

Abstract
Sudden unexpected death in epilepsy (SUDEP) is the most frequent epilepsy‐related cause of death and is characterized by an absence of any identifiable cause of death at post‐mortem, suggesting an underlying arrhythmogenic predisposition. This study sought to identify SUDEP cases in a review of post‐mortem records and to undertake genetic studies in key familial long QT syndrome (LQTS) genes. All autopsies performed from 1993‐2009 at a forensic centre in Sydney, Australia were reviewed and SUDEP cases identified. DNA was extracted from post‐mortem blood and the three most common LQTS genes, ie, KCNQ1, KCNH2 (HERG) and SCN5A, were amplified and analyzed. Sixty‐eight SUDEP cases were identified (mean age of 40 ± 16 years). Genetic analysis revealed 6 (13%) non‐synonymous (amino acid changing) variants in KCNH2 (n = 2) and SCN5A (n = 4), all previously reported in LQTS patients. Specifically, KCNH2 Arg176Trp and SCN5A Pro1090Leu were identified once in SUDEP cases and absent in control alleles. Both DNA variants have been previously identified in the pathogenesis of LQTS. The cause of SUDEP is currently unknown. Our results indicate that investigation of key ion channel genes should be pursued in the investigation of the relationship between epilepsy and sudden death.
Keywords: arrhythmias, genetics, ion channel, SUDEP
INTRODUCTION
Epilepsy is one of the most prevalent neurological conditions. People with epilepsy have a higher risk of mortality compared with healthy individuals 1, 2. While a proportion of these deaths have been attributed to suicide, accident or convulsive status epilepticus (3), a subgroup of patients with epilepsy die suddenly, in otherwise apparently good health, from an unknown cause of death. This phenomenon describes sudden unexpected death in epilepsy (SUDEP) and, with a reported occurrence of up to 18% of all deaths in people with epilepsy 4, 5, is the most frequent epilepsy‐related cause of death (6).
A number of risk factors for SUDEP have been proposed including young age, presence of generalized tonic‐clonic seizures, poor anti‐epileptic drug (AED) compliance, use of multiple AEDs, duration of the seizure disorder ranging from 15–20 years and early onset of epilepsy (7). The exact mechanisms underlying SUDEP remain unknown. Seizure‐related abnormalities of respiratory and cardiac function have both been implicated as possible contributors (8), and the most commonly suggested terminal event is a cardiac arrhythmia during and between seizures. Notably, SUDEP is characterized by an absence of any identifiable structural cause of death at post‐mortem, suggesting that an underlying arrhythmogenic predisposition may exist 9, 10.
The most common proposed pathogenic mechanism underlying sudden unexplained death is heritable arrhythmogenic syndromes, or cardiac channelopathies, such as familial long QT syndrome (LQTS). LQTS associated with syncope, seizures and sudden cardiac death (SCD) is caused by mutations in more than 10 genes, of which 8 encode ion channels and their subunits 11, 12, 13, 14, 15, 16, 17, 18, 19, 20. Over 80% of these LQTS mutations are identified in three genes encoding cardiac potassium or sodium ion channels; namely KCNQ1, KCNH2 (HERG) and SCN5A 21, 22. Alterations in these ion channel genes may result in a prolonged QT interval, ultimately leading to an increased susceptibility to ventricular arrhythmias and SCD. A genetic predisposition to the development of a cardiac arrhythmia may also represent a possible pathogenic mechanism in the sudden death of patients with epilepsy.
This study sought to characterize a large cohort of SUDEP cases from a review of post‐mortem reports and to identify variants in the three most common LQTS genes that may contribute to the development of cardiac arrhythmias and sudden death in SUDEP.
METHODS
Study cohort
Post‐mortem reports over a 16‐year period from 1993 to 2009 at the Department of Forensic Medicine, New South Wales (NSW), Australia, were reviewed in detail. The ISYS Query database system (ISYS Search Software, Sydney, NSW) was used to identify SUDEP cases by performing a search for all autopsy reports containing the words “epilepsy,”“sudden” and “death.” Records matching this word search were extracted and, in each case, all available demographic, clinical and autopsy data were collected. Police and ambulance reports were also examined. SUDEP cohort inclusion criteria included: a known history of epilepsy (evidence of AED therapy, reported in medical history), died suddenly and unexpectedly, and the post‐mortem examination revealed no structural, non‐cardiac or toxicological cause of death. Cases with “SUDEP” reported as the primary cause of death were considered definite SUDEP, while cases with a primary cause of death reported as either “epilepsy,”“complications of epilepsy,”“consistent with epilepsy,”“sudden death in association with seizure disorder” or “undetermined” were considered possible SUDEP. This study was approved by the Office of the NSW State Coroner and performed in accordance with institutional human ethics guidelines.
Genetic analysis
Genomic DNA was prepared from post‐mortem blood using the QIAamp DNA Midi Kit (Qiagen, Germantown, MD, USA). The coding regions and intron/exon boundaries of the three most common LQTS genes, i.e. the cardiac voltage‐gated potassium channels KCNQ1 and KCNH2, and the cardiac voltage‐gated sodium channel SCN5A were analyzed for DNA sequence variants. Primers flanking the intron/exon boundaries were designed to amplify protein‐encoding exons of KCNQ1, KCNH2 and SCN5A, in conjunction with previously published SCN5A primers 20, 23. Polymerase chain reaction (PCR) products were analyzed for sequence variants using high‐resolution melt (HRM) analysis (24) or DNA sequencing (25), as previously described. In brief, primers for HRM were designed using the LightScanner Design Software v1.0.R.84 (Idaho Technology, Inc., Salt Lake City, UT, USA). Genomic DNA was amplified using HRM Mastermix (TrendBio, Melbourne, Victoria, Australia) and PCR amplicons analyzed using the 96‐well LightScanner (Idaho Technology, Inc.) and LightScanner Software v2.0. Samples with abnormal HRM profiles were re‐amplified and sequenced to identify DNA variants. PCR products for sequencing were visualized on a 2% agarose gel, DNA sequenced, then analyzed using Sequencher v4.8 (Gene Codes Corp, Ann Arbor, MI, USA).
Genotyping of non‐synonymous sequence variants using direct DNA sequencing or restriction digestion was performed in at least 340 control alleles. The following criteria were investigated for each non‐synonymous variant to determine possible pathogenicity: absence of the variant in at least 340 healthy control alleles; conservation level of the amino acid residue among orthologous proteins; prediction of pathogenicity using the on‐line resource PolyPhen, which predicts the possible impact of an amino acid substitution on the structure and function of a human protein on the basis of three‐dimensional structure and multiple alignment of homologous sequences (http://genetics.bwh.harvard.edu/pph/).
RESULTS
A comprehensive review of post‐mortem reports from 1993 to 2009 identified a total of 68 SUDEP cases. Twenty‐two cases were reported as “definite SUDEP” and the remaining 46 were considered “possible SUDEP.” All these cases displayed features typical of SUDEP, including previous report of a healthy state prior to death, died suddenly and unexpectedly, normal toxicology and anatomical histopathology, and no cause of death identified at post‐mortem. The mean age of the SUDEP cohort was 40 ± 16 years (range 5–82 years) with a male predominance of 2:1. Forty cases (62%) were taking AED therapy and 64 cases (94%) were unwitnessed events, were in good health within 24 h of discovery and found deceased in bed. The specific cause of epilepsy was stated in four cases and attributed to motor vehicle accidents or head injuries. The characteristics of these 68 cases are summarized in Table 1.
Table 1.
Clinical characteristics of 68 SUDEP cases identified.
| Case | Age | Gender | Definite/possible SUDEP | Anti‐epileptic drug | Other |
|---|---|---|---|---|---|
| 1 | 5 | F | Possible | Sodium Valproate | Frequent seizure episodes with recent seizure requiring hospitalization |
| 2 | 6 | F | Possible | Epilepsy diagnosed at age 5 months; seizures difficult to control with medication; three seizures prior to death | |
| 3 | 7 | M | Possible | Diazepam | Cerebral palsy |
| 4 | 12 | M | Possible | Benign rolandic epilepsy; no prescribed anti‐epileptic therapy; seizures during sleep | |
| 5 | 17 | F | Possible | ||
| 6 | 19 | M | Definite | Lamotrigine | Epilepsy diagnosed at age 11 years; unwitnessed seizures every 2–3 months and often at night; last seizure 2 weeks prior to death |
| 7 | 21 | M | Possible | Lamotrigine | |
| 8† | 23 | M | Possible | Lamotrigine | History of poorly controlled epilepsy; complained of dizziness prior to death; heavy smoker |
| Sodium Valproate | |||||
| 9 | 25 | F | Possible | Sodium Valproate | Intellectual disability; schizophrenia |
| Topiramate | |||||
| 10 | 25 | F | Possible | Frequent seizure episodes; family history of seizures | |
| 11 | 25 | F | Possible | Sodium Valproate | |
| 12 | 25 | F | Definite | Sodium Valproate | Last seizure 4 months prior to death |
| 13 | 26 | M | Possible | Epilepsy diagnosed 3–4 years prior to death; no prescribed anti‐epileptic therapy; epilepsy controlled by diet | |
| 14 | 26 | M | Definite | Carbamazepine Sodium Valproate | Frequent seizure episodes; intellectual disability |
| 15 | 27 | F | Possible | Bipolar disease; alcoholic; insomnia; unwell prior to death | |
| 16 | 27 | F | Possible | Carbamazepine | |
| 17 | 28 | F | Possible | Carbamazepine | Grand mal epilepsy diagnosed at age 24 years; last seizure 2 days prior to death |
| 18 | 29 | M | Possible | Carbamazepine Lamotrigine | Obesity; hypertension; seizure prior to death |
| Sodium Valproate | |||||
| 19 | 30 | F | Possible | Carbamazepine | Unwell the evening prior to death |
| 20 | 31 | M | Definite | Carbamazepine Vigabratin | Mild asthma |
| 21 | 33 | M | Possible | Lamotrigine | Epilepsy diagnosed at age 17 years |
| Sodium Valproate | |||||
| 22 | 32 | M | Definite | Carbamazepine | Attention deficit disorder |
| Sodium Valproate | |||||
| 23 | 34 | M | Definite | Carbamazepine | Epilepsy diagnosed at age 6 years; frequent seizure episodes |
| Sodium Valproate | |||||
| 24 | 34 | M | Possible | Phenytoin | Slight mental disability |
| 25‡ | 34 | M | Possible | Carbamazepine | Post‐traumatic seizure disorder following motor vehicle accident |
| 26 | 34 | M | Definite | Phenytoin | Long history of epilepsy; asthma; marijuana user; non‐compliant with anti‐epileptic therapy and preferred herbal treatment |
| 27 | 35 | F | Possible | Sodium Valproate | Dysmorphia; mental disability; partially blind; type 2 diabetes |
| 28 | 36 | F | Definite | Lamotrigine | Epilepsy diagnosed at age 23 years; non‐compliant with anti‐epileptic therapy 6 months prior to death; fibromyalgia |
| 29 | 36 | F | Possible | ||
| 30 | 38 | M | Possible | Diabetes | |
| 31† | 39 | M | Definite | Phenytoin | Epilepsy diagnosed at age 34 years; 10 reported seizure episodes |
| 32 | 40 | F | Definite | Oxcarbazepine | Epilepsy diagnosed at age 12 years; partially deaf; frequent seizure episodes at night |
| 33 | 40 | M | Possible | Lamotrigine | Hypertension; hyperlipidemia |
| 34 | 41 | F | Possible | Phenytoin | Intellectual disability; cerebral palsy; blind |
| 35 | 41 | M | Definite | Lamotrigine | |
| Sodium Valproate | |||||
| 36‡ | 41 | M | Possible | Post‐traumatic seizure disorder following motor vehicle accident 7 years ago | |
| 37 | 42 | M | Definite | Phenytoin | Epilepsy diagnosed since birth; non‐compliant with anti‐epileptic therapy; consumed alcohol against medical advice |
| Vigabratin | |||||
| 38 | 42 | M | Definite | Sodium Valproate | Severe epilepsy; severe seizures once a month; schizophrenia; heavy smoker |
| 39 | 42 | M | Definite | Phenytoin | Seizures each week; alcohol abuse; heavy smoker |
| 40 | 42 | M | Possible | Carbamazepine | Hepatitis C positive; intravenous drug user |
| 41 | 43 | M | Possible | Carbamazepine | Headaches and vomiting prior to death |
| 42† | 43 | M | Possible | Seizure prior to death | |
| 43 | 44 | M | Possible | Lamotrigine | Frequent seizure episodes |
| Oxcarbazepine | |||||
| 44 | 44 | M | Possible | Carbamazepine | Epilepsy diagnosed at age 14 years; seizure prior to death |
| Phenytoin | |||||
| Sodium Valproate | |||||
| 45 | 44 | M | Definite | Frequent seizure episodes; heavy alcohol abuse | |
| 46 | 45 | M | Possible | Seizure and chest pain prior to death | |
| 47 | 45 | M | Possible | Carbamazepine | Grand mal epilepsy; non‐compliant with anti‐epileptic therapy over Christmas caused by alcohol consumption |
| 48 | 46 | M | Possible | Last seizure at age 14 years; intellectual impairment caused by brain damage at birth; sleep apnea; depression | |
| 49 | 47 | M | Definite | Sodium Valproate | Heavy smoker |
| 50 | 48 | M | Possible | Sodium Valproate | Last seizure 5 weeks prior to death; depression with suicidal ideation |
| 51 | 48 | M | Definite | Phenobarbitone | Grand mal epilepsy; viral encephalitis at age 14 years; obesity |
| Phenytoin | |||||
| 52‡ | 48 | M | Definite | Sodium Valproate | Post‐traumatic seizure disorder following motor vehicle accident at age 18 years; smoker |
| 53† | 51 | M | Possible | Phenytoin | Behavioral problems; moderate developmental disability |
| Lamotrigine | |||||
| 54† | 52 | F | Possible | Sodium Valproate | No seizure episode 12 months prior to death |
| Topiramate | |||||
| 55† | 52 | M | Possible | Epilepsy diagnosed at age 16 years; non‐compliant with anti‐epileptic therapy; frequent seizure episodes; last seizure 18 months prior to death | |
| 56 | 53 | M | Definite | Phenytoin | Epilepsy diagnosed at age 15 years |
| Sodium Valproate | |||||
| 57 | 53 | M | Possible | Carbamazepine | |
| Phenytoin | |||||
| 58‡ | 56 | M | Possible | Post‐traumatic seizure disorder following head injury; alcohol abuse; hepatitis C infection | |
| 59 | 56 | M | Definite | Sodium Valproate | Diagnosed with intracerebral hemorrhage after which developed seizure disorder; liver cancer |
| 60† | 59 | M | Possible | Epilepsy diagnosed at age 58 years; frequent seizure episodes; type 2 diabetes; hypertension; hypercholesterolemia | |
| 61 | 60 | F | Definite | Grand mal epilepsy; hypertension; severe encephalitis | |
| 62 | 60 | M | Possible | Carbamazepine | Controlled hypoglycemia by diet |
| 63 | 61 | F | Possible | Heavy alcohol consumption | |
| 64 | 63 | F | Possible | Carbamazepine | Epilepsy diagnosed at age 16 years |
| Phenytoin | |||||
| 65 | 70 | M | Definite | Carbamazepine | |
| Phenytoin | |||||
| 66 | 72 | M | Possible | Phenytoin | |
| 67 | 79 | F | Possible | Sodium Valproate | Hypertension |
| 68 | 82 | F | Possible | Sodium Valproate | Hypertension |
Non‐synonymous variants were found in these SUDEP cases.
The cause of epilepsy was known caused by a previous motor vehicle accident or head injury.
Post‐mortem blood was available for 48 of the 68 cases. Genetic analysis of the most common LQTS‐causing genes revealed; 11 synonymous variants in KCNQ1 (n = 2), KCNH2 (n = 3) and SCN5A (n = 6); and 6 non‐synonymous variants in KCNH2 (n = 2) and SCN5A (n = 4) (Table 2). The non‐synonymous variants included Arg176Trp and Arg1047Leu in KCNH2, found in one and four SUDEP cases, respectively, and Ala572Asp, Pro1090Leu and Pro2006Ala in SCN5A, each detected once in three separate SUDEP cases (Table 3). Interestingly, one case was heterozygous for both the KCNH2 Arg1047Leu and SCN5A Ala572Asp rare variants.
Table 2.
DNA variants in LQTS genes identified in SUDEP cases.
| Gene | SNP | Exon | Amino acid change | MAF (%) | ||
|---|---|---|---|---|---|---|
| SUDEP (n = 48) | Controls (n = 170) | dbSNP European controls† | ||||
| KCNQ1 | rs1057128 | 13 | Ser546Ser | 20.8 | 11.7–20.8 | |
| rs11601907 | 16 | Tyr662Tyr | 25.0 | 30.6 | ||
| KCNH2 | rs36210422 | 4 | Arg176Trp | 1.0 | 0 | 0–1.1 |
| rs740952 | 6 | Ile489Ile | 9.4 | 14.3–19.1 | ||
| rs33959111 | 7 | Leu564Leu | 45.8 | 27.7–33.3 | ||
| rs1137617 | 8 | Tyr652Tyr | 37.5 | 44.4–44.6 | ||
| rs36210421 | 13 | Arg1047Leu | 4.2 | 2.9 | 1.8 | |
| SCN5A | rs6599230 | 2 | Ala29Ala | 19.8 | 11.1–28.3 | |
| rs45533640 | 3 | His118His | 1.0 | NA | ||
| rs1805124 | 12 | His558Arg | 19.8 | 9.5–19.1 | ||
| rs45522138 | 12 | Leu561Leu | 1.0 | NA | ||
| rs36210423 | 12 | Ala572Asp | 1.0 | 0.6 | 0.4 | |
| rs7430407 | 17 | Glu1061Glu | 6.3 | 12.5–14.9 | ||
| rs1805125 | 18 | Pro1090Leu | 1.0 | 0 | NA | |
| rs41311123 | 23 | Gly1406Gly | 1.0 | 4.3 | ||
| rs1805126 | 28 | Asp1819Asp | 30.2 | 31.7–38.9 | ||
| rs45489199 | 28 | Pro2006Ala | 1.0 | 0.3 | NA | |
Multiple European control groups have been reported and are available in dbSNP. MAF shows the frequency range, ie. the lowest and highest frequency for each variant.
MAF, minor allele frequency. The percentage provided is the allelic frequency for the minor allele; NA, MAF is not available in a European control population.
Bold indicates likely pathogenic mutations.
Table 3.
Clinical characteristics of SUDEP cases with protein‐changing variants.
| Case | Gene variant | Age | Gender | Cause of death at autopsy | Anti‐epileptic drug | Other |
|---|---|---|---|---|---|---|
| 1 | KCNH2 Arg176Trp | 39 | M | SUDEP | Phenytoin | Epilepsy diagnosed at age 34 years |
| 10 reported seizure episodes | ||||||
| 2 | KCNH2 Arg1047Leu | 51 | M | Complications of epilepsy | Lamotrigine | Behavioral problems |
| Phenytoin | Moderate developmental disability | |||||
| 3 | KCNH2 Arg1047Leu | 52 | M | Undetermined | Epilepsy diagnosed at age 16 years | |
| Non‐compliant with anti‐epileptic therapy | ||||||
| Frequent seizure episodes | ||||||
| Last seizure 18 months prior to death | ||||||
| 4 | KCNH2 Arg1047Leu | 59 | M | Undetermined | Epilepsy diagnosed at age 58 years | |
| Frequent seizure episodes | ||||||
| Type 2 diabetes | ||||||
| Hypertension | ||||||
| Hypercholesterolemia | ||||||
| 5 | KCNH2 Arg1047Leu | 52 | F | Undetermined | Sodium Valproate | No seizure episode 12 months prior to death |
| SCN5A Ala572Asp | Topiramate | |||||
| 6 | SCN5A Pro1090Leu | 23 | M | Undetermined | Lamotrigine | History of poorly controlled epilepsy |
| Complained of dizziness prior to death | ||||||
| Sodium Valproate | Heavy smoker | |||||
| 7 | SCN5A Pro2006Ala | 43 | M | Undetermined | Seizure prior to death |
All non‐synonymous variants were genotyped in a control population. The KCNH2 Arg1047Leu variant was identified in 10 of 340 control alleles (2.9%), and was predicted to be “possibly damaging” by PolyPhen. The SCN5A Ala572Asp and Pro2006Ala variants were, respectively, found in 2 of 340 (0.6%) and 1 of 338 (0.3%) control alleles, and predicted to be “benign” and “possibly damaging” by PolyPhen. Of most significance, two variants, KCNH2 Arg176Trp (Figure 1) and SCN5A Pro1090Leu (Figure 2) were absent in at least 340 control alleles, respectively, and both were predicted to be “possibly damaging” by PolyPhen (Table 2).
Figure 1.

KCNH2 Arg176Trp mutation in SUDEP. A. Sequence chromatograms and B. amino acid conservation of Arg176Trp across species. C. Schematic representation of the linear topology of the KCNH2 protein shows both the location of critical domains and variants identified in cases of SUDEP. PAS = Per‐Arnt‐Sim domain; PAC = PAS‐associated C‐terminal domain; TSR = transmembrane spanning region; cNBD = cyclic nucleotide‐binding domain.
Figure 2.
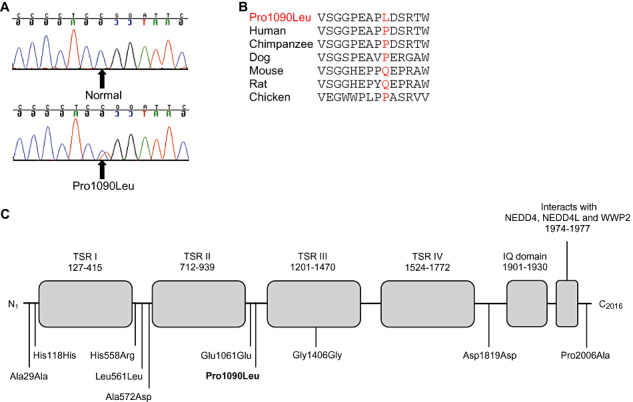
SCN5A Pro1090Leu mutation in SUDEP. A. Sequence chromatograms and B. amino acid conservation of Pro1090Leu across species. C. Linear topology of the SCN5A protein with the location of each variant found in SUDEP. TSR = transmembrane spanning region; IQ = calmodulin binding region; NEDD4 = E3 ubiquitin‐protein ligase NEDD4; NEDD4L = E3 ubiquitin‐protein ligase NEDD4‐like; WWP2 = NEDD4‐like E3 ubiquitin‐protein ligase WWP2.
DISCUSSION
This study describes a large cohort of SUDEP cases with genetic screening of the three most common genes known to cause familial LQTS, i.e. KCNQ1, KCNH2 and SCN5A. We identified 68 cases of SUDEP and, to our knowledge, this represents the largest SUDEP cohort reported to date. The high incidence of unwitnessed events with cases found deceased in bed (94%), as well as the frequent use of AED therapy (62%), supports previous reports that these are risk factors for SUDEP. Genetic analysis identified six non‐synonymous variants (13%), of which at least two, KCNH2 Arg176Trp and SCN5A Pro1090Leu, are likely to be involved directly in pathogenesis. These findings suggest a possible pathogenic link between SUDEP, mutations in ion channel genes and familial LQTS.
The exact mechanisms underlying the pathogenesis of SUDEP are currently unclear. A genetic susceptibility to the development of a cardiac arrhythmia, independent of (or related to) the epilepsy phenotype, could exist and increase the risk of SCD. One previous single case report has explored the possible role of LQTS gene mutations specifically in SUDEP. Mutation analysis of the LQTS‐associated genes KCNQ1, KCNH2, SCN5A, mink and MiRP1 revealed a novel SCN5A Arg523Cys variant in one of four SUDEP cases investigated (26). After genetic screening of our current larger SUDEP cohort, 17 variants were found in the KCNQ1, KCNH2 and SCN5A genes, of which 6 (13%) were rare non‐synonymous variants in KCNH2 (Arg176Trp and Arg1047Leu) and SCN5A (Ala572Asp, Pro1090Leu and Pro2006Ala). All of these six non‐synonymous variants identified in the current study have been reported previously in cases of LQTS 22, 27, 28, 29, 30, 31, 32.
The KCNH2 variant at codon 176 (Arg176Trp) is a non‐conservative substitution of a polar and positively charged arginine residue for a non‐polar and neutral tryptophan residue in a highly conserved region of the KCNH2 protein N‐terminus, and predicted to be probably damaging by the Polyphen prediction program (Figure 1b and 1c). The KCNH2176Trp allele was absent in 360 control alleles, and an online database of single nucleotide polymorphisms (SNPS) lists this allele as absent in the DNA of an additional 555 healthy individuals of European descent, but present once in a further cohort of 46 healthy Europeans. Interestingly, the KCNH2 Arg176Trp variant is reported as one of four common founder mutations associated with a prolonged QT interval in Finnish families with LQTS 29, 31, and was also reported in a case of sudden unexplained death (33). Moreover, functional studies have shown that the Arg176Trp substitution alters channel function in vitro, causing an acceleration in channel deactivation and reducing potassium current density, resulting in a prolonged QT interval (29). Collectively, these results implicate the KCNH2 Arg176Trp variant in prolongation of the QT interval, which may lead to a fatal arrhythmia. The SCN5A variant at codon 1090 (Pro1090Leu) is a conservative substitution of a proline to a leucine at a non‐conserved residue located between the TSRII and TSRII protein domains (Figure 2). The SCN5A 1090Leu allele was absent in 340 control alleles. This SCN5A 1090Leu variant has been previously reported in Japanese LQTS patients and was considered to be an Asian‐specific polymorphism 30, 34. Furthermore, functional studies showed that the 1090Leu allele caused a shift in channel function, which is dependent upon the SCN5A splice variant (35). The presence of this protein‐changing variant in the specific SUDEP case in the current study could be associated with an increased susceptibility to sudden death.
One SUDEP case was found to carry two rare non‐synonymous variants in KCNH2 Arg1047Leu and SCN5A Ala572Asp. Both variants have been reported in cases of LQTS and SIDS, and more importantly Ala572Asp has been identified in women with SCD 22, 27, 28, 31, 32, 36. This is consistent with the findings of our current SUDEP case (Table 1; Case #54), who was a 52 year old female with an undetermined cause of death. Although there has been no report of each variant acting in combination with a second genetic variant, one could speculate that the combined effect of these two protein‐changing variants may increase the risk of sudden death in this epilepsy patient. This “gene dose” effect caused by multiple mutations in the same patient leading to more severe clinical disease has now been reported in a number of other cardiac genetic diseases.
The identification of DNA variants in LQTS‐causing genes in this large SUDEP cohort supports the hypothesis that alterations in the QT interval may trigger life‐threatening ventricular arrhythmias specifically in epilepsy patients. An important recent study reported LQTS patients with a seizure phenotype, i.e. the presence of either a personal or family history of seizures or history of AED therapy were more common in LQTS type 2 compared with all the other subtypes of LQTS (38). In addition, significant prolongation of the QT interval has been reported during seizures in patients with epilepsy (39). The non‐synonymous variants in KCNH2 and SCN5A have been functionally shown to exhibit currents typical of mutations associated with LQTS 27, 29, 35, 36, 37.
Collectively, these findings support a common hypothesis that ion channels regulating the QT interval are likely to play an important role in the predisposition of epilepsy patients to sudden death. It remains to be determined whether these ion channel variants are the genetic cause or an accompanying risk factor in the sudden death of patients with epilepsy. The variants may act in isolation or require the presence of a second genetic factor or environmental influence, such as uncontrolled seizures, QT‐prolonging AEDs, and non‐compliance with AED therapy, to predispose epilepsy patients to malignant arrhythmias and sudden death.
Unfortunately, a limitation of our study was the inability to contact the surviving relatives of these SUDEP cases. All cases were studied in a de‐identified manner as per ethical guidelines and, therefore, there was no available ECG data in the deceased or in family members, and a family history of sudden death, epilepsy and/or LQTS could not be obtained. We were therefore unable to conduct genetic studies in other family members to assess co‐inheritance of disease. However, the positive findings of the current study have provided the proof‐of‐principle to conduct a more comprehensive prospective study of SUDEP cases.
SUDEP is a devastating complication in patients with epilepsy. There is no single risk factor common to all cases and the pathogenic mechanism underlying sudden death in those with epilepsy remains unclear. We report a large cohort of Australian cases of sudden unexpected death in epilepsy and potential pathogenic factors involved in the genetic predisposition to sudden death. Identification of genetic factors that predispose epilepsy patients to the development of cardiac arrhythmias and sudden death will be important in risk factor stratification and provide opportunities for therapeutic intervention to prevent sudden death in individuals with epilepsy.
DISCLOSURES
There are no conflicts of interest declared by the authors.
REFERENCES
- 1. Hauser WA, Hesdorffer DC (1990) Epilepsy: Frequency, Causes and Consequences, Demos: New York. [Google Scholar]
- 2. Nashef L, Fish DR, Garner S, Sander JW, Shorvon SD (1995) Sudden death in epilepsy: a study of incidence in a young cohort with epilepsy and learning difficulty. Epilepsia 36:1187–1194. [DOI] [PubMed] [Google Scholar]
- 3. Gaitatzis A, Sander JW (2004) The mortality of epilepsy revisited. Epileptic Disord 6:3–13. [PubMed] [Google Scholar]
- 4. Ficker DM (2000) Sudden unexplained death and injury in epilepsy. Epilepsia 41(Suppl. 2):S7–12. [DOI] [PubMed] [Google Scholar]
- 5. Walczak TS, Leppik IE, D'Amelio M, Rarick J, So E, Ahman P et al (2001) Incidence and risk factors in sudden unexpected death in epilepsy: a prospective cohort study. Neurology 56:519–525. [DOI] [PubMed] [Google Scholar]
- 6. Nashef L (1997) Sudden unexpected death in epilepsy: terminology and definitions. Epilepsia 38:S6–S8. [DOI] [PubMed] [Google Scholar]
- 7. Tomson T, Walczak T, Sillanpaa M, Sander JW (2005) Sudden unexpected death in epilepsy: a review of incidence and risk factors. Epilepsia 46(Suppl. 11):54–61. [DOI] [PubMed] [Google Scholar]
- 8. Stollberger C, Finsterer J (2004) Cardiorespiratory findings in sudden unexplained/unexpected death in epilepsy (SUDEP). Epilepsy Res 59:51–60. [DOI] [PubMed] [Google Scholar]
- 9. Nashef L, Ryvlin P (2009) Sudden unexpected death in epilepsy (SUDEP): update and reflections. Neurol Clin 27:1063–1074. [DOI] [PubMed] [Google Scholar]
- 10. Surges R, Adjei P, Kallis C, Erhuero J, Scott CA, Bell GS et al (2010) Pathologic cardiac repolarization in pharmacoresistant epilepsy and its potential role in sudden unexpected death in epilepsy: a case‐control study. Epilepsia 51:233–242. [DOI] [PubMed] [Google Scholar]
- 11. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW et al (1999) MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97:175–187. [DOI] [PubMed] [Google Scholar]
- 12. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT (1995) A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80:795–803. [DOI] [PubMed] [Google Scholar]
- 13. Medeiros‐Domingo A, Kaku T, Tester DJ, Iturralde‐Torres P, Itty A, Ye B et al (2007) SCN4B‐encoded sodium channel beta4 subunit in congenital long‐QT syndrome. Circulation 116:134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH et al (2003) Ankyrin‐B mutation causes type 4 long‐QT cardiac arrhythmia and sudden cardiac death. Nature 421:634–639. [DOI] [PubMed] [Google Scholar]
- 15. Plaster NM, Tawil R, Tristani‐Firouzi M, Canun S, Bendahhou S, Tsunoda A et al (2001) Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 105:511–519. [DOI] [PubMed] [Google Scholar]
- 16. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R et al (2004) Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119:19–31. [DOI] [PubMed] [Google Scholar]
- 17. Splawski I, Tristani‐Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT (1997) Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet 17:338–340. [DOI] [PubMed] [Google Scholar]
- 18. Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW et al (2006) Mutant caveolin‐3 induces persistent late sodium current and is associated with long‐QT syndrome. Circulation 114:2104–2112. [DOI] [PubMed] [Google Scholar]
- 19. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ et al (1996) Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12:17–23. [DOI] [PubMed] [Google Scholar]
- 20. Wang Q, Li Z, Shen J, Keating MT (1996) Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics 34:9–16. [DOI] [PubMed] [Google Scholar]
- 21. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL et al (2000) Spectrum of mutations in long‐QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102:1178–1185. [DOI] [PubMed] [Google Scholar]
- 22. Tester DJ, Will ML, Haglund CM, Ackerman MJ (2005) Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2:507–517. [DOI] [PubMed] [Google Scholar]
- 23. Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH, Keating MT (1998) Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics 51:86–97. [DOI] [PubMed] [Google Scholar]
- 24. Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA et al (2010) Mutations in alpha‐actinin‐2 cause hypertrophic cardiomyopathy: a genome‐wide analysis. J Am Coll Cardiol 55:1127–1135. [DOI] [PubMed] [Google Scholar]
- 25. Tu E, Bagnall RD, Duflou J, Lynch M, Twigg SM, Semsarian C (2010) Post‐mortem pathologic and genetic studies in “dead in bed syndrome” cases in type 1 diabetes mellitus. Hum Pathol 41:392–400. [DOI] [PubMed] [Google Scholar]
- 26. Aurlien D, Leren TP, Tauboll E, Gjerstad L (2009) New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure 18:158–160. [DOI] [PubMed] [Google Scholar]
- 27. Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C et al (2007) Prevalence of long‐QT syndrome gene variants in sudden infant death syndrome. Circulation 115:361–367. [DOI] [PubMed] [Google Scholar]
- 28. Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ (2004) Spectrum and frequency of cardiac channel defects in swimming‐triggered arrhythmia syndromes. Circulation 110:2119–2124. [DOI] [PubMed] [Google Scholar]
- 29. Fodstad H, Swan H, Laitinen P, Piippo K, Paavonen K, Viitasalo M et al (2004) Four potassium channel mutations account for 73% of the genetic spectrum underlying long‐QT syndrome (LQTS) and provide evidence for a strong founder effect in Finland. Ann Med 36(Suppl. 1):53–63. [DOI] [PubMed] [Google Scholar]
- 30. Iwasa H, Itoh T, Nagai R, Nakamura Y, Tanaka T (2000) Twenty single nucleotide polymorphisms (SNPs) and their allelic frequencies in four genes that are responsible for familial long QT syndrome in the Japanese population. J Hum Genet 45:182–183. [DOI] [PubMed] [Google Scholar]
- 31. Larsen LA, Andersen PS, Kanters J, Svendsen IH, Jacobsen JR, Vuust J et al (2001) Screening for mutations and polymorphisms in the genes KCNH2 and KCNE2 encoding the cardiac HERG/MiRP1 ion channel: implications for acquired and congenital long Q‐T syndrome. Clin Chem 47:1390–1395. [PubMed] [Google Scholar]
- 32. Paulussen A, Matthijs G, Gewillig M, Verhasselt P, Cohen N, Aerssens J (2003) Mutation analysis in congenital Long QT Syndrome—a case with missense mutations in KCNQ1 and SCN5A. Genet Test 7:57–61. [DOI] [PubMed] [Google Scholar]
- 33. Tester DJ, Ackerman MJ (2007) Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol 49:240–246. [DOI] [PubMed] [Google Scholar]
- 34. Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, Timothy KW et al (2004) Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm 1:600–607. [DOI] [PubMed] [Google Scholar]
- 35. Tan BH, Valdivia CR, Rok BA, Ye B, Ruwaldt KM, Tester DJ et al (2005) Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants. Heart Rhythm 2:741–747. [DOI] [PubMed] [Google Scholar]
- 36. Albert CM, Nam EG, Rimm EB, Jin HW, Hajjar RJ, Hunter DJ et al (2008) Cardiac sodium channel gene variants and sudden cardiac death in women. Circulation 117:16–23. [DOI] [PubMed] [Google Scholar]
- 37. Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M et al (2007) Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation 115:368–376. [DOI] [PubMed] [Google Scholar]
- 38. Johnson JN, Hofman N, Haglund CM, Cascino GD, Wilde AA, Ackerman MJ (2009) Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology 72:224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brotherstone R, Blackhall B, McLellan A (2010) Lengthening of corrected QT during epileptic seizures. Epilepsia 51:221–232. [DOI] [PubMed] [Google Scholar]