

Abstract
The Purkinje cell (PC) degeneration (pcd) phenotype results from mutation in nna1 gene and is associated with the degeneration and death of PCs during the postnatal life. Although the pcd mutation is a model of the ataxic mouse, it shares clinical and pathological characteristics of inherited human spinocerebellar ataxias. PC degeneration in pcd mice provides a useful neuronal system to study nuclear mechanisms involved in DNA damage‐dependent neurodegeneration, particularly the contribution of nucleoli and Cajal bodies (CBs). Both nuclear structures are engaged in housekeeping functions for neuronal survival, the biogenesis of ribosomes and the maturation of snRNPs and snoRNPs required for pre‐mRNA and pre‐rRNA processing, respectively. In this study, we use ultrastructural analysis, in situ transcription assay and molecular markers for DNA damage, nucleoli and CB components to demonstrate that PC degeneration involves the progressive accumulation of nuclear DNA damage associated with disruption of nucleoli and CBs, disassembly of polyribosomes into monoribosomes, ribophagy and shut down of nucleolar and extranucleolar transcription. Microarray analysis reveals that four genes encoding repressors of nucleolar rRNA synthesis (p53, Rb, PTEN and SNF2) are upregulated in the cerebellum of pcd mice. Collectively, these data support that nucleolar and CB alterations are hallmarks of DNA damage‐induced neurodegeneration.
Keywords: Cajal bodies, DNA damage, gene silencing, nucleolus, Purkinje cell degeneration, ribophagy
INTRODUCTION
Nuclear architecture includes the organization of chromosome territories and distinct nuclear bodies, such as nucleoli, Cajal bodies (CBs), nuclear speckles of splicing factors and promyelocytic leukemia (PML)‐nuclear bodies 6, 8, 14, 24, 41, 48. The nucleolus plays a fundamental role in the synthesis of rRNAs and their assembly into ribosomal particles 57, 65. Other non‐ribosomal functions include the biogenesis of several categories of ribonucleoprotein particles, cell cycle control and stress response 1, 36, 51.
In mammalian cells, the nucleolus has three basic structural components: (i) fibrillar centers (FCs), which concentrate components of the rDNA transcription machinery such as the RNA polymerase I and the upstream binding factor (UBF); (ii) the dense fibrillar component (DFC), the site of transcription of rRNA genes and early rRNA processing; and (iii) the granular component (GC), where the assembly of preribosomal particles takes place [for review, see 57, 65]. The structural configuration of the nucleolus is largely determined by its activity in ribosome biogenesis. Thus, mammalian cells that require a high level of ribosome biogenesis commonly have large nucleoli with numerous small FCs 5, 57.
Among nuclear bodies, the CB is an intriguing nuclear structure discovered by Santiago Ramón y Cajal in vertebrate neurons [for a review, see (40)]. Illustrating the importance and plurifunctionality of this structure, CBs play a fundamental role in the biogenesis, transport and recycling of small nuclear‐ (snRNPs) and nucleolar ribonucleoproteins (snoRNPs) involved in pre‐mRNA and pre‐rRNA processing, respectively [for a review, see 14, 24, 47, 48, 66]. Molecular analysis has shown that CBs are enriched in p80 coilin, a molecular marker of this structure 4, 56, spliceosomal snRNPs, the survival motor neuron protein (SMN) and CB‐specific RNA [scaRNAs; 10, 14, 16, 24]. Additionally, CBs share with the nucleolus the proteins fibrillarin, Nopp140, NAP57 and some snoRNPs (34). CBs are prominent structures in mammalian neurons and their number and size correlate with neuronal size and transcriptional activity 5, 39.
The Purkinje cell (PC) is one of the most spectacular neurons known because of its magnificent planar dendritic tree and enormous abundance of dendritic spines. Moreover, PCs are the sole efferent output of the cerebellar cortex and play a fundamental function in the cerebellar control of movement 30, 50. As a result, PC degeneration and death lead to a severe dysfunction of motor control 22, 42. At the cell nucleus level, the predominant euchromatic configuration, the presence of prominent nucleolus and CBs and the great abundance of nuclear pores correlate with a high activity in transcription and nuclear export of RNA 25, 50, which is required for the maintenance of strong synaptic activity in this cerebellar neuron. However, these cellular properties make PCs particularly vulnerable to oxidative stress and to other genotoxic agents that can induce DNA damage during lifespan and neurodegenerative diseases 9, 63.
The PC degeneration (pcd) mutant mouse is characterized by an early cerebellar ataxia. This clearly distinguishable neurological sign match with a massive loss of PCs between postnatal day 15 (P15) and P30 (42). A late degeneration of photoreceptors in the retina, mitral cells in the olfactory bulb, and a discrete population of thalamic neurons also occurs [for a review see 7, 26, 49, 70]. The early morphological features in predegenerative PCs include alterations of the rough endoplasmic reticulum (RER) and basal accumulation of polyribosomes (42). In the pcd mutant mice only the nna1 gene is affected, which encodes an ATP/GTP‐binding protein (Nna1) with a putative zinc carboxypeptidase domain that is essential for PC survival 13, 72, 73. Nna1 contains nuclear localization signals and is distributed in both the nucleus and cytoplasm, being particularly enriched in the outer mitochondrial membrane 13, 27. Although the primary function of Nna1 in nuclear physiology remains elusive, it has been suggested that it has a role in chromatin remodeling (27). Moreover, a recent proteomic analysis has revealed mitochondrial dysfunction and altered proteolytic processing of Nna1 interacting proteins in pcd mice (13).
Defects in DNA repair pathways have recently emerged as a new fundamental pathology associated with polyglutamine (polyQ) diseases, such as Huntington's disease and several spinocerebellar ataxias, as well as other neurodegenerative disorders 9, 20, 46, 55, 58. Indeed, in a previous work (70), we demonstrated that pcd mutation induces DNA damage during the predegenerative stage in mitral cells. Similarly, our preliminary studies demonstrate the presence of DNA damage foci in degenerating PCs of pcd mice. The rapid accumulation of DNA damage in PCs of this mutant mouse between 3 and 4 weeks of age, which ultimately leads to cell death, provides a relevant neuronal system for investigating nucleolar and CB mechanisms implicated in neurodegeneration. In this way, the nucleolus is involved in the biogenesis of ribosomes, an essential housekeeping function 8, 57. Similarly, CBs are functionally linked to the nucleolus through the biogenesis and supply of snoRNPs required for pre‐rRNA processing 14, 34, 48. In this study, we analyze how the accumulation of unrepaired DNA in pcd mice PCs correlates with a general inhibition of RNA synthesis, disruption of the nucleolus and CBs and severe dysfunction of ribosome biogenesis that ultimately leads to neuronal death. Therefore, the alterations of the nucleolus and CBs represent a fundamental component in PC neurodegeneration pathway.
MATERIALS AND METHODS
Mouse genotyping
C57BL/6J male mice heterozygous for the pcd 1J mutated gene were purchased from Jackson Laboratories (Bar Harbor, ME, USA). They were mated with DBA/2J females without the pcd mutation. The pcd1J allele was secluded with the genetic background of the C57BL/6J strain whereas the normal allele was associated with the genetic background of the DBA/2J strain. This differential segregation of both pcd and normal alleles allowed the control and heterozygous mice to be differentiated in order to expand the colony and genetically typify the experimental and control animals from the same litters. To investigate the genotype, DNA was extracted from the tail of the mice and the PCR was performed using the primers proposed for the markers D13Mit250 and D13Mit283 in the web resource of Jackson Laboratories: (http://www.informatics.jax.org/searches/probe.cgi?38700; http://www.informatics.jax.org/searches/probe.cgi?41581). PCR products of microsatellite regions D13Mit250 and D13Mit283, which have different sizes in both the C57BL/6J and DBA/2J strains, were resolved by electrophoresis in a 3% agarose gel. The animals were kept, handled and sacrificed in accordance with the Council of the European Communities and current Spanish legislation and the experiments were approved by the Bioethical Committee of the University of Salamanca.
Microarray analysis
To check the gene expression changes in pcd1J mice, we compared the gene expression profiles of control and pcd mice. Three control and three 20‐day‐old PCD mice were used. They were decapitated and the vermis of the cerebellum was rapidly removed. Total RNA from the vermis was extracted with TRIzol, and purified using RNeasy mini‐kit (Qiagen 74104, Qiagen, Valencia, CA, USA) and RNase‐free DNase I digestion. Once the samples of total RNA were obtained, the integrity and pureness of the RNA were determined using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The messengers (mRNA) were retrotranscripted to yield cDNA using a commercial kit from Gibco/BRL (Superscript Choice System for cDNA synthesis). Thereafter, the cDNA was put under in vitro transcription in the presence of biotin labeled nucleotides to generate cRNA using the IVT kit (Affymetrix, Charleroi, Belgium). cRNAs were degraded by alkaline digestion and used for hybridizations with commercial chips, but only after having been subjected to a second quality control with a biochips Affymetrix test (TestArray 3). The hybridizations were made with the GeneChip Mouse Genome 430 2.0 Array of Affymetrix. The level of signal was calculated using the Robust Microarray Analysis algorithm (29), whereas the differential expression was calculated using the significance analysis of microarrays that include an estimation of the error by means of the false discovery rate.
Tissue preparation
After deep anaesthesia with a mixture of xylazine (Rompum, Bayer, Kiel, Germany) and ketamine hydrochloride (Imalgene, Merial, Lyon, France), mice were perfused with heparinized saline for 1 minute, and fixative solution containing 4% paraformaldehyde and 0.2% saturated picric acid in 0.1 M phosphate buffer (PB), pH 7.4, for 15 minutes. After perfusion, the vermis of the cerebellum was dissected and postfixed in the same solution for 2 h at room temperature. Then, tissue blocks were washed in PB, and cryoprotected with 30% sucrose, overnight at 4°C until they sank. Thirty µm‐thick sagittal sections were cut using a freezing‐sliding microtome (Leica Frigomobil, Jung SM 2000, Leica, Nussloch, Germany), the slices being collected in PB and stored at −20°C in a freezing mixture containing 30% glycerol and 30% polyethylene glycol in PB, until required.
Immunofluorescence
Cryo‐cut sections from the vermis of control and mutant mice of 20 days of age were treated with 1% NaBH4 in PB for 20 minutes at room temperature, rinsed in PB (3 × 10 minutes), and incubated for 1 h in blocking serum (5% goat serum and 0.1% Triton X‐100 in PB). The primary rabbit polyclonal anti‐calbindin antibody (1:7.000; Swant, Switzerland) was incubated in the same solution overnight at 4°C. Cy2‐conjugated secondary anti‐rabbit antibody (1:500; Jackson Laboratories) was then applied for 2 h at room temperature. Finally, the sections were washed in phosphate‐buffered saline (PBS) pH 7.4 mounted, and coverslipped with antifade solution.
Squash preparations of tissue fragments from the vermis fixed by perfusion with 3.7% paraformaldehyde in PBS were processed as previously described (53). Briefly, small blocks containing the PC layer were isolated and cut into small fragments. Each tissue fragment was transferred to a drop of PBS on a siliconized slide (SuperFrostPlus, Menzel‐Gläser, Germany). A coverslip was then located at the top of the slide, and the tissue was squashed by mechanical percussion with a histologic needle to dissociate neuronal perikarya. The preparation was then frozen in dry ice, and the coverslip removed by using a razor blade. By using this procedure, most PCs remained adhered to the slide. Cell samples were then sequentially processed in 96% ethanol at 4°C for 10 minutes, and PBS at 4°C. Finally, the samples were stored at 4°C until used.
To perform the immunostaining on the dissociated PCs, the samples were sequentially treated with 0.1 M glycine in PBS for 15 minutes and 0.5% Triton X‐100 in PBS for 45 minutes. Then, they were incubated with the primary antibody overnight at 4°C, washed with 0.05% Tween 20 in PBS, incubated for 45 minutes in the specific secondary antibody conjugated with FITC or Texas‐red (Jackson Laboratories), rinsed in PBS, and counterstained with propidium iodide (1:2.000) for 15 minutes. Double inmunofluorescence experiments were also carried out. Finally, the samples were mounted with the antifade medium Vectashield (Vector, Burlingame, CA, USA). They were examined with a laser confocal microscope (Zeiss LSM 510) using a 63x plan‐apochromatic objective (1.4 NA), and argon ion (488 nm) and HeNe (543 nm) lasers.
The following primary antibodies were used. goat polyclonal anti‐B23 protein (Santa Cruz Laboratories, Santa Cruz, CA, USA), mouse monoclonal anti‐UBF (Santa Cruz Laboratories), rabbit polyclonal serum 204.3 anti‐coilin (kindly provided by Dr A.I. Lamond, University of Dundee, UK), rabbit polyclonal anti‐p53 (Santa Cruz Laboratories), mouse monoclonal anti‐fibrillarin (Abcam, Cambridge, MA, USA), mouse monoclonal antibody anti‐γH2AX (Upstate, Waltham, MA, USA), which recognizes the phosphorylated variant of histone H2AX on Ser 139, and a human autoimmune serum that recognizes the Sp100 protein of PML bodies (kindly provided by Dr Marcos del Hoyo, University Hospital “Marques de Valdecilla,” Spain).
The quantitative analysis of CBs and FCs was performed in cerebellar vermis samples from wild‐type (n = 3) and pcd (n = 3) mice. For the determination of the mean number of FCs per PC nucleolus, squash preparations were immunostained for UBF (FCs) and the number of UBF‐positive nucleolar spots was counted on serial confocal sections of the whole nucleolus. A minimum of 50 nucleoli for either wild‐type or pcd experimental groups were used. The quantification of CBs was performed in squash preparations immunolabeled with the anti‐coilin antibody and counterstained with propidium iodide. CBs were counted by direct examination of the different focal planes throughout neuronal nuclei and using a 63× oil immersion objective. A minimum of 200 PCs for each wild‐type and pcd experimental groups were examined. All quantitative data were analyzed by the Student t‐test to compare both experimental groups. Statistical significance was established at P < 0.01.
Run‐on transcription assay in situ
Active transcription sites were labeled by incorporation of 5′‐FU into nascent RNA as previously reported (11). Briefly, under anesthesia mice were given an i.p. injection of 5′‐FU (Sigma, Dorset, UK) at the doses of 5 µL/g of a stock solution of 0.4 M 5′‐FU in 0.9% saline. The mice were killed 60 minutes after the injection. The animals were fixed by perfusion with 3.7% paraformaldehyde in HPEM buffer [2X HPEM: Hepes, 60 mM; Pipes, 130 mM; ethylene glycol tetraacetic acid (EGTA), 20 mM; and MgCl2·6H2O, 4 mM] containing 0.5% Triton X‐100 for 10 minutes. The vermis were removed, postfixed in the same solution for 20 minutes, washed in HPEM buffer containing 0.5% Triton X‐100 for 10 minutes and cut into small fragments. Mechanical PC dissociation was performed as previously described. Then, the samples were sequentially treated with proteinase K (0.25 µg/mL in buffer Tris 1 M, pH 8) for 1 minute at 25°C, 0.1 M glycine in PBS containing 1% bovine serum albumin (BSA) for 15 minutes, and 0.01% Tween 20 in PBS for 5 minutes. The incorporation of the 5′‐FU into nascent RNA was detected with a mouse monoclonal (clone BU‐33) anti‐BrdU antibody (Sigma, Madrid, Spain), diluted 1:50 in PBS (overnight at 4°C). Then, the samples were washed with 0.01% Tween 20 in PBS, incubated for 45 minutes with an anti‐mouse FITC‐conjugated secondary antibody (Jackson Laboratories), washed in PBS, mounted with the antifade medium Vectashield and examined with the laser confocal microscope. Some samples were counterstained with propidium iodide or processed for a double immunofluorescence labeling.
Electron microscopy and immunogold labeling
Three controls and three pcd mice of 20 days of age were used in order to analyze the ultrastructure of PCs. Mice were perfused with 3% glutaraldehyde in 0.1 M PB, pH 7.4. The cerebellum was removed and the vermis was isolated. Five hundred‐micrometer‐thick sagittal sections were obtained using a vibratome (Leica). Then, they were rinsed in 0.1 M PB, postfixed in 2% osmium tetroxide diluted in double‐strength buffer (containing 3.5% dextrose in 0.2 M PB, pH 7.4), dehydrated in acetone, and embedded in Araldite (Durcupan, Fluka, Switzerland). Ultrathin sections stained with uranyl acetate and lead citrate were examined with a Philips EM‐208 electron microscope operated at 60 kV.
For immunoelectron microscopy the animals were perfused with 3.7% paraformaldehyde in 0.1 M cacodylate buffer for 10 minutes at room temperature. Small tissue fragments of the vermis were washed in 0.1 M cacodylate buffer, dehydrated in increasing concentrations of methanol at −20°C, embedded in Lowicryl K4 M at −20°C and polymerized with ultraviolet irradiation. Ultrathin sections were mounted on nickel grids and sequentially incubated with 0.1 M glycine in PBS for 15 minutes, 5% BSA in PBS for 30 minutes and the primary rabbit anti‐coilin antibody (1:50) diluted in 50 mM Tris–HCl, pH 7.6, containing 1% BSA and 0.1 M glycine for 2 h at 37°C. After washing, the sections were incubated with the goat anti‐rabbit IgG antibody coupled to 10‐nm gold particles (BioCell, Cardiff, UK; diluted 1:50 in PBS containing 1% BSA) for 1 h at room temperature. Following immunogold labeling, the grids were stained with lead citrate and uranyl acetate and examined with a Phillips EM208 electron microscope operated at 60 kV. As controls, ultrathin sections were treated as described above but with primary antibodies omitted.
Immunoblotting
For the preparation of nuclear extracts from cerebellar vermis of wild‐type and pcd mice, tissue samples were lisated using a pellet‐pestle motor (Sigma‐Aldrich) on ice in cold extraction buffer RSB (10 mM Tris pH 7.5, 10 mM NaCl, 3 mM MgCl2, NP40 1%) supplemented with protease and phosphatase inhibitor cocktail (Halt™ Protease and Phosphatase inhibitor single‐use cocktail, Thermo Scientific, Rockford, IL, USA) and incubated for 20 minutes on ice. After centrifugation (5 minutes at 1.000 rpm), pellets were washed with buffer RSB (without NP40) and centrifuged at 2.000 rpm for 5 minutes at 4°C and the supernatant eliminated. Pellet was resuspended with NB buffer [50 mM Tris pH 7.5, 0.4 M NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA)] supplemented with protease and phosphatase inhibitor cocktail (Halt™ Protease and Phosphatase inhibitor single use cocktail, Thermo Scientific) and incubated for 15 minutes at 4°C. Samples were sonicated with the Bioruptor UCD‐200‐ (Diagenode, Denville, NJ, USA) at 4°C and centrifuged at 11.000 rpm for 5 minutes at 4°C. Supernatant was taken as nuclear fraction. Nuclear fraction proteins were separated on SDS‐PAGE gels and transferred to nitrocellulose membranes by standard procedures. Protein bands were detected with the Odyssey™ Infrared‐Imaging System (Li‐Cor Biosciences, Lincoln, NE, USA) according to Odyssey™ Western‐Blotting Protocol. The following primary antibodies were used for immunoblotting: monoclonal mouse anti‐γH2AX (Millipore‐Upstate, Chicago, IL, USA) and polyclonal rabbit anti‐lamin A/C (Santa Cruz Biotechnology). Immunoblots were developed with anti‐mouse IRDye800DX or anti‐rabbit IRDye700DX (Rockland Immunochemicals, Gilbertsville, PA, USA) secondary antibodies.
RESULTS
Previous studies have reported that PC death in pcd mice starts at postnatal day 18 (P18) and results in a massive neuronal death at P30 (42). Thus, at P20, immunostaining for calbindin, a molecular marker of PCs, clearly demonstrated the normal organization of PCs in the cerebellar cortex of wild‐type mice and the neuronal loss in pcd mice (Figure 1A, B). Given that at P20 there coexists healthy PCs with different stages of neuronal degeneration in the cerebellar cortex of pcd mice, we have selected this postnatal day to study nucleolar and CBs alterations associated with this type of neurodegeneration.
Figure 1.
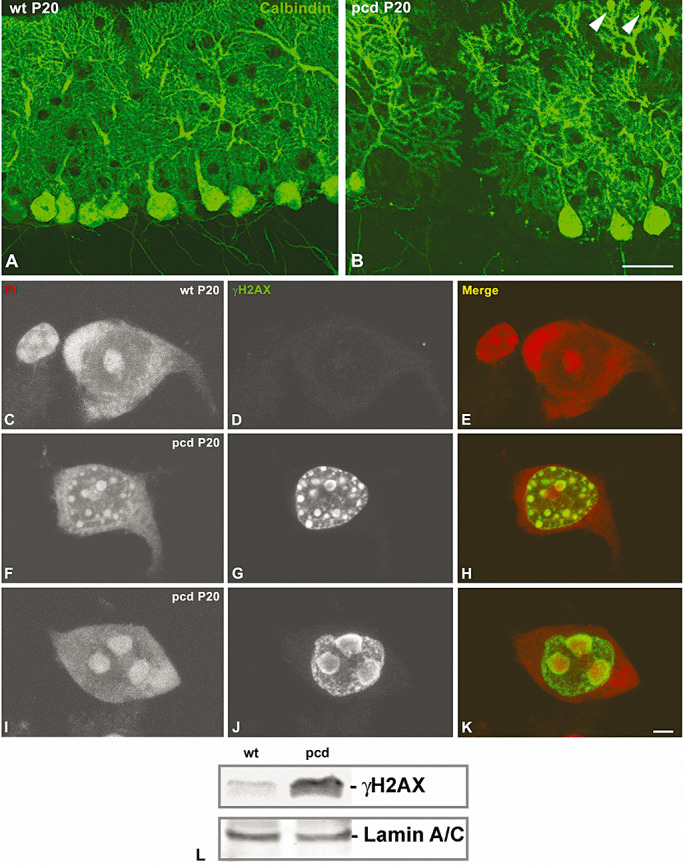
pcd mutation produces a rapid Purkinje cell (PC) degeneration associated to DNA damage. Confocal microscopy images of sagittal sections of the vermis of wild‐type (A) and pcd mutant mice (B) at P20 immunostained for calbindin D‐28k. Note in the pcd the loss of PCs as well as the swelling of dendritic terminals (arrowhead in B). Confocal microscopy images from squash preparations of PC from P20 wild‐type (C–E) and pcd mice (F–K), labeled for γH2AX (green) and counterstained with Propidium Iodide (PI). Note the absence of DNA damage nuclear foci immunoreactive for γH2AX in the control neuron (D). F.–H. The PC nucleus of the pcd mutant mouse contains numerous round‐shaped and variable sized γH2AX nuclear foci. I.–K. In advanced stages of PC degeneration DNA damage accumulates in a few large nuclear domains intensely contrasted with PI and identified as heterochromatin masses. Scale bar: (A–B) = 100 µm; (C–K) = 5 µm. L. Western blot analysis of phosphorylated histone H2AX at Ser 139 (γH2AX) in nuclear extracts from wild‐type and pcd mice cerebellar vermis. Phosphorylation of this histone H2AX variant is activated in pcd cerebellum. Expression of lamin A/C band was used as protein loading control.
To demonstrate the induction and progressive accumulation of DNA lesions in degenerating PCs of pcd mice, we performed immunolabeling for the phosphorylated histone H2AX at Ser 139 (γH2AX), a sensor for DNA damage (21), in combination with propidium iodide, a cytochemical staining for nucleic acids. In control PCs, γH2AX immunoreactivity was not detected, whereas the nucleolus and polyribosome‐rich cytoplasmic domains (Nissl substance) appeared counterstained with propidium iodide (Figure 1C–E). At early stages of PC degeneration, numerous nuclear foci of γH2AX immunoreactivity were detected throughout the nucleus. They were stained with propidium iodide, reflecting a process of chromatin compaction at sites of DNA damage (Figure 1F–H). As PC degeneration took place, the expression of γH2AX propagated to extensive genome domains, resulting in a few very large γH2AX‐positive nuclear domains in addition to other microfoci of DNA damage (Figure 1I–K). The activation of the expression of γH2AX in the pcd mice cerebellum was confirmed by Western blot analysis of nuclear extracts from cerebellar vermis (Figure 1L).
PC degeneration associates with a disruption of the nucleolus and cytoplasmic protein synthesis machinery
Having established that pcd mutation induces extensive DNA damage in PCs, we next investigated how this neuronal stress affects the organization and fine structure of protein synthesis machinery, particularly the nucleolus and polyribosomes. In wild‐type mice, PCs exhibited typical reticulated nucleoli composed of numerous small FCs surrounded by a shell of DFC and irregular areas of GC, in addition to the nucleolus‐attached heterochromatin (Figure 2A). However, several nucleolar alterations were detected in pcd mice PCs, ranging from a moderate reorganization of nucleolar components to a severe nucleolar disruption with macrosegregation of fibrillar and GCs (Figure 2B–F). Thus, at initial stages of PC degeneration, the most characteristic ultrastructural feature was the reorganization of FCs into a very few larger FCs, whereas the small FCs tended to disappear (Figure 2B). This alteration was accompanied by the formation of large nucleolus‐attached heterochromatin masses, reflecting the induction of gene silencing. As PC degeneration proceeded, nucleoli exhibited partial segregation of DFC and GC as well as the formation of nucleolar cavities containing electrondense granules of variable size, whereas FCs were rarely found (Figure 2C, D). At more advanced stages of neuronal degeneration, striking abnormalities were the macrosegregation of the DFC and GC in large separated masses and, less frequently, the formation of a very large mass of GC surrounded by small aggregates of DFC (Figure 2E, F).
Figure 2.
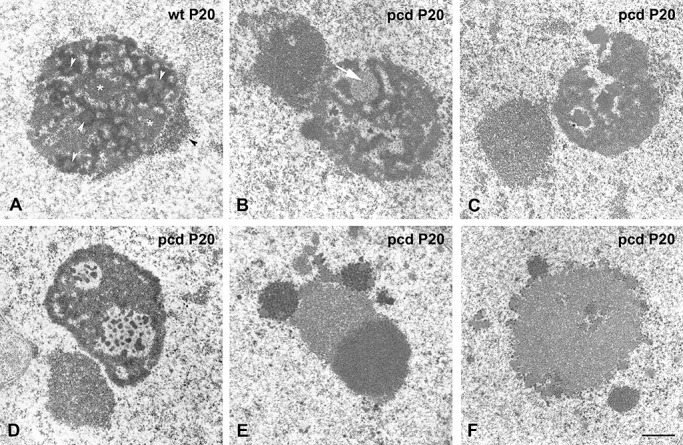
pcd mutation induces a disruption of the nucleolus in Purkinje cells (PCs). Electron microscopy images of PC nucleoli of wild‐type (A) and pcd mutant mice (B–F) at P20. A. Electron micrograph of a typical reticulated nucleolus composed of numerous small FCs (white arrow head) surrounded by a shell of dense fibrillar component (DFC) and intercalated masses of granular component (GC) (asterisks). Note the small aggregates of nucleolus‐attached heterochromatin (black arrowhead). At early stages of PC degeneration (B), the most noticeable ultrastructural features are the reorganization of FCs into a few large‐size fibrillar centers (FCs; white arrow) and the formation of prominent masses of nucleolus‐attached heterochromatin. As PC degeneration proceeds (C,D), the nucleolus shows a segregation of DFC and GC, disappearance of FCs, and formation of nucleolar cavities containing electrondense granules of variable sizes. At final stages of neuronal degeneration (E,F), nucleoli exhibit macrosegregation of DFC and GC in large separated masses (E), or the formation of a very large mass of GC together with small aggregates of DFC (F). Scale bar: 1 µm.
As the nucleolus plays a major role in biogenesis of ribosomes and maintaining protein synthesis machinery, we analyzed the reorganization of the RER and free polyribosomes associated with the nucleolar disruption in the same cells. PCs of wild‐type mice exhibited the typical organization of RER arrays of cisterns associated with polyribosomes, some attached to the membranes and some free interspersed among cisterns (Figure 3A), as previously reported in mature PCs 42, 50. At the initial stages of PC degeneration, the stacks of RER cisterns tended to disappear and most of neurons showed a prominent mass of densely packed free polyribosomes at the basal pole of cell soma (Figure 3B), as was reported in the initial description by Landis and Mullen (42). Interestingly, in PCs that showed macrosegregation of nucleolar components and massive heterochromatinization, indicating a severe dysfunction of nucleolar and extranucleolar transcription, free polyribosomes had been replaced by monoribosomes (Figure 3C). They were closely packed and frequently appeared sequestered into cytoplasmic compartments bounded by isolated RER cisterns and identified as autophagosomes (Figure 3C and insert). The selective autophagia of ribosomes has been reported in yeast and termed “ribophagy”(35). In conclusion, the accumulation of DNA damage in PCs of pcd mice causes disruption of nucleolus, disassembly of polyribosomes and autophagic degradation of monoribosomes.
Figure 3.
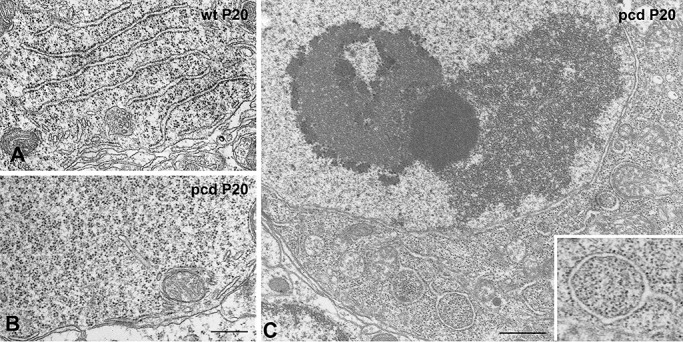
pcd mutation induces disorganization of rough endoplasmic reticulum (RER) and ribophagy. Electron microscopy images of Purkinje cells (PCs) from wild‐type (A) and pcd mice (B–C) at P20. Healthy PCs exhibit typical arrays of RER with polyribosomes attached to the membranes and distributed among cisterns (A). At the onset of PC degeneration, the RER cisterns tend to disappear and a prominent mass of closely packed free polyribosomes is observed at the basal cell body (B). As PC degeneration proceeds, free polyribosomes are replaced by densely packed monoribosomes (C, and insert in C). Cytoplasmic portions containing monoribosomes appear sequestered in autophagic vacuoles bounded by isolated RER cisternae (insert in C). Scale bars = 1 µm.
pcd mutation induces segregation of fibrillarin and B23, reorganization of UBF and inhibition of rRNA synthesis in PCs
In order to correlate the ultrastructural changes of the nucleolus with the reorganization of molecular markers of nucleolar components, we performed immunofluorescence with antibodies that recognized fibrillarin, the signature protein of the DFC, B23/Nucleophosmin, a marker of the GC, and the UBF for FCs [for a review, see 8, 57]. In control PCs, fibrillarin immunostaining of the nucleolus appeared as a cluster of closely packed nucleolar spherules (Figure 4A), corresponding to the shell of DFC that surrounded the FCs detected by electron microscopy. In contrast, fibrillarin was progressively segregated into irregular areas at the periphery of the nucleolus in PCs of pcd mice (Figure 4 B, C). Immunostaining for B23 revealed a non‐homogenous labeling of the nucleolar body in control PCs (Figure 4D), whereas B23 was relocalized diffusely throughout the nucleoplasm and concentrated in residual masses of GC in degenerating PCs showing accumulation of DNA damage (Figure 4E, F).
Figure 4.
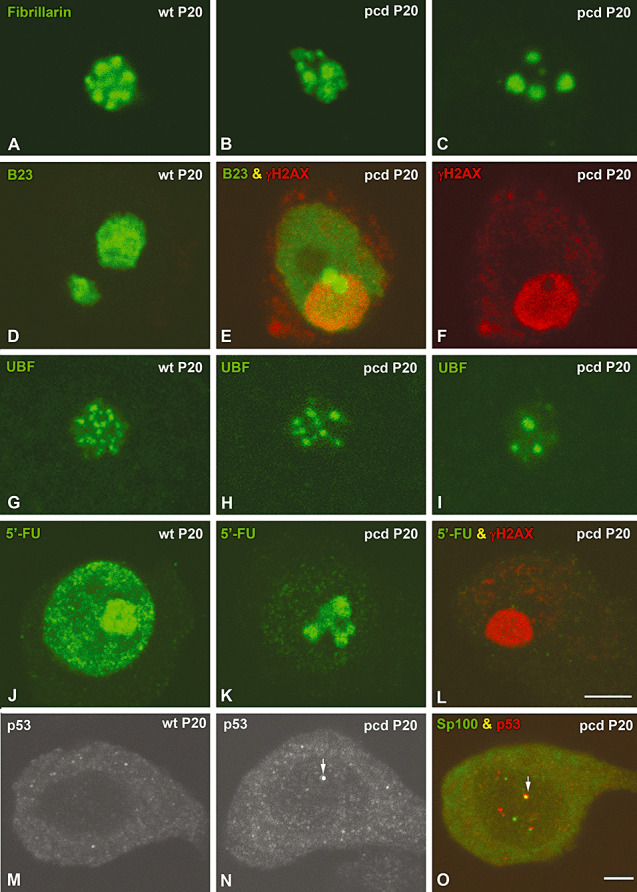
pcd mutation induces segregation of fibrillarin and B23, reorganization of upstream binding factor (UBF) and inhibition of rRNA synthesis. Confocal microscopy images from squash preparations of Purkinje cells (PCs) from wild‐type and pcd mice. In a control PC, staining for fibrillarin reveals a cluster of closely packed nucleolar spheres (A). In a mutant mice PC, fibrillarin appears segregated in irregular areas, which progressively aggregated into a few large masses mainly localized at the nuclear periphery (B–C). Immunostaining for B23 (green) shows a non‐homogenous labeling of the nucleolus in a control PC (D). E.–F. A PC of a mutant mouse with a prominent heterochromatin domain immunoreactive for γH2AX (red) shows nucleoplasmic localization of B23 (green) in addition to being concentrated in two masses attached to the heterochromatin. G. UBF immunolabeling reveals numerous small FCs in the nucleolus of a control PC. In pcd mice PCs, a decrease in the number of FCs and an increase in their size were observed (H,I). J. In situ transcription assay with 5′‐FU incorporation reveals the high nucleolar concentration of nascent RNA and the formation of numerous transcription foci throughout the nucleoplasm. In pcd mice PCs, 5′‐FU incorporation dramatically decreases in the nucleoplasm, whereas the reduced nucleolar signal of nascent rRNA appeared in separated masses of the segregated nucleolus (K). Co‐staining for 5′‐FU incorporation (green) and γH2AX (red) illustrates the absence of 5′‐FU incorporation in a degenerating PC with a prominent γH2AX‐positive heterochromatin mass (L). M. In control PC, staining for p53 showed a very weak diffusive signal both in the nucleous and the cytoplasm. N. By contrast, in mutant PC p53‐positive spots appear throughout the nucleous (arrow). O. Note that some of them colocalize with Sp100 protein (arrow). Scale bars: (A–L) = 5 µm; (M–O) = 10 µm.
To further investigate the relationships between nucleolar disruption and dysfunction of rRNA synthesis in pcd mice PCs, we analyzed the reorganization of UBF, a RNA polymerase I factor essential for the activation of rDNA transcription [for a review, see 57, 67], and the in situ transcriptional activity. Immunostaining for UBF displayed the nucleolar distribution of FCs, as previously demonstrated in other neuronal and non‐neuronal cell types 5, 60. Whereas control PCs exhibited numerous small FCs, an apparent reduction in their number was observed in neurons of pcd mice (Figure 4G–I). As the number of FCs in mammalian neuronal nucleoli positively correlated with transcriptional activity [for a review, see (5)], we estimated the number of UBF‐positive intranucleolar spots (FCs) by quantitative analysis. The mean number of FCs per nucleolus significantly decreased from 11.96 ± 2.26, in wild‐type PCs, to 5.96 ± 1.75, in pcd mice neurons (values are mean ± standard desviation of the mean (SD). Statistical analysis was carried out using the Student t‐test to compare both experimental groups, P < 0.01). Next, we investigated changes in transcriptional activity using the incorporation of 5′‐FU into nascent RNA after a pulse of 60 minutes of the intraperitoneal administration of this halogenated nucleotide (11). In control PCs, nascent RNA concentrated in the nucleolus and in numerous extranucleolar transcription foci, in addition to being diffusely distributed throughout the nucleoplasma (Figure 4J). In degenerating pcd mice PCs, the global transcription rate dropped dramatically, as was indicated by the reduction in both transcription foci and the diffuse nucleoplasmic signal of 5′‐FU incorporation into nascent RNA. Interestingly, these neurons had an altered nucleolar pattern of 5′‐FU labeling. It appeared as a few segregated areas of 5′‐FU incorporation probably corresponding to domains of segregated DFC of the nucleolus (Figure 4K). In advanced stages of PC degeneration with a great accumulation of DNA damage no signal of 5′‐FU incorporation was detected, indicating a complete silencing of gene expression (Figure 4L). In conclusion, nucleolar disruption in pcd mice PCs is another nuclear hallmark of neuronal degeneration that reflects the repression of rRNAs genes and the inhibition of ribosome biogenesis.
Microarray analysis revealed increased expression of some genes encoding negative regulators of rDNA transcription in pcd mice
Having established the existence of a nucleolar stress response to DNA damage with disruption of the structure and function of the nucleolus in pcd mice PCs, we next investigated whether these alterations correlate with changes in gene expression of rDNA transcription repressors. Microarray analysis of cerebellar mRNA expression revealed upregulation of certain genes encoding repressor factors of rDNA transcription in the pcd mice (Table 1). The transcripts of these genes that increase by two fold or more in pcd mice compared with wild‐type mice included those encoding (i) p53 and RB, two negative regulators of RNA Pol I activity 62, 71, 75, (ii) Snf2h, a component of the nucleolar remodeling complex (NoRC) involved in the epigenetic silencing of rRNA genes (64) and (iii) PTEN, a lipid phosphatase that downregulates the PI3K pathway and blocks recruitment of the basal transcription factor SL1 to the promotor of rRNA genes 18, 76. In the case of p53, we have observed by immunofluorescence a very weak or undetectable nuclear signal and a diffuse cytoplasmic staining in control PCs (Figure 4M). In contrast, the p53 nuclear staining increased and nuclear spots of higher intensity were frequently observed in pcd mice PCs (Figure 4N). The proportion of PCs containing p53‐positive spots significantly increased in the pcd mice (2.2% ± 0.1%, in the wild‐type mice, vs. 54% ± 0.3%, in the pcd mice, values are mean ± SD. Statistical analysis was carried out using the Student t‐test to compare both experimental groups P < 0.001). Co‐staining for p53 and Sp100, a typical constituent of PML bodies (6), revealed that most p53 spots colocalized with Sp100 in PML bodies (Figure 4O). This co‐localization is consistent with the role of PML bodies in the cellular stress response, which requires the recruitment and activation, through post‐translational modifications, of p53 in these nuclear structures (3). Taken together, these data suggest the transcriptional activation of repressors of the rDNA transcription machinery in the cerebellum of the mutant mice. Moreover, gene expression analysis is consistent with the present observations of a breakdown of nucleolar structure and disruption of protein synthesis machinery. However, it should be noted that these microarray data referred to the whole cerebellum, not only to PC mRNAs. Therefore, the possible involvement of other cerebellar cell populations, as a secondary response to the PC degeneration, cannot be excluded.
Table 1.
Differentially expressed transcripts of genes encoding repressors of nucleolar rRNA synthesis in vermis of pcd mutants compared with wild‐type mice.
| Gene | Gene designation | Fold | P value |
|---|---|---|---|
| Pten | Phosphatase and tensin homolog | +3.48 | 0.037 |
| Rb1 | Retinoblastoma 1 | +6.93 | 0.025 |
| Shprh | SNF2 histone linker PHD RING helicase | +8.89 | 0.010 |
| Trp53 | Transformation‐related protein 53 | +2.7 | 0.037 |
PC degeneration disassembles CBs and relocalizes coilin around segregated masses of fibrillarin
Because CBs have been linked to the organization and function of the nucleolus, particularly in mammalian neurons [for review, see 5, 31, 53], we investigated the reorganization of CBs that accompanied the nucleolar alterations detected in degenerating PCs in pcd mice. For this purpose, we performed double immunostaining for coilin and fibrillarin on dissociated PCs in order to visualize CBs and nucleoli, respectively. Control PCs exhibited typical CBs, free in the nucleoplasm or attached to the nucleolus, in which coilin and fibrillarin colocalized (Figure 5A–C). In contrast, the pcd mice PCs showed substantial changes in the number of CBs and in the nuclear distribution of coilin. The quantitative analysis at P20 of the mean number of CBs per nucleus revealed a significant reduction in PCs from pcd mice in comparison with wild‐type mice neurons (2.03 ± 0.39, in control, vs. 0.48 ± 0.16, in pcd mice, mean ± SD, P < 0.01). In PCs with initial segregation of nucleolar components, CBs with very irregular shape were found (Figure 5D, F). Notably, in nucleoli with macrosegregation of their components, coilin was redistributed as a thin shell around the large and small segregated masses of fibrillarin (Figure 5G–I). In more advanced stages of nucleolar disruption and fragmentation, the close association of coilin with the surface of the fibrillarin‐positive fragments was preserved (Figure 5J, L). At the final stage of PC degeneration, a coilin‐positive mass was frequently seen juxtaposed to a nucleolar remnant of fibrillarin at the periphery of the large heterochromatin domains (Figure 5M, O).
Figure 5.
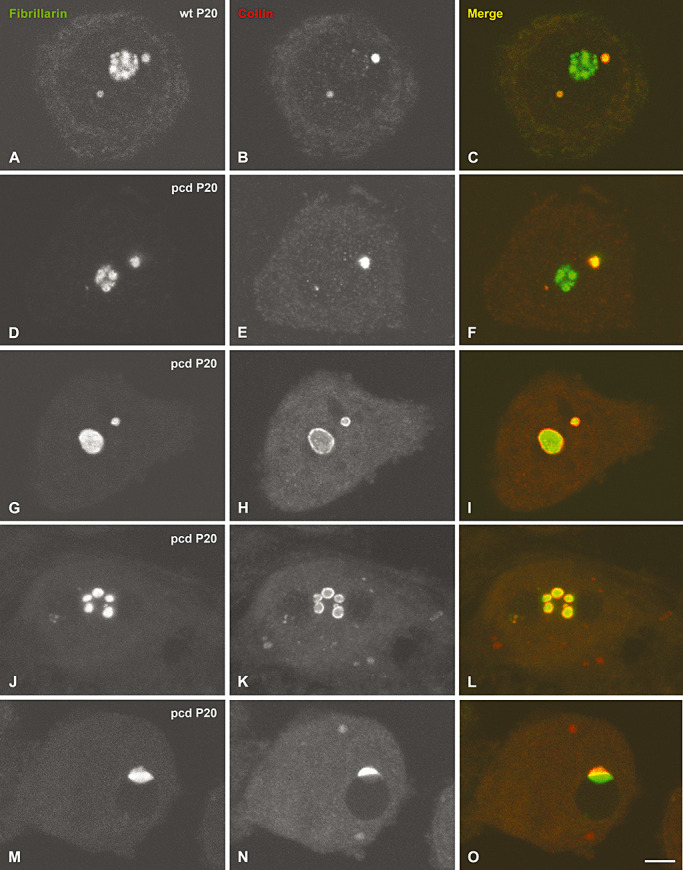
pcd mutation causes disassembly of Cajal bodies (CBs) and relocalization of coilin. Confocal microscopy images from squash preparations of Purkinje cell (PC) from wild‐type (A–C) and pcd mice (D–O) co‐stained for fibrillarin and coilin. Control PCs exhibit typical CBs free in the nucleoplasm or attached to the nucleolus, in which both proteins colocalized (A–C). D.–F. At early stages of degeneration, PCs display a segregation of the nucleolus (D) and CBs with an irregular morphology (E). In more advanced stages of PC degeneration with a severe segregation of the nucleolar components, coilin appears relocalized as a thin ring surrounding the segregated masses of fibrillarin (G–I). Note that as nucleolar disruption and fragmentation proceed, coilin redistributes as a thin shell around the segregated masses of fibrillarin (J–L). At the final stage of degeneration, a coilin‐positive mass is frequently seen juxtaposed to a nucleolar remnant of fibrillarin at the periphery of the large heterochromatin domains (M–O). Scale bar = 5 µm.
CB alterations and coilin relocalization in PCs of pcd mice were confirmed by immunogold electron microscopy for coilin detection. CBs in control neurons exhibited the typical configuration of round structures composed of closely packed coiled threads decorated with gold particles (Figure 6A). Interestingly, in degenerating PCs CBs frequently showed an irregular morphology with loosely arranged threads, which appeared to be in a process of disassembly (Figure 6B, C). Furthermore, small aggregates of coilin were observed at the nucleolar surface (Figure 6C). Finally, immunoelectron microscopy also revealed the presence of electrondense bodies surrounded by a shell of amorphous material decorated with gold particles of coilin immunoreactivity (Figure 6D). These structures clearly represent the ultrastructural counterpart of fibrillarin masses with their peripheral layer of coilin detected with immunofluorescence. In conclusion, the assembly/disassembly cycle of CBs is dramatically altered in degenerating PCs of pcd mice, parallel to the disruption of nucleolar structure and function. The disassembly of coiled threads of CBs may lead to the reduction in the number of these nuclear organelles. Interestingly, the close association of coilin and fibrillarin is preserved in all stages of PC degeneration: in CBs and around the nucleolus and nucleolar fragments upon disruption of CBs.
Figure 6.
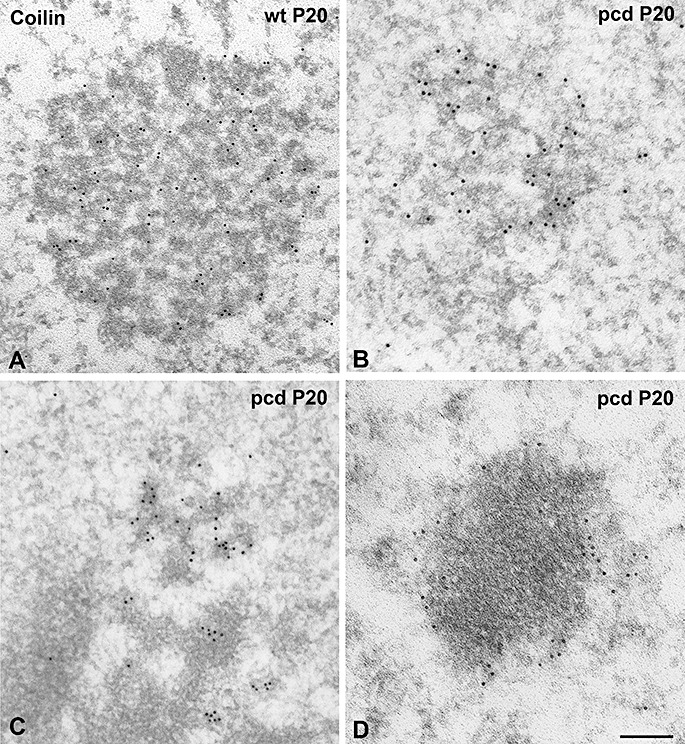
Ultrastructural analysis of Cajal bodies (CBs). In wild‐type PCs, CBs show the typical morphology of round bodies composed of closely packed coiled threads immunogold labeled for coilin (A). (B,C) Mutant PCs exhibit CBs with an irregular morphology and loosely arranged threads, in addition to the presence of small aggregates of coilin at the nucleolar surface (C). Gold particles labeling coilin immunoreactivity are also observed surrounding electrodense masses presumably corresponding to segregated portions of the dense fibrillar component (DFC) (D). Scale bar = 1 µm.
DISCUSSION
PCs of the pcd mouse provide a useful neuronal system to study nuclear mechanisms involved in DNA damage‐dependent neurodegeneration. A defective DNA repair has been involved in the molecular pathology of a growing list of neurodegenerative disorders [for review, see 20, 46, 58]. In the case of PCs, the inherited spinocerebellar ataxias represent an important group of human neurodegenerative diseases with PC degeneration phenotypes, some of them caused by mutations of DNA repair genes 17, 68, 69. Although the pcd mutation is a model of ataxic mouse, it shares clinical and pathological characteristics of inherited human spinocerebellar ataxias, such as difficulties in motor coordination and degeneration and loss of PCs [for review, see 19, 72]. A recent study on the protein‐protein interaction network for human ataxias and mouse models of PC degeneration, including the pcd mouse, has revealed that many ataxia‐causing proteins interact in an ataxia‐based network (44). Indeed, these human and mouse neurodegenerative disorders, although have different mutated genes, share molecular pathways that explain their phenotypic similarities 44, 61. Moreover, Gene Ontology analysis has revealed that nuclear compartments are the main cellular targets of the ataxia protein network (44). Taken together, these data point to the usefulness of the pcd mutant mouse as a model of PC degeneration and clinical cerebellar ataxia.
Our results indicate that PC degeneration in pcd mice involves a progressive accumulation of DNA damage, which is associated with the disruption of nucleoli and CBs and disassembly of protein synthesis machinery that ultimately leads to neuronal cell death. Nuclear hallmarks of PC degeneration are: (i) increased accumulation of the DNA‐damage signal γH2AX; (ii) disruption of the nucleolus with inhibition of rRNA synthesis and ribosome biogenesis; and (iii) disassembly and reduction of CBs. Our finding by microarray analysis that four genes encoding negative regulators of rDNA transcription (p53, Rb, PTEN and SNF2) are upregulated in the cerebellum of pcd mice suggests the activation of repressors of rRNA synthesis in PC neurodegeneration. This view is also consistent with previous Gene Ontology analysis illustrating that transcription corepressor activity is a molecular function with significantly enhanced representation in the ataxia network of protein interactions (44). Furthermore, our immunofluorescence data are consistent with the contribution of p53 to PC degeneration of pcd mice. It is well known that p53 is activated by DNA damage and can trigger DNA repair or apoptosis [for a review, see (46)]. The recruitment of p53 to PML bodies detected in pcd mice PCs may enhance p53 activity by inducing its phosphorylation and acetylation mediated by kinases (HIPK2, CK1) and the acetyl transferase CBP, two post‐tranlational modifications that occur in PML bodies in response to various cellular stresses 3, 6.
Under physiological conditions, the γH2AX signal was not detected by immunofluorescence in PCs of the P20 wild‐type mouse. In pcd mice PCs, however, DNA breaks labeled with the anti‐γH2AX antibody initially appear as numerous nuclear foci that propagate and coalesce into large nuclear domains of genome as PC degeneration proceeds. This finding, and our previous demonstration of DNA breaks in mitral cells (70), clearly support that the accumulation of DNA lesions is a key component in PC neurodegeneration of the pcd mouse. Although degenerating PCs are able to trigger the γH2AX signaling response to DNA damage, the persistence and progressive increase of this signal suggest that DNA lesions are refractory to repair in the mutant animals. This interpretation is consistent with the growing evidence that a defective DNA repair is engaged in the pathogenesis of several ataxias with PC degeneration phenotype 17, 20, 68, 69.
Nucleolar alterations associated with the progressive accumulation of DNA damage in PCs include a reduction in the number of FCs, segregation of nucleolar components, inhibition of rRNA synthesis and fragmentation of the nucleolus. Regarding FCs, nucleolar domains enriched in molecular components of the rRNA transcription machinery 60, 65, previous studies in other neuronal and non‐neuronal cells indicate that the number of FCs positively correlates with cell size and transcriptional activity 5, 41, 57. In this context, we envisage that the reduction of FCs detected in pcd mice PCs represents an early manifestation of the disruption of rRNA synthesis machinery. Subsequently, the segregation of DFC and GC in large separated masses is a typical morphological feature of a severe nucleolar dysfunction and it has been demonstrated in cells treated with drugs that interfere with transcription of the rDNA, such as actinomycin D and doxoburicin 12, 52, 54. Moreover, the recent observation that the inhibition of rRNA synthesis triggers apoptosis in cultured cortical neurons reflects the importance of nucleolar transcription for neuronal survival 28, 32. Similarly, our finding of the nucleoplasmic translocation of B23, a resident nucleolar chaperone involved in ribosome particle assembly and nucleocytoplasmic transport (23), has been associated with states of inhibition of rRNA synthesis in culture cell lines (74). In line with this, the existence of gene silencing in nucleolar and extranucleolar domains of the genome is clearly demonstrated by the dramatic reduction of 5′‐FU incorporation into nascent RNA detected here in degenerating PCs of pcd mice and also by the absence of detectable in situ transcriptional activity in advanced stages of PC degeneration. Collectively, these data suggest that the accumulation of unrepaired DNA induces a nucleolar stress response with severe and progressive inhibition of both rRNA synthesis and nucleolar assembly of preribosomal particles in pcd mice PCs. In support of this view, a previous study (37) has demonstrated a direct link between DNA breaks and block of rRNA synthesis in laser micro‐irradiated nucleoli of wild‐type mouse embryonic fibroblasts. These authors found a transient block in rRNA synthesis in irradiated nucleoli, whereas transcription was preserved in undamaged nucleoli of the same cell. Interestingly, the resumption of rRNA synthesis in irradiated cells correlated with the disappearance of γH2AX foci, suggesting that restoration of transcription is dependent on a successful repair of DNA damage (37).
What is noteworthy is the close relationship between nucleolar disruption and disassembly of protein synthesis machinery observed in pcd mice PCs. The disorganization of RER arrays and the persistent accumulation of free polyribosomes are well known ultrastructural features of PC degeneration in the pcd mouse (42). The initial disassembly of RER may contribute to the neuronal dysfunction by interfering with the vesicular transport of proteins, via the Golgi complex, to different cellular targets (2). An important additional finding in our results is the accumulation of free monoribosomes observed in PCs with macrosegregation of nucleolar components, suggesting that nucleolar disruption and disassembly of polyribosmes are linked cellular processes. In fact, the disassembly of polyribosomes into free monoribosomes lacking mRNA templates is a late cellular sign in degenerating PCs and reflects a blockade of protein synthesis. In nervous tissue, the formation of free monoribosomes correlates with a dramatic decay of 28S and 18S rRNAs (38) and has been associated with programmed cell death (33). Interestingly, in the case of PC degeneration in pcd mice, a fraction of free monoribosomes appear specifically sequestered into autophagic vesicles bounded by RER membranes, a process termed ribophagy (35). This observation suggests that autophagy‐related pathways are involved in the selective degradation of ribosomes in degenerating PCs of the pcd mouse.
Nucleolus and CBs are nuclear epicenters of the RNA world and their dysfunction could have important consequences for both protein synthesis and information processing and storage in neurons 31, 59. In our experimental system of PC degeneration in pcd mice, CBs are sensible nuclear targets of the general inhibition of RNA synthesis and processing, as shown by their structural disassembly, reduction in their number and redistribution of coilin to the periphery of nucleolar fragments. Although disruption of CBs has been reported in experimental models of transcriptional inhibition [for review, see 15, 39, 45], little information is available on the behavior of CBs in neurodegenerative disorders, with the exception of the spinal muscular atrophy, in which the reduction in the level of SMN protein alters the organization of CBs and associated SMN‐containing “gems”(43), and our previous observation in mitral cells of pcd mice (70). Unlike the experimental model of ultraviolet (UV)‐induced fragmentation of CBs, in which coilin relocalizes to nucleoplasmic microfoci (15), we have not observed nucleoplasmic accumulation of coilin in the DNA damage‐induced neurodegenation of PCs. Instead, coilin redistributes around both nucleoli and nucleolar fragments and ultimately coilin and fibrillarin form cap‐like structures apposed to heterochromatin clumps. Although these two cellular models of DNA damage, UV‐irradiation and pcd mutation, share a down‐regulation of transcription, our results suggest that the relocalization of coilin to different nuclear targets is not simply an indirect effect of transcription inhibition. Moreover, our immunocytochemical data support a cellular mechanism of disassembly of CBs mediated by the initial loosening of coiled threads followed by their fragmentation. Given the importance of CBs for the processing of pre‐mRNAs and pre‐rRNAs 14, 24, 48, the disassembly and loss of CBs may reflect the response of PCs in pcd mice to a reduced demand of RNA processing, and also supports the participation of these nuclear bodies in cellular stress‐response pathways.
Although the pcd mutation is a model of ataxic mouse with DNA damage, other perturbations of the ataxia protein network might cause nucleolar and CB alterations without the involvement of DNA damage, particularly those that interfere with global transcriptional activity and nuclear pre‐mRNA processing. Interestingly, three main hub proteins (RBM9, A2BP1 and RBPMS) in the ataxia network, which interact with several different ataxia‐causing proteins, are RNA‐binding proteins involved in pre‐mRNA splicing and maturation [for a review, see (44)]. However, in the case of the pcd ataxic mouse, the accumulation of DNA lesions profoundly impacts upon nuclear functions in PCs, as is indicated by the progressive transcriptional silencing. In this context, we propose that the disruption of the nucleolus and CBs are early and sensitive nuclear hallmarks of DNA damage‐induced neuronal degeneration in PCs and, presumably, in other neuronal types.
ACKNOWLEDGMENTS
The authors wish to thank Raquel García‐Ceballos and Saray Pereda for technical assistance. This work was supported by the following grants: Dirección General de Investigación (BFU2008‐00175); Instituto de Salud Carlos III (CIBERNED, CB06/05/0037), Ministerio de Ciencia y Tecnología (BFU2010‐18284), Ministerio de Sanidad, Política Social e Igualdad (Plan Nacional Sobre Drogas), Instituto de Formación e Investigación Marqués de Valdecilla (IFIMAV, FMV/UC09‐02), Junta de Castilla y León, Centro en Red de Medicina Regenerativa y Terapia Celular de Castilla y León and Fundación Memoria D. Samuel Solórzano‐Barruso, all of them from Spain.
REFERENCES
- 1. Abella N, Brun S, Calvo M, Tapia O, Weber JD, Berciano MT et al (2010) Nucleolar disruption ensures nuclear accumulation of p21 upon DNA damage. Traffic 11:743–755. [DOI] [PubMed] [Google Scholar]
- 2. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2008) Molecular Biology of the Cell, Garland Science. New York. [Google Scholar]
- 3. Alsheich‐Bartok O, Haupt S, Alkalay‐Snir I, Saito S, Appella E, Haupt Y (2008) PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene 27:3653–3661. [DOI] [PubMed] [Google Scholar]
- 4. Andrade LE, Chan EK, Raska I, Peebles CL, Roos G, Tan EM (1991) Human autoantibody to a novel protein of the nuclear coiled body: immunological characterization and cDNA cloning of p80‐coilin. J Exp Med 173:1407–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berciano MT, Novell M, Villagra NT, Casafont I, Bengoechea R, Val‐Bernal JF, Lafarga M (2007) Cajal body number and nucleolar size correlate with the cell body mass in human sensory ganglia neurons. J Struct Biol 158:410–420. [DOI] [PubMed] [Google Scholar]
- 6. Bernardi R, Pandolfi PP (2007) Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8:1006–1016. [DOI] [PubMed] [Google Scholar]
- 7. Blanks JC, Mullen RJ, Lavail MM (1982) Retinal degeneration in the pcd cerebellar mutant mouse: 2. Electron‐microscopic analysis. J Comp Neurol 212:231–246. [DOI] [PubMed] [Google Scholar]
- 8. Boisvert FM, van Koningsbruggen S, Navascués J, Lamond AI (2007) The multifunctional nucleolus. Nat Rev Mol Cell Biol 8:574–585. [DOI] [PubMed] [Google Scholar]
- 9. Bransjevic I, Hof PR, Steinbusch HW, Schmitz C (2008) Accumulation of nuclear DNA damage or neuron loss: molecular basis for a new approach to understanding selective neuronal vulnerability in neurodegenerative diseases. DNA Repair 7:1087–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carvalho T, Almeida F, Calapez A, Lafarga M, Berciano MT, Carmo‐Fonseca M (1999) The spinal muscular atrophy disease gene product, SMN: a link between snRNP biogenesis and the Cajal (coiled) body. J Cell Biol 147:715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Casafont I, Navascués J, Pena E, Lafarga M, Berciano MT (2006) Nuclear organization and dynamics of transcription sites in rat sensory ganglia neurons detected by incorporation of 5′‐fluororidine into nascent RNA. Neuroscience 140:453–462. [DOI] [PubMed] [Google Scholar]
- 12. Casafont I, Bengoechea R, Navascués J, Pena E, Berciano MT, Lafarga M (2007) The giant fibrillar center: a nucleolar structure enriched in upstream binding factor (UBF) that appears in transcriptionally more active sensory ganglia neurons. J Struct Biol 159:451–461. [DOI] [PubMed] [Google Scholar]
- 13. Chakrabarti L, Zhara R, Jackson SM, Kazemi‐Esfarjani P, Sopher BL, Mason AG et al (2010) Mitochondrial dysfunction in NnaD mutant flies and purkinje cell degeneration mice reveals a role for Nna proteins in neuronal bioenergetics. Neuron 66:835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cioce M, Lamond AI (2005) Cajal bodies: a long history of discovery. Annu Rev Cell Dev Biol 21:105–131. [DOI] [PubMed] [Google Scholar]
- 15. Cioce M, Boulon S, Matera AG, Lamond AI (2006) UV‐induced fragmentation of Cajal bodies. J Cell Biol 175:401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Darzacq X, Jády BE, Verheggen C, Kiss AM, Bertrand E, Kiss T (2002) Cajal body‐specific small nuclear RNAs: a novel class of 2′‐O‐methylation and pseudouridylation guide RNAs. EMBO J 21:2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S et al (2001) Early‐onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet 29:184–188. [DOI] [PubMed] [Google Scholar]
- 18. Deisenroth C, Zhang Y (2010) Ribosome biogenesis surveillance: probing the ribosomal protein‐Mdm2‐p53 pathway. Oncogene 29:4253–4260. [DOI] [PubMed] [Google Scholar]
- 19. Dusart I, Guenet JL, Sotelo C (2006) Purkinje cell death: differences between developmental cell death and neurodegenerative death in mutant mice. Cerebellum 5:163–173. [DOI] [PubMed] [Google Scholar]
- 20. Enokido Y, Tamura T, Ito H, Arumughan A, Komuro A, Shiwaku H et al (2010) Mutant huntingtin impairs Ku70‐mediated DNA repair. J Cell Biol 189:425–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fernández‐Capetillo O, Lee A, Nussenzweig M, Nussenzweig A (2004) H2AX: the histone guardian of genome. DNA Repair 3:959–967. [DOI] [PubMed] [Google Scholar]
- 22. Fernández‐González A, La Spada AR, Treadaway J, Higdon JC, Harris BS, Sidman RL et al (2002) Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy‐induced gene, Nna1. Science 295:1904–1906. [DOI] [PubMed] [Google Scholar]
- 23. Frehlick LJ, Eirín‐López JM, Ausió J (2007) New insights into the nucleophosmin/nucleoplasmin family of nuclear chaperones. Bioessays 29:49–59. [DOI] [PubMed] [Google Scholar]
- 24. Gall JG (2000) Cajal bodies: the first 100 years. Annu Rev Cell Dev Biol 16:273–300. [DOI] [PubMed] [Google Scholar]
- 25. García‐Segura LM, Lafarga M, Berciano MT, Hernández P, Andrés MA (1989) Distribution of nuclear pores and chromatin organization in neurons and glial cells of the rat cerebellar cortex. J Comp Neurol 290:440–450. [DOI] [PubMed] [Google Scholar]
- 26. Greer CA, Shepherd GM (1982) Mitral cell degeneration and sensory function in the neurological mutant mouse Purkinje‐cell degeneration (pcd) . Brain Res 235:156–161. [DOI] [PubMed] [Google Scholar]
- 27. Harris A, Morgan JI, Pecot M, Soumare A, Osborne A, Soares HD (2000) Regenerating motor neurons express Nna 1, a novel ATP/GTP‐binding protein related to zinc carboxypeptidases. Mol Cell Neurosci 16:578–596. [DOI] [PubMed] [Google Scholar]
- 28. Hetman M, Vashishta A, Rempala G (2010) Neurotoxic mechanisms of DNA damage: focus on transcriptional inhibition. J Neurochem 114:1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264. [DOI] [PubMed] [Google Scholar]
- 30. Ito M (1984) The Cerebellum and Neural Control. Raven Press: New York. [Google Scholar]
- 31. Jordan BA, Fernholz BD, Khatri L, Ziff EB (2007) Activity‐dependent AIDA‐1 nuclear signaling regulates nucleolar numbers and protein synthesis in neurons. Nat Neurosci 10:427–435. [DOI] [PubMed] [Google Scholar]
- 32. Kalita K, Makonchuk D, Gomes C, Zheng JJ, Hetman M (2008) Inhibition of nucleolar transcription as a trigger for neuronal apoptosis. J Neurochem 105:2286–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kinch G, Hoffman KL, Rodrigues EM, Zee MC, Weeks JC (2003) Steroid‐triggered programmed cell death of a motoneuron is autophagic and involves structural changes in mitochondria. J Comp Neurol 457:384–403. [DOI] [PubMed] [Google Scholar]
- 34. Kiss AM, Jády BE, Darzacq X, Verheggen C, Bertrand E, Kiss T (2002) A Cajal body‐specific pseudouridylation guide RNA is composed of two box H/ACA snoRNA‐like domains. Nucleic Acids Res 30:4643–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kraft C, Deplazes A, Sohrmann M, Peter M (2008) Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol 10:602–610. [DOI] [PubMed] [Google Scholar]
- 36. Krüger T, Scheer U (2010) p53 localizes to intranucleolar regions distinct from the ribosome production compartments. J Cell Sci 123:1203–1208. [DOI] [PubMed] [Google Scholar]
- 37. Kruhlak M, Crouch EE, Orlov M, Montaño C, Gorski SA, Nussenzweig A et al (2007) The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 447:730–734. [DOI] [PubMed] [Google Scholar]
- 38. Lafarga M, Lerga A, Andrés MA, Polanco JI, Calle E, Berciano MT (1997) Apoptosis induced by methylazoxymethanol in developing rat cerebellum. Cell Tissue Res 289:25–38. [DOI] [PubMed] [Google Scholar]
- 39. Lafarga M, Berciano MT, García‐Segura LM, Andrés MA, Carmo‐Fonseca M (1998) Acute osmotic/stress stimuli induce a transient decrease of transcriptional activity in the neurosecretory neurons of supraoptic nuclei. J Neurocytol 27:205–217. [DOI] [PubMed] [Google Scholar]
- 40. Lafarga M, Casafont I, Bengoechea R, Tapia O, Berciano MT (2009) Cajal's contribution to the knowledge of the neuronal cell nucleus. Chromosoma 118:437–443. [DOI] [PubMed] [Google Scholar]
- 41. Lamond AI, Spector DL (2003) Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 4:605–612. [DOI] [PubMed] [Google Scholar]
- 42. Landis SC, Mullen RJ (1978) The development and degeneration of Purkinje cells in pcd mutant mice. J Comp Neurol 177:125–144. [DOI] [PubMed] [Google Scholar]
- 43. Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A et al (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16:265–269. [DOI] [PubMed] [Google Scholar]
- 44. Lim J, Hao T, Shaw C, Patel AJ, Szabó G, Rual JF et al (2006) A protein‐protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 125:801–814. [DOI] [PubMed] [Google Scholar]
- 45. Malatesta M, Luchetti F, Marcheggiani F, Fakan S, Gazzanelli G (2001) Disassembly of nuclear bodies during arousal from hibernation: an in vitro study. Chromosoma 110:471–417. [DOI] [PubMed] [Google Scholar]
- 46. Martin LJ (2008) DNA damage and repair: relevance to mechanisms of neurodegeneration. J Neuropathol Exp Neurol 67:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matera AG, Izaguire‐Sierra M, Praveen K, Rajendra TK (2009) Nuclear bodies: random aggregates of sticky proteins or crucibles of macromolecular assembly? Dev Cell 17:639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nizami Z, Deryusheva S, Gall JG (2010) The Cajal body and histone locus body. Cold Spring Harb Perspect Biol 2:a000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. O'Gorman S, Sidman RL (1985) Degeneration of thalamic neurons in “Purkinje cell degeneration” mutant mice. I. Distribution of neuron loss. J Comp Neurol 234:277–297. [DOI] [PubMed] [Google Scholar]
- 50. Palay SL, Chan‐Palay V (1974) The Cerebellar Cortex. Cytology and Organization. Springer‐Verlag: Berlin‐Heidelberg‐New York. [Google Scholar]
- 51. Pederson T, Tsai RY (2009) In search of nonribosomal nucleolar protein function and regulation. J Cell Biol 184:771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pena E, Berciano MT, Fernández R, Crespo P, Lafarga M (2000) Stress‐induced activation of c‐Jun N‐terminal kinase in sensory ganglion neurons: accumulation in nuclear domains enriched in splicing factors and distribution in perichromatin fibrils. Exp Cell Res 256:179–191. [DOI] [PubMed] [Google Scholar]
- 53. Pena E, Berciano MT, Fernández R, Ojeda JL, Lafarga M (2001) Neuronal body size correlates with the number of nucleoli and Cajal bodies, and with the organization of the splicing machinery in rat trigeminal ganglion neurons. J Comp Neurol 430:250–263. [DOI] [PubMed] [Google Scholar]
- 54. Puvion‐Dutilleul F, Mazan S, Nicoloso M, Pichard E, Bachellerie JP, Puvion E (1992) Alterations of nucleolar ultrastructure and ribosome biogenesis by actinomycin D. Implications for U3 snRNP function. Eur J Cell Biol 58:149–162. [PubMed] [Google Scholar]
- 55. Qi ML, Tagawa K, Enokido Y, Yoshimura N, Wada Y, Watase K et al (2007) Proteome analysis of soluble nuclear proteins reveals that HMGB1/2 suppress genotoxic stress in polyglutamine diseases. Nat Cell Biol 9:402–414. [DOI] [PubMed] [Google Scholar]
- 56. Raska I, Andrade LE, Ochs RL, Chan EK, Chang CM, Roos G, Tan EM (1991) Immunological and ultrastructural studies of the nuclear coiled body with autoimmune antibodies. Exp Cell Res 195:27–37. [DOI] [PubMed] [Google Scholar]
- 57. Raska I, Shaw PJ, Cmarko D (2006) New insights into nucleolar architecture and activity. Int Rev Cytol 255:177–235. [DOI] [PubMed] [Google Scholar]
- 58. Rass U, Ahel I, West SC (2007) Defective DNA repair and neurodegeneration disease. Cell 130:991–1004. [DOI] [PubMed] [Google Scholar]
- 59. Richter JD, Fallon JR (2007) Synapses go nucle(ol)ar. Nat Neurosci 10:399–400. [DOI] [PubMed] [Google Scholar]
- 60. Roussel P, André C, Masson C, Géraud G, Hernandez‐Verdun D (1993) Localization of the RNA polymerase I transcription factor hUBF during the cell cycle. J Cell Sci 104:327–337. [DOI] [PubMed] [Google Scholar]
- 61. Rubinsztein DC (2006) Protein‐protein interaction networks in the spinocerebellar ataxias. Genome Biol 7:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ruggero D, Pandolfi PP (2003) Does the ribosome translate cancer? Nat Rev Cancer 3:179–192. [DOI] [PubMed] [Google Scholar]
- 63. Rutten BPF, Schmitz C, Gerlach OHH, Oyen HM, de Mesquita EB, Steinbusch HW, Korr H (2007) The aging brain: accumulation of DNA damage or neuron loss? Neurobiol Aging 28:91–98. [DOI] [PubMed] [Google Scholar]
- 64. Santoro R, Li J, Grummt I (2002) The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat Genet 32:393–396. [DOI] [PubMed] [Google Scholar]
- 65. Sirri V, Urcuqui‐Inchima S, Roussel P, Hernandez‐Verdun D (2008) Nucleolus: the fascinating nuclear body. Histochem Cell Biol 129:13–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stanek D, Neugebauer KM (2006) The Cajal body: a meeting place for spliceosomal snRNPs in the nuclear maze. Chromosoma 115:343–354. [DOI] [PubMed] [Google Scholar]
- 67. Stefanovsky V, Langlois F, Gagnon‐Kugler T, Rothblum LI, Moss T (2006) Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r‐chromatin remodeling. Mol Cell 21:629–639. [DOI] [PubMed] [Google Scholar]
- 68. Suraweera A, Becherel OJ, Chen P, Rundle N, Woods R, Nakamura J et al (2007) Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol 177:969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D et al (2002) Mutation of TDP1, encoding a topoisomerase I‐dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 32:267–272. [DOI] [PubMed] [Google Scholar]
- 70. Valero J, Berciano MT, Weruaga E, Lafarga M, Alonso JR (2006) Pre‐neurodegeneration of mitral cells in the pcd mutant mouse is associated with DNA damage, transcriptional repression, and reorganization of nuclear speckles and Cajal bodies. Mol Cell Neurosci 33:283–295. [DOI] [PubMed] [Google Scholar]
- 71. Voit R, Schäfer K, Grummt I (1997) Mechanism of repression of RNA polymerase I transcription by the retinoblastoma protein. Mol Cell Biol 17:4230–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang T, Morgan JI (2007) The Purkinje cell degeneration (pcd) mouse: an unexpected molecular link between neuronal degeneration and regeneration. Brain Res 1140:26–40. [DOI] [PubMed] [Google Scholar]
- 73. Wang T, Parris J, Leyi L (2006) The carboxypeptidase‐like substrate‐binding site in Nna1 is essential for the rescue of the Purkinje cell degeneration (pcd) phenotype. Mol Cell Neurosci 3:200–213. [DOI] [PubMed] [Google Scholar]
- 74. Yung BY, Bor AM, Chan PK (1990) Short exposure to actinomycin D induces “reversible” translocation of protein B23 as well as “reversible” inhibition of cell growth and RNA synthesis in HeLa cells. Cancer Res 50:5987–5991. [PubMed] [Google Scholar]
- 75. Zhai W, Comai L (2000) Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol Cell Biol 20:5930–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang C, Comai L, Johnson DL (2005) PTEN represses RNA Polymerase I transcription by disrupting the SL1 complex. Mol Cell Biol 25:6899–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]