence of rosettes in the periaqueductal neuropile. dw, dorsal wall of SA. Bar 100 µm. Insert: Detailed magnification of dorsal wall of SA (arrow). Same section shown in F. An overlay of a double‐immunofluorescence image to visualize βIV‐tubulin (red) and N‐cadherin (green). Cells with no or a few cilia are seen. Bar 30 µm. G. Detailed magnification of rectangle of Figure F. Double‐immunofluorescence for βIV‐tubulin (red) and N‐cadherin (green). Bar 23 µm. H. Detailed magnification of rectangle of Figure G showing cilia in red (c) and N‐cadherin‐based adherent junctions in green (arrow). Bar 12 µm.
Figure 4.
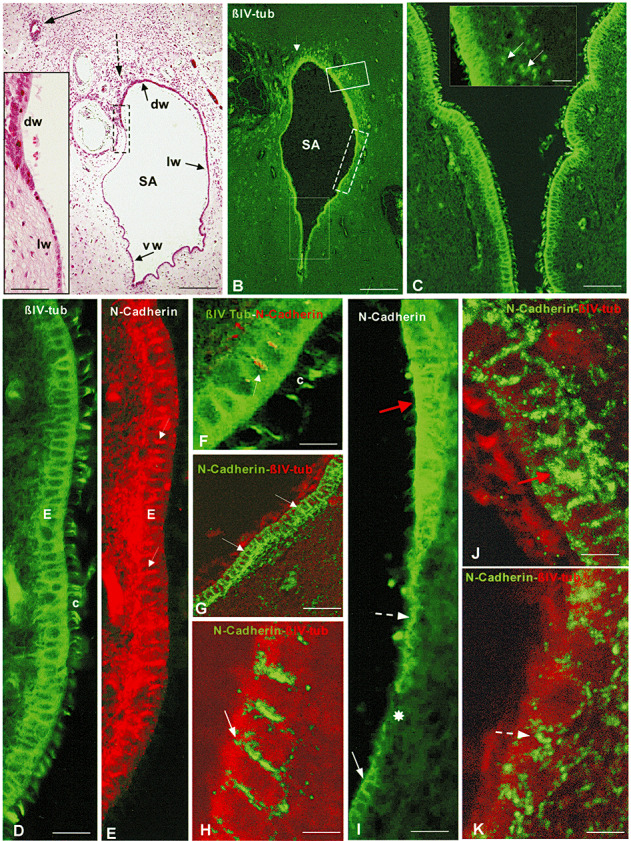
Cross‐section through the middle third of Sylvius aqueduct (SA) of a full‐term spina bifida aperta (SBA) foetus [40 weeks gestational age (GA)]. Immunofluorescence for βIV‐tubulin and N‐cadherin. A. Haematoxylin–eosin stain. Small (broken arrow) and large (full arrow) rosettes are shown. The dorsal (dw), lateral (lw) and ventral (vw) of the SA are lined by different ependymal cells. Bar 300 µm. Insert: Detailed magnification of area framed in A showing marked morphological differences between the ependyma of the dorsal (dw) and lateral (lw) walls of SA. Bar 60 µm. B. Immunostaining with the cilium marker βIV‐tubulin. The ependyma and small rosettes (arrow) are stained. Bar 350 µm. C. Detailed magnification of area framed by large rectangle in B. The ependyma (E) and their cilia are selectively stained. Bar 56µm. Insert: Detailed magnification of area framed by small rectangle in B. The ependyma and small rosettes (arrows) are selectively stained. Bar 37 µm. D,E. Detailed magnification of an area shown in C. Ventral ependyma with a normal appearance immunostained for βIV‐tubulin (D) and N‐cadherin (E). Ependyma (E) and cilia (c) in D and adherent junctions in E are strongly reactive. Bar 21 µm. F. Double immunolabeling. Ependymal cells and cilia (c) reacted with anti‐βIV‐tubulin (green) and adherent junctions (arrow) with anti‐N‐cadherin (red). Bar 9 µm. G. Ventral ependyma. Confocal microscopy using double immunostaining for N‐cadherin (green) and βIV‐tubulin (red). N‐cadherin localizes orderly at the lateral plasma membrane of the ependymal cells (arrows). Bar 36 µm. H. Detailed magnification of previous figure corresponding to an area similar to that indicated by white full arrow in I. Arrow points to N‐cadherin immunoreaction at the lateral plasma membrane. Bar 7µm. I. Detailed magnification of area framed by broken rectangle in B. There are areas with disruption of the ependymal lining (asterisk), or with abnormal location of the immunoreactive N‐cadherin (broken arrow), or with large amount of immunoreactive cadherin in the cytoplasm (red arrow). Full arrow points to normal ependyma. Bar 21 µm. J,K. Areas of abnormal ependyma similar to those indicated by red and broken arrows, respectively, in Figure I. Confocal microscopy using double immunostaining for N‐cadherin (green) and βIV‐tubulin (red). N‐cadherin localizes abnormally within the ependymal cells (red and broken arrows). Bar 7 µm.
Figure 5.

Sylvius aqueduct (SA; transverse section) of a full‐term spina bifida aperta (SBA) foetus (40 weeks GA). Immunofluorescence for connexin 43, βIV‐tubulin and S100 protein. A. Double Immunofluorescence for connexin 43 (green) and βIV‐tubulin (red). The multiciliated ependyma lining the ventral region of the SA displays a normal appearance. The area framed is shown in B and C using different channels at the fluorescence microscope. Bar 23 µm. Insert: Same section only visualized for connexin 43. Bar 78 µm. B. Detailed view of area framed in A. Immunofluorescence for connexin 43. Strongly reactive dots are mainly located in the apical cytoplasm (arrows). Bar 8 µm. Insert: Detailed view of an ependymal cell showing connexin 43 as elongated masses located in the supranuclear cytoplasm and at the lateral plasma membrane (arrow). Bar 5 µm. C. Same area shown in B. An overlay image of connexin 43 (green) and βIV‐tubulin (red). Bar 8 µm. D. Same section shown in C analysed with confocal microscopy. The normal subcellular distribution of connexin 43 is clearly visualized (arrow). Bar 8 µm. E. Section adjacent to that shown in Figure 2 I. A zone of the ependyma of the lateral wall of SA delimited by the two arrows appears disrupted. The area framed is shown in K. Bar 12 µm. Insert: Confocal microscopy of the same section showing the subcellular distribution of connexin 43 in a normal ependymal region (arrow). Bar 12 µm. F. Dorsal wall of SA. Double Immunofluorescence for connexin 43 (green) and βIV‐tubulin (red). The numerous small rosettes (arrows) express βIV‐tubulin. An area similar to that framed is shown in G. Bar 80 µm. G. Section adjacent to that shown in F processed for double Immunofluorescence for S100 (green) and βIV‐tubulin (red). Arrow points to transition between dorsal and lateral ependyma. In the dorsal ependyma there is a mixed population of green and red cells. Bar 15 µm. Top and middle inserts: Detail of some of the ependymal cells. Bar 10 µm. Bottom insert: Small rosettes express βIV‐tubulin but not S100. Bar 9 µm. H. Section adjacent to that shown in A. Ventral ependymal cells of normal appearance. Most cells express βIV‐tubulin and S100 (full arrow) and few express βIV‐tubulin only (broken arrow). Bar 11 µm. I, J. Areas of abnormal ependyma similar to those indicated by full and broken arrows, respectively, in figure K. Confocal microscopy using double immunostaining for connexin 43 (green) and βIV‐tubulin (red). Connexin 43 accumulates either intra‐cellular (I, full arrow) or is scarce or missing (J, broken arrow). Bar 5 µm. K. Detailed view of the region framed in E visualizing only the channel for connexin 43. Abnormal ependymal cells have either large masses of connexin 43 (full arrow) or little or none connexin 43 (broken arrow). Bar 4 µm.
All types of ependymal cells lining the SA were immunoreactive for NCAM, N‐cadherin, especially at the lateral plasma membrane (1, 4), and connexin 43, mainly as small cytoplasmic masses located at the apical cell pole and at the lateral plasma membrane (2, 5).
In one of the control foetuses (37 weeks GA) a few subependymal small rosettes were detected with anti‐βIV‐tubulin. These rosettes were circumscribed to a small region of the lateral SA wall (Figure 1B,E,F). In all other control foetuses rosettes were absent (2, 1).
Sequential ependymal SA alterations according to reconstructed pathology
The description of results will concentrate on findings occurring in the SA of full‐term SBA patients, and will follow what we think is the temporal sequence of events.
Abnormalities of junction proteins precede ependymal denudation
Areas that probably were about to undergo ependymal denudation were recognized by the abnormal subcellular distribution of junction proteins. In some of these areas, N‐cadherin appeared as large cytoplasmic masses (3, 4). This clearly contrasted with the dotted N‐cadherin arrangement at the lateral plasma membrane found in the ependyma of the control cases (Figure 1C,H) and in areas of the SA of SBA displaying a normal appearance (Figure 4B–H). In other abnormal ependymal areas, N‐cadherin was localized at the apical plasma membrane (Figure 4I) or around the cell nucleus (Figure 4K). In the SBA foetus of 37 weeks GA, marked SA abnormalities were observed (3, 7). In this foetus, ependymal patches with a normal N‐cadherin expression (Figure 3C,D) alternated with other ependymal patches with little or no N‐cadherin expression (Figure 3G) or displaying large masses of N‐cadherin Figure 3E,F). NCAM appeared as cytoplasmic masses of varying size and dots at the apical plasma membrane (Figure 3J,K). The SA of the 22‐week SBA case showed patches of neuroepithelium/ependyma devoid of N‐cadherin (Figure 2H).
Figure 7.
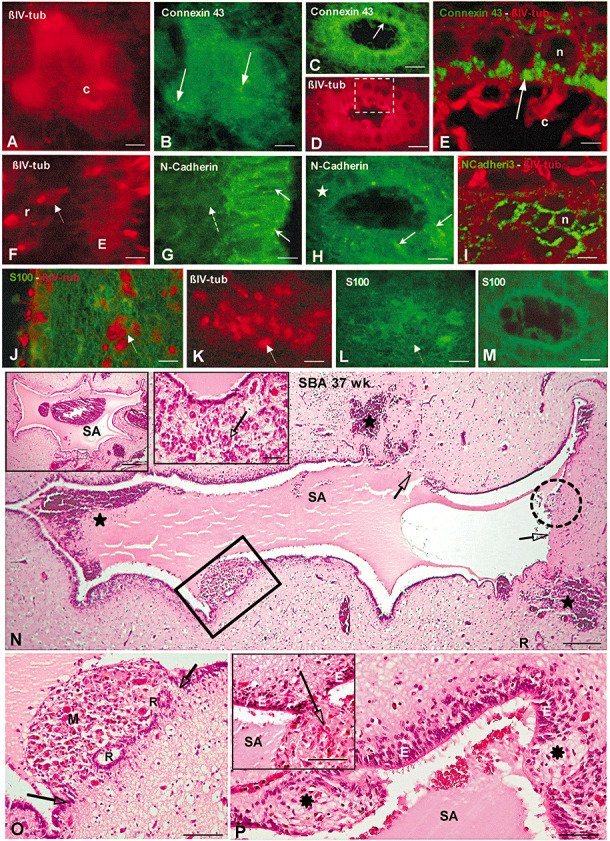
(A–M) Immunofluorescence properties of small and large rosettes present in the Sylvius aqueduct (SA) of spina bifida aperta (SBA) foetuses. A, B. Small rosette immunostained for βIV‐tubulin (A, red) and connexin 43 (B, green). The cells forming the small rosette display immunoreactive connexin 43 throughout the cytoplasm and as small dots (arrows). C cilia. Bar 4 µm. C,D. A large rosette immunostained for connexin 43 (C, green) and βIV‐tubulin (D, red). The cells forming the rosette display immunoreactive connexin 43 throughout the cytoplasm and as apical dots (arrow). The area framed is shown in E. Bar 10 µm. E. Confocal microscopy of area framed in D. Connexin 43 appears as apical dots (arrow). C cilia. Bar 3 µm. F, G. Dorsal wall of SA. Double‐immunofluorescence for βIV‐tubulin (F, red) and N‐cadherin (G, green). The ependyma (E) and small rosettes (r, arrow) react with anti‐βIV‐tubulin. The ependyma contains N‐cadherin (full arrows) but small rosettes do not (broken arrow). Bar 7 µm. H. Large rosettes express N‐cadherin abnormally, with some areas displaying immunoreactive N‐cadherin (arrows) and others being no reactive (asterisk). Bar 6 µm. I. Confocal microscopy. Large rosette immunostained for βIV‐tubulin (red) and N‐cadherin (green). N‐cadherin appears around the cell nucleus (n). Bar 5 µm. J. Lateral wall of SA immunostained for βIV‐tubulin (red) and S100 protein (green). Small rosettes express βIV‐tubulin (arrow) but not S100. Bar 10 µm. K, L. Double‐immunofluorescence for βIV‐tubulin (K, red) and S100 (L, green). Small rosettes express βIV‐tubulin (K, full arrow) but not S100 (L, broken arrow). Bar 4 µm. M. Large rosettes do not express S100. Bar 8 µm. N–P. The Sylvius aqueduct of full‐term SBA fetuses displays several abnormalities. Transverse section through the Sylvius aqueduct (SA) of SBA fetus (37 weeks GA) stained with Haematoxylin–eosin. N. The aqueduct has regions undergoing denudation (framed area), that had undergone denudation (arrows) and that are disrupted (broken circle). The fresh subependymal and intra‐aqueductal hemorrhages (indicated by stars) are attributed to acute, delivery‐related damage (perinatal cerebral insufficiency caused by asphyxia and incarceration). Bar 215 µm. Left insert: SA of the same patient sectioned at a different level. The small area framed is shown in right insert. Bar 450 µm. Right insert: Enlarged view of area framed in left insert. Numerous small rosettes are shown (arrow). Bar 35 µm. O. Detailed view of area framed in N showing a region of the ependyma undergoing denudation delimited by the two arrows, characterized by loss of ependyma, formation of large rosettes (R) and accumulation of macrophages (M). Bar 120 µm. P. Intraependymal nodular structures (stars) disrupt the ependyma (E). Bar 80 µm. Insert: Protrusion of neural tissue (arrow) through an area devoid of ependyma. Bar 160 µm.
The same ependymal regions that had an abnormal expression of N‐cadherin also displayed abnormal connexin 43 immunoreactivity (Figure 5E). In these areas there were cells containing large supranuclear clusters of immunoreactive granules whereas others were devoid of immunoreactive connexin 43 (Figure 5K). Confocal microscopy nicely confirmed these abnormal patterns (Figure 5I,J).
Ependymal denudation involves loss of ependymal cells, rosette formation and arrival of macrophages
Areas undergoing ependymal denudation were characterised by the following three histopathological features:
-
(i)
Absence of neuroepithelium/ependyma. The use of the ependymal markers, such as antibodies against βIV‐tubulin, caveolin 1, S100 and N‐cadherin made the denuded areas easy to recognize (Figure 4I). The extent of denudation ranged from a few ependymal cells (Figure 4I) to a few millimeters in length (Figure 7N).
-
(ii)
Accumulation of macrophages. Large clusters of macrophages were confined to those areas devoid of ependyma and endowed with rosettes of varying size (3, 7). This accumulation of macrophages was not observed in normal ependymal areas.
-
(iii)
Formation of subependymal rosettes. Two types of rosettes were distinguished, that are, large and small rosettes.
Large rosettes were present (one to five large rosettes per section) at the denuded areas (Figure 7A, B) and deep in the neuropile, at dorsolateral locations of the SA (Figure 4A). They ranged from 30 to 98 µm in diameter and were lined by cubic or cylindrical shaped multiciliated cells (6, 4). At the ventral wall of SA, large rosettes were absent (6, 4). Large rosettes expressed caveolin 1, βIV‐tubulin and connexin 43 in a normal pattern (ie, similar to intact ependymal SA areas (6, 7). This was contrasted by an abnormal N‐cadherin expression, as some cells displayed the protein around the cell nucleus and others displayed little or not protein (Figure 7H,I). Large rosettes were not immunoreactive for GFAP (Figure 6F) or S100 (Figure 7M). There were rosettes lined by ependyma displaying some normal morphological characteristics, that is, ordered arrangement of their cells and a strong reaction with anti‐βIV‐tubulin, whereas others were lined by cells with abnormal appearance and poorly reacting with anti‐βIV‐tubulin. These varying morphological appearances apparently reflect individual pathological alterations after rosette formation.
Small rosettes were numerous (a few hundreds per section) and located in the neuropile underlying the dorsolateral wall of the SA (6, 5). The small rosettes were about 18 µm in diameter. They were lined by a few flat cells that appeared to be multiciliated, as shown by the immunoreaction with anti‐βIV‐tubulin (Figure 6E, insert). Small rosettes expressed caveolin 1 (Figure 6E), βIV‐tubulin (6, 4, 5 insert, 7A,J,K) and connexin 43 (Figure 7B) and did not express N‐cadherin, S100, GFAP or Glut‐1 (6, 5, 7). Small rosettes were also observed at distance of the SA, in the surroundings of large rosettes. Rosettes were not found in the 22‐week SBA case (Figure 2G).
Astrocytes cover denuded areas
Newly denuded areas were devoid of a glial layer, allowing protrusion of neuropile into the SA (Figure 7P). Subependymal hemorrhages and bleeding into the SA also occurred at these areas (Figure 7N). A layer of GFAP‐reactive cells covered those areas that had earlier undergone denudation [see (10)].
Other malformations of the aqueduct, such as forking (Figure 6A–C) and nodular structures disrupting the ependyma (Figure 7P) co‐existed with areas of ependymal denudation.
All the phases described earlier 1, 2, 3 could be observed in the same foetus and even in the same section, irrespective of the age. This implies that there are several foci of ongoing ependymal denudation in the human foetal SA. Some foci reflected the initiation of ependymal denudation (ie, abnormal expression of junction proteins), other foci reflected ongoing ependymal denudation, and other foci reflected completed ependymal denudation. In the hyh mouse, this whole process may involve a time span of less than 1 day (42).
DISCUSSION
In human foetal SBA, we studied the pathogenesis and histology of ependymal denudation. SBA fetuses revealed various stages of ependymal denudation, whereas control foetuses did not. Areas with abnormal immunostaining for junction proteins of ependymal cells most likely corresponded to regions that were about to undergo ependymal denudation. Areas lacking ependyma and endowed with rosettes and macrophages indicated ongoing ependymal denudation. Areas devoid of ependyma but lined by an astrocyte layer indicated a more advanced phase of ependymal denudation. In full‐term foetal SBA, coexistent signs of pending, ongoing and recent ependymal denudation implicate that ependymal denudation is a continuous process which is likely to continue after birth.
The cerebral aqueduct of full‐term human foetuses is lined by several populations of ependymal cells that appear to be associated to different pathological events
At the dorsal, lateral and ventral walls of the SA, conventional light microscopy and immunocytochemistry revealed at least three populations of ependymal cells (present report). In the SA of wild‐type hyh mice, several populations of ependymal cells have been described (42). Interestingly, in mutant hyh mice, some of these ependymal populations undergo proliferation; others appear resistant to denudation whereas others undergo denudation 33, 40, 42. The functional significance of different ependymal populations found in control and SBA human foetuses is still unclear. However, the present findings suggest an association between ependymal lineages of SA and the observed SA pathology. For instance, ependymal cells lining the ventral SA walls of SBA foetuses display a normal subcellular distribution of N‐cadherin and connexin 43. These cells do not detach. Ependymal cells at the lateral SA walls display an abnormal intracellular location of junction proteins, and are likely to undergo denudation. Formation of large rosettes is mostly associated to this ependyma. The ependyma lining the dorsal SA is mostly associated with the formation of numerous small rosettes (see further discussion).
Alterations of cell junction proteins as a possible mechanism of ependymal denudation and HC in SBA
In former reports of human SBA foetuses, we indicated that neuroepithelial/ependymal denudation has been shown to occur in the SA, telencephalon 13, 14 and spinal cord (37). In these foetuses, ependymal denudation precedes Chiari type II malformation and HC. Loss of neuroepithelium/ependyma is associated with permanent neurodevelopmental abnormalities, as ependymal cells regenerate poorly (36). At the denuded SA, fusion with the opposing denuded neuropile would result in stenosis or obliteration and severe HC (42). In human and hyh mice, adhesion and junction proteins have been associated with the pathogenesis of SA stenosis and HC 4, 25, 26, 46. In this perspective, we investigated the expression of N‐cadherin and other junction molecules in normal and hydrocephalic SBA foetuses.
N‐cadherin‐based adherent junctions play a role in the maintenance of neuroepithelial/ependymal lining
Cadherins are known to play a key role during neural tube formation (22). After closure of the neural tube, N‐cadherin represents the major calcium‐dependent cell adhesion molecule in the neuroepithelium and later in the ependyma 7, 18. Blocking antibodies against chicken N‐cadherin disrupt the neuroepithelium/ependyma and leads to ependymal denudation, protrusion of the neuropile into the ventricle and formation of periventricular rosettes (16). In accordance with these findings, various animal models with a defect in cell–cell junctions undergo ependymal denudation, periventricular rosette formation, abnormal protrusion of neuropile into the SA and HC 4, 16, 24, 40.
According to the present investigation the ependymal cells of control foetuses and of preserved ependymal areas of SBA foetuses are joined together by N‐cadherin‐based adherent junctions and gap junctions. In SBA patients, areas with disorganized ependymal cells showed abnormal location of N‐cadherin, NCAM and connexin 43. Furthermore, similar to the pathological phenotype described in animal models 16, 29, 40, the SA of SBA foetuses carrying a defect in cell–cell junctions showed ependymal denudation associated with abnormal protrusion of the neuropile into the SA lumen and formation of large and small periventricular rosettes (present report). Some of these alterations have also been reported in different cases of human HC 14, 36.
Disruption of N‐cadherin‐based adherent junctions is associated to the formation of periventricular rosettes
Rosettes (large rosettes according to present description) consist of a halo or spoke‐wheel arrangement of cells surrounding a central cavity (45). In humans they have been associated to ependymomas (45) and different cases of HC 1, 14, 36. They have also been observed in cases of canine juvenile HC (47) and congenital HC induced by X‐radiation in rats (39). Furthermore, loss of function of different genes have also been associated to the development of ependymal rosettes in rodents 6, 8, 21, 27, 29. Interestingly, these studies suggest that rosette formation is associated with a defect in cell polarity and cell–cell adhesion of neuroepithelial/ependymal cells 16, 36. Indeed, N‐cadherin blockage in chicken embryos results in a disruption of the epithelial structure of the ependyma followed by the formation of ependymal rosettes (16). In SBA foetuses, the formation of periventricular rosettes appears to follow a similar pattern.
Many questions are still open with respect to the significance of rosettes. An intriguing aspect is that the ependyma forming the large rosettes shares some of the features of the normal ependyma (ie, normal subcellular distribution of connexin 43, caveolin 1 and βIV‐tubulin) and some of the characteristics of the defective ependyma (little or none N‐cadherin and S100). What is the actual pathological mechanism that leads to the formation of large rosettes? How to explain the presence of large rosettes at distance from the SA wall? What is the relation between the location of rosettes and the time of development? Are large rosettes occurring in pathologies different from HC also associated with ependymal denudation?
In addition to large subependymal rosettes associated with abnormal ependyma of SBA fetuses, we also observed numerous small subependymal rosettes in areas with intact ependyma. A few flat ependymal cells that do not express N‐cadherin and S100 form these small rosettes. The distinct phenotype of small rosettes and their presence underneath intact ependyma and the finding of a few of them in a control SA suggest that the origin and significance of small rosettes is different from that of large rosettes.
Abnormalities in the formation of adherent and gap junctions may be interrelated phenomena leading to alterations of cell–cell coupling
Gap junctions play an important role in the regulation of cell communication and cell–cell coupling 9, 35. For instance, intercellular communication through gap junctions may help to maintain synchronized ependymal ciliary beating 17, 34 and CSF flow (2). A number of studies indicate that the formation of gap junctions is interrelated with the formation of adherent junctions 12, 15, 23, 31, 32, 43. Indeed, recent studies suggested that connexin 43 and N‐cadherin may co‐assemble during their traffic to the plasma membrane (44). Accordingly, we observed that ependymal cells of SBA foetuses displayed an abnormal subcellular distribution of both connexin 43 and N‐cadherin.
A study by Xu et al (48) revealed that the association between connexins and cadherins is important for structural and functional purposes. In this context, defects of adherent junctions between ependymal cells in SBA foetuses could alter gap junction‐dependent ependymal physiology prior to, or in the absence of, ependymal disruption. Thus, an alteration of the CSF flow through the SA of SBA foetuses could be envisaged even if denudation is confined to small areas of the aqueduct wall.
In brief, we hypothesize that an abnormal expression of gap and adherent junction proteins could cause both, impaired CSF flow through the SA and ependymal denudation (Figure 8). It remains to be elucidated whether such phenomena could be attributed to underlying genetic defects, and/or to an altered ependymal microenvironment, such as changes in CSF composition.
Figure 8.
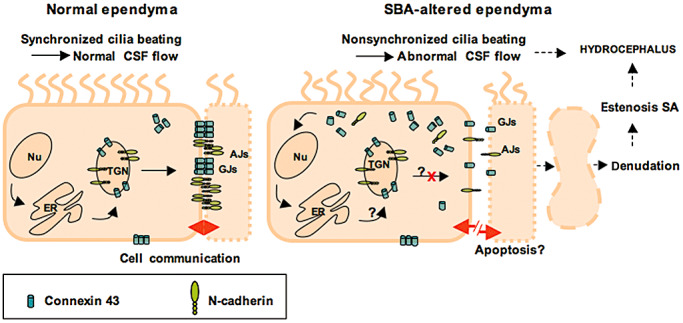
Line drawing of proposed mechanisms underlying ependymal denudation and abnormal CSF flow in the Sylvius aqueduct (SA) of spina bifida aperta (SBA) patients. The ependyma of the SA of control foetuses and the non‐altered regions of the SA of SBA patients display a normal expression and a normal transport to the lateral plasma membrane of the junction proteins N‐cadherin and connexin 43 (left panel). This results in normal gap and adherent junctions. In the altered SA regions of SBA patients, N‐cadherin and connexin 43 are expressed but their transport to the lateral plasma membrane appears impaired. Subsequently, N‐cadherin and connexin 43 may abnormally accumulate in the cytoplasm, whereas functional adherent and gap junctions fail (right panel). All together, this may induce: (i) ependymal denudation, aqueduct stenosis and CSF obstruction; (ii) nonsynchronized cilia beating, abnormal CSF flow, and may finally contribute to (iii) hydrocephalus. Abbreviations: AJ = adherent junction; GJ = gap junctions; ER = rough endoplasmic reticulum; Nu = cell nucleus; TGN = trans‐Golgi network.
In full‐term human foetal SBA, ependymal denudation is still an ongoing process
In full‐term SBA foetuses (37‐, 39‐ and 40‐week GA), early and late stages of ependymal denudation are concurrently present indicating that, at these stages, the ependymal denudation is still an ongoing process. In these foetuses, it may be suggested that such a process would have continued after birth. In the mutant hyh mice, the timely program of ependymal denudation starts during the foetal life and ends by the end of the first postnatal week 33, 42. Only when this program is completed, the obliteration of SA occurs, triggering a severe HC. Future histological investigations in human neonatal SBA patients may help to elucidate whether or not, similar mechanisms contribute to progressive high‐pressure HC.
ACKNOWLEDGMENTS
The authors whish to thank Mrs W. Kunst and Mrs den J. Dunnen‐Briggs for their administrative help and correction of the English grammar. DAS and WFAdD were supported by Jan Kornelis de Cock Stichting, the Netherlands; EMR was supported by Grants from Fondecyt 1070241, Chile. LFB was supported by Grants from Fondecyt 11090373, Chile. The monoclonal antibody against NCAM, developed by J. M. Hessel and J. Dodd, was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA 52242.
REFERENCES
- 1. Baker DW, Vinters HV (1984) Hydrocephalus with cerebral aqueductal dysgenesis and craniofacial anomalies. Acta Neuropathol (Berl) 63:170–173. [DOI] [PubMed] [Google Scholar]
- 2. Banizs B, Pike MM, Millican CL, Ferguson WB, Komlosi P, Sheetz J et al (2005) Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 132:5329–5339. [DOI] [PubMed] [Google Scholar]
- 3. Barf HA, Verhoef M, Jennekens‐Schinkel A, Gooskens RHJM, Prevo AJH (2003) Cognitive status of young adults with spina bifida. Dev Med Child Neurol 45:813–820. [DOI] [PubMed] [Google Scholar]
- 4. Bátiz LF, González CA, Ortloff A, Oliver C, Rodríguez S, Rodríguez EM (2009) Defective cell‐cell adhesion in hyh mutant mice: a pathogenic mechanism underlying cerebral ventricular surface disruption and hydrocephalus? Cerebrospinal Fluid Res 6(Suppl. 1):S24. [Google Scholar]
- 5. Cameron AH (1957) The Arnold‐Chiari and otherneuroanatomical malformations associated with spina bifida. J Pathol Bacteriol 73:195–211. [Google Scholar]
- 6. Cappello S, Attardo A, Wu X, Iwasato T, Itohara S, Wilsch‐Brauninger M et al (2006) The Rho‐GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat Neurosci 9:1099–1107. [DOI] [PubMed] [Google Scholar]
- 7. Chenn A, Zhang YA, Chang BT, McConnell SK (1998) Intrinsic polarity of mammalian neuroepithelial cells. Mol Cell Neurosci 11:183–193. [DOI] [PubMed] [Google Scholar]
- 8. Costa MR, Wen G, Lepier A, Schroeder T, Gotz M (2008) Par‐complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development 135:11–22. [DOI] [PubMed] [Google Scholar]
- 9. Dbouk HA, Mroue RM, El‐Sabban ME, Talhouk RS (2009) Connexins: a myriad of functions extending beyond assembly of gap junction channels. Cell Commun Signal 7:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Del Bigio MR (2010) Ependymal cells: biology and pathology. Acta Neuropathol 119:55–73. [DOI] [PubMed] [Google Scholar]
- 11. Del Bigio MR (2010) Neuropathology and structural changes in hydrocephalus. Dev Dis Res Rev 16:16–22. [DOI] [PubMed] [Google Scholar]
- 12. Derangeon M, Bozon V, Defamie N, Peineau N, Bourmeyster N, Sarrouilhe D et al (2009) 5‐HT4 and 5‐HT2 receptors antagonistically influence gap junctional coupling between rat auricular myocytes. J Mol Cell Cardiol 48:220–229. [DOI] [PubMed] [Google Scholar]
- 13. de Wit OA, den Dunnen WF, Sollie KM, Munoz RI, Meiners LC, Brouwer OF et al (2008) Pathogenesis of cerebral malformations in human fetuses with meningomyelocele. Cerebrospinal Fluid Res 5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dominguez‐Pinos MD, Paez P, Jimenez AJ, Weil B, Arraez MA, Perez‐Figares JM et al (2005) Ependymal denudation and alterations of the subventricular zone occur in human fetuses with a moderate communicating hydrocephalus. J Neuropathol Exp Neurol 64:595–604. [DOI] [PubMed] [Google Scholar]
- 15. Fujimoto K, Nagafuchi A, Tsukita S, Kuraoka A, Ohokuma A, Shibata Y (1997) Dynamics of connexins, E‐cadherin and alpha‐catenin on cell membranes during gap junction formation. J Cell Sci 110(Pt 3):311–322. [DOI] [PubMed] [Google Scholar]
- 16. Ganzler‐Odenthal SI, Redies C (1998) Blocking N‐cadherin function disrupts the epithelial structure of differentiating neural tissue in the embryonic chicken brain. J Neurosci 18:5415–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goodenough DA, Goliger JA, Paul DL (1996) Connexins, connexons, and intercellular communication. Annu Rev Biochem 65:475–502. [DOI] [PubMed] [Google Scholar]
- 18. Hatta K, Takagi S, Fujisawa H, Takeichi M (1987) Spatial and temporal expression pattern of N‐cadherin cell adhesion molecules correlated with morphogenetic processes of chicken embryos. Dev Biol 120:215–227. [DOI] [PubMed] [Google Scholar]
- 19. Hori A (1993) A review of the morphology of spinal cord malformations and their relation to neuro‐embryology. Neurosurg Rev 16:259–266. [DOI] [PubMed] [Google Scholar]
- 20. Humphreys P, Muzumdar DP, Sly LE, Michaud J (2007) Focal Cerebral Mantle Disruption in Fetal Hydrocephalus. Pediatr Neurol 36:236–243. [DOI] [PubMed] [Google Scholar]
- 21. Imai F, Hirai S, Akimoto K, Koyama H, Miyata T, Ogawa M et al (2006) Inactivation of λPKClambda results in the loss of adherens junctions in neuroepithelial cells without affecting neurogenesis in mouse neocortex. Development 133:1735–1744. [DOI] [PubMed] [Google Scholar]
- 22. Ivanov DB, Philippova MP, Tkachuk VA (2001) Structure and functions of classical cadherins. Biochemistry (Mosc) 66:1174–1186. [DOI] [PubMed] [Google Scholar]
- 23. Jongen WM, Fitzgerald DJ, Asamoto M, Piccoli C, Slaga TJ, Gros D et al (1991) Regulation of connexin 43‐mediated gap junctional intercellular communication by Ca2+ in mouse epidermal cells is controlled by E‐cadherin. J Cell Biol 114:545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kadowaki M, Nakamura S, Machon O, Krauss S, Radice GL, Takeichi M (2007) N‐cadherin mediates cortical organization in the mouse brain. Dev Biol 304:22–33. [DOI] [PubMed] [Google Scholar]
- 25. Kamiguchi H, Hlavin ML, Lemmon V (1998) Role of L1 in neural development: what the knockouts tell us. Mol Cell Neurosci 12:48–55. [DOI] [PubMed] [Google Scholar]
- 26. Kamiguchi H, Hlavin ML, Yamasaki M, Lemmon V (1998) Adhesion molecules and inherited diseases of the human nervous system. Annu Rev Neurosci 21:97–125. [DOI] [PubMed] [Google Scholar]
- 27. Klezovitch O, Fernandez TE, Tapscott SJ, Vasioukhin V (2004) Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes Dev 18:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lapras C, Guilburd JN, Patet JD (1988) [Spina bifida aperta myelomeningocele. Chiari's malformation type II]. Neurochirurgie 1:53–58. [PubMed] [Google Scholar]
- 29. Ma X, Bao J, Adelstein RS (2007) Loss of cell adhesion causes hydrocephalus in nonmuscle myosin II‐B‐ablated and mutated mice. Mol Biol Cell 18:2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Macfarlane A, Maloney AF (1957) The appearance of the aqueduct and its relationship to hydrocephalus in the Arnold‐Chiari malformation. Brain 80:479–491. [DOI] [PubMed] [Google Scholar]
- 31. Meyer RA, Laird DW, Revel JP, Johnson RG (1992) Inhibition of gap junction and adherens junction assembly by connexin and A‐CAM antibodies. J Cell Biol 119:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oka C, Matsuda H, Sarai N, Noma A (2006) Modeling the calcium gate of cardiac gap junction channel. J Physiol Sci 56:79–85. [DOI] [PubMed] [Google Scholar]
- 33. Paez P, Batiz LF, Roales‐Bujan R, Rodriguez‐Perez LM, Rodriguez S, Jimenez AJ et al (2007) Patterned neuropathologic events occurring in hyh congenital hydrocephalic mutant mice. J Neuropathol Exp Neurol 66:1082–1092. [DOI] [PubMed] [Google Scholar]
- 34. Perez Velazquez JL, Carlen PL (2000) Gap junctions, synchrony and seizures. Trends Neurosci 23:68–74. [DOI] [PubMed] [Google Scholar]
- 35. Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC (2003) Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev 83:1359–1400. [DOI] [PubMed] [Google Scholar]
- 36. Sarnat HB (1995) Ependymal reactions to injury. A review. J Neuropathol Exp Neurol 54:1–15. [DOI] [PubMed] [Google Scholar]
- 37. Sival DA, Verbeek RJ, Brouwer OF, Sollie KM, Bos AF, den Dunnen WF (2008) Spinal hemorrhages are associated with early neonatal motor function loss in human spina bifida aperta. Early Hum Dev 84:423–431. [DOI] [PubMed] [Google Scholar]
- 38. Takano T, Rutka JT, Becker LE (1996) Overexpression of nestin and vimentin in ependymal cells in hydrocephalus. Acta Neuropathol 92:90–97. [DOI] [PubMed] [Google Scholar]
- 39. Takeuchi IK, Murakami U (1979) Two types of congenital hydrocephalus induced in rats by X‐irradiation in utero: electron microscopic study on the telencephalic wall. J Anat 128:693–708. [PMC free article] [PubMed] [Google Scholar]
- 40. Tullio AN, Bridgman PC, Tresser NJ, Chan CC, Conti MA, Adelstein RS et al (2001) Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II‐B heavy chain. J Comp Neurol 433:62–74. [DOI] [PubMed] [Google Scholar]
- 41. Vinck A, Mullaart R, Rotteveel J, Maassen B (2009) Neuropsychological assessment of attention in children with spina bifida. Cerebrospinal Fluid Res 28:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wagner C, Batiz LF, Rodriguez S, Jimenez AJ, Paez P, Tome M et al (2003) Cellular mechanisms involved in the stenosis and obliteration of the cerebral aqueduct of hyh mutant mice developing congenital hydrocephalus. J Neuropathol Exp Neurol 62:1019–1040. [DOI] [PubMed] [Google Scholar]
- 43. Wang Y, Rose B (1997) An inhibition of gap‐junctional communication by cadherins. J Cell Sci 110:301–309. [DOI] [PubMed] [Google Scholar]
- 44. Wei CJ, Francis R, Xu X, Lo CW (2005) Connexin43 associated with an N‐cadherin‐containing multiprotein complex is required for gap junction formation in NIH3T3 cells. J Biol Chem 280:19925–19936. [DOI] [PubMed] [Google Scholar]
- 45. Wippold FJ, Perry A (2006) Neuropathology for the neuroradiologist: rosettes and pseudorosettes. Am J Neuroradiol 27:488–492. [PMC free article] [PubMed] [Google Scholar]
- 46. Wong EV, Kenwrick S, Willems P, Lemmon V (1995) Mutations in the cell adhesion molecule L1 cause mental retardation. Trends Neurosci 18:168–172. [DOI] [PubMed] [Google Scholar]
- 47. Wünschmann A, Oglesbee M (2001) Periventricular Changes Associated with Spontaneous Canine Hydrocephalus. Vet Pathol 38:67–73. [DOI] [PubMed] [Google Scholar]
- 48. Xu X, Li WE, Huang GY, Meyer R, Chen T, Luo Y et al (2001) N‐cadherin and Cx43alpha1 gap junctions modulates mouse neural crest cell motility via distinct pathways. Cell Commun Adhes 8:321–324. [DOI] [PubMed] [Google Scholar]