

Abstract
Infiltration of leukocytes into post‐ischemic cerebrum is a well‐described phenomenon in stroke injury. Because CD‐8+ T‐lymphocytes secrete cytotoxic proteases, including granzyme‐b (Gra‐b) that exacerbates post‐ischemic brain damage, we investigated roles of Gra‐b in human stroke. To study the role of Gra‐b in stroke, ischemic and non‐ischemic tissues (from post‐mortem stroke patients) were analyzed using immunoblotting, co‐immunoprecipitation, terminal deoxy uridine nick end labeling (TUNEL) and Annexin–V immunostaining, and in vitro neuron survival assays. Activated CG‐SH cells and supernatants were used to model leukocyte‐dependent injury. Non‐ischemic brain tissues were used as non‐pathological controls. Non‐activated CG‐SH cells and supernatants were used as controls for in vitro experiments. Human stroke (ischemic) samples contained significantly higher levels of Gra‐b and interferon‐gamma inducible protein‐10 (IP‐10/CXCL10) than non‐ischemic controls. In stroke, poly (ADP‐ribose) polymerase‐1 and heat shock protein‐70 were cleaved to canonical proteolytic “signature” fragments by Gra‐b. Gra‐b was also found to bind to Bid and caspase‐3. Gra‐b also co‐localized with Annexin‐V+/TUNEL+ in degenerating neurons. Importantly, Gra‐b inhibition protected both normal and ischemia‐reperfused neurons against in vitro neurotoxicity mediated by activated CG‐SH cells and supernatants. These results suggest that increased leukocyte infiltration and elevated Gra‐b levels in the post‐stroke brain can induce contact‐dependent and independent post‐ischemic neuronal death to aggravate stroke injury.
Keywords: Myeloid leukemia cell differentiation protein‐1, apoptosis‐inducing factor, heat shock protein‐70, poly (ADP‐ribose) polymerase‐1, terminal deoxy uridine nick end labeling
INTRODUCTION
Cerebral ischemia is the second leading cause of death and disability worldwide. The pathophysiology of stroke injury is highly complex, involving interactions between multiple cell types and signal systems 12, 45, 66, 69. Focal cerebral ischemia results in apoptosis (caspase dependent or independent), necrosis (calpains and/or cathepsins) and inflammation characterized by immune cell recruitment and infiltration of the post‐ischemic area 18, 69, 71. Interactions between immune cells and tissue in the post‐ischemic infarct increase injury beyond that mediated by reperfusion alone. While recent reports have shown that T‐cells can promote ischemic damage, the signaling intermediates and mechanisms involved are not yet known 37, 80. In our present study, we address the role of cytotoxic T‐cell (CTL) derived Gra‐b in neuronal death associated with ischemic damage. The functional role of Gra‐b in inducing neurotoxicity and aggravating human stroke injury was studied in vitro using human myeloid and neuronal cell lines. The results of our present study assign a pathological role of infiltrating CTLs and Gra‐b in the exacerbation and expansion of stroke injury in the post‐ischemic brain.
MATERIALS AND METHODS
Human stroke samples
Human post‐mortem stroke samples were obtained from Brain Bank‐National Institute of Mental Health and Neuro‐Sciences, Bangalore, India after informed consent from the close relatives allowing use of specimens for the research. The identity of the specimens was not disclosed. These protocols were also approved by Institutional Scientific Ethics Committee. The pathological details of the tissue samples are tabulated in Table 1.
Table 1.
Details of the post‐mortem human stroke samples. Abbreviations: CVT = Cerebrovenous thrombosis; MCA = Middle cerebral artery; PCA = Posterior cerebral artery.
| Sample ID | Age/sex/post‐mortem delay | Post‐stroke survival period | Recurrent strokes | Region of tissue removal | Other details |
|---|---|---|---|---|---|
| 08HBTR/T197 | 35 years/female/27 h/cerebrovenous thrombosis (CVT) | 5 days | None | Frontal cortex (control) and parieto‐occipital infarct (ischemic tissue) | Post‐partum CVT |
| 08HBTR/T198 | 65 years/female/12 h, 30 minutes/CVT | 11 days | None | Left frontal infarct (ischemic tissue) Left frontal away from infarct (control) | No other information |
| 08HBTR/T199 | 19 years/female/13 h/CVT | 8 days | None | Left fontal infarct (ischemic tissue) Left frontal away from infarct (control) | No other information |
| 08HBTR/T200 | 45 years/male/1 h, 30 minutes/no cortical infarcts | — | — | Frontal cortex (normal used as control), cerebellum | Death due to hypertension, systemic vasculitis, hyponatremia with pontine myelinolysis |
| 08HBTR/T201 | 55 years/female/2 h atherosclerotic cerebrovascular disease | 1 month | 1st episode left PCA, 1 week later—2nd episode in left MCA | Right MCA (control), left MCA infarct (used as ischemic tissue‐i1) and left MCA infarct edge (used as ischemic tissue‐i2) | Hypertensive (4 months), diffuse atherosclerosis of carotid vessels |
Antibodies
Gra‐b (Calbiochem, Cat: AM52, Darmstedt, Germany; Abcam, Cat: Ab53097, Temecula, CA, USA); Perforin (Cell Signaling and Technology, Cat: 3693, Danvers, MA, USA); poly (ADP‐ribose) polymerase‐1 (PARP‐1) (Cell Signaling and Technology, Cat: 9542, Danvers, MA, USA); Myeloid leukemia cell differentiation protein‐1 (MCL‐1) (BD Transduction, Cat: M54020, California, USA); FITC‐microtubule associated protein‐2 (MAP‐2) (Chemicon, Cat: MAB3418X, Temecula, CA, USA); neuron specific enolase (NSE) (Chemicon, Cat: AB951); Caspase‐3 (Cell Signaling Technology, Cat: 9664S, Danvers, MA, USA), apoptosis‐inducing factor (AIF) (Oncogene, Cat: PC536, Darmstadt, Germany), ApoAlert DNA fragmentation (TUNEL) kit (Clontech, Takara, CA, USA).
Immunoblotting of human post‐mortem stroke tissue
Post‐mortem brain tissues were divided into two parts. One part was fixed with 4% paraformaldehyde for staining, and the other part was used for immunoblot analysis. For immunoblot analysis samples were homogenized in modified radio immunoprecipitation assay (RIPA) buffer (+protease/phosphatase inhibitors). 50 µgms of protein (estimated by Lowry's method) was immunoblotted for Gra‐b (Calbiochem, 1:1000), PARP‐1, IP‐10/CXCL10, heat shock protein‐70 (HSP‐70), MCL‐1, AIF, KU‐70 and actin (1:1000) and developed using nitro blue tetrazolium/5‐bromo‐4‐chloro‐3‐indolyl phosphate (NBT/BCIP) or enhanced chemiluminiscence (ECL) detection methods.
Co‐immunoprecipitation (co‐IP)
Co‐IP was performed as described by Cao et al (11). Briefly after preclearing, 350 µg protein was incubated with Gra‐b antibody (0.5 µg) for 12 h at 4°C followed by incubation with 25 µl of protein A sepharose (90 minutes) at room temperature (RT). Beads were washed 3 times with PBS (pH 7.4), boiled in 2X sample buffer and immunoblotted for HSP‐70, MCL‐1, Caspase‐3 and PARP‐1 (1:1000). Ischemic tissue lysates without immunoprecipitation was used as positive control. Samples processed in the same way but without addition of primary antibodies to the tissue lysates were used as negative controls to identify non‐specific binding of proteins.
Immunofluorescent co‐localization
Paraffin embedded human post‐mortem stroke tissues (3 µm thick, n = 3) were processed, antigen‐retrieved in citrate buffer (pH −6.0) and incubated in 10% normal goat serum (NGS) (30 minutes). Double immunofluorescence analysis for Gra‐b: PARP‐1 was performed by incubating the sections in a primary antibody cocktail followed by incubation in cy3‐and cy5‐conjugated secondary antibodies. For Gra‐b: HSP‐70 and Gra‐b: MAP‐2, sequential staining was followed by an additional step of blocking with 5% NGS (30 minutes, RT). The sections were later washed, mounted (90% glycerol) and visualized by confocal microscopy (Leica confocal microscope, Heidelberg, Germany).
Immunofluorescent co‐localization of TUNEL with Gra‐b and NSE, Annexin‐V (Ann‐V) with Gra‐b and Neu‐N
Paraffin embedded human post‐mortem stroke tissues (3 µm thick, n = 3) were processed as described above and incubated with Gra‐b, NSE primary antibodies overnight at 4°C followed by cy‐3, cy‐5 secondary antibody incubation. TUNEL staining was performed according to manufacturer's instructions. A similar procedure was followed for Ann‐V: Neu‐N: Gra‐b except for additional incubation steps with diaminobenzidine (DAB). Briefly, sections were blocked (30 minutes with 5% NGS) and incubated in Neu‐N antibody at 4°C overnight followed by washes and incubating the sections in Vectastatin ABC elite kit secondary antibody kit (Burlingame, CA, USA) and developed with DAB. The sections were extensively washed and blocked again (for 30 minutes with 5% NGS) and incubated with Gra‐b (Abcam) antibody at 4°C overnight. Sections were incubated in cy‐3 conjugated secondary antibody followed by incubation in FITC‐Ann‐V (1:100) (90 minutes, RT). Sections were mounted with DAPI (4′, 6‐diamidino‐2‐phenylindole) and were visualized by fluorescent microscopy (Leica). All antibody dilutions were performed in Prohisto immunohistochemistry (IHC) amplification antibody dilution buffer (Columbia, SC, USA) and washed in Prohisto IHC amplification wash buffer for this staining procedure.
Cell culture
“SH‐SY5Y” cells (a human neuroblastoma cell line extensively used as an in vitro model of neurons) were cultured in dulbecco's modified eagle medium (DMEM) with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin/amphoptericin (PSA). Similarly CG‐SH cells (a human myeloid cell line, ∼82% CD‐3+) which secrete Gra‐b and perforin was used as model for CTL, Gra‐b and perforin induced cytotoxicity (provided by Dr. Reinhold Munker, Louisiana state university health sciences center‐Shreveport (LSUHSC‐S) (54), were cultured in Roswell Park Memorial Institute‐1640 medium (RPMI‐1640) with 10% FCS and 1% PSA at 37°C.
Activation of CG‐SH cells
2 × 106 CG‐SH cells were activated using 1 µM calcium ionophore (A23187, Sigma, St. Louis, MO, USA) + 50 ng/ml phorbol myristate (PMA) for 48 h 2, 16. Cells were re‐plated in fresh culture medium for additional 24 h. Culture medium was collected and stored at −80°C until use. Cells were centrifuged and re‐suspended in culture medium with or without Gra‐b inhibitor (50 µM). After this time point, activated CG‐SH cells ± Gra‐b inhibitor were added to normal or oxygen glucose deprived human neurons cultured at a 1:5 ratio (SH‐SY5Y: CG‐SH) in 96 well plates.
Oxygen glucose deprivation (OGD) and reperfusion of SH‐SY5Y cells
At confluence, neuronal culture media was replaced with OGD buffer (prepared using DMEM without glucose, supplemented with 3.7 g/L NaHCO3, pH −7.35) and incubated in an hypoxia chamber purged with 88% N2, 5% CO2, 1% O2. OGD was maintained for 3 h. Later, neurons were reperfused by transferring them back to normal culture media/air.
Neurotoxicity assay with activated leukocyte supernatants
SH‐SY5Y neurons were cultured to confluence in 96 well plates and culture media was replaced with control (1:1) or activated CG‐SH supernatants at various dilutions (1:1, 1:5, 1:10) with culture medium (CG‐SH supernatant: culture medium) 16, 76. In separate experiments SH‐SY5Y neurons are ischemic challenged by OGD for 3 h and reperfused with control or activated CG‐SH supernatants at the above‐mentioned dilutions. Twenty‐four hours post‐incubation with CG‐SH supernatants, thiazolyl blue tetrazolium bromide (MTT assay) was performed to assess the neuronal toxicity.
Gra‐b inhibition and neuronal survival assay
Briefly, SH‐SY5Y neurons were cultured to confluency in 96 well plates, and incubated with control or activated CG‐SH ± Gra‐b inhibitor I (50 µM, Calbiochem, cat. #368050) for 24 h at a 1:5 ratio (SH‐SY‐5Y: CG‐SH). In parallel experiments, neuronal ischemia was induced in SH‐SY5Y cells by depriving cells of oxygen/glucose and reperfusing with culture medium containing control or activated CG‐SH cells ± Gra‐b inhibitor (50 µM). Twenty‐four hours after treatment, cell viability was assessed using MTT assay.
Immunoblot analysis
Densitometric analysis of immunoblots was performed using NIH image J analysis (Bethesda, USA), and the values compared using Graphpad Instat3 software (CA, USA). Two‐tailed unpaired t‐test was used to check statistical significance between two groups. One‐way analysis of variance (ANOVA) with Dunnett's or Bonferroni post‐tests were used to determine statistical significance in MTT assays. A *P < 0.05 was considered to be significant, **P < 0.01 very significant, ***P < 0.001 extremely significant.
RESULTS
Increased cell death and tissue destruction in human stroke tissue
Hematoxylin and eosin tissue staining was used to distinguish signs of tissue deterioration in ischemic‐stroke and non‐ischemic control tissues. In ischemic stroke samples, increased numbers of cells showing disintegration of the neuropil, shrunken nuclei and extensive vacuolization were observed, consistent with extensive cell death (compared with non‐ischemic controls). Furthermore, sections were strongly stained with eosin indicating increased oncosis (cell swelling, increased water content). Basement membrane, neuropil and tissue architecture were also extensively deranged, revealing the necrotic features and edematous condition of ischemic stroke tissues compared with controls (Figure 1).
Figure 1.

Hematoxylin and eosin staining shows increased apoptosis and necrosis in the post‐mortem human stroke tissue. Human post‐mortem stroke samples were processed and stained with hematoxylin and eosin. Tissues obtained are from the infarct zone and away from the infarct (considered non‐ischemic tissue) which increased the homogeneity of the tissue to be used as control for the ischemic tissue. Deranged brain tissue with several degenerating cells (pointed with small arrows and arrow heads), shrunken nucleus, disrupted neuropil and extensive disruption of brain parenchyma and matrix (pointed with big arrows) is seen in the ischemic tissue. Moreover, most of the ischemic samples were heavily stained with eosin indicating the edematous condition of the tissue (indicated with *). Increased eosin staining was observed in all the ischemic samples over controls. Slight derangement of tissue was also observed occasionally in the control tissue but essentially looked similar to normal. Normal (non‐ischemic) brain tissue from patient sample ID 08/HBTR/T200 was obtained from cortical region. The regions and types of tissues obtained were outlined in the Table 1.
Increased IP‐10/CXCL10 levels correlate with increased Gra‐b levels secreted by infiltrating CD‐8+ T‐cells (CTLs) in human stroke samples
IP‐10/CXCL10, a CXC‐chemokine that functions as a potent chemo‐attractant for activated T cells, natural killer (NK) cells and blood monocytes, is an important mediator of several inflammatory conditions. Excessive production of IP‐10/CXCL10 by astrocytes has been observed in Japanese encephalitis (9) and inhibition of IP‐10/CXCL10 has shown to reduce pathology mediated by T‐cells in various experimental animal models, namely Theiler's virus, experimental autoimmune encephalitis (EAE) a model of multiple sclerosis (73). We observed a significant increase in IP‐10/CXCL10 levels in all post‐mortem ischemic stroke samples (Figure 2A). The elevation of IP‐10/CXCL10 in stroke tissues correlated with increase in tissue Gra‐b levels. Gra‐b, a cytotoxic serine protease secreted from CTLs and NK cells was elevated in all human post‐ischemic stroke samples along with IP‐10/CXCL10 levels in stroke tissues (Figure 2B). Double immunofluorescence analysis confirmed that CTLs infiltrating human stroke tissue were Gra‐b+. We observed a strong co‐reactivity of CD‐8+ T‐cells (CTLs) with Gra‐b in ischemic stroke samples (∼64% ± 4.4% of CD‐8+ T cells are Gra‐b+) compared with the non‐ischemic tissue controls, confirming that increased levels of Gra‐b in ischemic tissue match CTL infiltration in the stroke brain (Figure 3).
Figure 2.

A.Post‐mortem human stroke tissue contains elevated IP‐10/CXCL10 and Gra‐b levels. Immunoblot analysis of IP‐10/CXCL10 shows a significant increase in IP‐10/CXCL10 levels in the ischemic human stroke samples (ipsilateral) over the non‐ischemic controls (contralateral). B. Immunoblot analysis of Gra‐b also shows a significant increase in the Gra‐b levels in all ischemic human stroke samples (ipsilateral) over the non‐ischemic samples (contralateral). T200n‐normal brain tissue; T197c, T97i‐contralateral and ipsilateral tissue from HBTR‐197; T198c, T198i‐contralateral and ipsilateral tissue from HBTR‐198; T199c, T199i‐contralateral and ipsilateral tissue from HBTR‐199; T201c, T201‐i1, T201‐i2‐contralateral and ipsilateral tissue from HBTR‐201. Unpaired t‐test with two‐tail P value, bars standard error (SE) * denotes significance.
Figure 3.

Human ischemic infarcts contain increased numbers of Gra‐b+ CD‐8+ T‐cells. Double immunofluorescence analysis of CD‐8 and Gra‐b showed increased numbers of CD‐8+ T cells (FITC fluorescence‐ green) that were also Gra‐b+ (Cy3 fluorescence‐red) in human ischemic post‐mortem stroke samples compared with corresponding non‐ischemic controls. Representative immunofluorescent images are from the patient's samples with IDs’ T201c, T201‐i1.
Anti‐apoptotic molecules are proteolyzed by Gra‐b
HSP‐70 is a crucial anti‐apoptotic regulator 25, 26, which inhibits apoptosis by preventing the translocation of AIF to the nucleus, by blocking c‐Jun N‐terminal kinase (JNK) and lysosomal protease (cathepsin‐b and ‐d) activation and promoting stabilization of anti‐apoptotic MCL‐1 10, 30, 43, 44, 47, 56, 59, 68, 82. Co‐IP revealed an increase in binding of Gra‐b with HSP‐70 in ischemic samples over the controls (Figure 4A). Such binding interactions of Gra‐b might explain the appearance of 40‐kD HSP‐70 signature fragment in immunoblot analysis (Figure 4A). Immunoblot for AIF in nuclear samples revealed an increased level of AIF (Figure 4B). KU‐70, a nuclear protein was used to ensure the equal loading of protein. Gra‐b‐mediated breakdown of HSP‐70 might be one possible reason for increase in the nuclear translocation of AIF aggravating post‐ischemic damage in human ischemic brains correlating with our previous results (13).
Figure 4.
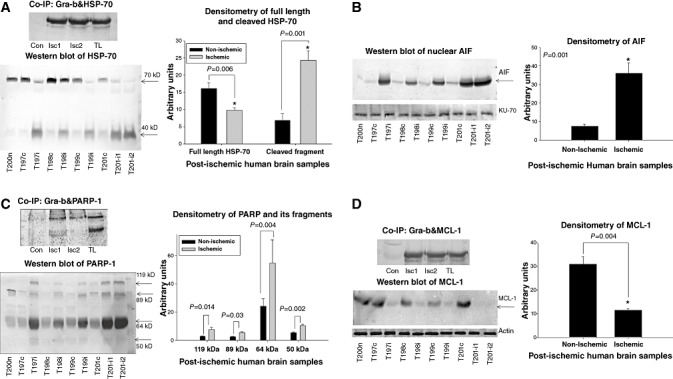
A. Gra‐b proteolyzes HSP‐70, PARP‐1 and MCL‐1 and facilitates the nuclear translocation of AIF. Co‐immunoprecipitation (co‐IP) of Gra‐b with HSP‐70. Increased binding of Gra‐b with HSP‐70 can be observed. Representative image is from the “T201” which has two separate regions of the ischemic infarct. Immunoblot of HSP‐70. Significantly decreased level of HSP‐70 correlated with increased 40 kD proteolytic signature fragment mediated by Gra‐b can be observed. B. Immunoblot of AIF on nuclear samples. A significant increase in the levels of nuclear AIF can be observed. KU‐70 was used as a loading control for nuclear samples. C. Co‐IP of Gra‐b with PARP‐1. Increased binding of Gra‐b with PARP‐1 can be observed (119 kD). Immunoblot of PARP‐1. Significant increase in the cleaved signature fragments 89 and 64 kD can be observed. Further faint increase in the levels of 50 kD cathepsin‐b and can also be observed. D. Co‐IP of Gra‐b with MCL‐1. Increased binding of Gra‐b with MCL‐1 can be observed. Immunoblot of MCL‐1. Significant decrease in the levels of MCL‐1 in all the ischemic samples over the contralateral was observed. Representative image for Co‐IP is from the “T201” which has two non‐overlapping regions of the ischemic infarct. Unpaired t‐test with two tailed P value was obtained between two specific groups. Con‐ Non ischemic tissue, Isc1, Isc2‐ Ischemic tissue lysates, TL‐ Non immunoprecipitated ischemic tissue lysate (used as positive control).
PARP‐1 is a nuclear enzyme with diverse influences on cell survival, inflammatory and apoptotic/necrotic cell death pathways, depending on the nature and intensity of insult 36, 51, 57. PARP‐1 is a preferred substrate for Gra‐b, which upon cleavage by Gra‐b generates two signature fragments of 64 and 72‐kD (22). Co‐IP of Gra‐b and PARP‐1 revealed increased binding of Gra‐b with PARP‐1 in ischemic samples (over the contralateral) (Figure 4C). Furthermore, we also found elevated levels of 64, 89 and 50‐kD fragments [signature fragments of Gra‐b, caspase‐3 (apoptotic) and cathepsin‐b (necrotic), respectively] evidence for apoptotic and necrotic cell death pathways (64) (Figure 4C).
MCL‐1 is also presumed to be an important anti‐apoptotic molecule that blocks apoptotic cell death (53). Gra‐b degrades MCL‐1 and disintegrates Bim‐MCL‐1 complexes, resulting in Bim‐mediated apoptosis 31, 32. Co‐IP for Gra‐b and MCL‐1 revealed an increased binding of Gra‐b with MCL‐1 resulting in decreased levels in ischemic samples (Figure 4D). We observed a significant decrease in the MCL‐1 levels in all the post‐ischemic human brain samples which correlated with the increased levels of Gra‐b. Furthermore, breakdown of HSP‐70 (which stabilizes MCL‐1) by Gra‐b may accelerate this process 46, 68.
Double immunofluorescent co‐localization of Gra‐b with HSP‐70 and Gra‐b with PARP‐1 revealed an increase in the co‐localization of Gra‐b with HSP‐70 and PARP‐1 in ischemic samples (vs. non‐ischemic controls) correlating with our co‐IP experiments (Figure 5A, B).
Figure 5.
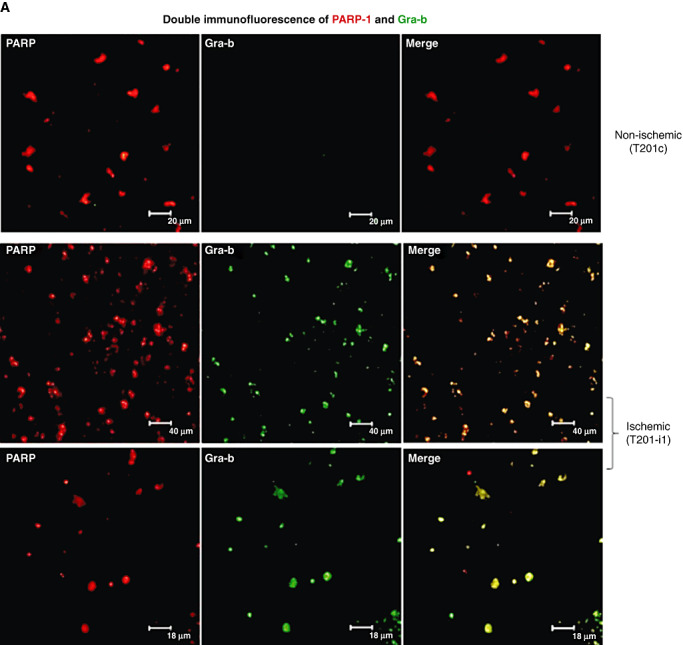

A. Gra‐b co‐localizes with PARP‐1 and HSP‐70 in the degenerating cells of human post‐mortem ischemic infarct samples. Double immunofluorescence of Gra‐b and PARP‐1. Increased immunoreactivity for cells stained positive with both Gra‐b and PARP‐1 can be observed in ischemic samples over the contralateral samples. B. Double immunofluorescence of Gra‐b and HSP‐70. Increased immunoreactivity for cells stained positive with both Gra‐b and HSP‐70 can be observed in ischemic samples over the contralateral. Unpaired test with two‐tail P value, bars SE. Representative immunofluorescent images are from the patient's samples with IDs’ T201c, T201‐i1.
Gra‐b interacts with pro‐apoptotic molecules
Gra‐b activates apoptotic cell death independent of caspases 35, 68, 70, 72. Bid, a pro‐apoptotic mediator, upon binding to Gra‐b and/ or caspase‐3 forms “truncated Bid” (t‐Bid). t‐Bid then translocates into mitochondria and activates intrinsic apoptosis pathways 3, 7, 29, 67, 77. Co‐IP experiments with Gra‐b and Bid showed increased binding between Gra‐b and Bid in the ischemic samples (compared with controls) and suggest an increase in the activation of the intrinsic apoptosis pathway by the formation of t‐Bid (Figure 6A).
Figure 6.

A. Gra‐b interacts with pro‐apoptotic Bid and caspase‐3 in the human post‐mortem ischemic infarcts. Co‐immunoprecipitation (co‐IP) of Gra‐b with Bid. Increased binding of Gra‐b with Bid can be clearly observed. B. Co‐IP of Gra‐b with caspase‐3. Increased binding of Gra‐b with caspase‐3 can be observed. Representative image for Co‐IP is from the “T201” which has two non‐overlapping regions of the ischemic infarct. Con‐ Non ischemic tissue, Isc1, Isc2‐ Ischemic tissue lysates, TL‐ Non immunoprecipitated ischemic tissue lysate (used as positive control).
Caspases are also known to contribute to post‐ischemic cerebral damage by apoptosis, a major mechanism for cell death in the penumbra of post‐ischemic infarcts 20, 21. We found increased binding of Gra‐b to caspase‐3 using co‐IP (Figure 6B). Gra‐b interactions with caspase‐3 would be anticipated to amplify activation of caspase‐3 and intensify post‐ischemic apoptosis in the infarct.
Gra‐b associates with increased numbers of degenerating neurons in human stroked samples
The precise roles of cytotoxic proteases in neuronal death during cerebral ischemia are not known. Double immunofluorescent co‐localization of Gra‐b with MAP‐2 clearly shows that Gra‐b is translocated into neurons within ischemic infarcts (Figure 7A). Gra‐b secreted from CTLs appears to bind, and penetrate into the neurons within the ischemic infarct resulting in neurons which are positive for Gra‐b. Therefore to address whether Gra‐b+ neurons were also going through cell death, we stained human brain sections with Ann‐V (an apoptotic marker)/Gra‐b/NSE and TUNEL (apoptotic marker)/Gra‐b/Neu‐N (Figure 7B, C). We found elevated numbers of Gra‐b+ and Ann‐V/ TUNEL+ neurons in post‐ischemic infarcts of human brain samples indicating that Gra‐b could contribute to neuronal death in the stroke brain. We observed approximately ∼31.3% ± 2.3% of the Ann‐V+ neurons were also Gra‐b+ and undergoing cell death in the ischemic stroke samples over the non‐ischemic controls (∼4.8 +/− 1.8%).
Figure 7.

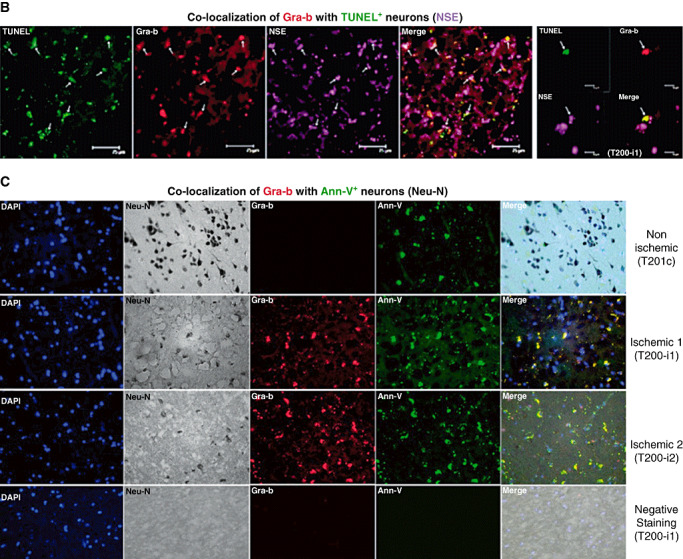
A. Apoptotic neurons in the human post‐mortem ischemic infarcts contain higher levels of translocated Gra‐b. Double immunofluorescence analysis of Gra‐b and MAP‐2. Increased presence of translocated Gra‐b (Cy3 fluorescence‐red) in the neurons stained positive for MAP‐2 (FITC fluorescence‐green) can be observed in the ischemic samples over the contralateral. Most of the cells positive for MAP‐2 appeared to be degenerating because of their shrunken shape, lost of cellular structure and disorganized neuropil. Representative immunofluorescent images are from the patient's samples with IDs’ T201c, T201‐i1. B. Double immunofluorescence analysis of Gra‐b and NSE with TUNEL. Increased number of cells positive for TUNEL (FITC fluorescence‐green), Gra‐b (Cy3 fluorescence‐red) and NSE (Cy5 fluorescence‐violet) can be observed in the ischemic samples. Representative immunofluorescent image is from the patient's sample with ID T200‐i1. C. Triple immunostaining of Gra‐b, Neu‐N and Annexin‐V (Ann‐V). Increased immunoreactivity of cells for Gra‐b (Cy3 fluorescence‐red), Neu‐N DAB stained and Ann‐V (FITC fluorescence‐green) can be observed in the human ischemic tissue compared with non‐ischemic human stroke samples. No immunoreactivity is observed in negative staining. Representative immunofluorescent images are from the patient's samples with IDs’ T201c, T201‐i1, T201‐i2.
Gra‐b inhibition protects SH‐SY5Y neurons from CG‐SH induced neurotoxicity
In order to assess whether activated lymphocytes mediated neuronal injury and if this injury was Gra‐b dependent, we performed in vitro neurotoxicity experiments using SH‐SY5Y‐human neuroblastoma cells as model of neurons. CG‐SH (human myeloid cell line) cells secrete Gra‐b and perforin and were used as a model for CTL‐mediated cytotoxicity. We observed that activated CG‐SH cells secrete significantly greater levels of Gra‐b and perforins compared with non‐activated CG‐SH cells (Figure 8A). Moreover, we observed a significant and dose‐dependent decrease in neuronal viability when incubated with activated CG‐SH supernatants. SH‐SY5Y neurons incubated with supernatants from non‐activated CG‐SH cells diluted at a 1:1 (CG‐SH supernatant: culture medium) ratio was used as controls. Activated CG‐SH supernatants diluted 1:1 with normal culture medium (which contain highest levels of Gra‐b and perforins) showed the highest neurotoxic effect (Figure 8B). Activated CG‐SH supernatants also induced neurotoxicity in ischemia‐challenged neurons in a dose dependent fashion. Controls included ischemia challenged SH‐SY5Y neurons reperfused with non‐activated CG‐SH supernatants at a 1:1 dilution (Figure 8C). We next assessed whether Gra‐b inhibition could attenuate the neurotoxic effects of activated CG‐SH cells. Normal or ischemia challenged SH‐SY5Y neurons were incubated with non‐activated (controls) or activated CG‐SH cells at 1:5 ratios (SH‐SY5Y: CG‐SH). Both cell types were incubated with Gra‐b inhibitor (50 µM) for 1 h prior to their co‐incubation. SH‐SY5Y neurons and activated or non‐activated CG‐SH cells were co‐incubated for 24 h in the presence of Gra‐b inhibitor. We observed that activated CG‐SH cells strongly induced neurotoxicity in both normal and ischemic challenged SH‐SY5Y neurons. This neurotoxic effect was significantly attenuated by the Gra‐b inhibitor when added either during normal or during OGD reperfused conditions, indicating that activated CG‐SH induce neurotoxicity via Gra‐b dependent mechanisms (Figure 8D).
Figure 8.
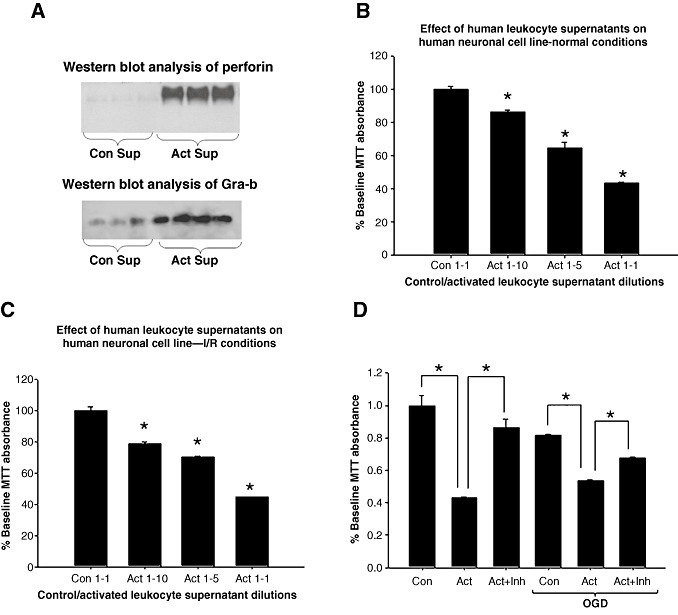
A. Activated CG‐SH cells secrete increased levels of Gra‐b and perforin and can induce Gra‐b dependent neurotoxicity. Immunoblots of perforin and Gra‐b. Immunoblot of non‐activated and activated CG‐SH supernatants showed higher levels of Gra‐b and perforin in activated CG‐SH supernatants than non‐activated controls. B. Activated CG‐SH cell supernatants induce neurotoxicity in human SH‐SY5Y neuronal cells in normal conditions in a dose dependent fashion compared with controls. MTT was significantly and dose dependently decreased with activated CG‐SH cell supernatants than non‐activated CG‐SH cell supernatants. C. Activated CG‐SH also induced neurotoxicity in ischemia challenged human SH‐SY5Y neurons in a dose dependent fashion assessed by decrease in MTT conversion. D. Gra‐b inhibition (50 µM) protected normal and ischemia challenged human SH‐SY5Y neurons from activated CG‐SH cells (leukocyte model) induced neurotoxicity mediated via Gra‐b.
DISCUSSION
Peripheral and local immune responses contribute to post‐ischemic brain injury. Cerebral ischemia results in intense cytokine and chemokine expression resulting in inflammation and a combined immune‐ischemic damage 4, 6, 38, 58, 62, 63. Though infiltration of immune (CTLs) cells into both ischemia‐reperfused (I/R) human and murine brain has been previously reported 38, 62, the mechanisms through which cytotoxic proteases contribute to CTL‐mediated pathology has not yet been fully explained 27, 33. As the pathological scenario in rodent models may not exactly match its human pathological counterpart, more relevant models will be required to compare injury mechanisms between human and rodent pathological conditions 1, 24.
Immune responses aggravate central nervous system (CNS) diseases like Alzheimer's disease, Parkinson's diseases and cerebral ischemia 19, 37, 45, 48, 80, 81. Though immune roles in neurodegenerative disorders are controversial and a matter of some debate, it is well accepted that acute immune responses can provoke post‐ischemic neurodegeneration. The CNS is normally impermeable to immune cells, and has far fewer T‐cells compared with other organs; their numbers are highly increased in the stroke‐inflamed CNS 16, 38, 62. CTLs infiltrating ischemic lesions have now been demonstrated to injure the post‐ischemic brain, as mice lacking CD‐8 T‐cells had smaller infarcts and injury scores than wild types 37, 80. The mechanisms by which T‐cells might contribute to I/R injury in the brain could involve contact‐dependent injury mechanisms or the secretion of TNF‐α, Fas‐ligand and cytotoxins like granzymes/ perforins which kill bystander cells near infiltrated CTLs 17, 39. Neurons have been shown to be sensitive to T‐ cell‐mediated injury as CTLs that infiltrate CNS induce neuronal injury via MHC dependent/independent mechanisms involving perforins and Gra‐b 28, 49, 50, 55, 60, 76. In this process, Gra‐b enters target cells by perforin dependent (pore formation in the target cells) or by endocytosis (via mannose 6‐phosphate receptor) mechanisms and induces neurotoxicity in target neurons via G‐protein coupled receptors 23, 74, 75, 76. Granzymes are serine protease family members sharing similar substrate specificity with caspases. However unlike caspases, granzymes/perforins are implicated in apoptotic as well as necrotic cell death (42). Gra‐b's ability to interact with most of the caspase‐3 substrates, and its ability to activate caspases makes Gra‐b an important integrator of cell death signaling (52). CTLs deficient in Gra‐b and perforin can still kill target cells; however, target cell killing is significantly delayed and the mode of cell death was found to be different from either perforin‐mediated target cell lysis or apoptosis 34, 65, 78. Under normal conditions the entry of these immune cells into the CNS is highly restricted by the blood brain barrier (BBB). However, cerebral ischemia results in the breakdown of BBB, with the concomitant induction of endothelial adhesion molecules that guide immune cells into the reperfused brain (79). Elevated levels of cytokines and chemokines released during ischemic stress strongly activate this process 4, 6, 69.
In this study we observed a significant elevation of Gra‐b in I/R brain along with elevations in chemokine IP‐10/CXCL10, evidence for inflammatory and immune responses in the brain post‐stroke. Previous reports have described T‐cell infiltration into the post‐ischemic infarct peaking at 7 days followed by gradual reduction (38). Our study found elevated Gra‐b levels were maintained for at least 1 month after I/R [one sample came from a patient with a relatively long survival time point (∼1 month) and was highly positive for elevated Gra‐b levels]. Elevated Gra‐b levels in the stroke brain at ∼1 month reflecting CTLs (and or NK cells) may suggest prolonged immune cell‐mediated injury which needs to be addressed. Despite evidence that acute infiltration of immune cells is injurious, chronic infiltration of immune cells has also been suggested as being protective by promoting neural reparative processes 40, 61. The elevated Gra‐b levels resulted in binding to and destruction of cellular substrates like HSP‐70, MCL‐1 and PARP‐1, well known as anti‐apoptotic mediators; (PARP‐1 exerts differential effects on survival depending on the length and intensity of insult) 36, 41. The presence of 89 and 50‐kD PARP‐1 fragments is evidence for apoptotic and necrotic forms of cell death 14, 22, 64 which involves multiple protease systems and their combined actions toward these substrates. Our current study points toward a requirement for a multiply‐targeted therapeutic approach that might simultaneously inhibit several proteases at different time points to lessen I/R injury.
HSP‐70 protects against apoptosis by preventing AIF nuclear translocation and stabilizing MCL‐1 against degradation. Recent reports have also identified MCL‐1 as a target of CTLs (5). We have observed that both MCL‐1 and HSP‐70 were proteolyzed, tipping the balance in the stroke brain toward cell death. Further support of this is found in the elevated levels of translocated AIF in nuclear fractions from post‐ischemic human brain samples consistent with compromised HSP‐70 functioning. Proteolysis of HSP‐70 by Gra‐b could facilitate nuclear translocation of AIF from the cytosol. Apart from these interactions, binding of Gra‐b with caspase‐3 is intriguing. Caspases can mediate ischemic cell death, and our observation that Gra‐b binds caspase‐3 suggests that Gra‐b also activates caspase‐3 to exacerbate cell death after cerebral ischemia. Although, ischemia leads to caspase activation, the role of Gra‐b‐mediated caspase activation to enhance apoptosis in the penumbra continues to grow in complexity. The temporal and spatial relationship between Gra‐b and caspase‐3 in human stroke needs to be further confirmed in animal models of stroke.
We have previously reported that Gra‐b levels were increased in spinal cord tissue from experimental spinal cord transection models and in focal ischemic infarcts from rat brain 15, 16. Even though Gra‐b is also elevated in human stroke brains and co‐localizes with degenerating neurons in the present study, whether Gra‐b is associated with ischemic pathology or if it aggravates post‐ischemic injury is not obvious. A more clearly defined understanding of Gra‐b in post‐ischemic injury is found in our in vitro experiments using neuron and lymphocyte model systems. Consistent with classical immunological observations, we found that activated CG‐SH cells secrete higher levels of granzymes and perforins and CTLs infiltrating stroke tissue are also Gra‐b+. These activated CG‐SH supernatants showed a strong neurotoxic effect when incubated with normal or ischemia challenged SH‐SY5Y neurons, significantly decreased neuronal viability in a dose dependent fashion. This phenomenon which is contact independent in vitro, shows that it is possible that CTLs infiltrating the ischemic infarct can induce neurotoxicity by secreting these proteases without direct contact with neurons. This process needs to be more thoroughly investigated to determine whether CTLs infiltrating ischemic infarct requires contact to whether secreted Gra‐b/perforins can provoke neurotoxicity in the vicinity of nearby neurons. Apart from this contact‐independent phenomenon of cell killing by activated CG‐SH supernatants, activated CG‐SH cells (co‐incubated with normal or ischemia challenged neurons) induced neurotoxicity which might be contact dependent.
Importantly the CG‐SH cell line used in this study to model lymphocyte function expresses markers found on several leukocyte subsets; therefore, the possibility of neuronal injury mediated by leukocyte subsets (other than activated lymphocytes) warrants further study. In accordance with this, Jander et al have previously reported that ischemic infarct is infiltrated with CD‐8+ and CD‐4+ macrophage‐like cells which were observed in the infarct for ∼1 week (CD‐8+ macrophage like cells) and from 1 week to ∼2 weeks (CD‐4+ macrophage like cells) (38). However, whether these cells exhibit only CD‐8 marker or whether they also have CTL‐like properties and the subset of leukocyte population they belong are not known. Taken together, these data indicate that an altered immune response with infiltration of different subsets of leukocytes that are CD‐4+ and CD‐8+ occurs in post‐ischemic infarcts. The specific participation of these cell types in the pathology of stroke deserves further attention. Furthermore, expression of Gra‐b and perforin by cell types other than those of lymphoid lineage is not well known. However, consistent with a previous report by Berthou et al, which indicated the expression of Gra‐b and perforin by KG1a (human acute myelogenous leukemia) cells (8), we too observed increased production of Gra‐b and perforins by CG‐SH (a human myeloid cell line), indicating that non‐lymphoid leukocytes can induce Gra‐b/perforin mediated injury in stroke brain. In the present study, the neurotoxic effect induced by activated CG‐SH cells was clearly mediated by Gra‐b, as Gra‐b inhibition was able to rescue target neurons from the effect activated CG‐SH cells and their secreted products. Regardless, this clearly indicates that Gra‐b/perforins could be key players of CTL‐mediated neuronal cell death. Our results indicate that lymphocytes infiltrating stroke brain secrete high levels of Gra‐b and perforins which induce neurotoxicity and aggravate stroke injury. These in vitro and human stroke data clearly suggest that the Gra‐b+ CTLs infiltrating I/R tissue magnify stroke pathology by inducing neuronal death and might enlarge infarct size and severity.
SUMMARY
In summary, this study indicates that Gra‐b released by CTLs can mediate neuronal death by inhibiting anti‐apoptotic molecules and by activating pro‐apoptotic molecules in the stroke brain. The involvement of other “suicidal” proteases and their interactions with Gra‐b would intensify post‐I/R damage. Therefore, future therapeutic targets for stroke might be enhanced by effectively inhibiting several cytotoxic proteases especially Gra‐b to mitigate the pathology of cerebral ischemia.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
GVC acknowledges a Senior Research Fellowship from UGC (India) and a post‐doctoral fellowship from the Feist‐Weiler Cardiovascular Research Institute–Shreveport, LA. The authors thank Dr. Shankar, Department of Neuropathology, “Human Brain Tissue Repository for Neurobiological Studies”, National Institute of Mental Health and Neurosciences, Bangalore, India for providing the post‐mortem human biological (stroke and normal brain) samples for research purposes. We are grateful to Prof. Pallu Reddanna, Department of Animal Sciences, University of Hyderabad, India for the HSP‐70 antibody; Dr. Anirban Basu, National Brain Research Center, Gurgoan, India for the IP‐10/CXCL10 antibody, Dr. Traci L. Testerman, LSU Health Sciences Center‐Shreveport, LA, for the kind gift of Neu‐N antibodies.
REFERENCES
- 1. Sherman DG, Bes A, Easton JD, Hacke W, Kaste M, Polmar SH et al (2001) Use of anti‐ICAM‐1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology 57:1428–1434. [DOI] [PubMed] [Google Scholar]
- 2. Alexander JS, Dayton T, Davis C, Hill S, Jackson TH, Blaschuk O et al (1998) Activated T‐lymphocytes express occludin, a component of tight junctions. Inflammation 22:573–582. [DOI] [PubMed] [Google Scholar]
- 3. Alimonti JB, Shi L, Baijal PK, Greenberg AH (2001) Granzyme B induces BID‐mediated cytochrome c release and mitochondrial permeability transition. J Biol Chem 276:6974–6982. [DOI] [PubMed] [Google Scholar]
- 4. Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT (2009) Post‐ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J 276:13–26. [DOI] [PubMed] [Google Scholar]
- 5. Andersen MH, Becker JC, Thor SP (2005) The antiapoptotic member of the Bcl‐2 family Mcl‐1 is a CTL target in cancer patients. Leukemia 19:484–485. [DOI] [PubMed] [Google Scholar]
- 6. Babcock AA, Kuziel WA, Rivest S, Owens T (2003) Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci 23:7922–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barry M, Heibein JA, Pinkoski MJ, Lee SF, Moyer RW, Green DR, Bleackley RC (2000) Granzyme B short‐circuits the need for caspase 8 activity during granule‐mediated cytotoxic T‐lymphocyte killing by directly cleaving Bid. Mol Cell Biol 20:3781–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berthou C, Marolleau JP, Lafaurie C, Soulie A, Dal Cortivo L, Bourge JF et al (1995) Granzyme B and perforin lytic proteins are expressed in CD34+ peripheral blood progenitor cells mobilized by chemotherapy and granulocyte colony‐stimulating factor. Blood 86:3500–3506. [PubMed] [Google Scholar]
- 9. Bhowmick S, Duseja R, Das S, Appaiahgiri MB, Vrati S, Basu A (2007) Induction of IP‐10 (CXCL10) in astrocytes following Japanese encephalitis. Neurosci Lett 414:45–50. [DOI] [PubMed] [Google Scholar]
- 10. Bivik C, Rosdahl I, Ollinger K (2007) Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis 28:537–544. [DOI] [PubMed] [Google Scholar]
- 11. Cao G, Minami M, Pei W, Yan C, Chen D, O'Horo C et al (2001) Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab 21:321–333. [DOI] [PubMed] [Google Scholar]
- 12. Chaitanya GV, Babu PP (2008) Activation of calpain, cathepsin‐b and caspase‐3 during transient focal cerebral ischemia in rat model. Neurochem Res 33:2178–2186. [DOI] [PubMed] [Google Scholar]
- 13. Chaitanya GV, Babu PP (2008) Multiple apoptogenic proteins are involved in the nuclear translocation of Apoptosis Inducing Factor during transient focal cerebral ischemia in rat. Brain Res 1246:178–190. [DOI] [PubMed] [Google Scholar]
- 14. Chaitanya GV, Babu PP (2009) Differential PARP cleavage: an indication of heterogeneous forms of cell death and involvement of multiple proteases in the infarct of focal cerebral ischemia in rat. Cell Mol Neurobiol 29:563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chaitanya GV, Kolli M, Babu PP (2009) Granzyme‐b mediated cell death in the spinal cord‐injured rat model. Neuropathology 29:270–279. [DOI] [PubMed] [Google Scholar]
- 16. Chaitanya GV, Schwaninger M, Alexander JS, Babu PP (2010) Granzyme‐b is involved in mediating post‐ischemic neuronal death during focal cerebral ischemia in rat model. Neuroscience 165:1203–1216. [DOI] [PubMed] [Google Scholar]
- 17. Chavez‐Galan L, Arenas‐Del Angel MC, Zenteno E, Chavez R, Lascurain R (2009) Cell death mechanisms induced by cytotoxic lymphocytes. Cell Mol Immunol 6:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cho BB, Toledo‐Pereyra LH (2008) Caspase‐independent programmed cell death following ischemic stroke. J Invest Surg 21:141–147. [DOI] [PubMed] [Google Scholar]
- 19. Engelhardt JI, Tajti J, Appel SH (1993) Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol 50:30–36. [DOI] [PubMed] [Google Scholar]
- 20. Ferrer I, Planas AM (2003) Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol 62:329–339. [DOI] [PubMed] [Google Scholar]
- 21. Ferrer I, Friguls B, Dalfo E, Justicia C, Planas AM (2003) Caspase‐dependent and caspase‐independent signalling of apoptosis in the penumbra following middle cerebral artery occlusion in the adult rat. Neuropathol Appl Neurobiol 29:472–481. [DOI] [PubMed] [Google Scholar]
- 22. Froelich CJ, Hanna WL, Poirier GG, Duriez PJ, D'Amours D, Salvesen GS et al (1996) Granzyme B/perforin‐mediated apoptosis of Jurkat cells results in cleavage of poly(ADP‐ribose) polymerase to the 89‐kDa apoptotic fragment and less abundant 64‐kDa fragment. Biochem Biophys Res Commun 227:658–665. [DOI] [PubMed] [Google Scholar]
- 23. Froelich CJ, Orth K, Turbov J, Seth P, Gottlieb R, Babior B et al (1996) New paradigm for lymphocyte granule‐mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J Biol Chem 271:29073–29079. [DOI] [PubMed] [Google Scholar]
- 24. Furuya K, Takeda H, Azhar S, McCarron RM, Chen Y, Ruetzler CA et al (2001) Examination of several potential mechanisms for the negative outcome in a clinical stroke trial of enlimomab, a murine anti‐human intercellular adhesion molecule‐1 antibody: a bedside‐to‐bench study. Stroke 32:2665–2674. [DOI] [PubMed] [Google Scholar]
- 25. Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G (2006) Heat shock proteins 27 and 70: anti‐apoptotic proteins with tumorigenic properties. Cell Cycle 5:2592–2601. [DOI] [PubMed] [Google Scholar]
- 26. Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G (2006) Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 13:1423–1433. [DOI] [PubMed] [Google Scholar]
- 27. Giulian D, Corpuz M, Chapman S, Mansouri M, Robertson C (1993) Reactive mononuclear phagocytes release neurotoxins after ischemic and traumatic injury to the central nervous system. J Neurosci Res 36:681–693. [DOI] [PubMed] [Google Scholar]
- 28. Giuliani F, Goodyer CG, Antel JP, Yong VW (2003) Vulnerability of human neurons to T cell‐mediated cytotoxicity. J Immunol 171:368–379. [DOI] [PubMed] [Google Scholar]
- 29. Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C et al (1999) Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL‐XL prevents this release but not tumor necrosis factor‐R1/Fas death. J Biol Chem 274:1156–1163. [DOI] [PubMed] [Google Scholar]
- 30. Gurbuxani S, Schmitt E, Cande C, Parcellier A, Hammann A, Daugas E et al (2003) Heat shock protein 70 binding inhibits the nuclear import of apoptosis‐inducing factor. Oncogene 22:6669–6678. [DOI] [PubMed] [Google Scholar]
- 31. Han J, Goldstein LA, Gastman BR, Froelich CJ, Yin XM, Rabinowich H (2004) Degradation of Mcl‐1 by granzyme B: implications for Bim‐mediated mitochondrial apoptotic events. J Biol Chem 279:22020–22029. [DOI] [PubMed] [Google Scholar]
- 32. Han J, Goldstein LA, Gastman BR, Rabinovitz A, Rabinowich H (2005) Disruption of Mcl‐1.Bim complex in granzyme B‐mediated mitochondrial apoptosis. J Biol Chem 280:16383–16392. [DOI] [PubMed] [Google Scholar]
- 33. Hanisch UK, Neuhaus J, Quirion R, Kettenmann H (1996) Neurotoxicity induced by interleukin‐2: involvement of infiltrating immune cells. Synapse 24:104–114. [DOI] [PubMed] [Google Scholar]
- 34. Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ (1994) Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell 76:977–987. [DOI] [PubMed] [Google Scholar]
- 35. Hoves S, Trapani JA, Voskoboinik I (2010) The battlefield of perforin/granzyme cell death pathways. J Leukoc Biol 87:237–243. [DOI] [PubMed] [Google Scholar]
- 36. Huang Q, Shen HM (2009) To die or to live: the dual role of poly(ADP‐ribose) polymerase‐1 in autophagy and necrosis under oxidative stress and DNA damage. Autophagy 5:273–276. [DOI] [PubMed] [Google Scholar]
- 37. Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H (2007) T‐ and B‐cell‐deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab 27:1798–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jander S, Schroeter M, D'Urso D, Gillen C, Witte OW, Stoll G (1998) Focal ischaemia of the rat brain elicits an unusual inflammatory response: early appearance of CD8+ macrophages/microglia. Eur J Neurosci 10:680–688. [DOI] [PubMed] [Google Scholar]
- 39. Jenkins MR, Griffiths GM (2010) The synapse and cytolytic machinery of cytotoxic T cells. Curr Opin Immunol 22:308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jones KJ, Serpe CJ, Byram SC, Deboy CA, Sanders VM (2005) Role of the immune system in the maintenance of mouse facial motoneuron viability after nerve injury. Brain Behav Immun 19:12–19. [DOI] [PubMed] [Google Scholar]
- 41. Kauppinen TM, Swanson RA (2007) The role of poly(ADP‐ribose) polymerase‐1 in CNS disease. Neuroscience 145:1267–1272. [DOI] [PubMed] [Google Scholar]
- 42. Ko YH, Park S, Jin H, Woo H, Lee H, Park C, Kim K (2007) Granzyme B leakage‐induced apoptosis is a crucial mechanism of cell death in nasal‐type NK/T‐cell lymphoma. Lab Invest 87:241–250. [DOI] [PubMed] [Google Scholar]
- 43. Kroemer G (2001) Heat shock protein 70 neutralizes apoptosis‐inducing factor. ScientificWorldJournal 1:590–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kumar Y, Tatu U (2003) Stress protein flux during recovery from simulated ischemia: induced heat shock protein 70 confers cytoprotection by suppressing JNK activation and inhibiting apoptotic cell death. Proteomics 3:513–526. [DOI] [PubMed] [Google Scholar]
- 45. Linfert D, Chowdhry T, Rabb H (2009) Lymphocytes and ischemia‐reperfusion injury. Transplant Rev (Orlando) 23:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Loeb CR, Harris JL, Craik CS (2006) Granzyme B proteolyzes receptors important to proliferation and survival, tipping the balance toward apoptosis. J Biol Chem 281:28326–28335. [DOI] [PubMed] [Google Scholar]
- 47. Lui JC, Kong SK (2007) Heat shock protein 70 inhibits the nuclear import of apoptosis‐inducing factor to avoid DNA fragmentation in TF‐1 cells during erythropoiesis. FEBS Lett 581:109–117. [DOI] [PubMed] [Google Scholar]
- 48. McCoy L, Tsunoda I, Fujinami RS (2006) Multiple sclerosis and virus induced immune responses: autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity 39:9–19. [DOI] [PubMed] [Google Scholar]
- 49. Medana IM, Gallimore A, Oxenius A, Martinic MM, Wekerle H, Neumann H (2000) MHC class I‐restricted killing of neurons by virus‐specific CD8+ T lymphocytes is effected through the Fas/FasL, but not the perforin pathway. Eur J Immunol 30:3623–3633. [DOI] [PubMed] [Google Scholar]
- 50. Medana I, Martinic MA, Wekerle H, Neumann H (2001) Transection of major histocompatibility complex class I‐induced neurites by cytotoxic T lymphocytes. Am J Pathol 159:809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Meli E, Pangallo M, Picca R, Baronti R, Moroni F, Pellegrini‐Giampietro DE (2004) Differential role of poly(ADP‐ribose) polymerase‐1in apoptotic and necrotic neuronal death induced by mild or intense NMDA exposure in vitro . Mol Cell Neurosci 25:172–180. [DOI] [PubMed] [Google Scholar]
- 52. Metkar SS, Wang B, Ebbs ML, Kim JH, Lee YJ, Raja SM, Froelich CJ (2003) Granzyme B activates procaspase‐3 which signals a mitochondrial amplification loop for maximal apoptosis. J Cell Biol 160:875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Michels J, Johnson PW, Packham G (2005) Mcl‐1. Int J Biochem Cell Biol 37:267–271. [DOI] [PubMed] [Google Scholar]
- 54. Munker R, Nordberg ML, Veillon D, Williams BJ, Roggero A, Kern W et al (2009) Characterization of a new myeloid leukemia cell line with normal cytogenetics (CG‐SH). Leuk Res 33:1405–1408. [DOI] [PubMed] [Google Scholar]
- 55. Nitsch R, Pohl EE, Smorodchenko A, Infante‐Duarte C, Aktas O, Zipp F (2004) Direct impact of T cells on neurons revealed by two‐photon microscopy in living brain tissue. J Neurosci 24:2458–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Park MA, Curiel DT, Koumenis C, Graf M, Chen CS, Fisher PB et al (2008) PERK‐dependent regulation of HSP70 expression and the regulation of autophagy. Autophagy 4:364–367. [DOI] [PubMed] [Google Scholar]
- 57. Pellicciari R, Camaioni E, Costantino G (2004) 3. Life or death decisions: the cast of poly(ADP‐ribose)polymerase (PARP) as a therapeutic target for brain ischaemia. Prog Med Chem 42:125–169. [DOI] [PubMed] [Google Scholar]
- 58. Planas AM, Gorina R, Chamorro A (2006) Signalling pathways mediating inflammatory responses in brain ischaemia. Biochem Soc Trans 34:1267–1270. [DOI] [PubMed] [Google Scholar]
- 59. Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N et al (2001) Heat‐shock protein 70 antagonizes apoptosis‐inducing factor. Nat Cell Biol 3:839–843. [DOI] [PubMed] [Google Scholar]
- 60. Rensing‐Ehl A, Malipiero U, Irmler M, Tschopp J, Constam D, Fontana A (1996) Neurons induced to express major histocompatibility complex class I antigen are killed via the perforin and not the Fas (APO‐1/CD95) pathway. Eur J Immunol 26:2271–2274. [DOI] [PubMed] [Google Scholar]
- 61. Sanders VM, Jones KJ (2006) Role of immunity in recovery from a peripheral nerve injury. J Neuroimmune Pharmacol 1:11–19. [DOI] [PubMed] [Google Scholar]
- 62. Schwab JM, Nguyen TD, Meyermann R, Schluesener HJ (2001) Human focal cerebral infarctions induce differential lesional interleukin‐16 (IL‐16) expression confined to infiltrating granulocytes, CD8+ T‐lymphocytes and activated microglia/macrophages. J Neuroimmunol 114:232–241. [DOI] [PubMed] [Google Scholar]
- 63. Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O'Banion MK (2007) Chronic interleukin‐1beta expression in mouse brain leads to leukocyte infiltration and neutrophil‐independent blood brain barrier permeability without overt neurodegeneration. J Neurosci 27:9301–9309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shah GM, Shah RG, Poirier GG (1996) Different cleavage pattern for poly(ADP‐ribose) polymerase during necrosis and apoptosis in HL‐60 cells. Biochem Biophys Res Commun 229:838–844. [DOI] [PubMed] [Google Scholar]
- 65. Shresta S, MacIvor DM, Heusel JW, Russell JH, Ley TJ (1995) Natural killer and lymphokine‐activated killer cells require granzyme B for the rapid induction of apoptosis in susceptible target cells. Proc Natl Acad Sci U S A 92:5679–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sizonenko SV, Camm EJ, Dayer A, Kiss JZ (2008) Glial responses to neonatal hypoxic‐ischemic injury in the rat cerebral cortex. Int J Dev Neurosci 26:37–45. [DOI] [PubMed] [Google Scholar]
- 67. Slee EA, Keogh SA, Martin SJ (2000) Cleavage of BID during cytotoxic drug and UV radiation‐induced apoptosis occurs downstream of the point of Bcl‐2 action and is catalysed by caspase‐3: a potential feedback loop for amplification of apoptosis‐associated mitochondrial cytochrome c release. Cell Death Differ 7:556–565. [DOI] [PubMed] [Google Scholar]
- 68. Stankiewicz AR, Livingstone AM, Mohseni N, Mosser DD (2009) Regulation of heat‐induced apoptosis by Mcl‐1 degradation and its inhibition by Hsp70. Cell Death Differ 16:638–647. [DOI] [PubMed] [Google Scholar]
- 69. Stoll G, Jander S, Schroeter M (1998) Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol 56:149–171. [DOI] [PubMed] [Google Scholar]
- 70. Sutton VR, Davis JE, Cancilla M, Johnstone RW, Ruefli AA, Sedelies K et al (2000) Initiation of apoptosis by granzyme B requires direct cleavage of bid, but not direct granzyme B‐mediated caspase activation. J Exp Med 192:1403–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Taoufik E, Probert L (2008) Ischemic neuronal damage. Curr Pharm Des 14:3565–3573. [DOI] [PubMed] [Google Scholar]
- 72. Trapani JA, Sutton VR (2003) Granzyme B: pro‐apoptotic, antiviral and antitumor functions. Curr Opin Immunol 15:533–543. [DOI] [PubMed] [Google Scholar]
- 73. Tsunoda I, Lane TE, Blackett J, Fujinami RS (2004) Distinct roles for IP‐10/CXCL10 in three animal models, Theiler's virus infection, EAE, and MHV infection, for multiple sclerosis: implication of differing roles for IP‐10. Mult Scler 10:26–34. [DOI] [PubMed] [Google Scholar]
- 74. Veugelers K, Motyka B, Frantz C, Shostak I, Sawchuk T, Bleackley RC (2004) The granzyme B‐serglycin complex from cytotoxic granules requires dynamin for endocytosis. Blood 103:3845–3853. [DOI] [PubMed] [Google Scholar]
- 75. Veugelers K, Motyka B, Goping IS, Shostak I, Sawchuk T, Bleackley RC (2006) Granule‐mediated killing by granzyme B and perforin requires a mannose 6‐phosphate receptor and is augmented by cell surface heparan sulfate. Mol Biol Cell 17:623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang T, Allie R, Conant K, Haughey N, Turchan‐Chelowo J, Hahn K et al (2006) Granzyme B mediates neurotoxicity through a G‐protein‐coupled receptor. FASEB J 20:1209–1211. [DOI] [PubMed] [Google Scholar]
- 77. Waterhouse NJ, Sedelies KA, Browne KA, Wowk ME, Newbold A, Sutton VR et al (2005) A central role for Bid in granzyme B‐induced apoptosis. J Biol Chem 280:4476–4482. [DOI] [PubMed] [Google Scholar]
- 78. Waterhouse NJ, Sutton VR, Sedelies KA, Ciccone A, Jenkins M, Turner SJ et al (2006) Cytotoxic T lymphocyte‐induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin‐dependent lysis. J Cell Biol 173:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yilmaz G, Granger DN (2008) Cell adhesion molecules and ischemic stroke. Neurol Res 30:783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yilmaz G, Arumugam TV, Stokes KY, Granger DN (2006) Role of T lymphocytes and interferon‐gamma in ischemic stroke. Circulation 113:2105–2112. [DOI] [PubMed] [Google Scholar]
- 81. Yong VW, Rivest S (2009) Taking advantage of the systemic immune system to cure brain diseases. Neuron 64:55–60. [DOI] [PubMed] [Google Scholar]
- 82. Zhu Q, Xu YM, Wang LF, Zhang Y, Wang F, Zhao J et al (2009) Heat shock protein 70 silencing enhances apoptosis inducing factor‐mediated cell death in hepatocellular carcinoma HepG2 cells. Cancer Biol Ther 8:792–798. [DOI] [PubMed] [Google Scholar]