

Abstract
Parkinson's disease (PD) is characterized by the accumulation of α‐synuclein aggregates and degeneration of melanized neurons. The tissue transglutaminase (tTG) enzyme catalyzes molecular protein cross‐linking. In PD brain, tTG‐induced cross‐links have been identified in α‐synuclein monomers, oligomers and α‐synuclein aggregates. However, whether tTG and α‐synuclein occur together in PD affected neurons remains to be established. Interestingly, using immunohistochemistry, we observed a granular distribution pattern of tTG, characteristic of melanized neurons in PD brain. Apart from tTG, these granules were also positive for typical endoplasmic reticulum (ER)‐resident chaperones, that is, protein disulphide isomerase, ERp57 and calreticulin, suggesting a direct link to the ER. Additionally, we observed the presence of phosphorylated pancreatic ER kinase (pPERK), a classical ER stress marker, in tTG granule positive neurons in PD brain, although no subcellular colocalization of tTG and pPERK was found. Our data therefore suggest that tTG localization to granular ER compartments is specific for stressed melanized neurons in PD brain. Moreover, as also α‐synuclein aggregates were observed in tTG granule positive neurons, these results provide a clue to the cellular site of interaction between α‐synuclein and tTG.
Keywords: endoplasmic reticulum, Parkinson's disease, neurodegeneration, tissue transglutaminase
INTRODUCTION
Parkinson's disease (PD) is characterized primarily by progressive degeneration of melanized neurons in the midbrain and brainstem, and accumulation in various brain areas of aggregated α‐synuclein protein in cytoplasmic neuronal inclusions known as Lewy bodies (LBs) (5). α‐Synuclein is abnormally processed in PD (7), and the conversion of its normal, monomeric form into mature β‐pleated fibrils is associated with neuronal loss not only in some autosomal‐dominant familial forms of PD, but also in the more common idiopathic form of PD (7). In addition to protein misfolding, a number of other factors including mitochondrial dysfunction and related oxidative stress have been strongly implicated in the pathogenesis of neurodegeneration in PD 32, 43, 53. Understanding the mechanisms that underlie neuronal cell loss and their interactions in PD is crucial for the development of effective neuroprotective strategies.
The transglutaminase (TG) protein family (EC 2.3.2.13) consists of nine members, among which transglutaminase 2, also known as tissue transglutaminase (tTG), is the best characterized. tTG plays an important role during development, cell differentiation and apoptosis (10). tTG is a calcium‐dependent enzyme that catalyzes several post‐translational modifications of proteins, including the formation of (γ‐glutamyl)polyamine bonds, the deamidation of protein substrates, and, perhaps most importantly, the formation of molecular cross‐links that result in covalent ε‐(γ‐glutamyl)lysine isopeptide bonds within or between peptide chains (31).
In various neurodegenerative diseases, evidence is accumulating that tTG contributes to the formation of protein aggregates (51). The presence of tTG, and its cross‐linking products, have been identified in aggregated protein deposits which are the pathological hallmarks of Alzheimer's disease (AD) 46, 50, other tauopathies 14, 38, 46 and Huntington's disease 25, 29, 55. Furthermore, the proteins that form the core of these accumulated protein deposits, that is, amyloid‐beta, tau and huntingtin, are all excellent substrates of tTG, and tTG is known to directly affect their aggregation state 13, 17, 23, 29, 35, 38, 46.
In PD brain, elevated levels of tTG protein have been observed (1). More importantly, tTG‐mediated cross‐links were identified in both soluble α‐synuclein monomers and oligomers, and LBs, suggesting that tTG plays an important role in α‐synuclein misfolding and the formation of α‐synuclein deposits 1, 22, 36. This notion is supported by in vitro studies revealing direct high affinity interactions of tTG with α‐synuclein, resulting in the formation of the various cross‐linked forms of α‐synuclein detected in PD brain 36, 44, 45. However, although there is compelling evidence that tTG plays a role in the misfolding and deposition of α‐synuclein in PD, Andringa and colleagues reported that tTG itself is not typically associated with LBs. In fact, they noted a fine granular staining pattern of tTG in melanized neurons in the Parkinsonian substantia nigra (SN), suggesting that under pathological conditions, tTG may be associated with and/or accumulate in a subcellular compartment (1).
Interestingly, although tTG is generally considered as a soluble cytosolic protein, it has also been described to exist intracellularly in a particulate form which has escaped further characterization (15). Recently, evidence has been provided for an association of tTG with mitochondria (41) and lysosomes (56), whereas a possible functional association of tTG with the endoplasmic reticulum (ER) has been hinted at (39). Therefore, given the emerging importance of tTG in PD pathogenesis, but uncertainty about its cellular localization and possible implications thereof, we set out to elaborate the findings of Andringa and co‐workers (1) and investigated the cellular distribution of tTG in melanized neurons in PD.
MATERIALS AND METHODS
Brain tissue
Paraffin‐embedded SN and locus coeruleus (LC) of PD cases (n = 12; 8 male, 4 female, age 72.5 ± 5) and control subjects (n = 5; 1 male, 4 female, age 61.3 ± 13) were obtained from the Born‐Bunge Institute, University of Antwerp, Campus “Drie Eiken,” Antwerp, Belgium. Paraffin‐embedded hypothalamus of PD (n = 4; 2 male, 2 female, age 76 ± 13.7) and control cases (n = 4; 2 male, 2 female, age 74.8 ± 12.7) were obtained from the Netherlands Institute for Neurosciences. All cases had a post‐mortem interval of less than 48 h. PD cases were clinically documented and neuropathologically confirmed to have idiopathic PD. Controls were age‐matched to PD and had no history of neuropsychiatric illness or neuropathological changes. Paraffin‐embedded SN of AD cases (n = 3, 2 male, 1 female, age 77 ± 5.6) was also obtained from Born‐Bunge Institute. The diagnosis of AD was based on a combination of neuropathological (both male patients Braak stage 5, female patient Braak stage 6) and clinical criteria, and all cases had a post‐mortem interval of less than 48 h. Tau pathology was observed in the midbrain of all AD patients. Substantia nigra tissue samples from frontal temporal dementia patients (n = 4; 1 male, 3 female, age 70.2 ± 6.8) were obtained after rapid autopsy and immediately frozen in liquid nitrogen (the Netherlands Brain Bank, Amsterdam, the Netherlands).
Immunohistochemistry
Serial coronal sections (6 µm) of the post mortem brain tissue were immunostained with primary antibodies listed in Table 1, as described previously 50, 52. In short, sections were pre‐incubated with 20% animal serum, the type of which was determined by the specific secondary antibody used. Secondary antibodies were all purchased from Jackson Immunoresearch Europe Ltd. (Suffolk, UK); biotin‐labeled donkey anti‐goat (1:200), biotin‐labeled goat anti‐mouse (1:200) and biotin‐labelled goat anti‐rabbit (1:200). Immunohistochemistry was performed according to the avidin‐biotin‐peroxidase method. Reaction products were visualized with the Vectastain elite Avidin Biotin kit (Vector Laboratories, Burlingame, CA, USA), using nickel enhanced 3,3'‐diaminobenzidine (DAB) as the chromogen (Sigma, St. Louis, MO, USA) to discriminate between antibody staining and neuromelanin present within neuromelanin‐containing neurons. Immunohistochemistry on the frontal temporal dementia (FTD) tissue was visualized using DAB as the chromogen. Endogenous peroxidase activity was blocked by incubation of the sections in 0.3% H2O2 with 0.1% sodium azide prior to immunolabeling. For each antibody, various concentrations were tested to determine the optimal immunoreactivity (ie, the highest intensity of specific labeling without significant background staining). Negative controls consisted of representative sections processed without the primary antibody.
Table 1.
Primary Antibodies Used in this Study for Immunohistochemistry. Abbreviations: PDI = anti‐protein disulfide isomerise; tTG = tissue transglutaminase; pPERK = phosphorylated pancreatic ER kinase; ERp57 = anti‐ER protein 57.
| Primary antibody | Antigen | Species raised in | Dilution | Source (reference) |
|---|---|---|---|---|
| Ab2792 | Rat PDI | Mouse | 1:100 | Abcam, Cambridge, UK |
| 06‐471 | Guinea pig tTG | Goat | 1:1500 | Upstate, Lake Placid, NY, USA |
| Ab‐1 | Guinea pig tTG | Mouse | 1:500 | Lab Vision, CA, USA |
| Ab4 | Mouse calreticulin | Rabbit | 1:1000 | Abcam, Cambridge, UK |
| Ab2907 | Human calreticulin | Rabbit | 1:200 | Abcam, Cambridge, UK |
| Z0458 | Human ubiquitin | Rabbit | 1:500 | Dako, Glostrup, Denmark |
| Ab15895 | Human VDAC1/porin | Rabbit | 1:1000 | Abcam, Cambridge, UK |
| Sc‐32577 | Human pPERK | Rabbit | 1:100 | Santa Cruz Biotechnology, CA, USA |
| Ab10287 | Human ERp57 | Rabbit | 1:1000 | Abcam, Cambridge, UK |
| 610786 | Human α‐synuclein | Mouse | 1:1000 | BD Biosciences, San Jose, CA, USA |
| AT8 | Human hyperphosphorylated tau | Mouse | 1:1000 | Thermoscientific , Rockford, IL, USA |
| MAB422 | Human cathepsin‐D | Mouse | 1:500 | Chemicon, Temecula, CA, USA |
Authors G.A and J.B. independently performed a semi‐quantitative analysis of both the anti‐tTG and anti‐calreticulin immunoreactivity in the melanized neurons of the SN, using a score from 0 to 4, with 0 indicating no immunoreactivity was observed in melanized neurons per microscopic field (magnification ×10), 1 indicating 0% to 30%, 2 indicating 30% to 60%, 3 indicating 60% to 90% and 4 indicating immunoreactivity was observed in more than 90% of melanized cells within a microscopic field. Inter‐observer scores were highly consistent (average inter‐observer consensus was 93%; differentially judged images were excluded).
Double immunofluorescence
For double immunostaining, tissue sections were fixed and pre‐incubated as described previously. Sections were simultaneously incubated with the primary antibodies of interest. Secondary antibodies used were: biotin‐labeled donkey anti‐goat antibody coupled to Alexa594 (1:400, Molecular Probes, Eugene, OR, USA) and biotin‐labeled donkey anti‐rabbit antibody coupled to Alexa488 (1:400, Molecular Probes) or biotin‐labeled donkey anti‐mouse antibody coupled to Alexa488 (1:400, Molecular Probes). Fluorescence was analyzed with a Leica TCS SP2 AOBS confocal laser scanning microscope (Leica Microsystems, Rijswijk, the Netherlands). A series of images was obtained separately in both channels through a 63× glycerin lens (zoom factor 4×, Z‐increment 0.12 µm, approximately 100 images of 1024 × 1024 pixels).
Statistical analysis
Statistical differences of the percentage of cells demonstrating tTG‐positive granules (Figure 1G) or calreticulin‐positive granules (Figure 4H) in melanized neurons of the SN between control and PD brain were performed using a Student's t‐test (analyzed by GraphPad Prism, version 4.03, La Jolla, CA, USA). The level of significance (P value) of the differences between groups and the control is indicated in the figures.
Figure 1.
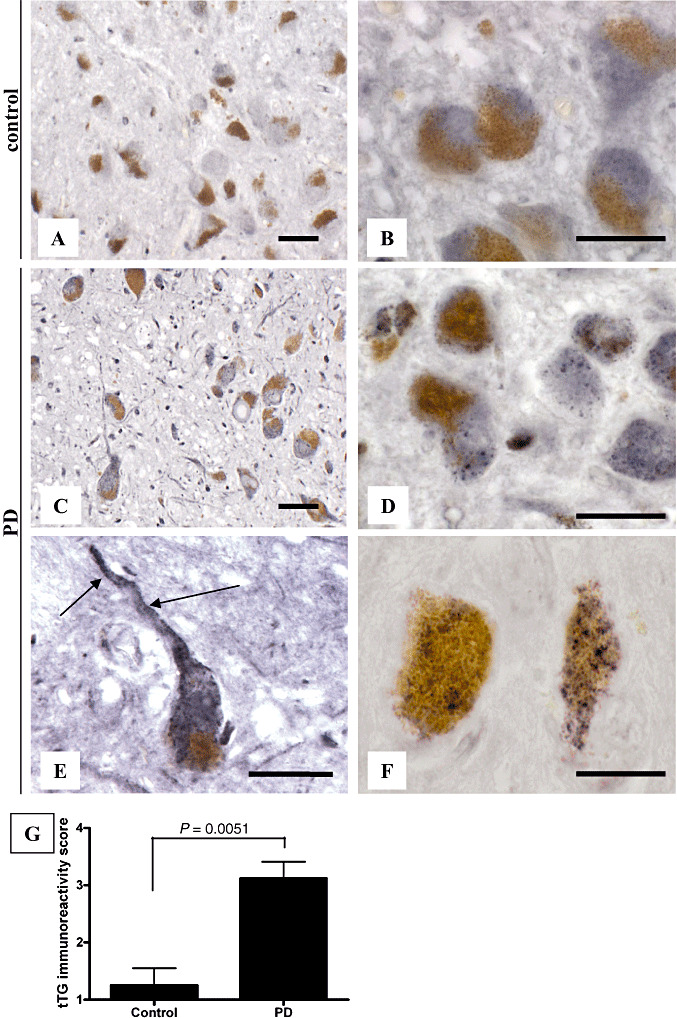
The tissue transglutaminase (tTG)‐immunoreactive granules are present in melanized neurons in Parkinson's disease (PD) brains. Immunohistochemical staining of tTG (antibody 06‐471, Table 1) in the substantia nigra (SN) of control and PD brains demonstrated tTG‐immunoreactive granules in melanized neurons of the SN in PD brains (C,D), whereas a weak homogeneous cytoplasmic tTG‐immunoreactivity was observed in control brains (A,B). tTG‐immunoreactive granules were also observed in neurites of melanized neurons in PD brains (E, arrow). Immunohistochemical staining of tTG with a monoclonal tTG‐specific antibody (antibody Ab‐1, Table 1) in the SN of PD brains demonstrated similar tTG‐immunoreactive granules in melanized neurons of the SN in PD brains (F) as observed with the polyclonal anti‐tTG antibody. Semi‐quantitative analysis, performed as described in the Materials and Methods section, of the presence of tTG‐immunoreactive granules in melanized neurons of the SN demonstrated a significant increase in tTG‐positive granule‐containing melanized neurons in PD brains compared with control brains (G). Statistical analysis was performed using a Student's t‐test. Mean ± standard deviation is shown. Scale bars: (A–F) 20 µm.
Figure 4.
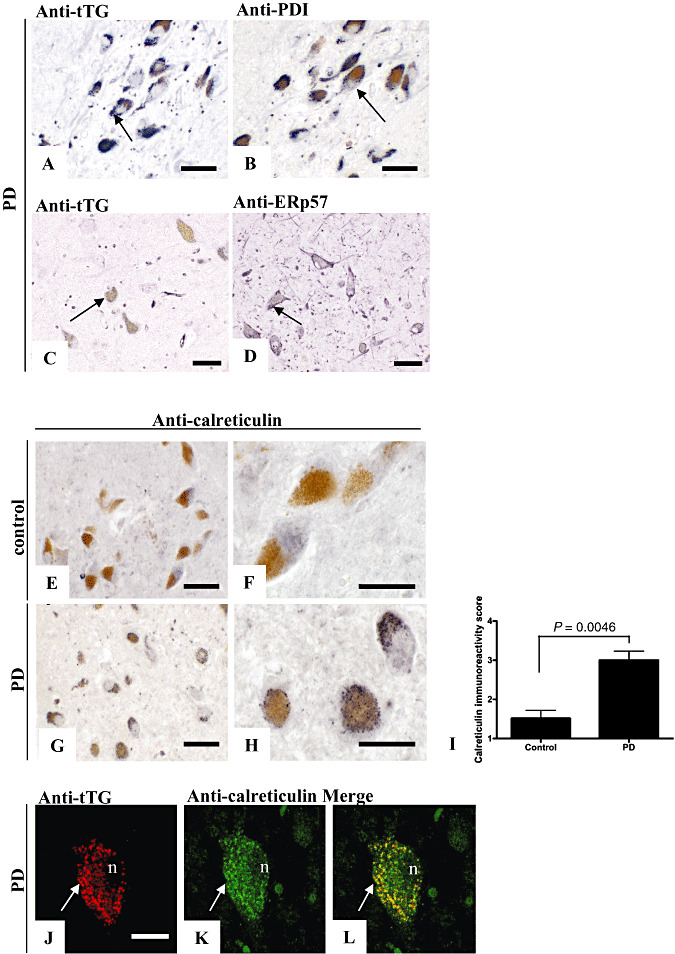
Anti‐protein disulfide isomerase (PDI), anti‐ER protein 57 (ERp57) and calreticulin staining are present in tissue transglutaminase (tTG)‐immunoreactive granules of melanized neurons in Parkinson's disease (PD) brains. Immunohistochemical staining of anti‐PDI (antibody Ab2792) or anti‐ERp57 (antibody Ab10287) antibodies in the substantia nigra (SN) of PD brains demonstrated both PDI‐immunoreactive granules (B, arrow) and ERp57‐immunopositive granules (D, arrow) in melanized neurons of the SN in PD brains, similar to the tTG staining (A and C, arrow). Immunohistochemical staining of calreticulin (antibody Ab2907) in the SN of control (E,F) brains demonstrated only weak and cytoplasmic immunoreactivity. In contrast, calreticulin‐positive granules were observed in melanized neurons of the SN in PD brains (G,H), similar to the observed tTG‐positive granules (A). Semi‐quantitative analysis, performed as described in the Materials and Methods section, of the presence of calreticulin‐positive granules in melanized neurons of the SN demonstrated a significant increase in calreticulin‐positive granule‐containing melanized neurons in PD brains compared with control brains (I). Double immunofluorescence of tTG (J, antibody 06‐471) with calreticulin (K, antibody Ab2907) staining demonstrated colocalization of calreticulin in tTG‐immunopositive granules in melanized neurons of the SN in PD brains (L). Statistical analysis was performed using a Student's t‐test. Mean ± standard deviation is shown. Scale bars: (A–L) 20 µm.
RESULTS
Granular tTG is specifically present in melanized neurons in PD brains
To investigate the staining pattern of tTG in melanized neurons in control and PD cases, we used immunohistochemistry. In both control (Figure 1A,B) and PD patients (Figure 1C,D),, a weak homogeneous cytoplasmic tTG immunoreactivity (polyclonal antibody 06‐471) of comparable intensity was observed in melanized neurons of the SN. Interestingly, in addition we observed the presence of abundant tTG‐immunoreactive granules in melanized neurons of SN in PD brains (Figure 1C,D), whereas these granules were only occasionally observed in melanized neurons of SN in control brains. Apart from the localization of these tTG‐immunoreactive granules in neuronal cell bodies, we also noticed these granules in neuronal processes of melanized neurons in PD brains (Figure 1E). Using a monoclonal antibody directed against tTG (Ab‐1), similar tTG‐immunoreactive granules were observed in melanized neurons of SN in PD brains (Figure 1F). In control cases, the monoclonal antibody demonstrated a weak homogeneous cytoplasmic tTG immunoreactivity, similar to the polyclonal anti‐tTG antibody described previously (not shown). Semi‐quantitative analysis of our results demonstrated a highly significant increase (P = 0.0051) in the number of melanized neurons demonstrating the tTG‐immunoreactive granules in SN in PD compared with control brains (Figure 1G).
In order to investigate possible colocalization of tTG with α‐synuclein, we double stained tTG and α‐synuclein in the SN of PD patients. In general, we observed no colocalization of tTG with α‐synuclein in both LBs and Lewy neurites (LNs) (Figure 2A–C). However, although both tTG‐positive granules and α‐synuclein positive inclusions coexisted within melanized neurons of the SN in PD brains, only minor subcellular colocalization of tTG and α‐synuclein was observed in an occasional LB (Figure 2D–F). To rule out the possibility that the tTG‐positive granules consisted of aggregated proteins, we investigated the staining pattern of ubiquitin, a general marker of protein accumulation (47). Although ubiquitin staining was abundantly present in LBs in melanized neurons of the SN in PD, we found no granular staining (Figure 2G). Furthermore, to study whether the observed tTG‐immunoreactive granules are also present in melanized neurons of other PD‐affected areas, we investigated tTG immunoreactivity in the LC. Indeed, in all PD patients, tTG‐immunoreactive granules, similar to those found in the SN (Figure 2H), were observed in the LC (Figure 2I). In contrast, although weak homogenous tTG immunoreactivity was found similar to that observed in the SN, in regions lacking typical α‐synuclein pathology, for example, the hypothalamus, only some tTG‐immunoreactive granules were observed within neurons of similar size as the melanized neurons of the SN and LC (Figure 2J). To exclude that these tTG‐immunoreactive granules are a general phenomenon of melanized neurons in other neurodegenerative diseases, we analyzed tTG staining in the SN of AD and FTD brain. However, in contrast to PD, no tTG‐immunoreactive granules were found in melanized neurons of the SN in AD brains (Figure 2K). In addition, earlier observations of our group demonstrated no tTG‐immunoreactive granules in neurons associated with senile plaques or in neurofibrillary tangles in AD brain (50). Furthermore, no tTG‐immunoreactive granules were observed in melanized neurons of the SN in FTD brain (Figure 2M, arrow), whereas hyperphosphorylated tau immunoreactivity (antibody AT8) was clearly present (Figure 2L, arrow).
Figure 2.
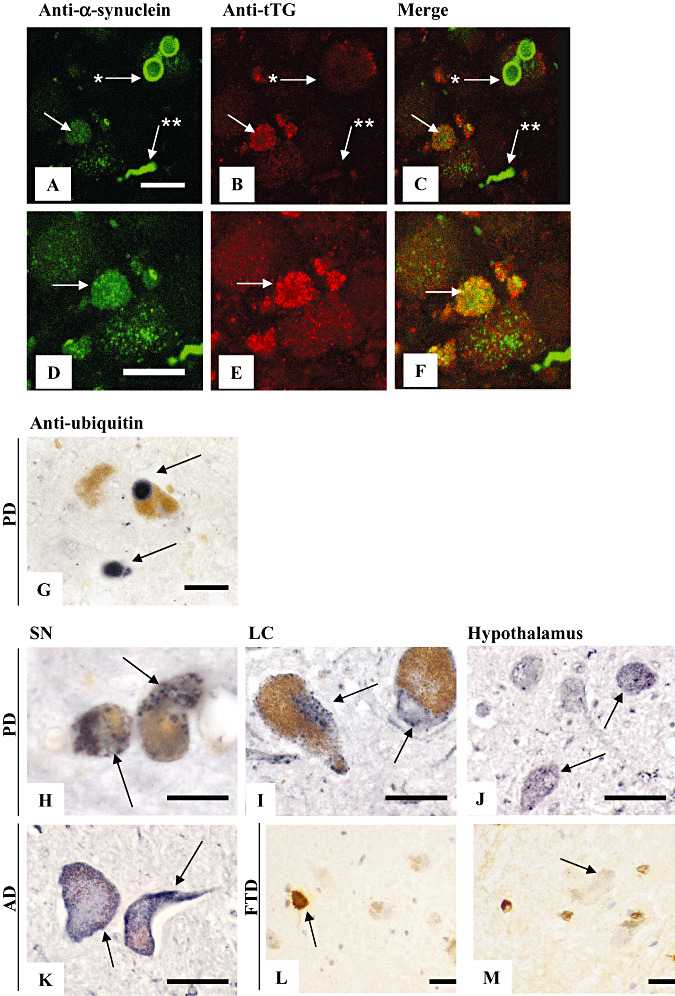
The tissue transglutaminase (tTG)‐immunoreactive granules localize to α‐synuclein containing neurons and are characteristic of substantia nigra (SN) and locus coeruleus (LC) of Parkinson's disease (PD) brain. Double immunofluorescence was performed using anti‐tTG (A,D, antibody 06‐471) and anti‐α‐synuclein (B,E, antibody 610786, Table 1) antibodies on SN of PD brain. In general, no spatial colocalization of tTG staining was observed with α‐synuclein in Lewy bodies (LBs) (arrow with asterisk, A–C) and Lewy neurites (arrow with double asterisk, A–C). However, although both tTG and α‐synuclein inclusions coexist within melanized neurons of the SN in PD brain, minor colocalization on the subcellular level was observed in occasional LBs (arrow with asterisk, A–F). Immunohistochemical staining of ubiquitin (antibody Z0458) demonstrated LBs in melanized neurons of the SN, but no granular staining pattern (G). Immunohistochemical staining of tTG (antibody 06‐471) in the SN, LC and hypothalamus of PD brains demonstrated tTG‐immunoreactive granules in both melanized neurons of the SN (H) and LC (I) in PD brains, whereas these tTG‐immunoreactive structures were present in a small number in hypothalamus (J). In addition, no tTG‐immunoreactive granules were observed in melanized neurons of the SN in Alzheimer's disease (AD) brain (K). Furthermore, no tTG‐immunoreactive granules were observed in melanized neurons of the SN in frontal temporal dementia (FTD) brain (M, arrow), whereas hyperphosphorylated tau immunoreactivity (antibody AT8) was present (L, arrow). Scale bars: (A–K) 20 µm.
No colocalization of granular tTG staining with mitochondria and lysosomes
So far, the subcellular localization of tTG is limited to its presence in the mitochondria and lysosomes, at least in some cell types (40). To elucidate whether the observed tTG immunoreactive granules represent either of these cellular organelles, we investigated the colocalization of tTG with proteins specifically present in these structures (40). We found no colocalization of tTG staining with the staining of antibodies directed against porin (specific for mitochondria, Figure 3A–C) or cathepsin D (specific for lysosomes, Figure 3D–F). Taken together, these data demonstrated that the observed tTG granules are not part of the subcellular structures known to possess tTG, at least not in the cell type studied.
Figure 3.
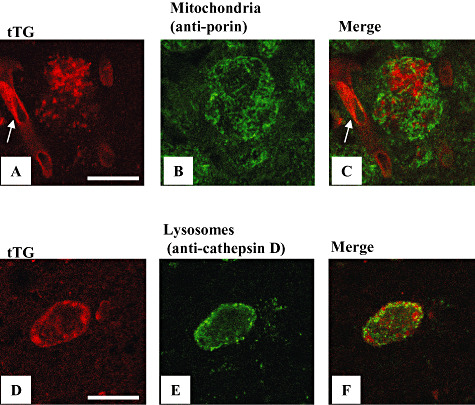
The tissue transglutaminase (tTG)‐immunoreactive granules do not colocalize with antibodies directed against proteins specific for mitochondria and lysosomes. Double immunofluorescence of tTG (A,D, antibody 06‐471) with either an anti‐porin antibody (B, antibody Ab15895, Table 1) or an anti‐cathepsin D antibody (E, antibody MAB422, Table 1) demonstrated no colocalization in melanized neurons of the substantia nigra in Parkinson's disease brains. Panels A and C also show a tTG positive capillary (arrow) using the 06‐471 antibody. Scale bars: (A–F) 20 µm.
Granular tTG is part of the ER in melanized neurons in PD brains
Apart from its connection to mitochondria and lysosomes (40), tTG has been suggested to be associated with the ER (39). Therefore, to investigate whether the tTG‐positive granules are of possible ER origin, we studied the colocalization of tTG with antibodies directed against ER‐specific proteins in melanized neurons in the SN of PD brains. Interestingly, we observed a staining pattern in serial sections of melanized neurons in the SN of PD brains for both the anti‐protein disulfide isomerase (PDI) (Figure 4B) and the anti‐ER protein 57 (ERp57) (Figure 4D) antibodies similar to that observed for tTG (Figure 4A,C). Unfortunately, however, both anti‐PDI and anti‐ERp57 antibodies were found to be unsuitable for double immunofluorescence in paraffin‐embedded tissue sections. We therefore studied the staining pattern of another ER resident protein, calreticulin (34). We found that calreticulin staining (antibody Ab2907, Table 1) demonstrated similar granules as observed with tTG staining in melanized neurons in PD brains, although no overlay in the staining pattern of calreticulin and tTG was observed in other parts of the SN (Figure 4E–H). Similar as to tTG, a semi‐quantitative analysis of our results for the SN demonstrated a highly significant increase in the number of calreticulin positive granule‐containing melanized neurons in PD compared with control brains (Figure 4I). Moreover, double immunofluorescence demonstrated that tTG (Figure 4J) colocalized with calreticulin (Figure 4K) in a significant number of the granular structures found in melanized neurons in the SN of PD brains (Figure 4J–L), indicating that only a specific part of the ER is positive for tTG under these pathological conditions. Furthermore, an alternative anti‐calreticulin (antibody Ab4, Table 1) antibody demonstrated both a similar staining pattern for calreticulin in immunohistochemistry, as well as colocalization of calreticulin with the tTG immunoreactive granules using double immunofluorescence in the melanized neurons in PD brains (not shown), as observed with the anti‐calreticulin antibody (antibody Ab2907).
tTG‐positive granules in melanized neurons are not part of the unfolded protein response
Finally, to establish whether tTG‐immunoreactive granules containing neurons are in a state of ER stress, we analyzed the presence of phosphorylated pancreatic ER kinase (pPERK), a critical mediator and indicator of the unfolded protein response (42), within melanized neurons of the SN in PD brain. Identical to our previous results in a different set of patients (20), we observed pPERK‐immunoreactive granules in melanized neurons of PD brains (Figure 5B–D), whereas no pPERK staining was observed in control patients (Figure 5A). Moreover, we found that melanized neurons, which demonstrated tTG‐immunoreactive granules (Figure 5E), also contained pPERK‐immunoreactive granules (Figure 5F). However, tTG and pPERK did not colocalize within the same set of granules (Figure 5G).
Figure 5.
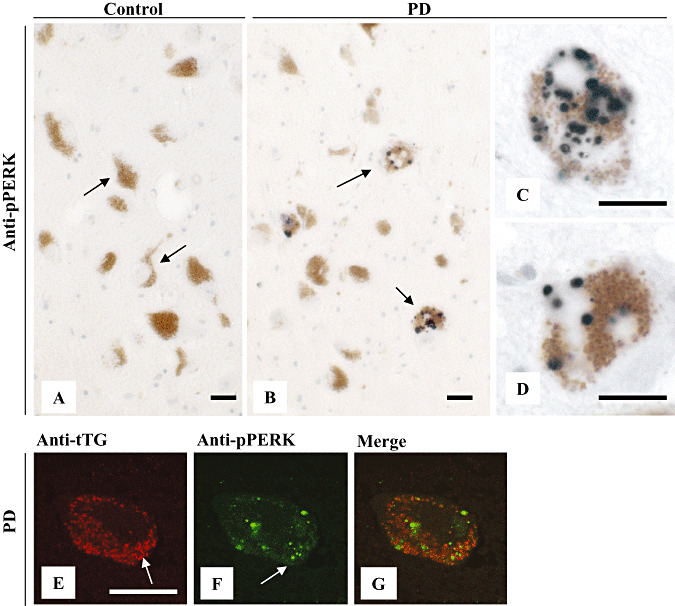
Phosphorylated pancreatic ER kinase (pPERK) staining does not colocalize with tissue transglutaminase (tTG)‐immunoreactive granules in melanized neurons in Parkinson's disease (PD) brains. Immunohistochemical staining of pPERK (antibody Sc‐32577) was absent in melanized neurons of the substantia nigra (SN) in control brain (A). In contrast, pPERK staining was observed in melanized neurons of the SN in PD brains (B–D). Double immunofluorescence of tTG (antibody 06‐471) (E, arrow) and pPERK staining (antibody Sc‐32577) (F, arrow) demonstrated no colocalization of pPERK in tTG‐immunoreactive granules in melanized neurons (G). Scale bars: (A–G) 20 µm.
DISCUSSION
tTG is increasingly implicated in PD pathogenesis. Confirming and extending earlier data by Andringa et al (1), in the current study we showed the presence of tTG immunoreactivity in melanized neurons in SN and LC of PD brain. Besides a faint diffuse cytoplasmic staining, not easily separated from background staining, tTG demonstrated a distinct intracellular granule‐like staining pattern in melanized neurons in PD brain, only scantly observed in controls. Besides cell bodies, these tTG‐immunoreactive structures were observed in neuronal extensions and localized to neurons showing α‐synuclein pathology, thereby implicating them in the PD disease process. Attempts to further characterize the granular tTG staining pattern led us to conclude that in melanized neurons in PD, tTG accumulates in an ER compartment, but does not associate with α‐synuclein containing aggregates. Although the fact that tTG‐induced cross‐links have been identified in LBs, there is no proof that tTG itself occurs in protein aggregates in the PD brain, or forms cross‐linked tTG‐polymers 1, 22. Together, therefore, our results make it highly unlikely that the granular tTG immunoreactivity observed in PD brain represents tTG containing cytoplasmic aggregates.
Inside cells, tTG is generally considered as a soluble cytosolic protein, but it is also observed in the nucleus in cultured cells (11) and in the nucleus of neurons in Huntington's disease brains (55). The association of tTG with mitochondria was the first evidence that tTG might also present in other specific cellular organelles (41). In addition, Zemskov and co‐workers recently demonstrated that the internalization of cell surface tTG results in the association of tTG with lysosomes, as it is routed through the endosomal‐lysosomal pathway toward degradation (56). Earlier data pointed already to the existence of a particulate form of tTG which, unfortunately, escaped further characterization (15). Our study represents the first report that describes a strong association of tTG with granular structures under pathological conditions. While characterizing the origin of these tTG‐immunoreactive structures, we ruled out the aforementioned subcellular compartments in which the presence of tTG has been described. Thus, the lack of immunoreactivity for mitochondria and lysosomal markers argued against these compartments being involved. On the other hand, the presence of immunoreactivity for the soluble ER‐chaperones PDI, ERp57 and calreticulin strongly argued in favor of an ER origin of the tTG‐immunopositive structures.
tTG does not contain a signal sequence targeting it to the ER, although association with membrane structures is known to occur 12, 16. This raises the question as to how and why tTG becomes linked to the ER. Within this context, it is of importance to note that tTG forms complexes with calreticulin inside cells (9). Considering the prevalent localization of calreticulin to the ER, this may be a way in which the tTG molecules are directed toward the ER (34). Furthermore, upon cellular stress, as occurs in melanized neurons of the SN in PD brain, the ER is reportedly capable of forming subcompartments containing ER‐chaperones, in particular calreticulin and PDI 24, 37. These chaperones are involved in protein folding and assembly steps, and are frequently upregulated following cellular stress (37). Among other functions, tTG is involved in the modification of proteins by inducing cross‐links that result in both the stabilization (33) and increased resistance of proteins toward proteolytic‐breakdown 17, 45, while at the same time ensuring continued protein solubility (27). Because almost all tTG positive granules colocalized with calreticulin immunoreactivity, but not vice versa, our data may suggest that the appearance of tTG immunoreactive ER granules reflects, at least part of, a protein‐quality control mechanism to prevent proteins from misfolding or destabilizing as a result of ER stress. As such, tTG redistribution and clustering with protein‐folding chaperones, together with the unfolded protein response (UPR), may initially act to relieve the burden on the ER. This interpretation of our data, albeit speculative at the present time, is supported by the fact that in melanized neurons in PD brain, tTG‐immunoreactive granules were detected in cells showing signs of UPR induction (presence of pPERK). In addition, Beck and colleagues recently demonstrated that tTG activation protects against toxicity induced by the Parkinsonian mimic 1‐methyl‐4‐phenylpyridinium (MPP+) in differentiated human neuroblastoma cells (2).
A number of other intriguing questions can be raised about the functional implications of our results. Under physiological conditions, intracellular cytoplasmic tTG is catalytically silent (36). An activation requires the elevation of Ca2+ to levels which are incompatible with cell survival and which originally formed the basis for the theory linking tTG‐mediated cross‐linking of cellular proteins to the execution phase of (apoptotic) cell death (21). In the Parkinsonian brain, tTG‐induced cross‐links have been identified in both soluble α‐synuclein monomers, small oligomers and in highly aggregated α‐synuclein fibrils in LBs 1, 22, 36. Although these data clearly suggest the presence of catalytically active tTG in PD neurons, it is not clear if and where in the cell the interaction between tTG and α‐synuclein occurs and how the Ca2+ concentrations necessary to activate cytoplasmic tTG can be reached. Here, we observed no apparent colocalization of tTG with highly aggregated α‐synuclein in LBs or LNs. However, melanized neurons in PD brain did show the presence of both granular tTG and aggregated α‐synuclein within the same set of neurons, suggesting that interaction may occur. Recently, Nemes and co‐authors demonstrated that only limited increases in Ca2+ levels are sufficient to activate cross‐linking activity when tTG is bound to membrane lipids (36). As soluble α‐synuclein binds avidly to membranes and the presence of α‐synuclein in ER has been noted (6), tTG associated with ER structures as observed by us, may provide an excellent location for α‐synuclein‐tTG interaction under the conditions prevalent in the Parkinsonian brain. Experiments evaluating this possibility are ongoing in our lab.
Another issue to be solved by future experiments is the possible contribution to PD pathogenesis of the other proteins identified by us in the tTG‐granules in melanized neurons of PD brains, that is, the folding chaperones PDI, ERp57 and calreticulin. The Ca2+‐binding chaperone calreticulin and the oxido‐reductases PDI and ERp57 are abundantly expressed in the ER, where they are primarily involved with (N‐linked) glycosylation of proteins and oxidative protein folding, respectively (37). Together, they play a crucial role in the assembly and transport of MHC class I complexes (4). Similar to other protein chaperones like Hsp70 and Hsp90, however, they are also known to be externalized through a process likely to involve vesicular exocytosis 8, 18, especially under conditions of cell stress (30). At the cell surface, calreticulin, PDI and ERp57 modulate cell adhesion, redox status and immune recognition 3, 4, 34, 49, 54. Only very little data are available about the possible role of these enigmatic proteins in PD pathogenesis. However, impaired function because of oxidative/nitrosative modifications (PDI) or increased expression (calreticulin, ERp57) in neuronal cells cultured in the presence of the dopaminergic neurotoxin 6‐hydroxydopamine has been described 19, 26, 28.
Finally, the prevalent association of tTG immunoreactivity with a granular ER compartment in affected neurons, as observed in the current study, adds to the growing list of reports describing altered ER function in PD (48). Whether an association of tTG with the ER is of a neuroprotective nature or forms part of the pathogenic process driving protein aggregation and neuronal cell death, remains to be established.
DISCLOSURE STATEMENT
None of the authors have any actual or potential conflicts of interest financially, or with other people or organizations that could influence this work.
ACKNOWLEDGMENTS
We would like to thank Dr Anne‐Marie van Dam for her helpful discussions. This work was supported by grants from the “Stichting Internationaal Parkinson Fonds” (to B.D., M.M.M.W and J.J.M.H.).
REFERENCES
- 1. Andringa G, Lam KY, Chegary M, Wang X, Chase TN, Bennett MC (2004) Tissue transglutaminase catalyzes the formation of alpha‐synuclein crosslinks in Parkinson's disease. FASEB J 18:932–934. [DOI] [PubMed] [Google Scholar]
- 2. Beck KE, De Girolamo LA, Griffin M, Billett EE (2006) The role of tissue transglutaminase in 1‐methyl‐4‐phenylpyridinium (MPP+)‐induced toxicity in differentiated human SH‐SY5Y neuroblastoma cells. Neurosci Lett 11:46–51. [DOI] [PubMed] [Google Scholar]
- 3. Bedard K, Szabo E, Michalak M, Opas M (2005) Cellular functions of endoplasmic reticulum chaperones calreticulin, calnexin, and ERp57. Int Rev Cytol 245:91–121. [DOI] [PubMed] [Google Scholar]
- 4. Blanchard N, Shastri N (2008) Coping with loss of perfection in the MHC class I peptide repertoire. Curr Opin Immunol 20:82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 6. Colla E, Liu Y, Stirling W, Jensen PH, Iwatsubo T, Lee MK (2009) Endoplasmic reticulum stress and mircosomes aggretated alpha‐synuclein are associated with the alpha‐synucleinopathies in vivo. Neuroscience Meeting, San Diego, CA: Society for Neuroscience, 532.11/K10.
- 7. Duda JE, Lee VM, Trojanowski JQ (2000) Neuropathology of synuclein aggregates. J Neurosci Res 61:121–127. [DOI] [PubMed] [Google Scholar]
- 8. Evdonin AL, Martynova MG, Bystrova OA, Guzhova IV, Margulis BA, Medvedeva ND (2006) The release of Hsp70 from A431 carcinoma cells is mediated by secretory‐like granules. Eur J Cell Biol 85:443–455. [DOI] [PubMed] [Google Scholar]
- 9. Feng JF, Readon M, Yadav SP, Im MJ (1999) Calreticulin down‐regulates both GTP binding and transglutaminase activities of transglutaminase II. Biochemistry 38:10743–10749. [DOI] [PubMed] [Google Scholar]
- 10. Fesus L (1993) Biochemical events in naturally occurring forms of cell death. FEBS Lett 328:1–5. [DOI] [PubMed] [Google Scholar]
- 11. Fesus L, Piacentini M (2002) Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci 27:534–539. [DOI] [PubMed] [Google Scholar]
- 12. Fesus L, Horvath A, Harsfalvi J (1983) Interaction between tissue transglutaminase and phospholipid vesicles. FEBS Lett 155:1–5. [DOI] [PubMed] [Google Scholar]
- 13. Gentile V, Sepe C, Calvani M, Melone MA, Cotrufo R, Cooper AJ et al (1998) Tissue transglutaminase‐catalyzed formation of high‐molecular‐weight aggregates in vitro is favored with long polyglutamine domains: a possible mechanism contributing to CAG‐triplet diseases. Arch Biochem Biophys 352:314–321. [DOI] [PubMed] [Google Scholar]
- 14. Halverson RA, Lewis J, Frausto S, Hutton M, Muma NA (2005) Tau protein is cross‐linked by transglutaminase in P301L tau transgenic mice. J Neurosci 25:1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hand D, Campoy FJ, Clark S, Fisher A, Haynes LW (1993) Activity and distribution of tissue transglutaminase in association with nerve‐muscle synapses. J Neurochem 61:1064–1072. [DOI] [PubMed] [Google Scholar]
- 16. Harsfalvi J, Arato G, Fesus L (1987) Lipids associated with tissue transglutaminase. Biochim Biophys Acta 923:42–45. [DOI] [PubMed] [Google Scholar]
- 17. Hartley DM, Zhao C, Speier AC, Woodard GA, Li S, Li Z et al (2008) Transglutaminase induces protofibril‐like amyloid beta‐protein assemblies that are protease‐resistant and inhibit long‐term potentiation. J Biol Chem 283:16790–16800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hegmans JP, Bard MP, Hemmes A, Luider TM, Kleijmeer MJ, Prins JB et al (2004) Proteomic analysis of exosomes secreted by human mesothelioma cells. Am J Pathol 164:1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holtz WA, O'Malley KL (2003) Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem 278:19367–19377. [DOI] [PubMed] [Google Scholar]
- 20. Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W (2007) Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun 354:707–711. [DOI] [PubMed] [Google Scholar]
- 21. Ientile R, Caccamo D, Griffin M (2007) Tissue transglutaminase and the stress response. Amino Acids 33:385–394. [DOI] [PubMed] [Google Scholar]
- 22. Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM (2003) Tissue transglutaminase‐induced aggregation of alpha‐synuclein: implications for Lewy body formation in Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 100:2047–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kahlem P, Green H, Djian P (1998) Transglutaminase action imitates Huntington's disease: selective polymerization of Huntingtin containing expanded polyglutamine. Mol Cell 1:595–601. [DOI] [PubMed] [Google Scholar]
- 24. Kamhi‐Nesher S, Shenkman M, Tolchinsky S, Fromm SV, Ehrlich R, Lederkremer GZ (2001) A novel quality control compartment derived from the endoplasmic reticulum. Mol Biol Cell 12:1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karpuj MV, Becher MW, Steinman L (2002) Evidence for a role for transglutaminase in Huntington's disease and the potential therapeutic implications. Neurochem Int 40:31–36. [DOI] [PubMed] [Google Scholar]
- 26. Kim‐Han JS, O'Malley KL (2007) Cell stress induced by the Parkinsonian mimetic, 6‐hydroxydopamine, is concurrent with oxidation of the chaperone, ERp57, and aggresome formation. Antioxid Redox Signal 9:2255–2264. [DOI] [PubMed] [Google Scholar]
- 27. Lai TS, Tucker T, Burke JR, Strittmatter WJ, Greenberg CS (2004) Effect of tissue transglutaminase on the solubility of proteins containing expanded polyglutamine repeats. J Neurochem 88:1253–1260. [DOI] [PubMed] [Google Scholar]
- 28. Lee YM, Park SH, Chung KC, Oh YJ (2003) Proteomic analysis reveals upregulation of calreticulin in murine dopaminergic neuronal cells after treatment with 6‐hydroxydopamine. Neurosci Lett 352:17–20. [DOI] [PubMed] [Google Scholar]
- 29. Lesort M, Chun W, Johnson GV, Ferrante RJ (1999) Tissue transglutaminase is increased in Huntington's disease brain. J Neurochem 73:2018–2027. [PubMed] [Google Scholar]
- 30. Li Z, Srivastava P (2004) Heat‐shock proteins. Curr Protoc Immunol; Appendix 1: Appendix 1T. Review. [DOI] [PubMed] [Google Scholar]
- 31. Lorand L, Graham RM (2003) Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 4:140–156. [DOI] [PubMed] [Google Scholar]
- 32. Martinez A, Portero‐Otin M, Pamplona R, Ferrer I (2010) Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol 20:281–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Melino G, Annicchiarico‐Petruzzelli M, Piredda L, Candi E, Gentile V, Davies PJ et al (1994) Tissue transglutaminase and apoptosis: sense and antisense transfection studies with human neuroblastoma cells. Mol Cell Biol 14:6584–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M (2009) Calreticulin, a multi‐process calcium‐buffering chaperone of the endoplasmic reticulum. Biochem J 417:651–666. [DOI] [PubMed] [Google Scholar]
- 35. Miller ML, Johnson GV (1995) Transglutaminase cross‐linking of the tau protein. J Neurochem 65:1760–1770. [DOI] [PubMed] [Google Scholar]
- 36. Nemes Z, Petrovski G, Aerts M, Seargent K, Devreese B, Fesus L (2009) Transglutaminase‐mediated intramolecular cross‐linking of membrane‐bound {alpha}‐synuclein promotes amyloid formation in Lewy bodies. J Biol Chem 40:27252–27264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ni M, Lee AS (2007) ER chaperones in mammalian development and human diseases. FEBS Lett 581:3641–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Norlund MA, Lee JM, Zainelli GM, Muma NA (1999) Elevated transglutaminase‐induced bonds in PHF tau in Alzheimer's disease. Brain Res 851:154–163. [DOI] [PubMed] [Google Scholar]
- 39. Orru S, Caputo I, D'Amato A, Ruoppolo M, Esposito C (2003) Proteomics identification of acyl‐acceptor and acyl‐donor substrates for transglutaminase in a human intestinal epithelial cell line. Implications for celiac disease. J Biol Chem 278:31766–31773. [DOI] [PubMed] [Google Scholar]
- 40. Park D, Choi SS, Ha KS (2010) Transglutaminase 2: a multi‐functional protein in multiple subcellular compartments. Amino Acids; doi 10.1007/s00726‐010‐0500‐z [E‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 41. Rodolfo C, Mormone E, Matarrese P, Ciccosanti F, Farrace MG, Garofano E et al (2004) Tissue transglutaminase is a multifunctional BH3‐only protein. J Biol Chem 279:54783–54792. [DOI] [PubMed] [Google Scholar]
- 42. Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J (2009) ER stress in Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer's pathology. J Neuroinflammation 6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 54:823–827. [DOI] [PubMed] [Google Scholar]
- 44. Schmid AW, Chiappe D, Pignat V, Grimminger V, Hang I, Moniatte M et al (2009) Dissecting the mechanisms of tissue transglutaminase‐induced cross‐linking of alpha‐synuclein: implications for the pathogenesis of Parkinson disease. J Biol Chem 284:13128–13142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Segers‐Nolten I, Wilhelmus MM, Veldhuis G, van RB, Drukarch B, Subramaniam V (2008) Tissue transglutaminase modulates {alpha}‐synuclein oligomerization. Protein Sci 17:1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Singer SM, Zainelli GM, Norlund MA, Lee JM, Muma NA (2002) Transglutaminase bonds in neurofibrillary tangles and paired helical filament tau early in Alzheimer's disease. Neurochem Int 40:17–30. [DOI] [PubMed] [Google Scholar]
- 47. Alves‐Rodrigues A, Gregori L, Figueiredo‐Pereira ME (1998) Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci 21:516–520. [DOI] [PubMed] [Google Scholar]
- 48. Wang HQ, Takahashi R (2007) Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson's disease. Antioxid Redox Signal 9:553–561. [DOI] [PubMed] [Google Scholar]
- 49. Wang J, Mayernik L, Armant DR (2002) Integrin signaling regulates blastocyst adhesion to fibronectin at implantation: intracellular calcium transients and vesicle trafficking in primary trophoblast cells. Dev Biol 245:270–279. [DOI] [PubMed] [Google Scholar]
- 50. Wilhelmus MM, Grunberg SC, Bol JG, van Dam AM, Hoozemans JJ, Rozemuller AJ et al (2008) Transglutaminases and transglutaminase‐catalyzed cross‐links colocalize with the pathological lesions in Alzheimer's disease brain. Brain Pathol 19:612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilhelmus MM, van Dam AM, Drukarch B (2008) Tissue transglutaminase: a novel pharmacological target in preventing toxic protein aggregation in neurodegenerative diseases. Eur J Pharmacol 585:464–472. [DOI] [PubMed] [Google Scholar]
- 52. Wilhelmus MM, Verhaar R, Bol JG, van Dam AM, Hoozemans JJ, Rozemuller AJ et al (2009) Novel role of transglutaminase 1 in corpora amylacea formation? Neurobiol Aging; doi.org/10.1016/j.neurobiolaging.2009.04.019 [E‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 53. Winklhofer KF, Haass C (2010) Mitochondrial dysfunction in Parkinson's disease. Biochim Biophys Acta 1802:29–44. [DOI] [PubMed] [Google Scholar]
- 54. Xiao G, Chung TF, Pyun HY, Fine RE, Johnson RJ (1999) KDEL proteins are found on the surface of NG108‐15 cells. Brain Res Mol Brain Res 72:121–128. [DOI] [PubMed] [Google Scholar]
- 55. Zainelli GM, Ross CA, Troncoso JC, Muma NA (2003) Transglutaminase cross‐links in intranuclear inclusions in Huntington disease. J Neuropathol Exp Neurol 62:14–24. [DOI] [PubMed] [Google Scholar]
- 56. Zemskov EA, Mikhailenko I, Strickland DK, Belkin AM (2007) Cell‐surface transglutaminase undergoes internalization and lysosomal degradation: an essential role for LRP1. J Cell Sci 120:3188–3199. [DOI] [PubMed] [Google Scholar]