

Abstract
Transmigration of neutrophil [polymorphonuclear neutrophil (PMN)] across the blood–brain barrier (BBB) is a critical event in the pathogenesis of bacterial meningitis. We have shown that IbeA is able to induce meningitic Escherichia coli invasion of brain microvascular endothelial cells (BMECs), which constitutes the BBB. In this report, we provide evidence that IbeA and its receptor, vimentin, play a key role in E. coli‐induced PMN transmigration across BMEC. In vitro and in vivo studies indicated that the ibeA‐deletion mutant ZD1 was significantly less active in stimulating PMN transmigration than the parent strain E44. ZD1 was fully complemented by the ibeA gene and its product. E. coli‐induced PMN transmigration was markedly inhibited by withaferin A, a dual inhibitor of vimentin and proteasome. These cellular effects were significantly stimulated and blocked by overexpression of vimentin and its head domain deletion mutant in human BMEC, respectively. Our studies further demonstrated that IbeA‐induced PMN migration was blocked by bortezomib, a proteasomal inhibitor and correlated with upregulation of endothelial ICAM‐1 and CD44 expression through proteasomal regulation of NFκB activity. Taken together, our data suggested that IbeA and vimentin contribute to E. coli K1‐stimulated PMN transendothelial migration that is correlated with upregulation of adhesion molecule expression at the BBB.
Keywords: BMEC, IbeA, meningitis, PMN migration, vimentin
INTRODUCTION
Escherichia coli is the most common gram‐negative bacterium causing neonatal sepsis and meningitis (NSM) 14, 17, 25. Most cases of bacterial meningitis in newborns develop as a result of hematogenous spread (17). It is accompanied by NFκB activation, pathogen penetration and polymorphonuclear leukocyte [polymorphonuclear neutrophil (PMN)] migration across the blood–brain barrier (BBB), three hallmark features of bacterial meningitis 14, 17, 22, 25, 51. NSM caused by E. coli usually begins with bacterial adhesion to intestinal epithelial cells and then pathogen penetration across the gut barrier into the blood stream (17). Human and animal studies suggest that the development of sepsis and meningitis is correlated with the magnitude of bacteremia, which is essential for E. coli crossing the BBB (17). Bacterial tropism for brain microvascular endothelial cells (BMECs) is the primary step in the pathogenesis of meningitis 14, 17, 25. Bacterial adhesion and penetration through the BBB initiates activation of brain endothelia and glia and leads to leukocyte recruitment and detrimental inflammatory mediator release 11, 25. While in vitro and in vivo models have been informative about E. coli invasion determinants and host factors that contribute to bacterial entry into the central nervous system (CNS) 14, 17, 25, little is known about the specific contribution of the BBB endothelium to PMN transmigration.
A number of virulence factors have been implicated in the pathogenesis of E. coli meningitis, including Ibe proteins (IbeA, IbeB, IbeC, IbeR and IbeT), AslA, FimH, TraJ and OmpA 5, 14, 17, 19, 25. Most of those virulence factors are present in both E. coli K‐1 and K‐12 strains 5, 14. Among these virulence factors, the GimA genetic island, which encodes IbeT, IbeA and IbeR, is unique to E. coli pathogens but not present in nonpathogenic E. coli K12 strains. IbeT, IbeA and IbeR contribute to endothelial adhesion, invasion and virulence regulation, respectively. IbeA has been identified as an important virulence factor that contributes to E. coli K1 invasion of both intestinal epithelial cells and BMEC in vitro and in vivo 18, 19, 37. We and others have recently demonstrated that IbeA is an outer membrane protein 20, 27. The ibeA gene is more significantly prevalent in the E. coli strains causing early infections of human neonates (42), suggesting that IbeA may play an important role in the early step of E. coli NSM. The specific IbeA‐BMEC surface protein interaction and subsequently induced signal transduction were shown to be essential for E. coli K1 invasion (8). Vimentin, a mesenchymal marker, has been identified as an IbeA‐binding protein that is present on the surface of human BMEC (HBMEC), but not on that of intestinal epithelial cells such as Caco‐2 (52). This suggests that different entry mechanisms may be responsible for E. coli K1 penetration across the gut barrier and the BBB. The extracellular vimentin can contribute to various novel functions (52). Vimentin has been recently found on the extracellular surface of various cells, including activated macrophages (32), viable Sezary T cells (21), apoptotic T lymphocytes (2), activated platelets (38), apoptotic neutrophils (29), endothelial cells (49) and skeletal‐muscle cells (3). A recent study has shown that vimentin is required for transmigration of peripheral blood T‐ and B‐lymphocytes across human umbilical vein endothelial cells (HUVECs) and skin (peripheral) endothelial cells (34). Vimentin on both endothelial cells and lymphocytes is able to form a highly dynamic anchoring structure at the site of contact between these two cell types. The initiation of this process was significantly decreased in vim‐deficient (vim −/−) lymphocytes and endothelial cells. Currently, it is unclear whether and how vimentin contributes to PMN transmigration across the BBB in response to meningitic infection in a manner substantially similar to the traversal of lymphocytes across peripheral endothelial cells. PMN transmigration into the CNS is a crucial feature of bacterial meningitis (51). It is a key aspect of the protective response against invading pathogens. But over recent years, evidence has accumulated that leukocytes also contribute importantly to the deleterious effects of inflammation on the brain in bacterial meningitis 47, 51. Whereas the paradigm for leukocyte migration to non‐BBB sites of infection has been extensively studied and fairly well defined 30, 31, 43, transmigration of neutrophils across the BBB endothelial surface appears to be mechanistically different. Studies on transmigration of PMN across the BBB have been limited to bacterial meningitis caused by Meningococcus, Pneumococcus and Group B Streptococcus 9, 10, 11, 39. E. coli‐induced adhesive interactions between transmigrating leukocytes and pulmonary endothelial cells are well understood 13, 31, 48. ICAM‐1 and CD44 play a role in the leukocyte transmigration process during E. coli pneumonia 13, 48. Meningitic E. coli K1 invasion has been extensively studied in both in vitro and in vivo systems of the BBB, BMEC and the neonatal murine models of E. coli meningitis 14, 15, 16, 17, 18, 19, 20, 25. However, there are no in vitro and in vivo studies to date that focus on the PMN‐endothelial cell interactions in response to meningitic E. coli K1 and its virulence factors. Such studies may provide new insights into the pathogenesis and therapeutics of neonatal E. coli meningitis.
In the previous studies, we have established and used both the in vitro and in vivo models of the BBB for dissecting the mechanisms of microbial invasion 5, 8, 14, 17, 18, 19, 52. The present study has focused specifically on interactions between the intact meningitic E. coli K1 and BMEC and the role of the virulence factor IbeA in eliciting neutrophil transendothelial migration. Using the in vitro and in vivo models of the BBB and molecular genetic approaches, we have demonstrated that IbeA and its receptor vimentin are required to induce the potent PMN transendothelial migratory response. IbeA is able to upregulate the endothelial adhesion molecules CD44 and ICAM‐1. The role of IbeA in E. coli K1‐induced neutrophil transmigration appears to require a complex interaction between the bacteria, the neutrophil and the endothelium.
MATERIALS AND METHODS
Isolation and preparation of PMN
PMN were isolated from heparin anticoagulated, human peripheral venous blood of healthy adult volunteers using the standard Ficoll–Hypaque method in accordance with a protocol approved by the Institutional Review Board for Human Subjects at Saban Research Institute of Childrens Hospital Los Angeles. Neutrophils were isolated by dextran sedimentation of leukocytes followed by Ficoll–Hypaque density gradient centrifugation of PMN and hypotonic lysis of erythrocytes as previously described 30, 31. PMN suspensions in normal saline were used for both adhesion and transmigration assays.
Isolation and culture of HBMEC
HBMEC were isolated and cultured as described previously 14, 17, 19, 45. HBMEC were routinely cultured in RPMI 1640 medium (Mediatech, Herndon, VA, USA) containing 10% heat inactivated fetal bovine serum, 10% Nu‐serum, 2 mM glutamine, 1 mM sodium pyruvate, essential amino acids, vitamins, penicillin G (50 µg/mL) and streptomycin (100 µg/mL) at 37°C in 5% CO2. Cells were passed from T‐75 flasks into experimental plates when ∼70%–80% confluent. Cells were detached by trypsin‐EDTA and subcultured on collagen‐coated Transwell (3‐µM pore size, 6.5‐mm diameter) (BD Biosciences, San Jose, CA, USA). BMEC monolayers on Transwell filters were monitored by measuring trans‐endothelial electrical resistance (TEER) changes across the endothelial cell monolayer using an End Ohm epithelial voltohmeter (World Precision Instruments, Sarasota, FL, USA) 7, 24. The confluent BMEC monolayer displays a cobblestone appearance when grown on collagen‐coated surfaces. The cells are positive for factor VIII and fluorescently labeled acetylated low‐density lipoprotein (Dil‐AcLDL) uptake, demonstrating their endothelial origin and also express gamma glutamyl transpeptidase (GGT) and carbonic anhydrase (CA) IV, indicating their brain origin (16). HBMEC are polarized and exhibit an average TEER value of 250–300 Ω/cm2 (7). The cells also exhibit the typical characteristics for brain endothelial cells expressing tight junctions and maintaining apical‐to‐basal polarity. BMEC monolayers grown to confluence on Transwell inserts were released from the Transwell filter by trypsin and counted for calculation of multiplicity of infection (MOI).
Bacterial strains, plasmids, protein expression/purification and reagents
E44 is a rifampin‐resistant derivative of E. coli K1 strain RS218 (018:K1: H7) isolated from the cerebrospinal fluid (CSF) of a neonate with meningitis (19). ZD1 is an ibeA in frame‐deletion mutant of the E44 strain (19). Plasmid pUC23A was used for ZD1 complementation, which was constructed with pUC13 carrying the ibeA gene (19). E. coli DH5α was used as the host strain for subcloning. All strains were grown at 37°C in brain–heart infusion (BHI) media supplemented with rifampin (100 µg/mL) and/or ampicillin (100 µg/mL) if required. The pET17A construct for expression of a His tagged‐IbeA protein was transformed into E. coli BL21 (DE3) (19). Purification of the His‐tagged IbeA was performed as described previously (19). The purified IbeA protein was incubated with polymyxin B resin (Sigma, St. Louis, MO, USA) to remove the potentially contaminated lipopolysaccharide (LPS). The IbeA protein was heat‐inactivated at 100°C for 5 minutes as a negative control. Construction and purification of glutathione transferase (GST)–vimentin head domain (VHD) and control protein GST were described previously (6). Restriction endonucleases and T4 DNA ligase were purchased from New England Biolabs (Beverly, MA, USA). Chemicals were purchased from Sigma unless otherwise noted. Plasmid DNA was extracted by using the MiniPrep kit (Qiagen Inc., Chatsworth, CA, USA).
PMN transmigration assay
PMN transmigration assays were performed as described by Moreland et al 30, 31 with modification. The confluence of the monolayer was confirmed by light microscopy before the start of the assay. Transwell filters (3.0‐µm pore size, 6.5‐mm diameter, BD Biosciences) with the confluent BMEC monolayers were transferred to clean 24‐well plates, and washed twice with experimental medium (EM; containing 49% M199, 49% Ham's F12, 1 mM sodium pyruvate and 2 mM L‐glutamine). For BMEC stimulation, different doses of E. coli strains, IbeA or BSA‐coated beads or purified protein were added to the lower chambers with 0.8 mL EM for 2 h. Then, PMN (1 × 106 cells in 0.2 mL of EM) were added to the upper chamber and allowed to migrate over for 4 h. At the end of the incubation, migrated PMN cells were collected from the lower chamber and counted in a blinded‐fashion using a hemacytometer. Final results of PMN transmigration were expressed as the percentage of PMN across the BMEC monolayers. For withaferin A (WFA) treatment, BMEC were incubated with WFA (ChromeDex, Irvine, CA, USA) in both upper and lower chambers for 0.5 h before stimulation, or PMN was incubated with WFA before adding to the upper chamber. After incubation, WFA was removed. The maximum concentration of WFA was 10 µM, which might evoke apoptosis of normal cells by 24 h 44, 50. To avoid interference by apoptosis the maximum incubation period was less than 6.5 h. For the bortezomib treatment, BMEC were incubated with the drug (LC laboratories, Woburn, MA, USA) in both upper and lower chambers for 1 h before stimulation. For the blocking assays with GST recombinant proteins, BMEC were incubated with different doses of GST‐VHD and GST (control) protein in both upper and lower chambers for 1 h before stimulation. For IbeA protein or LPS induction, BMEC were incubated with different concentrations of protein or LPS [Control Standard Endotoxin (CSE) purified from E. coli strain O55:B5, BioWhittaker, Walkersville, MD, USA] in the lower chambers before adding PMN.
PMN Adhesion assay
PMN adhesion assays were performed as described by Nieminen et al (34). Briefly, confluent HBMEC minolayers on 24‐well plates were stimulated with different concentrations of purified IbeA protein (0.01–1.0 µg/mL) for 6 h or at different time intervals (0–24 h) with a single dose of IbeA (0.5 µg/mL). After the incubation, the monolayers were washed with phosphate‐buffered saline (PBS) for four times. Each well was added with 1 × 106 PMN (0.5 mL) and incubated with 90 minutes at 37°C. Then, the cells were washed for 5 times and fixed with 4% paraformaldehyde in PBS. Assays were performed in triplicate wells. Fifteen microscope fields were randomly selected from three wells for each treatment to count the number of adherent leukocytes and the data were analyzed using analysis of variance (ANOVA).
IbeA coated beads and E. coli strains
The beads coating experiments were carried out as described previously (6). Briefly, a total amount of 80 µg of IbeA protein, heat‐inactivated IbeA and BSA (control) was mixed in an Eppendorf tube with 200 µL of a 2% aqueous suspension of carboxylate‐modified yellow‐green fluorescent (YGF) beads (1 µm in diameter; Molecular Probes, Sunnyvale, CA, USA). After the protein coating, the beads were centrifuged and washed three times in PBS and then suspended in PBS plus 1% BSA. For stimulation, 2 × 106 beads were added to the lower chamber for PMN transmigration. For coating of the ibeA deletion mutant ZD1 with IbeA, the stationary phase culture (grown in BHI medium for 15 h) of ZD1 was washed twice in PBS, pelleted and resuspended in 200 µL PBS containing IbeA protein or BSA (0.2 mg/mL). Proteins were allowed to bind to the bacterial surfaces for 30 minutes on a rocking wheel at room temperature. Bacteria were then washed five times with PBS to remove unbound material and resuspened in 200 µL PBS. For stimulation, E. coli (106 cfu/mL) were added to the lower chamber for PMN transmigration. An aliquot (100 µL) of suspension was centrifuged, sampled and subjected to SDS‐PAGE immunoblotting analysis.
Mouse meningitis model
This study was approved by the Institutional Animal Care and Use Committee of Childrens Hospital Los Angeles Saban Research Institute. All E. coli strains used in this work were derived from RS218, a CSF isolate from a neonate with E. coli meningitis 16, 19, 37. Before use, the bacteria were subcultured on blood‐agar plates, checked for purity, inoculated into BHI broth and incubated overnight at 37°C. Bacteria were collected and washed with PBS. Meningitis was induced in 10‐day‐old mice (C57BL/6J; Jackson Laboratory, ME) by injection of 10 µL of bacterial suspension containing 2 × 103 cfu of E. coli into the cisterna magna under short‐term anesthesia. For protein induction, 0.8 µg purified IbeA in 10 µL PBS was injected into mouse brain as mentioned earlier. Negative control mice were injected only with PBS or PBS containing the same amount of BSA. Twelve hours (injected with bacteria) or five hours (injected with protein) after injection, the mice were anesthetized with ketamine and lidocaine. After complete bleeding, the skull was opened and CSF was collected as described previously (26). The CSF was harvested with 200 µL of PBS and centrifuged in an Eppendorf tube. The resultant sediments were then suspended with PBS for erythrocyte and leukocyte counts. CSF samples containing more than 10 erythrocytes/µL were considered to be probably contaminated with blood (26) and discarded in this study.
E. coli translocation across the HBMEC monolayer
This study was carried out as described previously (30). E. coli K1 E44 or its mutant ZD1 (1 × 105 cfu) were inoculated into the lower chamber and incubated with the HBMEC monolayer at 37°C. Aliquots (10 µL) were taken from the upper and lower compartments at different time points (1, 2, 3, 4, 5 and 6 h) after the initial addition of bacteria and used for determination of cfu. Results for number of translocated E. coli into the upper chamber are expressed as a percentage of the cfu count in the lower chamber at the same time.
Lentivirus‐mediated overexpression of vimentin and VHD deletion mutant (VDM) in HBMEC
The construction of lentiviral vectors expressing GFP‐vimentin (VIM), GFP‐VDM or GFP, lentivirus packaging and production, and lentiviral transduction of HBMEC were performed as described previously (6). The GFP expression was examined under fluorescence microscope at 48 h incubation after lentiviral infection. The transduced HBMEC with more than 90% expression of GFP were subjected to Western blotting and PMN transmigration assays
Immunoblotting analysis
To assess IbeA‐induced expression of adhesion molecules in HBMEC, endothelial cell monolayers were grown on 60‐mm plates. Confluent HBMEC minolayers were incubated with different concentrations of purified IbeA protein (0.01–1.0 µg/mL) for 6 h or at different time intervals (0, 2, 6, 24 h) with a single dose of IbeA (0.5 µg/mL). For the blocking assays with inhibitors, endothelial cell monolayers were pre‐incubated with or without WFA (1–10 µM) or bortezomib (5–40 nM) for 0.5 h, and then simulated with IbeA (0.5 µg/mL) for 6 h. After the completion of the incubation, cytoplasm and nuclear proteins of BMEC cells were extracted as described previously (50). Both cytoplasm and nuclear proteins were mixed with SDS buffer, heated and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE). Separated proteins were transferred to nitrocellulose membrane by semi‐dry blotting. After overnight blocking with 5% milk in PBS with 0.1% Tween 20 (52), membranes with cytoplasm proteins were probed with rabbit anti‐ICAM‐1 polyclonal antibody (1 µg/mL, Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti‐CD44 monoclonal antibody (1 µg/mL, Santa Cruz Biotechnology), mouse anti‐Vimentin (V9) monoclonal antibody (0.2 µg/mL, Santa Cruz Biotechnology), and mouse anti‐β‐actin monoclonal antibody (0.1 µg/mL, Santa Cruz Biotechnology) for 2 h. The membranes with nuclear proteins were probed with rabbit anti‐NFκB (p65) polyclonal antibody (0.5 µg/mL, Santa Cruz Biotechnology) and mouse anti‐β‐actin monoclonal antibody (0.1 µg/mL, Santa Cruz Biotechnology) for 2 h. For proteasome activity examination, the membranes with cytoplasm proteins were incubated with polyubiquitinylated proteins mAb (FK1, 0.5 µg/mL, Endo Life Sciences, Plymouth Meeting, PA, USA) for 2 h. The washed membranes were incubated with horseradish peroxidase (HRP)‐conjugated secondary antibody for 1 h and then visualized using an enhanced chemiluminescence procedure (Roche Applied Science, Inidianapolis, IN, USA).
Assays of surface expression of endothelial cell adhesion molecules
The methods for this study were similar to those described previously (30). To assess HBMEC surface expression of adhesion molecules, monolayers were grown on 6.5‐mm Transwell filters to confluence. E. coli strains (106 cfu/mL) were added to the lower chamber of the transwell and incubated for 6 h. At the completion of the incubation, cells were washed twice with PBS, fixed with 4% paraformaldehyde and then blocked for 30 min with PBS containing 5% BSA. Primary antibodies for ICAM‐1 and CD44 were added immediately after the blocking step. Incubation was carried out for 1 h at room temperature on a rotating shaker. The primary antibodies used were rabbit anti‐ICAM‐1 antibody (2 µg/mL, Santa Cruz Biotechnology), and mouse anti‐CD44 antibody (1 µg/mL, Santa Cruz Biotechnology). Cells were washed five times with PBS/1% BSA and incubated with peroxidase‐conjugated secondary antibody for 1 h at room temperature. After the secondary antibody incubation, cells were washed five times and liquid TMB substrate (KPL, Gaithersburg, MD, USA) was added. The liquid was transferred to an enzyme‐linked immunosorbent assay (ELISA) plate after 5 minutes. Equal volume stop solution was added, and optical density at 450 nm was read. For each ELISA, an isotype‐matched control antibody was used in place of the primary antibody in three wells, and this background was subtracted from the signal.
Immunofluorescence microscopy
To observe the adhesion molecules expression on cell membrane, the endothelial cells were seeded on 8‐well chamber slides and incubated with E44 (25 MOIs) or stimulated by IbeA (0.5 µg/mL) or LPS (0.1 µg/mL). The incubation was carried out at 37°C for 6 h. Then, the cells were fixed with 4% paraformaldehyde, blocked with 5% BSA in PBS and stained with anti‐CD44 monoclonal antibody (2 µg/mL, Santa Cruz Biotechnology)/FITC or anti‐ICAM‐1 (CD54) monoclonal antibody Fluorescein (0.5 µg/mL, R&D Systems, Minneapolis, MN, USA). To determine colocalization of vimentin with CD44 or ICAM‐1, HBME transduced with VIM‐GFP, VDM‐GFP and GFP were seeded on 8‐well chamber slides and incubated with E44 (25 MOIs) for 2 h. Then the cells were fixed, blocked as mentioned earlier and stained with antibodies against CD44 (monoclonal) (2 µg/mL, Santa Cruz Biotechnology)/rhodamine or ICAM‐1 (polyclonal) (2 µg/mL, Santa Cruz Biotechnology)/rhodamine. Samples were examined under a Lecia fluorescence microscope (Wetzlar, Germany) at the Congressman Dixon Cellular Imaging Core Facility, Childrens Hospital Los Angeles. The same parameters were used for all the pictures to ensure the fluorescence strength of each treatment in a comparable manner. To study human PMN transmigration through HBMEC, transwell inserts with endothelial–PMN co‐cultures were washed, fixed with 4% paraformaldehyde and blocked with 5% BSA in PBS. Then, the transwell membranes were first exposed to an anti‐vimentin (V9) antibody (Santa Cruz Biotechnology) followed by Cy5‐conjugated anti‐mouse IgG. The samples were then incubated with CTxB‐FITC (10 µg/mL; Sigma, MO), a lipid raft marker, to mark the cell membranes for both BMEC and PMN. For confocal microscopy, the transwell filters were cut off from the inserts, immersed in 4'‐6‐diamidino‐2‐phenylindole (DAPI) solution and examined with a Leica TCS SP microscope (Wetzlar, Germany). Images were acquired with Leica confocal software. Three lasers with wavelengths of 488, 561 and 633 nm were used. Images were acquired with three fluorescence photomultiplier tube detectors, and the differential interference contrast (DIC) image was taken with a transmitted light photomultiplier tube detector. Images were acquired with exact same optimized parameters in the same set of experiments. The microscope was equipped with a Marzhauser motorized XY stage with joystick interface and internal “Wide Z” motorized focus drive. Z‐stacks of images were taken for some cells in order to confirm if the label was on the membrane or in an intracellular location. Orthogonal section function was used to show a side view of the z‐stack to demonstrate subcellular location of labeling in cells.
Statistical analysis
The statistical analysis of the data was performed as described previously (7). ANOVA and covariates followed by a multiple comparison test such as the Newmann–Keuls test were used to determine the statistical significance between the control and treatment groups. Nonparametric tests were conducted using PROC NPAR1WAY in SAS Version 9.2. (SAS Institute, Cary, NC, USA) P < 0.05 was considered to be significant.
RESULTS
PMN‐human BME C interactions in response to E. coli K1 and purified IbeA
To determine the role of IbeA in E. coli K1‐induced PMN‐BMEC interactions, we first tested effects of intact IbeA+ and IbeA‐deficient E. coli K1 on transendothelial migration at different inoculum sizes and time intervals. In the absence of any stimulus, only minimal migration of neutrophils across the intact HBMEC monolayer occurred (<1.0 % of the added PMN). E44, which was added to the basal site (the lower chamber) of a confluent HBMEC monolayer, dose‐dependently induced transmigration of PMN across the monolayer in the apical to basal direction. However, the ibeA‐deletion mutant (ZD1) was significantly less efficient in eliciting transendothelial migration of neutrophils than its parent strain E44. The mutant phenotype of ZD1 was fully restored by complementation of the mutant strain by pUC23A carrying the ibeA gene (Figure 1A). Kinetic analysis of PMN transmigration across BMEC in response to the induction by E44 and ZD1 at different time intervals was carried out to exclude the possibility that the decreased transendothelial migration by the IbeA‐deficient mutant was caused by a slow onset of the migratory response. The time‐course study demonstrated that PMN migration in response to the stimulation by 5 × 106 cfu of ZD1 (ibeA ‐) was kinetically reduced (Figure 1B), suggesting that the mutant phenotype of ZD1 in the induction of neutrophil migration across BMEC was not caused by a simple reflection of the slow initiation of PMN recruitment. To determine whether IbeA alone was sufficient for inducing PMN adhesion and migration, we tested the ability of purified protein to elicit PMN recruitment in the absence of intact bacteria. Purified IbeA was able to elicit neutrophil adhesion and migration in a dose‐(0.01 to 1 µg/mL for 6 h) and time‐(0, 2, 6 and 24 h at 0.5 µg/mL of protein) dependent manner (Figure 2A–D). These doses were much smaller than those (4–5 µg/mL) for inducing apoptosis of BMEC (20). There was an increase in the number of PMN adhesion and migration in response to higher concentrations of the purified IbeA. A maximum adhesion (about seven times over the basal level) (Fgure 2A and B) and a maximum migration of 25% of added PMN in the top chamber (Figure 2C and 2D) can be induced by at 6 h with 1 µg/mL of IbeA and at 24 h with 0.5 µg/mL of IbeA, which was added into the lower chambers. Inspection of the monolayer by immunofluorescence microscopy and apoptosis assays after incubation with different concentrations of purified IbeA (0.01–1.0 µg/mL) demonstrated no apoptosis within 6 h and no significant dose‐dependent destruction of the architecture of the endothelial monolayer that was seen in response to any of the concentrations of intact organisms studied. The PMN stimulating activity of IbeA was completely abolished after boiling at 100oC for 5 minutes (data not shown). In addition, electrical resistance across the monolayer declined to <10% of the starting value after incubation with these doses of IbeA and bacteria. These data suggest that IbeA is required for E. coli K1‐induced PMN transmigration across BMEC.
Figure 1.
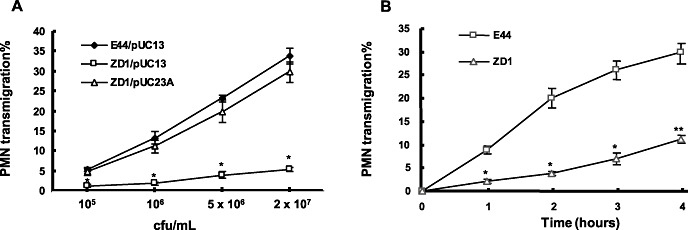
Dose‐dependent induction of human polymorphonuclear neutrophil (PMN) transmigration across brain microvascular endothelial cell (BMEC) monolayers by E. coli strains E44, ZD1 and the complemented ZD1. PMN transmigration assays were performed as described in the Materials and Methods section. A. Induction of PMN migration with different doses (1 × 105 to 2 × 107 cfu/mL) [corresponding to 2.5–500 multiplicity of infection (MOI)] of E. coli K1 strains. Bacteria E44/pUC13 (IbeA+), ZD1/pUC13 (IbeA‐) and the complemented ZD1 (ZD1/pUC23A)(IbeA+) were used in PMN migration assays. B. Time‐course study of E44‐ and ZD1‐induced PMN transmigration across human BMEC monolayers. PMN transmigration was triggered by 5 × 106 cfu/mL (corresponding to 125 MOI) of E. coli strain E44 or ZD1. PMN migration in response to ZD1 was significantly reduced at all time points between 1 and 4 h after addition of PMN to the human BMEC monolayer. The values represent the mean percent transmigrating PMN of triplicate samples and are representative of one experiment from three independent experiments showing similar data. *P < 0.05, **P < 0.01 compared with E44.
Figure 2.
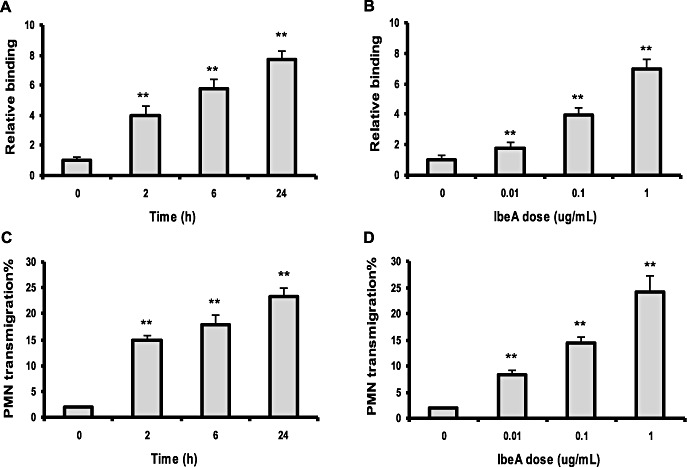
Transendothelial migration of polymorphonuclear neutrophil (PMN) across human brain microvascular endothelial cell (BMEC) in response to purified IbeA incubated in the abluminal compartment of the Transwell system. (A,C) There was a dose‐dependent induction of PMN transendothelial adhesion and migration in response to concentrations of IbeA (0.01 to 1 µg/mL) for 6 h. (B,D) Kinetic analysis of PMN adhesion and transendothelial migration in response to purified IbeA (0.5 µg/mL) for 0–24 h. IbeA significantly induced PMN adhesion and transendothelial migration at all time points between 2 and 24 h after addition of PMN to the monolayer when compared with the control (basal level). Assays were performed in triplicate wells. PMN adhesion was expressed as an n‐fold increase relative to the basal level (IbeA concentration: 0). *P < 0.05, **P < 0.01 compared with the control.
IbeA was sufficient to induce PMN transmigration in vitro and in vivo
To provide further evidence of the role of IbeA in PMN transmigration elicited by E. coli K1, we investigated PMN migration in vitro in response to IbeA‐coated beads or mutant strain and IbeA‐induced PMN recruitment into the CNS of mice. We first tested whether IbeA‐coated beads or mutants were able to mimic the natural setting for inducing PMN migration. YGF beads were covalently coupled to purified IbeA, which was treated with polymyxin B‐agarose to remove the contaminated LPS. YGF beads was also covalently coupled to boiled purified IbeA after polymyxin B‐agarose treatment, which is used to examine the LPS removing base on the fact that LPS is resistant to 100°C heat inactivation (12). IbeA‐coated beads and mutant were tested for efficiency of stimulation, using as control beads or bacteria coupled to similar amounts of BSA. The presence of IbeA in the ibeA‐deletion mutant ZD1 was confirmed by Western blotting (data not shown). As shown in Figure 3A, IbeA‐coated beads were able to significantly induce PMN transmigration while the heat‐inactivated IbeA‐beads were not able to induce PMN transmigration, suggesting that LPS was completely removed by polymyxin B‐agarose. The inactive phenotype of ZD1 was restored to the level of the parent strain E44 by IbeA protein (Figure 3B). To confirm the biological relevance of the above in vitro studies, we investigated the role of IbeA in bacteria‐induced PMN recruitment into the CNS of mice. As shown in Figure 3C,D, purified IbeA protein and IbeA+E. coli strains (E44 and the ibeA gene–complemented mutant ZD1/pUC23A) were able to significantly induce PMN recruitment into CSF compared with the controls (BSA and ZD1). These findings strongly suggest that IbeA is the important virulence factor to promote PMN transmigration across the BBB in vitro and in vivo.
Figure 3.
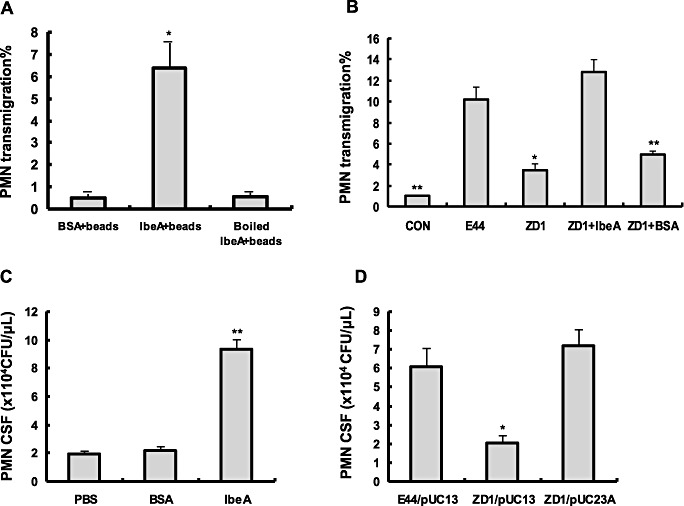
In vitro and in vivo comparative studies of transendothelial migration of polymorphonuclear neutrophil (PMN) induced by E44, ZD1 and complemented strains. (A) Induction of PMN transmigration with YGF beads coated with IbeA protein. PMN transmigration assays were performed with stimulation of YGF beads coated with IbeA, heat‐inactivated IbeA, or BSA (2 × 106 beads/mL). (B) Induction of PMN transmigration with ZD1 coated with IbeA protein or BSA. E44 and ZD1 [(1 × 106 cfu/mL = 25 multiplicities of infection (MOIs)] were included as the positive and negative controls, respectively. In vitro assays were performed in triplicates (A,B). C. IbeA protein induced PMN transmigration in vivo. Meningitis was induced by injecting 10 µL of PBS with or without 0.8 µg IbeA or BSA protein into the cisterna magna of 10‐day‐old mice. After 5 h, CSF was harvested and PMN numbers were counted under microscope. (D) PMN recruitment into the CNS of mice in response to the wildtype (E44/pUC13), mutant (ZD1/pUC13) and complemented mutant (ZD1/pUC23A) strains. Meningitis was induced by injection of different E. coli strains into the cisterna magna of 10‐day‐old mice. After 12 h, CSF was harvested and PMN numbers were counted in a blinded‐fashion under microscope. Results are expressed as PMN numbers per microliters of CSF. *P < 0.05; **P < 0.01. Eight animals were used for each group.
Effects of conditioned media on PMN transmigration
While IbeA is an outer membrane protein, it is not clear whether it depends on release of the virulence factor from the intact bacterial cell. The effects of conditioned media (CM) from each of the two strains (E44 and ZD1) on the stimulation PMN transmigration were also determined. E44 and ZD1 were cultured in the EM to log phase. The cultures were centrifuged and the supernatants were filtered through 0.2 µm polycarbonate filters to trap bacterial particles. The stimulating activities of the prepared CM were tested alone and in combination for transmigration studies by addition of the media to the lower chamber (basal surface) of the Transwell system. To determine whether there were soluble/secreted products released from the wild‐type strain, we tested the effects of the CM from E44 (CM‐E) on ZD1 (ibeA ‐)‐induced transendothelial migration of neutrophils across HBMEC. No significant differences were observed in the growth rates of all bacterial strains (Figure 4A). No stimulating effects on PMN migration were observed either in the conditioned medium from E44 (CM‐E) or in that from ZD1 (CM‐Z) as compared with the inducing activity of the intact E44 (Figure 4B). The mutant phenotype of ZD1 in the induction of transendothelial migration of neutrophils was also not affected by the conditioned medium from E44 as compared with that seen with the mutant strain alone (Figure 4B). These results suggest that IbeA is an unsecreted surface protein, which contributes to meningitic E. coli K1‐induced neutrophil transmigration across BMEC through a more complex and intricate mechanism.
Figure 4.
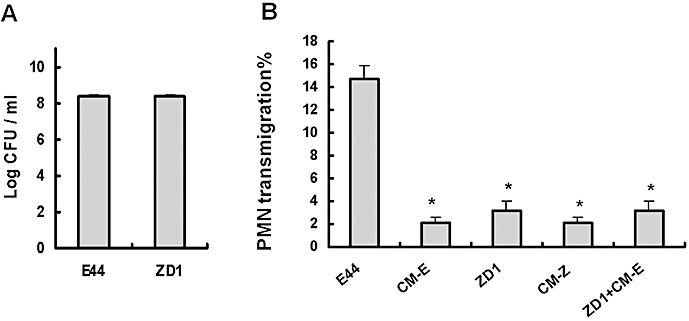
Effects of conditioned media (CM) from E44 and ZD1 on polymorphonuclear neutrophil (PMN) transmigration across human brain microvascular endothelial cell (BMEC) monolayers. E. coli strains were cultured in the experimental medium for 6 h. The supernatants (CM) were filtered through 0.2 µm polycarbonate filters to trap bacterial particles. A. Growth rates of bacteria. B. PMN transmigration induced by E. coli[25 multiplicities of infection (MOIs)], CM or CM plus ZD1 (25 MOIs). CM from both strains E44 (CM‐E) and ZD1 (CM‐Z) were unable to induce significant PMN migration compared with E44. CM‐E was also unable to restore or enhance the ability of ZD1 to induce neutrophil migration. The values represent the mean % transmigrating PMN of triplicate samples and are representative of one experiment from three independent experiments showing similar data. *P ≤ 0.05 compared with E44.
Translocation rates of E44 and its ibeA deletion mutant ZD1
As IbeA plays an important role in meningitic E. coli K1 invasion of BMEC, there is a possibility that E. coli K1 to infect and translocate across the BMEC monolayers are required for IbeA‐induced PMN migration. In order to address this issue, the rates of E44 (wild‐type) and ZD1 (mutant) translocation across the monolayer were compared. The samples were taken from the upper and lower chambers at 1, 2, 3, 4, 5 and 6 h after the initial inoculation of E. coli into the lower chamber at time 0. As shown in Supporting Information Figure S1, the translocation rates of E44 were significantly higher than that of ZD1 at 1, 2, 3 and 4 h. These data suggest that the direct interaction of E. coli K1 at the abluminal surface of the BMEC monolayer and subsequent bacterial translocation in an abluminal to luminal direction contribute to IbeA‐induced PMN migration.
IbeA‐induced PMN transmigration was suppressed by cleavage of vimentin
As vimentin is an IbeA‐binding protein, we tested the role of vimentin in E. coli K1‐induced PMN transmigration across BMEC. First, we examined whether IbeA‐induced leukocyte transmigration was blocked by inhibition of vimentin. WFA is one of the most pharmacologically active Withanolide compounds that are present in roots and leaves of the medicinal plant Withania somnifera. It has been demonstrated that WFA is a specific inhibitor of vimentin by protein cleavage 1, 6. To determine whether the effect of WFA on vimentin cleavage was correlated with inhibition of PMN migration, IbeA+E. coli K1‐induced transendothelial migration of leukocytes was examined in HBMEC treated with different doses (1–10 µM) of WFA. As shown in Figure 5, WFA was able to induce cleavage of vimentin (Figure 5A) and to block E. coli K1‐induced PMN migration across BMEC (Figure 5B) in a dose‐dependent manner, suggesting that vimentin is required for IbeA+E. coli K1‐induced leukocyte migration during inflammation. Furthermore, when either BMEC or PMN were pretreated with WFA before the assay, the leukocyte transmigration markedly decreased. Figure 5C showed that addition of WFA to either of the two cell types significantly reduced the leukocyte transmigration to less than 15% when compared with the untreated cells. A similar result was obtained when both cell types were pretreated with WFA.
Figure 5.
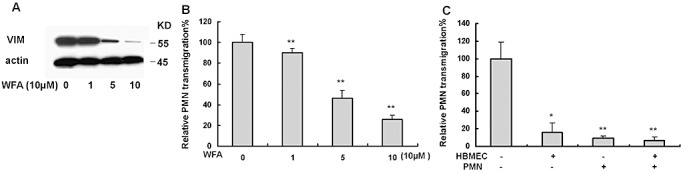
Blockage of meningitic E. coli K1 induced polymorphonuclear neutrophil (PMN) transmigration by withaferin A (WFA)‐induced cleavage of vimentin. A. Immunoblotting analysis of vimentin (VIM) in human brain microvascular endothelial cell (BMEC) treated with different doses of WFA. β‐actin was used as an internal loading control. B. Inhibition of PMN transmigration across human BMEC after preincubation with different doses of WFA at the indicated concentration at 37°C for 0.5 h. C. Suppression of PMN transmigration by WFA (10 µM) using BMEC/PMN after preincubation with (+) or without (−) WFA. In both experiments, PMN transmigration was triggered by E44 (25 MOIs). Assays were performed in triplicates. Results are expressed as relative PMN migration compared with the positive control (100%). The significant differences with regard to the control were marked by asterisks (*P < 0.05; **P < 0.01).
VHD is required for IbeA induced PMN transmigration and endothelial expression of adhesion molecules
To further confirm the role of vimentin in BMEC in the process of leukocyte transmigration and adhesion molecule expression, we tested whether overexpression of full‐length vimentin and VDM in HBMEC would affect PMN transmigration triggered by IbeA+ E. coli and expression of ICAM‐1 and CD44. HBMEC were transduced with lentivirus expressing a GFP‐tagged human vimentin (VIM) and GFP‐tagged VDM (6). Expression of GFP‐VIM and GFP‐VDM was verified by fluorescence microscopy and Western blot as described previously (6). Effects of vimentin overexpression on upregulation of adhesion molecules were detected by immunoblotting with antibodies against human ICAM‐1 and CD44 (Figure 6A), suggesting that adhesion molecules may be involved in the PMN recruitment process. The adhesion molecules ICAM‐1 and CD44 were downregulated on VDM‐GFP transduced BMEC, suggesting the VHD is required for vimentin‐mediated upregulation of adhesion molecules. Vimentin and VDM‐overexpressed BMEC were tested in PMN transmigration assays in response to IbeA+ E. coli K1 (E44) or IbeA‐ E. coli K1 (ZD1). As shown in Figure 6B, when stimulated with IbeA+ E. coli K1, the PMN transmigration across BMEC expressing the GFP‐VIM was a twofold value of the control with the GFP‐transduced BMEC. This indicated that vimentin played a significant role in leukocyte transmigration across BMEC. However, when stimulated with IbeA‐ E. coli K1 (ZD1), there were no significant differences between the leukocyte transmigration rates with either GFP‐VIM‐transduced or GFP‐transduced BMEC (Figure 6B). When stimulated with IbeA+ E. coli K1, the rate of PMN transmigration was significantly reduced in BMEC overexpressing GFP‐VDM compared with the control with the GFP‐transduced BMEC. It suggests that the VHD is required for IbeA induced PMN transmigration. After treatment of BMEC with WFA, a vimentin inhibitor, the leukocyte transmigration decreased to the similar level (30% of the standard positive control) on either GFP‐VIM‐transduced or GFP‐transduced BMEC (Figure 6C). Apparently, WFA abolished the stimulating effect of the overexpressed vimentin on leukocyte transmigration in transduced BMEC. This further supported that the PMN transmigration depended on the function of vimentin, and also implied that at least most of transmigration occurred via the vimentin‐network of BMEC. To further disset the role of the VHD in this process, we next performed a protein blocking study with a recombinant protein GST‐VHD, which was made as described previously (6). This study showed that VHD could block IbeA+E. coli stimulated PMN transmigration in a dose dependent manner, while no significant difference was found in different doses of the control protein GST (Figure 6D). This result further confirmed the important role of the VHD in IbeA induced PMN transmigration across BMEC.
Figure 6.
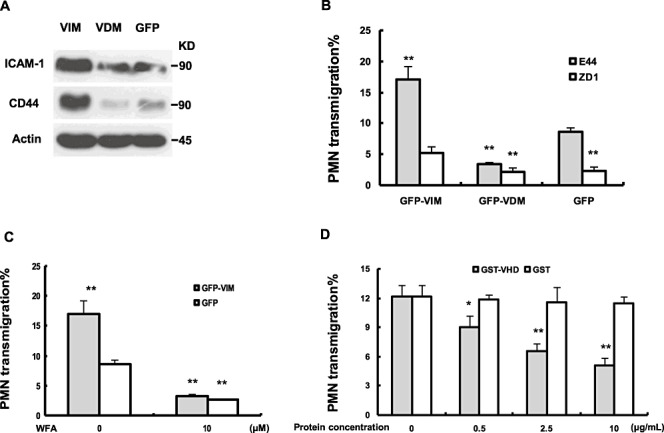
Vimentin head domain is required for IbeA enhanced polymorphonuclear neutrophil (PMN) transmigration across human brain microvascular endothelial cell (HBMEC). A. Protein expression in HBMEC with vimentin overexpression. HBMEC transduced with the GFP‐VIM (HBMEC‐VIM), GFP‐VDM (vimentin head domain deletion mutant) (HBMEC‐VDM) and GFP (HBMEC‐GFP) lentivirus constructs were immunoblotted with antibodies against human ICAM‐1, CD44 and β‐actin (internal loading control). B. Transendothelial migration of PMN was significantly enhanced and reduced in HBMEC‐VIM and HBMEC‐VDM, respectively. HBMEC‐VIM and HBMEC‐VDM were tested for E44‐ and ZD1‐induced PMN transmigration [25 multiplicities of infection (MOIs)] with the in vitro migration assay. C. Increased PMN transmigration across HBMEC‐VIM was blocked by WFA. E44‐induced PMN migration across BMEC‐GFP was taken as the control in both (B) and (C), and the significant differences between the treatment groups and control were marked with asterisks (*P < 0.05, **P < 0.01). D. E44 (IbeA+)‐ induced PMN transmigration was blocked by the recombined vimentin head domain (GST‐VHD) in a dose‐dependent manner. GST protein was taken as a negative control in different doses (0–25 µg/mL). PMN transmigration without treatment was taken as a positive control and the significant differences between the treated and untreated (control) cells were marked with asterisks (*P < 0.05, **P < 0.01). All the invasion assays were performed in triplicates.
PMN transcellular migration across BMEC is mediated by vimentin.
It has been shown that leukocytes use both paracellular and transcellular routes for transmigration across the endothelium, and that the transcellular migration process depends on vimentin in both skin endothelial cells and lymphocytes (34). In order to examine whether PMN emigrate between BMEC (paracellular pathway) or transmigrate through BMEC (transcellular pathway), a PMN transmigration study was carried out using Transwell (San Jose, CA, USA)31, 45. Migration assays were performed as described above. The transwell membranes with cells were cut off and used for immunostaining and confocal microscopy analysis (see Supporting Information Figure S2 legend). DAPI, CTx‐B and an anti‐vimentin antibody were used to stain the nuclear regions and overall structures of cells, respectively. As shown in Supporting Information Figure S2A, most PMN were topologically distributed along or through the endothelial cells. Confocal images showed that the leukocytes were clearly inside the endothelial cells, as they were obviously entangled by the vimentin network (Supporting Information Figure S2B). Interestingly, the vimentin intermediate‐filament polymers seemed to be actively reorganized towards the migrating PMN—the filamentous network was clustered towards the site of contact of the endothelial cell and the leukocyte, and formed an anchoring structure at the contact area between these two cell types (Supporting Information Figure S2B). Concurring with the prior finding on transcellular migration of lymphocytes across skin endothelial cells, E. coli K1‐stimulated PMN could migrate across the BBB through the transcellular pathway.
IbeA‐ and bacteria‐induced expression of endothelial cell adhesion molecules and vimentin
After demonstrating the role of IbeA/vimentin in PMN transmigration across BMEC and the effects of vimentin overexpression on the upregulation of ICAM‐1 and CD44, we focused on the role of IbeA+E. coli K1 and purified IbeA in upregulation of endothelial adhesion molecules that might be involved in neutrophil movement across the monolayer. Surface expression of endothelial adhesion molecules was studied by using two approaches, whole‐cell ELISA for quantitative data and immunofluorescence microscopy. Following either 6‐h exposure of HBMEC to ZD1, a slight increase in surface expression of ICAM‐1 and CD44 was observed by intact‐cell ELISA. The parent strain E44 elicited a more than twofold upregulation of ICAM‐1 and CD44 surface expression (Figure 7A–B). Similar observations were made by examination of the treated HBMEC monolayers using immunofluorescence microscopy, showing surface ICAM‐1 and CD44 were upregulated after exposure of the endothelial monolayer to intact E. coli (Figure 7C). Monolayers exposed to purified IbeA and LPS had further increases in the total number of cells displaying ICAM‐1 expression as compared with the monolayers exposed to intact E. coli. Enhanced and decreased colocalization of vimentin with CD44 or ICAM‐1 was observed in HBMEC with overexpression of vimentin and VDM, respectively (Supporting Information Figure S3A,B), suggesting that there are direct interactions between vimentin and adhesion molecules. To further examine IbeA‐induced expression of endothelial adhesion molecules, HBMEC stimulated with IbeA at different time points and dose levels were subjected to immunoblotting analysis of ICAM‐1, CD44 and vimenitn. Our studies indicated that IbeA was able to significantly upregulate expression of vimenitn, ICAM‐1 and CD44 in a dose (0–1 µg/mL for 6 h)‐ and time (0–24 h/0.5 µg/mL of IbeA)‐dependent manner (Figure 7D,E). These studies suggest that both ICAM‐1 and CD44 are upregulated by IbeA.
Figure 7.
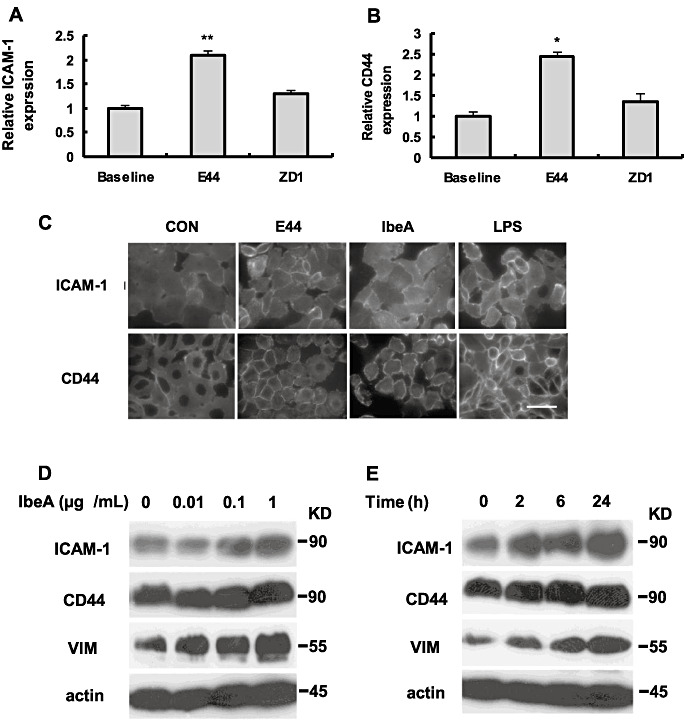
Expression of endothelial adhesion molecules was induced by IbeA+E. coli and purified IbeA protein. Surface expression of ICAM‐1 (A) and CD44 (B) was analyzed by enzyme‐linked immunosorbent assays (ELISAs). Human brain microvascular endothelial cell (BMEC) was incubated with E44 or ZD1, which was added in the lower chamber with 25 multiplicities of infection (MOIs) at 37°C for 6 h. In both experiments, untreated human BMEC were used as controls. Assays were performed in triplicates. Results were expressed as an n‐fold increase of protein expression, taking the control as 1. The significant differences between the treatment and control were marked with asterisks (*P < 0.05, **P < 0.01). C. Surface expression of ICAM‐1 and CD44 was further detected by immunofluorescence microscopy. Human BMEC were infected by E44 (25 MOIs) or stimulated by IbeA (0.5 µg/mL) or LPS (0.1 µg/mL). The incubation was carried out at 37°C for 6 h. (D‐E) Immunoblotting analysis of ICAM‐1, CD44 and vimentin (VIM) expression in human BMEC that were induced with different doses of IbeA (0 to 1 g/mL) for 6 h (D) and at different time points (0–24 h) with 0.5 g/mL IbeA (E). β‐actin was used as an internal loading control.
Role of the NFκB and ubiquitin‐proteasome pathways in IbeA/vementin‐mediated PMN transmigration
It has been shown that expression of vimentin and adhesion molecules ICAM‐1 and CD44 is regulated via the NFκB pathway, which is a key player in the inflammatory response and is controlled through ubiquitin proeasome‐mediated proteolysis of phosphorylated IκBα protein (28). In order to further dissect the molecular mechanism responsible for IbeA‐stimulated PMN transmigration, we tested whether expression of vimentin, ICAM‐1 and CD44 upon stimulation by IbeA was correlated with NFκB activation and nuclear translocation. WFA is a dual inhibitor of vimentin 1, 5 and the ubiquitin proteasome pathway 36, 50. In order to test whether the proteasome‐regulated NFκB played a role in IbeA/vimentin‐induced expression of adhesion molecules and transmigration of PMN across HBMEC, effects of WFA and bortezomib (Velcade), a proteasome inhibitor, on IbeA‐induced cellular response, were investigated. As shown in Figure 8A (WFA) and Figure 8B (bortezomib), both drugs were able to block IbeA‐induced expression of ICAM‐1 and CD44, activation/translocation of NFκB (p65) and proteasomal activity in a dose‐dependent manner. Bortezomib could also dose‐dependently block E. coli K1‐induced PMN migration across HBMEC (Figure 8C). However, IbeA‐induced expression of vimentin was inhibited by WFA but not by bortezomib. These data suggest that both WFA and bortezomib could block IbeA‐induced upregulation of ICAM‐1/CD44 and activation/translocation of NFκB through vimentin‐dependent and independent mechanisms to destabilize adhesion molecules via the cleavage of vimentin and to downregulate gene expression through the inhibition of proteasomal activity.
Figure 8.
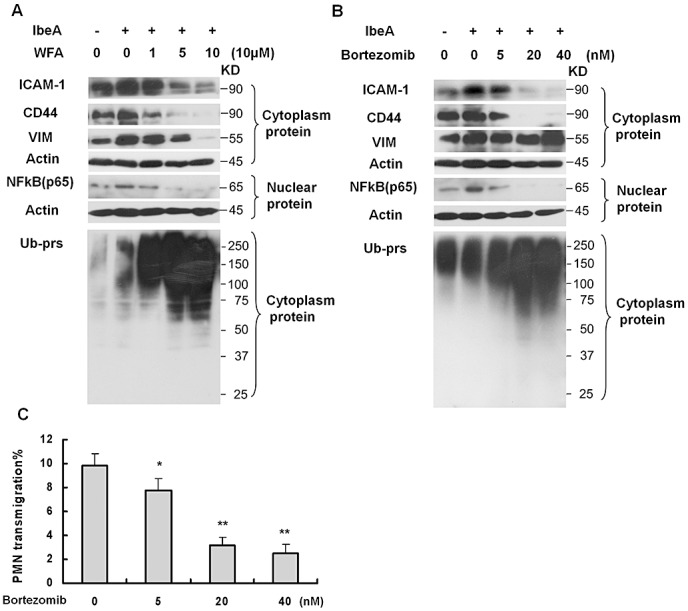
Regulation of adhesion molecule (ICAM‐1 and CD44) expression that is associated with IbeA‐induced polymorphonuclear neutrophil (PMN) transmigration. (A,B) Immunoblotting analysis of adhesion molecules (ICAM‐1 and CD44), VIM, nuclear NFκB (p65) and polyubiquitinylated proteins. Human brain microvascular endothelial cell (HBMEC) treated with WFA (1–10 µM) (A) or brothemib (5–40 nM) (B) for 0.5 h and then incubated with the IbeA protein (0.5 µg/mL) for 6 h. In both (A) and (B), the cytoplasmic and nuclear proteins were extracted as described in the Methods section. To determine the proteasome activity, cytoplasmic polyubiquitinylated proteins (Ub‐prs) were immunoblotted with polyubiquitinylated proteins mAb. Cytoplasmic‐actin and nuclear β‐actin were used as internal loading controls, respectively. In all experiments, untreated HBMEC were used as controls. C. Inhibition of PMN transmigration across HBMEC after pre‐incubation with different doses of brothemib at the indicated concentration (5–40 nM) at 37°C for 1 h. PMN transmigration was triggered by E44 (25 MOIs). The values represent the mean % transmigrating PMN of triplicate samples. The significant differences between treated and untreated cells were marked by asterisks (*P < 0.05; **P < 0.01).
DISCUSSION
The concurrent pathogen invasion with PMN transmigration across the BBB is the hallmark of bacterial meningitis 47, 51. As a “double‐edge sword,” the recruitment of PMN into the CNS by transmigration across the BBB is not only crucial for host defense against meningitic bacterial pathogens, but it is also responsible for significant CNS tissue damage, which results in devastating neurologic sequelae. The role of IbeA in the pathogenesis of E. coli meningitis has primarily been studied with its virulence effects on bacterial penetration across the BBB in vitro and in vivo 5, 14, 17, 18, 19, 25. Vimentin on HBMEC surface has been identified as a primary receptor for IbeA (52). It concurs with a number of recent studies that vimentin contributes to various novel cellular functions through its extracellular location 2, 3, 21, 29, 32, 38, 49. The IbeA‐deficient mutant has been shown to be impaired in penetrating the BBB and invading into the CNS (19). A recent study shows that vimentin is required for transmigration of lymphocytes across HUVEC and skin endothelial cells (34). Despite these findings, whether IbeA plays a role in one of the most critical aspects of pathogenesis of E. coli meningitis, neutrophil recruitment, remains to be defined. In this report, we demonstrate that IbeA mediates a potent induction of PMN transmigration across brain endothelial cells in the in vitro and in vivo models of the BBB. Vimentin and endothelial expression of adhesion molecules are required for the induction process.
To investigate the role of IbeA in PMN transmigration across the BBB in vitro and in vivo, the ibeA‐deletion mutant (ZD1) (18), the mutant complemented with the IbeA protein or the ibeA gene, IbeA‐coated beads and the parent strain (E44) were tested in HBMEC and the mouse model of E. coli meningitis for the E. coli‐induced PMN transmigration into the CNS. It was found that the ibeA mutant ZD1 was significantly less active in stimulation of PMN transmigration than the parent strain E44. Both the wildtype strain E44‐ and the complemented mutant ZD1‐induced transendothelial migration of PMN was increased in a dose‐dependent manner, with 105 IbeA+ bacteria inducing significant migration and with 2 × 107 bacteria eliciting the highest migration rate (35%). In response to a similar inoculum size of the IbeA‐deficient mutant ZD1 only 5% of PMN transmigrated across the BMEC monolayers. These results demonstrate that the IbeA‐deficient mutant was unable to significantly induce PMN transmigration. Interestingly, addition of the recombinant protein IbeA to the mutant ZD1 and IbeA‐coated beads were able to induce PMN migration. These findings suggest that IbeA‐coated mutant and beads could activate the HBMEC monolayer and then induce PMN migration in a manner similar to that of the wildtype bacteria. Complementation of ZD1 with the ibeA gene was tested using the same in vitro assay system. The ability to induce leukocyte transmigration by the mutant ZD1 was restored by expressing IbeA from the plasmid pUC23A. This further supported that IbeA played an important role in induction of PMN transmigration. To examine whether IbeA played a similar role in vivo, a mouse meningitis model was exploited. It was found that the rate of PMN migration across the BBB induced by the mutant strain ZD1 was much lower than the wild‐type strain in 10‐day old mice, and the plasmid pUC23A carrying the ibeA gene complemented the mutant in PMN transmigration. The purified IbeA was able significantly induce PMN recruitment into the CNS. The significant reduction in the IbeA‐deficient mutant‐induced PMN transmigration further suggests that IbeA is an important virulence factor contributing to the recruitment of neutrophils into the CNS and hence the severity of neural injury by neuroinflammation.
While it is not exactly clear how IbeA contributes to meningitic E. coli K1‐induced PMN transmigration across BMEC, our studies clearly show that IbeA does not act as a secreted protein: (i) IbeA is present in E. coli K1 as an outer membrane protein but not a secreted toxin (20); and (ii) the conditioned medium from the wild‐type E44 was unable to restore or enhance the ability of the IbeA‐deficient mutant to induce PMN migration. These findings indicate that the role of IbeA in E. coli K1‐endothelial cell interactions in our in vitro and in vivo model systems is not simply a result of secretion of the toxin during autolysis of the bacteria. In addition, concurring with the observations that the noninvasive phenotype of ZD1 was fully restored by the complementation with the ibeA gene in our in vitro and in vivo model systems and purified IbeA was able to efficiently compete with the wild‐type bacteria for its receptor‐binding on the surface of HBMEC 8, 18, purified IbeA or IbeA‐coated beads were sufficient to induce transendothelial migration of PMN. Taken together, these findings suggest a complex interaction between E. coli K1, BMEC and the PMN.
The molecular participants contributing to E. coli K1‐endothelial cell interactions may be highly complex and may employ multiple endothelial cell surface receptors. Recently, two IbeA‐binding proteins have been identified: vimentin, which is constitutively present in the surface of HBMEC, and polypyrimidine tract‐binding protein (PTB)‐associated splicing factor (PSF), which is inducibly expressed on the surface of both mesenchymal (BMEC) and nonmesenchymal (epithelium) cells 52, 53. As PSF is inducibly expressed on the surface of both mesenchymal and nonmesenchymal cells (6), this protein may be a common co‐receptor that contributes to IbeA+E. coli K1 penetration across epithelial cells and HBMEC. Various novel functions of vimentin in cell adhesion, migration and signaling have recently received considerable attention (23). The presence or absence of vimentin markedly affects the organization and expression of surface molecules critical for adhesion and migration (23). Although these studies focused predominantly on the role of vimentin in endothelial cell migration and microbial invasion, the current data suggest that vimentin might also function in the recruitment of PMN into the CNS in response to meningitic E. coli K1 infection. IbeA+E. coli K1‐elicited PMN migration across HBMEC was dose‐dependently blocked by WFA‐induced cleavage of vimentin in both BMEC and PMN. Overexpression of vimentin and its head domain deletion mutant (VDM) in HBMEC resulted in a significant increase and reduction in neutrophil transmigration, respectively. Concurring with the role of vimentin in transcellular migration of lymphocytes across peripheral endothelial cells (34) and E. coli K1 invasion, vimentin contributed to IbeA+E. coli K1‐induced PMN transmigration across the BBB at least partially through the transcellular pathway. The exact role of vimentin in E. coli K1‐induced transcellular or paracellular transmigration of PMN remains further to be defined. Several studies have shown that vimentin is a common receptor for multiple microbial virulence factors. E. coli K1 CglE is able to bind to vimentin (4). Salmonella virulence protein SptP directly interacts with vimentin, which is recruited to the membrane ruffles (33). The toxin protein from Pasteurella multocida is able to bind the head domain of vimentin (41). Vimentin may serve as the common receptor that contributes to pathogen‐induced leukocyte recruitment. We postulated that recognition of E. coli K1 by the microvascular endothelial cell and subsequent activation of the endothelium might require more distinct receptors that act sequentially with IbeA‐mediated signaling through vimentin as the initial step in this process. As our previous studies demonstrated that PSF and downstream signaling molecules [eg, PI3 kinase and extracellular‐signal‐regulated kinase (ERK)] contribute to IbeA‐mediated bacterial invasion of BMEC 6, 8, 53, these molecules may also play a role in transendothelial migration of PMN in a coordinated manner with the primary receptor vimentin. Upregulation and/or activation of endothelial cell adhesion molecules have been postulated as the mechanism(s) by which bacteria induce PMN recruitment into the CNS. The molecular determinants of cell adhesion for neutrophil‐endothelial cell interactions in response to E. coli K1 in the CNS are not well defined. It has been shown that E. coli significantly increased endothelial surface expression of ICAM‐1 but did not affect expression of ICAM‐2, VCAM‐1 or E‐selectin in pulmonary endothelial cells (31). PMN transmigration and lung injury in E. coli pneumonia were significantly enhanced in CD44‐deficient mice (48). LPS has been shown to potently induce upregulation of multiple endothelial adhesion molecules including ICAM‐1. Whereas LPS may play a significant role in the leukocyte recruitment process, intact E. coli were 10 times more potent in inducing transmigration of PMN than a corresponding amount of purified LPS (31). Using a rat model of gram‐negative pneumonia, and several E. coli mutant strains, the LPS component has been shown to be only one of several inflammatory mediators in gram‐negative infection (40). Here we demonstrated that intact E. coli K1 and LPS‐free IbeA were able to potently induce ICAM‐1 and CD44 expression in HBMEC. IbeA was able to significantly induce PMN adherence to HBMEC in a dose‐ and time‐dependent manner. The induction activity of purified IbeA protein was not affected by LPS decontamination using polymyxin B‐agarose beads but abolished by heating treatment at 100oC, indicating that IbeA‐induced adhesion molecule expression was not caused by contamination with LPS, which is resistant to heat (12). It concurs with our recent study that IbeA‐induced bacterial entry is protein‐dependent but not affected by LPS decontamination using polymyxin B‐agarose (6). In order to exclude the possibility that PMN migration elicited was caused by the destruction of BMEC, the monolayer was inspected by immunofluorescence microscopy and apoptosis assays after incubation with the intact bacteria or their virulence factors. No significant dose‐dependent destruction of the architecture of the endothelial monolayer was seen in response to microbial stimulation during the PMN migration assays, suggesting that IbeA‐induced PMN migration was not simply caused by the destruction of the BMEC monolayer. Interestingly, upregulation and downregulation of ICAM‐1 and CD44 expression are correlated with overexpression of vimentin and its head domain deletion mutant (VDM) in HBMEC, respectively. This observation has been further confirmed with ELISA assays and immunofluorescence microscopy. The IbeA+ strain E44 is able to significantly enhance expression of ICAM‐1 and CD44 when compared with its mutant ZD1. E44 and IbeA could remarkably induce surface expression of ICAM‐1 and CD44 as demonstrated with immunofluorescence staining. Furthermore, IbeA‐induced upregulation of ICAM‐1/CD44 and transmigration of PMN across HBMEC are blocked by WFA, a dual inhibitor of vimentin and proteasome and bortezomib (Velcade), a proteasome inhibitor. Our data suggest that both the drugs could block IbeA‐induced upregulation of adhesion molecules and activation/translocation of NFκB through vimentin‐dependent and independent mechanisms. As it has been recently shown that vimentin could stabilize the Scribble polarity protein by protecting it from proteasomal degradation (36), it is most likely that the vimentin cleavage by WFA could decrease the protective role of vimentin in the stabilization of adhesion molecules. This notion is further supported by the direct interactions between vimentin and adhesion molecules. Alternatively, WFA could block the NFκB activity through reduced proteasomal degradation of phosphorylated IκBα protein, which might result in downregulation of adhesion molecules. Therefore, WFA‐mediated inhibition of IbeA‐induced cellular effects on adhesion molecules clearly involves two different mechanisms: destabilization via the cleavage of vimentin and downregulation of gene expression through the inhibition of the NFκB activity. Our data are in agreement with the published literature that expression of ICAM‐1 is inhibited by WFA and significantly reduced in vimentin‐deficient mice 34, 35 and that vimentin is required for CD44‐induced spreading of B lymphocytes (46). IbeA is different from pneumolysin, which is able to induce PMN transmigration across pulmonary endothelial cells without eliciting upregulation of the endothelial adhesion molecules (30). Together with our finding that PMN adherence to BMEC was strongly enhanced by IbeA, and that endothelial expression of ICAM‐1 and CD44 was activated by IbeA+E. coli, these data suggest that the molecular determinants of neutrophil transendothelial migration elicited by meningitic E. coli K1 may be unique from those defined for other meningitic bacteria such as S. pneumoniae. The mechanism by which IbeA directly or indirectly (through vimentin) regulates endothelial expression of adhesion molecules remains to be defined.
The present study demonstrates that the meningitic virulence factor IbeA and its primary receptor vimentin are necessary to elicit neutrophil transendothelial migration. Vimentin on both BMEC and PMN may be necessary in this process. Whatever precise mechanism(s), our findings indicate a novel role for IbeA and its receptor vimentin in meningitic E. coli K1‐induced migration of PMN across BMEC in a coordinate manner with upregulation of the two major endothelial adhesion molecules ICAM‐1 and CD44. The in vitro and in vivo models described in this report should help, in the future, to better define the molecular basis of bacteria, endothelial cells and PMN interactions needed to induce maximal recruitment of PMN from the vascular space to the CNS.
DISCLOSURE
The authors have no conflicting financial interests.
Supporting information
Figure S1. Translocation of E44 and its ibeA deletion mutant ZD1 across the HBMEC monolayers in the abluminal to luminal direction. The translocation rates of E. coli strains were expressed as percentages of the bacteria numbers in the upper chamber/lower chamber at different time points (0–6 h). Assays were performed in triplicates, and the mean values were shown with bars. The significant differences between the two strains were marked with asterisks (*P < 0.05, **P < 0.01).
Figure S2. Confocal microscopy analysis of human PMN migrating through human BMEC after stimulation with IbeA + E. coli K1. (A) Confocal laser‐scanning micrographs of PMN migrating through human BMEC. Left panel: BMEC (large cells) and PMN (smaller cells with multiple nuclei) were stained with DAPI (4’, 6‐diamidino‐2’‐phenylindole, dihydrochloride). A neutrophil with 4 nuclei inside BMEC is indicated with an arrow. Middle panel: Cells were stained with anti‐Vim antibody followed by FITC‐labeled second antibody. Right panel: Digitally combining two images (left and middle), showing PMN present inside human BMEC during transmigration (indicating with an arrow). Bar = 25 µm. The micrograph was one of the middle pictures from a Z‐series, indicating that the neutrophil nuclei were inside the BMEC. (B) Vimentin is involved with intracellular PMN transmigration. Left panel: Cells were stained for the lipid raft marker GM1 with FITC‐labeled CTx‐B (green). A neutrophil migrating into human BMEC was indicated with an arrow. Middle panel: Cells were stained for VIM with V9 antibody followed by Cy5‐labeled second antibody. Right panel: An overlapping image was generated by combining two images (left and middle). The orthogonal projections of the optical section were viewed from XZ and YZ angles at a point where the two broken white lines intersect in each panel. The leukocyte marked with CTx‐B was indicated with arrows in each XY, YZ and XZ view image, showing the link between vimentin of human BMEC and the neutrophil in migrating process. Bar = 25 µm.
Figure S3. Colocalization of vimentin with ICAM‐1 or CD44. Colocalizations of vimentin with CD44 (A) and ICAM‐1 (B) were detected by immunofluorescence microscopy. HBMEC transduced with VIM‐GFP, VDM‐GFP and GFP were infected by E44 (25 MOIs) for 2 h. The HBMEC without infection were used as a control (CON). The scale bar is 25 µm.
Supporting info item
ACKNOWLEDGMENTS
We thank Lina He for technical assistance and Dr. Fred Dorey for his valuable advice on statistic analysis. This project was financially supported by Public Health Service grants R01‐AI40635 and R21‐AT003207 (S.H.H.), China Natural Science Foundation of Guangdong province grant 3251064101000021 (X.N.W) and China Scholarship Council (X.C).
REFERENCES
- 1. Bargagna‐Mohan P, Hamza A, Kim YE, Khuan Abby Ho Y, Mor‐Vaknin N, Wendschlag N et al (2007) The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem Biol 14:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boilard E, Bourgoin SG, Bernatchez C, Surette ME (2003) Identification of an autoantigen on the surface of apoptotic human T cells as a new protein interacting with inflammatory group IIA phospholipase A2. Blood 102:2901–2909. [DOI] [PubMed] [Google Scholar]
- 3. Bryant AE, Bayer CR, Huntington JD, Stevens DL (2006) Group A streptococcal myonecrosis: increased vimentin expression after skeletal‐muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis 193:1685–1692. [DOI] [PubMed] [Google Scholar]
- 4. Cao H, Chen LD, Yang J, Gong SJ, Zhou H, Huang SH (2007) CglE's role in meningitic E. coli K1 infection and energy metabolism defined by bioinformatic and molecular characterization. Front Converg Biosci Inform Technol (FBIT) 1:251–255. [Google Scholar]
- 5. Chi F, Wang Y, Gallaher TK, Wu CH, Jong A, Huang SH (2009) Identification of IbeR as a stationary‐phase regulator in meningitic Escherichia coli K1 that carries a loss‐of‐function mutation in rpoS . J Biomed Biotechnol 2009:520283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chi F, Jong T, Wang L, Ouyang Y, Wu C, Li W, Huang SH (2010) Vimentin‐mediated signaling is required for IbeA+ E. coli K1 invasion of human brain microvascular endothe lial cells. Biochem J 427:79–90. [DOI] [PubMed] [Google Scholar]
- 7. Chen SH, Stins M, Huang SH, Chen YH, Kwon‐Chung KJ, Chang Y et al (2003) Cryptococcus neoformans induces alterations in the cytoskeleton of human brain microvascular endothelial cells. J Med Microbiol 52:961–970. [DOI] [PubMed] [Google Scholar]
- 8. Chen YH, Chen SH, Zhou ZY, Li W, Jong A, Suzuki K, Huang SH (2002) Enhanced Escherichia coli invasion of human brain microvascular endothelial cells is associated with alterations in cytoskeleton induced by nicotine. Cell Microbiol 4:503–514. [DOI] [PubMed] [Google Scholar]
- 9. Doran KS, Liu GY, Nizet V (2003) Group B streptococcal beta‐hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J Clin Invest 112:736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doulet N, Donnadieu E, Laran‐Chich MP, Niedergang F, Nassif X, Couraud PO, Bourdoulous S (2006) Neisseria meningitidis infection of human endothelial cells interferes with leukocyte transmigration by preventing the formation of endothelial docking structures. J Cell Biol 173:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Echchannaoui H, Frei K, Letiembre M, Strieter RM, Adachi Y, Landmann R (2005) CD14 deficiency leads to increased MIP‐2 production, CXCR2 expression, neutrophil transmigration, and early death in pneumococcal infection. J Leukoc Biol 78:705–715. [DOI] [PubMed] [Google Scholar]
- 12. Gao B, Tsan MF (2003) Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor release by murine macrophages. J Biol Chem 278:174–179. [DOI] [PubMed] [Google Scholar]
- 13. Gao X, Xu N, Sekosan M, Mehta D, Ma SY, Rahman A, Malik AB (2001) Differential role of CD18 integrins in mediating lung neutrophil sequestration and increased microvascular permeability induced by Escherichia coli in mice. J Immunol 167:2895–2901. [DOI] [PubMed] [Google Scholar]
- 14. Huang SH, Jong A (2001) Cellular mechanisms of microbial proteins contributing to invasion of the blood‐brain barrier. Cell Microbiol 3:277–287. [DOI] [PubMed] [Google Scholar]
- 15. Huang SH, Wass CA, Fu Q, Prasadarao NA, Stins MF, Kim KS (1995) Escherichia coli invasion of brain microvascular endothelial cells in vitro and in vivo: molecular cloning and characterization of invasion gene ibe10 . Infect Immun 63:4470–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang SH, Chen YH, Fu Q, Stins M, Wang Y, Wass C, Kim KS (1999) Identification and characterization of an Escherichia coli invasion gene locus, ibeB, required for penetration of brain microvascular endothelial cells. Infect Immun 67:2103–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang SH, Stins M, Kim KS (2000) Bacterial penetration across the blood‐brain barrier during the development of neonatal meningitis. Microbes Infect 2:1237–1244. [DOI] [PubMed] [Google Scholar]
- 18. Huang SH, Chen YH, Kong G, Chen SH, Besemer J, Borodovsky M, Jong A (2001) A novel genetic island of meningitic Escherichia coli K1 containing the ibeA invasion gene (GimA): functional annotation and carbon‐source‐regulated invasion of human brain microvascular endothelial cells. Funct Integr Genomics 1:312–322. [DOI] [PubMed] [Google Scholar]
- 19. Huang SH, Wan ZS, Chen YH, Jong AY, Kim KS (2001) Further characterization of Escherichia coli brain microvascular endothelial cell invasion gene ibeA by deletion, complementation, and protein expression. J Infect Dis 183:1071–1078. [DOI] [PubMed] [Google Scholar]
- 20. Huang SH, Zhou YH, Wang Y, Chen SH, Jong A (2009) Meningitic E. coli K1 IbeA is an outer membrane protein involved in inducing apoptosis in human brain endothelial cells. J Pure Appl Microbiol 3:1–10. [Google Scholar]
- 21. Huet D, Bagot M, Loyaux D, Capdevielle J, Conraux L, Ferrara P et al (2006) SC5 mAb represents a unique tool for the detection of extracellular vimentin as a specific marker of Sezary cells. J Immunol 176:652–659. [DOI] [PubMed] [Google Scholar]
- 22. Ichiyama T, Isumi H, Yoshitomi T, Nishikawa M, Matsubara T, Furukawa S (2002) NF‐ B activation in cerebrospinal fluid cells from patients with meningitis. Neurol Res 24:709–712. [DOI] [PubMed] [Google Scholar]
- 23. Ivaska J, Pallari HM, Nevo J, Eriksson JE (2007) Novel functions of vimentin in cell adhesion, migration, and signaling. Exp Cell Res 313:2050–2062. [DOI] [PubMed] [Google Scholar]
- 24. Jong AY, Stins MF, Huang SH, Chen SH, Kim KS (2001) Traversal of Candida albicans across human blood‐brain barrier in vitro . Infect Immun 69:4536–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim KS (2003) Pathogenesis of bacterial meningitis: from bacteraemia to neuronal injury. Nat Rev Neurosc 4:376–385. [DOI] [PubMed] [Google Scholar]
- 26. Lee JD, Tsai LY, Chen CH, Wang JJ, Hsiao JK, Yen CM (2006) Blood‐brain barrier dysfunction occurring in mice infected with Angiostrongylus cantonensis . Acta Trop 97:204–211. [DOI] [PubMed] [Google Scholar]
- 27. Mendu DR, Dasari VR, Cai M, Kim KS (2008) Protein folding intermediates of invasin protein IbeA from Escherichia coli . FEBS J 275:458–469. [DOI] [PubMed] [Google Scholar]
- 28. Mohan R, Hammers HJ, Bargagna‐Mohan P, Zhan XH, Herbstritt CJ, Ruiz A et al (2004) Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis 7:115–122. [DOI] [PubMed] [Google Scholar]
- 29. Moisan E, Girard D (2006) Cell surface expression of intermediate filament proteins vimentin and lamin B1 in human neutrophil spontaneous apoptosis. J Leukoc Biol 79:489–498. [DOI] [PubMed] [Google Scholar]
- 30. Moreland JG, Bailey G (2006) Neutrophil transendothelial migration in vitro to Streptococcus pneumoniae is pneumolysin dependent. Am J Physiol Lung Cell Mol Physiol 290:833–840. [DOI] [PubMed] [Google Scholar]
- 31. Moreland JG, Bailey G, Nauseef WM, Weiss JP (2004) Organism‐specific neutrophil‐endothelial cell interactions.in response to Escherichia coli, Streptococcus pneumoniae, and Staphylococcus aureus . J Immunol 172:426–432. [DOI] [PubMed] [Google Scholar]
- 32. Mor‐Vaknin N, Punturieri A, Sitwala K, Markovitz DM (2003) Vimentin is secreted by activated macrophages. Nat Cell Biol 5:59–63. [DOI] [PubMed] [Google Scholar]
- 33. Murli S, Watson RO, Galan JE (2001) ) Role of tyrosine kinases and the tyrosine phosphatise SptP in the interaction of Salmonella with host cells. Cell Microbiol 3:795–810. [DOI] [PubMed] [Google Scholar]
- 34. Nieminen M, Henttinen T, Merinen M, Marinen M, Martila‐Ichihara F, Eriksson JE, Jalkanen S (2006) Vimentin function in lymphocyte adhesion and transcellular migration. Nat Cell Biol 8:156–162. [DOI] [PubMed] [Google Scholar]
- 35. Oh JH, Kwon TK (2009) Withaferin A inhibits tumor necrosis factor‐alpha‐induced expression of cell adhesion molecules by inactivation of Akt and NF‐kappaB in human pulmonary epithelial cells. Int Immunopharmacol 9:614–619. [DOI] [PubMed] [Google Scholar]
- 36. Phua DC, Humbert PO, Hunziker W (2009) Vimentin regulates scribble activity by protecting it from proteasomal degradation. Mol Biol Cell 20:2841–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pietzak MM, Badger J, Huang SH, Thomas DW, Shimada H, Kim KS, Inderlied C (2001) Escherichia coli K1 IbeA is required for efficient intestinal epithelial invasion in vitro and in vivo in neonatal rats. J Pediatr Gastroenterol Nutr 33:400. [Google Scholar]
- 38. Podor TJ, Singh D, Chindemi P, Foulon DM, McKelvie R, Weitz JL et al (2002) Vimentin exposed on activated platelets and platelet microparticles localizes vitronectin and plasminogen activator inhibitor complexes on their surface. J Biol Chem 277:7529–7539. [DOI] [PubMed] [Google Scholar]
- 39. Rowin ME, Xue V, Irazuzta J (2000) Integrin expression on neutrophils in a rabbit model of Group B Streptococcal meningitis. Inflammation 24:157–173. [DOI] [PubMed] [Google Scholar]
- 40. Russo TA, Davidson BA, Priore RL, Carlino UB, Helinski JD, Knight PR 3rd (2000) Capsular polysaccharide and O‐specific antigen divergently modulate pulmonary neutrophil influx in an Escherichia coli model of gram‐negative pneumonitis in rats. Infect Immun 68:2854–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shime H, Ohnishi T, Nagao K, Oka K, Takao T, Horiguchi Y (2002) Association of Pasteurella multocida toxin with vimentin. Infect Immun 70:6460–6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Soto SM, Bosch J, Jimenez de Anta MT, Vila J (2008) Comparative study of virulence traits of Escherichia coli clinical isolates causing early and late neonatal sepsis. J Clin Microbiol 46:1123–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Springer T (1994) Traffic signals for lymphocyte recicirculation and leukocyte emigration: the multistep paradigm. Cell 76:301–314. [DOI] [PubMed] [Google Scholar]
- 44. Stan SD, Hahm ER, Warin R, Singh SV (2008) Withaferin A causes FOXO3a‐ and Bim‐dependent apoptosis and inhibits growth of human breast cancer cells in vivo . Cancer Res 68:7661–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stins MF, Shen Y, Huang SH, Gilles F, Kalra VK, Kim KS (2001) Gp120 activates children's brain endothelial cells via CD4. J Neurovirol 7:125–134. [DOI] [PubMed] [Google Scholar]
- 46. Sumoza‐Toledo A, Santos‐Argumedo L (2004) The spreading of B lymphocytes induced by CD44 cross‐linking requires actin, tubulin, and vimentin rearrangements. J Leukoc Biol 75:233–239. [DOI] [PubMed] [Google Scholar]
- 47. van der Flier M, Geelen SP, Kimpen JL, Hoepelman IM, Tuomanen EI (2003) Reprogramming the host response in bacterial meningitis: how best to improve outcome? Clin Microbiol Rev 16:415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Q, Teder P, Judd NP, Noble PW, Doerschuk CM (2002) CD44 deficiency leads to enhanced neutrophil migration and lung injury in Escherichia coli pneumonia in mice. Am J Pathol 161:2219–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu B, deWaal RM, Mor‐Vaknin N, Hibbard C, Markovitz DM, Kahn ML (2004) The endothelial cell‐specific antibody PAL‐E identifies a secreted form of vimentin in the blood vasculature. Mol Cell Biol 24:9198–9206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang H, Shi G, Dou QP (2007) The tumor proteasome is a primary target for the natural anticancer compound Withaferin A isolated from “Indian winter cherry”. Mol Pharmacol 71:426–437. [DOI] [PubMed] [Google Scholar]
- 51. Zwijnenburg PJ, van der Poll T, Roord JJ, van Furth AM (2006) Chemotactic factors in cerebrospinal fluid during bacterial meningitis. Infect Immun 74:1445–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zou Y, He L, Huang SH (2006) Identification of a surface protein on human brain microvascular endothelial cells as vimentin interacting with Escherichia coli invasion protein IbeA. Biochem Biophys Res Commun 351:625–630. [DOI] [PubMed] [Google Scholar]
- 53. Zou Y, He L, Wu CH, Cao H, Xie ZH, Ouyang Y et al (2007) PSF is an IbeA‐binding protein contributing to meningitic Escherichia coli K1 invasion of human brain microvascular endothelial cells. Med Microbiol Immunol 196:135–131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Translocation of E44 and its ibeA deletion mutant ZD1 across the HBMEC monolayers in the abluminal to luminal direction. The translocation rates of E. coli strains were expressed as percentages of the bacteria numbers in the upper chamber/lower chamber at different time points (0–6 h). Assays were performed in triplicates, and the mean values were shown with bars. The significant differences between the two strains were marked with asterisks (*P < 0.05, **P < 0.01).
Figure S2. Confocal microscopy analysis of human PMN migrating through human BMEC after stimulation with IbeA + E. coli K1. (A) Confocal laser‐scanning micrographs of PMN migrating through human BMEC. Left panel: BMEC (large cells) and PMN (smaller cells with multiple nuclei) were stained with DAPI (4’, 6‐diamidino‐2’‐phenylindole, dihydrochloride). A neutrophil with 4 nuclei inside BMEC is indicated with an arrow. Middle panel: Cells were stained with anti‐Vim antibody followed by FITC‐labeled second antibody. Right panel: Digitally combining two images (left and middle), showing PMN present inside human BMEC during transmigration (indicating with an arrow). Bar = 25 µm. The micrograph was one of the middle pictures from a Z‐series, indicating that the neutrophil nuclei were inside the BMEC. (B) Vimentin is involved with intracellular PMN transmigration. Left panel: Cells were stained for the lipid raft marker GM1 with FITC‐labeled CTx‐B (green). A neutrophil migrating into human BMEC was indicated with an arrow. Middle panel: Cells were stained for VIM with V9 antibody followed by Cy5‐labeled second antibody. Right panel: An overlapping image was generated by combining two images (left and middle). The orthogonal projections of the optical section were viewed from XZ and YZ angles at a point where the two broken white lines intersect in each panel. The leukocyte marked with CTx‐B was indicated with arrows in each XY, YZ and XZ view image, showing the link between vimentin of human BMEC and the neutrophil in migrating process. Bar = 25 µm.
Figure S3. Colocalization of vimentin with ICAM‐1 or CD44. Colocalizations of vimentin with CD44 (A) and ICAM‐1 (B) were detected by immunofluorescence microscopy. HBMEC transduced with VIM‐GFP, VDM‐GFP and GFP were infected by E44 (25 MOIs) for 2 h. The HBMEC without infection were used as a control (CON). The scale bar is 25 µm.
Supporting info item