

Abstract
The vacuolation, neuronal loss and gliosis that characterize human prion disease pathology are accompanied by the accumulation of an aggregated, insoluble and protease‐resistant form (termed PrPSc) of the host‐encoded normal cellular prion protein (PrPC). In variant Creutzfeldt‐Jakob disease the frontal cortex and cerebellum exhibit intense vacuolation and the accumulation of PrPSc in the form of amyloid plaques and plaque‐like structures. In contrast the posterior thalamus is characterized by intense gliosis and neuronal loss, but PrPSc plaques are rare and vacuolation is patchy. We have used sucrose density gradient centrifugation coupled with conformation dependent immunoassay to examine the biochemical properties of the PrPSc that accumulates in these different brain regions. The results show a greater degree of PrPSc polydisperal in thalamus compared with frontal cortex or cerebellum, including a subpopulation PrPSc molecules in the thalamus that have sedimentation properties resembling those of PrPC. Much effort has focused on identifying aspects of PrPSc biochemistry that distinguish between different forms of human prion disease and contribute to differential diagnosis. Here we show that PrPSc sedimentation properties, which can depend on aggregation state, correlate with, and may underlie the distinct neurodegenerative processes occurring in different regions of the variant Creutzfeldt‐Jakob disease brain.
Keywords: aggregation state, biochemistry, polydispersal, prion protein, thalamus, variant Creutzfeldt‐Jakob disease
INTRODUCTION
The human transmissible spongiform encephalopathies or human prion diseases are group of fatal neurodegenerative conditions that currently include Creutzfeldt‐Jakob disease (CJD), Gerstmann‐Sträussler‐Scheinker disease (GSS), kuru, fatal familial insomnia (FFI) and protease sensitive prionopathy (PSPr). What unifies these diseases is a clinical phenotype, typically involving dementia and movement disorders and a neuropathology that involves neuronal loss, gliosis and spongiform change. Certain of these diseases, such as GSS, have a genetic origin; others, such as kuru, are acquired, whereas CJD can occur in genetic, acquired or sporadic forms. A more fundamental commonality between these diseases is the proposal that they are all caused by the accumulation of PrPSc, a self‐perpetuating conformationally altered and aggregated form of the host‐encoded prion protein, PrPC (39). This hypothesis provides a link between prion diseases and other neurological conditions in which abnormally folded or aggregated protein spreads and accumulates within the brain, but a key difference remains that some prion diseases are quite clearly acquired and most are also fairly readily transmissible 1, 50.
Variant Creutzfeldt‐Jakob disease (vCJD) results from infection with the bovine spongiform encephalopathy agent (57) and is transmissible by blood transfusion (24). vCJD has a highly stereotyped neuropathology, by which it can be readily distinguished from other prion diseases, including multiple florid and cluster plaques in the cerebral and cerebellar cortex (Cb), severe spongiform change in the caudate nucleus and putamen, and marked astrocytosis and neuronal loss in the posterior thalamus (Th) 15, 16. The thalamic pathology corresponds to the “pulvinar sign” evident on magnetic resonance imaging (MRI) (60).
Considerable progress has been made in the identification of biochemical aspects of PrPSc that might account for the differing disease characteristics and these have been exploited diagnostically. This has largely focused on the size and glycosylation state of the N‐, and sometimes N‐ and C‐terminally truncated prion protein that survives digestion with proteinase K (PK), so called PrPres 14, 35. vCJD, for example, is characterized by the PrPres with a mobility of ∼19 kDa and dominated by the diglycosylated isoform, termed type 2B [see for example reference (12)]. However, this approach is likely to give an incomplete description of the populations of PrP molecules present for the following reasons. Firstly, PrPSc may naturally exist as a mixture of abnormal conformers 38, 59. Secondly, there is evidence for the existence of PrPSc that is sensitive to proteolysis under the conditions generally used (46) and thirdly, the Western blot method does not directly address the property of aggregation state.
The relationship between misfolded protein aggregate size and biological activity in neurodegenerative diseases is a subject of particular current interest and speculation. Relatively small soluble oligomeric forms have been implicated as the molecular species of Aβ associated with neurotoxicity in Alzheimer's disease 11, 52 and as the molecular species of PrPSc most closely associated with infectivity in animal models of prion disease (48). Surprisingly little attention has been paid to this aspect of human prion diseases. Sucrose density gradient centrifugation has been used to compare PrP sedimentation properties in prion disease and non‐prion disease brain (58), in sporadic CJD with long and short durations (4) and in PSPr (7). However, there has been no systematic study comparing sedimentation profiles in cohorts representing the full‐range of human prion diseases. Nor crucially has there been any indication of whether there is regional variation within the brain, perhaps associated with differing cellular populations or pathological processes.
We address the last of these issues here by comparing three different regions of the vCJD brain using sucrose density gradient centrifugation to effect a separation of the population of PrP molecules present, followed by conformation‐dependent immunoassay (CDI) to operationally distinguish between the normal (PrPC) and abnormal (PrPSc) forms.
MATERIALS AND METHODS
Human brain materials
Brain tissue from five control (non‐CJD) cases and seven vCJD cases were analyzed in this study. Brain tissue from each case had been examined both histologically and biochemically, and a definite diagnosis has been made by established criteria (15). Cases were selected based on the availability of autopsy‐collected frozen half‐brain with consent for research use. The use of brain tissue in this study was covered by LREC 2000/4/157 (Prof. J.W. Ironside). All vCJD cases were of the methionine homozygous (MM) genotype at PRNP codon 129. The five control cases were referred to the National CJD Surveillance Unit suspected of being CJD, but received an alternative neuropathological diagnosis. Among the five neurological control cases, three cases were MM at codon 129 (with diagnoses of vascular dementia, motor neuron disease and B‐cell lymphoma) and the remaining two cases were methionine/valine (MV) (with a diagnosis of vascular dementia) and valine/valine (VV) (with a diagnosis of dementia with Lewy bodies). The tissue analyzed was gray matter‐enriched frontal cortex (FC), and in the vCJD cases, FC was compared with Cb and Th taken from frozen half‐brain specimens.
Brain histology and immunohistochemistry
The neuropathology of selected cases was reviewed in terms of the presence of plaques and spongiform change in hematoxylin and eosin stained sections, deposition of abnormal prion protein by immunohistochemistry using pretreatments and the monoclonal antibody KG9 (TSE Resource Centre, Edinburgh, UK), and immunostaining for glial fibrillary acidic protein as a maker of reactive gliosis, as previously described 15, 16.
Preparation of brain tissue for biochemical assays
Brain homogenates were prepared in nine volumes (w/v) of phosphate‐buffered saline (PBS), pH 7.4 by two cycles of homogenization in the FastPrep instrument (Qbiogene®, Cambridge, UK). Each cycle was run for 45 s at the speed of 6.5 m/s with the interval of 10 minutes between the runs. The homogenized brain samples were stored at −80°C until use.
Velocity sedimentation in sucrose step gradients
Fractionation in sucrose step gradients was performed as described previously (53) with minor modifications. The 10% brain homogenate in PBS was mixed with equal volume of lysis buffer (4% n‐octyl‐β‐D‐glucopyranoside in PBS) and then incubated for 30 minutes in ice. After clarifying the sample for 1 minute at 3800 × g to remove insoluble material, the supernatant was incubated for 30 minutes on ice in the presence of 1% sarcosyl, followed by careful loading on top of a 10%–60% sucrose step gradient. The gradient was prepared in a 5 mL ultracentrifuge tube (Beckman Coulter, High Wycombe, UK) from 745 µL of each of the following sucrose concentration: 10%, 15%, 20%, 25%, 30%, 60% in Tris, NaCl, Sarcosyl (TNS) buffer (20 mM Tris‐HCl, pH 7.5, 150 mM NaCl, 1% sarcosyl). The samples were centrifuged for 1 h at 4°C at 50 000 rpm (g average = 200 000 × g) in a MLS‐50 rotor in the Optima MAX ultracentrifuge (Beckman Coulter). Eleven fractions of 450 µL were then collected from the top of the tube and were stored at −80°C. Samples were prepared and separated in sucrose gradients in pairs. Our practice was to prepare dissimilar samples in parallel (eg, FC or Cb samples with a Th sample) thus minimizing the potential for any day‐to‐day variation in sucrose gradient preparation and centrifugation conditions from influencing the separation obtained and invalidating comparisons.
CDI
The CDI method used in this study was based on an Enzyme‐Linked Immunosorbent Assay (ELISA)‐formatted, dissociation‐enhanced lanthanide fluoroimmunoassay 3, 45. For CDI analysis, each fraction from the sucrose gradient was prepared in two parts. On some occasions, samples were assayed by CDI after digestion with 2.5 µg/mL PK for 1 h at 37°C. One part was mixed with same volume of PBS containing Complete EDTA‐free® (Roche, Basle, Switzerland) protease inhibitors (native sample, N) and the other part was mixed with the same volume of 4 M GdnHCl and incubated for 6 minutes at 81°C (denatured sample, D). Both D and N samples were adjusted using distilled water containing EDTA‐free protease inhibitors to a final concentration of GdnHCl of 0.35 M in 435 µL final volume. A 96‐well black polystyrene plate (Fisher, Loughborough, UK) was coated with 2 µg/well anti‐PrP antibody MAR‐1 (CSL Behring, Marburg, Germany) overnight at room temperature. After saturating the plate with 0.5% (w/v) bovine serum albumin and 6% Sorbitol (w/v) in wash buffer (PerkinElmer, Cambridge, UK), prepared samples were loaded in duplicate into the plate (200 µL/well). After incubating the plate for 2 h, Europium‐labeled PrP antibody 3F4 diluted in assay buffer (PerkinElmer) at 50 ng/mL was added to each well, followed by incubation for 2 h. The plate was developed after six washes by 5 minutes incubation in enhancement solution (PerkinElmer) at room temperature. The time‐resolved fluorescence (TRF) signal, measured as cps (counts per seconds), was counted using a Victor 2 fluorometer (PerkinElmer). The CDI D/N ratios were obtained by dividing TRF counts of denatured samples (D) by the counts of the corresponding native samples (N; No GdnHCl).
RESULTS
Neuropathology of vCJD
The neuropathology of vCJD is highly characteristic involving spongiform change, amyloid plaque formation (visible in hematoxylin and eosin stained section) and the presence of cluster plaques (immunostained for PrP) in both FC and in Cb (Figure 1A–D). The characteristic neuropathology of the Th differs, in that few plaques are present, but neuronal loss is extensive and astrogliosis is intense (Figure 1E–H). The morphological appearance of disease‐associated PrP in cortical regions and Th differ qualitatively. In order to investigate the possible biochemical basis of these differences, we chose to examine the sedimentation properties of PrP in sucrose gradients and to use CDI to differentiate between PrPC and PrPSc in these separations.
Figure 1.

Characteristic neuropathology of variant Creutzfeldt‐Jakob disease (vCJD). Frontal cortex (A,B,H), cerebellum (C,D) and posterior thalamus (pulvinar) (E,F,G) from one case of vCJD are shown stained with hematoxylin and eosin (A,C,E), immunostained for the prion protein using the monoclonal antibody KG9 (B,D,F) or immunostained for glial acidic fibrillary protein (G,H).
Sedimentation profile of PrPC
As the epitope of the mAb 3F4, employed as a detector in CDI, is not accessible in PrPSc and becomes exposed only after denaturation, the CDI fluorescence signals obtained from native samples are taken to represent PrPC 37, 45. Accordingly, the sedimentation profiles of PrPC after ultracentrifugation in the sucrose step gradient were investigated by CDI using native aliquots of the fractionated samples. When fractions from FC of five neurological (non‐CJD) disease cases were examined, PrP fluorescence signals were mostly detected in upper fractions of the gradient (Figure 2A). When the 11 fractions from the gradient were categorized into three groups (top fractions: fractions 1–3; intermediate fractions: fractions 4–8; bottom fractions: fractions 9–11), the amount of PrP recovered in the top fractions was around 90% of total PrPC (Figure 2B). Very much lower CDI signals were found in the intermediate and bottom fractions of the gradient.
Figure 2.
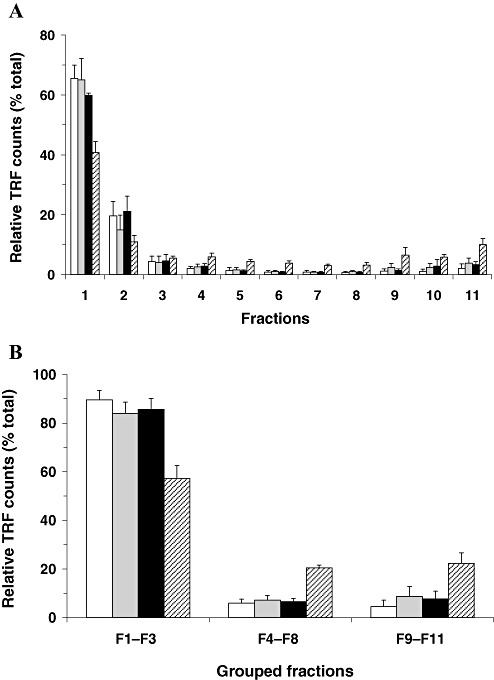
Sedimentation profiles of host‐encoded prion protein (PrPC) derived from different regions of variant Creutzfeldt‐Jakob disease (vCJD) brains after ultracentrifugation in the sucrose step gradient analyzed by conformation‐dependent immunoassay (CDI) using native conditions. Brain lysates were fractionated in the sucrose step gradients by ultracentrifugation and 11 fractions from top to bottom were collected. The amount of PrPC in each fraction was measured by CDI using native samples. A. The amount of PrPC in each fraction was expressed as a percentage (%) to the sum of 11 fractions. B. Eleven fractions were categorized into three groups (top fractions: fractions 1–3; intermediate fractions: fractions 4–8; bottom fractions: fractions 9–11) and the distribution of PrP between groups was expressed as a percentage (%) to the sum of the three groups. Data shown represent the average± SD obtained from five cases of FC of control (white bars) and seven cases of vCJD FC (gray bars). In vCJD Cb (black bars) and vCJD Th (striped bars), data shown represent the average± SD obtained from three cases. The result in every individual was an average for duplicate wells. Abbreviations: FC = frontal cortex; Cb = cerebellum; Th = posterior thalamus; TRF = time‐resolved fluorescence; SD = standard deviation.
Next, the sedimentation profiles of PrPC in vCJD brains were investigated by CDI in the native state. When brain lysates from FC or Cb of vCJD cases were centrifuged in the sucrose gradient, the sedimentation profiles of PrPC were not distinguishable from those of non‐CJD controls (Figure 2). Although PrPC from the posterior thalamic samples of vCJD cases was also predominantly recovered in the light upper fractions of the sucrose gradient, a higher proportion of PrPC appeared to migrate to the heavier lower fractions when compared with the two other regions of vCJD (Figure 2A). Around 90% of PrPC from FC and the Cb of vCJD cases was recovered in the top fractions, but only around 60% of PrPC from the Th was present in the top fractions, with ∼20% of PrPC being recovered in the intermediate and bottom fractions, respectively (Figure 2B).
Sedimentation profile of total PrP
In order to examine the distribution of total PrP in the sucrose gradient after velocity sedimentation, aliquots of fractionated samples were assayed by CDI after denaturation. In neurological controls, similar to the results obtained from native aliquots, more than 90% of total PrP was identified in the upper factions of the sucrose gradient (fractions 1–3) and only trace CDI signals were also found in the intermediate and bottom fractions (3, 4). Thus, in non‐CJD brains, the sedimentation profiles of PrP measured in the two different folding states were very similar to each other. In contrast, samples from FC or Cb of vCJD cases showed clearly distinct results in CDI measurement between native and denatured states. While fluorescence signals obtained from native samples were mostly present in the top three fractions, CDI signals identified in those top fractions after denaturation were reduced to less than 50% of the total recovered in all fractions (Figure 3). The remainder was mostly detected in the bottom three fractions in which only a trace CDI signal was detected in the native state (3, 4). Given that the recognition site of mAb 3F4 employed as a detector in the CDI is hidden in native PrPSc, but becomes accessible after denaturation, the newly detected CDI signals after denaturation (in bottom fractions) can be attributable to PrPSc present in those bottom fractions. When the fractionated samples from the Th of vCJD cases were analyzed in the same manner, a relatively higher proportion of the PrP signal was detected in the intermediate fractions of the gradient (3, 4). Although the levels of PrP molecules detected in the intermediate fractions (fractions 4–8) from FC or Cb were less than 10%, they were greater than 20% in the intermediate fractions from the Th of vCJD cases (Figure 3B).
Figure 3.
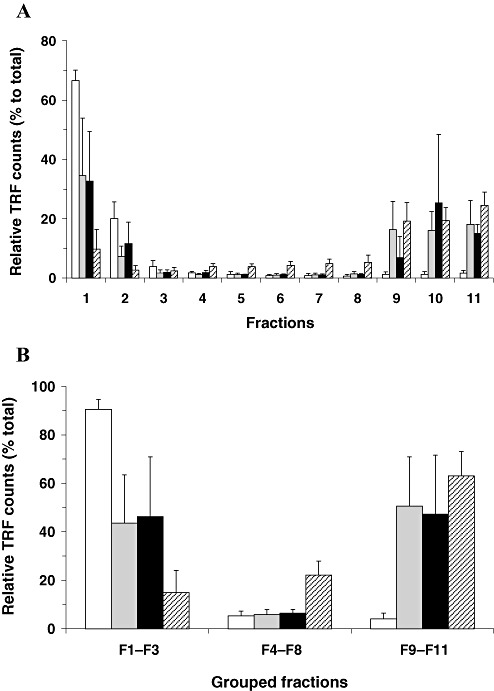
Sedimentation profiles of total prion protein (PrP) derived from different regions of variant Creutzfeldt‐Jakob disease (vCJD) brains after ultracentrifugation in the sucrose step gradient analyzed by conformation‐dependent immunoassay (CDI) using denaturing conditions. Brain lysates were fractionated in the sucrose step gradients by ultracentrifugation and 11 fractions from top to bottom were collected. The amount of total PrP present in each fraction was determined by CDI using denatured samples. A. The amount of total PrP present in each fraction was expressed as a percentage (%) to the sum of 11 fractions. B. Eleven fractions were categorized into three groups (top fractions: fractions 1–3; intermediate fractions: fractions 4–8; bottom fractions: fractions 9–11) and the distribution of PrP between groups was expressed as a percentage (%) to the sum of the three groups. Data shown represent the average± SD obtained from five cases of FC of control (white bars) and seven cases of vCJD FC (gray bars). In vCJD Cb (black bars) and vCJD Th (striped bars), data shown represent the average± SD obtained from three cases. The result in every individual was an average for duplicate wells. Abbreviations: FC = frontal cortex; Cb = cerebellum; Th = posterior thalamus; TRF = time‐resolved fluorescence; SD = standard deviation.
Figure 4.
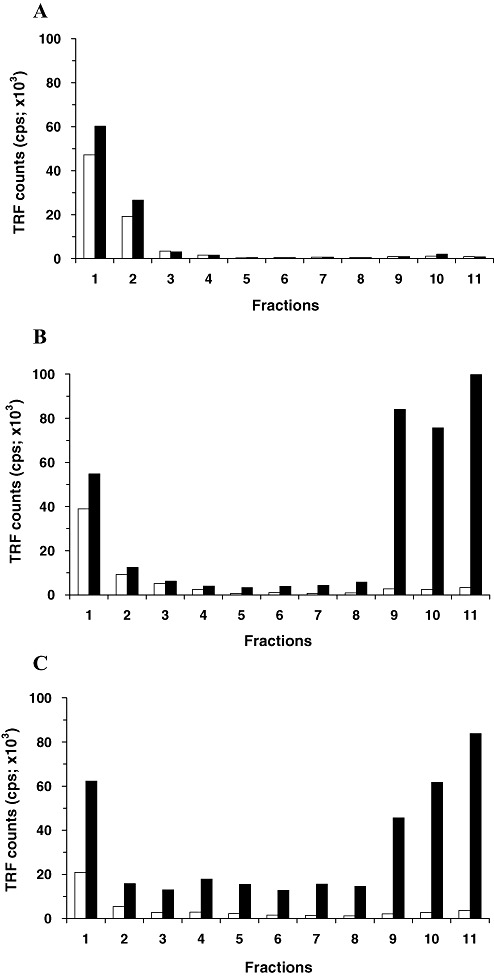
Comparison of conformation‐dependent immunoassay (CDI) results between native and denatured states. CDI fluorescence signals from fractions of FC tissue of a control case (A), from fractions of FC tissue of a variant Creutzfeldt‐Jakob disease (vCJD) case (B) and from fractions of Th tissue of another vCJD case (C) were directly compared between the native (white bars) and denatured (black bars) folding states. Data shown represent the average for duplicate wells. Abbreviations: FC = frontal cortex; Th = posterior thalamus; TRF = time‐resolved fluorescence; CPS = counts per seconds.
Sedimentation profile of PrPSc
The ratio of mAb 3F4 binding to denatured/native PrP (D/N ratio) has been used as an indicator for the presence of PrPSc in scrapie‐affected hamster brains and in human CJD brains 3, 45. Based on these previous studies, the sedimentation profile of PrPSc in the sucrose gradient was inferred from CDI D/N ratios, which were generated by dividing the TRF counts of the denatured aliquots by those of corresponding native aliquots. D/N ratios for the fractions derived from the non‐CJD control cases were usually lower than 1.5 (Figure 5), except for three fractions each from three different cases. D/N ratios for those three fractions were in the range of 1.5–1.8. It is noteworthy that D/N ratios for PrPC from normal hamster brains were lower or equal to 1.8 and the ratios higher than 1.8 were considered to indicate the presence of PrPSc in hamster brains (45).
Figure 5.
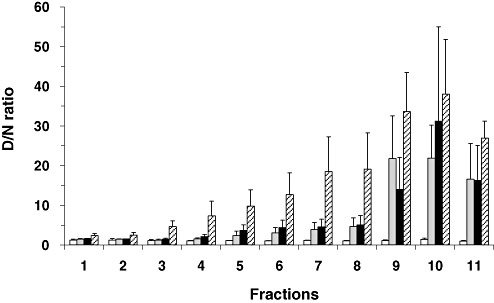
Conformation‐dependent immunoassay (CDI) denatured/native (D/N) ratios for fractions derived from different regions of vCJD brains. The CDI D/N ratios for the fractions obtained by ultracentrifugation in the sucrose gradient were calculated by dividing TRF counts of denatured samples by the counts of the corresponding native samples. Data shown represent the average± SD obtained from five cases of FC of control (white bars) or from seven cases of vCJD FC (gray bars). In variant Creutzfeldt‐Jakob disease (vCJD) Cb (black bars) and vCJD Th (striped bars), data shown represent the average ± SD obtained from three cases. Abbreviations: FC = frontal cortex; Cb = cerebellum; Th = posterior thalamus; TRF = time‐resolved fluorescence; SD = standard deviation.
To investigate the sedimentation profiles of PrPSc from FC and Cb of vCJD in the sucrose gradient, D/N ratios for the fractions from the two regions were generated and compared with those of non‐CJD controls. The D/N ratios for the upper three fractions from FC or Cb of vCJD were in the range of 1.0–1.7, which were indistinguishable from their non‐CJD counterparts (Figure 5). While the D/N ratios in the intermediate fractions (fractions 4–8) were slightly higher than the non‐CJD counterparts, the ratios in the bottom three fractions increased dramatically and were noticeably higher than those of controls (Figure 5). In comparison, the pattern of D/N ratios for the fractions from the Th of vCJD cases was distinct from those of FC or Cb. Although the D/N ratios in the bottom three fractions were higher than intermediate fractions (fractions 4–8), the difference between the two groups appeared less distinct in the Th when compared with the FC and Cb (Figure 5). Moreover, the D/N ratios in the top fractions of vCJD Th were slightly, but measurably higher than those of the corresponding fractions from the control brains or from the two other regions of vCJD brains (Figure 5). In two of the three vCJD cases in which thalamic region was analyzed, the D/N ratios were higher than 2 in fraction 1 and reached to 4.6 and 6.1 in fraction 3, respectively. In the remaining case, the ratio was 1.8 in fractions 1 and 2 but increased to 3.1 in fraction 3. While a majority of PrPSc in the FC and Cb of vCJD cases migrated to the bottom fractions of the sucrose gradient after ultracentrifugation, PrPSc in the Th appeared to be distributed more evenly throughout the sucrose gradient (even including the top fraction of the gradient).
Sedimentation profile of PrPSc: CDI analysis after mild proteolysis
As shown in Figure 2, PrPC from both neurological control and vCJD brains was mostly (or largely in the posterior Th) recovered in the top few fractions. If low levels of PrPSc are present in the top fractions of the sucrose gradient, the increase of CDI fluorescence counts after denaturation may not be sufficient for D/N ratios to be distinguishable from those of non‐CJD controls. As PrPC was found to be highly susceptible to the digestion with 2.5 µg/mL PK for 1 h at 37°C, the fractionated samples from FC and Th of vCJD were further explored by CDI after mild proteolysis. In the FC, the high D/N ratios in the bottom three fractions were extreme when compared with the intermediate fractions with the ratios usually lower than 5 or to the top fractions with the ratios lower than 1.5 (Figure 6). Therefore, in the FC, the application of mild proteolysis did not cause any major change in the overall D/N ratios, showing once more the absence of detectable PrPSc in the top three fractions and the migration of the major fraction of PrPSc to the bottom three fractions. In contrast, the application of mild proteolysis to the fractions from vCJD Th caused a great change in the CDI results of the top fractions. While the overall profiles of the D/N ratios in the intermediate and bottom fractions were similar before and after the proteolysis, the D/N ratios in the top fractions were greatly increased (Figure 6). The increase of the D/N ratios was particularly clear in fraction 1 from all three vCJD cases investigated; the ratios of fraction 1 were in the range of 1.8–3 before the proteolysis, but they reached the range of 20–57 after the proteolysis. Thus, the introduction of the mild proteolysis to the fractions prior to the CDI measurement clearly revealed the presence of readily detectable amount of PrPSc in the upper most fraction of the sucrose gradient separation of vCJD Th. Independent Western blot results conducted on FC and Th samples from a single case of vCJD were in good agreement with the results obtained using CDI, and confirmed that PrPSc from vCJD Th were distributed throughout the sucrose gradient after ultracentrifugation, including the topmost fractions (data not shown).
Figure 6.
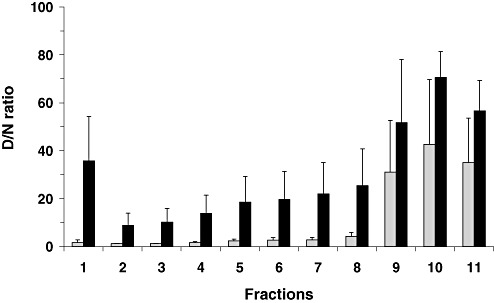
Comparison of conformation‐dependent immunoassay (CDI) denatured/native (D/N) ratios for proteinase K (PK)‐treated fractions between frontal cortex (FC) and thalamus (Th) of variant Creutzfeldt‐Jakob disease (vCJD) brains. Eleven fractions from FC or Th of vCJD brains were digested with 2.5 µg/mL PK for 1 h at 37°C and then analyzed by CDI. The D/N ratios for individual fractions were calculated by dividing TRF counts of denatured samples by the counts of the corresponding native samples. Data shown represent the average ± SD obtained from six cases of vCJD FC (gray bars) and three cases of vCJD Th (black bars). The result in every individual was an average for duplicate wells. Abbreviations: TRF = time‐resolved fluorescence; SD = standard deviation.
DISCUSSION
Disease‐associated prion protein has been traditionally defined on the basis of its protease‐resistance. The PK‐resistant core fragment of PrPSc (or PrPres) derived from the vCJD brain is predominantly truncated at serine 97 following proteolysis and is characterized by high occupancy of the two potential glycosylation sites. Despite marked differences in the neuropathology between distinct anatomical regions in the vCJD brain 15, 16, no regional variation in PrPres type has been identified 12, 23, 29. In this study, we report that the brain regions showing distinct neuropathology (FC and Cb, as compared with the posterior Th) can be distinguished by a biochemical parameter: specifically, by the sedimentation property of prion protein in the sucrose gradient.
We investigated the sedimentation properties of the prion protein after ultracentrifugation in sucrose density gradients using CDI to obtain as full a picture as possible of the PrP species involved. The PrP measured by CDI is operationally defined in this study as PrPC if detectable under native conditions, total PrP (PrPC and PrPSc) when detected under denaturing conditions, whereas the presence of PrPSc is indicated by an increase in fluorescence signal following denaturation (D/N ratio).
The analysis of FC of neurological controls showed PrPC predominantly in the upper few fractions, with much lower levels in the bottom fractions. The recovery of PrPC from the top fractions of such gradients is a consistent finding in normal mouse 31, 32, 33, hamster (53) and human 7, 58 brains. Yuan and colleagues also reported very small amounts of PrP, which resembled PrPSc in the heavier fractions (58). Certain of our findings could be used to support this view, however technical differences between the approach of Yuan et al and our study preclude a direct comparison.
The sedimentation profiles of PrPC from vCJD FC and Cb were indistinguishable from those of controls, with the vast majority of PrPC signals remaining in the top few fractions. In contrast, a substantial proportion of PrPC from the vCJD Th was found to migrate to the intermediate and bottom fractions of the sucrose gradient. The PrPC species detected in the heavier fractions therefore appear to represent aggregated forms of PrPC. As PrPC was defined in this study on the basis of accessibility of the 3F4 epitope in CDI, not only secreted forms of PrPC, but also topological variants such as cytosolic PrP could be classified as “PrPC”. Given that the accumulation of PrPSc during prion infection may favor the accumulation of newly synthesized PrP into the cytosol in association with Endoplasmic reticulum (ER) stress 13, 17, 30, 43 and that cytosolic PrP is known to form aggregates 25, 26, the PrPC species found in the heavier fractions of the gradient may be at least partly associated with cytosol PrP. Alternatively, given that the formation of PrPC/PrPSc complexes proceeds conversion 28, 42, the PrPC molecules detected in these heavier fractions may represent PrPC partly recruited into PrPSc aggregates, but not yet converted into final PrPSc isoform and therefore identified as PrPC by CDI criteria. Lastly, they might represent PrPC bound to massive or dense structures other than PrP, which are not disrupted by the detergent conditions used. Our data cannot distinguish between these alternatives.
The sedimentation profiles of PrPSc (as defined by CDI criteria) from the Th of vCJD were also clearly distinguishable from those of FC and Cb. While PrPSc from the vCJD Th was readily detectable in all fractions of the sucrose gradient, PrPSc from the FC and Cb was largely recovered in the bottom three fractions with only very low levels of PrPSc in the intermediate fractions. As the separation of prion protein in the sucrose step gradient will be a function of distinct molecular densities, sizes and shapes, it is not certain whether the separation of PrPSc species shown in this study reflects their size or is influenced by additional factors such as density and intermolecular interaction. However, if PrP is the only component in PrPSc aggregates in the vCJD brain and density and aggregate shape are homogeneous, then the sedimentation profiles of PrPSc should be a direct reflection of aggregate size. If so, then the size range of PrPSc in vCJD brains, as judged from relevant literature, would range from less than 600 kDa (oligomeric form in the intermediate fractions) to more than 20 000 kDa (large aggregates in the bottom fraction) or that of prion rods containing as many as 1000 PrP molecules 41, 48, 53. Aggregate sizes ranging from 1000 kDa to more than 20 000 kDa have been reported for scrapie on the basis of an agarose gel electrophoresis study (40). The thalamic PrPSc molecules identified in the very topmost fraction of our gradients may be the size of PrP dimers or trimers, on the basis of ionizing radiation studies (2) and could correspond to the recently proposed trimeric PrPSc 6, 8. However, it should be noted that PrPSc has been reported to have strong affinity for lipids (47) and sphingolipids are detectable in highly purified prion rods (20). Moreover, lipids may facilitate the conversion of PrPC into a PrPSc‐like conformation 19, 56 and act as a cofactor facilitating the formation of prions (56). In hamster scrapie, the acquisition of protease resistance in PrPSc was shown to be associated with their size 36, 53. It therefore remains possible that the appearance of PrPSc in the top fractions of vCJD Th might relate to the buoyant density of particular thalamic detergent‐stable lipid/PrPSc complexes. It would be of interest to know whether other methods of estimating aggregate size such as size exclusion chromatography, filtration and field flow fractionation 5, 48, 53 would confirm our observations using sucrose gradient sedimentation.
This last technique (field flow fractionation) has been used to implicate relatively small PrPSc aggregates as those associated with the highest infectivity in rodent scrapie brain (48). Tixador et al (51) have gone on to suggest that PrPSc sedimentation characteristics are a strain‐determining property, speculating that so‐called fast strains are so by virtue of their rapidly replicating oligomeric forms of PrPSc. In this context, it is interesting to note that although numerous studies have identified a single strain of agent in vCJD [most recently in reference (44)], the only published transmission from vCJD Th reported the derivation of two distinct strains in mice expressing a mouse‐human chimeric PrP transgene (21). However, contrary to what one might infer from the work of Silveira et al (48) and Tixador et al (51), the additional strain that was derived from Th, had a longer incubation period than those derived from FC.
The Th (particularly the pulvinar) in vCJD is pathologically characterized by marked astrocytosis and neuronal loss showing only very rare PrPSc plaques, even in PrP immunostaining, whereas the FC and Cb display multiple florid plaques in histology with numerous small cluster plaques in PrP immunostaing (16). Thus, in the vCJD posterior Th, the abundance of the PrPSc species migrating to the intermediate fractions might reflect the size of PrPSc deposits as revealed by neuropathology. The neuronal loss and gliosis in diseased human brains were reported not to be directly related with the local burden of PrPres alone (35) although it is our observation that both Western blotting and CDI identify slightly higher levels of PrPSc in vCJD Th as compared with FC. As in other neurodegenerative diseases 18, 27, 54, a growing body of evidence supports the proposal that oligomeric forms of PrPSc may be more relevant to neurotoxicity in prion diseases than large aggregates 22, 49. It is also possible that certain population of neurons, perhaps abundant in the Th may be more vulnerable to certain species of PrPSc such as oligomeric forms. Evidence for such selective neuronal vulnerability has been described previously for other human prion diseases 9, 10. It is not known when during the disease course neuronal dysfunction and loss occurs in the Th in vCJD, but is tempting to speculate on the basis of the MRI pulvinar sign and the basis of some of the early clinical symptoms that these pathological changes occur very early and it may be that a combination of highly cytotoxic forms of PrP and selective vulnerability of thalamic neurons, result in the severe posterior thalamic pathology evident at end stage disease.
In summary, we have shown that the sedimentation profiles of prion protein (both PrPC and PrPSc) are distinguishable between brain regions in vCJD that exhibit pathologically distinct features, but are indistinguishable on the basis of PrPres type. The PrPSc found in the Th was more polydispersed. We speculate that these differences are indicative of small oligomeric PrP species in the Th and that it is these oligomers that are responsible for the highly characteristic thalamic pathology. Such an interpretation will require confirmation using complementary biochemical assays and would benefit from a more extensive survey of neuroanatomical regions from the whole spectrum of human prion diseases, including cases of PSPr and FFI. A close examination of FFI (and its sporadic counterpart, sFI) using our combined sucrose gradient and CDI methodology could be particularly informative. Not only do these diseases selectively target the thalamus, albeit more anterior regions, but a proportion of FFI cases have PrPSc that is particularly difficult to detect by conventional Western blotting for protease‐resistant forms of PrP. Hence, in common with PSPr, such cases of FFI may offer insights into forms of PrPSc that are relevant to pathophysiology, but which may not figure in the most commonly employed laboratory tests for the prion disease.
ACKNOWLEDGMENTS
This is an independent report commissioned and funded by the Policy Research Programme in the Department of Health, UK. The views expressed in the publication are those of the authors and not necessarily those of the Department of Health. Dr Albrecht Gröner is an employee of CSL Behring. We would like to thank Diane Ritchie for supplying the histological images and Alexander Peden and Michael Jones for their help and advice. The National CJD Surveillance Unit Brain & Tissue Bank is supported by the Medical Research Council (UK) and the National CJD Surveillance Unit as a whole is funded by the Department of Health, UK, and the Scottish Government. We are indebted to UK neuropathologists and their staff for providing tissue and to patients' relatives for generously giving their consent to conduct research on these specimens.
REFERENCES
- 1. Aguzzi A (2009) Cell biology. Beyond the prion principle. Nature 459:924–925. [DOI] [PubMed] [Google Scholar]
- 2. Bellinger‐Kawahara CG, Kempner E, Groth D, Gabizon R, Prusiner SB (1988) Scrapie prion liposomes and rods exhibit target sizes of 55 000 Da. Virology 164:537–541. [DOI] [PubMed] [Google Scholar]
- 3. Bellon A, Seyfert‐Brandt W, Lang W, Baron H, Gröner A, Vey M (2003) Improved conformation‐dependent immunoassay: suitability for human prion detection with enhanced sensitivity. J Gen Virol 84:1921–1925. [DOI] [PubMed] [Google Scholar]
- 4. Cali I, Castellani R, Yuan J, Al‐Shekhlee A, Cohen ML, Xiao X et al (2006) Classification of sporadic Creutzfeldt‐Jakob disease revisited. Brain 129:2266–2277. [DOI] [PubMed] [Google Scholar]
- 5. Caughey B, Kocisko DA, Raymond GJ, Lansbury PT (1995) Aggregates of scrapie‐associated prion protein induce the cell‐free conversion of protease‐sensitive prion protein to the protease‐resistant state. Chem Biol 2:807–817. [DOI] [PubMed] [Google Scholar]
- 6. DeMarco ML, Daggett V (2004) From conversion to aggregation: protofibril formation of the prion protein. Proc Natl Acad Sci U S A 101:2293–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A et al (2008) A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 63:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Govaerts C, Wille H, Prusiner SB, Cohen FE (2004) Evidence for assembly of prions with left‐handed beta‐helices into trimers. Proc Natl Acad Sci U S A 101:8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guentchev M, Hainfellner JA, Trabattoni GR, Budka H (1997) Distribution of parvalbumin‐immunoreactive neurons in brain correlates with hippocampal and temporal cortical pathology in Creutzfeldt‐Jakob disease. J Neuropathol Exp Neurol 56:1119–1124. [DOI] [PubMed] [Google Scholar]
- 10. Guentchev M, Wanschitz J, Voigtlander T, Flicker H, Budka H (1999) Selective neuronal vulnerability in human prion diseases. Fatal familial insomnia differs from other types of prion diseases. Am J Pathol 155:1453–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta‐peptide. Nat Rev Mol Cell Biol 8:101–112. [DOI] [PubMed] [Google Scholar]
- 12. Head MW, Bunn TJ, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS et al (2004) Prion protein heterogeneity in sporadic but not variant Creutzfeldt‐Jakob disease: UK cases 1991–2002. Ann Neurol 55:851–859. [DOI] [PubMed] [Google Scholar]
- 13. Hetz CA, Soto C (2006) Stressing out the ER: a role of the unfolded protein response in prion‐related disorders. Curr Mol Med 6:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hill AF, Joiner S, Wadsworth JD, Sidle KC, Bell JE, Budka H et al (2003) Molecular classification of sporadic Creutzfeldt‐Jakob disease. Brain 126:1333–1346. [DOI] [PubMed] [Google Scholar]
- 15. Ironside JW, Head MW, Bell JE, McCardle L, Will RG (2000) Laboratory diagnosis of variant Creutzfeldt‐Jakob disease. Histopathology 37:1–9. [DOI] [PubMed] [Google Scholar]
- 16. Ironside JW, Ghetti B, Head MW, Piccardo P, Will RG (2008) Prion diseases. In: Greenfield's Neuropathology, 8th edn. Love S, Louis DN, Ellison DW (eds), Chapter 16 pp. 1197–1274. Edward Arnold: London. [Google Scholar]
- 17. Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS (2006) Substrate‐specific translocational attenuation during ER stress defines a pre‐emptive quality control pathway. Cell 127:999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaylor J, Bodner N, Edridge S, Yamin G, Hong DP, Fink AL (2005) Characterization of oligomeric intermediates in alpha‐synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F alpha‐synuclein. J Mol Biol 353:357–372. [DOI] [PubMed] [Google Scholar]
- 19. Kazlauskaite J, Sanghera N, Sylvester I, Venien‐Bryan C, Pinheiro TJ (2003) Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry 42:3295–3304. [DOI] [PubMed] [Google Scholar]
- 20. Klein TR, Kirsch D, Kaufmann R, Riesner D (1998) Prion rods contain small amounts of two host sphingolipids as revealed by thin‐layer chromatography and mass spectrometry. Biol Chem 379:655–666. [DOI] [PubMed] [Google Scholar]
- 21. Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J et al (2003) Abbreviated incubation times for human prions in mice expressing a chimeric mouse‐human prion protein transgene. Proc Natl Acad Sci U S A 100:4784–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H et al (2007) Disease‐associated prion protein oligomers inhibit the 26S proteasome. Mol Cell 26:175–188. [DOI] [PubMed] [Google Scholar]
- 23. Levavasseur E, Laffont‐Proust I, Morain E, Faucheux BA, Privat N, Peoc'h K et al (2008) Regulating factors of PrP glycosylation in Creutzfeldt‐Jakob disease—implications for the dissemination and the diagnosis of human prion strains. PLoS ONE 3:e2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Llewelyn CA, Hewitt PE, Knight RK, Amar K, Cousins S, Mackenzie J, Will RG (2004) Possible transmission of variant Creutzfeldt‐Jakob disease by blood transmission. Lancet 363:417–421. [DOI] [PubMed] [Google Scholar]
- 25. Ma J, Lindquist S (2001) Wild‐type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci U S A 98:14955–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma J, Lindquist S (2002) Conversion of PrP to a self‐perpetuating PrPSc‐like conformation in the cytosol. Science 298:1785–1788. [DOI] [PubMed] [Google Scholar]
- 27. McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K et al (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46:860–866. [DOI] [PubMed] [Google Scholar]
- 28. Meier P, Genoud N, Prinz M, Maissen M, Rulicke T, Zurbriggen A et al (2003) Soluble dimeric prion protein binds PrP(Sc) in vivo and antagonizes prion disease. Cell 113:49–60. [DOI] [PubMed] [Google Scholar]
- 29. Notari S, Moleres FJ, Hunter SB, Belay ED, Schonberger LB, Cali I et al (2010) Multiorgan detection and characterization of protease‐resistant prion protein in a case of variant CJD examined in the United States. PLoS ONE 5:e8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Orsi A, Fioriti L, Chiesa R, Sitia R (2006) Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J Biol Chem 281:30431–30438. [DOI] [PubMed] [Google Scholar]
- 31. Pan T, Li R, Kang SC, Wong BS, Wisniewski T, Sy MS (2004) Epitope scanning reveals gain and loss of strain specific antibody binding epitopes associated with the conversion of normal cellular prion to scrapie prion. J Neurochem 90:1205–1217. [DOI] [PubMed] [Google Scholar]
- 32. Pan T, Chang B, Wong P, Li C, Li R, Kang SC et al (2005) An aggregation‐specific enzyme‐linked immunosorbent assay: detection of conformational differences between recombinant PrP protein dimers and PrP(Sc) aggregates. J Virol 79:12355–12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pan T, Wong P, Chang B, Li C, Li R, Kang SC et al (2005) Biochemical fingerprints of prion infection: accumulations of aberrant full‐length and N‐terminally truncated PrP species are common features in mouse prion disease. J Virol 79:934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parchi P, Giese A, Capellari S, Brown P, Schulz‐Schaeffer W, Windl O et al (1999) Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. [PubMed] [Google Scholar]
- 35. Parchi P, Capellari S, Gambetti P (2000) Intracerebral distribution of the abnormal isoform of the prion protein in sporadic Creutzfeldt‐Jakob disease and fatal insomnia. Microsc Res Tech 50:16–25. [DOI] [PubMed] [Google Scholar]
- 36. Pastrana MA, Sajnani G, Onisko B, Castilla J, Morales R, Soto C, Requena JR (2006) Isolation and characterization of a proteinase K‐sensitive PrPSc fraction. Biochemistry 45:15710–15717. [DOI] [PubMed] [Google Scholar]
- 37. Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB et al (1997) A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273:614–622. [DOI] [PubMed] [Google Scholar]
- 38. Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A (2005) Coexistence of multiple PrPSc types in individuals with Creutzfeldt‐Jakob disease. Lancet Neurol 4:805–814. [DOI] [PubMed] [Google Scholar]
- 39. Prusiner SB (1998) Prions. Proc Natl Acad Sci U S A 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prusiner SB, Groth DF, Bildstein C, Masiarz FR, McKinley MP, Cochran SP (1980) Electrophoretic properties of the scrapie agent in agarose gels. Proc Natl Acad Sci U S A 77:2984–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG (1983) Scrapie prions aggregate to form amyloid‐like birefringent rods. Cell 35:349–358. [DOI] [PubMed] [Google Scholar]
- 42. Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C et al (1990) Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63:673–686. [DOI] [PubMed] [Google Scholar]
- 43. Rane NS, Kang SW, Chakrabarti O, Feigenbaum L, Hegde RS (2008) Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev Cell 15:359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ritchie DL, Boyle A, McConnell I, Head MW, Ironside JW, Bruce ME (2009) Transmissions of variant Creutzfeldt‐Jakob disease from brain and lymphoreticular tissue show uniform and conserved bovine spongiform encephalopathy‐related phenotypic properties on primary and secondary passage in wild‐type mice. J Gen Virol 90:3075–3082. [DOI] [PubMed] [Google Scholar]
- 45. Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M et al (1998) Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 4:1157–1165. [DOI] [PubMed] [Google Scholar]
- 46. Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H et al (2005) Diagnosis of human prion disease. Proc Natl Acad Sci U S A 102:3501–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Safar JG, Wille H, Geschwind MD, Deering C, Latawiec D, Serban A et al (2006) Human prions and plasma lipoproteins. Proc Natl Acad Sci U S A 103:11312–11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B (2005) The most infectious prion protein particles. Nature 437:257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simoneau S, Rezaei H, Sales N, Kaiser‐Schulz G, Lefebvre‐Roque M, Vidal C et al (2007) In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 3:e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soto C, Estrada L, Castilla J (2006) Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem Sci 31:150–155. [DOI] [PubMed] [Google Scholar]
- 51. Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le DA et al (2010) The physical relationship between infectivity and prion protein aggregates is strain‐dependent. PLoS Pathog 6:e1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tomic JL, Pensalfini A, Head E, Glabe CG (2009) Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol Dis 35:352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G et al (2002) Protease‐sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 41:12868–12875. [DOI] [PubMed] [Google Scholar]
- 54. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS et al (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long‐term potentiation in vivo. Nature 416:535–539. [DOI] [PubMed] [Google Scholar]
- 55. Wang F, Yang F, Hu Y, Wang X, Wang X, Jin C, Ma J (2007) Lipid interaction converts prion protein to a PrPSc‐like proteinase K‐resistant conformation under physiological conditions. Biochemistry 46:7045–7053. [DOI] [PubMed] [Google Scholar]
- 56. Wang F, Wang X, Yuan CG, Ma J (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327:1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A et al (1996) A new variant of Creutzfeldt‐Jakob disease in the UK. Lancet 347:921–925. [DOI] [PubMed] [Google Scholar]
- 58. Yuan J, Xiao X, McGeehan J, Dong Z, Cali I, Fujioka H et al (2006) Insoluble aggregates and protease‐resistant conformers of prion protein in uninfected human brains. J Biol Chem 281:34848–34858. [DOI] [PubMed] [Google Scholar]
- 59. Yull HM, Ritchie DL, Langeveld JP, van Zijderveld FG, Bruce ME, Ironside JW, Head MW (2006) Detection of type 1 prion protein in variant Creutzfeldt‐Jakob disease. Am J Pathol 168:151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zeidler M, Sellar RJ, Collie DA, Knight R, Stewart G, Macleod MA et al (2000) The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt‐Jakob disease. Lancet 355:1412–1418. [DOI] [PubMed] [Google Scholar]