

Abstract
Cerebral cavernous malformations (CCMs) are vascular lesions of the CNS characterized by abnormally enlarged capillary cavities. CCMs can occur as sporadic or familial autosomal dominant form. Familial cases are associated with mutations in CCM1[K‐Rev interaction trapped 1 (KRIT1)], CCM2 (MGC4607) and CCM3 (PDCD10) genes. In this study, a three‐gene mutation screening was performed by direct exon sequencing, in a cohort of 95 Italian patients either sporadic or familial, as well as on their at‐risk relatives. Sixteen mutations in 16 unrelated CCM patients were identified, nine mutations are novel: c.413T > C; c.601C > T; c.846 + 2T > G; c.1254delA; c.1255‐4delGTA; c.1681‐1682delTA in CCM1; c.48A > G; c.82‐83insAG in CCM2; and c.396G > A in CCM3 genes. The samples, negative to direct exon sequencing, were investigated by MLPA to search for intragenic deletions or duplications. One deletion in CCM1 exon 18 was detected in a sporadic patient. Among familial cases 67% had a mutation in CCM1, 5.5% in CCM2, and 5.5% in CCM3, whereas in the remaining 22% no mutations were detected, suggesting the existence of either undetectable mutations or other CCM genes. This study represents the first extensive research program for a comprehensive molecular screening of the three known genes in an Italian cohort of CCM patients and their at‐risk relatives.
Keywords: brain vascular pathologies, CCM genes, cerebral cavernous malformation, molecular genetic analysis in Italian patients
INTRODUCTION
Cerebral cavernous malformations (CCM; OMIM 116860) are discrete multi‐lobed vascular malformations that consist of a cluster of thin‐walled vascular sinusoids. Lined by a single layer of endothelium, they lack an intervening neural parenchyma or identifiable mature vessel‐wall elements (8). Histological analysis shows the lesions lack an arterial wall smooth muscle layer and frequently reveals a peripheral hemosiderin deposition suggestive of chronic hemorrhage. Electron microscopy analyses have implicated defective endothelial tight junctions as a potential explanation for the propensity for hemorrhage seen in these lesions (8) (Figure 1).
Figure 1.
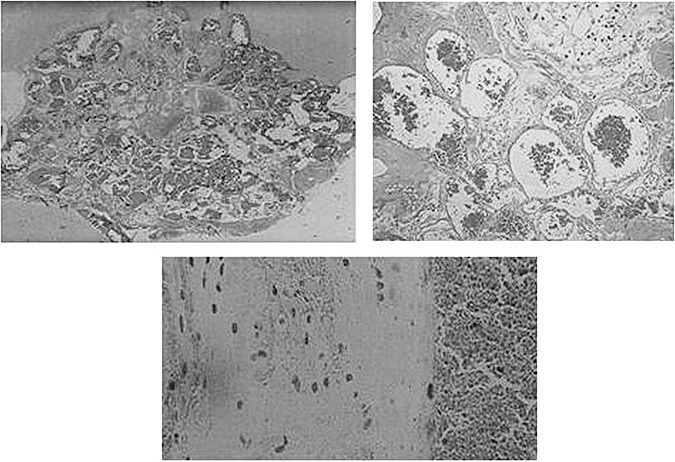
Histological features of cerebral cavernous malformations (CCMs), clusters of markedly dilated sinusoids filled with blood and lined only with a single layer endothelium without intervening parenchyma.
Cavernous malformations are most commonly found in the cerebral cortex, although they may also occur in the brainstem, skin, spinal cord, retina, cranial nerves and cerebral ventricles (22). They occur as single or multiple lesions and, depending on size and location, can be clinically silent or show clinical symptoms ranging from headaches to focal neurological deficits, seizures and fatal intra‐cerebral hemorrhage. As a result of a series of magnetic resonance imaging (MRI) and/or autopsy studies, this disease has been recognized as a common clinical entity: its prevalence in world population has been estimated to range from 0.1% to 0.5%, although only 20%–30% of affected individuals develop symptomatic disease 33, 38. CCMs have been reported in infants and children, but the majority of patients present with symptoms between the second and fifth decades of life.
CCM can arise sporadically or may be inherited as an autosomal dominant condition with incomplete penetrance and variable clinical expression. The proportion of familial cases has been estimated to be as high as 50% in Hispanic‐American patients of Mexican descent, and close to 10%–40% in other populations. The familial form is usually characterized by a high presence of multiple lesions, whereas sporadic cases often present a single lesion 22, 36) (Figure 2). The presence of multiple lesions on cerebral MRI is one of the main features of familial CCM, which is an evolutive condition as assessed by the strong correlation between patient age and the number of lesions. However, multiple lesions have been found in a patient with no positive family history for CCM (9).
Figure 2.
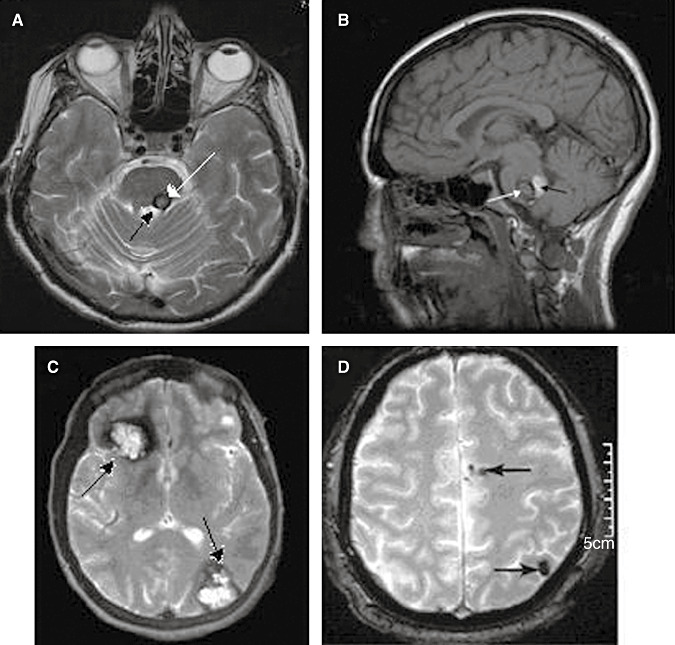
Radiological features of cerebral cavernous malformations (CCMs). At the top, two patients whit single cavernous malformation; at bottom, two patients whit multiple cavernous malformation. A. T2‐weighted axial magnetic resonance imaging (MRI) image shows a typical reticulated “popcorn‐like” cavernous malformation in the left dorsal pons adjacent to the floor of the fourth ventricle. B. T1 sagittal MRI shows a brainstem (pontine) cavernous malformation. The white arrow indicated the cavernous malformation itself, the black arrow indicated a blood cavity immediately behind the cavernoma. C. Gradient‐echo axial MRI shows a large right frontal and a left occipital cavernous malformations (arrows). D. Axial gradient‐echo shows at least two cavernomas in the left hemisphere (arrows).
Genetic linkage studies mapped three CCM loci to chromosome bands 7q21‐22 (CCM1), 7p13‐15 (CCM2) and 3q25.2‐27 (CCM3), and the proportion of families linked to these loci was estimated to be close to 40% (CCM1), 20% (CCM2) and 40% (CCM3), respectively 10, 13. So far, three genes lying at these loci have been identified: CCM1[K‐Rev interaction trapped 1 (Krit1)] includes 19 exons, 16 of them encoding for Krit1, a 736 amino acid protein with three ankyrin repeats, a band 4.1/ezrin/radixin/moesin (FERM) domain and multiple NPXY sequences, which are essential for integrin cytoplasmic domain‐associated protein‐1α (ICAP1α) (43) and CCM2 (MGC4607) 44, 46 binding, as well as for intramolecular interactions (18); CCM2 contains 10 coding exons and encodes a 444 amino acid protein, MGC4607, also called malcavernin, containing a phosphotyrosine‐binding domain (PTB), by which it binds to CCM1 in a ternary complex with mitogen‐activated protein kinase kinase kinase 3 (MEKK3), affecting the p38 mitogen‐activated protein kinase (MAPK) signaling cascade (15)—when disturbed, this signaling cascade causes abnormal vascular morphogenesis in the brain, leading to CCM formation (44); and CCM3 (PDCD10) contains 10 exons, seven of them encoding for Programmed Cell Death 10 (PDCD10), a 212 amino acid protein that might play a role in apoptotic pathways (3) Recently, it has been shown that the CCM3 product interacts in vitro with malcavernin as part of the Krit1/malcavernin complex, thereby participating in CCM1‐dependent modulation of β1‐integrin‐mediated signaling and CCM2‐mediated p38 MAPK signaling in response to cellular stress (42).
Recently, more has been known about the structural domain organization of CCM proteins, their association as a ternary complex and their subcellular localization (15). CCM proteins regulate diverse aspects of endothelial cell morphogenesis and blood vessel stability such as cell–cell junctions, cell shape and polarity, or cell adhesion to the extracellular matrix. Animal models contributed to growing understanding of the biology of cavernous malformations, including the elucidation of the cellular context of CCM protein actions and the in vivo confirmation of abnormal endothelial cell–cell interactions (7). Comprehensive molecular screenings of these genes in CCM patients have led to the identification of more than 150 different mutations, which are characterized by an even distribution over the whole genes and a very low degree of redundancy among different families. An exception is the c.742T > C transition in CCM1 gene, the prevalent mutation in Hispano‐Mexican families (23); more recently, a founder effect in four Sardinia families was observed for the transversion c.987C > A in CCM1 gene, and the c.554del14 bp in CCM2 gene was found in 11 families from the Iberian Peninsula 5, 32.
To date, little is known about the Italian population: genetic analysis allowed identification in 12 unrelated Italian families with CCM, eight different heterozygous CCM1 mutations and one deletion in CCM2 2, 5, 31.
Almost all germ line mutations reported so far result in a premature termination codon (PTC) and are predicting truncated proteins or are leading to a nonsense‐mediated mRNA decay (NMD), suggesting that the function of the relative proteins must be severely impaired.
Nevertheless, it is still debated whether the pathogenetic mechanism occurs through the haploinsufficiency or the two‐hit model.
The haploinsufficiency is as a condition that arises when the normal phenotype requires the protein product of both alleles, and reduction of 50% of gene function results in an abnormal phenotype.
The three CCM genes appear to cause haploinsufficiency in a respective protein predisposing to CCM lesion genesis. It was seen, for example, that KRIT1 protein haploinsufficiency could result in a deregulated pool of ICAP‐1A and in a perturbation of β1 integrin‐mediated angiogenesis 43, 45.
In a molecular analysis of the 10 exons and intron–exon boundaries of CCM2, five mutations have been identified that lead to a premature stop codon, and one of them altered the ATG initiator codon, strongly suggesting that CCM2 lesions are the consequence of CCM2 haploinsufficiency (11).
In another study, in eight unrelated families included on the basis of a negative CCM1 and CCM2 mutation screening, seven distinct mutations were found. The nature of some of these mutations, particularly the deletion of the whole gene observed in one family, strongly suggests that one of the mechanisms that lead to cavernous angiomas might be CCM3 haploinsufficiency (3).
The presence of multiple lesion in most familial cases of CCMs and single lesions in the majority of sporadic cases, besides the fact that familial cases show a more aggressive phenotype, led to the hypothesis that the basis of CCM pathogenesis may be a two‐hit molecular mechanism originally described for tumor suppressor genes and inherited cancers. In addition, the lack of immunoreactivity of respective CCM protein in tissues from individuals with CCM1, CCM2 and CCM3 germline mutations supports that the abnormal endothelial cells lining the caverns form the basis of CCM disease and are completely lacking in the mutated CCMs protein (34). Furthermore, bi‐allelic germ‐line and somatic CCM1 mutations have been thus far only identified in a singular lesion (20). Recently, biallelic germline and somatic mutations were identified in CCM lesions from all three forms of inherited CCMs. These data suggest that CCM lesion genesis requires complete loss of function for one of the CCM genes (1), whereas two previous screenings failed to detect somatic mutations in 21 and 72 patients 30, 35. The failure to detect somatic mutations may have been due to the heterogeneous nature of cells in CCM lesions and very limited number of endothelial cells lining the cavity of the capillaries. Direct sequencing of an amplified product effectively identifies heterozygous germline mutations. However, if a somatic mutation is present in substantially ≤50% of the alleles, it may not be easily detected. This question would, however, require additional investigations. It would also be important to analyze several lesions from a given patient to test for the presence of the somatic mutation in multiple lesions (37).
Interestingly, a significant discrepancy has recently emerged between the locus linkage data and the proportion of CCM families with mutations in the three identified genes. In particular, molecular screenings showed disease‐gene frequencies of 72% for CCM1, 18% for CCM2 and 10% for CCM3. With the exception of CCM3 mutations, which may confer a high risk for cerebral hemorrhage during childhood, not known genotype–phenotype correlations (especially the prognosis) for CCM1 and CCM2 mutation carriers were observed; moreover, in 22%–30% of CCM cases with multiple lesion and/or an affected relative, no mutations were detected 12, 16. It emphasizes the need for further molecular analysis of large CCM cohorts in order to establish the real percentage of the hereditary forms linked to the three CCM genes and whether additional genes are involved, as well as to provide a detailed framework for genetic counseling. In this study, a cohort of unrelated CCM patients originating from various Italian regions and their at‐risk relatives were enrolled and screened for mutations in CCM1, CCM2 and CCM3 genes.
MATERIALS AND METHODS
Patients and families
Neuroscience and Molecular Genetics Centers from northern, central and southern regions of Italy participated in this study, which was approved by the local ethics committee. A total of 95 unrelated, clinically affected CCM probands were consecutively enrolled on the basis of neuroradiologic diagnosis of CCM by MRI. This cohort was composed of 58 women and 37 men from different Italian regions. Clinical and neuroimaging information on the number and localization of CCM lesions were collected for all probands through direct interview and review of medical records. Pedigrees were established systematically with the help of each proband. Patients with single or multiple cavernoma but without known clinically affected relatives were classified as apparently sporadic patients, whereas patients with at least one affected relative were considered familial cases. About the familial cases, the study was extended to 18 of their at‐risk relatives.
Written informed consent for clinical investigations and molecular analysis were obtained from all patients enrolled in this study.
Molecular analyses
CCM genes mutation analysis was also performed on a control group comprising 100 unrelated, randomly selected, healthy individuals (53 female and 47 male, aged 20–79 years), from same geographical areas of the probands.
The variants not reported in the SNP database and with a frequency, in healthy control group, less than 1%, were classified as “mutation.”
DNA extraction, polymerase chain reaction and sequencing
Genomic DNA was extracted from peripheral blood using standard salting out procedures. All coding exons and intron–exon boundaries of CCM1, CCM2 and CCM3 genes were screened using the DNA direct sequencing with an ABI Prism TM Genetic analyzer (Applied Biosystems, Foster City, CA, USA). To design each primer pair the OLIGO'S (version 4.0, MedProbe A.S.‐Oslo, Norway, Europe) and AMPLIFY (version 2.539) programs were used. The specificity for the three genes was tested by NCBI/Blast program [detailed polymerase chain reaction (PCR) conditions and sequences of primer sets are available upon request].
Reverse transcriptase PCR (RT‐PCR) and protein truncation test (PTT)
RT‐PCR of specific cDNA fragments was carried out on total RNA isolated from peripheral leukocytes and the relative RT‐PCR products, separated on gel electrophoresis, were analyzed by direct sequencing. Amplification of proband and control cDNAs was conducted with sets of primers (available on request) annealing two exons upstream and downstream of the site where the sequence variant was located. PTT was performed as previously described (29).
Mutation numbering is based on the cDNA sequences obtained from GenBank (accession number: AF310133 for CCM1, NM_031443 for CCM2, and NM_007217.3 for CCM3) with +1 corresponding to the A of the ATG initiation codon. Mutations are described according to http://www.genomic.unimelb.edu.au/mdi/mutnomen/. SNP database http://www.ncb.nlm.org were also utilized. Homologous sequences of the above CCM genes were compared using database: http://www.ncbi.nlm.nih.gov/sites/homologene.
Multiplex ligation‐dependent probe amplification (MLPA) assay
MLPA was performed on those patients who were negative, by direct sequencing, for mutations in CCM1, CCM2 and CCM3 genes.
MLPA analyses were performed using two MLPA kits (SALSA MLPA Kits, P130 and P131 CCM, MRC Holland, Amsterdam, The Netherlands, Europe). The P130 probe mix contains MLPA probes for nine of the 19 exons of CCM1 and all 10 exons of CCM2 gene (two probes for exon 2), as well as one probe located 0.7 kb upstream from CCM2. For reference, 20 probes for other human genes located on different chromosomes are included. The P131 probe mix contains probes for 10 of the 19 exons of CCM1 and for all nine exons of CCM3 gene (two probes for exon 1). For reference, 18 probes for other human genes located on different chromosomes are included. MLPA was performed according to the protocol supplied, by use of 100–300 ng DNA sample per reaction, using FAM‐labeled primers. Samples were run on ABI PRISM 377, and the data were analyzed with Gene Scan® version 3.1.2 (Applied Biosystems, Foster City, CA, USA) to size the PCR products and to obtain peak areas.
For the visual inspection, peak heights were compared between the sample and controls, to find any alteration in relative peak heights within the test sample.
For the normalized peak‐area calculations, each peak area was normalized by dividing the individual peak area by the total peak area of all peaks for that sample.
Exon numbering is based on GenBank reference sequences can be found on the website: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Nucleotide (accession number NG_012964.1).
RESULTS
Clinical features
A total of 95 CCM probands belonging to unrelated Italian families were collected. On the basis of the pedigree analysis, 79 of these probands were considered sporadic probands and 16 familial cases. Lesions were mainly located within the brain; however, spinal, orbital, retinal and cutaneous cavernous malformations were also found. Sixty‐seven cerebral, six orbital and four spinal single lesions, and 15 cerebral, two cerebral/cutaneous and one retinal multiple lesion were detected; it is worth nothing that the multiple retinal cavernoma were present in the absence of any associated brain lesion. Multiple lesions were observed mainly in familial cases, whereas single lesions were found mainly in sporadic cases (Table 1).
Table 1.
Clinical features in 95 cerebral cavernous malformation (CCM) Italian patients. Abbreviations: S = sporadic; F = familial; MRI = magnetic resonance imaging.
| Variables | *At recruitment | ∧ After genetic test | ||
|---|---|---|---|---|
| S | §F | S | §F | |
| No. of patients | 79• | 16 | 77 | 18• |
| Single lesions | 73 | 4 | 72 | 5 |
| Cerebral | 64 | 3 | 64 | 3 |
| Orbital | 5 | 1 | 4 | 2 |
| Spinal | 4 | — | 4 | — |
| Multiple lesion | 6 | 12 | 5 | 13 |
| Cerebral | 6 | 9 | 5 | 10 |
| Cerebral/cutaneous | — | 2 | — | 2 |
| Retinal | — | 1 | — | 1 |
Patients with at least one CCM affected relative.
Families classified on the basis of MRI and medical records.
Families classified on the basis of molecular analysis results.
After genetic test two of the 79 sporadic cases were included in the familial cases, so that they become 18.
CCM1 gene analysis
Twelve different germ‐line heterozygous mutations were identified in familial cases: six novels and six previously described 5, 6, 23, 29, 39. Nine mutations were exonic: c.268 C > T, c.413T > C, c.601C > T, c.987C > A, c.1204‐1208delAACAA, c.1254delA, c.1277‐1280delGAAT, c.1362‐1363delTC and c.1681‐1682delTA; of these eight leading to PTC through nucleotide substitution or small deletions, one was a nucleotide transition (c.413T > C) apparently leading to a missense mutation. Three mutations: c.846 + 2T > G, c.1147‐13C > G and c.1255‐4delGTA were intronic in splicing control regions (Table 2).
Table 2.
CCM1, CCM2 and CCM3 mutations and cDNA, PTT analysis. Abbreviations: wt = wild type; mt = mutated; nd = non‐determined.
| Family | N° of lesions | Occurrence | Gene | Exon | Nucleotide changes | Predicted effect | cDNA expression (bp) | Truncated protein | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 35 | Multiple | Familial | CCM1 | 6 | c.268C > T | p.R90X | 761:wt/mt | p.89 | (6) |
| 36 | Multiple | Familial | CCM1 | 8 | c.601C > T | p.Q201X | 601:wt | NMD? | This report |
| 38 | Multiple | Familial | CCM1 | 9 | c.846 + 2T > G | Aberrant splicing? | nd | — | this report |
| 52 | Multiple | Familial | CCM1 | 10 | c.987C > A | p.C329X | 978:wt/mt | p.328 | 5 |
| 14 | Multiple | Familial | CCM1 | 12 | c.1147‐13C > G | †Cryptic splice site | 400:wt;412:mt | p.385—PTT:42 kDa | (29) |
| 37 | Multiple | Familial | CCM1 | 12 | c.1254delA | 419fs436X | 614: wt/mt | p.435 | This report |
| 11 | Multiple | Familial | CCM1 | 13 | c.1277_1280delGAAT | 427fs435X | 614:wt/mt | p.434—PTT:47 kDa | (23) |
| 58 | Multiple | Familial | CCM1 | 13 | c.1362_1363delTC | 454fs478X | 614:wt/mt | p.477 | (39) |
| 27 | Multiple | Familial | CCM1 | 15 | c.1681_1682delTA | 561fs566X | 572:wt/mt | p.565—PTT:62 kDa | This report |
| 22 | Single | Familial | CCM1 | 7 | c.413T > C | p.I138T | 580:wt;450:mt;350:mt | p.201; p.162 | This report |
| 48 | Single | Familial | CCM1 | 12 | c.1204_1208delAACAA | 402fs414X | nd | — | (39) |
| 57 | Single | Familial | CCM1 | 13 | c.1255‐4delGTA | Aberrant splicing? | nd | This report | |
| 39 | Multiple | Familial | CCM2 | 2 | c.169_172delAGAC | 57fs58X | nd | — | (24) |
| 29 | Multiple | Familial | CCM3 | 7 | c.396G > A | p.K132K | nd | — | This report |
| 56 | Single | Sporadic | CCM2 | 2 | c.48A > G | p.P16P | nd | — | This report |
| 28 | Single | Sporadic | CCM2 | 2 | c.82_83insAG | 29fs37X | nd | — | This report |
Cryptic intronic acceptor splice site leading to a truncated protein of 42 kDa. (29)
To identify the mutated transcripts and assess aberrant splicing events, expression analysis at cDNA level was performed when RNA was available. In six mutations predicting a PTC (c.268C > T → p.R90X, c.987C > A → p.C329X, c.1254delA → p.436X, c.1277‐1280delGATT → p.435X, c.1362‐1363delTC → p.478X and c.1681‐1682delTA → p.566X) the mutated cDNA was detected on sequencing chromatogram; on the contrary in the c.601C > T → Q201X mutation only the wt transcript was detected (Table 2). The c.413T > C → I138T missense mutation showed, along with the normal transcript, two shorter electrophoretic bands, absent in healthy controls; the relative sequence analysis of the two abnormal products showed the partial and complete skipping of exon 8 (Figure 3 and Table 2). The PTT assay performed for p.435X and p.566X mutations, to verify the presence of the truncated protein, showed a 47 kDa and a 62 kDa truncated product, respectively (Figure 3 and Table 2).
Figure 3.
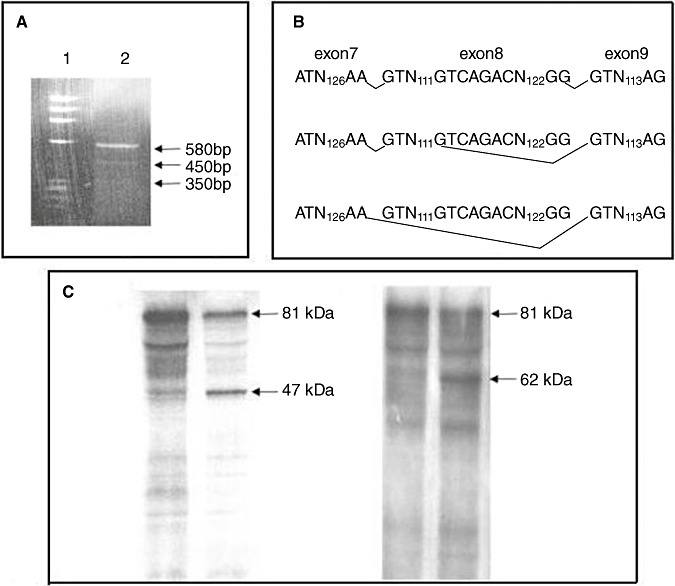
cDNA analysis of c.413T ≥ C CCM1 mutation and protein truncation test (PTT) analysis of p.435X and p.566X CCM1 mutations. A. Reverse transcriptase polymerase chain reaction electrophoresis analysis: lane 1 = molecular weight marker; lane 2 = cDNA of proband number 22 carrying the c.413T ≥ C CCM1 mutation. Along with the normal transcript of 580 bp, there are two shorter bands of 450 and 350 bp, absent in healthy controls, and originated by the partial (450 bp) and complete skipping (350 bp) of exon 8, respectively. B. At the top: Scheme of normal splicing; in the middle and at the bottom: scheme of two aberrant splicing showing partial and complete skipping of exon 8, respectively. C. PTT analysis: (Left) c.1277‐1280del GAAT → p.435X CCM1 mutation; lane 1 = control subject; Lane 2 = proband number 11 carrying the mutation. The arrows indicate the wt protein of 81 kDa and the truncated product of 47 kDa. (Right) c.1681‐1682del Ta → p.566X CCM1 mutation; lane 1 = control subject; lane 2 = proband number 27 carrying the mutation. The arrows indicate the wt protein of 81 kDa and the truncated product of 62 kDa.
By MLPA assay we have also identified one deletion, which we are going to characterize, in exon 18 in a sporadic patient (Figure 5).
Figure 5.
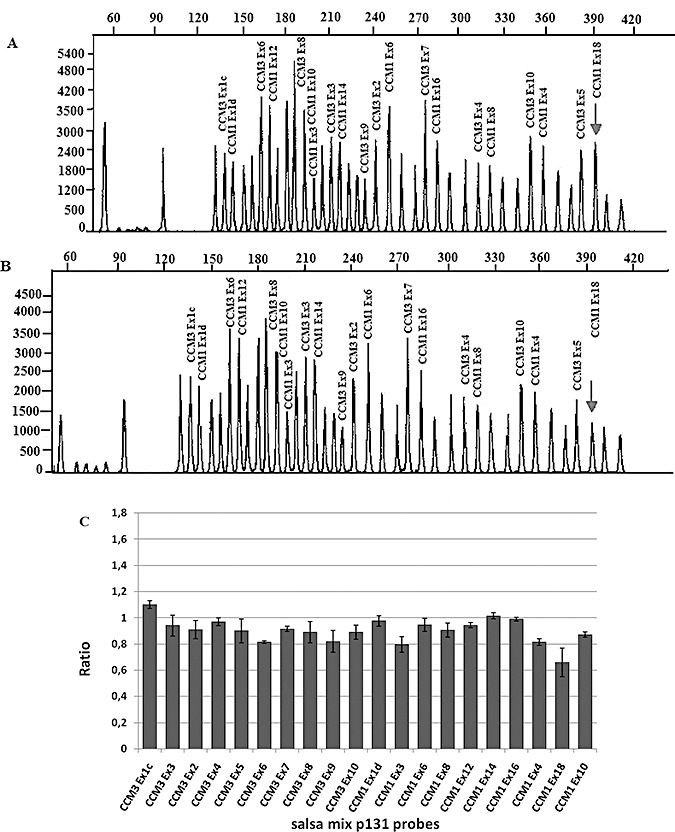
Upper: representative chromatograms from MLPA analysis of CCM1 and CCM3 (salsa MLPA P131 CCM probe mix). A, Results for control individual with peaks corresponding to CCM1 and CCM3 exonic probes. All unlabeled peaks represent the control peaks resulting from the amplification of probes located on different chromosomes. B, Chromatogram from CCM affected proband; Arrow marks the peak corresponding to exon 18 of CCM1, which shows a relative reduction in the peak area in the proband compared with the control. Lower: C. A quantitative analysis demonstrates that the relative peak area has decreased to ∼50%. CCM1 exon 18 deletion is represented, in the graph, by smaller bar marked with arrow. In this graph, the probes located on different chromosomes are not shown and the order of CCM1 and CCM3 exonic probes in C (bars) does not coincide with that shown in A and B (peaks).
CCM2 gene analysis
CCM1‐negative patients were screened for CCM2 gene mutations. Two exonic germ‐line heterozygous mutations, both leading to frame shift and predicting a PTC, were identified: one in a familial case, the already described c.169‐172delAGAC (23) (Table 2) and the other in a sporadic case c.82‐83insAG not previously described (Table 2). Moreover a novel germ‐line transition c.48A > G not predicting amino acid change (p.P16P), nor reported in the SNP database nor observed in the 100 healthy controls, was identified in a sporadic case (Table 2). The Proline at position 16 is conserved in all known sequences across the major evolutionary phyla.
CCM3 gene analysis
Molecular analysis of the CCM3 gene in non‐CCM1 and non‐CCM2 probands revealed, in a familial case, a novel germ‐line transition c.396G > A theoretically predicting a wobble codon (p.K132K), not reported in the SNP database nor observed in 100 healthy controls (Table 2). The structural importance of Lysine 132 is also shown by its conservation in all known homolog sequences.
Family pedigrees
The pedigrees of the 14 families harboring mutations in CCM1, CCM2 or CCM3 genes are shown in Figure 4. All probands had multiple lesion, except the probands of families 22, 48 and 57 presenting a single lesion (Table 2). Besides the 14 probands, 28 affected, several asymptomatic and/or obligate carrier relatives were identified. Mutation analysis performed on the 14 probands was also performed on 18 at‐risk relatives, and allowed to identify nine mutated and nine wild‐type subjects in this group. Interestingly, molecular analysis allowed to identify the CCM1 gene mutation in the respective asymptomatic and MRI‐negative fathers of the probands 22 and 38 (Figure 4); moreover, two individuals of the 22 family presented hepatic cavernomas in the absence of the family's mutation. No pedigrees were available for three sporadic cases: two with the mutations in CCM2 gene (patients: 56 and 28) and one patient with the deletion in CCM1 gene.
Figure 4.
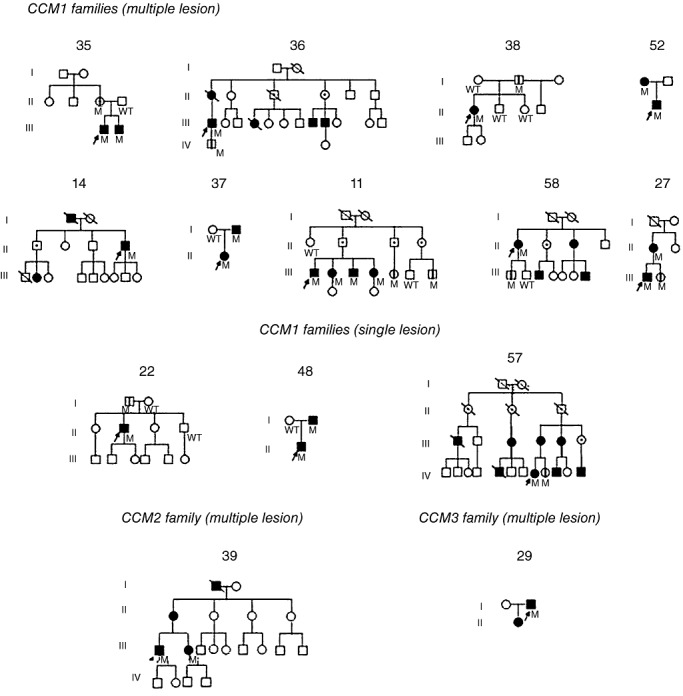
Pedigrees of families with CCM1, CCM2 and CCM3 mutations. Pedigrees are arranged as reported in Table 2; no pedigrees were available for probands 56 and 28.  = proband;
= proband; 
 = affected;
= affected; 
 = asymptomatic;
= asymptomatic; 
 = obligate carrier;
= obligate carrier; 
 = not known to be affected; M = mutation; WT = no mutation;
= not known to be affected; M = mutation; WT = no mutation; 
 = deceased.
= deceased.
DISCUSSION
The presence of multiple lesions is a hallmark of the familial form of the CCM disease. Indeed, the hereditary nature of the disorder has been overlooked in some patients who presented as sporadic cases with multiple lesions (22). In addition, a negative MRI has been observed in some mutation carriers suggesting that not only clinical but also neuroimaging penetrance is incomplete, precluding therefore the use of MRI to firmly exclude the possibility that an asymptomatic family member is a CCM mutation carrier and might later develop lesions and/or have an affected child. The identification of CCM1, CCM2 and CCM3 genes provided a unique opportunity to perform comprehensive genetic screenings and build up nationwide genetic counseling and medical surveillance of patients and their at‐risk relatives 16, 22. So far, more than 150 different mutations have been identified in the three CCM genes, and genotype–phenotype correlation studies have suggested that CCM3 mutations may confer a high risk for cerebral hemorrhage during childhood, whereas no genotype–phenotype correlations were observed for CCM1 and CCM2 mutation carriers 12, 21.
Mutational screening of CCM1, CCM2 and CCM3 genes
This study represents the first comprehensive molecular screening of the three genes in an Italian cohort of CCM patients. All 95 CCM patients were screened for mutations in CCM1, CCM2 and CCM3 genes. This screening led to the identification of 16 subjects carrying a mutation in a CCM gene (Table 2). Interestingly, the family's mutation was identified in the MRI negative fathers of 22 and 38 probands, recruited as sporadic cases (Figure 4). In conclusion 14 mutations were identified in familial cases and two in two sporadic cases with a single lesion and no available relatives for molecular analysis (Table 2). 12 CCM1, three CCM2 and one CCM3 germline heterozygous mutations were identified, nine not previously described; out of self‐interest, two mutation previously described 23, 29 were found only in Italian families (Table 2). Among the identified mutations, 13 were exonic and three intronic in critical splice site regions; 10 of the exonic mutations resulted in a PTC, two were silent mutations and one was a missense (Table 2).
CCM1 molecular analyses
Expression analysis at cDNA level was carried out in eight patients with CCM1 mutation. Mutated cDNA were detected in six mutation carriers (c.268 C > T, c.987C > A, c.1254delA, c.1277‐1280delGATT, c.1362‐1363delTC and c.1681‐1682delTA), but in c.268 C > T cDNA abundance of mutated allele was lower compared to the wt (as judged on the respective heights of sequence chromatogram peaks) suggesting that aberrant mRNA might be more unstable. In the cDNA of the c.601C > T transition, predicting a PTC (Q201X), only the wt transcript was detected, suggesting that the mutated transcript might be degraded by NMD (Table 2) 4, 28; it should be noted that the C > G transversion at the same position (c.601), apparently leading to missense mutation (p.Q201E), resulted in premature splicing of exon 8 predicting a truncated protein of 201 amino acids (39).Two altered mRNA along with the wt allele were identified in the cDNA of the c.413T > C → p.I138T missense mutation, predicting a p.201 and a p.162 truncated proteins (Figure 3 and Table 2); accordingly other four CCM1 missense mutations have been previously shown to lead to aberrant splicing 6, 40. The structural importance of Isoleucine 138 is also shown by the high degree of conservation observed across the major evolutionary phyla. In particular, all known sequences, except that of Caenorhabditis elegans, where a methionine is found, display a conserved Isoleucine at this position. PTT analysis, performed for p.435X and p.566X mutations, where the mutated transcript was observed on sequence chromatogram, confirmed the presence of the truncated protein of 47 and 62 kDa, respectively, comparing to the wt protein of 81 kDa (Figure 3 and Table 2) (29).
The two novel intronic mutations identified in the CCM1 gene are located in splicing control regions; the c.846 + 2T > G transversion is located in the invariant splice donor consensus sequence where a different substitution at the same nucleotide (c.846 + 2T > C) has been previously reported to lead to abnormal splicing products (14). The c.1255‐4delGTA mutation alters the canonic AG acceptor splice site and might lead to utilization of either a cryptic or a new splice site. A deletion in exon 18 was detected in a sporadic case with a single lesion and no family history.
CCM2 mutational screening
Two novel hereditable variants in the CCM2 gene were identified in two sporadic probands (56 and 28) with a single lesion and no familial history for CCM; unfortunately in these cases no family members for genetic testing, nor RNA to evaluate expression at cDNA level were available. The first one was the c.48A > G transition leading to a wobble codon (p.P16P); the second one was the c.82‐83insAG insertion predicting a putative truncated protein of 36 amino acids (Table 2). Although we did not have the possibility to demonstrate the causative value of the two above variants, it should be noted that CCM2 gene mutations have not been previously reported in a cohort of 31 true sporadic patients, 21 with a single lesion and 10 with multiple malformation (41).
CCM3 mutational screening
In a familial case with multiple lesion the AAG → AAA transition, affecting the last nucleotide of exon 7 in CCM3 gene, appearing to be a silent mutation (p.K132K) was observed (Figure 4 and Table 2); nonetheless, this variant might cause a splicing defect, because it results in a less favorable nucleotide at the donor splice junction and this position is not infrequently a site of mutations affecting splicing. Consistently, the previously described transitions AAG → AAA (K30K) and AAG → AAA (K203K) affecting the last nucleotide of exon 1 and 5, respectively, in the CCM2 gene has been analyzed at cDNA level: the first one revealed only the wt allele (24), the second one produced an aberrant transcript (11).
Mutations' frequency of the three CCM genes in a cohort of 95 Italian patients
Taken together, on the basis of the CCM positive family history and of the mutational screening, 18 unrelated Italian patients have been considered as familial cases, thus familial CCM recurs with a frequency of 19% (Table 1). Almost all sporadic cases (72/77) show one lesion, whereas 13 out of 18 familial cases harbor multiple lesions. Both in familial and sporadic cases the lesions are mostly located in the brain (Table 1). Affected individuals belonging to the same family showed variable clinical symptoms, indicating the absence of genotype–phenotype correlation. Molecular screening yielded a mutation detection rate of 78% in the familial patients; in particular, mutations in the CCM1, CCM2 and CCM3 genes accounted for 67%, 5.5% and 5.5% of the hereditary cases, respectively. Consistently with previous papers 12, 16, in close to 22% of the familial cases we failed to identify any mutation. Large genomic deletions and duplications reported for all three CCM genes 17, 19, 26, 27 have not been detected by us by MLPA in the familial cases but only one deletion was found in a sporadic case. Furthermore, in agreement with previous reports we also noticed that the proportion (5.5%) of families linked to mutations in the CCM3 gene was much lower than expected on the basis of the locus linkage data (40%) 3, 25. This might be due to the occurrence of mutations within the regulatory regions or to the existence of a fourth yet unidentified CCM gene, putatively located in close proximity to the CCM3 gene 3, 22.
CONCLUSIONS
Genetic testing in CCM families is essential for an adequate counseling as well as for disease management. In this context, the nine novel mutations identified and one deletion extend the genetic knowledge to assess testing and counseling of CCM patients and their families. The finding of a significant proportion of families with no mutations in the three CCM genes points to further and new molecular biology techniques able to discover the mutations undetectable so far, but at the same time focus the attention toward the identification of other possible involved genes, to better understand the pathogenesis of CCM disease.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors are grateful to the patients and their families for their participation in this study. We also thank Dr. Alfredo Brusco (Department of Genetics, Biology and Biochemistry, University of Torino) for critical reading of the manuscript and helpful discussion. This work was supported by grants from the MIUR (PRIN‐National Project 2004 to C Garrè and SF Retta), and the Telethon Foundation (grant n° GGP06222 to SF Retta).
REFERENCES
- 1. Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA (2009) Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two‐hit mechanism of CCM pathogenesis. Hum Mol Genet 18:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Battistini S, Rocchi R, Cerase A (2007) Clinical, magnetic resonance imaging, and genetic study of 5 Italian families with cerebral cavernous malformation. Arch Neurol 64:843–848. [DOI] [PubMed] [Google Scholar]
- 3. Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M et al; Société Française de Neurochirurgie (2005) Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet 76:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cartegni L, Chew SL, Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3:285–298. [DOI] [PubMed] [Google Scholar]
- 5. Cau M, Loi M, Melis M, Congiu R, Loi A, Meloni C et al (2009) C329X in KRIT1 is a founder mutation among CCM patients in Sardinia. Eur J Med Genet 52:344–348. [DOI] [PubMed] [Google Scholar]
- 6. Cavé‐Riant F, Denier C, Labauge P, Cécillon M, Maciazek J, Joutel A et al (2002) Spectrum and expression analysis of KRIT1 mutations in 121 consecutive and unrelated patients with Cerebral Cavernous Malformations. Eur J Hum Genet 10:733–740. [DOI] [PubMed] [Google Scholar]
- 7. Chan AC, Li DY, Berg MJ, Whitehead KJ (2010) Recent insights into cerebral cavernous malformations: animal models of CCM and the human phenotype. FEBS J 277:1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clatterbuck E, Eberhart CG, Crain BJ, Rigamonti D (2001) Ultrastructural and immunocytochemical evidence that an incompetent blood‐brain barrier is related to the pathophysiology of cavernous malformations. J Neurol Neurosurg Psychiatry 71:188–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coban A, Gurses C, Bilgic B, Sencer S, Karasu A, Bebek N et al (2008) Sporadic multiple cerebral cavernomatosis report of case and review of literature. Neurologist 14:46–49. [DOI] [PubMed] [Google Scholar]
- 10. Craig HD, Günel M, Cepeda O, Johnson EW, Ptacek L, Steinberg GK et al (1998) Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15‐13 and 3q25.2‐27. Hum Mol Genet 7:1851–1858. [DOI] [PubMed] [Google Scholar]
- 11. Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A et al; Société Française de Neurochirurgie (2004) Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet 74:326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M et al; Société Française de Neurochirurgie (2006) Genotype‐phenotype correlations in cerebral cavernous malformations patients. Ann Neurol 60:550–556. [DOI] [PubMed] [Google Scholar]
- 13. Dubovsky J, Zabramski JM, Kurth J, Spetzler RF, Rich SS, Orr HT, Weber JL (1995) A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum Mol Genet 4:453–458. [DOI] [PubMed] [Google Scholar]
- 14. Eerola I, Plate KH, Spiegel R, Boon LM, Mulliken JB, Vikkula M (2000) KRIT1 is mutated in hyperkeratotic cutaneous capillary‐venous malformation associated with cerebral capillary malformation. Hum Mol Genet 9:1351–1355. [DOI] [PubMed] [Google Scholar]
- 15. Faurobert E, Albiges‐Rizo C (2010) Recent insights into cerebral cavernous malformations: a complex jigsaw puzzle under construction. FEBS J 277:1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Felbor U, Sure U, Grimm T, Bertalanffy H (2006) Genetics of cerebral cavernous angioma. Zentralbl Neurochir 67:110–116. [DOI] [PubMed] [Google Scholar]
- 17. Felbor U, Gaetzner S, Verlaan DJ, Vijzelaar R, Rouleau GA, Siegel AM (2007) Large germline deletions and duplication in isolated cerebral cavernous malformation patients. Neurogenetics 8:149–153. [DOI] [PubMed] [Google Scholar]
- 18. Francalanci F, Avolio M, De Luca E, Longo D, Menchise V, Guazzi P, Sgrò F, Marino M, Goitre L, Balzac F, Trabalzini L, Retta SF (2009) Structural and functional differences between KRIT1A and KRIT1B isoforms: a framework for understanding CCM pathogenesis. Exp Cell Res 315:285–303. [DOI] [PubMed] [Google Scholar]
- 19. Gaetzner S, Stahl S, Sürücü O, Schaafhausen A, Halliger‐Keller B, Bertalanffy H et al (2007) CCM1 gene deletion identified by MLPA in cerebral cavernous malformation. Neurosurg Rev 30:155–160. [DOI] [PubMed] [Google Scholar]
- 20. Gault J, Shenkar R, Recksiek P, Awad IA (2005) Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke 36:872–874. [DOI] [PubMed] [Google Scholar]
- 21. Gianfrancesco F, Cannella M, Martino T, Maglione V, Esposito T, Innocenzi G et al (2007) Highly variable penetrance in subjects affected with cavernous cerebral angiomas (CCM) carrying novel CCM1 and CCM2 mutations. Am J Med Genet B Neuropsychiatr Genet 144:691–695. [DOI] [PubMed] [Google Scholar]
- 22. Labauge P, Denier C, Bergametti F, Tournier‐Lasserve E (2007) Genetics of cavernous angiomas. Lancet Neurol 6:237–244. [DOI] [PubMed] [Google Scholar]
- 23. Laberge‐le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M et al (1999) Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet 23:189–193. [DOI] [PubMed] [Google Scholar]
- 24. Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T et al (2003) Mutations in a gene encoding a novel protein containing a phosphotyrosine‐binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet 73:1459–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liquori CL, Berg MJ, Squitieri F, Ottenbacher M, Sorlie M, Leedom TP et al (2006) Low frequency of PDCD10 mutations in a panel of CCM3 probands: potential for a fourth CCM locus. Hum Mutat 27:118. [DOI] [PubMed] [Google Scholar]
- 26. Liquori CL, Berg MJ, Squitieri F, Leedom TP, Ptacek L, Johnson EW, Marchuk DA (2007) Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am J Hum Genet 80:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liquori CL, Penco S, Gault J, Leedom TP, Tassi L, Esposito T et al (2008) Different spectra of genomic deletions within the CCM genes between Italian and American CCM patient cohorts. Neurogenetics 9:25–31. [DOI] [PubMed] [Google Scholar]
- 28. Maquat LE (2004) Nonsense‐mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5:89–99. [DOI] [PubMed] [Google Scholar]
- 29. Marini V, Ferrera L, Dorcaratto A, Viale G, Origone P, Mareni C, Garrè C (2003) Identification of a novel KRIT1 mutation in an Italian family with cerebral cavernous malformation by the protein truncation test. J Neurol Sci 212:75–78. [DOI] [PubMed] [Google Scholar]
- 30. Marini V, Ferrera L, Pigatto F, Origone P, Garre C, Dorcaratto A et al (2004) Search for loss of heterozygosity and mutation analysis of KRIT1 gene in CCM patients. Am J Med Genet A 130:98–101. [DOI] [PubMed] [Google Scholar]
- 31. Nannucci S, Pescini F, Poggesi A, Ciolli L, Patrosso MC, Marocchi A et al (2009) Familial cerebral cavernous malformation: report of afurther Italian family. Neurol Sci 30:143–147. [DOI] [PubMed] [Google Scholar]
- 32. Ortiz L, Costa AF, Bellido ML, Solano F, García‐Moreno JM, Gamero MA et al (2007) Study of cerebral cavernous malformation in Spain and Portugal High prevalence of a 14 bp deletion in exon 5 of MGC4607 (CCM2 gene). J Neurol 254:322–326. [DOI] [PubMed] [Google Scholar]
- 33. Otten P, Pizzolato GP, Rilliet B, Berney J (1989) 131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24 535 autopsie. Neurochirurgie 35:82–83.32. [PubMed] [Google Scholar]
- 34. Pagenstecher A, Stahl S, Sure U, Felbor U (2009) A two‐hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet 18:911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reich P, Winkler J, Straube A, Steiger HJ, Peraud A (2003) Molecular genetic investigations in the CCM1 gene in sporadic cerebral cavernomas. Neurology 60:1135–1138. [DOI] [PubMed] [Google Scholar]
- 36. Revencu N, Vikkula M (2006) Cerebral cavernous malformation: new molecular and clinical insights. J Med Genet 43:716–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Riant F, Bergametti F, Ayrignac X, Boulday G, Tournier‐Lasserve E (2010) Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J 277:1070–1075. [DOI] [PubMed] [Google Scholar]
- 38. Robinson JR, Awad IA, Little JR (1991) Natural history of the cavernous angioma. J Neurosurg 75:709–714. [DOI] [PubMed] [Google Scholar]
- 39. Sahoo T, Goenaga‐Diaz E, Serebriiskii IG, Thomas JW, Kotova E, Cuellar JG et al (2001) Computational and experimental analyses reveal previously undetected coding exons of the KRIT1 (CCM1) gene. Genomics 71:123–126. [DOI] [PubMed] [Google Scholar]
- 40. Verlaan DJ, Siegel AM, Rouleau GA (2002) Krit1 missense mutations lead to splicing errors in cerebral cavernous malformation. Am J Hum Genet 70:1564–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Verlaan DJ, Laurent SB, Rouleau GA, Siegel AM (2004) No CCM2 mutations in a cohort of 31 sporadic cases. Neurology 63:1979. [DOI] [PubMed] [Google Scholar]
- 42. Voss K, Stahl S, Schleider E, Ullrich S, Nickel J, Mueller TD, Felbor U (2007) CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 8:249–256. [DOI] [PubMed] [Google Scholar]
- 43. Zawistowski JS, Serebriiskii IG, Lee MF, Golemis EA, Marchuk DA (2002) KRIT1 association with the integri‐binding protein ICAP‐1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum Mol Genet 11:389–396. [DOI] [PubMed] [Google Scholar]
- 44. Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, Marchuk DA (2005) CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet 14:2521–2531. [DOI] [PubMed] [Google Scholar]
- 45. Zhang J, Clatterbuck RE, Rigamonti D, Chang DD, Dietz HC (2001) Interaction between krit1 and icap1α infers perturbation of integrin β1‐mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum Mol Genet 10:2953–2960. [DOI] [PubMed] [Google Scholar]
- 46. Zhang J, Rigamonti D, Dietz HC, Clatterbuck RE (2007) Interaction between krit1 and malcavernin: implications for the pathogenesis of cerebral cavernous malformations. Neurosurgery 60:353–359. [DOI] [PubMed] [Google Scholar]