

Abstract
Several lines of evidence support protective as well as deleterious effects of oleic acid (OA) on Alzheimer's disease (AD) and other neurological disorders; however, the bases of these effects are unclear. Our investigation demonstrates that amyloid precursor protein (APP) 695 transfected Cos‐7 cells supplemented with OA have reduced secreted amyloid‐beta (Aβ) levels. An early‐onset AD transgenic mouse model expressing the double‐mutant form of human APP, Swedish (K670N/M671L) and Indiana (V717F), corroborated our in vitro findings when they were fed a high‐protein, low‐fat (18% reduction), cholesterol‐free diet enriched with OA. These mice exhibited an increase in Aβ40/Aβ42 ratio, reduced levels of beta‐site APP cleaving enzyme (BACE) and reduced presenilin levels along with reduced amyloid plaques in the brain. The decrease in BACE levels was accompanied by increased levels of a non‐amyloidogenic soluble form of APP (sAPPα). Furthermore, the low‐fat/+OA diet resulted in an augmentation of insulin‐degrading enzyme and insulin‐like growth factor‐II. These results suggest that OA supplementation and cholesterol intake restriction in a mouse model of AD reduce AD‐type neuropathology.
Keywords: Alzheimer's disease, amyloid, Cos‐7 cells, oleic acid, plaques, transgenic mice
INTRODUCTION
The brains of Alzheimer's disease (AD) patients are characterized by the deposition of amyloid plaques (26). The amyloid plaques consist of different forms of the amyloid‐beta (Aβ) derivatives of the amyloid precursor protein (APP) (28), which are produced by the cleavage of APP by two proteolytic enzymes, β‐ and γ‐secretases. Out of various Aβ peptides produced, products of 40 (Aβ40) and in particular 42 (Aβ42) residues are the most common constituents of amyloid plaques (37). Moreover, soluble form of oligomeric Aβ are widely accepted as the prime trigger for AD pathogenesis, resulting in neuronal loss with consequential dementia and death [reviewed in (27)].
There is a substantial amount of data that conferred a protective 11, 32, 33 as well as deleterious effects 21, 25, 43 of oleic acid (OA; 18:1 n‐9) on AD and other neurological disorders. For instance, a significant decrease in monounsaturated fatty acids (FAs), including OA, has been reported in the frontal cortex and hippocampus of AD brains 11, 33. OA has also been shown to significantly inhibit the activity of prolyl endopeptidase (32); prolyl endopeptidase levels are significantly increased in AD brains, suggesting their functional role in brain amyloidogenesis (3). However, the biochemical and pathological bases whereby OA affects AD remain unclear. In addition, none of the previous studies had examined cholesterol‐free diets in combination with low‐fat intake and enriched OA.
In this study for the first time, we show that the brains of AD mice on low‐fat/+OA diet show multifactorial protective effects in pathways important to the development of amyloid plaques. These results suggest that regulation of dietary OA intake may offer a new tool to reduce the risk of AD.
MATERIALS AND METHODS
Cell culture
Cos‐7 cells were stably transfected with pCEP‐APP695 (encoding human APP695) plasmid maintained in DMEM (Sigma, Germany) with 10% fetal calf serum (FCS, PAN, Germany) and 200 µg/mL hygromycin (14). Cells were cultured in 10‐cm dishes, washed three times with FCS‐free media (to get rid of the lipids present in the FCS) and incubated for 12 h with delipidated‐FCS DMEM containing 0.01–25 µM of OA–bovine serum albumin (BSA) complexes. OA containing DMEM were renewed every 4 h to maintain steady concentration of lipid. The 8‐ to 12‐h conditioned media were analyzed. Cells grew equally well as determined by cell protein concentration and maintained normal morphology at all concentrations of OA used.
OA preparation
OA (Nu‐Chek‐Prep, Inc., USA), organic solvents and BSA solution were purged with argon extensively, prior to use. The protocols to form a fresh OA–BSA (2:1) complex are based on established procedures (2).
Mice, treatment and tissue preparation
All experimental procedures were performed according to the animal care guidelines of the University of Western Ontario (approval ID: 2004‐065‐06). All studies were performed on 5‐month‐old congenic C57BL/6J male mice heterozygous for TgCRND8, which express a double‐mutant form of human APP695: Swedish (K670N/M671L) and Indiana (V717F) (6). TgCRND8 mice demonstrate severe AD‐like amyloid pathogenesis, including A brain deposits as early as 3 months of age (6). Mice used in these studies were housed on 12 h light/dark cycles. At 3 weeks of age, TgCRND8 mice were provided ad libitum access to water and one of three diets (n = 6 for each diet) until 20–21 weeks of age, at which time they were sacrificed by cervical decapitation. Brains were rapidly removed and sectioned sagitally into hemibrains. One hemibrain was used for either biochemical [protein, RNA and enzyme‐linked immunosorbent assays (ELISAs)] or lipid (OA) analyses. The other hemibrain was fixed in 10% neutral buffered formalin and 0.1 M Phosphate buffered saline (PBS) for 48 h and then stored at 4°C in PBS and 1% sodium azide until it is used for immunohistochemistry.
Dietary manipulations
The experimental diets involved the following three groups: (i) conventional mouse chow (PicoLab Mouse Chow Diet 20, Purina Mills, St. Louis, MO, USA); (ii) soybean oil (1%), high‐protein, cholesterol‐free control diet (low fat/−OA; L10047, Research Diets, New Brunswick, NJ, USA) and (iii) soybean oil (1%), high‐protein, cholesterol‐free diet supplemented with 2% OA (low fat/+OA). The compositions of ii and iii diets were as follows: alcohol extracted casein (304.7 g/kg), DL‐methionine (4.6 g/kg), sucrose (279.3 g/kg), maltodextrin (Fro‐Dex) (284.4 g/kg), vitamin mix (V10001, AIN‐76A) (10.2 g/kg), mineral mix (S10001, AIN‐76A; Research Diets, New Brunswick, NJ, USA) (35.0 g/kg), cellulose (40.6 g/kg), xanthan gum (8.6 g/kg) and soybean oil (10.2 g/kg) with and without OA (20.0 g/kg). Chow diet was used to compare the effects of low‐fat/−OA diet alone on AD parameters.
Quantitative Western blotting
Western blot to analyze proteins has been described previously (5). Blots were probed with anti‐APP (A‐8717, 1:15 000) and anti‐nicastrin (NCT, N‐1660) from Sigma, anti‐β site APP cleaving enzyme (BACE)–C‐terminal fragment (CTF) (1:1000, a gift from Dr Michael Willem), anti‐β‐actin (1:5000) from Santa Cruz (sc‐1616), anti‐insulin‐degrading enzyme (IDE)‐1 (1:4000) and anti‐soluble APPα (sAPPα) (1:5000, gifts from Dr Dennis Selkoe), anti‐presenilin 1‐N terminal fragment (PS1‐NTF) (1:2500, a gift from Dr Sam Gandy), anti‐transthyretin (TTR) (1:500, a gift from Dr Sancia Gaetani), anti‐insulin‐like growth factor‐II (IGF‐II) (1:300, a gift from Dr Victor Han) and anti‐prion protein (PrP) (1:5000, a gift from Dr Alex Strom). Secondary antibodies were from Amersham, USA (1:15 000 for goat anti‐rabbit, 1:5000 for goat anti‐mouse). Data analysis was performed using the Molecular Analyst II densitometric software (Bio‐Rad, Hercules, CA, USA).
Real‐time RT‐PCR
RNA isolation, reverse transcription and PCR conditions have been described previously (5). The primers used are: TgAPP, forward: 5′‐GAT GAC GTC TTG GCC AAC ATG‐3′ and reverse: 5′‐CGG AAT TCT GCA TCC AGA TTC AC‐3′ (308‐bp product); murine APP, forward: 5′‐CCG ACG ATG TCT TGG CCA AC‐3′ and reverse: 5′‐CCG AAT TCT GCA TCC ATC TTC AC‐3′ (310‐bp product); PrP, forward: 5′‐GGG GAC AAC CTC ATG GTG GTA GT‐3′ and reverse: 5′‐TCC ACT GGC CTG TAG TAC ACT TGG‐3′ (283‐bp product) and β‐actin, forward: 5′‐TCG TGG GCC GCT CTA GGC ACC A‐3′ and reverse: 5′‐GTT GGC CTT AGG GTT CAG GGG GG‐3′ (256‐bp product). Data analysis was performed using the BioRad GeneX (Bio‐Rad, Hercules, CA, USA).
Aβ sandwich ELISAs
Quantification of Aβ40 and Aβ42 production in conditioned media (18) and in brain (Signet Laboratories, Dedham, MA, USA) was carried out by standard ELISA methods.
Fatty acid analysis
FAs in the brain were isolated and methylated according to Moser and Moser (29). The fatty acid methyl ester (FAME) mixture was then re‐suspended in hexane and analyzed by gas chromatography–mass spectroscopy (GC‐MS). GC‐MS analysis was performed on a Hewlett‐Packard Series II 5890 gas chromatograph (Hewlett‐Packard, Wilmington, DE, USA). coupled to an HP‐5971 mass spectrometer equipped with a Supelcowax SP‐10 capillary column (Supelco, Bellefonte, PA, USA) . FAME mass was determined by comparing areas of unknown FAMEs to those of a fixed concentration of 17:0 internal standard.
Immunohistochemistry and image analysis
Plaque density was measured using immunohistochemistry with a biotinylated‐4G8 monoclonal antibody (Signet Laboratories), directed against amino acids 17–24 of the β‐amyloid peptide. The mean area of the field (0.307 mm2) was examined in three regions of neocortex, hippocampus and amygdala: 0.5–0.9 mm, 0.9–1.35 mm, and 1.35–1.8 mm mediolateral from the bregma (Mouse Brain Atlas, Franklin and Paxinos 2007) on 40 μm thick rostral (cross) sections of each hemisphere. Plaque density was calculated by dividing the total area of Aβ‐positive structures by the total area of the regions analyzed (in square micrometres).
Statistics
All values were presented as mean ± standard error of the mean (S.E.M.). Protein densitometric, Aβ40 and 42 levels, and plaque denisty were compared by Student's unpaired two‐tailed t‐tests. In vitro Aβ levels and ratios of CTFs to full‐length APP were analyzed using one‐way analysis of variance. The significance level was chosen to be 0.05 (P ≤ 0.05). Correlations between parameters were tested by linear regression analysis.
RESULTS
Effects on CNS lipid profile
Central nervous system (CNS) lipid profile showed a very significant increase in the levels of OA up to twofold (12.01 ± 2.0% distribution or 2842.35 ± 514.59 nmol/g, P < 0.01) compared to low‐fat/−OA control (6.4 ± 2.3% distribution or 1403.28 ± 474.59 nmol/g) and chow mice (8.2 ± 1.1% distribution or 2037.4 ± 6012 nmol/g).
Effects on in vitro Aβ levels
OA supplementation resulted in lower levels of both Aβ peptides in the conditioned media of APP695 transfected Cos‐7 cells starting at as low as 0.01 µM compared to the cells supplemented with BSA (Figure 1A).
Figure 1.
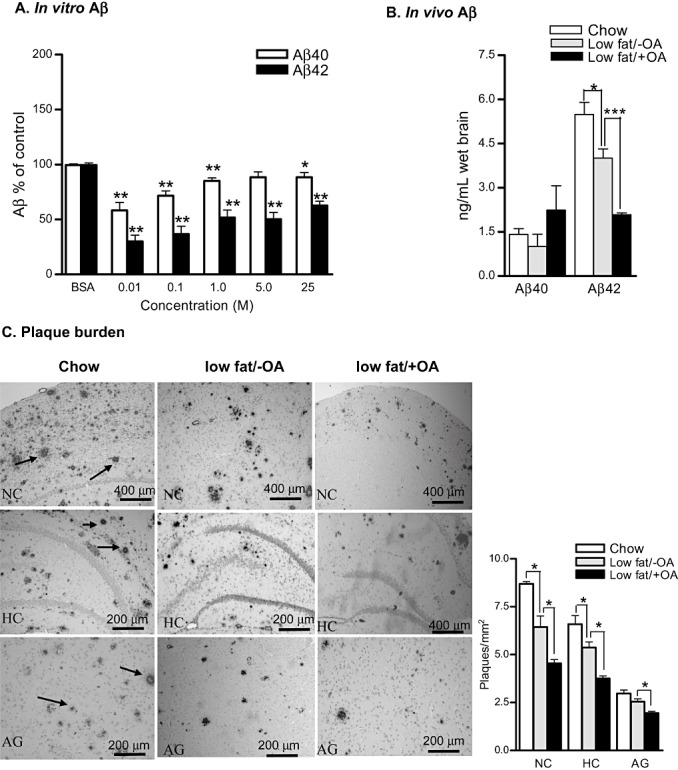
In vitro and in vivo effects of oleic acid (OA) supplement on amyloid‐beta (Aβ) levels and amyloid plaque burden. A. BSA control normalized Aβ levels in the conditioned media after 12‐h incubation with OA (n = 3). B. Enzyme‐linked immunosorbent assay results for Aβ40 and Aβ42 levels in the hippocampus and adjacent cortices of chow, low‐fat/−OA and low‐fat/+OA mice. C. Photomicrographs of Aβ stained amyloid plaque burden (number and size) in the neocortex (NC), hippocampus (HC) and amygdala (AG) of chow, low‐fat/−OA and low‐fat/+OA mice. Arrows indicate Aβ stained plaques. The plot shows the quantitative analysis of plaque density. The mean ± standard error of the mean are shown for all plots (n = 6 for each experiment). ***P < 0.001, **P < 0.01 and *P < 0.05.
Effects on in vivo Aβ levels
The hippocampus and the adjacent cortices of low‐fat/+OA animals demonstrated a non‐significant increase in the Aβ40 levels (2.241 ± 0.83, 1.00 ± 0.41, 1.4±0.19, ng/mL) and a very significant >30% decrease in the Aβ42 levels (2.08 ± 0.14, 4.0 ± 0.7, 5.5±0.40, ng/mL, P < 0.001) compared to low‐fat/−OA mice and chow mice, respectively (Figure 1B).
Effects on plaque pathology
The decrease in Aβ levels following dietary OA enrichment also led to histopathological changes, specifically that of (>30%) decreased amyloid plaque number in the neocortical (4.21 ± 0.23, 6.22 ± 0.63, 8.7 ± 0.11, P < 0.05), hippocampal (3.88 ± 0.20, 5.65 ± 0.22, 6.6 ± 0.43, P < 0.05) and amygdala (1.89 ± 0.09, 2.75 ± 0.15, 2.96 ± 0.18, P < 0.05) regions of low‐fat/+OA mice brains when compared to low‐fat/−OA and chow mice, respectively (Figure 1C).
Effects on in vivoα‐secretase pathway
Western blot showed over 20% decrease in the levels of mature membrane‐bound amyloid precursor protein (APPm; 6.25 ± 0.59 vs. 8.12 ± 0.62, P < 0.05) and ∼50% increase in the levels of neuroprotective sAPPα (9.4 ± 0.31 vs. 6.0 ± 0.65, P < 0.05) in the cortices of low‐fat/+OA mice compared to low‐fat/−OA mice, respectively (Figure 2A).
Figure 2.
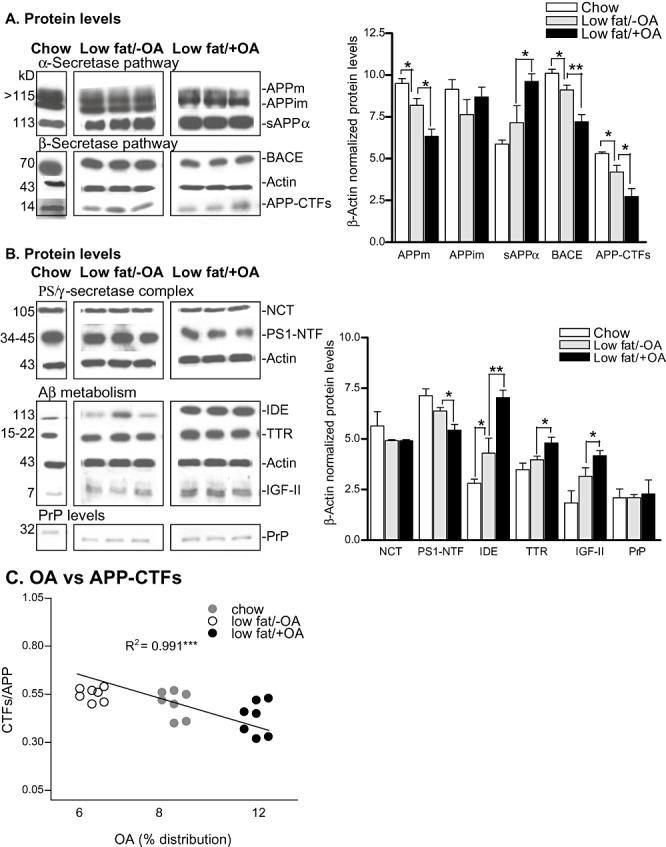
In vivo effects of oleic acid (OA) supplement on brain protein levels. Western blots of mature amyloid precursor protein (APPm), immature amyloid precursor protein (APPim), soluble APPα (sAPPα), β‐site APP cleaving enzyme (BACE), β‐actin and APP–C‐terminal fragments (CTFs) (A) nicastrin (NCT), presenilin 1‐N terminal fragment (PS1‐NTF), insulin‐degrading enzyme (IDE), transthyretin (TTR), insulin‐like growth factor‐II (IGF‐II), prion protein (PrP) and β‐actin protein levels (B) in the frontal cortices of chow, low‐fat/−OA and low‐fat/+OA mice, (n= 6 for each experiment; out of six, only one animal is shown for chow, and three animals are shown for low‐fat/‐OA and low‐fat/+OA mice). Respective molecular weights (kD) are shown on the left. Plots on the right show the quantitative analysis of Western blots. C. Plot shows secretase‐generated APP‐C‐terminal fragment (α/β) ratios vs. OA percentage distribution, The mean +/− standard error for the mean are shown for all plots. **P < 0.01 and *P < 0.05. Abbreviation: Aβ = amyloid‐beta.
Effects on in vivoβ‐secretase pathway
The levels of BACE were >20% lowered in low‐fat/+OA mice (6.83 ± 0.39 vs. 9.0 ± 0.4, P < 0.01) compared to low‐fat/−OA mice (Figure 2A). APP–CTFs levels (2.56 ± 0.49 vs. 3.85 ± 0.29, P < 0.05) were >30% lowered in low‐fat/+OA mice.
Effects on in vivo PS/γ‐secretase enzyme complex
The PS/γ‐secretase enzyme complex generates Aβ from APP–CTFβ. PS1‐NTF levels (5.59 ± 0.28 vs. 6.29 ± 0.18, P < 0.05) were >10% lowered in low‐fat/+OA brains compared to low‐fat/−OA controls (Figure 2B). There was no significant change in NCT levels in either low‐fat/−OA or low‐fat/+OA groups of mice.
Effects on in vivo Aβ metabolism
We next checked the levels of IDE and TTR (proteins responsible for the clearance of Aβ) as well as IGF‐II as a function of diet. Low‐fat/+OA mice showed >50% increases in the IDE levels (6.8 ± 0.40 vs. 4.37 ± 0.81, P < 0.01), >20% increases in IGF‐II levels (4.01 ± 0.40 vs. 3.1 ± 0.22, P < 0.05) and >40% increases in the TTR levels (4.81 ± 0.31 vs. 3.25 ± 0.02, P < 0.05) compared to low‐fat/−OA controls, respectively (Figure 2B).
Effects on PrP levels
The CRND8 transgenic APP (Tg) expression is driven by the PrP promoter; to support the absence of the effect of the different diets on PrP activity, the protein levels of PrP among different groups were investigated. As shown in Figure 2B, the different dietary regimens did not cause any significant change in PrP levels, thereby supporting that both the low‐fat/−OA and low‐fat/+OA diets did not influence the levels of transgene expression.
Effects on secretase products
Significant direct correlation was found between increasing levels of OA (percentage distribution) and CTFs/APP ratios (R 2 = 0.991, P < 0.001) (Figure 2C). Together, these results suggest that there is an association between OA levels and secretase activity.
Effects on WT and transgene expressions
Both the low‐fat/−OA and low‐fat/+OA diets did not have any effect on the expression levels of either CRND8 transgene‐derived APP or endogenous murine APP (Figure 3A). Similar to PrP protein levels, the levels of PrP gene were comparable across the groups (Figure 3B).
Figure 3.
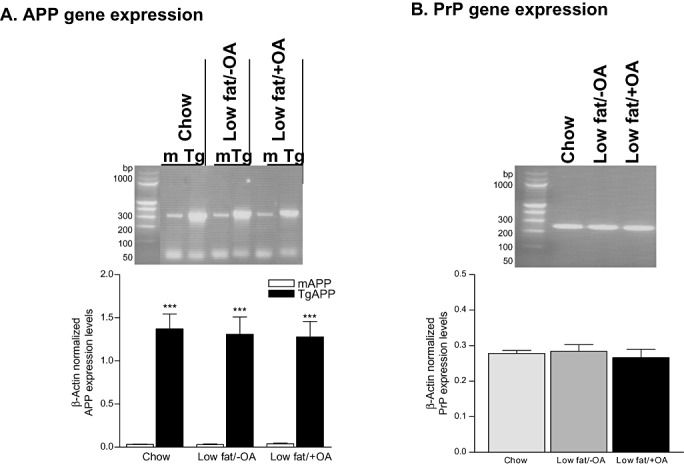
Effects of oleic acid (OA) supplement on gene expression levels. Real‐time RT‐PCR of endogenous murine (m), CRND8 transgene‐specific (Tg) amyloid precursor protein (A) and prion protein (B) in chow, low‐fat/−OA and low‐fat/+OA mice, (n=6 for each experiment, out of 6 only 1 animal is shown for each group). Plots show the quantitative analysis (mean ± S.E.M.) of β‐actin‐normalized gene expression levels, ***p < 0.001.
DISCUSSION
As cholesterol‐enriched diets have been shown to increase BACE level and activity, APP processing, and Aβ42 levels (reviewed in reference 44), we therefore designed a diet depleted in cholesterol and enriched in OA to find out if the protective effects of cholesterol depletion could be enhanced by OA supplementation. Instead of doing a blinded, full‐blown animal study, in pilot experiments we screened OA in the cell culture AD model to generate the pilot data as well as to set up the direction for future in vivo experiments and to do the power analysis. In the cell culture model that produces high levels of Aβ in the medium to mimic transgene‐derived Aβ overproduction in TgCRND8 mice, OA tends to have inhibitory effects on the levels of both Aβ species in the conditioned media. However, as the supplemental concentration of OA is increased, the inhibition of Aβ levels is decreased, most probably attributed to the increased rigidity of the membrane bilayer or altered cellular pH as a result of OA enrichment. Membrane‐rigidifying properties of OA at higher concentrations (when it exceeded 20% of the total fat) have been shown to impair the transport of Aβ from the membranes and to be positively correlated with reference memory errors in AD rats (43). Furthermore, higher concentrations of OA are expected to increase the acidic pH of the cells, and the highest β‐secretase activity is detected at acidic pH 12, 13, 22, 46. In addition, Aβ degradation is mediated by several proteases; given the intracellular localizations and pH optima, these proteases are unlikely to operate at highly acidic pHs (30). We therefore speculate that at a higher OA concentration, the cellular pH is altered, which then exerts a significant biological effect on the intracellular processing of APP, resulting in increased Aβ levels. Correspondingly, OA has been shown to promote the aggregate formation of superoxide dismutase 1 mutants in vitro (21) and caused an elevation in PS1, γ‐secretase and consequently, Aβ levels in Chinese hamster ovary (CHO) PSwt‐1 cells (25) when administered at 2500 and 400 µM, respectively. It is important to mention that transfected Cos‐7 cells are well established as models for examining the effects of AD proteins/genes on a number of parameters 24, 36, 40. Our results also demonstrate that APP protein could be effectively expressed in Cos‐7 cells as already shown 14, 17, 24, 36, 40 and that the transfected Cos‐7 cells could be used as a model of APP protein overexpression to examine the function of APP in mammalian cells 24, 36, 40. In line with our in vitro data, we decided to maintain a lower concentration of OA in the experimental mice diet to enhance its membrane‐fluidizing effects. Here, we show that dietary OA enrichment indeed provides protection against AD‐type neuropathology in the brains of TgCRND8 mice, provided fat intake is lowered (approximately 18% reduction) and cholesterol is depleted from the diet. This outcome appears to be the result of a combination of several underlying factors.
Low‐fat/+OA diet resulted in an overall increase of the alpha (sAPPα) and a decrease in beta (combined APP–CTFs, although the predominant form lowered is CTFβ and not CTFα as a result of >50% elevated sAPPα) proteolytic processing of the membrane‐bound mature APP and corresponding Aβ42 peptides, implying that OA may downregulate Aβ generation either by altering APP trafficking to secretase‐containing compartments of the membrane or the secretase enzymatic activity itself. Although the cause of this shift is unclear, a decrease in primary brain BACE levels is likely to contribute. Though not investigated here, it would be of interest to assess whether the OA‐mediated regulation of BACE levels is stearoyl CoA desaturase (SCD‐1) dependent. SCD‐1 is the enzyme that catalyzes the conversion of long‐chain saturated FA to oleate 39, 41. There have been reports stating that the induction of SCD‐1 activity leads to an increase in cholesterol, plasma lipids and lipoproteins 20, 31. As a feedback mechanism, high OA may down‐regulate SCD‐1 and may consequently lower cholesterol and corresponding diminished BACE levels and activity. In this regard, SCD‐1 has been shown to be upregulated by Aβ treatment (41). Alternatively, the shift could be explained by the altered processing of APP, where the alpha pathway is augmented, resulting in a reciprocal diminishment of the beta pathway.
The decrease in the levels of BACE (and consequently Aβ42 peptides and amyloid plaque burden) also reflects a compensatory or regulatory feedback mechanism stemming from the lower levels of its substrates, APP (47). Cholesterol reduction also appears to augment OA's direct effect on APP processing, as cholesterol reduction has been shown to promote the α‐secretase pathway, elevate sAPPα(23) and reduce 12‐kDa CTFβ(10). Conversely, cholesterol retention enhances β‐secretase 19, 42 and γ‐secretase activities and corresponding Aβ production 9, 45. While the overall reduction of Aβ42 peptides following OA enrichment should be enough to diminish the brain amyloid plaque burden, the concurrent decrease in the levels of PS1‐NTF observed following OA enrichment also supplemented this outcome. In addition to the effects of OA on decreasing PS1‐NTF levels, a previous study showed that lysosomal cholesterol levels are the strong inducers of PS2 transcription (7); therefore, lower dietary cholesterol in a low‐fat/+OA diet may also have suppressed the levels of PS1‐NTF. Nevertheless, as NCT is likely the limiting factor in functional PS/γ‐secretase complex assembly rather than PS1 (which could be present in excess without any effect on γ‐secretase activity) (5), this suggests that the activity of the PS/γ‐secretase complex should not be necessarily reduced with a decrease in PS1 levels. Although no significant change was observed on the levels of NCT, nevertheless, OA's effects on other members of the γ‐secretase complex, such as Pen2 or Aph‐1, cannot be ruled out.
In order to find out the involvement of additional factors in decreasing Aβ levels and amyloid plaques, we next checked the proteins responsible for Aβ degradation, such as IDE, insulin‐like growth factors (IGFs) and TTR. A reduction in Aβ levels is likely to be partially enhanced by an increased degradation of these peptides as a consequence of an over 50% increment in brain IDE levels following OA enrichment. This agrees well with the diminishment in the levels of IDE that is inversely correlated with increased levels of Aβ42 in transgenic mice (16) and human mutant PS2 and PS1 (V97L) expressing cells (34), and perhaps has resulted from the reduced levels of its substrate (35), insulin caused by a low‐fat diet (1) in low‐fat/+OA mice.
Elevated levels of circulating insulin (such as in diabetes) provoke amyloid accumulation by directly competing with Aβ for the IDE, thereby limit Aβ degradation by IDE(15). The significant elevation in IGF‐II levels in low‐fat/+OA mice supports and extends earlier findings, where IGFs (IGF‐1 & IGF‐II) have been demonstrated to protect against Aβ toxicity in vitro (8). In addition, OA, along with LA, has been shown to enhance the growth‐promoting effects of IGF‐I (4). Alternatively, or in conjunction with OA's direct effect on IGF‐II, increased levels of sAPPα have also been shown to drive the expression of IGF‐II and to protect against Aβ‐induced neuronal death (38).
Finally, an interesting and important observation from these studies was that cholesterol depletion (low fat/−OA) alone imparted significant protective effects on the levels of factors directly or indirectly related to APP processing or metabolism. The AD brains on a low‐fat/−OA diet had dramatic decreases of BACE, Aβ42 peptides and amyloid plaque burden, which may reflect a compensatory or regulatory feedback mechanism stemming from the lower levels of its substrates, APP. Along similar lines, the AD brains on a low‐fat/−OA diet showed significantly higher levels of sAPPα and IDE. While the exact underlying basis for this particular effect is unknown, cholesterol deficiency and a low‐fat diet are most likely the factors responsible for the shift in the non‐amyloidogenic processing of APP (reviewed in reference 44) and elevated IDE levels (1), respectively.
We argue that the OA metabolism either directly or indirectly caused by SCD‐1 led to a decrease in the levels of BACE and consequently, CTFβ and Aβ42. At the same time, OA, along with a low‐fat diet, decreased circulating insulin levels and increased the availability of IDE to metabolize Aβ. Therefore, the major factor leading to decreased brain levels of Aβ in low‐fat/+OA transgenic mice is not only its decreased production (APP, BACE, PS1 levels) but also the decreased tissue accumulation (increased IDE and IGF‐II). Dietary strategies aimed at reducing Aβ levels should take into account interactions of dietary components and the metabolic outcomes, in particular, levels of protein, fat, total calories and enrichment of beneficial FAs.
ACKNOWLEDGMENTS
Grants from the London and Middlesex Alzheimer's Association (Marion and Chester Fish Alzheimer's Disease Research Grant), the Canadian Institutes of Health Research (MOP49546) to R. F. R., Alberta Heritage Foundation for Medical research to D.W. and an Ontario Mental Health Foundation award to Z. A. funded this research. The authors declare that they have no conflict of interest.
REFERENCES
- 1. Ahren B (1999) Plasma leptin and insulin in C57BI/6J mice on a high‐fat diet: relation to subsequent changes in body weight. Acta Physiol Scand 165:233–240. [DOI] [PubMed] [Google Scholar]
- 2. Amtul Z, Uhrig M, Supino R, Beyreuther K (2010) Phospholipids and a phospholipid‐rich diet alter the in vitro amyloid‐beta peptide levels and amyloid‐beta 42/40 ratios. Neurosci Lett 481:73–77. [DOI] [PubMed] [Google Scholar]
- 3. Aoyagi T, Wada T, Nagai M, Kojima F, Harada S, Takeuchi T et al (1990) Deficiency of kallikrein‐like enzyme activities in cerebral tissue of patients with Alzheimer's disease. Experientia 46:94–97. [DOI] [PubMed] [Google Scholar]
- 4. Askari B, Carroll MA, Capparelli M, Kramer F, Gerrity RG, Bornfeldt KE (2002) Oleate and linoleate enhance the growth‐promoting effects of insulin‐like growth factor‐I through a phospholipase d‐dependent pathway in arterial smooth muscle cells. J Biol Chem 277:36338–36344. [DOI] [PubMed] [Google Scholar]
- 5. Brijbassi S, Amtul Z, Newbigging S, Westaway D, St George‐Hyslop P, Rozmahel RF (2007) Excess of nicastrin in brain results in heterozygosity having no effect on endogenous APP processing and amyloid peptide levels in vivo . Neurobiol Dis 25:291–296. [DOI] [PubMed] [Google Scholar]
- 6. Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J et al (2001) Early‐onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276:21562–21570. [DOI] [PubMed] [Google Scholar]
- 7. Crestini A, Napolitano M, Piscopo P, Confaloni A, Bravo E (2006) Changes in cholesterol metabolism are associated with PS1 and PS2 gene regulation in SK‐N‐BE. J Mol Neurosci 30:311–322. [DOI] [PubMed] [Google Scholar]
- 8. Dore S, Kar S, Quirion R (1997) Insulin‐like growth factor I protects and rescues hippocampal neurons against beta‐amyloid‐ and human amylin‐induced toxicity. Proc Natl Acad Sci U S A 94:4772–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghribi O, Larsen B, Schrag M, Herman MM (2006) High cholesterol content in neurons increases BACE, beta‐amyloid, and phosphorylated tau levels in rabbit hippocampus. Exp Neurol 200:460–467. [DOI] [PubMed] [Google Scholar]
- 10. Grimm MO, Grimm HS, Tomic I, Beyreuther K, Hartmann T, Bergmann C (2008) Independent inhibition of Alzheimer disease beta‐ and gamma‐secretase cleavage by lowered cholesterol levels. J Biol Chem 283:11302–11311. [DOI] [PubMed] [Google Scholar]
- 11. Guan Z, Wang Y, Cairns NJ, Lantos PL, Dallner G, Sindelar PJ (1999) Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J Neuropathol Exp Neurol 58:740–747. [DOI] [PubMed] [Google Scholar]
- 12. Haass C, Hung AY, Schlossmacher MG, Teplow DB, Selkoe DJ (1993) Beta‐amyloid peptide and a 3‐kDa fragment are derived by distinct cellular mechanisms. J Biol Chem 268:3021–3024. [PubMed] [Google Scholar]
- 13. Haass C, Capell A, Citron M, Teplow DB, Selkoe DJ (1995) The vacuolar H(+)‐ATPase inhibitor bafilomycin A1 differentially affects proteolytic processing of mutant and wild‐type beta‐amyloid precursor protein. J Biol Chem 270:6186–6192. [DOI] [PubMed] [Google Scholar]
- 14. Hartmann T, Bergsdorf C, Sandbrink R, Tienari PJ, Multhaup G, Ida N et al (1996) Alzheimer's disease betaA4 protein release and amyloid precursor protein sorting are regulated by alternative splicing. J Biol Chem 271:13208–13214. [DOI] [PubMed] [Google Scholar]
- 15. Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z et al (2004) Diet‐induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J 18:902–904. [DOI] [PubMed] [Google Scholar]
- 16. Hwang DY, Cho JS, Kim CK, Shim SB, Jee SW, Lee SH et al (2005) Aging‐related correlation of insulin‐degrading enzyme with gamma‐secretase‐generated products involving insulin and glucose levels in transgenic mice. Neurochem Res 30:1171–1177. [DOI] [PubMed] [Google Scholar]
- 17. Iizuka T, Shoji M, Kawarabayashi T, Sato M, Kobayashi T, Tada N et al (1996) Intracellular generation of amyloid beta‐protein from amyloid beta‐protein precursor fragment by direct cleavage with beta‐ and gamma‐secretase. Biochem Biophys Res Commun 218:238–242. [DOI] [PubMed] [Google Scholar]
- 18. Jensen M, Hartmann T, Engvall B, Wang R, Uljon SN, Sennvik K et al (2000) Quantification of Alzheimer amyloid beta peptides ending at residues 40 and 42 by novel ELISA systems. Mol Med 6:291–302. [PMC free article] [PubMed] [Google Scholar]
- 19. Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, Simons K (2005) Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro . J Biol Chem 280:36815–36823. [DOI] [PubMed] [Google Scholar]
- 20. Kim YC, Ntambi JM (1999) Regulation of stearoyl‐CoA desaturase genes: role in cellular metabolism and preadipocyte differentiation. Biochem Biophys Res Commun 266:1–4. [DOI] [PubMed] [Google Scholar]
- 21. Kim YJ, Nakatomi R, Akagi T, Hashikawa T, Takahashi R (2005) Unsaturated fatty acids induce cytotoxic aggregate formation of amyotrophic lateral sclerosis‐linked superoxide dismutase 1 mutants. J Biol Chem 280:21515–21521. [DOI] [PubMed] [Google Scholar]
- 22. Knops J, Suomensaari S, Lee M, McConlogue L, Seubert P, Sinha S (1995) Cell‐type and amyloid precursor protein‐type specific inhibition of A beta release by bafilomycin A1, a selective inhibitor of vacuolar ATPases. J Biol Chem 270:2419–2422. [DOI] [PubMed] [Google Scholar]
- 23. Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F (2001) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha‐secretase ADAM 10. Proc Natl Acad Sci U S A 98:5815–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lichtenthaler SF, Ida N, Multhaup G, Masters CL, Beyreuther K (1997) Mutations in the transmembrane domain of APP altering gamma‐secretase specificity. Biochemistry 36:15396–15403. [DOI] [PubMed] [Google Scholar]
- 25. Liu Y, Yang L, Conde‐Knape K, Beher D, Shearman MS, Shachter NS (2004) Fatty acids increase presenilin‐1 levels and [gamma]‐secretase activity in PSwt‐1 cells. J Lipid Res 45:2368–2376. [DOI] [PubMed] [Google Scholar]
- 26. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82:4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mattson MP (2004) Pathways towards and away from Alzheimer's disease. Nature 430:631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K (1993) Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch Biochem Biophys 301:41–52. [DOI] [PubMed] [Google Scholar]
- 29. Moser HW, Moser AB (1991) Measurement of Saturated Very Long Chain Fatty Acids in Plasma, pp. 177–191. Wiley‐Liss Inc.: New York. [Google Scholar]
- 30. Nixon RA (2007) Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120:4081–4091. [DOI] [PubMed] [Google Scholar]
- 31. Ntambi JM (1999) Regulation of stearoyl‐CoA desaturase by polyunsaturated fatty acids and cholesterol. J Lipid Res 40:1549–1558. [PubMed] [Google Scholar]
- 32. Park YS, Jang HJ, Lee KH, Hahn TR, Paik YS (2006) Prolyl endopeptidase inhibitory activity of unsaturated fatty acids. J Agric Food Chem 54:1238–1242. [DOI] [PubMed] [Google Scholar]
- 33. Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR (1998) Regional membrane phospholipid alterations in Alzheimer's disease. Neurochem Res 23:81–88. [DOI] [PubMed] [Google Scholar]
- 34. Qin W, Jia J (2008) Down‐regulation of insulin‐degrading enzyme by presenilin 1 V97L mutant potentially underlies increased levels of amyloid beta 42. Eur J Neurosci 27:2425–2432. [DOI] [PubMed] [Google Scholar]
- 35. Roth RA, Mesirow ML, Yokono K, Baba S (1984) Degradation of insulin‐like growth factors I and II by a human insulin degrading enzyme. Endocr Res 10:101–112. [DOI] [PubMed] [Google Scholar]
- 36. Smedman M, Potempska A, Rubenstein R, Ju W, Ramakrishna N, Denman RB (1997) Effects of cadmium, copper, and zinc and beta APP processing and turnover in COS‐7 and PC12 cells. Relationship to Alzheimer disease pathology. Mol Chem Neuropathol 31:13–28. [DOI] [PubMed] [Google Scholar]
- 37. St George‐Hyslop PH (2000) Molecular genetics of Alzheimer's disease. Biol Psychiatry 47:183–199. [DOI] [PubMed] [Google Scholar]
- 38. Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA (2004) Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J Neurosci 24:7707–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun Y, Yao J, Kim TW, Tall AR (2003) Expression of liver X receptor target genes decreases cellular amyloid beta peptide secretion. J Biol Chem 278:27688–27694. [DOI] [PubMed] [Google Scholar]
- 40. Tagawa K, Maruyama K, Ishiura S (1992) Amyloid beta/A4 precursor protein (APP) processing in lysosomes. Ann N Y Acad Sci 674:129–137. [DOI] [PubMed] [Google Scholar]
- 41. Uryu S, Tokuhiro S, Oda T (2003) Beta‐amyloid‐specific upregulation of stearoyl coenzyme A desaturase‐1 in macrophages. Biochem Biophys Res Commun 303:302–305. [DOI] [PubMed] [Google Scholar]
- 42. Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T et al (2002) Cholesterol‐dependent gamma‐secretase activity in buoyant cholesterol‐rich membrane microdomains. Neurobiol Dis 9:11–23. [DOI] [PubMed] [Google Scholar]
- 43. Wilson DM, Binder LI (1997) Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer's disease. Am J Pathol 150:2181–2195. [PMC free article] [PubMed] [Google Scholar]
- 44. Wolozin B (2004) Cholesterol and the biology of Alzheimer's disease. Neuron 41:7–10. [DOI] [PubMed] [Google Scholar]
- 45. Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG et al (2008) Cholesterol retention in Alzheimer's brain is responsible for high beta‐ and gamma‐secretase activities and Abeta production. Neurobiol Dis 29:422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A et al (2000) Nicastrin modulates presenilin‐mediated notch/glp‐1 signal transduction and betaAPP processing. Nature 407:48–54. [DOI] [PubMed] [Google Scholar]
- 47. Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T et al (2007) Beta‐site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci 27:3639–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]