

Abstract
Glioblastoma (GBM) prognosis remains dismal, with most patients succumbing to disease within 1 or 2 years of diagnosis. Recent studies have suggested that many solid tumors, including GBM, are maintained by a subset of cells termed cancer stem cells (CSCs). It has been shown that these cells are inherently radio‐ and chemotherapy resistant, and may be maintained in vivo in a niche characterized by reduced oxygen tension (hypoxia). This review examines the recently described effects of hypoxia on CSC in GBM, and the potential promise in targeting the hypoxic pathway therapeutically.
Keywords: cancer stem cells, CD133, digoxin, HIF, hypoxia, glioma, side population
INTRODUCTION
The key role played by the local microenvironment in the initiation and progression of tumors is becoming increasingly clear. One element of the cellular milieu that modulates tumor behavior is oxygen. Locally reduced oxygen levels are a feature of many malignancies, particularly those that grow rapidly. One cancer in which hypoxia‐induced necrosis and neovascularization are central to pathological diagnosis is glioblastoma (GBM), the most common adult malignant brain tumor. Hypoxic regions are frequent in GBM, and increased levels of tumor hypoxia have been associated with worse clinical outcomes 47, 100, 118. Hypoxia is also known to support non‐neoplastic neural stem cells, raising the possibility that “cancer stem cells” (CSCs) may also be affected by oxygen levels [recently reviewed in (86)].
Recent work published by a number of groups 5, 65, 74, 93, 101, 106 highlight these issues. This article reviews the recent literature about the effects of hypoxic conditions on the biology of GBM CSC, and discusses how this new knowledge may lead to improvements in treatment of GBM.
THE CELLULAR RESPONSE TO HYPOXIA
The cellular response to hypoxia is controlled by hypoxia‐inducible factors 1α, 2α and 3α (HIF‐1–3α) 36, 87, which heterodimerize with the constitutively expressed HIF‐1β[(aryl hydrocarbon receptor nuclear translocator (ARNT)]. Under atmospheric oxygen concentration, HIF‐1α is hydroxylated on specific proline residues (Pro‐402, Pro‐564) by prolyl hydroxylase domain (PHD) proteins. This modification is a prerequisite step for its binding to the Von Hippel–Lindau (VHL) tumor suppressor and subsequent ubiquitination by an ubiquitin ligase complex containing Elongin C. Following ubiquitination, HIF‐1α is rapidly degraded by the 26S proteasome 43, 49. Under hypoxia, the activity of the prolyl‐hydroxylases is inhibited and the affinity of VHL to HIF‐1α is reduced, resulting in rapid accumulation of the HIF‐1α protein [reviewed in (103)]. Hypoxia is known to control transcription of many genes that are pivotal in many aspects of cancer biology: angiogenesis, cell survival, chemotherapy and radiation resistance, genomic instability, invasion and metastasis, glucose metabolism and more [recently reviewed (102)].
HYPOXIA IN NORMAL AND NEOPLASTIC BRAIN
All high‐grade gliomas contain centers of hypoxia and necrosis. The extensive hypoxia in these tumors may appear paradoxical, especially as histological assessment suggests they are highly vascularized. However, the vasculature arising in a rapidly proliferating tumor is often torturous and poorly organized, showing severe abnormalities in vessel formation, including many that are either close ended or with slow and inefficient blood flow 17, 117.
From the studies of Evans et al on normal brain and glioma oxygenation, we learn that physiological oxygen concentrations in healthy brain tissue range between 12.5% and 2.5% (pO2 = 100 to 20 mm Hg). The majority of GBMs examined showed mild to moderate/severe hypoxia, with oxygen concentrations ranging between 2.5% and 0.5% (pO2 = 20 to 4 mm Hg) for mild hypoxia and 0.5%–0.1% (pO2 = 4 to 0.75 mm Hg) for moderate/severe hypoxia 19, 20, 21. Using polarographic measurements of oxygen tension, Collingridge et al showed that most high‐grade gliomas they tested fall into the category of moderate hypoxia based on Evans classification (13). In studying the hypoxic environment and its effects on cancer cells, one must consider these ranges of oxygen concentration appropriate for the process being studied. For example, an environment of 5% oxygen or more will probably be appropriate to model normal, physiological oxygen tension but not necessarily the hypoxic tumor microenvironment. On the other hand, oxygen tension of 0.5%–2.5% will likely be appropriate to model the hypoxic microenvironment.
HYPOXIA, DEVELOPMENT AND NEURAL STEM CELLS
The first examination of the effects of oxygen on development was done by Morriss and New, who showed that low oxygen tension was required for proper morphogenesis of the cranial neural folds and closure of the brain tube in cultured rat embryos (80). In a subsequent study, it was found that 9.5‐day‐old rat embryos developed optimally if they were cultured for 24 h in 5% oxygen (78).
In cultured cells, oxygen levels play an important role in regulating cellular differentiation [recently reviewed in (86)]. Growth under low oxygen concentrations is known to maintain pluripotency, and to inhibit the differentiation of embryonic stem cell (ESC) (22). Studer et al have shown that embryonic day‐12 rat mesencephalic precursor cells grown in a 3% oxygen environment exhibited increased proliferation and reduced apoptosis (109). Morrison et al found that hypoxia may also regulate cell fate in isolated neural crest stem cells (79). Finally, Zhang et al have demonstrated that in rat mesencephalic neural stem cells, hypoxia dictates cell fate decision through HIF‐1α(125).
In the brain, oxygen sensing is suggested to be integrated into normal signaling pathways controlling neural stem cell (NSC) proliferation and cell fate choice in their niche, and this control may be disrupted in gliomas and other brain cancers (86). For example, Li et al have elegantly shown that HIF‐1α promotes the transcription of cell cycle genes such as nucleophosmin (NPM), a nucleolar protein that positively modulates cell cycle progression and can inhibit p53 activity (63). Taken together these studies underscore the importance of oxygen control during neural development.
GLIOBLASTOMA AND CANCER STEM CELLS
Glioblastoma is classified by the World Health Organization (WHO) as the most advanced grade (IV) of astrocytic tumors. A key feature of this aggressive disease is the presence of small foci of hypoxic necrosis, often associated with surrounding microvessel proliferation [Figure 1 and (28)]. The ability of GBM cells to infiltrate normal brain tissue makes them impossible to resect using conventional surgery techniques, and patients have a median survival of 14–15 months even with aggressive multimodality treatment by surgery, radiation and chemotherapies 40, 110. Thus new therapeutic approaches are desperately needed.
Figure 1.
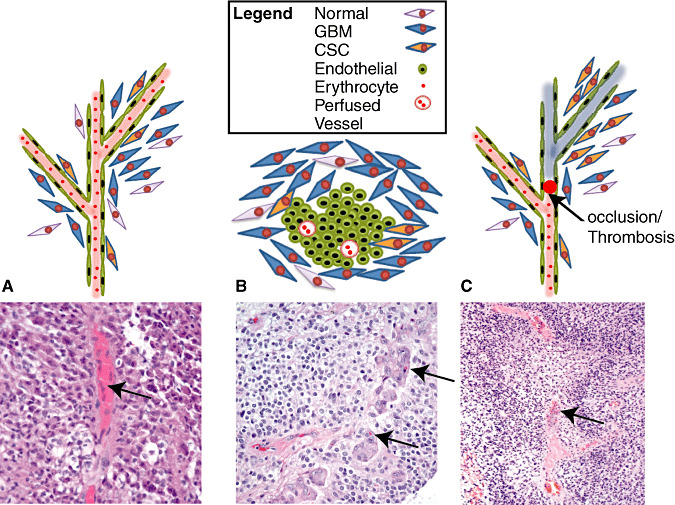
Potential cancer stem cell niches. GHliblastoma cancer stem cells (GBM CSCs) are suggested to localize to either the vascular (A) or hypoxic (B–C) niches. These microenvironments are distinct not only by the presence of oxygen (vascular) or lack thereof (hypoxic), but also their cellular composition. A. GBM CSC associated with the vasculature. Interaction between endothelial cells and GBM CSC maintains CSC in an undifferentiated state. Physical as well as diffusible factors may be involved in this process (in the bottom panel, arrow points to a well perfused vessel as indicated by the presence of numerous erythrocytes). B. Hypoxic niche, which is often found adjacent to necrosis, appears as glomeruloid tufts. CSC may contact proliferating endothelial cells directly and reciprocal signaling may promote endothelial cell proliferation as well as maintenance of the CSC identity (on the bottom panel, arrows point at single erythrocytes, indicating poor perfusion). C. Thrombosis as a potential mechanism for hypoxic niche formation (top). Pseudopalisades forming around a central vessel containing a thrombus. Note the early stage of necrosis and how the pseudopalisades take on the contour of the occluded vessel (marked with an arrow, bottom) (p. vessel—perfused vessel).
Cancers have long been thought to posses features associated with stem cells, and cells with stem‐like properties have been isolated from freshly resected human GBM 29, 41, 105. This subset of neoplastic cells, which appears to be resistant to standard therapies and endowed with increased clonogenic potential, are generally called “cancer stem cells” (CSC), although other terms such as “tumor initiating cells” are also used. It has been suggested that the persistence of CSC after treatment may explain why tumors recur and that only by their eradication can a neoplasm be successfully treated 15, 64, 85. Therefore, therapies causing CSC to differentiate or die represent a novel therapeutic avenue with great promise. To achieve this, several groups, including ours, have investigated if factors affecting normal stem cells can be targeted in cancer 4, 23, 24, 89, 131.
Over recent years, several markers have been utilized to study CSC in GBM. These markers include CD133 (105), side population (53), Olig2 (66), ALDH 4, 96 and more. The Dirks laboratory was first to report that only CD133‐positive cells, isolated from GBM, were capable of xenograft initiation in vivo (105). Many studies have since been published where CD133 was utilized to mark CSC in GBM and other tumor types. However, the relevance of CD133 in defining a putative CSC population in GBM is still the center of much debate, with several studies suggesting that cells that do not express this marker also have the capacity to self‐renew and engraft 6, 12, 46, 48, 52, 82, 83, 88, 111, 120. Moreover, Griguer et al have shown that CD133 expression was upregulated in the adherent GBM cell line U251 in response to reduced oxygen concentration (34). However, in their studies they also found that CD133 was induced by various stress conditions, suggesting it being a non specific “cellular stress” indicator rather than a CSC marker. These studies highlight the importance of validation via accepted CSC assays such as clonogenic assays (growth in semisolid medium such as methyl cellulose, single‐cell growth frequencies, etc.), and most importantly, testing the capacity of cells to initiate tumors in vivo, which is still undoubtedly the gold standard in any CSC frequency determination.
HYPOXIA AND CANCER STEM CELLS
Expression of CD133 and other stem cell markers
Expression of CD133 has been reported by several groups to be upregulated under hypoxic conditions. McCord et al found that in two out of three GBM neurosphere lines they tested, CD133 percentage increased approximately twofold when cells were incubated at 7% oxygen (74). Importantly, additional stem cell markers such as Sox2, Oct4, and Nestin were also found to be upregulated. Siedel et al have similarly shown that incubation of primary GBM cultures in 1% oxygen resulted in CD133 mRNA and protein induction, an observation that was confirmed by flow cytometry (101). Similarly, using flow cytometry, Soeda et al have also showed that the percentage of CD133‐positive cells increases over time when GBM neurospheres are treated with 1% oxygen, and further characterization of the expanded population demonstrated that hypoxia treatment results in preferential expansion of the CD133/CXCR4/CD44low/A2B5/CD24‐positive subpopulation of neurosphere cells. Interestingly, pretreatment of GBM neurospheres with either LY294002 (PI3K inhibitor), PD98059 (ERK inhibitor) or rapamycin (mTOR inhibitor), independently attenuated the hypoxia‐dependent increase in HIF‐1α protein levels and CD133‐positive cell fraction (106). These observations may be clinically significant as they suggest that inhibiting these growth factor pathways may be a useful approach to inhibit the cellular response to hypoxia.
One of the defining features of GBM in vivo is that they are extremely heterogeneous. The cytoarchitecture showing mostly normoxic cells in their periphery, hypoxic cells in their centers and necrotic/dead cells in their inner most cores. It is conceivable therefore, that CSC residing in these various environments may differ in their tumor‐initiating capacity, the markers they express, and their susceptibility to therapy. Indeed, three recent studies compared properties of cells isolated from core and periphery of matched GBM samples. Pistollato et al have shown that more immature cells are found in the inner core and intermediate layers, whereas more mature cells (as indicated by neuronal and glial marker expression) are distributed in the periphery (92). In addition, core cells expressed increased levels of O6‐methylguanine‐DNA‐methyltransferase (MGMT), a known factor involved in chemotherapy resistance (40). Similarly, when comparing the growth characteristics of neurospheres established from core vs. the periphery, Piccirillo et al showed that the former are more clonogenic, proliferate faster under neurosphere growth conditions and have increased capacity to initiate tumors in vivo (88). Most recently, Glas et al have shown that while “residual tumor cells”, which are cells found at the margins after bulk tumor resection, are more invasive and proliferative and posses reduced clonogenic capacity as compared with cells isolated from the core (33). Interestingly, while the studies by Pistollato et al and Glas et al highlight increased expression of CD133 mRNA and percentage of CD133‐positive cells (respectively) in core vs. periphery, Piccirillo et al reported similar percentages in cultures established from these regions. One possibility for this potential discrepancy is the length of time the cells were grown in culture prior to CD133 determination as well as different culture conditions (growth medium, etc.).
To investigate the effects of hypoxia on clonogenic CSC in GBM, we exposed cells isolated from various sources (freshly resected tumors or xenografts, as well as neurosphere models of this disease) to moderate hypoxia (1% oxygen) for varying lengths of time (5). Within 9 h of hypoxic exposure (the earliest time we tested) cells showed increased expression of hypoxia signature genes, including transcripts involved in glycolysis, angiogenesis, growth factor response, etc. Importantly, we observed that genes that are associated with stem cells, such as Krupple‐like‐4 (Klf4), a transcription factor that may contribute to induction of pluripotency (84) and the neural stem cell marker CD133, were induced. In our studies, increases in CD133 percentages in response to hypoxia were accompanied by increases in its mRNA and protein levels as measured by quantitative PCR and Western blot analyses, respectively. The induction of CD133 mRNA and protein was observed within 24–48 h of hypoxic treatment, and increased over time. Importantly, we observed similar magnitudes of change in terms of fold induction of CD133 whether we used established neurospheres or cells that were freshly isolated from human patients or flank xenografts in mice. However, the absolute percentage of CD133‐positive cells, as assessed by flow cytometric analysis, was always lower in cells isolated from in vivo tumors, suggesting that either in vitro growth can enrich for cells expressing this stem cell marker, or that the reduced percentage of CD133‐positive cells in freshly resected tissues is because of the fact that these cultures contain mixed populations of neoplastic and non‐neoplastic cells.
In addition to induction of CD133 and Klf4, we noticed a marked increase in the side population, another marker often used to identify stem/progenitor stem cells. Interestingly, Seidle et al have identified a set of genes that are enriched in side population cells from three adherent GBM cell lines. They found that these side population signature genes are expressed in vascular and hypoxic niches in GBM in areas where HIF‐1α and HIF‐2α are expressed (101).
GBM and the cancer stem cell niche
Several distinct niches have been proposed for CSC in GBM, including perivascular (Figure 1A) and hypoxic (Figure 1B) regions. Stem cell niches provide a specialized microenvironment that maintains and regulates the properties of the residing cells (reviewed in (27)). For a recent review of the perivascular niche in GBM, the reader is encouraged to refer to (31). As to the hypoxic niche, pseudopalisading necrosis, a fundamental feature of primary and secondary GBMs (57), represents one microscopic correlate. With the advent of immunohistochemical and molecular markers these regions have been confirmed to be hypoxic, and surviving tumor cells strongly express HIF‐1a and VEGF (129). It has been suggested that CSC survive and thrive in these hypoxic environments potentially via upregulation of HIF and other factors [reviewed in (116)]. The mechanism by which foci of pseudopalisading necrosis arise is not entirely clear, although vascular thrombosis causing occlusion likely accounts for at least a portion (97). In these, as the surroundings of the thrombosed vessel become hypoxic, glioma cells migrate away and form the characteristic cellular rim.
While the space around blood vessels is generally thought to be well oxygenated, perivascular and hypoxic niches are not necessarily mutually exclusive. Microvascular proliferations with a “glomeruloid” appearance represent another cardinal feature of GBM, and are commonly positioned near areas of necrosis. These glomeruloid tufts consist of mitotically‐active endothelial cells and of smooth muscle cells/pericytes 38, 81, 123. They therefore contain the cellular elements of a perivascular CSC niche. However, vascular channels and red blood cells are generally attenuated or absent within the microvascular proliferations, suggesting the possibility of low oxygen levels and a unique “perivascular/hypoxic” niche.
Hypoxia and clonogenicity
The functional significance of the relative increase in the fraction of CD133‐positive cells was also studied by several groups. Soeda et al reported that growth in 1% oxygen increased the total number and size of neurospheres. Moreover, 1% oxygen inhibited the pro differentiative effects of fetal calf serum on GBM neurospheres as indicated by reduced expression of lineage markers GFAP (astrocytic) and Tuj1 (neuronal) (106). Pistollato et al reported that normal SVZ and high‐grade glioma precursors proliferated optimally in reduced oxygen concentrations (5% and 2%). In these studies, hypoxia was shown to inhibit the expression of multiple bone morphogenic proteins, leading to reduction in canonical SMAD activation. Importantly, reperfusion resulted in rapid loss of HIF‐1α, activation of SMAD, and induced differentiation within 48 h of reoxygenation (93).
Examining the effects of more physiological (7%) oxygen levels, McCord et al reported that larger neurospheres are formed when cells are grown under reduced oxygen concentrations. Importantly, under these conditions, CD133‐positive cells proliferated faster as compared with CD133‐positive cells grown under 20% oxygen. In addition, colony forming efficiency increased almost twofold. The effect of low oxygen levels was observed in both neurosphere lines that showed hypoxia‐dependent increase in the CD133‐positive fraction as well as in one neurosphere line, which did not but started off with a baseline CD133‐positive cells of over 95% (74). Heddleston et al have recently shown that culturing CD133‐negative cells in 2% oxygen for 9 days resulted in increased clonogenicity of these cells, again demonstrating increased number and size of neurospheres. Moreover, these CD133‐negative cells appeared to proliferate faster from day 5 of hypoxic treatment. However, we do not know the percentage of CD133‐positive cells at the end of the hypoxic treatment, and an increased ratio of positive to negative cells could explain the relative increase in clonogenicity seen. The CD133‐negative cells showed marked increases in both HIF‐1α and HIF‐2α, both of which were shown to control CD133 expression. It is therefore a possibility that induction of CD133 expression in these CD133‐negative cells may account for the increased clonogenic capacity (39).
We compared the capacity of single CD133‐positive and CD133‐negative cells to form large (>100 µm) neurospheres in semisolid media (methyl cellulose). Growth under similar conditions has been recently described as a rigorous assay for identification of non‐neoplastic neural stem cells (68). We found that steady‐state CD133‐positive GBM cells formed large, stem‐cell driven, spheres twice as frequent as CD133‐negative cells did. In addition, hypoxia appeared to preferentially increase the capacity of CD133‐positive cells to form large spheres. These observations led us to suggest that clonogenic GBM cells are found in both CD133‐negative and CD133‐positive subpopulations. However, the former's clonogenic capacity is not affected by hypoxia as that of the CD133‐positive cells. Taken together, these observations suggest that hypoxia inhibits differentiation and increases the percentage of CD133‐positive cells, which are more clonogenic than CD133‐negative cells.
This raises the issues of how the relative increase in CD133‐positive cells is achieved and if this is a transient phenotype or a more stable one. We started to tackle these questions and found that at least in the first week of growth under conditions of moderate hypoxia, positive and negative cells proliferation is roughly equivalent. Similar results were reported previously (39). It is possible that longer incubation will result in apoptotic induction and cell death, as indeed chronic hypoxia in tumors result in cell death and necrosis. However, the effect on clonogenicity is manifested within 2 or 3 days at a time in which neither reduction in cell growth or increased apoptosis are detected. Our interpretation of these results is that hypoxia augments the clonogenic capacity of existing (short term) and newly formed (longer term) CD133‐positive cells, which under high oxygen levels will posses reduced clonogenic capacity. As to the question of reversibility, McCord et al have shown that the increase in clonogenicity seen following exposure of neurospheres to physiologic oxygen tension (7%) is reversed when cells are reperfused in 20% oxygen for 7 days. In addition, expression of the stem cell markers Nestin and Oct4 were also found to be reduced to baseline levels. In cultured neurospheres that were exposed to hypoxia (1%) we found that the effect on CD133 expression remained relatively stable even after 7 days in normoxia. We however have not examined the clonogenic capacity of reperfused cells. One speculative possibility is that hypoxia increases the percentage of CD133‐positive cells and augments their and existing CD133‐positive cells clonogenic capacity, however, once reperfused, the clonogenic capacity of these hypoxia responsive CD133 cells is reduced to baseline. Further studies are required to get further insight into the long term effects of hypoxia on clonogenicity.
Differential effects of HIF‐1α and HIF‐2α: selective roles or differences stemming from tumor heterogeneity?
As mentioned earlier, HIF‐1α and HIF‐2α are master regulators of the transcriptional response to hypoxia, and a growing body of work from several labs suggests that there are functional differences between HIF‐1α and HIF‐2α in regards to response of neoplastic cells to hypoxia. We found that the hypoxia‐dependent increase in the CD133‐positive fraction could be recapitulated simply by over expression of an oxygen stable form of HIF‐1α under normoxic conditions, suggesting that HIF‐1α is sufficient to mediate these effects. A similar approach was utilized by Heddleston et al who used an oxygen stable form of HIF‐2α. They found that HIF‐2α increased the percentage of CD133‐positive cells in a sorted population of CD133‐negative cells maintained in serum containing medium and switched to neurosphere growth medium prior to analysis. This HIF‐2α expression also resulted in concomitant increases in the mRNA levels of the stem‐cell associated genes cMyc, Nanog and Oct4 (39).
Examining cells extracted from primary human brain tumor specimens or xenografts of established human GBM cell lines, Li et al have shown that HIF‐2α mRNA is expressed to a higher degree in CD133‐positive than in CD133‐negative cells. Also, HIF‐2α mRNA levels were shown to increase in response to hypoxia in CD133‐positive cells, whereas HIF‐1α mRNA was equally expressed in both CD133‐positive and CD133‐negative cells and its levels were unaffected by hypoxia. When protein levels were quantitated by Western blot, HIF‐1α and HIF‐2α both responded to deferoxamine, 1% or 2% oxygen in both populations (albeit with different kinetics and degree of change). Similar observations were reported by McCord et al with respect to the induction of HIF‐2α protein in response to more physiological oxygen levels (7%). However, under these non‐hypoxic conditions, HIF‐1α levels were barely detected (74) providing further support for HIF‐2α specific effects on CD133‐positive GBM cells at physiologic oxygen levels. Additional differences between HIF‐1α and HIF‐2α were reported by Seidel et al. They showed that expression of HIF‐2α but not HIF‐1α was sufficient to induce the expression of the side population signature gene‐panel. Moreover, knocking down the expression of HIF‐2α but not HIF‐1α was sufficient to reduce the hypoxia induced expression of CD133 as well as ASPHD2 and MAML3, two genes belonging to the side population signature panel (101). However, as previously shown by Li et al (65), reduction of both HIF factors resulted in reduced clonogenicity.
In our studies of GBM neurospheres, we could not detect HIF‐2α protein expression in normoxia or hypoxia, while HIF‐2α mRNA was widely expressed. When CD133‐positive and CD133‐negative populations were FACS–sorted and exposed to hypoxia, HIF‐1α mRNA level was unchanged, whereas HIF‐2α expression appeared to increase only in the CD133‐negative fraction. Importantly, this increase in HIF‐2α did not result in translation of detectable protein. This raises the possibility that in some GBMs a regulation mechanism might be present to prevent HIF‐2α protein from being expressed. Additional studies will be required to assess if such mechanism exists. Further emphasizing the differences between HIF‐1α and HIF‐2α, Soeda et al have shown that in three neurosphere lines they tested, HIF‐1α but not HIF‐2α protein was responsive to hypoxia (1% O2). Furthermore, knocking down the expression of HIF‐1α but not HIF‐2α, PHD2 (prolyl‐hydroxylase promoting HIF degradation), or Notch1 (see below) resulted in reduced clonogenicity in vitro (106).
When a given cell type is experiencing a reduced oxygen environment HIF‐1α and HIF‐2α will induce (and indirectly inhibit) the expression of hundred of genes with some target overlap between these two factors. Therefore the biological consequences of HIF activation are dependent on which factor is active at any given time and what the targets it regulates are. In recent studies of GBM neurospheres, HIF‐1α and HIF‐2α mRNAs where shown to be expressed anywhere between physiologic oxygen levels (5%–7% range in reported literature) to moderate hypoxia (1%–2% range in reported literature). While HIF‐2α protein is present in a wide range of oxygen levels, HIF‐1α protein is more restricted to cells experiencing moderate hypoxia. Despite these differences, both HIF‐1α and HIF‐2α are crucial when it comes to the test of clonogenicity and tumor propagation in xenografts, suggesting that pharmacological inhibition of these factors holds great promise in targeting neoplastic cells.
Hypoxia and Notch
The Notch signaling pathway is involved in many cell fate decisions, and in cellular processes that are implicated in gliomagenesis (most recently reviewed by Stockhausen et al (107)). Gustafsson et al have shown that HIF‐1α physically interacts with the activated notch intracellular domain (NICD), thereby blocking terminal differentiation of neural precursors. Under increased oxygen concentrations, such interaction is abolished, allowing neural precursors to differentiate (37). Furthermore, increased O2 derepresses p53, which can lead to p21cip1‐mediated cell differentiation or apoptosis 98, 115.
From the therapeutic stand point, Fan et al have recently shown that Notch pathway blockade depletes CD133‐positive GBM cells and inhibits growth of tumor neurospheres and xenografts (23). We found that hypoxia regulates the expression of the Notch ligands Jag1 and/or Jag2 in multiple neurosphere and adherent, high serum, cell lines, suggesting that hypoxia may modulate Notch signaling. Indeed we observed a modest induction of the downstream target genes Hes1 and Hey2 grown under hypoxic conditions. Similar results have been previously described by Sahlgren et al who showed that growth in 1% oxygen induced Notch signaling in various cells lines, including the human‐derived GBM cell line U87‐MG, in a Notch ligand‐specific manner (99).
Using microarray gene‐expression analysis of CD133‐positive GBM cells exposed to physiological oxygen concentration (7%), McCord et al have reported that the Notch signaling pathway, among several other pathways, was upregulated. These observations may have therapeutic implications, as Wang et al have recently shown that inhibition of Notch signaling either by the gamma‐secretase inhibitors (DAPT or L685,458) or siRNA against Notch1 or Notch2 result in increased sensitivity to ionizing radiation, whereas overexpression of either Notch1 or Notch2 intracellular domains (activated receptors) resulted in increased resistance to radiation (121). Earlier data from the same group has shown that GBM CD133‐positive cells are inherently more resistant to ionizing radiation because of higher baseline activation level of the DNA‐damage response (3). However, the mechanism controlling resistance to radiation in Notch‐inhibited cells appears to rely on AKT and Mcl‐1 rather on alterations of the DNA‐damage response (121). While the relative radio‐resistance of CD133+ GBM cells as compared with CD133‐negative cells has been validated by at least two other groups in additional tumor types 8, 11, it is clear that this relative resistance is regulated by the culture conditions. Supporting this view is the recent publication comparing the radio‐resistance of established GBM cell lines to that of CD133‐positive cells from low passage GBM neurospheres (75) showing that neurosphere‐derived CD133‐positive cells are more sensitive to ionizing radiation than established high serum adherent cultures. Despite these differences, taken together, these reports suggest that inhibition of Notch signaling may be a promising approach to deplete tumor propagating cells and increase GBM sensitivity to ionizing radiation.
Can we take advantage of tumor cell dependency on HIF factors to design better therapies?
The dependency of tumor cells on HIF factor activity in GBMs suggests that these factors are prime candidates for pharmacological intervention. Indeed, several studies have been published in recent years exploring this. Moreover, it has been long recognized that HIF‐α factors are active in most, if not all, solid tumors and that HIF stabilization is regulated by additional conditions or factors (other than oxygen levels). This point is important as any potential therapy targeting HIF activity may not only target hypoxic cancer cells but may target normoxic cancer cells as well. Some of these conditions/factors are illustrated in Figure 2, and recently reviewed (104). For example, Zhao et al have recently shown that the R132H mutation in the gene encoding iso‐citrate dehydrogenase (IDH1R132H) dominantly inhibits its catalytic activity and induces HIF‐1α activity under normoxia (127). More recently, Dang et al reported that IDH1 mutations result in a gain of function phenotype, the ability to catalyze the reduction of α‐ketoglutarate to 2‐hydroxyglutaric acid (14). Additional examples of hypoxia‐independent HIF‐1α stabilization include: Oncogenic transformation, associated with activation of the Ras/RAF/mitogen‐activated protein kinase, phosphoinositide 3‐kinase (PI3K), phosphatase and tensin homolog or Akt pathways, can also cause HIF‐1α accumulation 73, 133. Succinate, which is produced during hydroxylation of HIF‐1α, has also been shown to slow the rate of HIF‐1α hydroxylation leading to its increased stability. Prostaglandins and certain nitric oxide donors can also induce HIF‐1α under non‐hypoxic conditions (42). Finally, transforming viruses have also been shown to result in HIF‐1α activation [reviewed in (102)]. Taken together these studies provide compelling evidence that HIF‐1α and HIF‐2α activation promote oncogenesis and/or tumor progression and that both represent excellent targets for pharmacological intervention [recently reviewed 16, 47].
Figure 2.
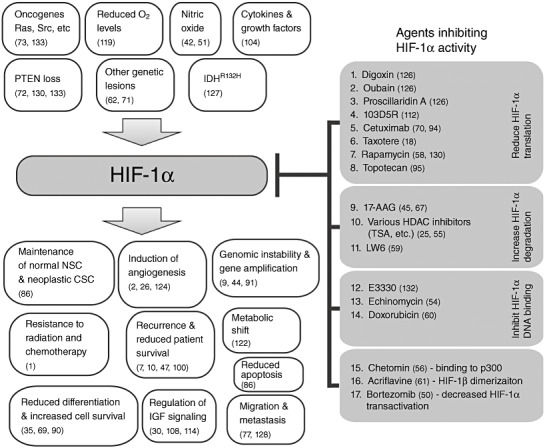
HIF‐1α plays a pivotal role in many physiological and pathological processes (shown on the left side). Agents that inhibit hypoxia inducible factor 1α (HIF‐1α) activity have been reported to control various downstream processes (shown on the right side). The majority of these agents inhibit HIF‐1α translation 1, 2, 3, 4, 5, 6, 7, 8. However, agents that increase HIF‐1α degradation 9, 10, 11 or reduce its ability to bind DNA 12, 13, 14 have also been reported. Fewer agents were identified as inhibitors of HIF‐1α transactivation (17) or binding to other proteins such as the transcription activator p300 (15) and to its heterodimeric partner, HIF‐1β(16). HDAC = Histone deacetylase.
As mentioned earlier, there are significant similarities between normal neural stem cells and GBM CSC in regards to their response to hypoxia. This raises concerns about whether there is a therapeutic window in which therapies inhibiting HIF‐1α and/or HIF‐2α can be tailored in such a way to prevent them from affecting non‐neoplastic neural stem cells. Keeping these concerns in mind, inhibition of HIF factors in GBMs has been shown to be extremely effective. Gillespie et al have shown that reducing HIF‐1α, using siRNA, inhibited the hypoxic response and attenuated GBM growth in vitro and in vivo (32). Similarly, Heddleston et al have shown that targeting either HIF‐1α or HIF‐2α, resulted in reduced VEGF expression in GBM CSC cells, proliferation of HMVEC (co‐cultured with these GBM CSC cells), and most importantly reduced the clonogenicity of CD133‐positive cells in vitro and in vivo (39).
We have taken advantage of the common heart glycoside, digoxin, which has been recently shown by the Semenza group to inhibit the translation of HIF‐1α(126). We found that inhibiting HIF‐1α translation in GBM using digoxin result in significant reduction in growth, which can partially be rescued by expressing the oxygen stable form of this protein. Interestingly, we found that HIF‐1α protein was present in some GBM cells even under 20% oxygen, and that targeting HIF‐1α in these cells, using shRNA, inhibited the growth of these cultures even at these elevated oxygen levels, suggesting that in some GBMs HIF‐1α may act as a survival factor. Importantly, in our studies, we found that digoxin reduced the hypoxia‐dependent increase in CD133‐positive fraction, xenograft implantation and growth of pre‐established GBM flank xenografts, again pointing the finger towards HIF‐1α, as the driving force for tumor initiation and growth. Previously, Zhang et al have reported successful inhibition of tumor cell growth in response to digoxin in other tumor types. Specifically, they found that in addition to digoxin, other glycosides such as ouabain, and proscillaridin A, could reduce HIF‐1α protein levels and its downstream signaling targets. Digoxin appeared to inhibit both HIF‐1α and HIF‐2α translation (although with a higher IC50 for HIF‐2α). Digoxin or siRNAs against HIF‐1α or HIF‐2α were also shown to significantly reduce growth of PC3 (prostate) and P493‐Myc (B‐cell) tumor xenografts (126). Other agents targeting HIF‐1α or its downstream signaling pathways are currently under development. A selection of some of these inhibitors is illustrated in Figure 2. For a more in depth review of these agents, the reader is encouraged to read the following recent reviews 47, 76, 113.
Taken together, these studies underscore the promise of this approach of targeting HIF factors in GBM, as well as other solid tumors, a very valuable area of research that no doubt will attract increased attention in the coming years.
ACKNOWLEDGMENTS
I would like to thank Dr Daniel Brat for his kind contribution of an image of a thrombosed vessel shown in Figure 1C.
REFERENCES
- 1. Amberger‐Murphy V (2009) Hypoxia helps glioma to fight therapy. Curr Cancer Drug Targets 9:381–390. [DOI] [PubMed] [Google Scholar]
- 2. Argyriou AA, Giannopoulou E, Kalofonos HP (2009) Angiogenesis and anti‐angiogenic molecularly targeted therapies in malignant gliomas. Oncology 77:1–11. [DOI] [PubMed] [Google Scholar]
- 3. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760. [DOI] [PubMed] [Google Scholar]
- 4. Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W et al (2007) Cyclopamine‐mediated hedgehog pathway inhibition depletes stem‐like cancer cells in glioblastoma. Stem Cells 25:2524–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG (2010) Hypoxia increases the expression of stem‐cell markers and promotes clonogenicity in glioblastoma neurospheres. 1491–1502. [DOI] [PMC free article] [PubMed]
- 6. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ et al (2007) CD133(+) and CD133(−) glioblastoma‐derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 67:4010–4015. [DOI] [PubMed] [Google Scholar]
- 7. Birner P, Piribauer M, Fischer I, Gatterbauer B, Marosi C, Ambros PF et al (2003) Vascular patterns in glioblastoma influence clinical outcome and associate with variable expression of angiogenic proteins: evidence for distinct angiogenic subtypes. Brain Pathol 13:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blazek ER, Foutch JL, Maki G (2007) Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133‐ cells, and the CD133+ sector is enlarged by hypoxia. Int J Radiat Oncol Biol Phys 67:1–5. [DOI] [PubMed] [Google Scholar]
- 9. Bristow RG, Hill RP (2008) Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer 8:180–192. [DOI] [PubMed] [Google Scholar]
- 10. Chi JT, Wang Z, Nuyten DS, Rodriguez EH, Schaner ME, Salim A et al (2006) Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med 3:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiou SH, Kao CL, Chen YW, Chien CS, Hung SC, Lo JF et al (2008) Identification of CD133‐positive radioresistant cells in atypical teratoid/rhabdoid tumor. PLoS ONE 3:e2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clement V, Dutoit V, Marino D, Dietrich PY, Radovanovic I (2009) Limits of CD133 as a marker of glioma self‐renewing cells. Int J Cancer 125:244–248. [DOI] [PubMed] [Google Scholar]
- 13. Collingridge DR, Piepmeier JM, Rockwell S, Knisely JP (1999) Polarographic measurements of oxygen tension in human glioma and surrounding peritumoural brain tissue. Radiother Oncol 53:127–131. [DOI] [PubMed] [Google Scholar]
- 14. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al (2009) Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature 462:739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dean M, Fojo T, Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5:275–284. [DOI] [PubMed] [Google Scholar]
- 16. Denko NC (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8:705–713. [DOI] [PubMed] [Google Scholar]
- 17. Dewhirst MW, Kimura H, Rehmus SW, Braun RD, Papahadjopoulos D, Hong K, Secomb TW (1996) Microvascular studies on the origins of perfusion‐limited hypoxia. Br J Cancer 27:S247–S251. [PMC free article] [PubMed] [Google Scholar]
- 18. Escuin D, Kline ER, Giannakakou P (2005) Both microtubule‐stabilizing and microtubule‐destabilizing drugs inhibit hypoxia‐inducible factor‐1alpha accumulation and activity by disrupting microtubule function. Cancer Res 65:9021–9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang WT, Nelson PT et al (2004) Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res 10:8177–8184. [DOI] [PubMed] [Google Scholar]
- 20. Evans SM, Judy KD, Dunphy I, Jenkins WT, Nelson PT, Collins R et al (2004) Comparative measurements of hypoxia in human brain tumors using needle electrodes and EF5 binding. Cancer Res 64:1886–1892. [DOI] [PubMed] [Google Scholar]
- 21. Evans SM, Jenkins KW, Jenkins WT, Dilling T, Judy KD, Schrlau A et al (2008) Imaging and analytical methods as applied to the evaluation of vasculature and hypoxia in human brain tumors. Radiat Res 170:677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ezashi T, Das P, Roberts RM (2005) Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA 102:4783–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N et al (2010) NOTCH pathway blockade depletes CD133‐positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, Eberhart CG (2006) Notch pathway inhibition depletes stem‐like cells and blocks engraftment in embryonal brain tumors. Cancer Res 66:7445–7452. [DOI] [PubMed] [Google Scholar]
- 25. Fath DM, Kong X, Liang D, Lin Z, Chou A, Jiang Y et al (2006) Histone deacetylase inhibitors repress the transactivation potential of hypoxia‐inducible factors independently of direct acetylation of HIF‐alpha. J Biol Chem 281:13612–13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fischer I, Gagner JP, Law M, Newcomb EW, Zagzag D (2005) Angiogenesis in gliomas: biology and molecular pathophysiology. Brain Pathol 15:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fuchs E, Tumbar T, Guasch G (2004) Socializing with the neighbors: stem cells and their niche. Cell 116:769–778. [DOI] [PubMed] [Google Scholar]
- 28. Fuller GN (2008) The WHO Classification of Tumours of the Central Nervous System, 4th edition. Arch Pathol Lab Med 132:906. [DOI] [PubMed] [Google Scholar]
- 29. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S et al (2004) Isolation and characterization of tumorigenic, stem‐like neural precursors from human glioblastoma. Cancer Res 64:7011–7021. [DOI] [PubMed] [Google Scholar]
- 30. Gariboldi MB, Ravizza R, Monti E (2010) The IGFR1 inhibitor NVP‐AEW541 disrupts a pro‐survival and pro‐angiogenic IGF‐STAT3‐HIF1 pathway in human glioblastoma cells. Biochem Pharmacol 80:455–462. [DOI] [PubMed] [Google Scholar]
- 31. Gilbertson RJ, Rich JN (2007) Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7:733–736. [DOI] [PubMed] [Google Scholar]
- 32. Gillespie DL, Whang K, Ragel BT, Flynn JR, Kelly DA, Jensen RL (2007) Silencing of hypoxia inducible factor‐1alpha by RNA interference attenuates human glioma cell growth in vivo . Clin Cancer Res 13:2441–2448. [DOI] [PubMed] [Google Scholar]
- 33. Glas M, Rath BH, Simon M, Reinartz R, Schramme A, Trageser D et al (2010) Residual tumor cells are unique cellular targets in glioblastoma. Ann Neurol 68:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Griguer CE, Oliva CR, Gobin E, Marcorelles P, Benos DJ, Lancaster JR Jr, Gillespie GY (2008) CD133 is a marker of bioenergetic stress in human glioma. PLoS ONE 3:e3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gu JF, Shi YL (1994) [Effect of hypoxia on the growth and differentiation of NG108‐15 cells]. Sheng Li Xue Bao 46:244–248. [PubMed] [Google Scholar]
- 36. Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA (1998) Molecular characterization and chromosomal localization of a third alpha‐class hypoxia inducible factor subunit, HIF3alpha. Gene Expr 7:205–213. [PMC free article] [PubMed] [Google Scholar]
- 37. Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J et al (2005) Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 9:617–628. [DOI] [PubMed] [Google Scholar]
- 38. Haddad SF, Moore SA, Schelper RL, Goeken JA (1992) Vascular smooth muscle hyperplasia underlies the formation of glomeruloid vascular structures of glioblastoma multiforme. J Neuropathol Exp Neurol 51:488–492. [DOI] [PubMed] [Google Scholar]
- 39. Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN (2009) The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8:3274–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. [DOI] [PubMed] [Google Scholar]
- 41. Hemmati HD, Nakano I, Lazareff JA, Masterman‐Smith M, Geschwind DH, Bronner‐Fraser M, Kornblum HI (2003) Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA 100:15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hirota K, Fukuda R, Takabuchi S, Kizaka‐Kondoh S, Adachi T, Fukuda K, Semenza GL (2004) Induction of hypoxia‐inducible factor 1 activity by muscarinic acetylcholine receptor signaling. J Biol Chem 279:41521–41528. [DOI] [PubMed] [Google Scholar]
- 43. Huang LE, Gu J, Schau M, Bunn HF (1998) Regulation of hypoxia‐inducible factor 1alpha is mediated by an O2‐dependent degradation domain via the ubiquitin‐proteasome pathway. Proc Natl Acad Sci USA 95:7987–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang LE, Bindra RS, Glazer PM, Harris AL (2007) Hypoxia‐induced genetic instability—a calculated mechanism underlying tumor progression. J Mol Med 85:139–148. [DOI] [PubMed] [Google Scholar]
- 45. Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM (2002) Hsp90 regulates a von Hippel Lindau‐independent hypoxia‐inducible factor‐1 alpha‐degradative pathway. J Biol Chem 277:29936–29944. [DOI] [PubMed] [Google Scholar]
- 46. Jaksch M, Munera J, Bajpai R, Terskikh A, Oshima RG (2008) Cell cycle‐dependent variation of a CD133 epitope in human embryonic stem cell, colon cancer, and melanoma cell lines. Cancer Res 68:7882–7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jensen RL (2009) Brain tumor hypoxia: tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a therapeutic target. J Neurooncol 92:317–335. [DOI] [PubMed] [Google Scholar]
- 48. Joo KM, Kim SY, Jin X, Song SY, Kong DS, Lee JI et al (2008) Clinical and biological implications of CD133‐positive and CD133‐negative cells in glioblastomas. Lab Invest 88:808–815. [DOI] [PubMed] [Google Scholar]
- 49. Kallio PJ, Wilson WJ, O’Brien S, Makino Y, Poellinger L (1999) Regulation of the hypoxia‐inducible transcription factor 1alpha by the ubiquitin‐proteasome pathway. J Biol Chem 274:6519–6525. [DOI] [PubMed] [Google Scholar]
- 50. Kaluz S, Kaluzova M, Stanbridge EJ (2006) Proteasomal inhibition attenuates transcriptional activity of hypoxia‐inducible factor 1 (HIF‐1) via specific effect on the HIF‐1alpha C‐terminal activation domain. Mol Cell Biol 26:5895–5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG (2005) Hypoxia and the hypoxia‐inducible‐factor pathway in glioma growth and angiogenesis. Neuro-Oncol 7:134–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kelly JJ, Stechishin O, Chojnacki A, Lun X, Sun B, Senger DL et al (2009) Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells 27:1722–1733. [DOI] [PubMed] [Google Scholar]
- 53. Kondo T, Setoguchi T, Taga T (2004) Persistence of a small subpopulation of cancer stem‐like cells in the C6 glioma cell line. Proc Natl Acad Sci USA 101:781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kong D, Park EJ, Stephen AG, Calvani M, Cardellina JH, Monks A et al (2005) Echinomycin, a small‐molecule inhibitor of hypoxia‐inducible factor‐1 DNA‐binding activity. Cancer Res 65:9047–9055. [DOI] [PubMed] [Google Scholar]
- 55. Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J (2006) Histone deacetylase inhibitors induce VHL and ubiquitin‐independent proteasomal degradation of hypoxia‐inducible factor 1alpha. Mol Cell Biol 26:2019–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A et al (2004) Small molecule blockade of transcriptional coactivation of the hypoxia‐inducible factor pathway. Cancer Cell 6:33–43. [DOI] [PubMed] [Google Scholar]
- 57. Lantos PLVS, Kleihues P (1996) Tumours of the nervous system. In: Greenfield's Neuropathology. Graham DILP (ed.), pp. 583–879, Edward Arnold: London. [Google Scholar]
- 58. Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL (2001) HER2 (neu) signaling increases the rate of hypoxia‐inducible factor 1alpha (HIF‐1alpha) synthesis: novel mechanism for HIF‐1‐mediated vascular endothelial growth factor expression. Mol Cell Biol 21:3995–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee K, Kang JE, Park SK, Jin Y, Chung KS, Kim HM et al (2010) LW6, a novel HIF‐1 inhibitor, promotes proteasomal degradation of HIF‐1alpha via upregulation of VHL in a colon cancer cell line. Biochem Pharmacol 80:982–989. [DOI] [PubMed] [Google Scholar]
- 60. Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL (2009) Anthracycline chemotherapy inhibits HIF‐1 transcriptional activity and tumor‐induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci USA 106:2353–2358. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61. Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL (2009) Acriflavine inhibits HIF‐1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA 106:17910–17915. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62. Lehtonen HJ, Makinen MJ, Kiuru M, Laiho P, Herva R, van Minderhout I et al (2007) Increased HIF1 alpha in SDH and FH deficient tumors does not cause microsatellite instability. Int J Cancer 121:1386–1389. [DOI] [PubMed] [Google Scholar]
- 63. Li J, Zhang X, Sejas DP, Bagby GC, Pang Q (2004) Hypoxia‐induced nucleophosmin protects cell death through inhibition of p53. J Biol Chem 279:41275–41279. [DOI] [PubMed] [Google Scholar]
- 64. Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF et al (2008) Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 100:672–679. [DOI] [PubMed] [Google Scholar]
- 65. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S et al (2009) Hypoxia‐inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15:501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL et al (2004) The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol 63:499–509. [DOI] [PubMed] [Google Scholar]
- 67. Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL (2007) RACK1 competes with HSP90 for binding to HIF‐1alpha and is required for O(2)‐independent and HSP90 inhibitor‐induced degradation of HIF‐1alpha. Mol Cell 25:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Louis SA, Rietze RL, Deleyrolle L, Wagey RE, Thomas TE, Eaves AC, Reynolds BA (2008) Enumeration of neural stem and progenitor cells in the neural colony‐forming cell assay. Stem Cells 26:988–996. [DOI] [PubMed] [Google Scholar]
- 69. Lu H, Li Y, Shu M, Tang J, Huang Y, Zhou Y et al (2009) Hypoxia‐inducible factor‐1alpha blocks differentiation of malignant gliomas. FEBS J 276:7291–7304. [DOI] [PubMed] [Google Scholar]
- 70. Luwor R, Lu Y, Li X, Mendelsohn J, Fan Z (2005) The anti‐epidermal growth factor receptor monoclonal antibody cetuximab/C225 reduces hypoxia‐inducible factor‐1 alpha, leading to transcriptional inhibition of vascular endothelial growth factor expression. 1192‐b. [DOI] [PubMed]
- 71. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME et al (1999) The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature 399:271–275. [DOI] [PubMed] [Google Scholar]
- 72. Maynard MA, Ohh M (2007) The role of hypoxia‐inducible factors in cancer. Cell Mol Life Sci 64:2170–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mazure NM, Chen EY, Yeh P, Laderoute KR, Giaccia AJ (1996) Oncogenic transformation and hypoxia synergistically act to modulate vascular endothelial growth factor expression. Cancer Res 56:3436–3440. [PubMed] [Google Scholar]
- 74. McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, Tofilon PJ (2009) Physiologic oxygen concentration enhances the stem‐like properties of CD133+ human glioblastoma cells in vitro . Mol Cancer Res 7:489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McCord AM, Jamal M, Williams ES, Camphausen K, Tofilon PJ (2009) CD133+ glioblastoma stem‐like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin Cancer Res 15:5145–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Melillo G (2007) Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev 26:341–352. [DOI] [PubMed] [Google Scholar]
- 77. Mendez O, Zavadil J, Esencay M, Lukyanov Y, Santovasi D, Wang SC et al (2010) Knock down of HIF‐1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol Cancer 9:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Miki A, Fujimoto E, Ohsaki T, Mizoguti H (1988) Effects of oxygen concentration on embryonic development in rats: a light and electron microscopic study using whole‐embryo culture techniques. Anat Embryol (Berl) 178:337–343. [DOI] [PubMed] [Google Scholar]
- 79. Morrison SJ, Csete M, Groves AK, Melega W, Wold B, Anderson DJ (2000) Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J Neurosci 20:7370–7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Morriss GM, New DA (1979) Effect of oxygen concentration on morphogenesis of cranial neural folds and neural crest in cultured rat embryos. J Embryol Exp Morphol 54:17–35. [PubMed] [Google Scholar]
- 81. Nagashima T, Hoshino T, Cho KG (1987) Proliferative potential of vascular components in human glioblastoma multiforme. Acta Neuropathol (Berl) 73:301–305. [DOI] [PubMed] [Google Scholar]
- 82. Nishide K, Nakatani Y, Kiyonari H, Kondo T (2009) Glioblastoma formation from cell population depleted of Prominin1‐expressing cells. PLoS ONE 4:e6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA et al (2008) Identification of A2B5+CD133‐ tumor‐initiating cells in adult human gliomas. Neurosurgery 62:505–514. discussion 14–5. [DOI] [PubMed] [Google Scholar]
- 84. Okita K, Ichisaka T, Yamanaka S (2007) Generation of germline‐competent induced pluripotent stem cells. Nature 448:313–317. [DOI] [PubMed] [Google Scholar]
- 85. Oliver TG, Wechsler‐Reya RJ (2004) Getting at the root and stem of brain tumors. Neuron 42:885–888. [DOI] [PubMed] [Google Scholar]
- 86. Panchision DM (2009) The role of oxygen in regulating neural stem cells in development and disease. J Cell Physiol 220:562–568. [DOI] [PubMed] [Google Scholar]
- 87. Park SK, Dadak AM, Haase VH, Fontana L, Giaccia AJ, Johnson RS (2003) Hypoxia‐induced gene expression occurs solely through the action of hypoxia‐inducible factor 1alpha (HIF‐1alpha): role of cytoplasmic trapping of HIF‐2alpha. Mol Cell Biol 23:4959–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Piccirillo SG, Combi R, Cajola L, Patrizi A, Redaelli S, Bentivegna A et al (2009) Distinct pools of cancer stem‐like cells coexist within human glioblastomas and display different tumorigenicity and independent genomic evolution. Oncogene 28:1807–1811. [DOI] [PubMed] [Google Scholar]
- 89. Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G et al (2006) Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour‐initiating cells. Nature 444:761–765. [DOI] [PubMed] [Google Scholar]
- 90. Pietras A, Johnsson AS, Pahlman S (2010) The HIF‐2alpha‐driven pseudo‐hypoxic phenotype in tumor aggressiveness, differentiation, and vascularization. Curr Top Microbiol Immunol 345:1–20. [DOI] [PubMed] [Google Scholar]
- 91. Pires IM, Bencokova Z, Milani M, Folkes LK, Li JL, Stratford MR et al (2010) Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res 70:925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pistollato F, Abbadi S, Rampazzo E, Persano L, Della Puppa A, Frasson C et al (2010) Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 28:851–862. [DOI] [PubMed] [Google Scholar]
- 93. Pistollato F, Chen HL, Rood BR, Zhang HZ, D’Avella D, Denaro L et al (2009) Hypoxia and HIF1alpha repress the differentiative effects of BMPs in high‐grade glioma. Stem Cells 27:7–17. [DOI] [PubMed] [Google Scholar]
- 94. Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A (2006) EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia‐inducible factor (HIF)‐1‐independent and HIF‐1‐dependent mechanisms. Cancer Res 66:3197–3204. [DOI] [PubMed] [Google Scholar]
- 95. Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, Melillo G (2002) Identification of small molecule inhibitors of hypoxia‐inducible factor 1 transcriptional activation pathway. Cancer Res 62:4316–4324. [PubMed] [Google Scholar]
- 96. Rasper M, Schafer A, Piontek G, Teufel J, Brockhoff G, Ringel F et al (2010) Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro-Oncol 12:1024–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rong Y, Durden DL, Van Meir EG, Brat DJ (2006) “Pseudopalisading” necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol 65:529–539. [DOI] [PubMed] [Google Scholar]
- 98. Roy S, Khanna S, Bickerstaff AA, Subramanian SV, Atalay M, Bierl M et al (2003) Oxygen sensing by primary cardiac fibroblasts: a key role of p21(Waf1/Cip1/Sdi1). Circ Res 92:264–271. [DOI] [PubMed] [Google Scholar]
- 99. Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U (2008) Notch signaling mediates hypoxia‐induced tumor cell migration and invasion. Proc Natl Acad Sci USA 105:6392–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sathornsumetee S, Cao Y, Marcello JE, Herndon JE, 2nd , McLendon RE, Desjardins A et al (2008) Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol 26:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K et al (2010) A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 133:983–995. [DOI] [PubMed] [Google Scholar]
- 102. Semenza GL (2010) Defining the role of hypoxia‐inducible factor 1 in cancer biology and therapeutics. Oncogene 29:625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Semenza GL (2003) Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 3:721–732. [DOI] [PubMed] [Google Scholar]
- 104. Semenza GL (2009) Regulation of oxygen homeostasis by hypoxia‐inducible factor 1. Physiology (Bethesda) 24:97–106. [DOI] [PubMed] [Google Scholar]
- 105. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401. [DOI] [PubMed] [Google Scholar]
- 106. Soeda A, Park M, Lee D, Mintz A, Androutsellis‐Theotokis A, McKay RD et al (2009) Hypoxia promotes expansion of the CD133‐positive glioma stem cells through activation of HIF‐1alpha. Oncogene 28:3949–3959. [DOI] [PubMed] [Google Scholar]
- 107. Stockhausen MT, Kristoffersen K, Poulsen HS (2010) The functional role of Notch signaling in human gliomas. Neuro-Oncol 12:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Stoeltzing O, Liu W, Reinmuth N, Fan F, Parikh AA, Bucana CD et al (2003) Regulation of hypoxia‐inducible factor‐1alpha, vascular endothelial growth factor, and angiogenesis by an insulin‐like growth factor‐I receptor autocrine loop in human pancreatic cancer. Am J Pathol 163:1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay R (2000) Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci 20:7377–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. [DOI] [PubMed] [Google Scholar]
- 111. Sun Y, Kong W, Falk A, Hu J, Zhou L, Pollard S, Smith A (2009) CD133 (Prominin) negative human neural stem cells are clonogenic and tripotent. PLoS ONE 4:e5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tan C, de Noronha RG, Roecker AJ, Pyrzynska B, Khwaja F, Zhang Z et al (2005) Identification of a novel small‐molecule inhibitor of the hypoxia‐inducible factor 1 pathway. Cancer Res 65:605–612. [PubMed] [Google Scholar]
- 113. Tennant DA, Duran RV, Gottlieb E (2010) Targeting metabolic transformation for cancer therapy. Nat Rev Cancer 10:267–277. [DOI] [PubMed] [Google Scholar]
- 114. Thomas R, Kim MH (2009) A HIF‐1alpha‐dependent autocrine feedback loop promotes survival of serum‐deprived prostate cancer cells. Prostate 69:263–275. [DOI] [PubMed] [Google Scholar]
- 115. Tomasevic G, Shamloo M, Israeli D, Wieloch T (1999) Activation of p53 and its target genes p21(WAF1/Cip1) and PAG608/Wig‐1 in ischemic preconditioning. Brain Res Mol Brain Res 70:304–313. [DOI] [PubMed] [Google Scholar]
- 116. Tuettenberg J, Friedel C, Vajkoczy P (2006) Angiogenesis in malignant glioma—a target for antitumor therapy? Crit Rev Oncol Hematol 59:181–193. [DOI] [PubMed] [Google Scholar]
- 117. Vaupel P, Kallinowski F, Okunieff P (1989) Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 49:6449–6465. [PubMed] [Google Scholar]
- 118. Vaupel P, Mayer A (2007) Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 26:225–239. [DOI] [PubMed] [Google Scholar]
- 119. Wang GL, Semenza GL (1993) Characterization of hypoxia‐inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem 268:21513–21518. [PubMed] [Google Scholar]
- 120. Wang J, Sakariassen PO, Tsinkalovsky O, Immervoll H, Boe SO, Svendsen A et al (2008) CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer 122:761–768. [DOI] [PubMed] [Google Scholar]
- 121. Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR et al (2010) Notch promotes radioresistance of glioma stem cells. Stem Cells 28:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Weljie AM, Jirik FR (2010) Hypoxia‐induced metabolic shifts in cancer cells: moving beyond the Warburg effect. Int J Biochem Cell Biol; doi:10.1016/j.biocel.2010.08.009 [E‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 123. Wesseling P, Schlingemann RO, Rietveld FJ, Link M, Burger PC, Ruiter DJ (1995) Early and extensive contribution of pericytes/vascular smooth muscle cells to microvascular proliferation in glioblastoma multiforme: an immuno‐light and immuno‐electron microscopic study. J Neuropathol Exp Neurol 54:304–310. [DOI] [PubMed] [Google Scholar]
- 124. Wong ET, Brem S (2008) Antiangiogenesis treatment for glioblastoma multiforme: challenges and opportunities. J Natl Compr Canc Netw 6:515–522. [DOI] [PubMed] [Google Scholar]
- 125. Zhang CP, Zhu LL, Zhao T, Zhao H, Huang X, Ma X et al (2006) Characteristics of neural stem cells expanded in lowered oxygen and the potential role of hypoxia‐inducible factor‐1Alpha. Neuro-Signals 15:259–265. [DOI] [PubMed] [Google Scholar]
- 126. Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR et al (2008) Digoxin and other cardiac glycosides inhibit HIF‐1alpha synthesis and block tumor growth. Proc Natl Acad Sci USA 105:19579–19586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P et al (2009) Glioma‐derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF‐1alpha. Science 324:261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zheng X, Jiang F, Katakowski M, Kalkanis SN, Hong X, Zhang X et al (2007) Inhibition of ADAM17 reduces hypoxia‐induced brain tumor cell invasiveness. Cancer Sci 98:674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D et al (1999) Overexpression of hypoxia‐inducible factor 1alpha in common human cancers and their metastases. Cancer Res 59:5830–5835. [PubMed] [Google Scholar]
- 130. Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM et al (2000) Modulation of hypoxia‐inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3‐kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 60:1541–1545. [PubMed] [Google Scholar]
- 131. Zhou J, Zhang H, Gu P, Bai J, Margolick JB, Zhang Y (2008) NF‐kappaB pathway inhibitors preferentially inhibit breast cancer stem‐like cells. Breast Cancer Res Treat 111:419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Zou GM, Maitra A (2008) Small‐molecule inhibitor of the AP endonuclease 1/REF‐1 E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Ther 7:2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zundel W, Schindler C, Haas‐Kogan D, Koong A, Kaper F, Chen E et al (2000) Loss of PTEN facilitates HIF‐1‐mediated gene expression. Genes Dev 14:391–396. [PMC free article] [PubMed] [Google Scholar]