

ABSTRACT
Brain‐derived neurotrophic factor (BDNF) was shown to produce its neuroprotective effect through extracellular signal‐regulated kinase 1/2 (ERK1/2) and phosphatidylinositol‐3 kinase (PI3‐K) signaling. But whether other pathways also mediate the neuroprotective effect of BDNF is less known. In this study, we found that direct administration of BDNF to rat hippocampal CA1 area dose‐dependently increased the mRNA and protein levels of Bcl‐xL. BDNF also increased protein kinase casein kinase II (CK2) activity and NF‐κB phosphorylation at Ser529 dose‐dependently. Further, transfection of the wild‐type CK2α DNA to CA1 neurons increased nuclear factor kappa B (NF‐κB) phosphorylation and Bcl‐xL mRNA expression, whereas transfection of CK2α156A, the catalytically inactive mutant of CK2α, decreased these measures. Moreover, transfection of CK2α small interfering RNA (siRNA) blocked the enhancing effect of BDNF on NF‐κB phosphorylation and Bcl‐xL expression. These results were further confirmed by treatment of 4,5,6,7‐tetrabromobenzotriazole (TBB), a specific CK2 inhibitor. Transfection of NF‐κBS529A, the dominant negative mutant of NF‐κB, prevented the enhancing effect of BDNF on Bcl‐xL expression. More importantly, BDNF activation of CK2 is not affected by co‐administration of the ERK1/2 inhibitor, PD98059, and the PI3‐K inhibitor, LY294002. These results demonstrate a novel BDNF signaling pathway and provide an alternative therapeutic strategy for the protective effect of BDNF on hippocampal neurons in vivo.
Keywords: Bcl‐xL, brain‐derived neurotrophic factor, casein kinase 2, hippocampus, neuroprotection, NF‐κB
INTRODUCTION
Brain‐derived neurotrophic factor (BDNF) is a member of the neurotrophin family that has been shown to play an important role in hippocampal synaptic plasticity (27) and memory function 21, 31. In addition, BDNF was earlier found to increase the survival of acetylcholine neurons and dopamine neurons 3, 24. In studying the signaling pathway that mediates the neuroprotective effect of BDNF, it is found that BDNF protects neonatal cortical and hippocampal neurons from hypoxia‐ischemia injury through the mediation of extracellular signal‐regulated kinase 1/2 (ERK1/2) (18). BDNF also protects immortalized hippocampal neurons from serum‐deprivation‐induced cell death through both ERK1/2 and phosphatidylinositol‐3 kinase (PI3‐K) pathways (39). Further, BDNF increases the expression of Bcl‐2 and protects hippocampal neurons from glutamate‐induced neuronal death through the mediation of ERK1/2 and PI3‐K signaling (4). However, in addition to the role of ERK1/2 and PI3‐K, whether BDNF also activates other protein kinase to mediate its neuroprotective effect is less known.
Casein kinase II (CK2) is a constitutively active and ubiquitous serine/threonine protein kinase that plays a multifunctional role and is highly conserved during evolution 30, 36. The CK2 holoenzyme consists of subunits α, α′ (catalytic) and β (regulatory), which associate to form α2β2, α′2β2 and αα′β2 heterotetramers. There appears to be more than 300 substrates of CK2 that are involved in various cellular functions including DNA synthesis, gene transcription, signal transduction, cytoskeleton structure and cell‐cell adhesion (34). Further, CK2 also functions as a key suppressor of apoptosis (2). Inhibition of CK2 activity was shown to induce apoptosis in cancer cells (40). CK2 was also found to directly phosphorylate the pro‐apoptotic protein Bid and decrease the cleavage of Bid, thereby reducing Bid‐induced cell death (15). CK2 also phosphorylates the ARC protein (an apoptosis repressor) to increase its inhibitory effect on caspase activity (29). Moreover, CK2 was shown to enhance cell survival through activation of Akt and nuclear factor kappa B (NF‐κB)‐mediated pathways 16, 32. However, despite of this anti‐apoptotic effect of CK2, it is not known whether CK2 also mediates the neuroprotective effect of BDNF. In the present study, we aimed to examine whether CK2 mediates the neuroprotective effect of BDNF in rat hippocampus in vivo by measuring the expression of Bcl‐xL, an anti‐apoptotic gene belongs to the Bcl‐2 family (45). We also examined the CK2 signaling pathway that mediates the protective effect of BDNF on hippocampal neurons.
MATERIALS AND METHODS
Animals
Adult male Sprague‐Dawley rats (250–350 g) bred at the Animal Facility of the Institute of Biomedical Sciences, Academia Sinica in Taiwan were used. They were maintained on a 12/12 h light/dark cycle (light on at 6:30 am) with food and water continuously available. Experimental procedures follow the Guidelines of Animal Use and Care of the National Institute of Health.
Plasmid DNA construction and DNA/polyethyleneimine (PEI) complex preparation
For construction of the HA‐tagged CK2α plasmid, full‐length CK2α was cloned by amplifying the rat hippocampal CK2α cDNA with primers 5′‐CGGAATTCTCGGGACCCGTGCCAAGCAG‐3′ (an EcoRI cutting site is underlined and the ATG start coden was removed) and 5′‐GCTCTAGATTACTGCTAGCGCCAGCGG‐3′ (an XbaI cutting site is underlined and the TAA stop coden was maintained) as described previously (12). The PCR product was subcloned between the EcoRI and XbaI sites of the mammalian expression vector pcDNA3. For construction of the Flag‐tagged NF‐κB plasmid, full‐length NF‐κB was cloned by amplifying the rat hippocampal NF‐κB cDNA with primers 5′‐CAGGAATTCGATGGACGATCTGTTTCCC‐3′ (an EcoRI cutting site is underlined) and 5′‐GACCTCGAGTTAGGAGCTGATCTGACTA‐3′ (an XhoI cutting site is underlined) (41). The PCR product was subcloned into the pCMV‐Tag2A expression vector with EcoRI and XhoI sites. The CK2α mutant plasmid (CK2α156A) and the NF‐κB mutant plasmid (NF‐κBS529A) were generated by using the QuickChange Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). PEI was used as the transfection agent. The PEI molecule is a highly branched organic cationic polymer that is able to introduce genes into the mammalian brain in vivo (1) and we have found that PEI is non‐toxic to hippocampal neurons (Figure 1). Therefore, we have used PEI as a transfection reagent for the present study. Before injection, plasmid DNA was diluted in 5% glucose to a stock concentration of 2.77 µg/µL. Branched PEI of 25 kDa (Sigma, St. Louis, MO, USA) was diluted to 0.1 M concentration in 5% glucose and added to the DNA solution (41). Immediately before injection, 0.1 M PEI was added to reach a ratio of PEI nitrogen per DNA phosphate equal to 6 for CK2 and equals to 10 for NF‐κB. The mixture was subjected to vortex for 30 s and allowed to equilibrate for 15 minutes.
Figure 1.
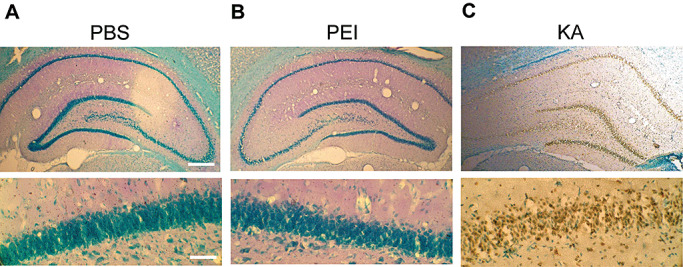
Polyethyleneimine (PEI) does not produce toxicity to hippocampal neurons. Apoptotic nucleus in CA1 area was evaluated by TUNEL staining after (A) phosphate‐buffered saline (PBS), (B) PEI (25 kDa) and (C) kainic acid (KA; 4 µg) was infused to the CA1 area in rats. Animals were sacrificed 48 h after infusion. Their brains were removed and the paraformaldehyde‐fixed brain sections (30‐µm thickness) were subjected to TUNEL assay and examined under a light microscope. The apoptotic nuclei stained with DAB showed brown color under KA treatment. The lower panel is the magnification of CA1 layer for the corresponding upper panel. Scale bar equals 300 µm for the upper panel and scale bar equals 100 µm for the lower panel. n = 2 each group.
TUNEL assay
TUNEL assay was carried out using the Apoptag plus peroxidase in situ apoptosis detection kit (Chemicon, Temecula, CA, USA). Apoptotic cells in the hippocampus were detected according to the procedures described previously (23). Animals were sacrificed 48 h after PBS, PEI or kainic acid (KA) injection in CA1 area. Brain sections (30‐µm thickness) were fixed in 4% paraformaldehyde for 10 minutes and were permeabilized with pre‐cold EtOH/CH3COOH (2:1) followed by reacting with 3% H2O2 to remove the endogenous peroxidase activity. The sections were then incubated with the TdT enzyme and digoxigenin‐dNTP for 1 h at 37°C followed by incubation with anti‐digoxigenin antibody‐conjugated peroxidase for 30 minutes. The apoptotic nuclei showed brown color after staining with DAB. The slides were then counterstained with methyl green for visualization of total cells. Cells were examined under a light microscope.
RNA interference
The rat CK2α small interfering RNA (siRNA) was used to knock down CK2α expression in CA1 neurons. Two sets of CK2α siRNA were used. The sense strand sequence for the first set is: 5′‐CAAACUAUAAUCGUACAUCdTdT‐3′ and the antisense strand sequence for the first set is: 5′‐GAUGUACGAUUAUAGUUUGdTdT‐3′. The sense strand sequence for the second set is: 5′‐AAAUCCCUGACAUCAUAUUdTdT‐3′ and the antisense strand sequence for the second set is: 5′‐AAUAUGAUGUCAGGGAUUUdTdT‐3′(43). The reason for using two sets of CK2α siRNA is because our preliminary results showed that a mixture of these siRNA has a better effect in knocking down CK2α expression (Supporting Information Figure S1). Both sets of CK2α siRNA were synthesized and conjugated with Cy3 by MDBio Inc. (Taipei, Taiwan). The Silencer Negative Control number 1 siRNA was used as a control (Ambion, Austin, TX, USA).
Intra‐hippocampal drug administration, DNA and siRNA transfection
Rats were anesthetized with pentobarbital (40 mg/kg, i.p.) and subjected to stereotaxic surgery. Cannulae were implanted bilaterally to dorsal hippocampal CA1 area at the following coordinates: 3.5 mm posterior to the bregma; 2.5 mm lateral to the midline; and 3.4 mm ventral to the skull surface. For BDNF administration experiment, PBS, 0.2 µg, 0.4 µg and 1.2 µg of recombinant human BDNF (Pepro Tech Ltd., London, UK) were injected to CA1 area in different animals and rats were sacrificed 4 h later. This time interval was adopted from a previous study (5). For BDNF and kinase inhibitor co‐injection experiment, PD98059 (1.4 µg) or LY294002 (1.5 µg) was injected 15 minutes before BDNF (0.4 µg) injection and animals were sacrificed 1 h after BDNF injection. This short time interval was used because we aimed to also examine BDNF activation of ERK1/2 and PI3‐K that occurs soon after BDNF administration (39). For plasmid transfection and BDNF co‐injection experiment, plasmid DNA (1.5 µg/µL) was transfected 48 h before BDNF (0.4 µg) injection and animals were sacrificed 4 h after BDNF injection. Similar procedure was adopted for CK2α siRNA mixture (4 pmol each) and BDNF (0.4 µg) co‐injection experiment except that CK2α siRNA was given 60 h before BDNF injection. The siRNA was transfected by using the cationic polymer transfection reagent jetSITM (Polyplus‐Transfection Inc., New York, NY, USA). For plasmid DNA co‐transfection experiment, both plasmids (0.75 µg each) were mixed and injected together and animals were sacrificed 48 h later. The injection volume was 0.7 µL each side and the injection rate was 0.1 µL/min. For co‐injections at the same time or with 15 minutes apart, 0.35 µL of each molecule was injected. The injection needle was left in place for 5 minutes to limit the diffusion of injected molecule. Animal's brain was removed and the hippocampal tissue slices (2‐mm thickness each slice, two slices in all) were dissected out by using a brain slicer. The hippocampal tissue containing the major CA1 area and the transfected area was further dissected out by using a punch with 2 mm in outer diameter and 1.4 mm in inner diameter with the lower part of the punch located at the dentate gyrus. The injection site was recognized by the position of the needle tip and/or the needle track during tissue dissection and that was covered by the punch (Supporting Information Figure S2). This punched area with 1.4 mm in diameter and 4 mm in thickness approximates 6 mm3 in volume. We then removed subarea “A” located above the alveus hippocampus layer from the punched tissue, which is easily detachable, and used the remaining tissue for mRNA determination and Western blot (Supporting Information Figure S2). Subarea “A” counts about 13% of the total punched area in volume. Thus, the tissue volume used for biochemical assays was approximately 5.2 mm3. But this area presumably contains more fibers than cells (see Discussion for further information).
CK2 activity assay
CK2 activity was determined as described previously (12). Briefly, CK2 activity was measured in 5 µg protein aliquots from each sample using the protein kinase CK2 assay kit (Upstate, Lake Placid, NY, USA). The assay was carried out at 30°C for 10 minutes by using a specific synthetic peptide as substrate and a kinase inhibitor that blocks the activity of other serine/threonine kinases. An amount of 5 µCi of [γ‐32P] ATP was added to each reaction mixture. The reaction was stopped by the addition of 20 µL of 40% TCA. A volume of 25 µL of the reaction mixture was spotted onto P81 papers, and washed five times with 400 mL of 0.75% phosphoric acid. Paper pieces were then rinsed with acetone, dried and transferred to scintillation vials for radioactivity counting. The enzyme activity was calculated by subtracting the blank (determined in identical assays from which the peptide substrate was omitted) from the 32P radioactivity incorporated in the presence of the substrate.
Quantitative real‐time PCR
Total RNA from CA1 tissue was isolated by using the RNAspin mini kit (GE Healthcare, Barrington, IL, USA). The cDNA was generated from 0.5 µg total RNA with 0.5 µg of oligo‐dT by Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Real‐time PCR analysis was performed by using the Applied Biosystem 7500 Real‐Time PCR System (Foster City, CA, USA) with the TaqMan method. The primers and probe of Bcl‐xL, an Assays‐on‐Demand Gene Expression product (Rn00580568_g1), were purchased from Applied Biosystems. The house‐keeping enzyme hypoxanthine phosphoribosyl transferase (HPRT) was used as an internal control. The primers and TaqMan probe of HPRT were synthesized by Applied Biosystems with the following sequences: forward primer 5′‐GCCGACCGGTTCTGTCAT‐3′ and reverse primer 5′‐TCATAACCTGGTTCATCATCACTAATC‐3′; TaqMan probe 5′‐VIC‐TCGACCCTCAGTCCCAGCGTCG‐TARMA‐3′. The amount of Bcl‐xL and HPRT mRNA from each sample was analyzed simultaneously but in separate tubes. The thermal conditions were: 2 minutes at 55°C; and 10 minutes at 95°C; followed by 40 cycles at 95°C for 15 s and 60°C for 1 minute.
Western blot
Hippocampal CA1 tissue lysate was lysed in RIPA buffer (50 mM Tris‐HCl (pH 7.4), 150 mM NaCl, 2 mM ethylenediaminetetraacetic acid (EDTA), 1% IGEPAL CA‐630, 1 mM phenymethylsulfonyl fluoride (PMSF), 20 µg/mL pepstatin A, 20 µg/mL leupeptin, 20 µg/mL aprotinin, 50 mM NaF and 1 mM Na3VO4). The lysate was resolved by 8% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE). The proteins resolved by SDS‐PAGE were transferred to the polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA, USA). The PVDF membrane was pre‐incubated with 0.05% TBS‐T containing 2% BSA followed by incubation with different antibodies: anti‐pS529NF‐κB (1:1000; Acris, Herford, Germany), anti‐NF‐κB (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐Bcl‐xL (1:2000; Chemicon), anti‐ERK1/2 (1:4000; Upstate), anti‐pERK1/2 (1:2000; Cell Signaling, Beverly, MA, USA), anti‐Akt (1:4000; Cell Signaling), anti‐pS308Akt (1:1000; Cell Signaling), anti‐CK2α (1:4000, Abcam, Cambridge, MA, USA), anti‐NeuN (1:1000, Chemicon) and anti‐β‐actin (1:10 000; Chemicon) antibody and then with HRP‐conjugated secondary antibody (Amersham, Piscataway, NJ, USA). Membrane was developed by reacting with chemiluminescence HRP substrate and exposed to the LAS‐3000 image system (Fujifilm, Tokyo, Japan) for visualization of protein bands. The density of each band was quantified by using the NIH Image J software (NIH, Bethesda, MD).
Immunohistochemistry
At appropriate times after drug (KA) injection or DNA, siRNA transfection, rats were anesthetized with pentobarbital (100 mg/kg, i.p.) and perfused with ice‐cold phosphate‐buffered saline followed by 4% paraformaldehyde. Brain were removed and post‐fixed in 30% sucrose/4% paraformaldehyde solution for 20–48 h. Brains were then frozen, cut into 30‐µm sections on a cryostat and mounted on gelatin‐coated slides. Brain sections were rinsed with 1xPBS for 10 minutes and permeabilized with pre‐cold EtOH/CH3COOH (95%: 5%) for 10 minutes followed by 1xPBS for 10 minutes three times. The sections were preincubated in a blocking solution containing 3% normal goat serum, 3% BSA, and 0.2% Triton X‐100 in 1×PBS for 2 h followed by 1×PBS for 10 minutes three times. For immunofluorescence detection, tissue sections were incubated with a mouse monoclonal anti‐HA antibody (1:100; Roche Products, Welwyn Garden City, Hertfordshire, UK) in blocking buffer at 4°C overnight. Sections were washed three times in 1×PBS and incubated in goat anti‐mouse fluorescein isothiocyanate (FITC)‐conjugated IgG antibody (1:100, Sigma) in 1×PBS for 1 h. Sections were washed three times in 1×PBS and mounted with mounting medium.
Statistics
Data from CK2 enzyme activity, Western blot and real‐time PCR were analyzed with Student's t‐test or one‐way analysis of variance (ANOVA) followed by post‐hoc Dunnett's t‐test (represented by tD value) or Newman–Keuls multiple comparison test (represented by q‐value).
RESULTS
PEI does not produce toxicity to hippocampal neurons
Because PEI was used as a reagent for transfection in the present study, we first examined whether PEI itself produces toxicity to hippocampal neurons in vivo. KA was used as a positive control. Animals received bilateral injections of PBS, PEI (25 kDa) or KA (4 µg) to hippocampal CA1 area (n = 2 each) and were sacrificed 48 h later for examination of the apoptotic cells by using the TUNEL assay. The apoptotic nuclei were shown in brown color after staining with DAB. Results showed that KA dramatically increased the density of apoptotic cells in both the CA1 area and dentate gyrus of the hippocampus (Figure 1C vs. Figure 1A). But PEI injection did not cause any apoptotic cell death in either subregion examined (Figure 1B vs. Figure 1A). The lower panels are magnifications of the CA1 layer for the corresponding upper panels.
BDNF increased Bcl‐xL expression, CK2 activity and NF‐κB phosphorylation
In this experiment, we first examined the effect of BDNF on Bcl‐xL expression and CK2 enzyme activity. In addition, because CK2 was shown to phosphorylate NF‐κB at Ser‐529 and upregulate its transcriptional activity in controlling tumor necrosis factor alpha signaling (44), and NF‐κB plays an important role in neuronal survival (33), we also examined the effect of BDNF on NF‐κB phosphorylation at this residue. Animals were randomly divided into four groups (n = 4–8 each) to receive different concentrations (PBS, 0.2, 0.4 and 1.2 µg) of BDNF injection to hippocampal CA1 area. Results revealed that BDNF dose‐dependently increased Bcl‐xL mRNA level (F 3,22 = 37.49, P < 0.001; Figure 2A) and Bcl‐xL protein level (F 3,14 = 18.63, P < 0.001; Figure 2B). Meanwhile, the same BDNF injection elevated CK2 enzyme activity (F 3,14 = 23.33, P < 0.001; Figure 2C) and NF‐κB phosphorylation at Ser‐529 (F 3,14 = 92.7, P < 0.001; Figure 2D) in a dose‐dependent manner. Figure 2E is a representative illustration showing the location of needle placement and dye distribution in CA1 area.
Figure 2.
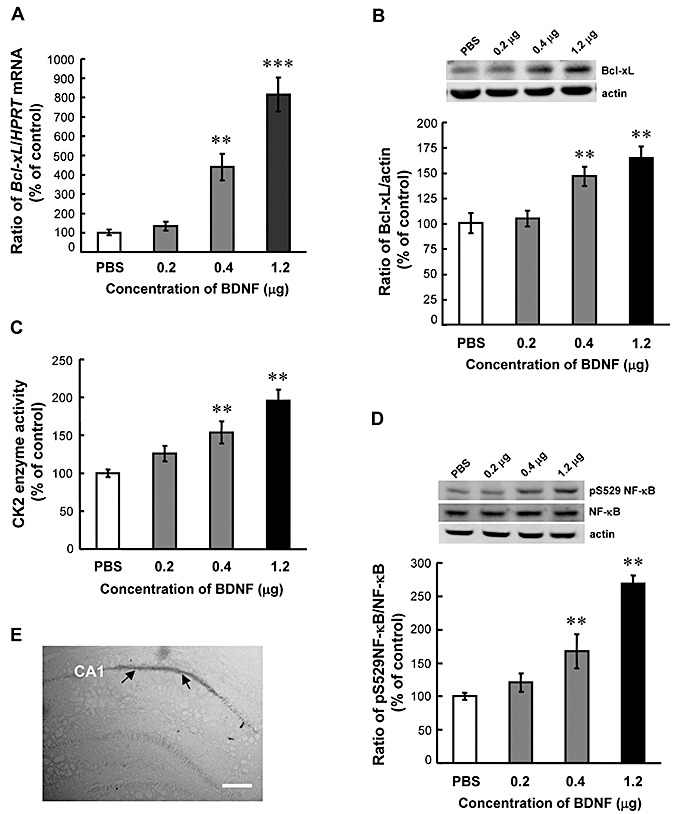
Brain‐derived neurotrophic factor (BDNF) increases Bcl‐xL expression, casein kinase II (CK2) enzyme activity and nuclear factor kappa B (NF‐κB) phosphorylation in rat hippocampus. Dose‐response effect of intra‐hippocampal BDNF injection on (A) Bcl‐xL mRNA level, (B) Bcl‐xL protein level, (C) CK2 enzyme activity and (D) NF‐κB phosphorylation at Ser529 in CA1 area. Animals received bilateral infusions of PBS, 0.2, 0.4 or 1.2 µg BDNF in CA1 area and were sacrificed 4 h later. n = 4–8 each group. Data are expressed as mean ± standard error of the mean. Statistical significance was evaluated by one‐way analysis of variance followed by Dunnett's t‐test. **P < 0.01, ***P < 0.001. E. A representative illustration showing the location of needle placement and dye distribution (0.3% methylene blue) in CA1 area. Arrows indicate the area of dye distribution. Scale bar equals 300 µm.
CK2 activation enhanced NF‐κB phosphorylation and Bcl‐xL expression
We next examined whether CK2 regulates NF‐κB activity and Bcl‐xL expression in rat hippocampus. Because the phosphorylation of NF‐κB also enhances the transcriptional activity of NF‐κB (14), we have measured the level of NF‐κB phosphorylation in this experiment. Animals were randomly divided into three groups (n = 6–10 each) and the CK2α wild‐type DNA (CK2αWT) or the catalytically inactive CK2α mutant DNA (CK2α156A) was transfected to hippocampal CA1 area. Control animals received pcDNA3‐HA transfection. NF‐κB phosphorylation and Bcl‐xL mRNA expression were examined 48 h after transfection. Results revealed that transfection of CK2αWT increased NF‐κB phosphorylation at Ser‐529 (F 2,20 = 69.91, P < 0.001; q = 8.12, P < 0.01), whereas transfection of CK2α156A decreased NF‐κB phosphorylation at this residue (q = 4.71, P < 0.01) (Figure 3A). Similarly, CK2αWT transfection increased Bcl‐xL mRNA level (F 2,20 = 116.74, P < 0.001; q = 10.18, P < 0.01), but CK2α156A transfection decreased this measure (q = 6.48, P < 0.01) (Figure 3B). To confirm the transfection and expression of CK2αWT plasmid in CA1 area, immunohistochemistry was carried out by using antibody against the HA‐tagged protein and FITC‐conjugated IgG secondary antibody. Results showed apparent immunostaining in the CA1 area (Figure 3C, left panel). The area of transfection is approximately 14% of the total CA1 area (marked in dotted lines) viewed from a single plane according to the quantification method we adopted before (42). Immunohistochemistry at a higher magnification revealed that HA‐CK2αWT plasmid was indeed transfected and expressed in individual cells in CA1 area (Figure 3C, right panel). To further estimate the percentage of cells transfected with this plasmid and the type of cells been transfected, we have examined the expression of CK2α and DAPI as well as the expression of CK2α and NeuN by using a confocal microscope. Results from Figure 3D revealed that CK2α is co‐expressed with DAPI in the same neurons, but CK2α is mainly distributed in the cytoplasm area (upper panels). Further cell count revealed that the number of HA‐positive cells is 160 in the area selected (indicated by arrow in the upper left panel of Figure 3D) and the number of DAPI‐positive cells is 155 (The nucleus of some cells may not be seen under the same tissue plane). Thus, the transfection efficiency is 100% in the selected area. The total number of cells been transfected in the CA1 area at least doubles in this single plane because the area adjacent to the selected area (left to the arrow, counts for about 50% of total transfected area) showed more intense staining, which made the exact cell count more difficult. Results from confocal images also showed that CK2α is co‐expressed with NeuN (Figure 3D, lower panels) in most of the cells examined. This result suggests that CK2α is expressed in neurons in CA1 area.
Figure 3.
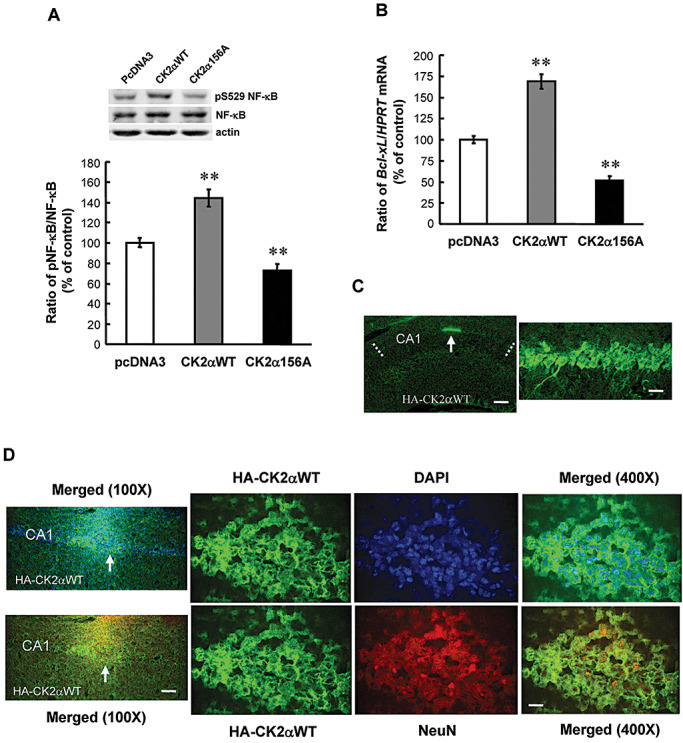
Casein kinase 2 (CK2) increases nuclear factor kappa B (NF‐κB) phosphorylation and Bcl‐xL mRNA expression. Effects of CK2αWT DNA transfection and CK2α156A mutant DNA transfection on (A) NF‐κB phosphorylation at Ser529 and (B) Bcl‐xL mRNA expression in CA1 area. Animals received plasmid DNA transfection (1.5 µg) in CA1 area bilaterally and were sacrificed 48 h later. n = 6–10 each group. Statistical significance was evaluated by one‐way analysis of variance followed by Dunnett's t‐test. **P < 0.01. C. Immunohistochemical staining showing CK2αWT DNA transfection and expression in CA1 area (left panel, arrow indicates the area of plasmid expression). The anti‐HA‐tagged antibody and fluorescein isothiocyanate (FITC)‐conjugated IgG secondary antibody were used for visualization of plasmid transfection and expression in individual cells in CA1 area at a higher magnification (right panel). Dotted lines indicate the CA1 area. Scale bar equals 250 µm for the left panel and scale bar equals 25 µm for the right panel. D. Confocal images showed the co‐expression of CK2α and 4′,6‐diamidino‐2‐phenylindole (DAPI) in the same cells in CA1 area (upper panels). Arrow indicates the right half of the tissue showing coexpression and was used for viewing at a higher magnification (right three panels). Confocal images also showed the co‐expression of CK2α and NeuN in the same neurons in CA1 area (lower panels). Similarly, arrow indicates the right half of the tissue showing co‐expression and was further viewed at a higher magnification (right three panels). Scale bar of the lower left panel equals 100 µm and scale bar of the lower right panel equals 25 µm.
CK2 mediates the effect of BDNF on NF‐κB phosphorylation and Bcl‐xL expression
We next examined whether CK2 mediates the effect of BDNF on NF‐κB phosphorylation and Bcl‐xL expression. The CK2α siRNA (mixture of two sets, 4 pmol each) was used to knock down CK2 protein expression. Results revealed that CK2α siRNA transfection decreased CK2α protein level in the hippocampus (t1,6 = 3.64, P = 0.01, n = 4 each) (Figure 4A). Immunofluorescence staining against Cy3 and DAPI indicated that CK2α siRNA was indeed transfected to the CA1 area (pink color, Figure 4B upper left panel). The area of transfection is approximately 18% of the total CA1 area (marked in dotted lines) viewed from a single plane. Confocal image at a higher magnification revealed that CK2α siRNA was indeed transfected in individual cells in CA1 area (Figure 4B, upper right panel). The lower left panel of Figure 4B indicated DAPI staining of the same tissue session and the lower right panel of Figure 4B showed that CK2α siRNA is coexpressed with DAPI in the same neurons. Further cell count revealed that the number of CK2α siRNA‐positive cells is 103 in the area selected (indicated by arrow in the upper left panel of Figure 4B) and the number of DAPI‐positive cells is 117 in the same area selected. Thus, the transfection efficiency is approximately 88%. Similar to the situation of HA‐CK2αWT transfection, the total number of cells been transfected with CK2α siRNA is at least 206 viewed from a single plane. The CK2α siRNA was therefore used for further experiments. Animals were randomly divided into four groups (n = 4–5 each) to receive intra‐hippocampal negative siRNA + PBS injection, negative siRNA + BDNF injection, CK2α siRNA + PBS injection and CK2α siRNA + BDNF injections. Results revealed that BDNF consistently increased NF‐κB phosphorylation at Ser‐529 (F 3,13 = 21.99, P < 0.001; q = 7.17, P < 0.01) and CK2α siRNA decreased NF‐κB phosphorylation at this residue (q = 4.18, P = 0.01). But CK2α siRNA further antagonized the effect of BDNF on NF‐κB phosphorylation (q = 6.89, P < 0.01 comparing the BDNF + CK2α siRNA group with BDNF group) (Figure 4C). In analyzing the Bcl‐xL mRNA and protein levels, we have found that BDNF consistently increased Bcl‐xL mRNA expression (F 3,13 = 96.38, P < 0.001; q = 18.3, P < 0.001). CK2α siRNA alone decreased Bcl‐xL mRNA level (q = 3.2, P < 0.05), but it antagonized the effect of BDNF on Bcl‐xL mRNA expression (q = 8.98, P < 0.01 comparing the BDNF + CK2α siRNA group with BDNF group) (Figure 4D). Similar results were found with Bcl‐xL protein expression. BDNF increased Bcl‐xL protein level (F 3,13 = 25.2, q = 6.88, P < 0.01). CK2α siRNA alone decreased this measure (q = 4.43, P < 0.05), but it further blocked the effect of BDNF on Bcl‐xL protein expression (q = 10.05, P < 0.01 comparing the BDNF + CK2α siRNA group with BDNF group) (Figure 4E).
Figure 4.
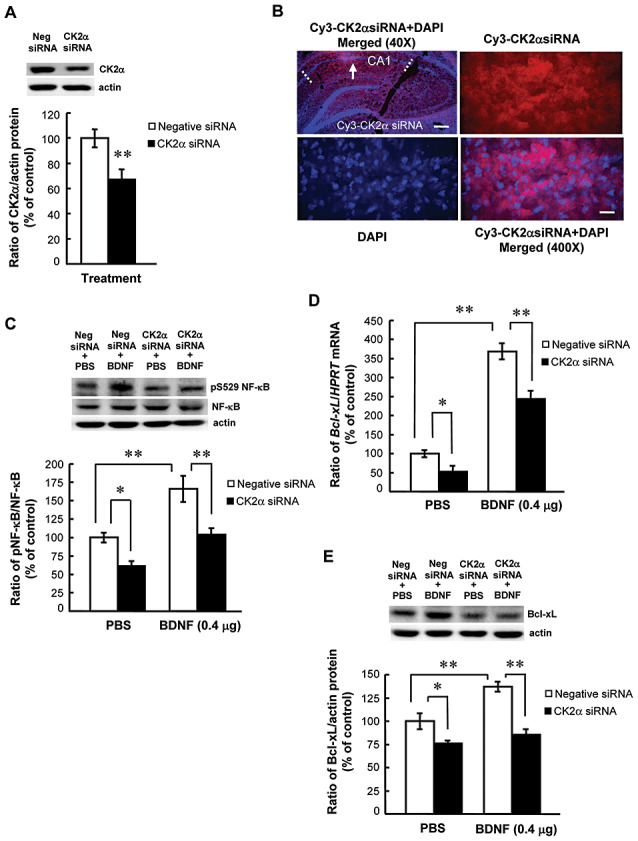
CK2α small interfering RNA (siRNA) antagonized the effects of brain‐derived neurotrophic factor (BDNF) on nuclear factor kappa B (NF‐κB) phosphorylation and Bcl‐xL expression. A. Effect of CK2α siRNA transfection on basal casein kinase II (CK2) protein level in CA1 area (n = 4 each group). B. Representative immunohistochemical staining against Cy3 and confocal image showing the co‐expression of CK2α siRNA and 4′,6‐diamidino‐2‐phenylindole (DAPI) in the same neurons in CA1 area (color in pink). Arrow indicates the right half of the CA1 tissue showing the co‐expression and was further viewed at a higher magnification. Dotted lines indicate the CA1 area. CK2α siRNA (upper right) and DAPI (lower left) staining alone at a higher magnification was shown. The merged image at a higher magnification showed the co‐expression of CK2α siRNA and DAPI in the same neurons in CA1 area. Scale bar equals 360 µm for the upper left panel and scale bar equals 36 µm for the lower right panel. Effects of prior CK2α siRNA transfection (8 pmol) on BDNF‐induced (C) NF‐κB phosphorylation at Ser529 (D) Bcl‐xL mRNA level and (E) Bcl‐xL protein level in CA1 area. Animals received bilateral transfections of CK2α siRNA (or negative siRNA) 60 h prior to 0.4 µg BDNF infusion in CA1 area and were sacrificed 4 h after BDNF infusion. N = 4–5 each group. Data are expressed as mean ± standard error of the mean. Statistical significance was evaluated by Student's t‐test or one‐way analysis of variance followed by Newman–Keul's comparison. *P < 0.05, **P < 0.01.
Inhibition of CK2 activity blocks the effect of BDNF on NF‐κB phosphorylation and Bcl‐xL expression
To further assess the role of CK2 in mediating the effect of BDNF on NF‐κB phosphorylation and Bcl‐xL mRNA expression, different concentrations (PBS, 10 ng, 20 ng and 40 ng) of the CK2 inhibitor 4,5,6,7‐tetrabromobenzotriazole (TBB) was injected to the CA1 area in different animals (n = 4 each). Animals were sacrificed 30 minutes after TBB injection and their CA1 tissue was subjected to CK2 enzyme activity assay. Results revealed that TBB decreased CK2 activity in a dose‐dependent manner (F 3,12 = 8.11, P < 0.01) with 20 ng and 40 ng of TBB both producing a significant effect (tD = 3.02, P < 0.05 and tD = 4.59, P < 0.01, respectively) (Figure 5A). In the next experiment, animals were randomly divided into three groups (n = 6 each) to receive PBS + PBS infusions, PBS + BDNF (0.4 µg) infusions and TBB (10 ng) + BDNF (0.4 µg) infusions. TBB was given 30 minutes before BDNF infusion and animals were sacrificed 4 h after BDNF infusion. The CA1 tissue from these animals was subjected to NF‐κB phosphorylation level determination and Bcl‐xL mRNA level determination. Results revealed that BDNF administration consistently increased the level of NF‐κB phosphorylation (F 2,15 = 13.87, P < 0.001; q = 7.29, P < 0.01), but this effect was blocked by prior TBB injection (q = 4.95, P < 0.01 when comparing the TBB + BDNF group with BDNF group) (Figure 5B). In addition, BDNF consistently increased Bcl‐xL mRNA level (F 2,15 = 20.84, P < 0.001; q = 9.02, P < 0.01), and this effect was also antagonized by prior TBB injection (q = 5.75, P < 0.01 when comparing the TBB + BDNF group with BDNF group) (Figure 5C).
Figure 5.
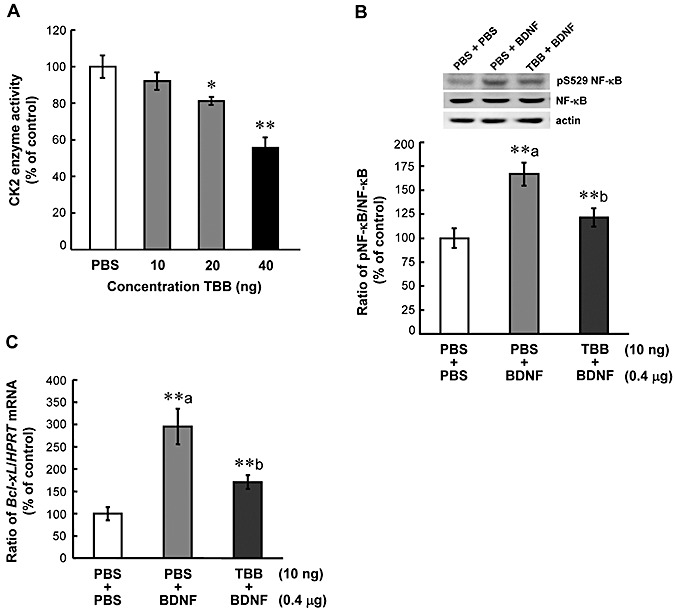
Casein kinase II (CK2) inhibitor blocks the effects of brain‐derived neurotrophic factor (BDNF) on nuclear factor kappa B (NF‐κB) phosphorylation and Bcl‐xL expression. A. Dose–response effect of 4,5,6,7‐tetrabromobenzotriazole (TBB), a specific CK2 inhibitor, on CK2 activity in rat hippocampus. Animals received bilateral hippocampal infusions of phosphate‐buffered saline (PBS) or TBB (10, 20 and 40 ng) and were sacrificed 30 minutes after the injection. n = 4 each group. Effects of prior TBB injection on BDNF‐induced (B) NF‐κB phosphorylation at Ser529 and (C) Bcl‐xL mRNA expression. Animals received bilateral hippocampal infusions of 10 ng TBB (or PBS) followed by 0.4 µg BDNF in CA1 area and were sacrificed 4 h after BDNF infusion. The interval between the two infusions was 30 minutes. N = 6 each group. Data are expressed as mean ± standard error of the mean. Statistical significance was evaluated by one‐way analysis of variance followed by Newman–Keul's comparison. *P < 0.05, **P < 0.01; a = compared with the PBS + PBS group, b = compared with the PBS + BDNF group.
NF‐κB mediates the effect of BDNF and CK2α on Bcl‐xL expression
After establishing the relationship among BDNF, CK2 and Bcl‐xL expression, we then examined whether NF‐κB plays a role in this signaling pathway. This is proposed because CK2 was shown to enhance cell survival through activation of NF‐κB (32). Animals were randomly divided into four groups (n = 5–7 each). The kinase‐dead mutant of NF‐κB, NF‐κBS529A, was transfected to hippocampal CA1 area and its effect on BDNF‐induced or CK2‐activated Bcl‐xL mRNA expression was determined. Control animals received pCMV vector transfection + PBS injection (or pCMV transfection + pcDNA3 transfection). Results revealed that NF‐κBS529A transfection alone significantly decreased Bcl‐xL mRNA level in CA1 area (F 3,17 = 101.67, P < 0.001; q = 6.14, P < 0.01). BDNF consistently increased Bcl‐xL mRNA level (q = 20.14, P < 0.01), but NF‐κBS529A antagonized this effect of BDNF (q = 11.01, P < 0.01 comparing the NF‐κBS529A + BDNF group with BDNF group) (Figure 6A). Further, NF‐κBS529A transfection alone consistently decreased Bcl‐xL mRNA level (F 3,17 = 27.13, P < 0.001; q = 7.9, P < 0.01). CK2αWT transfection increased Bcl‐xL mRNA level (q = 7.08, P < 0.01), but this effect was similarly blocked by NF‐κB S529A co‐transfection (q = 5.67, P < 0.01 comparing the NF‐κBS529A+CK2αWT group with CK2αWT group) (Figure 6B).
Figure 6.
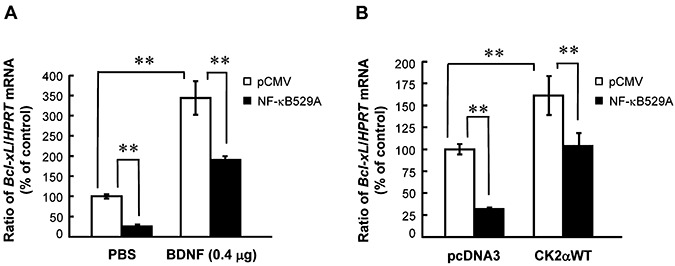
NF‐κB mediates the effect of brain‐derived neurotrophic factor (BDNF) and casein kinase II (CK2) α on Bcl‐xL expression. A. Effect of prior NF‐κBS529A transfection (1.5 µg) on BDNF‐induced Bcl‐xL mRNA expression. Animals received bilateral NF‐κBS529A DNA transfection (1.5 µg) and BDNF infusion (0.4 µg) to CA1 area 48 h apart and were sacrificed 4 h after BDNF infusion. B. Effect of prior NF‐κBS529A transfection (1.5 µg) on CK2αWT DNA‐induced Bcl‐xL mRNA expression. Animals received bilateral NF‐κBS529A and CK2αWT DNA co‐transfection (0.75 µg each) in CA1 area and were sacrificed 48 h later. n = 5–7 each group. Data are expressed as mean ± standard error of the mean. Statistical significance was evaluated by one‐way analysis of variance followed by Newman–Keul's comparison. **P < 0.01.
BDNF activation of CK2 is independent of BDNF activation of ERK1/2 and PI3‐K
In this experiment, inhibitors for ERK1/2 (PD98059) and PI3‐K (LY294002) were injected to CA1 area 15 minutes before BDNF injection in separate groups of animals (n = 5 each) and CK2 activity was determined 1 h after BDNF injection. Results revealed that BDNF increased CK2 activity (F 3,16 = 5.23, P < 0.01; q = 3.84, P < 0.05), but prior PD98059 treatment did not alter this effect of BDNF (q = 0.55, P > 0.05 comparing the BDNF + PD98059 group with BDNF group) (Figure 7A, upper). The effectiveness of PD98059 injection was confirmed by an apparent reduction of pERK1/2 level in Western blot (Figure 7A, lower). Similarly, BDNF increased CK2 activity in another assay (F 3,16 = 4.51, P < 0.05; q = 3.93, P < 0.05), but prior LY294002 treatment did not alter this effect of BDNF either (q = 0.01, P > 0.05 comparing the BDNF + LY294002 group with BDNF group) (Figure 7B upper). The effectiveness of LY294002 injection was confirmed by an apparent reduction of pS308Akt level in Western blot (Figure 7B, lower).
Figure 7.
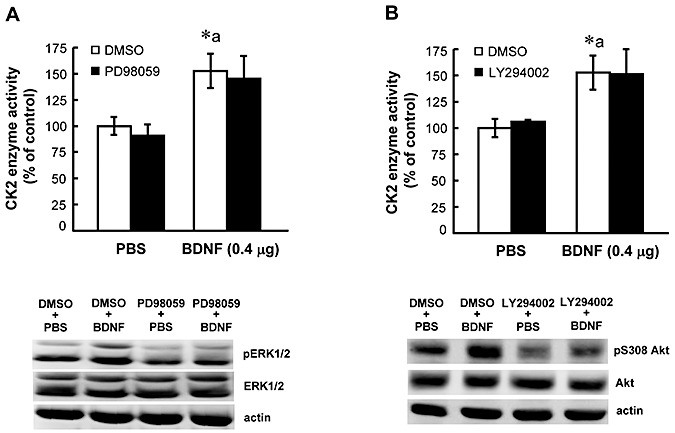
Brain‐derived neurotrophic factor (BDNF) activation of casein kinase II (CK2) is independent of BDNF activation of ERK1/2 and PI3‐K. A. Effects of prior infusion of PD98059 on BDNF‐induced CK2 enzyme activity (upper panel) and pERK1/2 as well as ERK1/2 levels (lower panel) in CA1 area. Animals received BDNF infusion alone (0.4 µg) or in combination with the ERK1/2 inhibitor PD98059 (1.4 µg). PD98059 was given 15 minutes before BDNF infusion and animals were sacrificed 1 h after BDNF infusion. B. Effects of prior infusion of LY294002 on BDNF‐induced CK2 enzyme activity (upper panel) and pS308Akt as well as Akt levels (lower panel) in CA1 area. Animals received BDNF infusion alone (0.4 µg) or in combination with the PI3‐K inhibitor LY294002 (1.5 µg). LY294002 was given 15 mintues before BDNF infusion and animals were sacrificed 1 h after BDNF infusion. N = 5 each group. Data are expressed as mean ± standard error of the mean. Statistical significance was evaluated by one‐way analysis of variance followed by Newman–Keul's comparison. *P < 0.05, a: compared with the corresponding control group.
DISCUSSION
The present results demonstrate that BDNF upregulated Bcl‐xL expression through activation of the CK2‐NF‐κB signaling pathway in rat hippocampus. This result is consistent with a previous report showing that BDNF enhances CK2 activity in rat hippocampal slice (6) and that CK2 is an important enzyme in the nervous system (7). It is also consistent with the notion that CK2 plays an important role in anti‐apoptosis (2). On the other hand, BDNF was previously found to increase anti‐apoptotic gene expression and produce its neuroprotective effect through the mediation of ERK1/2 and P13‐K signaling. Here, we showed an alternative pathway that BDNF could produce its effect through CK2 signaling. Furthermore, BDNF activation of CK2 seems to be independent of BDNF activation of ERK1/2 and PI3‐K. In another study, BDNF was found to enhance bone morphogenetic protein 7‐dependent Smad phosphorylation through activation of CK2 that is also independent of BDNF activation of ERK1/2 and PI3‐K (13). The present result is also congruent with a previous report that CK2 mediates the neuroprotective effect of glial cell line‐derived neurotrophic factor on dopamine neurons (12). These results together suggest that CK2 may play an important role in conveying the information of neurotrophic factors other than ERK1/2 and PI3‐K. However, the present result is incongruent with the finding that CK2 is activated by ERK1/2 possibly through protein phosphatase 1 (PP1) or PP2A under BDNF treatment (6). On the other hand, CK2 was shown to be a component of the kinase suppressor of Ras (KSR1) complex and KSR1/CK2 interaction facilitates KSR1 to activate Raf kinase and ERK1/2 signaling (38). Moreover, CK2 was shown to modulate ERK1/2 signaling through phosphorylation of MAP kinase phosphatase 3 (MKP3) (9). These latter results suggest that CK2 may even be a molecule upstream of ERK1/2 that regulates ERK1/2 activity. Yet, despite the fact that CK2 is constitutively active in the cell, the mechanism for BDNF activation of CK2 remains to be elucidated.
The transcription factor NF‐κB was shown to play an important role in cell survival 8, 33. Decrease in NF‐κB/RelA phosphorylation by ERK1/2 inhibitor or PI3‐K inhibitor was shown to increase cell death in cancer cells or HTLV‐1‐transfected cells 26, 37. It is suggested that NF‐κB mediates cell survival through transcriptional regulation of various anti‐apoptotic genes, although NF‐κB also regulates few pro‐apoptotic gene expression (8). Further, DNA damage was shown to activate NF‐κB and NF‐κB activation is believed to mediate cell survival (25). In studying the relationship between CK2 and NF‐κB, it is found that CK2 directly phosphorylates NF‐κB/RelA at Ser529 in mediating tumor necrosis factor α signaling (44). CK2 also phosphorylates NF‐κB at Ser529 to upregulate nitric oxide synthase II (NOSII) gene expression (11). Here, our result is consistent with a model in which CK2 activation of NF‐κB at Ser529 mediates the enhancing effect of BDNF on Bcl‐xL expression. But it is possible that CK2 may also indirectly upregulate Bcl‐xL expression through activation of other unknown molecules to mediate the effect of BDNF or that CK2 may be permissively required for the upregulation of Bcl‐xL by BDNF. Further, activity‐dependent signaling pathway among different cells may also account for the effect of BDNF on Bcl‐xL expression. This result is consistent with the observation that inhibition of CK2 phosphorylation on NF‐κB/RelA increases the sensitivity of apoptosis in constitutively NF‐κB‐expressing cells (32). It is also congruent with the finding that inhibition of CK2 activity by TGF‐β1 increases the stabilization of IκBα and promotes apoptosis in immortalized hepatocytes (10). In analyzing the promoter of the Bcl‐xL gene, two functional NF‐κB DNA‐binding sites were identified (17). This report suggests that NF‐κB directly regulates Bcl‐xL expression and it is supported by our finding that NF‐κBS529A transfection decreased Bcl‐xL mRNA level in the hippocampus. In addition, over‐expression of Inhibitor of apoptotic protein hRFI reduces apoptosis in cancer cells through activation of NF‐κB and upregulation of Bcl‐2 and Bcl‐xL (28). Further, platelet activating factor upregulates Bcl‐xL expression and this is also mediated through NF‐κB (22). These results support our finding that transfection of NF‐κBS529A blocked the effects of BDNF and CK2α on Bcl‐xL expression. As mentioned previously, in addition to NF‐κB, CK2 also phosphorylates other proteins to result in anti‐apoptosis. Whether these CK2 signaling cascades also mediate the effect of BDNF on Bcl‐xL expression requires further investigation.
In the present study, we found that BDNF dose‐dependently increased Bcl‐xL expression in rat hippocampus. This result is congruent with the notion that one major mechanism for BDNF to promote cell survival is mediated through upregulation of anti‐apoptotic gene expression of the Bcl‐2 family (4). Indeed, our results showed that BDNF also increased Bcl‐2 mRNA level except that the magnitude of increase was not as dramatic as that of Bcl‐xL mRNA (Supporting Information Figure S3). Further, BDNF at higher concentrations increased Bcl‐xL mRNA level for several folds whereas it increased Bcl‐xL protein level for only 1.5‐ to twofold. This is probably caused by a strong effect of BDNF in regulation of Bcl‐xL mRNA expression, rapid degradation of the Bcl‐xL mRNA and low stability of the Bcl‐xL protein because Bcl‐xL protein was found to degrade more rapidly than any other protein of the Bcl‐2 family when interleukin‐3 stimulation was withdrawn (35). Other translational regulation mechanisms may also account for this difference.
In the present study, transfection was made only to a limited area in CA1 neurons (about 350–450 µm viewed from a single plane and 150 µm in thickness; that counts for only 3.6% of the total volume of the punched tissue), but significant biochemical changes were observed. One possible explanation is that the protein extraction method we used is suitable for extraction of proteins in the cell. The proteins present in the fiber are not easily extracted. Besides, there are presumably more fibers (such as the neuronal processes of CA1 neurons and dentate gyrus neurons as well as part of the corpus callosum) than cells in the punched area, and the protein density is much higher in cells than in fibers. When biochemical assays were performed, the amount of protein, instead of tissue volume, was used as a criterion. That is probably why significant biochemical changes were still observed in a limited transfection area. There are other possible explanations. For example, in another study, Han et al have found that transfection of the cyclic AMP‐responsive element‐binding protein (CREB) plasmid was made to only approximately 16% to 20% of lateral amygdala cells, but the CREB plasmid transfection to this limited area successfully rescues fear memory deficit in CREB‐deficient mice (19). Moreover, selective depletion of the neurons overexpressing CREB after behavioral training blocks that fear memory (20). These results suggest that activation of a specific subpopulation of neurons and the neuronal activity as well as the establishment of neuronal circuits among these neurons is sufficient to mediate behavioral changes. The same mechanism may take place in hippocampal neurons in the present study. But the exact mechanism of how do these neurons communicate with other untransfected neurons and the neuronal network among these neurons mediating the observed changes remains to be studied. In addition, the present results showed that CK2α plasmid DNA and CK2α siRNA are effectively transfected and expressed in neurons adjacent to the needle tip. But they are not easily diffused to the neighboring cells such as the injected dye does (Figure 2E). This probably relates to the property of the transfection reagents we used because both PEI (for plasmid transfection) and jetSITM (for siRNA transfection) have a higher viscosity than PBS does. Future experiments adjusting the concentrations of PEI and jetSITM or using an infusion needle of a relatively larger inner diameter should help to increase the transfection area.
In summary, the present results demonstrate that BDNF could produce its anti‐apoptotic effect in vivo through activation of a CK2‐mediated pathway other than the originally identified ERK1/2 and PI3‐K pathways. Further, BDNF activation of CK2 is independent of BDNF activation of ERK1/2 and PI3‐K (Figure 8). These results may provide an alternative therapeutic strategy for protection of hippocampal neurons against injury.
Figure 8.
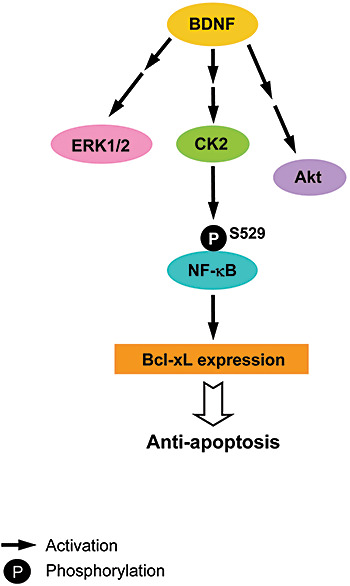
Brain‐derived neurotrophic factor (BDNF) increases Bcl‐xL expression through casein kinase II (CK2) activation and NF‐κB mediation. BDNF enhances Bcl‐xL expression through a CK2‐activated and NF‐κB‐mediated pathway in rat hippocampus. Further, BDNF activation of CK2 is independent of BDNF activation of ERK1/2 and PI3‐K.
Supporting information
Figure S1. CK2α siRNA decreased the level of CK2 expression. A. Representative gel pattern of the first set of CK2α siRNA on CK2 protein level in PC12 cells. PC12 cells were transfected with 50 pmol of the first set CK2α siRNA (or negative siRNA) and CK2 protein level was determined 48 h later by Western blot. Experiments are in duplicates. B. Representative gel pattern of the second set of CK2α siRNA or the combination of the first and second sets of CK2α siRNA on CK2 protein level in hippocampal CA1 area. Animals received bilateral infusions of the negative siRNA (8 pmol), second set of CK2α siRNA (8 pmol) or equal mixture of the first set and second set of CK2α siRNA (4 pmol each) in CA1 area and were sacrificed 60 h later. CK2 protein level was determined by Western blot. N = 2–3 each group.
Figure S2. An illustration showing the area of plasmid transfection, needle position and the hippocampal area punched out for mRNA and protein determination. An infusion needle with 0.31 mm of inner width was used for CK2α plasmid transfection and CK2α siRNA transfection. The transfected area is marked in red lines. The infusion needle protrudes the cannula for 1.5 mm. The tissue been punched out is marked by the inner yellow circle. The outer diameter of the punch is 2 mm and the inner diameter is 1.4 mm with the wall thickness of 0.3 mm each side. The subarea “A” marked in white lines is the area that is further removed from the punched tissue before biochemical assays (It is about 1/7 in volume of the punched tissue). The area marked in purple color around the inner boundary of the punch is the true area that was used for mRNA determination and Western blot. The dotted lines in white color mark the boundary of CA1 layer.
Figure S3. BDNF increased Bcl‐2 mRNA expression. Animals received bilateral infusions of BDNF in the CA1 area (PBS, 0.2, 0.4 and 1.2 µg) and were sacrificed 4 h later. Their brain tissues containing the CA1 area were subjected to quantitative real‐time PCR determination of the Bcl‐2 mRNA level. N = 4–8 each group. Data are expressed as mean ± SEM. Statistical significance was evaluated by one‐way ANOVA followed by Dunnett’s t‐test. **P < 0.01 (F 3,22 = 14.52, P < 0.001).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
This work was supported a Grant from the National Science Council of Taiwan (NSC 96‐2320‐B‐320‐012‐MY2) and by research fund from the Institute of Biomedical Sciences (IBMS), Academia Sinica in Taiwan. Part of this work was done when C.C. Chao was a postdoctoral fellow at IBMS, Academia Sinica. The authors do not have conflicts of interest in this research.
REFERENCES
- 1. Abdallah B, Hassan A, Benoist C, Goula D, Behr JP, Demeneix BA (1996) A powerful nonviral vector for in vivo gene transfer into the adult mammalian brain: polyethylenimine. Hum Gene Ther 7:1947–1954. [DOI] [PubMed] [Google Scholar]
- 2. Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K (2008) Protein kinase CK2α—a key suppressor of apoptosis. Adv Enzyme Regul 48:179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alderson RF, Alterman AL, Barde YA, Lindsay RM (1990) Brain‐derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron 5:297–306. [DOI] [PubMed] [Google Scholar]
- 4. Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Grãos MM et al (2005) Neuroprotection by BDNF against glutamate‐induced apoptotic cell death is mediated by ERK and PI3‐kinase pathways. Cell Death Differ 12:1329–1343. [DOI] [PubMed] [Google Scholar]
- 5. Blanquet PR (1998) Neurotrophin‐induced activation of casein kinase 2 in rat hippocampal slices. Neuroscience 86:739–749. [DOI] [PubMed] [Google Scholar]
- 6. Blanquet PR (2000) Identification of two persistently activated neurotrophinregulated pathways in rat hippocampus. Neuroscience 95:705–719. [DOI] [PubMed] [Google Scholar]
- 7. Blanquet PR (2002) Casein kinase 2 as a potentially important enzyme in the nervous system. Prog Neurobiol 60:211–246. [DOI] [PubMed] [Google Scholar]
- 8. Burstein E, Duckett CS (2003) Dying for NF‐kappaB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr Opin Cell Biol 15:732–737. [DOI] [PubMed] [Google Scholar]
- 9. Castelli M, Camps M, Gillieron C, Leroy D, Arkinstall S, Rommel C et al (2004) MAP kinase phosphatase 3 (MKP3) interacts with and is phosphorylated by protein kinase CK2alpha. J Biol Chem 279:44731–44739. [DOI] [PubMed] [Google Scholar]
- 10. Cavin LG, Romieu‐Mourez R, Panta GR, Sun J, Factor VM, Thorgeirsson SS et al (2003) Inhibition of CK2 activity by TGF‐beta1 promotes IkappaBalpha protein stabilization and apoptosis of immortalized hepatocytes. Hepatology 38:1540–1551. [DOI] [PubMed] [Google Scholar]
- 11. Chantome A, Pance A, Gauthier N, Vandroux D, Chenu J, Solary E et al (2004) Casein kinase II mediated phosphorylation of NF‐kappaB p65 subunit enhances inducible nitric‐oxide synthase gene transcription in vivo . J Biol Chem 279:23953–23960. [DOI] [PubMed] [Google Scholar]
- 12. Chao CC, Chiang CH, Ma YL, Lee EH (2006) Molecular mechanism of the neurotrophic effect of GDNF on DA neurons: role of protein kinase CK2. Neurobiol Aging 27:105–118. [DOI] [PubMed] [Google Scholar]
- 13. Chaverneff F, Barrett J (2009) Casein kinase II contributes to the synergistic effects of BMP7 and BDNF on Smad 1/5/8 phosphorylation in septal neurons under hypoglycemic stress. J Neurochem 109:733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L et al (2005) NF‐κB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol 25:7966–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Desagher S, Osen‐Sand A, Montessuit S, Magnenat E, Vilbois F, Hochmann A et al (2001) Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol Cell 8:601–611. [DOI] [PubMed] [Google Scholar]
- 16. Di Maira G, Salvi M, Arrigoni G, Marin O, Sarno S, Brustolon F et al (2005) Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ 12:668–677. [DOI] [PubMed] [Google Scholar]
- 17. Glasgow JN, Qiu J, Rassin D, Grafe M, Wood T, Perez‐Pol JR (2001) Transcriptional regulation of the BCL‐X gene by NF‐kappaB is an element of hypoxic responses in the rat brain. Neurochem Res 26:647–659. [DOI] [PubMed] [Google Scholar]
- 18. Han BH, Holtzman DM (2000) BDNF protects the neonatal brain from hypoxicischemic injury in vivo via the ERK pathway. J Neurosci 20:5775–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA et al (2007) Neuronal competition and selection during memory formation. Science 316:457–460. [DOI] [PubMed] [Google Scholar]
- 20. Han JH, Kushner SA, Yiu AP, Hsiang HL, Buch T, Waisman A et al (2009) Selective erasure of a fear memory. Science 323:1492–1495. [DOI] [PubMed] [Google Scholar]
- 21. Heldt SA, Stanek L, Chhatwal JP, Ressler KJ (2007) Hippocampus‐specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry 12:656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heon SK, Ko HM, Kim HA, Choi JH, Jun Park S, Kim KJ et al (2006) Platelet‐activating factor induces up‐regulation of antiapoptotic factors in a melanoma cell line through nuclear factor‐kappaB activation. Cancer Res 66:4681–4686. [DOI] [PubMed] [Google Scholar]
- 23. Hsu WL, Chiu TH, Tai JC, Ma YL, Lee EHY (2009) A novel defense mechanism that is activated on amyloid‐β insult to mediate cell survival: role of SGK1‐STAT1/STAT2 signaling. Cell Death Differ 16:1515–1529. [DOI] [PubMed] [Google Scholar]
- 24. Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP et al (1991) BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 350:230–232. [DOI] [PubMed] [Google Scholar]
- 25. Janssens S, Tschopp J (2006) Signals from within: the DNA‐damage‐induced NFkappaB response. Cell Death Differ 13:773–784. [DOI] [PubMed] [Google Scholar]
- 26. Jeong SJ, Pise‐Masison CA, Radonovich MF, Park HU, Brady JN (2005) Activated AKT regulates NF‐kappaB activation, p53 inhibition and cell survival in HTLV‐1‐transformed cells. Oncogene 24:6719–6728. [DOI] [PubMed] [Google Scholar]
- 27. Kang H, Schuman EM (1995) Long‐lasting neurotrophin‐induced enhancement of synaptic transmission in the adult hippocampus. Science 267:1658–1662. [DOI] [PubMed] [Google Scholar]
- 28. Konishi T, Sasaki S, Watanabe T, Kitayama J, Nagawa H (2006) Overexpression of hRFI inhibits 5‐fluorouracil‐induced apoptosis incolorectal cancer cells via activation of NF‐kappaB and upregulation of BCL‐2 and BCL‐XL. Oncogene 25:3160–3169. [DOI] [PubMed] [Google Scholar]
- 29. Li PF, Li J, Muller EC, Otto A, Dietz R, von Harsdorf R (2002) Phosphorylation by protein kinase CK2: a signaling switch for the caspase‐inhibiting protein ARC. Mol Cell 10:247–258. [DOI] [PubMed] [Google Scholar]
- 30. Litchfield DW (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma YL, Wang HL, Wu HC, Wei CL, Lee EH (1998) Brain‐derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long‐term potentiation in rats. Neuroscience 82:957–967. [DOI] [PubMed] [Google Scholar]
- 32. Manna SK, Manna P, Sarkar A (2007) Inhibition of RelA phosphorylation sensitizes apoptosis in constitutive NF‐kappaB‐expressing and chemoresistant cells. Cell Death Differ 14:158–1570. [DOI] [PubMed] [Google Scholar]
- 33. Mattson MP, Meffert MK (2006) Roles for NF‐kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ 13:852–860. [DOI] [PubMed] [Google Scholar]
- 34. Meggio F, Pinna LA (2003) One‐thousand‐and one‐substrates of protein kinase CK2. FASEB J 17:349–368. [DOI] [PubMed] [Google Scholar]
- 35. Packham G, White EL, Eischen CM, Yang H, Parganas E, Ihle JN et al (1998) Selective regulation of Bcl‐xL by a Jak kinase‐dependent pathway is bypassed in murine hematopoietic malignancies. Genes Dev 12:2475–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pinna LA, Meggio F (1997) Protein kinase CK2 (“casein kinase‐2”) and its implication in cell division and proliferation. Prog Cell Cycle Res 3:77–97. [DOI] [PubMed] [Google Scholar]
- 37. Rengifo‐Cam W, Umar S, Sarkar S, Singh P (2007) Antiapoptotic effects of progastrin on pancreatic cancer cells are mediated by sustained activation of nuclear factor‐{kappa}B. Cancer Res 67:7266–7274. [DOI] [PubMed] [Google Scholar]
- 38. Ritt DA, Zhou M, Conrads TP, Veenstra TD, Copeland TD, Morrison DK (2007) CK2 is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr Biol 17:179–184. [DOI] [PubMed] [Google Scholar]
- 39. Rossler OG, Giehl KM, Thiel G (2004) Neuroprotection of immortalized hippocampal neurones by brain‐derived neurotrophic factor and Raf‐1 protein kinase: role of extracellular signal‐regulated protein kinase and phosphatidylinositol 3‐kinase. J Neurochem 88:1240–1252. [DOI] [PubMed] [Google Scholar]
- 40. Ruzzene M, Penzo D, Pinna LA (2002) Protein kinase CK2 inhibitor 4,5,6,7‐tetrabromobenzotriazole (TBB) induces apoptosis and caspase‐dependent degradation of haematopoietic lineage cell‐specific protein 1 (HS1) in Jurkat cells. Biochem J 364:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tai DJC, Su CC, Ma YL, Lee EHY (2009) SGK1 phosphorylation of IκB kinase and p300 upregulates NF‐κB activity and increases N‐methyl‐D‐aspartate receptor NR2A and NR2B expression. J Biol Chem 284:4073–4089. [DOI] [PubMed] [Google Scholar]
- 42. Tsai KJ, Chen SK, Ma YL, Hsu WL, Lee EHY (2002) sgk, a primary glucocorticoid‐induced gene, facilitates memory consolidation of spatial learning in rats. Proc Natl Acad Sci USA 99:3990–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ulery PG, Nestler EJ (2007) Regulation of DeltaFosB transcriptional activity by Ser27 phosphorylation. Eur J Neurosci 25:224–230. [DOI] [PubMed] [Google Scholar]
- 44. Wang D, Westerheide SD, Hanson JL, Baldwin AS Jr (2000) Tumor necrosis factor alpha‐induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem 275:32592–32597. [DOI] [PubMed] [Google Scholar]
- 45. Youle RJ, Strasser A (2008) The BCL‐2 protein family: opposing activities that mediate cell death. Nat Rev 9:47–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CK2α siRNA decreased the level of CK2 expression. A. Representative gel pattern of the first set of CK2α siRNA on CK2 protein level in PC12 cells. PC12 cells were transfected with 50 pmol of the first set CK2α siRNA (or negative siRNA) and CK2 protein level was determined 48 h later by Western blot. Experiments are in duplicates. B. Representative gel pattern of the second set of CK2α siRNA or the combination of the first and second sets of CK2α siRNA on CK2 protein level in hippocampal CA1 area. Animals received bilateral infusions of the negative siRNA (8 pmol), second set of CK2α siRNA (8 pmol) or equal mixture of the first set and second set of CK2α siRNA (4 pmol each) in CA1 area and were sacrificed 60 h later. CK2 protein level was determined by Western blot. N = 2–3 each group.
Figure S2. An illustration showing the area of plasmid transfection, needle position and the hippocampal area punched out for mRNA and protein determination. An infusion needle with 0.31 mm of inner width was used for CK2α plasmid transfection and CK2α siRNA transfection. The transfected area is marked in red lines. The infusion needle protrudes the cannula for 1.5 mm. The tissue been punched out is marked by the inner yellow circle. The outer diameter of the punch is 2 mm and the inner diameter is 1.4 mm with the wall thickness of 0.3 mm each side. The subarea “A” marked in white lines is the area that is further removed from the punched tissue before biochemical assays (It is about 1/7 in volume of the punched tissue). The area marked in purple color around the inner boundary of the punch is the true area that was used for mRNA determination and Western blot. The dotted lines in white color mark the boundary of CA1 layer.
Figure S3. BDNF increased Bcl‐2 mRNA expression. Animals received bilateral infusions of BDNF in the CA1 area (PBS, 0.2, 0.4 and 1.2 µg) and were sacrificed 4 h later. Their brain tissues containing the CA1 area were subjected to quantitative real‐time PCR determination of the Bcl‐2 mRNA level. N = 4–8 each group. Data are expressed as mean ± SEM. Statistical significance was evaluated by one‐way ANOVA followed by Dunnett’s t‐test. **P < 0.01 (F 3,22 = 14.52, P < 0.001).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item