

Abstract
Integrative genomics approaches associating DNA structure and transcriptomic analysis should allow the identification of cascades of events relating to tumor aggressiveness. While different genome alterations have been identified in pituitary tumors, none have ever been correlated with the aggressiveness. This study focused on one subtype of pituitary tumor, the prolactin (PRL) pituitary tumors, to identify molecular events associated with the aggressive and malignant phenotypes. We combined a comparative genomic hybridization and transcriptomic analysis of 13 PRL tumors classified as nonaggressive or aggressive. Allelic loss within the p arm region of chromosome 11 was detected in five of the aggressive tumors. Allelic loss in the 11q arm was observed in three of these five tumors, all three of which were considered as malignant based on the occurrence of metastases. Comparison of genomic and transcriptomic data showed that allelic loss impacted upon the expression of genes located in the imbalanced region. Data filtering allowed us to highlight five deregulated genes (DGKZ, CD44, TSG101, GTF2H1, HTATIP2), within the missing 11p region, potentially responsible for triggering the aggressive and malignant phenotypes of PRL tumors. Our combined genomic and transcriptomic analysis underlines the importance of chromosome allelic loss in determining the aggressiveness and malignancy of tumors.
Keywords: aggressiveness of tumors, integrative genomics, loss of 11p, prolactinomas
INTRODUCTION
Cancer is widely accepted as a clonal disease that arises from a single normal cell and progresses because of the accumulation of alterations. The model proposed by Peter C. Nowell in 1976 (29) hypothesized that neoplasia is the consequence of a single primary genetic alteration [mutation of specific genes, loss of heterozygosity, chromosomal rearrangement, copy number alteration (CNA)] which confers a selective proliferative advantage to the cell. The progression from neoplasia to metastasis could then be a result of the stepwise accumulation of alterations (acquired randomly or not) impacting downstream specific signaling pathways. Despite our knowledge of the common occurrence of DNA alterations in different forms of cancers, the driving role of this remains controversial (11). One way of deciphering the events cascade involves an integrative genomic approach performed in the same samples associating DNA structure and transcriptomic analyses.
The prolactin (PRL) pituitary tumor is an interesting model for studying tumor development and progression because it is a slow‐growing tumor and thus allows analysis of the first steps of tumoral development. It originates from a single cell (1) and presents well‐defined phenotypes. Furthermore, the phenotypes, noninvasive, invasive, and invasive–aggressive, are well characterized clinically, histologically, and molecularly 33, 42. The diagnosis of malignancy is still based on the presence of metastasis (26), although the molecular events responsible for tumor progression toward the malignant phenotype remain unknown.
Comparative genomic hybridization (CGH) or microsatellite analysis, performed on all subtypes of pituitary tumors [8, 22, 36, 37, 41 and for review (7)], has revealed alterations on many chromosomes but failed to highlight any specific candidate loci. A more recent CGH analysis (32) performed on 24 sporadic pituitary adenomas showed that the most considerable changes observed occurred on chromosome 11. Despite these interesting descriptive findings, the functional role of these DNA alterations in tumor progression was not assessed.
Comparative transcriptomic studies performed on all types of pituitary tumor have highlighted different deregulated genes and pathways [review in (43)]. However, as all tumor types were combined, the comparison of gene expression was not appropriate. A recent comparative transcriptomic analysis focusing on one subtype of pituitary tumor (PRL tumors) revealed the deregulation of specific genes (42) connected in a signaling pathway associated specifically with aggressiveness (43).
To date, no genome‐wide exploration of DNA structure, coupled with a transcriptomic analysis of benign and aggressive pituitary tumors, has been performed. In this study we compared the genomic DNA alterations in aggressive (including malignant) PRL tumors (A) with those in nonaggressive PRL tumors (NA). Simultaneously, within the same samples, we analyzed the relationship existing between the transcriptomic activity and the DNA CNAs, and proposed a strategy of data filtering to find candidate genes involved in malignancy.
MATERIALS AND METHODS
Tumor samples
The 13 PRL tumors were surgically harvested from seven male and six female patients. All tumors were immunostained with PRL antibody and classified into two pathological groups: nonaggressive (NA: n = 7), including noninvasive and invasive tumors33, 42, and aggressive (A: n = 6), including invasive–aggressive and tumors that were considered as malignant 33, 42. The NA showed no proliferation or invasion markers and were totally removed by surgery. Those patients showed no signs of recurrence after a long follow‐up. The invasive PRL macroadenomas sometimes showed signs of proliferation (few mitoses and low Ki‐67 index) and polysialic acid neural cell adhesion molecule (PSA‐NCAM) expression and were not completely surgically removed: the PRL plasma levels remained high and a long time to recurrence was often observed. The A were characterized by a high proliferation rate [numerous mitoses, high Ki‐67 index, nuclear labeling of pituitary transforming tumor gene (PTTG) and P53], frequent dopamine resistance and a short time to recurrence. Malignancy was based on the occurrence of metastases during clinical follow‐up. Genetic analysis was performed on frozen fragments that had been stored at −80°C (Neurobiotec Bank, Lyon, France). The main clinical, pathological and genetic data are shown in Table 1. The studies were approved by the ethics committee of Lyon, and informed consent was obtained from each patient according to French law.
Table 1.
Pathological and genetic data from patients with PRL pituitary tumors. Abbreviations: A = aggressive PRL tumors; NA = nonaggressive PRL tumors; PRL = prolactin.
| Tumor number | Sex | Age | Tumor size (mm3) | Invasion | Pathological group | Postoperative events | Chromosome gains | Chromosome losses |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 66 | 4620 | Yes | A | Recurrence, death | 3p, 5, 8, 14q, 19p | 11p |
| 2 | F | 54 | 4000 | Yes | A | Recurrence | 4q | 1p, 11p |
| 3 | M | 68 | 5000 | Yes | A carcinoma | Recurrences, metastasis, death | 1q, 3p, 8q, 9, 14q, 19p | 1q, 11 |
| 4 | M | 54 | 6912 | Yes | A carcinoma | Recurrences, metastasis, death | 1q, 5, 15q, 19q | 11, 17p |
| 5 | F | 31 | 864 | Yes | A carcinoma | Recurrences, metastasis | 1q, 8q | 1, 4, 11, 13q, 15q |
| 6 | F | 43 | 4600 | Yes | A | Recurrence | — | — |
| 7 | M | 40 | 8000 | Yes | NA | Persistence | Y | — |
| 8 | M | 41 | 500 | Yes | NA | Persistence | — | 15q, 2p |
| 9 | M | 41 | 2450 | No | NA | Remission | 7, 9 | — |
| 10 | F | 44 | 365 | No | NA | Remission | 3, 5p, 7, 9 | 15q |
| 11 | M | 39 | 13282 | No | NA | Remission | 8, Y | — |
| 12 | F | 30 | 216 | No | NA | Remission | 9 | — |
| 13 | F | 38 | 1000 | No | NA | Remission | 7p, 20 | 13q |
CGH analysis
Genomic DNA was isolated from frozen tumoral and normal pituitary fragments with the QIAamp® DNA micro kit (Qiagen, Hilden, Germany) and was quantified with The Nanodrop® ND‐1000 (NanoDrop, Wilmington, DE, USA); the quality was verified on agarose gel. Genotyping and CNA analysis were performed using the Affymetrix® Genome‐wide human single nucleotide polymorphism (SNP) array 6.0 chip (Affymetrix, Santa Clara, CA, USA) according to the manufacturer's instructions. Arrays were washed using the GeneChip® Fluidics Station 450 (Affymetrix) station and scanned using the GeneChip® scanner 3000 7G (Affymetrix). All pituitary samples and reference genomic DNA, 103 (Affymetrix) were processed in the same batch and hybridized at ProfileXpert platform (Bron, France).
CGH and genotyping data have been deposited in Gene Expression Omnibus under the accession number GSE 22615.
CNAs
Affymetrix CEL files were extracted using the Genotyping Console™ software version 3.0 (Affymetrix). For SNP genotyping, we used the Birdseed (v2.2) analysis algorithm. Accuracy of genotyping was checked by performing a concordance test between processed reference 103 and a preprocessed reference 103 (Affymetrix). The test of concordance showed a 99.79% homology between the two genotypes, indicating consistent platform performance. Moreover, only samples showing a call rate >96% and a Median Absolute Pairwise Difference (MAPD) metric <4 were considered for further analysis. Copy number analysis was performed using Partek® Genomics Suite version 6.4 (Partek, St Louis, MO, USA) following normalization by the invariant set normalization procedure, and signal intensities were computed using PM/MM model‐based expression. Raw copy number data were computed using a batch of 270 normal external control samples from the International HapMap project as a reference. To remove alterations not associated with tumor phenotype, copy number variation was also analyzed on a normal pituitary sample processed simultaneously in the same batch as tumor samples. Inferred copy numbers were predicted using a genomic segmentation algorithm. Only CNAs with cut‐offs at >2.7 copies for gain and <1.3 for loss were considered.
Transcriptomic analysis
Total RNA was extracted from the same human pituitary tumors using Trizol® (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol. For q‐RTPCR, total RNA was subjected to DNAse treatment using an RNeasy® minikit (Qiagen) according to the manufacturer's protocol.
Total RNA yield was measured by spectrophotometry at OD260, the purity confirmed by an A260/A280 ratio of 1.9–2.1 using the Nanodrop® ND‐1000 (NanoDrop), and the sample quality evaluated on nanochips with the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) according to the manufacturer's protocol.
Total RNA (2 µg) was amplified and biotin labeled by a round of in vitro transcription (IVT) with a MessageAmp™ aRNA kit (Ambion, Austin, TX, USA) following the manufacturer's instructions. To confirm process efficiency, all samples were spiked with various concentrations of control synthetic mRNA before amplification. aRNA yield was measured with NanoDrop and the quality assessed on nanochips with the Agilent 2100 Bioanalyzer (Agilent).
Ten micrograms of biotin‐labeled aRNA were fragmented using 5 µL of fragmentation buffer in a final volume of 25 µL, mixed with 235 µL of Amersham hybridization solution (Applied Microarrays, Tempe, AZ, USA) and injected onto CodeLink™ Uniset Human Whole Genome bioarrays (Applied Microarrays, Tempe, AZ, USA) containing 55 000 human oligonucleotide gene probes. Arrays were hybridized overnight at 37°C at 250 rpm in a shaking incubator The slides were washed in stringent Tris‐NaCl‐Tween (TNT) buffer at 46°C for 1 h before incubation for 30 min in 3.4 mL of streptavidin‐cy5 solution (GE Healthcare) as described previously (10). Slides were then washed four times in 240 mL of TNT buffer, rinsed twice in 240 mL of water containing 0.2% Triton® X‐100 (Sigma‐Aldrich®, St Louis, MO, USA), and dried by centrifugation at 640 g before being scanned using a Genepix® 4000B scanner (Axon, Union City, CA, USA) and Genepix® software (Axon), with the laser set at 635 nm, 100% power, and 60% photomultiplier tube voltage. The scanned image files were analyzed using CodeLink™ Expression Analysis software, version 4.0 (Applied Microarrays), which produces both a raw and normalized hybridization signal for each spot on the array.
To account for the difference in relative intensity of the raw hybridization signal on arrays between experiments, CodeLink™ software was used to normalize the signal on each array to the median (median intensity is 1 after normalization), thus allowing better cross‐array comparison. The threshold of detection was calculated using the normalized signal intensity of the 100 negative control probes in the array. Spots with signal intensities below this threshold were considered as “absent.”
The quality of processing was evaluated by generating scatter plots of positive signal distribution. Statistical comparison and filtering were performed using Partek® Genomics Suite version 6.4.
Transcriptomic data have been deposited in Gene Expression Omnibus under the accession number GSE 22812.
Q‐RTPCR
Total RNA (0.5 µg) was reverse transcribed using Moloney murine leukemia virus (MMLV) reverse transcriptase (RT) (Invitrogen). The absence of contaminating genomic DNA in the RT reactions was checked by q‐PCR directly on total RNA.
The cDNA synthesized was measured using q‐RTPCR (SYBR® Green PCR, LightCycler®, Roche Diagnostics Indianapolis, IN, USA) following the manufacturer's recommendations. The LightCycler experimental protocol consisted of an initial Taq activation at 95°C for 10 minutes followed by a touch‐down PCR step of 12 cycles consisting of 15 s at 95°C, 5 s at 68°C and 8 s at 72°C, followed by 43 cycles consisting of 15 s at 95°C, 5 s at 60°C, and 10 s at 72°C, with a single fluorescence measurement. The specificity of PCR amplification was analyzed with a melting curve program (69°C–95°C), with a heating rate of 0.1°C/s and continuous fluorescence measurement. Primers were designed with Primer3 software (Whitehead Institute, Cambridge, MA, USA) and purchased from Eurogentec (Seraing, Belgium). All primers had melting temperatures between 60°C and 62°C, and all the products were 100–150 bp. The primer sequences were forward CCG TAA ACA GTT CCA GCT GAG GGC, reverse GCT CAA CCT CCA GCT GGT ATC AGA GAA (TSG 101); forward AGG GAC ATC CCC GGC AAT CCA, reverse GGG TCT GCC AGA GGC CCA GA (CD44); forward CGG AGG GAT TTG TTC GTG TTG, reverse AGC TCC TTT AGA GGA TAG CAA GT (HTATIP2); forward CAT CGT GGA TAA GGT GCT CA, reverse GAC ACC CTC CTC CTC ATT CA (DGKZ var 4); forward CAC ATG GCA GTC ACG GCG TCT, reverse TCT GCT TGT CTG TCA GGC CAG GT (GTF2H1); forward AG GCA AGA AGC CTG TGG TAG, reverse AG GGG CTG GTT TCT TGG TAG (RPL4); and forward AGC CAC ATG GGG ACC TGC CT, reverse AAC CTC ACT GCT TCC CAC CCC A (SOX6). RPL4 mRNA was used as an internal standard to quantify variations caused by differences in starting mRNA concentrations. The relative mRNA levels for each tissue were computed from the Ct values obtained for the gene of interest, the efficiency of the primer set and RPL4 mRNA levels in samples using RealQuant software (Roche Diagnostics).
Data analysis
Data analysis and filtering was performed upon the microarray data using Partek® Genomics Suite version 6.4. Statistical comparisons were made between A and NA tumor groups. mRNA transcript was considered as differentially expressed when a comparison between the two groups yielded a P‐value < 0.05 in the one‐way ANOVA parametric test and if a variation of 1.5‐fold was observed.
RESULTS
Common imbalanced region in the p arm of chromosome 11 in aggressive and malignant PRL tumors
PRL tumors (n = 13) were classified by radiological, histological, and molecular data into two groups: nonaggressive (NA: n = 7), including noninvasive and invasive tumors 33, 42, and aggressive (A: n = 6), including invasive–aggressive and malignant tumors 33, 42. DNA CNAs were assessed at the genomic level using Genome‐Wide Human SNP 6.0 arrays (Affymetrix) and analyzed with the Partek® Genomics Suite version 6.4 software. For quality monitoring of the analysis, genomic DNA from normal pituitary tissue and from a human genome reference (Genome 103 from Affymetrix) were analyzed in the same batch. As expected, only a small number of discrete CNA occurred in normal or reference samples in comparison with numerous alterations observed in the tumors (Figure 1 and Table 1). The main alterations in NA tumors were found in chromosomes 9, 7, Y, 8, 3 (gains in order of decreasing frequency) and 15q, 2p (losses in order of decreasing frequency). Higher numbers of extensive alterations were detected in the A tumors, principally in chromosomes 8, 19p, 1q, 3p, 5, 14q, 15q, 4q, 9 (gains in order of decreasing frequency) and 11p, 11q, 1p, 4, 13q, 15q (losses in order of decreasing frequency) (Figure 1 and Table 1). Moreover, we found a common imbalanced region in the p arm of chromosome 11 corresponding to an allelic loss in five out of the six A tumors. This region, extending from position 14.9 to position 46.5 Mb, harbors the cytobands 11p15.2, 11p15.1, 11p14.3, 11p14.2, 11p14.1, 11p13, 11p12 and 11p11.2 (Figure 2). Three of these five tumors were carcinomas and presented additional allelic loss in the 11q arm, with a common region extending from 109.5 to 134.1 Mb, covering the cytobands q22.3, q23.1, q23.2, q23.3, q24.1, q24.2, q24.3 and q25 (Figure 2). Finally, we did not observe any accumulation of alterations between nonaggressive and aggressive tumors.
Figure 1.

Summary of genome copy number alterations (CNAs) detected by comparative genomic hybridization (CGH) analysis. A. Nonaggressive (n = 7) PRL tumors. B. Aggressive PRL tumors (n = 6). Gains are indicated by black bars and losses by grey bars. PRL = prolactin.
Figure 2.
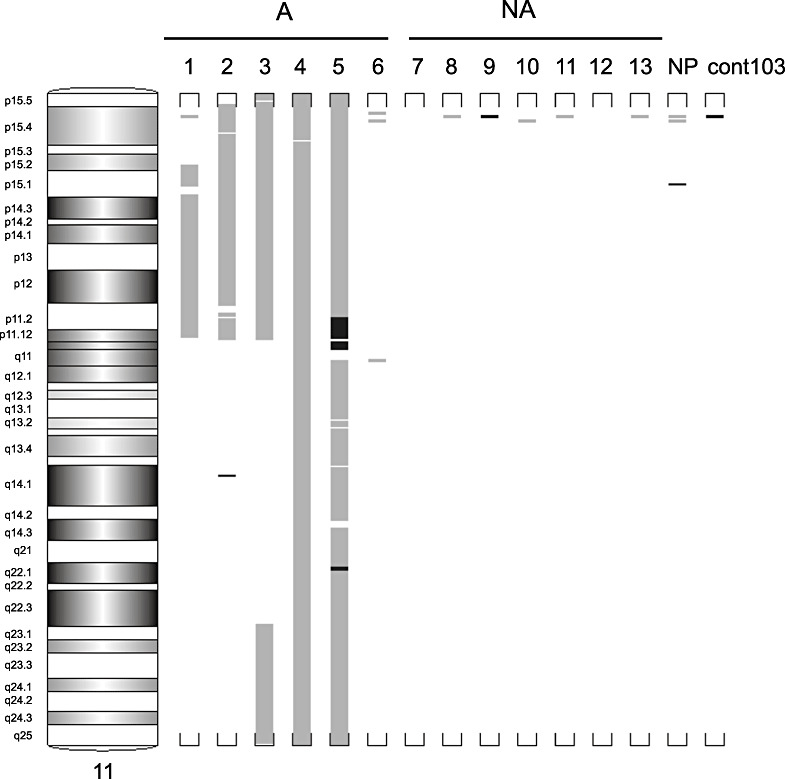
Details of regions lost or gained in chromosome 11 in the six aggressive and seven nonaggressive tumors. Gains are indicated by black bars and losses by grey bars. NA = nonaggressive PRL tumors; A = aggressive PRL tumors; NP = normal pituitary, cont103, genomic DNA reference (Affymetrix); PRL = prolactin.
The common 11p region loss included 139 coding genes (Appendix S1). Only one tumor (#6) presented a low number of DNA alterations, with no detectable alteration in chromosome 11.
A major signaling pathway involved in cell cycle is deregulated in aggressive PRL tumors
Transcriptomic analysis was performed on the five A tumors with 11p arm loss (A 11pdel) and on the seven NA tumors. Signature comparison revealed an upregulation of 572 genes and a downregulation of 842 in the A 11pdel when compared with the NA tumors (Appendix S2). Using Ingenuity Pathway Analysis (IPA) software, we deciphered a main signaling pathway that included genes involved in the cell cycle (Figure 3) that were specifically upregulated in tumors presenting an aggressive phenotype.
Figure 3.
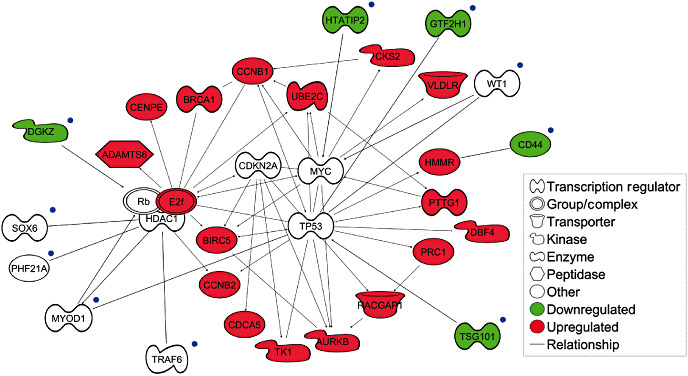
Relationship between the molecular pathway of genes linked to aggressive PRL tumor phenotype and genes located in the deleted region of chromosome 11p in aggressive prolactin (PRL) tumors. Candidate genes located on the deleted 11p region and linked to the molecular pathway associated with aggressive tumors are indicated with blue dots. Genes in red are those found to be upregulated by transcriptomic comparison in aggressive tumors compared with nonaggressive tumors. The green color represents genes in the 11p deleted region found to be downregulated by transcriptomic analysis. The signaling pathway has been deciphered using Ingenuity Pathway Analysis software.
Loss of p arm of chromosome 11 impacts transcriptome activity
To elucidate if the 11p chromosome loss could be associated with the deregulation of downstream specific genes involved in the aggressive phenotype, we compared the behavior of genes located in the chromosome intervals described above with the transcriptomic signature obtained from the same A 11pdel samples. The 139 genes located in the 11p deleted region were matched with the list of genes investigated by transcriptomic analysis. Among these 139, only 122 were interrogated by the transcriptome analysis (Appendix S1). The remaining 17 genes included those for which no corresponding DNA probes are readily available, or those that are not appropriately interrogated by the DNA chip.
We compared the transcriptome activity of these 122 genes between the A 11pdel group and the NA tumor group and found that 29 out of the 122 genes (23%) were significantly downregulated, 3 were upregulated (2%), and 91 showed no deregulation (75%). To confirm the specificity of this effect, we analyzed a region belonging to chromosomes 2p and 21q without common alterations and including the same number of genes as the 11p deleted region. Results failed to show a similar enrichment of downregulated genes. Thus, these results show unequivocally the impact of the 11pdel region on global transcriptome activity (Figure 4).
Figure 4.
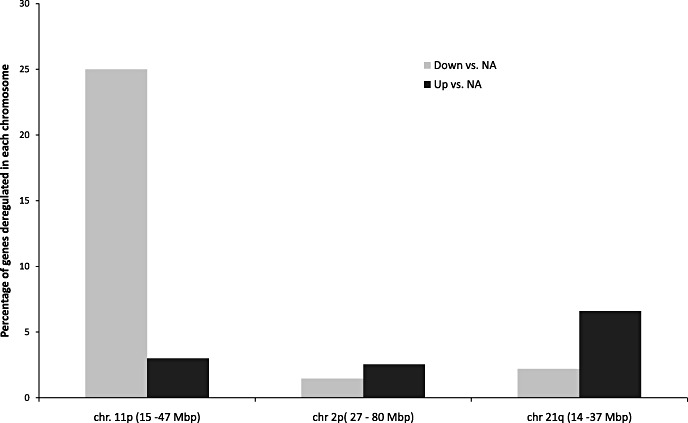
Impact of 11p loss on transcriptomic activity. Percentage of genes, located in the 11p deleted region and in different regions chosen in chromosomes 2p and 21q, found to be deregulated by transcriptomic analysis in aggressive vs. nonaggressive tumors. NA = nonaggressive PRL tumours (n = 7); A = aggressive 11p deleted PRL tumors (n = 5); PRL = prolactin.
CD44, DGKZ, TSG101, GTF2H1, and HTATIP2 are good candidates for inducing aggressiveness and malignancy of PRL tumors
Among genes located in the 11p deleted region in aggressive tumors (Appendix S1), 10 genes (DGKZ, SOX6, PHF21A, MYOD1, TRAF6, TSG101, CD44, WT1, GTF2H1 and HTATIP2), present associations with components of the signaling pathway found deregulated in the A tumors (Figure 3).
The results of the cross‐comparison with transcriptomic analysis showed that six of these genes (CD44, DGKZ, TSG101, GTF2H1, HTATIP2 and SOX6) were also downregulated and located within cytobands 11p11, 11p12, 11p13, and 11p15 suspected to contain tumor suppressor genes (see discussion). The significant downregulation of five (CD44, DGKZ, TSG101, GTF2H1 and HTATIP2) out of these six genes in A 11pdel tumors when compared with NA tumors was confirmed by q‐RTPCR (Figure 5).
Figure 5.
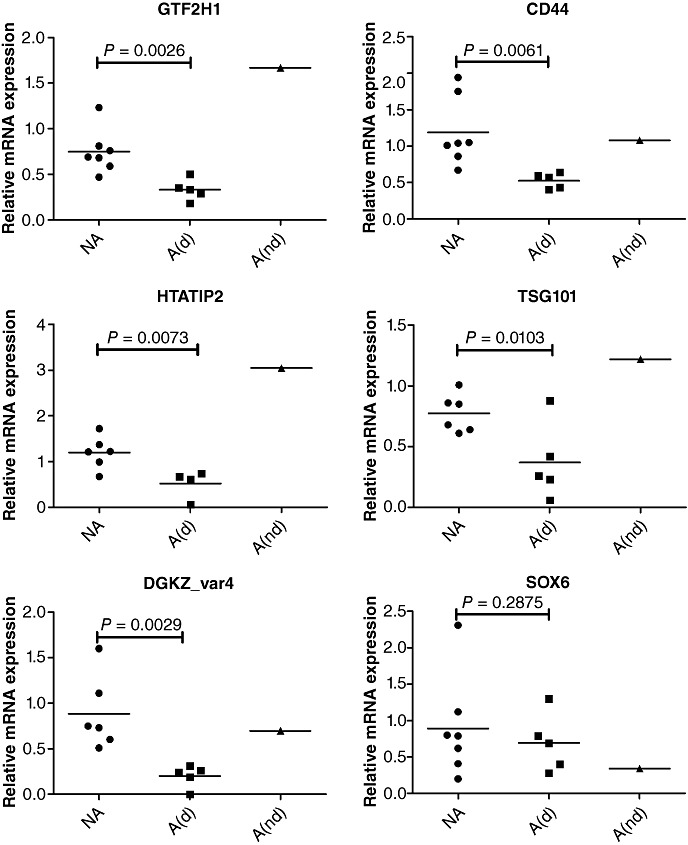
QRT‐PCR verification of microarray data. mRNA levels corresponding to the six candidate genes were measured in aggressive 11p deleted tumors, in aggressive non‐11p deleted tumors and in nonaggressive tumor groups. NA = nonaggressive tumors (n = 7); A(d) = aggressive deleted tumors (n = 5); A(nd) = aggressive nondeleted tumors (n = 1).
DISCUSSION
In this study, we aimed to find specific genomic alterations in aggressive and malignant PRL tumors and to test the hypothesis that an alteration of the genome structure may impact transcriptomic activity and drive tumoral progression to an aggressive phenotype. To our knowledge, this is the first work on one subtype of pituitary tumor coupling whole genome alteration study and whole transcriptomic analysis of the same six aggressive and seven nonaggressive PRL tumors.
As previously described when considering all types of pituitary tumors, we found that genomic imbalance occurred frequently in pituitary tumors 31, 32, 41. The most frequent and extensive alteration observed in aggressive tumors (five out of six) corresponded to the allelic loss of the p arm region of chromosome 11. Among the aggressive tumors studied, 23% of interrogated genes located in the deleted region were underexpressed while only 2% of genes within the same region were upregulated. This disproportion does not exist for the expression of genes in unaltered genomic regions, thus confirming the notion proposed by other groups 5, 14, 16, 27 that allelic imbalance impacts upon the transcriptomic activity of the genome. However, a relatively low correlation existed between the region of allelic loss and the downregulated gene expression, as already observed by other groups (16), despite a better correlation in our case (23% vs. 8%). This observation could be explained by the existence of an imprinting phenomenon in chromosome 11, specifically in the 11p15 and 11p13 cytobands (9), leading to a lower correlation with transcriptomic activity depending on allelic utilization. Furthermore, we cannot exclude that the deletion of one allele may also induce a compensation level of gene expression by the second allele by as yet undetermined molecular mechanisms.
The loss of the 11q arm was also observed in the three aggressive tumors that are considered as malignant. The rarity of malignant PRL tumors does not allow us to draw any conclusions on the role of the 11q region in the progression to malignant/metastatic PRL tumors. However, we can speculate that the failure of increased genomic stability of the 11q region in some of the PRL tumors in association with the 11p loss can lead to the malignant phenotype. Allelic loss of the 11q region in Growth hormone (GH) and PRL tumors has been reported by different authors, leading to the search for the presence of putative tumor‐suppressor genes. Among the potential candidates (for review see 7, 12, genes encoding multiple endocrine neoplasia (MEN‐1) and aryl hydrocarbon receptor interacting protein (AIP), both located in the 11q13 cytoband, are thought to be responsible for pituitary tumorigenesis 15, 30. As only two out of the five 11pdel aggressive tumors also showed an 11q13 loss, our conclusion is that the 11q, MEN‐1 or AIP losses are neither necessary nor sufficient to induce PRL tumor progression toward an aggressive phenotype.
In contrast, our findings may suggest the presence of an important putative tumor suppressor gene on the 11p area. Loss of the 11p region has already been described in several hereditary and sporadic tumors [eg, ovarian cancer (20), bladder tumors (39), breast cancer (18) or thyroid carcinoma (21))]. Some of these studies showed that the loss of the 11p arm was frequently associated with tumor progression and metastasis 19, 20. Cell manipulation and the introduction of chromosome 11 or part of its short arm suppresses the tumorigenesis of several cell lines 28, 34. Altogether, these data indicate the presence of genes associated with tumorigenicity and malignancy in the 11p regions. We hypothesized that genes included in the deleted 11p region and downregulated may control signaling pathways specifically found altered in aggressive tumors 33, 42, 43. Using IPA, we deciphered one such altered signaling pathway that contains upregulated genes implicated in the cell cycle and represents the main molecular pathway associated with the aggressive phenotype (Figure 3). Second, we searched for genes (i) located in the 11p area previously described by other groups as containing potential tumor suppressors; (ii) related to our molecular pathway associated with the aggressive phenotype; and (iii) downregulated in all five 11p deleted aggressive tumors. Five genes (CD44, TSG101, DGKZ, HTATIP2 and GTF2H1) met our three criteria and may consequently be involved in the aggressive behavior of PRL tumors.
The TSG101 gene, involved in p53 gene repression (24), has a complex and controversial role. Indeed, some studies have demonstrated a positive correlation between protein expression and tumor proliferation (40), while others suggest a tumor suppressive function (25). A functional inhibition of this gene by antisense RNA in rodent fibroblasts resulted in cellular transformation and metastatic tumor induction in nude mice (23). Furthermore, a downregulation of TSG101 was observed in metastatic breast cancer (45), thus supporting a malignancy‐suppressing role of this gene rather than a tumorigenesis‐suppressing role. The GTF2H1 gene is a general transcription factor and component of the basal transcription machinery and has a DNA repair helicase activity. This gene is also found downregulated in metastatic hepatocellular carcinoma (2). The CD44 gene encodes a transmembrane glycoprotein expressed on lymphocytes and macrophages that serves as a homing receptor that mediates binding to high endothelial venules. The presence of this receptor on tumoral cells has been reported to support tumor growth, and signal transduction through CD44 can induce proto‐oncogenes such as Ras (4). A downregulation of CD44 in ovarian tumors has been found associated with aggressive tumoral behavior and poor prognosis (35).
The HIV Tat‐interacting protein (TIP30), also called CC3 or HTATIP2, is encoded by Tip30, a putative suppressor gene. Recent studies demonstrated that a downregulation of this gene promotes metastatic progression of lung cancer, hepatocellular carcinoma, and breast carcinoma by enhancing expression of osteopontin, as well as matrix metalloproteinase‐2 and vascular endothelial growth factor 38, 44. Upon induction by the liganded estrogen receptor, Tip30, interacting with CIA (an ERalpha interacting coactivator), regulates c‐myc transcription activation. This process suggests a role for TIP30 in the regulation of tumorigenesis and tissue development in ERalpha‐targeted organs (17). Finally, the DGKZ gene encodes a diacylglycerol kinase that could act as a negative regulator of the cell cycle by binding to Rb and altering its phosphorylation status 6, 13. Analysis of transcriptomic data revealed that the DGKZ was not found to be statistically deregulated in aggressive tumors. However, PCR analysis of the different gene variants revealed a strong downregulation of the DGKZ variant 4 in aggressive tumors. This variant may therefore be considered as a candidate trigger of pathway deregulation.
Only one tumor described as aggressive presented no alteration of the genome in the region 11p. This tumor presented a relatively low hormonal expression of PRL compared with the other aggressive tumors (data not shown). Moreover, clinical signs of aggressiveness of this tumor were lower (few mitoses, low labeling index of Ki‐67and no labeling of the P53). These characteristics suggest that this tumor was either less aggressive or misclassified, or a subtype different from the five other tumors.
From this study combining DNA and transcriptomic analysis of the same PRL tumor samples, we proposed a model of PRL tumor progression involving an allelic deletion of the 11p region that impacts on transcriptome activity via the deregulation of key genes (Figure 6). The deregulation of these genes could be the upstream event responsible for the subsequent deregulation of specific signaling pathways leading to the aggressive and malignant PRL phenotypes. The remaining challenge at this level of analysis is to identify the mechanisms, such as DNA hypomethylation, involved in the 11p deletion (18). Moreover, in this study we observed no accumulation of chromosomal alterations between nonaggressive and aggressive tumors, contrary to that portrayed by the clonal genetic model of tumor progression. Thus, we suggest that nonaggressive and aggressive PRL tumors may arise via different mechanisms and follow two distinct tumor progression routes (Figure 6).
Figure 6.
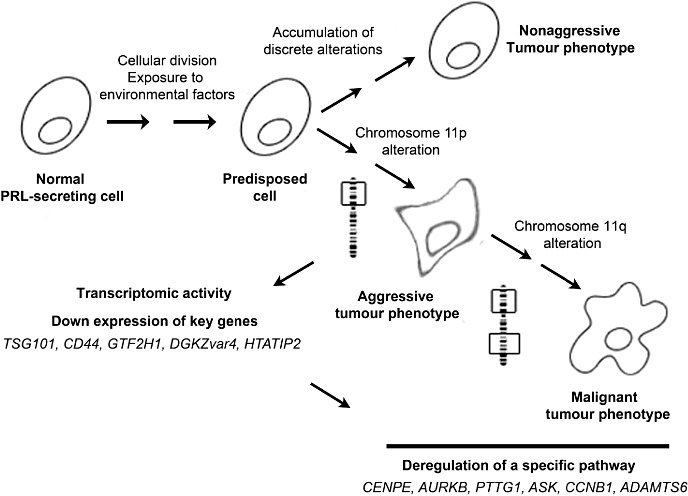
Proposed model of prolactin (PRL) tumor progression. The structural alterations of genomes seem to be an early event in PRL tumor development. Discrete and sparse alterations led to a nonaggressive phenotype. Specific loss of the p arm of chromosome 11 impacts the gene expression, leading to deregulation of cell growth and inducing the aggressive phenotype. Afterward, loss of the q arm of chromosome 11 occurs in the aggressive tumor group and may contribute to malignant phenotype of PRL tumors.
The study of aggressiveness and malignancy of pituitary tumors is difficult to implement because of these tumors' rarity (33). However, it seems essential to work only on one subtype of tumor 7, 43, even if it means reducing the number of cases in a study. Thus, the statistical analysis of gene expression requiring a large number of tumors cannot be applied. The combination of various parameters such as genetic alteration and transcriptome activity with our current knowledge in a multidimensional approach can increase the detection and understanding of mechanisms involved in tumor progression from a small cohort (3). This strategy allowed us to find candidate genes that could be involved in the progression of PRL pituitary tumors. While we demonstrated its usefulness in rare tumors, the use of multiparametric measurements could also be applied to other types of tumor to find relevant candidate genes implicated in tumor progression from a large panel of deregulated genes and may be expanded to include other measurements such as DNA methylation and miRNA.
CONFLICT OF INTEREST
The authors declare having no conflict of interest that would prejudice the impartiality of this scientific work.
Supporting information
Appendix S1. List of genes located in the deleted 11 p arm region. In the table are indicated (i) the cytobands corresponding to the deleted 11p region; (ii) the transcriptomic behavior of genes located in the deleted region; and (iii) the genes linked to the specific pathway deciphered in aggressive (A) tumors. X indicates when genes are both downregulated and linked to the signaling pathway.
Appendix S2. List of genes found statistically deregulated in aggressive (A) vs. nonaggressive (NA) tumors. GE, CodeLink™ microarray identifiers (General Electrics); FC, fold change between NA and the A 11p deleted tumor.
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
We are greatly indebted to the Neurobiotec Bank for tissues provided, AngloScribe for help with the English translation and Programme Hospitalier de Recherche Clinique National 2006–2008 (no 27–43), Institut National de la Santé et de la Recherche Médicale (INSERM), the Ligue Contre le Cancer (comités Puy de Dôme and Rhône‐Alpes), and the Région Rhône‐Alpes (France) for financial support.
REFERENCES
- 1. Asa SL, Ezzat S (2009) The pathogenesis of pituitary tumors. Annu Rev Pathol 4:97–126. [DOI] [PubMed] [Google Scholar]
- 2. Budhu A, Jia HL, Forgues M, Liu CG, Goldstein D, Lam A et al (2008) Identification of metastasis‐related microRNAs in hepatocellular carcinoma. Hepatology 47:897–907. [DOI] [PubMed] [Google Scholar]
- 3. Chari R, Coe BP, Vucic EA, Lockwood WW, Lam WL (2010) An integrative multi‐dimensional genetic and epigenetic strategy to identify aberrant genes and pathways in cancer. BMC Syst Biol 4:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng C, Yaffe MB, Sharp PA (2006) A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev 20:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cifola I, Spinelli R, Beltrame L, Peano C, Fasoli E, Ferrero S et al (2008) Genome‐wide screening of copy number alterations and LOH events in renal cell carcinomas and integration with gene expression profile. Mol Cancer 7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Evangelisti C, Tazzari PL, Riccio M, Fiume R, Hozumi Y, Fala F et al (2007) Nuclear diacylglycerol kinase‐zeta is a negative regulator of cell cycle progression in C2C12 mouse myoblasts. FASEB J 21:3297–3307. [DOI] [PubMed] [Google Scholar]
- 7. Farrell WE (2006) Pituitary tumours: findings from whole genome analyses. Endocr Relat Cancer 13:707–716. [DOI] [PubMed] [Google Scholar]
- 8. Farrell WE, Simpson DJ, Bicknell JE, Talbot AJ, Bates AS, Clayton RN (1997) Chromosome 9p deletions in invasive and noninvasive nonfunctional pituitary adenomas: the deleted region involves markers outside of the MTS1 and MTS2 genes. Cancer Res 57:2703–2709. [PubMed] [Google Scholar]
- 9. Feinberg AP (1999) Imprinting of a genomic domain of 11p15 and loss of imprinting in cancer: an introduction. Cancer Res 59(7 Suppl.):1743s–1746s. [PubMed] [Google Scholar]
- 10. Fèvre‐Montange M, Champier J, Szathmari A, Wierinckx A, Mottolese C, Guyotat J et al (2006) Microarray analysis reveals differential gene expression patterns in tumors of the pineal region. J Neuropathol Exp Neurol 65:675–684. [DOI] [PubMed] [Google Scholar]
- 11. Fischer M, Bauer T, Oberthur A, Hero B, Theissen J, Ehrich M et al (2010) Integrated genomic profiling identifies two distinct molecular subtypes with divergent outcome in neuroblastoma with loss of chromosome 11q. Oncogene 29:865–875. [DOI] [PubMed] [Google Scholar]
- 12. Gurlek A, Karavitaki N, Ansorge O, Wass JA (2007) What are the markers of aggressiveness in prolactinomas? Changes in cell biology, extracellular matrix components, angiogenesis and genetics. Eur J Endocrinol 156:143–153. [DOI] [PubMed] [Google Scholar]
- 13. Hasegawa H, Nakano T, Hozumi Y, Takagi M, Ogino T, Okada M et al (2008) Diacylglycerol kinase zeta is associated with chromatin, but dissociates from condensed chromatin during mitotic phase in NIH3T3 cells. J Cell Biochem 105:756–765. [DOI] [PubMed] [Google Scholar]
- 14. Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F et al (2009) Segmental copy number variation shapes tissue transcriptomes. Nat Genet 41:424–429. [DOI] [PubMed] [Google Scholar]
- 15. Herman V, Drazin NZ, Gonsky R, Melmed S (1993) Molecular screening of pituitary adenomas for gene mutations and rearrangements. J Clin Endocrinol Metab 77:50–55. [DOI] [PubMed] [Google Scholar]
- 16. Jarvinen AK, Autio R, Haapa‐Paananen S, Wolf M, Saarela M, Grenman R et al (2006) Identification of target genes in laryngeal squamous cell carcinoma by high‐resolution copy number and gene expression microarray analyses. Oncogene 25:6997–7008. [DOI] [PubMed] [Google Scholar]
- 17. Jiang C, Ito M, Piening V, Bruck K, Roeder RG, Xiao H (2004) TIP30 interacts with an estrogen receptor alpha‐interacting coactivator CIA and regulates c‐myc transcription. J Biol Chem 279:27781–27789. [DOI] [PubMed] [Google Scholar]
- 18. Kanai Y (2010) Genome‐wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci 101:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karnik P, Paris M, Williams BR, Casey G, Crowe J, Chen P (1998) Two distinct tumor suppressor loci within chromosome 11p15 implicated in breast cancer progression and metastasis. Hum Mol Genet 7:895–903. [DOI] [PubMed] [Google Scholar]
- 20. Kiechle‐Schwarz M, Bauknecht T, Wienker T, Walz L, Pfleiderer A (1993) Loss of constitutional heterozygosity on chromosome 11p in human ovarian cancer. Positive correlation with grade of differentiation. Cancer 72:2423–2432. [DOI] [PubMed] [Google Scholar]
- 21. Kitamura Y, Shimizu K, Tanaka S, Ito K, Emi M (2000) Allelotyping of anaplastic thyroid carcinoma: frequent allelic losses on 1q, 9p, 11, 17, 19p, and 22q. Genes Chromosomes Cancer 27:244–251. [PubMed] [Google Scholar]
- 22. Kontogeorgos G, Kapranos N, Orphanidis G, Rologis D, Kokka E (1999) Molecular cytogenetics of chromosome 11 in pituitary adenomas: a comparison of fluorescence in situ hybridization and DNA ploidy study. Hum Pathol 30:1377–1382. [DOI] [PubMed] [Google Scholar]
- 23. Li L, Cohen SN (1996) Tsg101: a novel tumor susceptibility gene isolated by controlled homozygous functional knockout of allelic loci in mammalian cells. Cell 85:319–329. [DOI] [PubMed] [Google Scholar]
- 24. Li L, Liao J, Ruland J, Mak TW, Cohen SN (2001) A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control. Proc Natl Acad Sci U S A 98:1619–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin SF, Lin PM, Liu TC, Chang JG, Sue YC, Chen TP (2000) Clinical implications of aberrant TSG101 transcripts in acute myeloblastic leukemia. Leuk Lymphoma 36:463–466. [DOI] [PubMed] [Google Scholar]
- 26. Lloyd RV, Kovacs K, Young WF Jr, Farrel WE, Asa SL, Trouillas J et al (2004) Pituitary tumors: introduction. In: World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Endocrine Organs. DeLellis RA, Lloyd RV, Heitz PU, Eng C (eds), pp. 10–13. IARC Press: Lyon, France. [Google Scholar]
- 27. Lo KC, Rossi MR, LaDuca J, Hicks DG, Turpaz Y, Hawthorn L, Cowell JK (2007) Candidate glioblastoma development gene identification using concordance between copy number abnormalities and gene expression level changes. Genes Chromosomes Cancer 46:875–894. [DOI] [PubMed] [Google Scholar]
- 28. Negrini M, Sabbioni S, Possati L, Rattan S, Corallini A, Barbanti‐Brodano G, Croce CM (1994) Suppression of tumorigenicity of breast cancer cells by microcell‐mediated chromosome transfer: studies on chromosomes 6 and 11. Cancer Res 54:1331–1336. [PubMed] [Google Scholar]
- 29. Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194:23–28. [DOI] [PubMed] [Google Scholar]
- 30. Ozfirat Z, Korbonits M (2010) AIP gene and familial isolated pituitary adenomas. Mol Cell Endocrinol 326:71–79. [DOI] [PubMed] [Google Scholar]
- 31. Pack SD, Kirschner LS, Pak E, Zhuang Z, Carney JA, Stratakis CA (2000) Genetic and histologic studies of somatomammotropic pituitary tumors in patients with the “complex of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J Clin Endocrinol Metab 85:3860–3865. [DOI] [PubMed] [Google Scholar]
- 32. Pack SD, Qin LX, Pak E, Wang Y, Ault DO, Mannan P et al (2005) Common genetic changes in hereditary and sporadic pituitary adenomas detected by comparative genomic hybridization. Genes Chromosomes Cancer 43:72–82. [DOI] [PubMed] [Google Scholar]
- 33. Raverot G, Wierinckx A, Dantony E, Auger C, Chapas G, Villeneuve L et al (2010) Prognostic factors in prolactin pituitary tumors: clinical, histological, and molecular data from a series of 94 patients with a long postoperative follow‐up. J Clin Endocrinol Metab 95:1708–1716. [DOI] [PubMed] [Google Scholar]
- 34. Sabbioni S, Negrini M, Possati L, Bonfatti A, Corallini A, Sensi A et al (1994) Multiple loci on human chromosome 11 control tumorigenicity of BK virus transformed cells. Int J Cancer 57:185–191. [DOI] [PubMed] [Google Scholar]
- 35. Saegusa M, Hashimura M, Machida D, Okayasu I (1999) Down‐regulation of CD44 standard and variant isoforms during the development and progression of uterine cervical tumours. J Pathol 187:173–183. [DOI] [PubMed] [Google Scholar]
- 36. Simpson DJ, Bicknell EJ, Buch HN, Cutty SJ, Clayton RN, Farrell WE (2003) Genome‐wide amplification and allelotyping of sporadic pituitary adenomas identify novel regions of genetic loss. Genes Chromosomes Cancer 37:225–236. [DOI] [PubMed] [Google Scholar]
- 37. Tanaka C, Kimura T, Yang P, Moritani M, Yamaoka T, Yamada S et al (1998) Analysis of loss of heterozygosity on chromosome 11 and infrequent inactivation of the MEN1 gene in sporadic pituitary adenomas. J Clin Endocrinol Metab 83:2631–2634. [DOI] [PubMed] [Google Scholar]
- 38. Tong X, Li K, Luo Z, Lu B, Liu X, Wang T et al (2009) Decreased TIP30 expression promotes tumor metastasis in lung cancer. Am J Pathol 174:1931–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Voorter CE, Ummelen MI, Ramaekers FS, Hopman AH (1996) Loss of chromosome 11 and 11 p/q imbalances in bladder cancer detected by fluorescence in situ hybridization. Int J Cancer 65:301–307. [DOI] [PubMed] [Google Scholar]
- 40. Wagner KU, Krempler A, Qi Y, Park K, Henry MD, Triplett AA et al (2003) Tsg101 is essential for cell growth, proliferation, and cell survival of embryonic and adult tissues. Mol Cell Biol 23:150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weil RJ, Vortmeyer AO, Huang S, Boni R, Lubensky IA, Pack S et al (1998) 11q13 allelic loss in pituitary tumors in patients with multiple endocrine neoplasia syndrome type 1. Clin Cancer Res 4:1673–1678. [PubMed] [Google Scholar]
- 42. Wierinckx A, Auger C, Devauchelle P, Reynaud A, Chevallier P, Jan M et al (2007) A diagnostic marker set for invasion, proliferation, and aggressiveness of prolactin pituitary tumors. Endocr Relat Cancer 14:887–900. [DOI] [PubMed] [Google Scholar]
- 43. Wierinckx A, Raverot G, Nazaret N, Jouanneau E, Auger C, Lachuer J, Trouillas J (2010) Proliferation markers of human pituitary tumors: contribution of a genome‐wide transcriptome approach. Mol Cell Endocrinol 326:30–39. [DOI] [PubMed] [Google Scholar]
- 44. Zhao J, Lu B, Xu H, Tong X, Wu G, Zhang X et al (2008) Thirty‐kilodalton Tat‐interacting protein suppresses tumor metastasis by inhibition of osteopontin transcription in human hepatocellular carcinoma. Hepatology 48:265–275. [DOI] [PubMed] [Google Scholar]
- 45. Zhu G, Reynolds L, Crnogorac‐Jurcevic T, Gillett CE, Dublin EA, Marshall JF et al (2003) Combination of microdissection and microarray analysis to identify gene expression changes between differentially located tumour cells in breast cancer. Oncogene 22:3742–3748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. List of genes located in the deleted 11 p arm region. In the table are indicated (i) the cytobands corresponding to the deleted 11p region; (ii) the transcriptomic behavior of genes located in the deleted region; and (iii) the genes linked to the specific pathway deciphered in aggressive (A) tumors. X indicates when genes are both downregulated and linked to the signaling pathway.
Appendix S2. List of genes found statistically deregulated in aggressive (A) vs. nonaggressive (NA) tumors. GE, CodeLink™ microarray identifiers (General Electrics); FC, fold change between NA and the A 11p deleted tumor.
Supporting info item
Supporting info item