

Abstract
Excitotoxicity is a widely studied mechanism underlying motoneuron degeneration in amyotrophic lateral sclerosis (ALS). Synaptic alterations that produce an imbalance in the ratio of inhibitory/excitatory synapses are expected to promote or protect against motoneuron excitotoxicity. In ALS patients, motoneurons suffer a reduction in their synaptic coverage, as in the transition from the presymptomatic (2‐month‐old) to early‐symptomatic (3‐month‐old) stage of the hSOD1G93A mouse model of familial ALS. Net synapse loss resulted from inhibitory bouton loss and excitatory synapse gain. Furthermore, in 3‐month‐old transgenic mice, remaining inhibitory but not excitatory boutons attached to motoneurons showed reduction in the active zone length and in the spatial density of synaptic vesicles in the releasable pool near the active zone. Bouton degeneration/loss seems to be mediated by bouton vacuolization and by mechanical displacement due to swelling vacuolated dendrites. In addition, chronic treatment with a nitric oxide (NO) synthase inhibitor avoided inhibitory loss but not excitatory gain. These results indicate that NO mediates inhibitory loss occurring from the pre‐ to early‐symptomatic stage of hSOD1G93A mice. This work contributes new insights on ALS pathogenesis, recognizing synaptic re‐arrangement onto motoneurons as a mechanism favoring disease progression rather than as a protective homeostatic response against excitotoxic events.
Keywords: hypoglossal motoneurons, myosin light chain phosphorylation, nitric oxide, SOD1G93A mice, synaptic alterations
INTRODUCTION
The imbalance between inhibitory and excitatory synaptic neurotransmission could be a pivotal event for motoneuron degeneration in amyotrophic lateral sclerosis (ALS) 4, 11, 45, 58. Excitatory signaling gain on motoneurons and/or their disinhibition will increase Ca2+ influx and depolarization, supporting excitotoxicity, one of the most studied mechanisms contributing to motoneuron death in ALS 15, 43. The post‐synaptic changes underpinning excitotoxicity involve an increase in the GluR1/GluR2 subunits ratio in excitatory alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole propionic acid (AMPA) receptors (AMPARs), which then permit Ca2+ influx (28) to motoneurons, in the hSOD1G93A mouse model of familial ALS 16, 69. Furthermore, GluR1 enrichment favors excitatory synaptogenesis on cultured motoneurons (46), which strongly suggests that a gain in excitatory synapses on motoneurons would be expected in ALS progression, but this required further investigation.
In addition, N‐methyl‐D‐aspartate (NMDA) receptors (NMDARs) in spinal interneurons are a potential source for overexcitation of sacral hSOD1G93A motoneurons as symptoms develop; synchronous bursting, evoked by inhibitory receptors blockage or spontaneously occurring, was more common in hSOD1G93A mice at early and late stages of disease, respectively (32). These findings also suggest changes in spinal circuits involving disinhibition, leading to motoneuron overexcitation in ALS. Alterations in inhibitory neurotransmission have been reported in ALS patients and hSOD1G93A mice 11, 19, 37, 44. Therefore, the possibility of a change in the motoneuron synaptic array to a net excitatory gain merits exploration as a feasible pathogenic mechanism contributing to disease progression.
Neonatal lumbar motoneurons from hSOD1G85R mice displayed lower input resistance than controls, but similar amplitude of evoked excitatory post‐synaptic potentials (9), suggesting a rise in synaptic current. Increased frequency, but not amplitude, of spontaneous excitatory transmission in neonatal hSOD1G93A hypoglossal motoneurons (HMNs) provides evidence of presynaptic alterations (65). These data point to a gain in excitatory neurotransmission on motoneurons at presymptomatic stages that might exacerbate excitotoxic events. In contrast, a reduction in the number of boutons occurs around chromatolytic and normal‐appearing anterior horn neurons of ALS patients 31, 41, 53, 54, 55 and in early‐symptomatic hSOD1G93A mice (68). Clarifying the nature of lost boutons will contribute information on its compensatory homeostatic properties, protecting motoneurons from excitotoxic death or, on the contrary, on the synaptic mechanisms contributing to disease progression.
The present study had two primary aims: (i) to determine whether synaptic alterations, occurring on motoneurons at early‐symptomatic stage in hSOD1G93A mice, involve the imbalance of the inhibitory/excitatory synapse ratio; and (ii) to scrutinize feasible mechanisms involved in synaptic alterations, including the possible role of the highly reactive gas nitric oxide (NO) as a mediator in synaptic alterations occurring in disease progression, given that NO has anti‐synaptotrophic and anti‐synaptogenic properties in adult motoneurons under pathological conditions 60, 61.
METHODS
Animals were obtained from an authorized supplier (Animal Supply Services, University of Cádiz, Spain), and were cared for and handled in accordance with the guidelines of the European Union Council (86/609/UE) and Spanish regulations (BOE 67/8509‐12; BOE 1201/2005) on the use of laboratory animals. Experimental procedures were approved by the local Animal Care and Ethics Committee. Experiments were performed in hSOD1G93A transgenic mice and in their non‐transgenic wild‐type (WT) littermates taken as controls. TgSOD1G93A high copy mice [Tg(SOD1‐G93A)1Gur] were obtained from the Jackson Laboratory (Bar Harbor, ME, USA), with B16XSJL background. These mice were bred and maintained as hemizygotes by mating transgenic males with F1 hybrid (B6SJLF1/J) females. Transgenic mice were identified by polymerase chain reaction amplification of DNA extracted from the tail. We used previously described primer sequences for exon 4 (49).
Studies were performed in 2‐ and 3‐month‐old male mice. At 90 days postnatal (P90), we observed that transgenic animals manifested an early symptomatic phenotype similar to that described by others 5, 21, 32, 35, 40, 42, characterized by fine shaking or tremor in one or more limbs 14, 27. The hanging wire test was performed on the hSOD1G93A mice and their non‐transgenic littermates (n = 10 animals per group) to assess muscular strength, as previously described (42). No animal in either group fell down before the arbitrary cut‐off time of 180 s at 60 days postnatal (P60). Thus, 2‐month‐old hSOD1G93A mice were considered to be in an adult presymptomatic stage of disease. One month later, a subtle difference was already observed between the transgenic and non‐transgenic groups [WT: 180.0 ± 0.0 s; hSOD1G93A: 158.8 ± 11.1 s; mean ± standard error of the mean (SEM); P < 0.05, unpaired one‐tailed Student's t‐test]. Therefore, 3‐month‐old hSOD1G93A mice were considered to be at the early‐symptomatic stage of disease. To achieve a homogeneous experimental group and minimize the number of animals used per experimental condition, only animals that performed the hanging wire test in ±5 s around the mean value were selected for histological procedures.
Retrograde labeling of HMNs
Adult male mice (∼83 days old; 30–35 g) were anesthetized with diethyl ether. Next, 50 µL of the retrograde tracer aminostilbamidine methanesulfonate [FluoroGold (FG); Molecular Probes, Eugene, OR, USA], 1% in phosphate‐buffered saline (PBS), was injected into the tongue, distributing the total volume at three injection points. Animals were then allowed to survive for at least 7 days before perfusion.
Histological procedures
For confocal microscopy
Animals were anesthetized with chloral hydrate (0.5 g/kg, i.p.), injected intraventricularly with heparin and perfused transcardially with PBS, followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB), pH 7.4, at 4°C. Brains were removed and postfixed for 2 h in the same fixative solution, to be subsequently cryoprotected by overnight immersion in 30% sucrose in 0.1 M PB at 4°C. Serial coronal sections (30 µm thick) were obtained using a cryostat and stored at −20°C in a cryoprotectant solution (glycerol : PBS, 1:1 v/v).
Immunohistochemistry was performed against synaptophysin (syn), vesicular glutamate (VGLUT2) and/or GABA (VGAT) transporters as synaptic markers and/or against the non‐phosphorylated form of neurofilament H (SMI32) as a motoneuron marker. Sections were rinsed in PBS and immersed in 2.5% (w/v) bovine serum albumin, 0.25% (w/v) sodium azide and 0.1% (v/v) Triton X‐100 in PBS for 30 minutes, followed by overnight incubation at 4°C with different combinations of up to three antisera. Polyclonal primary antibodies used in this study were: anti‐syn (1:200; Zymed Laboratories, San Francisco, CA, USA), anti‐VGAT (1:2000; Chemicon, Temecula, CA, USA) and anti‐phosphorylated‐myosin light chain (p‐MLC; 1:125; Cell Signaling, Danvers, MA, USA), all developed in rabbit; anti‐VGLUT2 (1:2500; Chemicon) developed in guinea pig; and anti‐SMI32 (1:8000; Covance, Princeton, NJ, USA) developed in mouse. Subsequently, the tissue was rinsed three times with PBS for 5 minutes each and incubated for 2 h at room temperature with the secondary antibodies, developed in donkey: anti‐guinea pig, anti‐mouse or anti‐rabbit IgGs labeled with cyanine 2, 3 or 5 (Cy2, Cy3, Cy5; 1:400; Jackson ImmunoResearch, West Grove, PA, USA). Finally, sections were washed with PBS and mounted on slides with a solution containing propyl gallate (0.1 mM in PBS : glycerol, 1:9 v/v). Omission of the primary antibodies resulted in no detectable staining.
Slides were analyzed using a Leica (Nussloch, Germany) confocal microscope for fluorescence. All motoneurons were analyzed in a z‐plane containing the nucleus. The pinhole opening was the same for all experimental conditions. Immunohistochemistry and confocal image acquisition were performed in parallel for the experimental groups to be compared.
For electron microscopy
Mice were deeply anesthetized with chloral hydrate (as above). The animals then received intraventricular heparin injection and transcardial perfusion with PBS, followed by 1%:1.25% PFA : glutaraldehyde in 0.1 M PB, pH 7.4, at 4°C. Brainstems and lumbar spinal cords were removed and postfixed for 2 h in the same fixative solution. Next, coronal sections (200 µm thick) were cut in a vibratome. Slices were stored in PB with 0.05% sodium azide at 4°C until electron microscopy processing.
For ultrastructural studies
Sections were washed in 0.1 M PB and postfixed with 2% osmium tetroxide in 0.1 M PB for 90 minutes. Slices were then rinsed with distilled water; dehydrated in graded series of ethanol, 30% (5 minutes), 50% (5 minutes) and 70% (10 minutes); and stained for 2 h and 30 minutes in 2% uranyl acetate in 70% ethanol at 4°C. Sections were dehydrated again in graded series of ethanol, 70% (5 minutes), 96% and 100%; then washed with propylene oxide, embedded in Araldite resin and left to polymerize for at least 72 h at 70°C. Ultrathin sections (70–80 nm thick) obtained with an ultramicrotome (Leica EM UC6) were analyzed under a FEI Tecnai Spirit (Tecnai Spirit G2, FEI, OR, USA) electron microscope at high magnification (43 000×) attached to a digital camera (Morada, Soft Imaging System, Olympus, Japan). Captured images were analyzed off‐line using the Image Tool software (University of Texas, San Antonio, Texas, USA). HMNs were analyzed at the level of the nucleolus.
For light microscopy
Tissue embedded in resin for electron microscopy studies was also used for light microscopy inspection. Semi‐thin sections (1.5 µm thick), cut in the ultramicrotome, were collected on a slide and lightly stained with a solution containing 1% toluidine blue and 1% borax in distilled water. The slices were covered with the toluidine blue solution, incubated for 2–5 minutes on a heater at 60°C, rinsed with distilled water and air dried. Tissue was observed under a light microscope (Nikon Eclipse E800, Melville, NY, USA) and images were acquired using a digital camera attached to the microscope (Nikon FDX 35) using the ACT‐1 software (Nikon).
Western blotting
Two‐month‐old mice were deeply anesthetized (chloral hydrate), their brainstems were quickly removed and sliced using a vibroslicer. Slicing and microdissection of hypoglossal nuclei (HNs) were both performed in ice‐cold (<4°C) sucrose artificial cerebrospinal fluid (S‐aCSF) bubbled with 95% O2 and 5% CO2 supplemented with protease (1 mM phenylmethylsulfonylfluoride, 10 mg/mL leupeptin, 10 mg/mL pepstatin A, 10 mg/mL aprotinin) and phosphatase inhibitors. S‐aCSF composition was (in mM): 26 NaHCO3; 10 Glucose; 3 KCl; 1.25 NaH2PO4; 2 MgCl2; and 218 sucrose. Microdissected HNs were homogenized in lysis buffer (50 mM Tris/HCl, pH 7.4, 1% (v/v) Triton X‐100, 0.5% (w/v) sodium deoxycholate), supplemented with protease and phosphatase inhibitors using a 1 mL syringe. Equal amounts of protein were processed for SDS‐PAGE and immunoblotting, using a specific antibody against p‐MLC and MLC developed in goat and rabbit, respectively (1:200; Santa Cruz Biotechnologies, Santa Cruz, CA, USA). Membranes were also probed with anti‐β‐actin antibody (1:250 000; Sigma, St. Louis, MO, USA) as control for the total amount of protein contained in each well. Analysis was performed using the ImageJ 1.36b software from the National Institutes of Health (NIH, Bethesda, Maryland, USA).
Drugs and treatments
Nω‐nitro‐L‐arginine methyl ester (L‐NAME, 1 mg/mL; Sigma) or its inactive steroisomer, Nω‐nitro‐D‐arginine methyl ester (D‐NAME; 1 mg/ml; Sigma), was added to the drinking water from P60 to P90 available ad libitum. The solution was freshly prepared every 2–3 days. A mean consumption of 4 mL water/mouse/day was estimated, yielding an approximate L‐NAME intake rate of 200 mg/kg/day per animal. After drug treatment, histological analyses were performed in blind‐coded preparations.
Statistics
Data are expressed as the mean ± SEM. Two‐way analysis of variance (ANOVA) with post hoc Tukey's test was used to compare groups, taking as factors mice and genotype/treatment. Statistical significance was placed at P < 0.05. No statistical difference was detected between the mice grouped per genotype or treatment in any case. The number of analyzed HMNs and number of animals per experimental condition are indicated in the results section, as well as in the table and in the figure legends. The non‐parametric Mann–Whitney U‐test was applied in immunoblot studies.
RESULTS
Net synapse loss of HMNs in early‐symptomatic hSOD1G93A mice
The linear density of syn‐immunoreactive (syn‐ir) puncta on spinal cord motoneurons drops in 3‐month‐old (early‐symptomatic) but not in 2‐month‐old (presymptomatic) hSOD1G93A mice (68). This result suggested that during the course of the disease synaptic alterations occur in the transition period from pre‐ to early‐symptomatic stages and might influence disease progression. We first investigated whether synaptic dysfunction also takes place at this time in brainstem HMNs. The frequency of syn‐ir puncta apposed to SMI32‐identified HMNs at P60 was similar in the genetic mouse model of ALS (16.2 ± 0.6 puncta/100 µm of membrane perimeter; n = 38 HMNs from three animals) and in control mice (15.9 ± 0.6 puncta/100 µm; n = 49 HMNs from three animals), which is congruent with findings in spinal motoneurons (68). However, the linear density of syn‐ir puncta of FG‐backlabeled motoneurons in 3‐month‐old hSOD1G93A mice was significantly lower (−32.5 ± 3.0%) than in their non‐transgenic littermates (Figure 1A, B, G). Results were not explained by changes in motoneuron size (WT: 82.8 ± 1.7 µm; hSOD1G93A: 82.7 ± 2.2 µm in perimeter; n = 50 HMNs from three mice per genotype). Therefore, HMNs followed a time course similar to lumbar motoneurons with respect to synaptic alterations.
Figure 1.

Net synapse loss on hypoglossal motoneurons (HMNs) from early‐symptomatic hSOD1G93A mice. A. B. Synaptophysin‐immunoreactive (syn‐ir) puncta around FluoroGold (FG; Molecular Probes, Eugene, OR, USA)‐backlabeled HMNs from non‐transgenic wild‐type (WT) littermates (A) and transgenic (B) control mice at 90 days postnatal (P90). Secondary antibody was labeled with cyanine 5. C, D. Electron microscopy photomicrographs showing a HMN (C) and a hypoglossal interneuron (D) obtained from a control mouse. Outlined arrow signals the nuclear invagination characteristic of interneurons from the hypoglossal nucleus. E, F. Segments of plasma membrane of HMNs obtained from the indicated mice at P90. Solid arrows point to synaptic boutons attached to the plasma membrane. Note the low frequency of boutons attached to the HMN from the hSOD1G93A mouse. G. Table showing the average number of Syn‐ir puncta (confocal microscopy analysis) and synaptic boutons (electron microscopy analysis) relative to 100 µm of membrane perimeter of HMNs from 3‐month‐old mice with the indicated genotypes. The number of analyzed HMNs from three different mice per genotype is indicated in parentheses. **P < 0.001, two‐way analysis of variance, post hoc Tukey's test. Scale bars, A–D, 10 µm; E, F, 1 µm.
Changes in synaptic density could be the consequence of a disorganization of the synaptic machinery rather than the actual loss of boutons. To address this question we studied the synaptic coverage of HMNs under electron microscopy. The HN consists of 90% motoneurons and 10% interneurons (59). Under electron microscopy, motoneurons are easily distinguishable from interneurons because the latter present a noticeable invaginated nucleus [(62); Figure 1C, D]. Our analysis showed that the synaptic array of HMNs was altered in 3‐month‐old transgenic mice relative to controls. We first measured the synaptic coverage of motoneurons by calculating the percentage of the cell body perimeter taken up by boutons. In control motoneurons, 53.9 ± 4.3% (n = 20 HMNs from three mice) of the perimeter was covered by boutons. Nevertheless, HMNs from hSODG93A mice displayed a significant reduction in their synaptic coverage (32.9 ± 3.5%; P < 0.05, two‐way ANOVA; post hoc Tukey's test; n = 20 HMNs from three mice). The frequency of boutons attached to the membrane of motoneurons in hSOD1G93A was significantly lower (−34.9 ± 7.2%) than in WT mice (Figure 1E–G). As in immunohistochemical studies, these results were not based on changes in the size of analyzed motoneurons (WT: 76.7 ± 2.6 µm; hSOD1G93A: 74.3 ± 2.7 µm in perimeter; n = 20 HMNs from three mice per genotype). These data demonstrate that a reduction in synaptic coverage is an early step in the course of motoneuron degeneration, which occurred concomitantly with early symptoms in the genetic mouse model of familial ALS 5, 14, 32, 35, 40, 42.
Excitatory and inhibitory synaptic alterations on HMNs in hSOD1G93A mice
We analyzed the excitatory and inhibitory nature of the synaptic loss undergone by HMNs from 3‐month‐old hSOD1G93A mice in an attempt to clarify their relevance in motoneuron degeneration. Immunohistochemistry was performed against VGLUT2, to identify a subtype of excitatory synaptic inputs, and VGAT, to label inhibitory terminals. In age‐matched WT mice, the linear density of inhibitory (19.5 ± 0.7 VGAT‐ir puncta/100 µm) was higher than excitatory (10.8 ± 0.5 VGLUT2‐ir puncta/100 µm) puncta apposed to FG‐identified HMNs (Figure 2A, C, E). A notable finding was that the frequency of VGLUT2‐ir and VGAT‐ir puncta apposed to hSOD1G93A HMNs was significantly increased (36.3 ± 6.6%) and reduced (−19.5 ± 4.5%), respectively (Figure 2B, D, E). Thus, the calculated VGAT (inhibitory)/VGLUT2 (excitatory) puncta ratio per motoneuron was reduced in hSOD1G93A mice (1.1 ± 0.1; P < 0.001, two‐way ANOVA, post hoc Tukey's test) relative to their non‐transgenic littermates (2.0 ± 0.1). This outcome suggests a gain in excitatory inputs and a loss of inhibitory ones on motoneurons in the early‐symptomatic transgenic mice.
Figure 2.
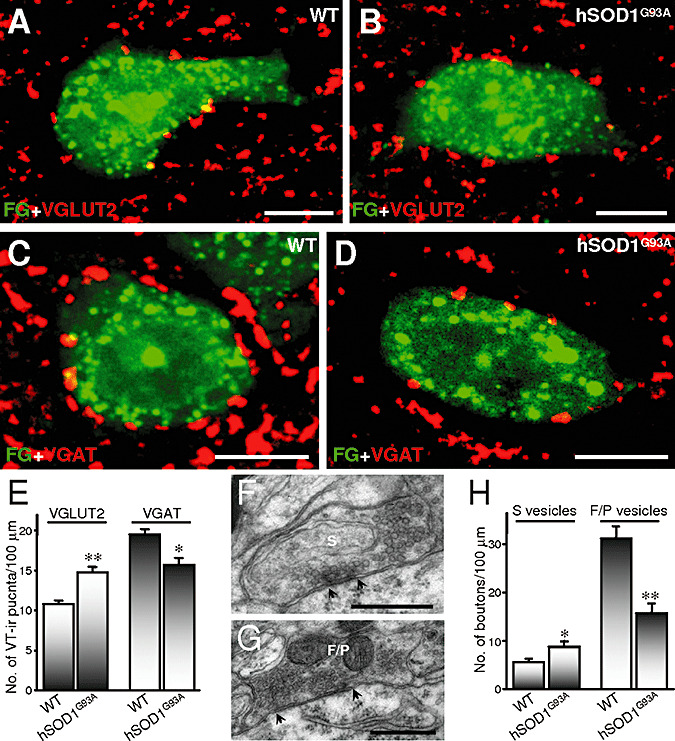
Inhibitory loss and excitatory gain on hypoglossal motoneurons (HMNs) in early‐symptomatic hSOD1G93A mice. A–D. Vesicular glutamate (VGLUT2)‐ (A, B) and GABA (VGAT) transporters‐immunoreactive (ir) (C, D) puncta around FluoroGold (FG; Molecular Probes, Eugene, OR, USA)‐backlabeled HMNs from wild‐type (WT) non‐transgenic (A, C) and transgenic (B, D) mice at 90 days postnatal. Secondary antibodies were labeled with cyanines 3 and 5 for immunolabeling of VGLUT2 and VGAT, respectively. E. Average number of vesicular transporters (VT)‐ir puncta per 100 µm of HMN perimeter from mice with the indicated genotypes (WT, n = 50 HMNs from three animals; hSOD1G93A, n = 25 HMNs from three animals). F, G. Illustrative examples of synaptic boutons attached to the plasma membrane of HMNs with spherical (S; F) or flat/pleomorphic (F/P; G) vesicles. Arrows indicate the borders of active zones. H. Average linear density of boutons, characterized by the type of vesicles, attached to motoneurons from mice with the indicated genotypes (WT, n = 20 HMNs from three animals; hSOD1G93A, n = 20 HMNs from three animals). *P < 0.05, **P < 0.001, two‐way analysis of variance, post hoc Tukey's test. Error bars indicate standard error of the mean. Scale bars: A–D, 10 µm; F, G, 0.5 µm.
An increase in the quantity of VGLUT2‐ir puncta, similar to the reduction in the number of VGAT‐ir puncta, could explain why a net decrease in the syn‐ir puncta was not observed at the presymptomatic stage (P60) in hSOD1G93A mice. Nevertheless, the linear density of VGAT‐ir puncta apposed to SMI32‐positive HMNs was similar in transgenic (18.8 ± 0.9 puncta/100 µm; n = 22 HMNs from three animals) and in non‐transgenic mice (18.4 ± 0.5 puncta/100 µm; n = 38 HMNs from three animals). In the same way, changes were not observed in the frequency of VGLUT2‐ir puncta in presymptomatic hSOD1G93A mice (9.4 ± 0.6 puncta/100 µm; n = 38 HMNs from three animals) relative to age‐matched control WT mice (9.8 ± 0.5 puncta/100 µm; n = 51 HMNs from three animals). Thus, both inhibitory puncta loss and excitatory puncta gain take place in the transition period from the pre‐ to early‐symptomatic phase in the mouse model of ALS.
Additional electron microscopy analysis was performed to study actual synaptic excitatory gain and/or inhibitory loss on hSOD1G93A motoneurons. In our preparations, synaptic contacts did not show pre‐ and/or post‐synaptic densities (Figure 2F, G) as thick as those described originally in the cerebral cortex (24) or in organotypic slice cultures obtained from embryonic spinal cords (4). Therefore, classification was done by type of synaptic vesicle, assisted by synaptic densities when these were evident. We distinguished between boutons containing spherical (S‐type; Figure 2F) and flat/pleomorphic (F/P‐type; Figure 2G) vesicles, since they are presumably excitatory and inhibitory, respectively 8, 24, 63. In support of immunohistochemistry results, a significant increase was observed in the perimeter covered by S‐type boutons (66.2 ± 18.9%; Table 1) and in their frequency (51.8 ± 19.6%) on transgenic motoneurons (8.5 ± 1.1 vs. 5.6 ± 0.7 boutons/100 µm in WT; Figure 2H). In WT motoneurons, most boutons contained clear F/P vesicles (31.2 ± 2.5 boutons/100 µm). The frequency of this type of boutons was significantly reduced (−49.4 ± 6.4%) in hSOD1G93A HMNs (15.8 ± 2.0 boutons/100 µm; Figure 2H), together with the percentage of motoneuron membrane that they covered (‐56.3 ± 5.3%; Table 1). Furthermore, the ratio of F/P‐type (inhibitory) to S‐type (excitatory) boutons per motoneuron was, as expected, reduced in transgenic mice (2.4 ± 0.4; P < 0.05) as compared to control WT mice (7.5 ± 1.8). Altogether, these data strongly demonstrate that HMNs underwent a decrease in the ratio of inhibitory to excitatory synapses. This finding supports a relationship between an imbalance toward excitatory signals received on motoneurons and the appearance of the early‐symptomatic phenotype in hSOD1G93A mice. It is reasonable to hypothesize that this could underpin a net overexcitation of motoneurons, which would exacerbate excitotoxic events and accelerate motoneuron degeneration.
Table 1.
Ultrastructural characterization of synaptic boutons attached to hypoglossal motoneurons. Abbreviations: WT = wild‐type; S = spherical; F/P = flat/pleomorphic.
| WT | hSOD1G93A | |||
|---|---|---|---|---|
| S‐type | F/P‐type | S‐type | F/P‐type | |
| Covered perimeter (in percentage)† | 7.4 ± 0.8 (20) | 45.1 ± 4.0 (20) | 12.3 ± 1.4 (20)* | 19.7 ± 2.4 (20)** |
| Apposed membrane/bouton (µm)‡ | 1.11 ± 0.1 (85) | 1.42 ± 0.1 (492) | 1.38 ± 0.1 (128)* | 1.27 ± 0.0 (242)* |
| Detached membrane/bouton (in percentage)§ | 3.5 ± 1.4 (62) | 2.9 ± 1.1 (70) | 6.3 ± 1.7 (49) | 11.0 ± 2.0 (63)** |
| Boutons with active zone (in percentage) | 43.2 ± 7.3 | 48.8 ± 2.6 | 48.1 ± 5.7 | 46.3 ± 3.5 |
| No. of active zones/bouton | 1.32 ± 0.1 (31) | 1.38 ± 0.1 (41) | 1.26 ± 0.1 (27) | 1.17 ± 0.1 (29) |
| Active zone length (nm) | 179.0 ± 16.2 (41) | 160.6 ± 15.4 (58) | 149.6 ± 9.7 (34) | 121.0 ± 10.5 (33)* |
| No. of vesicles/µm2 near the active zone | 380.6 ± 34.7 (41) | 659.1 ± 40.1 (58) | 422.9 ± 23.3 (34) | 452.6 ± 59.2 (33)* |
| Vacuolated boutons (in percentage) | — | — | 10.8 ± 3.8 | 24.2 ± 3.8 |
P < 0.05;
P < 0.001, two‐way analysis of variance, post hoc Tukey's test.
Percentage of the motoneuron cell body perimeter covered by synaptic boutons.
Average length per bouton apposed to the membrane motoneuron.
Percentage of the apposed bouton length detached from motoneuron membrane.
Number of sampled motoneurons, boutons or active zones is indicated in parentheses.
In contrast to the anticipated results, ultrastructural analysis revealed that synaptic frequency was altered to a greater extent than puncta, as detected by immunofluorescence. The reasons for this apparent discrepancy may lie in the fact that: (i) under confocal microscopy, we only analyzed puncta for VGLUT2, but not for VGLUT1; (ii) a single punctum does not necessarily represent an individual synaptic bouton; and (iii) it is likely that our quantitative analysis of puncta on confocal images underestimates synaptic stripping because it does not allow us to discriminate between boutons contacting the neuronal membrane and those that have been detached but remain in proximity to the soma (61).
Ultrastructural analysis of boutons still attached to hSOD1G93A HMNs
We analyzed several ultrastructural parameters of boutons attached to HMNs from hSOD1G93A mice as indicators of synaptic functional capacity. In control motoneurons, boutons appeared in intimate apposition over the post‐synaptic membrane (Figure 3A). The average profile per bouton apposed to the plasma membrane of WT motoneurons was higher for F/P‐type than for S‐type boutons (Table 1). However, in age‐matched transgenic mice the mean apposition length of presynaptic membrane covering HMNs was significantly reduced for F/P‐type (−10.6 ± 2.1%) and increased for S‐type (24.3 ± 7.2%) boutons (Table 1). These results partially account for the changes in synaptic coverage suffered by impaired motoneurons. However, in hSOD1G93A animals we found that the space between the pre‐ and post‐synaptic elements was enlarged (Figure 3B). Bouton separation was quantified as the percentage of the detached length relative to the total apposed length of the bouton. Detachment was significantly higher for F/P‐type boutons in transgenic mice than in non‐transgenic littermates, but no significant difference was detected for S‐type boutons (Table 1). So far, these data support a correlation between the decay of motoneuron synaptic coverage and an increase in the proportion of bouton membrane detached from the post‐synaptic membrane. This could comprise the initial step for bouton detachment.
Figure 3.
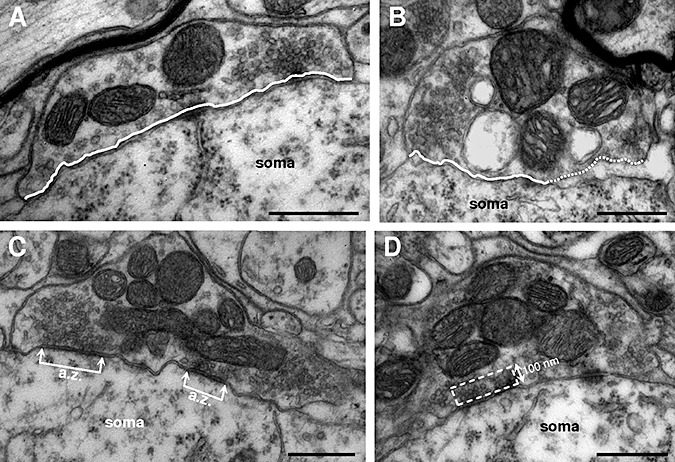
Ultrastructural characterization of hypoglossal motoneurons‐attached boutons. A, B. Two boutons with flat/pleomorphic (F/P) vesicles attached to the motoneuron soma from wild‐type (WT) non‐transgenic littermates (A) and hSOD1G93A (B) mice. Note the enlarged subsynaptic space observed in the bouton from the transgenic mouse. Continuous and dotted lines indicate the segment of bouton attached and detached, respectively, from the motoneuron cell body. C. A F/P‐type bouton with two active zones (a.z.) is shown. The distance between connected arrows represents the length of each a.z. Note that an end of the bouton is detached from the motoneuron. D. Picture illustrating the rectangular area considered in counting the number of vesicles in the releasable pool near the a.z. One side of the rectangle was taken as the length of the active zone and the other side had a fixed length of 100 nm. Scale bars: 0.5 µm.
We also sampled the percentage of S‐ and F/P‐type boutons with evident active zones (a.z.), the number of a.z. per bouton, the average length of a.z. (Figure 3C) and the number of synaptic vesicles in the releasable pool near a.z. (Figure 3D) in control and transgenic mice. No ultrastructural alterations were found in the percentage of boutons with apparent a.z. nor in the number of a.z. per bouton for either F/P‐ or S‐type boutons (Table 1). However, the length of the a.z., represented by the length of the post‐synaptic density, was significantly reduced (−24.7 ± 6.5%) in F/P‐, but not in S‐type, boutons in hSOD1G93A relative to control animals (Table 1). Furthermore, the spatial density of synaptic vesicles in the releasable pool near to a.z. (Figure 3D) decreased (−31.3 ± 9.0%) in presumably inhibitory, but not in excitatory, boutons (Table 1). Taken together, these ultrastructural alterations strongly support a possible functional impairment of the still attached F/P‐type boutons on hSOD1G93A HMNs in transgenic mice, as compared to controls. However, no ultrastructural evidence points to functional alterations in the presumably excitatory boutons.
Evidence for the involvement of vacuolization in bouton loss
Next, we looked for feasible mechanisms underlying synapse loss. One of the most prominent features in the course of the disease is the large number of vacuoles, derived from dilated mitochondria and endoplasmic reticulum, in hSOD1G93A and hSOD1G37R mice 5, 17, 34, 40, 66. We conducted light microscopy studies of semi‐thin sections of the HN, which revealed numerous vacuoles widely spread along the neuropil of 3‐month‐old hSOD1G93A mice, but absent in age‐matched WT mice (Figure 4A, B). The nuclei of all the cells were always euchromatic and morphologically unaltered (Figure 4B). At this stage, only microvesiculation was observed in the cell body of hSOD1G93A HMNs (Figure 4C), while all‐sized vacuoles were already invading motoneurons at the lumbar spinal cord (Figure 4D). This finding agrees with the caudocranial spread of degeneration and inflammatory reactions in this animal model (35).
Figure 4.
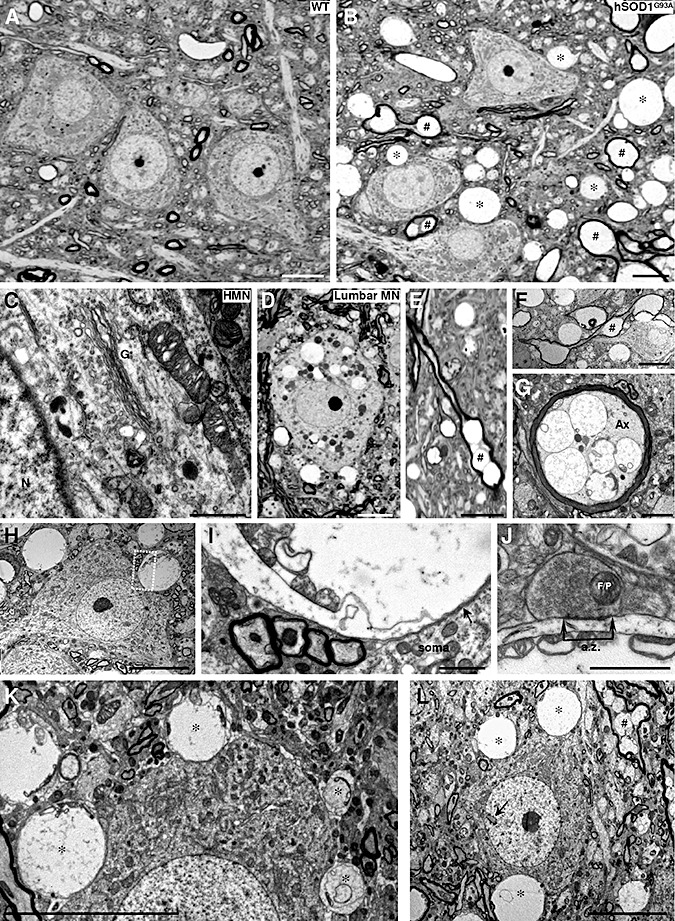
Neuropathological alterations in the hypoglossal nucleus (HN) of 3‐month‐old hSOD1G93A mice. A. B. Photomicrographs of semi‐thin sections from the HN of a 3‐month‐old wild‐type (WT) non‐transgenic (A) mouse compared to an age‐matched hSOD1G93A transgenic mouse (B). Vacuolated and swollen myelin‐wrapped axons (#) and feasible dendrites (*) are evidenced in the transgenic mouse model of amyotrophic lateral sclerosis. C. Details of the soma of a hypoglossal motoneuron (HMN) taken with electron microscopy, showing microvacuoles into mitochondrion and cytoplasm, as well as disorganization of the Golgi apparatus (G). N, nucleus. D. Vacuolated perikarya of a lumbar motoneuron (lumbar MN) in the same mouse as in C. E–G. Photomicrographs from semi‐ (E) and ultra‐thin (F, G) sections showing longitudinally (E, F; #) and transversally (G) transected myelin‐wrapped axons. Note the vacuoles containing remnants of mitochondria observed in the axon (Ax) shown in G. H, I. A HMN from a SOD1G93A mouse at 90 days postnatal with a vacuolated structure apposed to its plasma membrane. The boxed area in H is presented at higher magnification in I. Note that the membrane of the vacuolated structure containing mitochondria vestiges is apposed to the plasma membrane of the motoneuron (arrow). J. Detail of a bouton containing flat/pleomorphic (F/P) vesicles showing an active zone (a.z.) apposed to a post‐synaptic density on a vacuolated structure with mitochondria remnants. This supports the dendritic nature of the degenerating structure. K. In this photomicrography four different‐sized vacuolated structures (*), containing mitochondria residues, appear close to the soma membrane of a HMN. This could represent the pathological progression for the formation of a large vacuolated structure. Note that the larger the vacuole, the greater the deformation that is induced in the apposed motoneuron. L. Example of a hypoglossal interneuron identified by its invaginated nucleus (arrow) surrounded by vacuolar structures. Scale bars: A, B, D–F, H, K, L, 10 µm; G, I, 2 µm; C, J, 1 µm.
Prominent vacuolated structures involved myelin‐wrapped axons (Figure 4E–G). In addition, non‐myelinated vacuolated structures apposed to the plasma membrane of the cell body of HMNs were usually observed (Figure 4H, I, K), some of them still carrying remnants of mitochondria and receiving synaptic contacts (Figure 4J) indicative of dendritic origin. These vacuolated and swollen dendrites in most cases distorted the profile of adjacent neurons (Figure 4H, K). This scenario was also noticed around hypoglossal interneurons (Figure 4L). The fact that vacuolated structures consist of axons and dendrites, but not of cell bodies, robustly signals a centripetal progression of motoneuron degeneration. Vacuolated structures found near or apposed to HMNs seem to be crushing (Figure 5A) and/or laterally displacing and compressing synaptic boutons (Figure 5B, C), which could also impair synaptic function. This drawing suggests that a mechanism of action by which synapse loss occurs could involve mechanical crushing and displacement of boutons by swelling vacuolated structures (4, 5).
Figure 5.
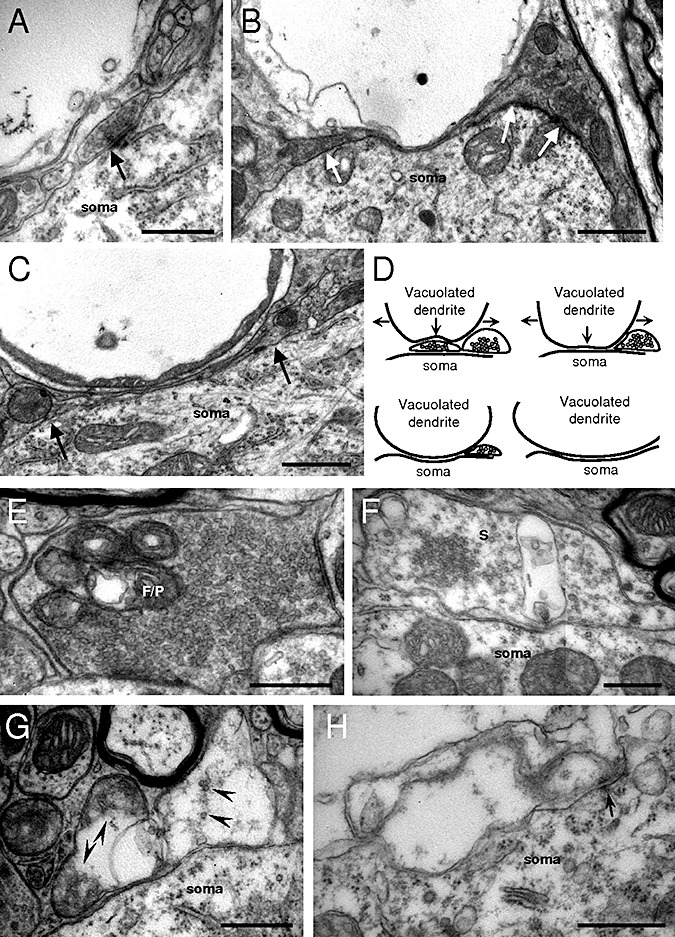
Evidence for vacuolization‐induced synapse loss/degeneration on hypoglossal motoneurons (HMNs) in hSOD1G93A mice. A. A vacuolated structure, possibly a dendrite, seems to compress a bouton that is making a synaptic contact (arrow) with a motoneuron. B, C. These types of structures deform both motoneuron plasma membrane and laterally synaptic boutons (arrows), which could disrupt synaptic functionality and move boutons away from their original place. D. Drawing summarizing the mechanical process by which vacuolization and swelling of dendrites near motoneurons could induce synapse detachment from HMNs in the genetic mouse model of amyotrophic lateral sclerosis. Arrows signal the expanding direction of the vacuolated structure. E. Example of a bouton containing flat/pleomorphic (F/P) vesicles with vacuolated mitochondria, impairing energy support for synaptic functionality. F. Bouton containing spherical (S) vesicles showing a vacuole of unknown origin. G, H. Illustrations of degenerated boutons containing remnants of synaptic‐like vesicles (arrowheads in G) and vestige of an active zone (arrow in H). Scale bars: B, C, 1 µm; A, E–H 0.5 µm.
We scrutinized for a “vacuolar degeneration” of boutons underlying synapse degeneration/loss. Vacuole formation was observed within mitochondria and in the axoplasm of either F/P‐ (Figure 5E) or S‐type (Figure 5F) boutons in hSOD1G93A mice. Strikingly, the percentage of vacuolated F/P‐type boutons doubled the frequency of vacuolated S‐type inputs on HMNs (Table 1). In addition, bouton‐like structures attached to HMNs presented a more advanced stage of degeneration, in which typical synaptic mitochondria were absent and some synaptic‐like vesicles were evident (Figure 5G). Finally, vacuoles similar in size to a bouton and preserving membrane densities (likely vestiges of a.z.) were found attached to motoneurons (Figure 5H). These findings suggest that vacuolar degeneration is a mechanism involved in synapse degeneration/loss in the course of ALS. In any case, at the early‐symptomatic stage of disease, presumably inhibitory terminals, still attached, are proportionally more affected than excitatory boutons.
NO mediates inhibitory synapse loss, but not excitatory input gain
We also looked for a synaptic alteration regulatory mechanism mediated by the gaseous molecule NO, given more than a decade of findings in our laboratory and others: (i) neuronal and/or inducible NO synthase (n/iNOS) upregulation occurs in motoneurons and reactive astrocytes in ALS patients 3, 12, 56 and in hSOD1G93A mice 1, 13, 57 and (ii) de novo expression of nNOS in motoneurons is sufficient to reduce the synaptic coverage of HMNs in pathological conditions 60, 61. Bouton detachment is preceded by NO‐induced phosphorylation of MLC, which is known to trigger actomyosin contraction and neurite outgrowth retraction (61).
We first analyzed the level of p‐MLC in the HN of 2‐month‐old (presymptomatic) animals, an age when synapse loss had not yet occurred on lumbar (68) and HMNs (current results). Immunoblot analysis of microdissected HNs revealed a significant increase (29.0 ± 6.0%) in the ratio of p‐MLC/MLC in hSOD1G93A relative to WT mice (Figure 6A, B). Subsequently, we analyzed syn‐ir puncta co‐expressing p‐MLC (Figure 6C, D). The percentage of syn‐ir puncta apposed to SMI32‐ir HMNs co‐localizing with p‐MLC‐ir structures increased fivefold in transgenic (25.9 ± 3.8%; P < 0.001, two‐way ANOVA, post hoc Tukey's test) relative to control (5.3 ± 1.5%; n = 20 HMNs from three animals per genotype) mice (Figure 6C, D). These results suggest that phosphorylation of MLC in synaptic puncta could precede bouton loss in the mouse model of ALS, signaling NO as a feasible factor involved in synapse withdrawal suffered by mSOD1G93A motoneurons.
Figure 6.

Increased phosphorylation of myosin light chain (MLC) in synaptic puncta apposed to hypoglossal motoneurons (HMNs) preceding synapse loss. A. Immunoblots for phosphorylated‐MLC (p‐MLC), MLC and β‐actin (β‐act) performed from microdissected hypoglossal nuclei (HNs) of 2‐month‐old non‐transgenic and age‐matched hSOD1G93A mice. B. Table showing the average ratios for p‐MLC/MLC and p‐MLC/β‐act in arbitrary units (a.u.) obtained from transgenic mice, compared to control (wild‐type, WT) animals taken as 1. n = 3 Western blots from six animals per genotype. *P < 0.05, non‐parametric Mann–Whitney U‐test. C, D. Synaptophysin‐immunoreactive (syn‐ir) colocalizing with p‐MLC puncta (arrowheads) apposed to SMI32‐immunolabeled HMNs in non‐transgenic (C) and transgenic (D) mice at 2 months of age. Secondary antibodies were labeled with cyanines 2, 3 and 5 for immunolabeling of SMI32, p‐MLC and Syn, respectively. Scale bars, 10 µm.
Next, we analyzed the synaptic coverage of HMNs in 3‐month‐old hSOD1G93A mice that had received in their drinking water, from P60, either the broad spectrum NOS inhibitor L‐NAME or its inactive steroisomer D‐NAME. The linear density of syn‐ir puncta apposed to SMI32‐identified HMNs was not altered by D‐NAME treatment of WT mice (16.5 ± 1.2 syn‐ir puncta/100 µm) relative to non‐treated non‐transgenic littermates (16.6 ± 0.4 syn‐ir puncta/100 µm). As expected, a reduction in the motoneuron synaptic coverage (−22.7 ± 7.5%) was observed in D‐NAME‐treated hSOD1G93A mice relative to D‐NAME‐treated control mice (Figure 7A, B, D). However, L‐NAME treatment prevented syn‐ir puncta reduction in transgenic mice (18.2 ± 1.3 syn‐ir puncta/100 µm; Figure 7C, D). These data point to NO as a feasible mediator in synapse loss by hSOD1G93A motoneurons from pre‐ to early‐symptomatic stages.
Figure 7.

Chronic administration of a nitric oxide synthase (NOS) inhibitor prevents inhibitory puncta loss, but not excitatory gain, on hypoglossal motoneurons (HMNs) in hSOD1G93A mice. A–C. Synaptophysin‐immunoreactive (syn‐ir) puncta around SMI32‐immunolabeled HMNs from 3‐month‐old animals with the indicated genotypes and receiving the indicated drugs in the drinking water from 60 days postnatal. Secondary antibodies were labeled with cyanines 2 and 5 for immunolabeling of SMI32 and syn, respectively. D. Average number of syn‐ir puncta per 100 µm of SMI32‐identified HMN perimeter at the indicated conditions. n = 20 HMNs from 3 mice per condition. E–H. Vesicular glutamate (VGLUT2)‐ and GABA (VGAT) transporters‐immunoreactive (ir) puncta around FluroGold (FG; Molecular Probes, Eugene, OR, USA)‐backlabeled HMNs obtained from 3‐month‐old wild‐type (WT) non‐transgenic (E, F) or age‐matched hSOD1G93A (G, H) mice non‐treated or after receiving Nω‐nitro‐L‐arginine methyl ester (L‐NAME) in the drinking water. Secondary antibodies were labeled with cyanines 5 and 3 for immunolabeling of VGAT and VGLUT2, respectively. I, J. Average number of the indicated vesicular transporter‐ir puncta per 100 µm of HMN perimeter in FG‐identified motoneurons at the indicated conditions. The number of analyzed motoneurons was as follows: WT, non‐treated, n = 50 HMNs from three mice; WT, L‐NAME, n = 42 HMNs from three mice; hSOD1G93A, non‐treated, n = 25 HMNs from three mice; hSOD1G93A, L‐NAME, n = 50 HMNs from three mice. *P < 0.05, two‐way analysis of variance, post hoc Tukey's test. Error bars indicate standard error of the mean. Scale bars, 10 µm.
Nevertheless, systemic NOS inhibition could induce synaptic reordering, compensating for synapse loss rather than preventing it. We looked for a feasible regulation on synaptic arrangement of physiologically‐synthesized NO in WT HMNs. The frequency of vesicular transporter‐ir puncta apposed to FG‐backlabeled HMNs of 3‐month‐old WT receiving L‐NAME from P60 was similar (11.2 ± 0.5 VGLUT2‐ir puncta/100 µm; 19.2 ± 0.6 VGAT‐ir puncta/100 µm) to the untreated ones (Figure 7E, F, I, J). In addition, L‐NAME‐supplemented drinking water prevented VGAT‐ir puncta loss, maintaining the linear density of inhibitory puncta of hSOD1G93A motoneurons (19.1 ± 0.7 VGAT‐ir puncta/100 µm) at the levels of control mice (Figure 7G–I). Nevertheless, L‐NAME did not prevent the occurrence of gain in excitatory puncta on hSOD1G93A HMNs (14.1 ± 0.5 VGLUT2‐ir puncta/100 µm; Figure 7G, H, J).
Immunohistochemical results were supported by electron microscopy studies. Under L‐NAME treatment, the linear density of synaptic boutons attached to the plasma membrane of hSOD1G93A HMNs did not differ (32.8 ± 2.5 boutons/100 µm) from the control condition (Figure 8A, B). The gain of S‐type boutons was not avoided by NOS inhibition (9.4 ± 0.9 boutons/100 µm) but the loss of F/P‐type boutons was partially prevented (22.0 ± 1.9 boutons/100 µm). Altogether, these results strongly support that NO mediates synapse loss suffered by motoneurons during the third month of life in hSOD1G93A mice. L‐NAME did not distort the percentage of vacuolated F/P‐ (27.2 ± 2.3%; n = 20 HMNs) and S‐type (18.5 ± 3.9%) boutons. The study results indicate that NO induced synapse loss by a mechanism not mediated by vacuolar degeneration and that vacuole formation, at least in boutons, was NO independent.
Figure 8.
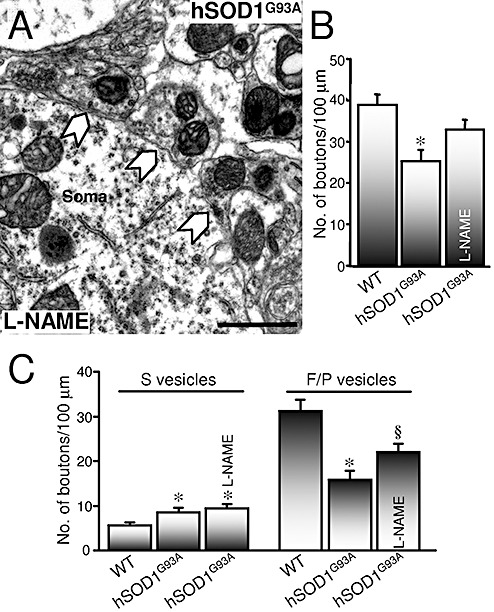
Chronic treatment with a nitric oxide synthase (NOS) inhibitor partially prevents flat/pleomorphic (F/P)‐type bouton loss, but not spherical (S)‐type bouton gain, on hypoglossal motoneurons (HMNs) in hSOD1G93A mice. A. Segment of the plasma membrane of a HMN obtained from the indicated animal at 90 days postnatal that received Nω‐nitro‐L‐arginine methyl ester (L‐NAME) in the drinking water from 60 days postnatal. The solid arrows point to synaptic boutons attached to the plasma membrane. B. Average number of attached boutons per 100 µm of HMN perimeter at the indicated conditions. *P < 0.001, two‐way analysis of variance (ANOVA), post hoc Tukey's test, relative to non‐transgenic wild‐type (WT) mice. C. Average bouton frequency, characterized by the type of vesicles, attached to motoneurons obtained from the indicated conditions. n = 20 HMNs from three mice per condition. *,§P < 0.05, two‐way ANOVA, post hoc Tukey's test, relative to WT or to WT and hSOD1G93A mice, respectively. Error bars indicate standard error of the mean. Error bar, 1 µm.
DISCUSSION
Reduction in synaptic coverage around spinal motoneurons in ALS patients 31, 41, 53, 54, 55 and in hSOD1G93A mice (68) could be an event contributing to motoneuron degeneration. It could also be a compensatory homeostatic mechanism attenuating excitotoxicity induced by interference with glutamatergic neurotransmission 47, 50, 64. Our results indicate profound alterations in the synaptic coverage of motoneurons in the early‐symptomatic stage of a genetic mouse model of ALS. Changes involved loss of inhibitory terminals and gain of excitatory boutons, together with a higher deterioration of inhibitory than excitatory still‐attached terminals. Overexcitation of motoneurons exacerbates excitotoxic events and contributes to motoneuron degeneration in the early‐symptomatic stage of the disease. Taken together, these findings support a conclusion that net synapse loss results in a lower ratio of inhibitory/excitatory inputs, which could influence the onset of the disease. Motoneuron disinhibition together with overexcitation could explain initial hyperreflexia and tremor in the hSOD1G93A mouse model 14, 25, 27, as well as disinhibition of spinal reflex in ALS patients (25).
Symptom onset in ALS patients may occur in the muscles of the limbs (spinal onset) or of the head and neck (bulbar onset). Bulbar ALS is clinically characterized by degeneration of brainstem motoneurons (52). The HN is among the most strongly involved brainstem nuclei in ALS; this underlies the deficits in speech, chewing, swallowing and breathing (18). In the transgenic hSOD1G93A mouse model, a caudocortical disease progression has been reported (35), but some degenerative characteristics of the disease, such as neuropil vacuolation and gliosis, followed a parallel course in the HN and spinal cord 14, 35. Further, MRI studies have pointed to motor nuclei in the brainstem as valuable indicators of disease progression, comparable to the condition of motoneurons within the spinal cord (2). Although we did not detect differences in pathological characteristics at the neuropil between HN and the lumbar ventral horn of the spinal cord, we observed less marked vacuolar invasion in HMNs than in lumbar motoneurons in 3‐month‐old hSOD1G93A mice. In one study, a 12% reduction in the number of HMNs was quantified at this stage (26), but another study detected no HMN death in 102‐day‐old transgenic mice (14). Finally, we found a higher reduction in the synaptic coverage of HMNs (33%–35%), under confocal and electron microscopy inspection, than previously reported, by epifluorescence analysis, for lumbar motoneurons (∼14%) in 3‐month‐old hSOD1G93A mice (68). It remains unclear whether an actual difference exists between the synaptic pathology of brainstem and spinal motoneurons or the conflicting results are due to differences in analytical methods. However, our data strongly agree with the 30%–33% reduction in corticospinal and bulbospinal projections previously found by the same group (67).
Since inhibitory synapses onto HMNs were more abundant than excitatory ones, proportionally similar loss of inhibitory inputs and gain in excitatory boutons resulted in a net synapse loss at the early‐symptomatic stage of hSOD1G93A mice. This agrees with the marked inhibitory loss reported in patients with ALS 19, 37, 44. Synapse deterioration/loss was induced by dendritic vacuolation and swelling and by vacuolar degeneration of synaptic boutons. On the other hand, an increase in synaptic GluR1 levels and a decrease of synaptic GluR2 occur in presymptomatic hSOD1G93A mice (69). Presymptomatic increase in the GluR1/GluR2 ratio could be the basis for the increase in glutamatergic boutons detected at the early‐symptomatic stage, since overexpression of the GluR1 subunit increases excitatory synaptogenesis in mixed spinal cord cultures (46). Excitatory input gain could be a homeostatic attempt to keep the motor output steady despite motoneuron loss. A gain in glutamate vesicular transporters has also been observed in hippocampus after hypoxic insults to induce a hyperexcitability that may play a role in reconstruction of neuronal circuits after injury (33). However, further studies are needed to check this hypothesis. Additionally, we found ultrastructural changes in synaptic boutons still contacting with motoneurons that support a functional disruption of F/P‐type, but not S‐type, boutons. Taken together, these data strongly point to synaptic imbalance contributing to overexcitation of motoneurons in early‐symptomatic hSOD1G93A mice. Given that gain in VGLUT2 puncta did not occur in presymptomatic mice, presynaptic alterations supported by the increased frequency, but not amplitude, in spontaneous excitatory transmission in neonatal hSOD1G93A HMNs (65) must be the result of presynaptic changes other than the increase in excitatory boutons at neonatal stages.
We have recently reported that traumatic injury of the XIIth nerve induced synaptic withdrawal from HMNs, which underlies profound physiological alterations of motoneurons (23). These outcomes were triggered by the synthesis of NO from de novo‐expressed nNOS in motoneurons in response to axon injury 60, 61. Similarly, n/iNOS upregulation occurs in motoneurons and reactive astrocytes in ALS patients 3, 12, 56 and in hSOD1G93A mice 1, 13, 57, indicating a feasible involvement of NO in synaptic rearrangement onto motoneurons in the course of the disease. In support of this possible mechanism, treatment with the broad spectrum NOS inhibitor L‐NAME, beginning at P60 (presymptomatic) and analyzed at P90 (early‐symptomatic), prevented inhibitory synapse loss but not excitatory gain in this genetic mouse model of ALS. Whereas presymptomatic transgenic mice occasionally presented nNOS immunoreactive ventral horn neurons, a significantly large increase in the proportion of nNOS‐positive neurons was observed in early‐symptomatic mice (57). Taken together, we suggest that nNOS upregulation during the third month of life underpins, at least in part, the loss of inhibitory synapses. However, we have no evidence for NO involvement in excitatory gain. Strikingly, no animal treated with the NOS inhibitor fell down before the arbitrary cut‐off time of 180 s at P90 in the hanging wire test. This suggests beneficial effects of NOS inhibition also at spinal level and in the beginning of the first motor symptoms in this mouse model.
An increase in p‐MLC was measured in the HN and in syn‐ir puncta preceding synapse loss, as we previously observed after traumatic XIIth nerve injury (61). This cascade involved Rho kinase (ROCK), which mediates neurite retraction, preventing axon growth initiation and dendrite retraction in different neuronal types in culture 38, 39. Remarkably, ROCK is upregulated in the progression of the disease in late symptomatic hSOD1G93A mice (29). All these findings argue for a NO‐ROCK‐pMLC‐mediated synapse loss in the progression of the disease in hSOD1G93A mice. Activation of this molecular cascade could be a hallmark in diverse neuropathological conditions coursing with synaptic deterioration (61).
Chronic treatment of transgenic mice with the NOS inhibitor prevented inhibitory loss in ∼40% from pre‐ to early‐symptomatic stages in hSOD1G93A mice. This could be explained by either an insufficient dose of L‐NAME or an additional NO‐independent mechanism, such as vacuolation, inducing synapse degeneration. However, systemic treatment with L‐NAME, in a dose (90 mg/kg/day) lower than that calculated for this work (200 mg/kg/day), fully prevented synapse loss induced after axotomy of HMNs 60, 61. Therefore, we could disregard the dose hypothesis. The remaining 60% of synapse loss taking place in these mice could be the result of inhibitory synapse displacement induced by growing vacuolated dendrites and/or of bouton degeneration by vacuolation. In this way, L‐NAME had no effect on the proportion of vacuolated inhibitory synapses. Furthermore, macroscopic inspection of neuropil vacuolation of the HN from L‐NAME‐treated transgenic mice did not report qualitative changes (data not shown) relative to non‐treated hSOD1G93A mice. Therefore, the mechanism of action by which NO mediates inhibitory loss seems to not involve synaptic “vacuolar degeneration.” These observations suggest that vacuolation is a NO‐independent process, at least in this animal model of ALS.
Glutamatergic‐mediated excitotoxicity is one of the most studied mechanisms mediating motoneuron degeneration in ALS 47, 50, 64. The clinical effectiveness of anti‐glutamatergic agents such as riluzole, which has anti‐excitotoxic properties (7), adds credibility to a possible involvement of excitotoxicity in ALS. It is generally thought that excitotoxicity arises from excessive Ca2+ entry into motoneurons, which have a low Ca2+‐buffering capacity. Glutamatergic synaptic inputs evoke Ca2+ entry through NMDARs and AMPARs, although AMPARs‐mediated Ca2+ influx occurs in the absence of the GluR2 subunit (28). In this way, two major mechanisms have been proposed to mediate excitotoxic degeneration of motoneurons in ALS: (i) glutamate accumulation due to decreased clearance resulting from selective loss of the astroglial glutamate transporter, EAAT2/GLT1 (64), which has been observed in both genetic mouse models 6, 10 and ALS patients 22, 51; and (ii) an increase in the number of Ca2+‐permeable AMPARs, as documented in spinal cord motoneurons of hSOD1G93A mice (69). Surprisingly, presynaptic input re‐organization onto motoneurons has not been a subject of much attention so far. Our study contributes new elements that could impair Ca2+ homeostasis in motoneurons. Increased linear density of glutamatergic synapses can lead to the enhancement of Ca2+ influx into motoneurons as a result of overactivation of NMDARs and Ca2+‐permeable AMPARs. Additionally, it is logical to think that disinhibition resulting from the loss of GABAergic synapses by hSOD1G93A HMNs produces membrane depolarization, which relieves NMDARs from the normal blocking by Mg2+. This facilitates the opening of NMDA channels by glutamate, potentiating Ca2+ influx into the motoneuron (36). These mechanisms might exacerbate excitotoxic stress in motoneurons, which underlies disease progression. Glutamate‐coupled NO synthesis must be enhanced in the process; this has been shown to contribute to FAS‐triggered motoneuron death in ALS mouse models (48). Accordingly, a selective nNOS inhibitor significantly prolonged survival of hSOD1G93A mice (20) and delayed spinal motoneuron degeneration in the wobbler mouse (30). The observations drawn from our recent results provide new insights into the synaptic mechanisms contributing to ALS pathogenesis.
ACKNOWLEDGMENTS
This work was supported by grants from Spain's Ministerio de Ciencia e Innovación (SAF2008‐01415) and Consejería de Innovación, Ciencia y Empresa of the Junta de Andalucía (PAI2007‐CTS‐02606); Mutua Madrileña Foundation (B.M.‐L.); Ministerio de Ciencia e Innovación (SAF2008‐01274), Instituto de Salud Carlos III (RD06/0010/0022), and Conselleria de Sanitat–Centro de Investigación Príncipe Felipe (J.M.G.‐V.); Instituto de Salud Carlos III (PI071133) and Action Cost B30 of the European Union (R.O.). We thank Elaine Lilly, PhD (Writer's First Aid), for English language revision of this manuscript.
REFERENCES
- 1. Almer G, Vukosavic S, Romero N, Przedborski S (1999) Inducible nitric oxide synthase up‐regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem 72:2415–2425. [DOI] [PubMed] [Google Scholar]
- 2. Angenstein F, Niessen HG, Goldschmidt J, Vielhaber S, Ludolph AC, Scheich H (2004) Age‐dependent changes in MRI of motor brain stem nuclei in a mouse model of ALS. Neuroreport 15:2271–2274. [DOI] [PubMed] [Google Scholar]
- 3. Anneser JM, Cookson MR, Ince PG, Shaw PJ, Borasio GD (2001) Glial cells of the spinal cord and subcortical white matter up‐regulate neuronal nitric oxide synthase in sporadic amyotrophic lateral sclerosis. Exp Neurol 171:418–421. [DOI] [PubMed] [Google Scholar]
- 4. Avossa D, Grandolfo M, Mazzarol F, Zatta M, Ballerini L (2006) Early signs of motoneuron vulnerability in a disease model system: characterization of transverse slice cultures of spinal cord isolated from embryonic ALS mice. Neuroscience 138:1179–1194. [DOI] [PubMed] [Google Scholar]
- 5. Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M et al (2001) Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci 191:25–33. [DOI] [PubMed] [Google Scholar]
- 6. Bendotti C, Tortarolo M, Suchak SK, Calvaresi N, Carvelli L, Bastone A et al (2001) Transgenic SOD1 G93A mice develop reduced GLT‐1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J Neurochem 79:737–746. [DOI] [PubMed] [Google Scholar]
- 7. Bensimon G, Lacomblez L, Meininger V (1994) A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 330:585–591. [DOI] [PubMed] [Google Scholar]
- 8. Bodian D (1966) Electron microscopy: two major synaptic types on spinal motoneurons. Science 151:1093–1094. [DOI] [PubMed] [Google Scholar]
- 9. Bories C, Amendola J, Lamotte d'Incamps B, Durand J (2007) Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci 25:451–459. [DOI] [PubMed] [Google Scholar]
- 10. Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG et al (1997) ALS‐linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1‐containing inclusions. Neuron 18:327–338. [DOI] [PubMed] [Google Scholar]
- 11. Carunchio I, Mollinari C, Pieri M, Merlo D, Zona C (2008) GAB(A) receptors present higher affinity and modified subunit composition in spinal motor neurons from a genetic model of amyotrophic lateral sclerosis. Eur J Neurosci 28:1275–1285. [DOI] [PubMed] [Google Scholar]
- 12. Catania MV, Aronica E, Yankaya B, Troost D (2001) Increased expression of neuronal nitric oxide synthase spliced variants in reactive astrocytes of amyotrophic lateral sclerosis human spinal cord. J Neurosci 21:RC148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cha CI, Kim JM, Shin DH, Kim YS, Kim J, Gurney ME, Lee KW (1998) Reactive astrocytes express nitric oxide synthase in the spinal cord of transgenic mice expressing a human Cu/Zn SOD mutation. Neuroreport 9:1503–1506. [DOI] [PubMed] [Google Scholar]
- 14. Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME (1995) Age‐dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci 6:349–362. [DOI] [PubMed] [Google Scholar]
- 15. Cleveland DW, Rothstein JD (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2:806–819. [DOI] [PubMed] [Google Scholar]
- 16. Corona JC, Tapia R (2007) Ca2+‐permeable AMPA receptors and intracellular Ca2+ determine motoneuron vulnerability in rat spinal cord in vivo . Neuropharmacology 52:1219–1228. [DOI] [PubMed] [Google Scholar]
- 17. Dal Canto MC, Gurney ME (1994) Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol 145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- 18. DePaul R, Abbs JH, Caligiuri M, Gracco VL, Brooks BR (1988) Hypoglossal, trigeminal, and facial motoneuron involvement in amyotrophic lateral sclerosis. Neurology 38:281–283. [DOI] [PubMed] [Google Scholar]
- 19. Enterzari‐Taher M, Eisen A, Stewart H, Nakajima M (1997) Abnormalities of cortical inhibitory neurons in amyotrophic lateral sclerosis. Muscle Nerve 20:65–71. [DOI] [PubMed] [Google Scholar]
- 20. Facchinetti F, Sasaki M, Cutting FB, Zhai P, MacDonald JE, Reif D et al (1999) Lack of involvement of neuronal nitric oxide synthase in the pathogenesis of a transgenic mouse model of familial amyotrophic lateral sclerosis. Neuroscience 90:1483–1492. [DOI] [PubMed] [Google Scholar]
- 21. Ferrucci M, Spalloni A, Bartalucci A, Cantafora E, Fulceri F, Nutini M et al (2009) A systematic study of brainstem motor nuclei in a mouse model of ALS, the effects of lithium. Neurobiol Dis 37:370–383. [DOI] [PubMed] [Google Scholar]
- 22. Fray AE, Ince PG, Banner SJ, Milton ID, Usher PA, Cookson MR, Shaw PJ (1998) The expression of the glial glutamate transporter protein EAAT2 in motor neuron disease: an immunohistochemical study. Eur J Neurosci 10:2481–2489. [DOI] [PubMed] [Google Scholar]
- 23. Gonzalez‐Forero D, Portillo F, Sunico CR, Moreno‐Lopez B (2004) Nerve injury reduces responses of hypoglossal motoneurones to baseline and chemoreceptor‐modulated inspiratory drive in the adult rat. J Physiol (Lond) 557:991–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gray EG (1959) Axo‐somatic and axo‐dendritic synapses of the cerebral cortex: an electron microscope study. J Anat 93:420–433. [PMC free article] [PubMed] [Google Scholar]
- 25. Gurney ME (1997) Transgenic animal models of familial amyotrophic lateral sclerosis. J Neurol 244(Suppl. 2):S15–S20. [DOI] [PubMed] [Google Scholar]
- 26. Haenggeli C, Kato AC (2002) Differential vulnerability of cranial motoneurons in mouse models with motor neuron degeneration. Neurosci Lett 335:39–43. [DOI] [PubMed] [Google Scholar]
- 27. Hayworth CR, Gonzalez‐Lima F (2009) Pre‐symptomatic detection of chronic motor deficits and genotype prediction in congenic B6.SOD1(G93A) ALS mouse model. Neuroscience 164:975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hollmann M, Hartley M, Heinemann S (1991) Ca2+ permeability of KA‐AMPA – gated glutamate receptor channels depends on subunit composition. Science 252:851–853. [DOI] [PubMed] [Google Scholar]
- 29. Hu JH, Chernoff K, Pelech S, Krieger C (2003) Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over‐expressing mice. J Neurochem 85:422–431. [DOI] [PubMed] [Google Scholar]
- 30. Ikeda K, Iwasaki Y, Kinoshita M (1998) Neuronal nitric oxide synthase inhibitor, 7‐nitroindazole, delays motor dysfunction and spinal motoneuron degeneration in the wobbler mouse. J Neurol Sci 160:9–15. [DOI] [PubMed] [Google Scholar]
- 31. Ince PG, Slade J, Chinnery RM, McKenzie J, Royston C, Roberts GW, Shaw PJ (1995) Quantitative study of synaptophysin immunoreactivity of cerebral cortex and spinal cord in motor neuron disease. J Neuropathol Exp Neurol 54:673–679. [DOI] [PubMed] [Google Scholar]
- 32. Jiang M, Schuster JE, Fu R, Siddique T, Heckman CJ (2009) Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J Neurosci 29:15031–15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim DS, Kwak SE, Kim JE, Won MH, Choi HC, Song HK et al (2005) Bilateral enhancement of excitation via up‐regulation of vesicular glutamate transporter subtype 1, not subtype 2, immunoreactivity in the unilateral hypoxic epilepsy model. Brain Res 1055:122–130. [DOI] [PubMed] [Google Scholar]
- 34. Kong J, Xu Z (1998) Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci 18:3241–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leichsenring A, Linnartz B, Zhu XR, Lubbert H, Stichel CC (2006) Ascending neuropathology in the CNS of a mutant SOD1 mouse model of amyotrophic lateral sclerosis. Brain Res 1096:180–195. [DOI] [PubMed] [Google Scholar]
- 36. Lipton SA (2004) Failures and successes of NMDA receptor antagonists: molecular basis for the use of open‐channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 1:101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lloyd CM, Richardson MP, Brooks DJ, Al‐Chalabi A, Leigh PN (2000) Extramotor involvement in ALS: PET studies with the GABA(A) ligand [(11)C]flumazenil. Brain 123:2289–2296. [DOI] [PubMed] [Google Scholar]
- 38. Luo L (2000) Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci 1:173–180. [DOI] [PubMed] [Google Scholar]
- 39. Luo L (2002) Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol 18:601–635. [DOI] [PubMed] [Google Scholar]
- 40. Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, Golden WC (2007) Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase‐1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol 500:20–46. [DOI] [PubMed] [Google Scholar]
- 41. Matsumoto S, Goto S, Kusaka H, Ito H, Imai T (1994) Synaptic pathology of spinal anterior horn cells in amyotrophic lateral sclerosis: an immunohistochemical study. J Neurol Sci 125:180–185. [DOI] [PubMed] [Google Scholar]
- 42. Miana‐Mena FJ, Munoz MJ, Yague G, Mendez M, Moreno M, Ciriza J et al (2005) Optimal methods to characterize the G93A mouse model of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 6:55–62. [DOI] [PubMed] [Google Scholar]
- 43. Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7:710–723. [DOI] [PubMed] [Google Scholar]
- 44. Petri S, Krampfl K, Hashemi F, Grothe C, Hori A, Dengler R, Bufler J (2003) Distribution of GABAA receptor mRNA in the motor cortex of ALS patients. J Neuropathol Exp Neurol 62:1041–1051. [DOI] [PubMed] [Google Scholar]
- 45. Plaitakis A (1990) Glutamate dysfunction and selective motor neuron degeneration in amyotrophic lateral sclerosis: a hypothesis. Ann Neurol 28:3–8. [DOI] [PubMed] [Google Scholar]
- 46. Prithviraj R, Kelly KM, Espinoza‐Lewis R, Hexom T, Clark AB, Inglis FM (2008) Differential regulation of dendrite complexity by AMPA receptor subunits GluR1 and GluR2 in motor neurons. Dev Neurobiol 68:247–264. [DOI] [PubMed] [Google Scholar]
- 47. Rao SD, Weiss JH (2004) Excitotoxic and oxidative cross‐talk between motor neurons and glia in ALS pathogenesis. Trends Neurosci 27:17–23. [DOI] [PubMed] [Google Scholar]
- 48. Raoul C, Estevez AG, Nishimune H, Cleveland DW, DeLapeyriere O, Henderson CE et al (2002) Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS‐linked SOD1 mutations. Neuron 35:1067–1083. [DOI] [PubMed] [Google Scholar]
- 49. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62. [DOI] [PubMed] [Google Scholar]
- 50. Rothstein JD (2009) Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol 65(Suppl. 1):S3–S9. [DOI] [PubMed] [Google Scholar]
- 51. Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT‐1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84. [DOI] [PubMed] [Google Scholar]
- 52. Rowland LP, Shneider NA (2001) Amyotrophic lateral sclerosis. N Engl J Med 344:1688–1700. [DOI] [PubMed] [Google Scholar]
- 53. Sasaki S, Iwata M (1996) Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci Lett 204:53–56. [DOI] [PubMed] [Google Scholar]
- 54. Sasaki S, Iwata M (1996) Ultrastructural study of the synapses of central chromatolytic anterior horn cells in motor neuron disease. J Neuropathol Exp Neurol 55:932–939. [DOI] [PubMed] [Google Scholar]
- 55. Sasaki S, Maruyama S (1994) Synapse loss in anterior horn neurons in amyotrophic lateral sclerosis. Acta Neuropathol 88:222–227. [DOI] [PubMed] [Google Scholar]
- 56. Sasaki S, Shibata N, Iwata M (2001) Neuronal nitric oxide synthase immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol 101:351–357. [DOI] [PubMed] [Google Scholar]
- 57. Sasaki S, Warita H, Abe K, Iwata M (2002) Neuronal nitric oxide synthase (nNOS) immunoreactivity in the spinal cord of transgenic mice with G93A mutant SOD1 gene. Acta Neuropathol 103:421–427. [DOI] [PubMed] [Google Scholar]
- 58. Schutz B (2005) Imbalanced excitatory to inhibitory synaptic input precedes motor neuron degeneration in an animal model of amyotrophic lateral sclerosis. Neurobiol Dis 20:131–140. [DOI] [PubMed] [Google Scholar]
- 59. Sturrock RR (1991) Stability of motor neuron and interneuron number in the hypoglossal nucleus of the ageing mouse brain. Anat Anz 173:113–116. [PubMed] [Google Scholar]
- 60. Sunico CR, Portillo F, Gonzalez‐Forero D, Moreno‐Lopez B (2005) Nitric oxide‐directed synaptic remodeling in the adult mammal CNS. J Neurosci 25:1448–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sunico CR, Gonzalez‐Forero D, Dominguez G, Garcia‐Verdugo JM, Moreno‐Lopez B (2010) Nitric oxide induces pathological synapse loss by a protein kinase G‐, Rho kinase‐dependent mechanism preceded by myosin light chain phosphorylation. J Neurosci 30:973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Takasu N, Hashimoto PH (1988) Morphological identification of an interneuron in the hypoglossal nucleus of the rat: a combined Golgi‐electron microscopic study. J Comp Neurol 271:461–471. [DOI] [PubMed] [Google Scholar]
- 63. Uchizono K (1965) Characteristics of excitatory and inhibitory synapses in the central nervous system of the cat. Nature 207:642–643. [DOI] [PubMed] [Google Scholar]
- 64. Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W (2006) The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta 1762:1068–1082. [DOI] [PubMed] [Google Scholar]
- 65. Van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH et al (2008) Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult‐onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 28:10864–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA et al (1995) An adverse property of a familial ALS‐linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105–1116. [DOI] [PubMed] [Google Scholar]
- 67. Zang DW, Cheema SS (2002) Degeneration of corticospinal and bulbospinal systems in the superoxide dismutase 1(G93A G1H) transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett 332:99–102. [DOI] [PubMed] [Google Scholar]
- 68. Zang DW, Lopes EC, Cheema SS (2005) Loss of synaptophysin‐positive boutons on lumbar motor neurons innervating the medial gastrocnemius muscle of the SOD1G93A G1H transgenic mouse model of ALS. J Neurosci Res 79:694–699. [DOI] [PubMed] [Google Scholar]
- 69. Zhao P, Ignacio S, Beattie EC, Abood ME (2008) Altered presymptomatic AMPA and cannabinoid receptor trafficking in motor neurons of ALS model mice: implications for excitotoxicity. Eur J Neurosci 27:572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]