

Abstract
The expression of hypoxia‐inducible factor‐1‐alpha (HIF‐1α) is upregulated in ischemic stroke, but its function is still unclear. In the present study, biphasic expression of HIF‐1α was observed during 1–12 h and after 48 h in neurons exposed to ischemic stress in vitro and in vivo. Treating neurons with 2‐methoxyestradiol (2ME2) 0.5 h after ischemic stress or pre‐silencing HIF‐1α with small interfering RNA (siRNA) decreased brain injury, brain edema and number of apoptotic cell, and downregulates Nip‐like protein X (Nix) expression. Conversely, applying 2ME2 to neurons 8 h after ischemic stress or silencing the HIF‐1α with siRNA 12 h after oxygen–glucose deprivation (OGD) increased neuron damage and decreased vascular endothelial growth factor (VEGF) expression. Taken together, these results demonstrate that HIF‐1α induced by ischemia in early and late times leads cellular apoptosis and survival, respectively, and provides a new insight into the divergent roles of HIF‐1α expression in neurons after ischemic stroke.
Keywords: 2ME2, cultured cortical neurons, HIF‐1, MCAO, OGD, VEGF
INTRODUCTION
Ischemic stroke makes up 80% of all strokes and results from a thrombotic or embolic occlusion of a major cerebral artery (most often the middle cerebral artery, MCA) or its branches. Converging evidence indicates that ischemic damage is initiated by energy failure (25) and excitotoxicity. Excitotoxic mechanisms can cause a dynamic process that evolves over a period of hours to several days, including necrosis or apoptosis, inflammation and activation of endogenous adaptive and regenerative mechanisms. Regulation of these processes occurs at the transcriptional level and involves the activation of various transcription factors, including hypoxia‐inducible factor‐1‐alpha (HIF‐1α).
HIF‐1, a key regulator of oxygen homeostasis, is a transcription factor composed of an oxygen‐regulated subunit known as HIF‐1α and a constitutively expressed HIF‐1β subunit. In most cases, HIF‐1 activity is controlled by the availability and activity of the HIF‐1α subunit (22). Under normoxic conditions, the proline 402 and 564 residues in HIF‐1α are hydroxylated by O2‐dependent proline hydroxylases, following which they undergo ubiquitination and proteasomal degradation. In hypoxia, when proline hydroxylases are not active, HIF‐1α accumulates and binds to HIF‐1β, thus forming a heterodimer. This heterodimer plays a role in the transcription regulation of hypoxia‐inducible genes involved in angiogenesis, glucose transport and metabolism, erythropoiesis, inflammation, apoptosis and cellular stress 17, 36. In addition to hypoxia, HIF‐1α is also activated by environmental, physiological and pathological stimuli, including iron chelators (28), hormones (1) and bacteria infection at normoxia (24). Lipopolysaccharide treatment of macrophages augments hypoxia‐regulated genes via activation of HIF‐1α(6). Furthermore, insulin‐like growth factor‐1 is necessary to prolonged neuron HIF‐1α expression after global ischemia (8). In neuroblastoma cells, brain‐derived neurotrophic factor (BDNF) induces vascular endothelial growth factor (VEGF) expression via HIF‐1α activation (27).
The role of HIF‐1α in the pathophysiology of neurons under hypoxia or stroke is still not well established. Several studies suggest that HIF‐1α plays a protective role. Inducing HIF‐1α expression by prolyl 4‐hydroxylase inhibition and hypoxic preconditioning can protect neurons from damage after focal cerebral ischemia 18, 30. HIF‐1α also rescues neurons from nerve growth factor (NGF) deprivation (33). In contrast, other studies indicate that HIF‐1α is involved in hypoxia‐induced cell death events by activating the expression of various pro‐death genes (32). In a murine hippocampal cell line, pro‐survival and pro‐death effects of HIF‐1α depend on the type of cellular stress (2). Furthermore, neuron‐specific knockdown of HIF‐1α increases brain injury in transient focal cerebral ischemia (3) but reduces hypoxic–ischemic damage in the global ischemia model (15). A hypothesis is suggested that events other than hypoxia occur during a stroke that also regulate HIF‐1α expression and result in opposite effects of HIF‐1α in neurons. 2‐methoxyestradiol (2ME2) is a naturally occurring derivative of estradiol and a known HIF‐1α inhibitor 14, 19. It has been reported that 2ME2 can provide neuroprotection but also can reduce cell survival after experimental stroke (9, 38). It's controversial about the role of 2ME2 treatment after stroke.
In the present results, there was a biphasic increased expression in HIF‐1α in neurons after ischemic stroke. Furthermore, brain injury was remitted in groups with inhibition of early phase of HIF‐1α expression. In contrast, the infarct area was served when the late phase of HIF‐1α was attenuated. These results indicated a divergent effect of HIF‐1α after recovery of experimental stroke and provide potent leads for therapy.
METHODS
Experimental animals and surgical preparation
Male Sprague–Dawley rats weighing 280–320 g were used. They were housed in group cages of two or three rats each in an air‐conditioned vivarium with free access to food and water. Throughout the study, a 12:12‐h light/dark cycle was maintained with lights on at 8 am. All procedures adhere to the Guidelines for Care and Use of Experimental Animals of the National Cheng‐Kung University (Tainan, Taiwan). For surgical preparation, rats were anesthetized by intraperitoneal (ip) injection with 10% chloral hydrate. Then, a 23‐gauge stainless steel guide cannula was stereotaxically implanted into the piriform cortex 2 mm dorsal to the left MCA. The stereotaxic coordinates were modified for this rat strain (0.2 mm anterior, −5.2 mm lateral and −8 mm ventral, from the bregma) 20, 29. The cannula was secured with dental cement, and two small screws were inserted into the skull. Animals were housed individually and allowed to recover for 3 days before induction of stroke. Rats were divided into drug or vehicle groups before induction of stroke. Focal ischemia was induced in conscious rats by administration of endothelin‐1 (ET‐1, 240 pmol in 10 µL of saline over 5 minutes) via a 30‐gauge injector that protruded 2 mm beyond the end of the previously implanted guide cannula. The injector was held in place by a poly tubing cuff, and the animal was placed in a clear Plexiglas box (Sunpoint, Taoyuan, Taiwan) for observation during ET‐1 injection. To determine that the MCA was successful occluded, regional cerebral blood flow was measured in the cortex supplied by the MCA by continuous laser Doppler flowmetry (Moor Instruments, Devon, England). At 1 day before stroke induction, a small hole (2 mm posterior and 3.5 mm lateral to bregma) was drilled, and a cannula was fixed to the skull with dental cement. Laser Doppler flowmetry probe (0.5 mm diameter) was placed on the dura using the cannula, and cortical regional cerebral blood flow was assessed during all surgery time to ensure MCA occlusion. Only animals with a regional cerebral blood flow reduction of more than 75% were included (n = 50). It had been reported that the alteration of cerebral blood flow after ET‐1 injection could be recovered within 1 h (7). Sham‐operated rats were subjected to the same surgical procedure without the occlusion of the MCA.
Neurological evaluation
To determine motor activity, rats were assessed by a scoring system using a modification of the postural reflex test (4). Rats were evaluated before middle cerebral artery occlusion (MCAO) to establish a baseline and at various time points during recovery. To determine sensory activity by using an adhesive‐removal patch test, two small pieces of adhesive‐backed paper dots were used as bilateral tactile stimuli occupying the distal–radial region on the wrist of each forelimb. The time to remove each stimulus from forelimbs was recorded at five trials per day. Individual trials were separated by at least 5 minutes. Before surgery, the animals were trained for 3 days. Once the rats were able to remove the dots within 10 s, they were subjected to MCAO.
Cultured cortical neurons
Rat cortical neurons were cultured from postnatal day 0 (P0) to P1 rat pups. The cortex was dissected and digested in 7 mL of trypsin (10 U/mL) in phosphate buffered saline (PBS) at 37°C for 30 minutes. Cells were plated at a density of 0.5 × 106 cells/cm2 onto a plastic culture plate that was pre‐coated with poly‐L‐lysine (50 µg/mL, Sigma, St. Louis, MO, USA) and maintained in the Neurobasal™‐A medium (Invitrogen, Carlsbad, CA, USA) supplemented with 100 U/mL penicillin, 0.1 mg/mL streptomycin, 0.5 mM L‐glutamine and 5% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA). On the next day, 8 µM of cytosine arabinoside (Invitrogen) was added to prevent glial cell growth.
Oxygen glucose deprivation
Primary cortical neurons were transferred into serum‐free medium containing 5 mM of 2‐deoxy‐D‐glucose (Merck, Darmstadt, Germany) for 1 h. The cells were then placed in an anaerobic chamber that was flushed with a mixture of 95% N2 and 5% CO2. After incubation for 30 minutes, cells were removed from the anaerobic chamber to the normal conditions. Having determined that 60‐minute oxygen–glucose deprivation (OGD) caused 40% of the neurons to become terminal deoxyuridine‐triphosphate nick‐end labeling (TUNEL) positive by 24 h and 60% of the neurons lose by 96 h (Figure S4B), this duration was used for all further experimentation, except when time course experiments were conducted.
RNA interference
To suppress the expression of HIF‐1α, Nix or VEGF, either 20‐nM HIF‐1α siRNA (Ambion, Austin, TX, USA), or Nix or VEGF shRNA (short hairpin RNA) (Institute of Molecular Biology, Academia Sinica, Taipei, Taiwan) was transfected to cortical neurons with Lipofectamine™ 2000 (Invitrogen). Thirty hours after transfection, neurons were subjected to OGD for various periods of time. The transfect efficiency was confirmed using Alexa Fluor® 555‐labeled Red Fluorescent Oligo (Invitrogen). About 40% of neurons were transfected with RNA oligo without significant morphology change (Figure S5).
Western blotting
Primary cortical neurons were lysed briefly in homogenizing buffer [1% Triton X‐100 (Sigma, St. Louis, MO, USA), 50 mM Tris‐HCl, pH 7.5, 0.3 M sucrose, 5 mM ethylenediaminetetraacetic acid (EDTA), 2 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 20 µg/mL leupeptin and 4 µg/mL aprotinin]. Equal amount of the samples was loaded per lane. For detection of HIF‐1α, Nix, VEGF and actin, blots were incubated with antibodies against HIF‐1α (1:1000, Novus Biological, Littleton, CO, USA), Nix (1:2000, Santa Cruz Biotech, Santa Cruz, CA, USA), VEGF (1:2000, Santa Cruz Biotech), caspase‐3 (1:2000, Santa Cruz Biotech), nuclear factor‐kB (NF‐κB) p65 (1:2000, Santa Cruz Biotech), c‐fos (1:1000, Santa Cruz Biotech) or actin (1:5000, Santa Cruz Biotech). An enhanced chemiluminescence kit was used for detection. Western blotting was developed in the linear range used for densitometry. The density of the immunoblots was determined by an image analysis system installed with BIO‐ID software (Vilber Lourmat, Marne la Vallée, France).
Immunohistochemistry
Briefly, rat brain was fixed in 4% paraformaldehyde in PBS for 16 h at 4°C. Transverse slices of 20‐µm thickness were cut, and the appropriate slices were placed on a microscope slide. After a subsequent wash in PBS, slices were permeabilized and blocked for 60 minutes in 0.1% Triton X‐100/5% goat serum/PBS, and incubated for 12 h at 4°C. Brain slices were incubated with rabbit anti‐HIF‐1α antibodies (1:200, Novus Biological), anti‐mouse IgG1 monoclonal antibody (MAb1) antibodies (1:50, Chemicon, Littleton, CO, USA) and mouse anti‐Neuronal Nuclei (NeuN) antibody (1:50, Chemicon) overnight. After three washes with PBS, cells were incubated with Alexa 488‐conjugated anti‐rabbit antibody (1:200; Invitrogen), Alexa 568‐conjugated anti‐mouse antibody (1:200; Invitrogen) and 4′,6‐diamidino‐2‐phenylindole. The slides were washed three times with PBS and mounted with glycerol. Images were analyzed under a confocal fluorescence microscope (Olympus FluoViewTMFV 1000, Melville, NY, USA). Hypoxyprobe™‐1 (pimonidazole hydrochloride; Chemicon, Littleton, CO, USA) was intravenously injected into the rat (60 mg/kg) 30 minutes before MCAO. IgG staining used for detecting blood‐brain barrier (BBB) breakdown was performed at 24 h after hypoxia‐ischemia (HI), and conjugated goat anti‐rat IgG biotin (Santa Cruz Biotech) was used. Apoptosis was determined by detecting DNA strand breaks with a TUNEL assay, using the in situ death detection kit (Roche Diagnostics GmbH, Mannheim, Germany).
Methanethiosulfonate assay
Primary cortical neurons were plated in 6 × 105 cells/cm2 in 24‐well plates 2 days before treatment. Forty‐eight hours after OGD challenge, 20 µL of methanethiosulfonate (5 mg/mL) was added to each well, followed by incubation for an additional 30 minutes. The media was collected, and the absorbance was measured at 510 nm using a Rainbow Spectra ELISA microplate reader (Tecan, San Jose, CA, USA). Cell viability was defined relative to corresponding control cells (ie, relative cell viability = absorbance of treated sample/absorbance of control sample ×100%).
Determination of infarct volume and edema ratio
Rats were deeply anesthetized and decapitated, and the brain was cut into 2‐mm thick coronal block slices. The brain slices were immersed in 1% solution of 2,3,5‐triphenyltetrazolium chloride (TTC) in normal saline at 37°C for 30 minutes and then fixed in 4% paraformaldehyde at 4°C. Brain edema (brain swelling) was quantified by the ratio of the weight (dry/wet) of the hemisphere. The hemispheres were separated and weighed immediately after removal and again after drying in an oven at 105°C for 24 h.
Quantification of cell number and volume
Stereological volume estimations of the injury area were based on Cavalieri's principle by using a transparent counting grid on the slices:
Where V is the total volume of the injury area, t is the average slice thickness, a(p) is the area per point, and Σp is the total number of points hitting the structure of interest.
The total cell number in each region, N(cell,reg), was estimated according to Cavalieri principle (13):
Where ΣQ ‐ is the total number of cells counted in all dissectors in a region, Σv(dis) is the total volume of these dissectors equal to the area of the counting frame, and V(reg) is the regional cortical reference volume. Five serial 10‐µm‐thick sections, spaced 50 µm apart from bregma level 2 to 4 mm, were counted under high‐power microscope field (200×).
Statistical analysis
All data from three separate experiments were expressed as mean (standard deviation). Comparisons among multiple groups were performed using one‐way ANOVA with appropriate post hoc tests. P ≤ 0.05 was considered statistically significant.
RESULTS
Focal cerebral ischemia and oxygen glucose deprivation induce time‐dependent expression of HIF‐1α
Brain tissues taken from ipsilateral and contralateral hemisphere were removed at various time points after MCAO and homogenized to determine the expression of HIF‐1α. As shown in Figure 1A and 1C, a biphasic increase in HIF‐1α expression occurred in the ipsilateral hemisphere. ANOVA showed a main effect for group (F 13,42 = 39.47, P < 0.0001), and Newman–Keuls post hoc comparison revealed differences between the naive‐control and the 4‐h, 8‐h, 48‐h and 72‐h time points (P < 0.01). No significant difference was observed between the naive‐control and the 16‐h and 24‐h time points (P > 0.05). Immunoblots of full‐length recombinant HIF‐1α and endogenous HIF‐1α from brain lysates confirm the specificity of the HIF‐1α antibody (Figure 1B). Tissues taken from an ischemic rat were fixed and double‐labeled with antibodies against HIF‐1α and NeuN to investigate whether the biphasic expression of the former occurs in neurons. As illustrated in Figure S1A and S1B, the expression of HIF‐1α was increased in the ipsilateral hemisphere and colocalized with NeuN. These results indicate that biphasic expression of HIF‐1α mainly occurred in NeuN‐positive neurons.
Figure 1.
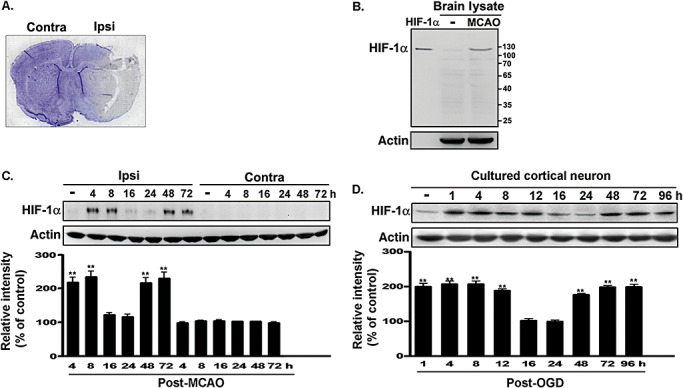
Biphasic expression of HIF‐1 α was induced by in vivo and in vitro ischemic challenges. A. The labeled area in a cresyl violet‐stained section indicates the dissected section from the ischemic brain. B. HIF‐1α protein expression in brain lysates obtained from naive and MCAO rats was detected by performing Western blotting with anti‐HIF‐1α antibody. Full‐length recombinant HIF‐1α was used as a positive control. C. Rats were decapitated at various time points after MCAO, and the level of HIF‐1α expression in the ischemic or non‐ischemic hemisphere was determined by performing immunoblotting with anti‐HIF‐1α antibody, using actin as an internal control. D. Total lysates of the primary neurons were obtained at various time points following OGD challenge. The HIF‐1α expression levels were quantified in three independent experiments and were expressed as a percentage of the levels in the naive‐controls. Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; **P < 0.01 vs. naive‐control group. Abbreviations: HIF‐1α = hypoxia‐inducible factor‐1‐alpha; MCAO = middle cerebral artery occlusion; OGD = oxygen–glucose deprivation; Contra = contralateral side; Ipsi = ipsilateral side.
OGD was performed on cultured cortical neurons to establish an in vitro model of the ischemic brain environment. Figure 1D shows biphasic expression of HIF‐1α in primary neurons within 4 days of OGD. ANOVA showed a main effect for group (F 9,30 = 51.42, P < 0.0001). Post hoc tests revealed that the expression level was significantly greater in the OGD group at 1 h, 4 h, 8 h, 12 h, 48 h, 72 h and 96 h than in the naive‐control group (P < 0.01); no difference was observed between the naive‐control and the 16 h and 24 h of OGD groups (P > 0.05). These data demonstrate time‐dependent, biphasic regulation of HIF‐1α expression in in vivo and in vitro models of ischemia.
Different effects of early‐ and late‐phase induction of HIF‐1α following MCAO‐induced brain damage
We determined the role of biphasic HIF‐1α expression after ischemic stroke by intravenously injecting 2ME2 (2.5 mg/kg) into the rats at various time points after MCAO to inhibit HIF‐1α expression. We then measured the degree of damage using TTC 4 days after MCAO. Cerebral infarction in the rat brains from each group is shown in Figure 2A. The white‐colored areas represent infarction. Quantification of infarcted areas in brains taken from the five groups is shown in Figure 2B. Severe infarction was observed in all rats of the MCAO group compared with that in the sham‐control group. 2ME2 treated 0.5 h after MCAO significantly decreased infarct volume but not when given 8 h after MCAO (Figure 2A). ANOVA showed a main effect for group (F 4,10 = 103.2, P < 0.0001). Newman–Keuls post hoc comparison revealed differences between the sham‐control and the MCAO, MCAO plus 2ME2 (0.5 h) and MCAO plus 2ME2 (8 h) groups (P < 0.05). No significant difference was observed between the sham‐control and 2ME2 group (P > 0.05). However, immunohistochemical analysis revealed that NeuN‐positive neurons increased following 2ME2 given 0.5 h after MCAO in ipsilateral hemispheres. However, the number of labeled neurons was lower in rats given 2ME2 8 h after MCAO than in the MCAO group (F 4,20 = 90.69, P < 0.0001; Figure 2B). Newman–Keuls post hoc comparison revealed differences between the sham‐control and the MCAO, MCAO plus 2ME2 (0.5 h) and the MCAO plus 2ME2 (8 h) groups (P < 0.05). No significant difference was observed between the sham‐control and 2ME2 group (P > 0.05). Furthermore, IgG staining was used to demonstrate BBB disruption because BBB leakage allows the IgG to penetrate the brain parenchyma. IgG staining was performed at 24 h after injury. The IgG‐positive region in the MCAO group correlated well with the infarct area. 2ME2 administered 0.5 h, but not 8 h, after MCAO reduced IgG staining (Figure 2C). Finally, brain edema, as indicated by increased brain/water content, was seen at 48 h after MCAO. The water content in the MCAO ipsilateral hemispheres increased significantly compared with that seen in the sham‐control group (F 9,20 = 12.94, P < 0.0001; Figure 2D). 2ME2 administered 0.5 h, but not 8 h, after MCAO significantly influenced the mean water content of the ipsilateral hemispheres relative to that determined in the MCAO group (P < 0.05). Brain water content in the contralateral hemispheres did not differ in the MCAO and 2ME2 groups (P > 0.05). Immunostaining with a Hypoxyprobe™‐1 (pimonidazole hydrochloride) indicated that the groups had similar hypoxic areas (data not shown), which implies that the difference in brain injury did not result from distinct hypoxia challenges.
Figure 2.
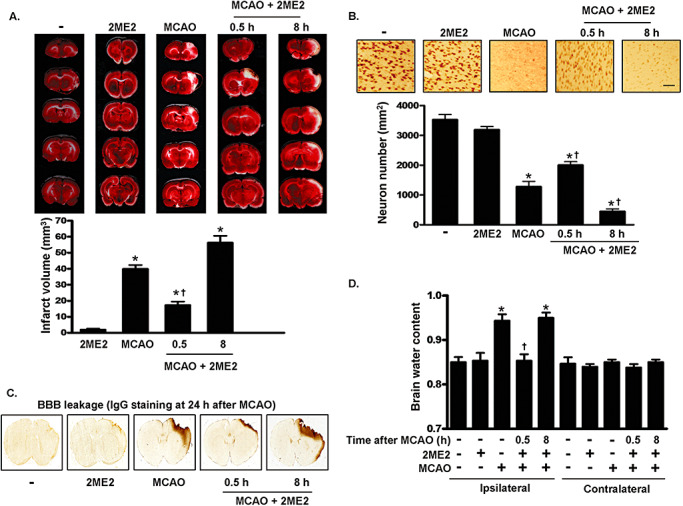
MCAO‐induced brain damage is regulated by 2ME2. A. Infarct area evaluated by TTC staining. Representative sections from the indicated treatments after 4 days of recovery are shown. The infarct area was measured in 12 sequential sections. B. Quantification of NeuN‐positive cells using immunohistochemistry after the indicated treatments. Brain sections were obtained 4 days later and immunostained with anti‐NeuN antibody. Scale bar, 100 µm. C. IgG staining in sections of the rat brain of sham‐control, MCAO and MCAO plus 2ME2 groups, respectively. Brain sections were obtained 24 h later and immunostained with goat anti‐rat IgG biotin. D. The hemispheres were separated and weighed immediately after removal and again after drying in an oven at 105°C for 24 h. In panels A, B and D, data are presented as mean (SD) (n = 4). Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. the sham‐control group. †P < 0.05 vs. MCAO group. Abbreviations: MCAO = middle cerebral artery occlusion; 2ME2 = 2‐methoxyestradiol; TTC = 2,3,5‐triphenyltetrazolium chloride; NeuN = Neuronal Nuclei; IgG = immunoglobulin; SD = standard deviation; BBB = blood‐brain barrier.
Time‐dependent effects of 2ME2 on neurological function following MCAO
To test whether 2ME2 regulates neurologic function in a time‐dependent manner, muscle tension was first measured using the swing test. Figure 3A shows that fewer rats given 2ME2 0.5 h after MCAO swing to the side opposite the ischemic region than the untreated, MCAO only rats. In contrast, the highest percentage of rats exhibiting this activity was seen 8 and 12 days after MCAO in the group given 2ME2 8 h after MCAO. ANOVA showed a main effect for group (F 11,60 = 35.51, P < 0.0001). Newman–Keuls post hoc comparison revealed differences between the MCAO group, the MCAO plus 2ME2 (0.5 h) and the MCAO plus 2ME2 (8 h) groups at 8 and 12 days (P < 0.05). No significant difference was observed between these groups at 0 or 4 days (P > 0.05). However, the unilateral somatosensory dysfunction was determined using the adhesive‐removal patch test. Treatment at 8 h after MCAO produced the longest removal time at 4, 8 and 12 days relative to that seen in the MCAO group. No significant difference was observed in rats treated with 2ME2 0.5 h after MCAO compared with the untreated MCAO group (Figure 3B). ANOVA showed a main effect for group (F 11,60 = 74.58, P < 0.0001). Newman–Keuls post hoc comparison revealed differences between the MCAO group and the MCAO plus 2ME2 (8 h) group at 4, 8 and 12 days (P < 0.05). No significant difference was observed between the MCAO group and the MCAO plus 2ME2 (0.5 h) group (P > 0.05). Furthermore, the effects of MCAO and 2ME2 on motor function were also determined. As shown in Figure 3C, 2ME2 treatment 0.5 h after MCAO significantly reduced motor function impairment compared with that seen following MCAO without treatment, but impairment worsened in rats given 2ME2 8 h after MCAO (F 23,168 = 30.42, P < 0.0001). Newman–Keuls post hoc comparison revealed differences among the MCAO group, the MCAO plus 2ME2 (0.5 h) and the MCAO plus 2ME2 (8 h) groups at 8, 12 and 16 days (P < 0.05). No significant difference was observed between these groups at 0 or 4 days (P > 0.05). Finally, Kaplan–Meier analysis revealed that the survival time for rats treated with 2ME2 8 h after MCAO was less than that for untreated rats; however, the survival time increased when they were treated 0.5 h after MCAO (Figure 3D). When normal rats were treated with 2ME2, there was no behavioral deficit or effect on survival rate (Figure S3). These results demonstrate that treatment with 2ME2 0.5 h after MCAO reduced neurologic damage and death following MCAO in rats, but that treatment 8 h after the insult exacerbated neurologic dysfunction and reduced survival.
Figure 3.
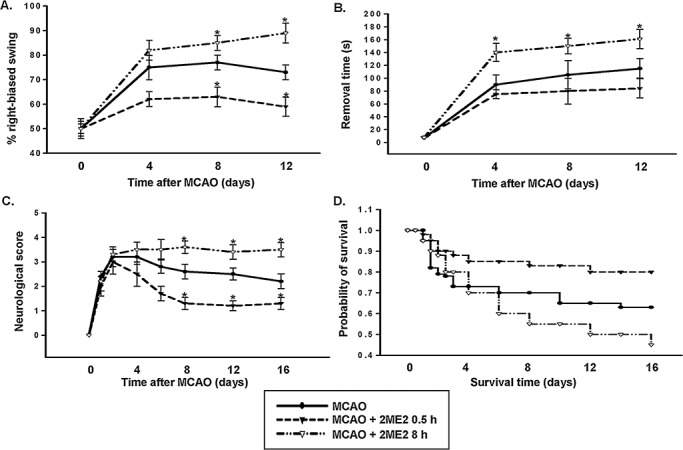
Role of early‐ and late‐phase HIF‐1αexpression in ischemic brain injury. A–C. Line graphs show temporal profiles of functional recovery from rats in each treatment. The distinct effects of rats that received 2ME2 0.5 h or 8 h after subjected to MCAO were detected using the swing test (A), adhesive‐removal patch test (B) and scoring of neurological deficit (C). D. Kaplan–Meier survival analysis after MCAO in the indicated 2ME2 treatment. In panels A–C, statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. MCAO group. HIF‐1α = hypoxia‐inducible factor‐1‐alpha; MCAO = middle cerebral artery occlusion; 2ME2 = 2‐methoxyestradiol.
Biphasic HIF‐1α differentially regulates protein expression following focal cerebral ischemia
MCAO induces various HIF‐1α‐regulated genes, including glycolytic enzymes and growth factors such as VEGF and erythropoietin. Moreover, pro‐survival and pro‐death mediators were downregulated by neuron‐specific knockdown of HIF‐1α in a mouse model of transient, focal cerebral ischemia (3). We investigated the role of biphasic HIF‐1α expression in the regulation of pro‐survival (VEGF) and pro‐death (Nix) mediators after MCAO. For this, cortical lysates obtained at different time points after MCAO were analyzed using immunoblotting with anti‐VEGF and anti‐Nix antibodies. As shown in Figure 4A, Nix expression increased immediately after the ischemic insult and returned to basal levels within 3 days. ANOVA showed a main effect for group (F 6,14 = 22.25, P < 0.0001). Newman–Keuls post hoc comparison revealed differences between the naive‐control group and the 4‐h, 8‐h, 16‐h, 24‐h and 48‐h (P < 0.01) time points after MCAO. Expression was similar in the naive‐control group and the 72‐h time point (P > 0.05). VEGF expression, however, increased at 24 h, and this was sustained for at least 3 days (F 6,14 = 58.48, P < 0.0001). Expression was also different in the naive‐control group compared with the 24‐h, 48‐h and 72‐h time points (P < 0.01), but it was similar in the naive‐control group and the 4‐h, 8‐h and 16‐h time points after MCAO (P > 0.05; Figure 4A). Similar results were seen in primary neurons exposed to OGD (Figure 4B). Nix expression increased immediately after the ischemic insult and returned to basal levels within 3 days. ANOVA showed a main effect for group (F 9,20 = 39.16, P < 0.0001). Newman–Keuls post hoc comparison showed significant differences between the naive‐control and at 4 h, 8 h, 12 h, 16 h, 24 h and 48 h after MCAO (P < 0.01). No significant difference was observed between the naive‐control and 0.5‐h, 72‐h and 96‐h time points (P > 0.05). In contrast, VEGF expression increased at 24 h and was sustained for 3 days (F 9,20 = 27.87, P < 0.0001). Expression of VEGF was significantly different between the naive‐control and at 24 h, 48 h and 72 h after MCAO (P < 0.01); however, the expression in the naive‐control was similar to that at 0.5 h, 4 h, 8 h, 12 h, 16 h and 96 h (P > 0.05; Figure 4B). Primary neurons were treated with 2ME2 0.5 h or 8 h after OGD to evaluate HIF‐1α regulation of Nix and VEGF expression. 2ME2 (5 µM) alone had no effect on the expression of HIF‐1α, Nix and VEGF (Figure 4C). In contrast, 2ME2 treatment, 0.5 h or 8 h after MCAO, attenuated VEGF expression (Figure 4D: F 7,16 = 0.7218, P > 0.05; Figure 4E: F 7,16 = 1.13, P > 0.05). 2ME2 treatment 0.5 h, but not 8 h, after OGD eliminated OGD‐induced Nix expression (Figure 4D: F 7,16 = 0.979, P > 0.05; Figure 4E: F 7,16 = 42.26, P < 0.0001). Expression of Nix in neurons treated with 2ME2 0.5 h after OGD was not significantly different between the naive‐control and at 1 h, 4 h, 8 h, 16 h, 24 h, 48 h and 72 h (P > 0.05; Figure 4D). Newman–Keuls post hoc comparison revealed differences between the naive‐control and the OGD plus 2ME2 (8 h) group at 4 h, 8 h and 16 h (P < 0.01; Figure 4E). Treatment with 2ME2 was specific for HIF‐1α; it did not have any effect on the expression of other transcription factors, such as NF‐κB p65 and c‐fos (Figure 4F), which can be activated by OGD 16, 26.
Figure 4.
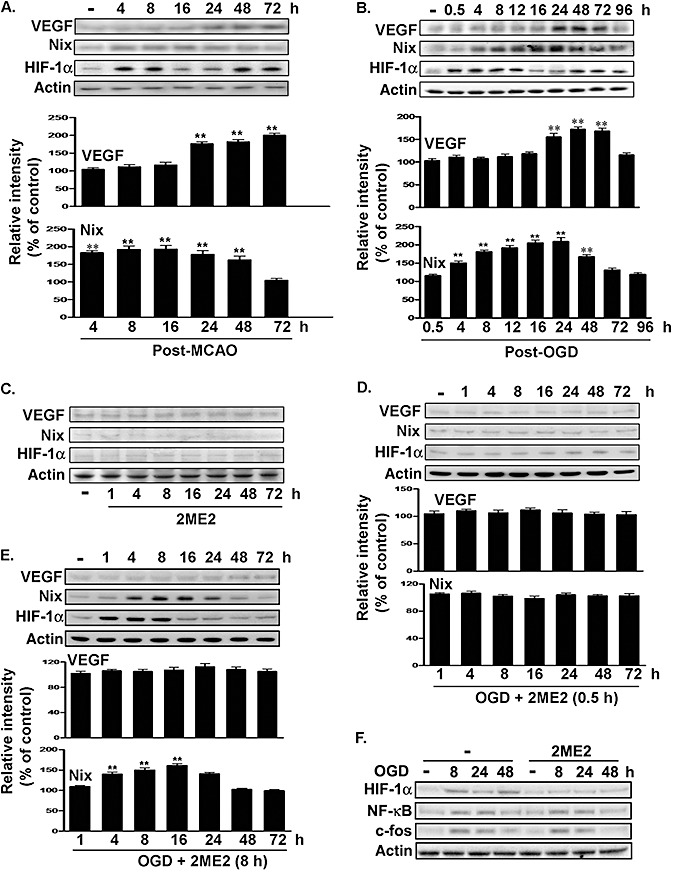
Effects of 2ME2 on pro‐death or pro‐survival gene expression after ischemic challenge. A. and B. Time course expression of VEGF and Nix in the primary neurons and ischemic hemispheres. Total lysates were prepared from ischemic hemispheres (A) and primary neurons (B). The levels of VEGF, Nix and HIF‐1α were determined by performing immunoblotting with anti‐VEGF antibodies, anti‐Nix antibody and anti‐HIF‐1α antibody, using actin as an internal control. C–F. Total lysates of the primary neurons were obtained at various time points following 2ME2 treatment (C,F), 2ME2 treatment 0.5 h (D) or 8 h (E) after OGD challenge. VEGF, Nix or HIF‐1α expression was detected by performing immunoblotting with anti‐VEGF antibodies, anti‐Nix antibody, anti‐HIF‐1α antibody, anti‐NF‐κB p65 antibodies and anti‐c‐fos antibodies, using actin as an internal control. All of the experiments were quantified in three independent experiments and were expressed as a percentage of the levels in the naive‐controls. Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; **P < 0.01 vs. the naive‐control group. Abbreviations: 2ME2 = 2‐methoxyestradiol; VEGF = vascular endothelial growth factor; Nix = Nip‐like protein X; HIF‐1α = hypoxia‐inducible factor‐1‐alpha; OGD = oxygen–glucose deprivation; NF‐κB = Nuclear factor‐kB; MCAO = middle cerebral artery occlusion.
Furthermore, 2ME2 was replaced with siRNA targeting HIF‐1α to determine the role of early‐ and late‐phase HIF‐1α on the expression of related genes. Figure 5A shows that HIF‐1α siRNA transfected into primary neurons 30 h before OGD exposure downregulated both early‐ and late‐phase HIF‐1α expression (F 4,10 = 2.366, P > 0.05). Conversely, only late‐phase HIF‐1α expression was attenuated when HIF‐1α siRNA was applied 12 h after OGD (F 4,10 = 88.66, P < 0.0001; Figure 5B). Expression differed in the naive‐control group compared with the 4‐h and 8‐h time points (P < 0.01), but no differences were seen 24 h and 48 h after OGD (P > 0.05). HIF‐1α siRNA application 30 h before OGD downregulated expression of VEGF (F 4,10 = 0.8221, P > 0.05; Figure 5D). However, HIF‐1α siRNA treatment 12 h after OGD also downregulated VEGF at 48 h compared with the naive‐control group, but expression was still elevated at 24 h (F 4,10 = 45.53, P < 0.0001; Figure 5D). Newman–Keuls post hoc comparison revealed differences between the naive‐control and the 240h time point (P < 0.01). No significant difference was observed between the naive‐control and the 4‐h, 8‐h and 48‐h time points (P > 0.05). In contrast, Nix was only downregulated when HIF‐1α siRNA was applied 30 h before OGD (Figure 5C). Expression of Nix differed in the naive‐control group compared with 4 h and 8 h after HIF‐1α siRNA application 12 h after OGD (F 4,10 = 71.69, P < 0.0001; Figure 5C). No significant difference was detected between the naive‐control group, 24‐h (P > 0.05) and 48‐h (P > 0.05) time points (Figure 5C). No differences in expression were also seen following HIF‐1α siRNA application 30 h before OGD in the naive‐control group compared with the 4‐h, 8‐h, 24‐h and 48‐h time points (F 4,10 = 1.225, P > 0.05; Figure 5C). Next, we investigated the effects of HIF‐1α on neuron viability after OGD. OGD for 48 h significantly reduced cell viability (F 7,16 = 36.49, P < 0.0001; Figure 5E). Newman–Keuls post hoc comparison revealed significant differences between the naive‐control group and the OGD, OGD plus HIF‐1α siRNA (12 h), OGD plus luciferase siRNA (−30 h) and OGD plus luciferase siRNA (12 h) groups (P < 0.05). Cell viability in the naive‐control group was similar to that in the HIF‐1α siRNA, luciferase siRNA and OGD plus HIF‐1α siRNA (−30 h) groups (P > 0.05). Taken together, the results suggest that the pro‐death and pro‐survival effects of HIF‐1α are because of regulation of the expression of adaptive and pro‐death modulators.
Figure 5.
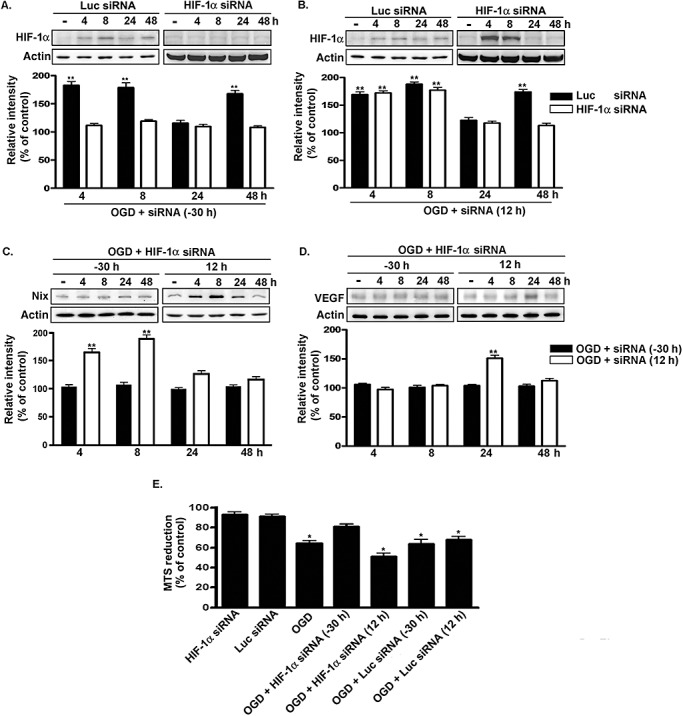
Effects of siRNA targeting HIF‐1αon Nix and VEGF expression. A. and B. Time course of HIF‐1α expression in primary neurons after been transfected with siRNA into cultured cortical neurons 30 h before (A) or 12 h after (B) being subjected to OGD. HIF‐1α expression was detected by immunoblotting with anti‐HIF‐1α antibodies, using actin as an internal control. C. and D. Primary neurons were transfected with siRNA either 30 h before or 12 h after being subjected to OGD. Total lysates were immunoblotted with either anti‐Nix antibody or anti‐VEGF antibodies, using actin as an internal control. E. The viability of the cultured cortical neurons was assessed by using the MTS assay. All experiments were performed in triplicate, and the results were expressed as a percentage of the levels in the naive‐controls. Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05; **P < 0.01 vs. the naive‐control group. Abbreviations: siRNA = Small interfering RNA; HIF‐1α = hypoxia‐inducible factor‐1‐alpha; Nix = Nip‐like protein X; VEGF = vascular endothelial growth factor; OGD = oxygen–glucose deprivation; MTS = methanethiosulfonate; Luc = luciferase.
The effect of biphasic HIF‐1α expression on neuronal survival after ischemic challenge
We investigated the effects of 2ME2 on neuronal viability following OGD. Treatment with 2ME2 0.5 h after an ischemic insult recovered neuron viability (F 4,15 = 68.01, P < 0.0001; Figure 6A). Newman–Keuls post hoc comparison revealed significant differences between the naive‐control group and the OGD, OGD plus 2ME2 (0.5 h) and OGD plus 2ME2 (8 h) groups (P < 0.05). Cell viability in the naive‐control group was similar to that in the 2ME2 groups (P > 0.05). Apoptosis underlies the majority of delayed neuronal death after ischemic stroke (11). We used the TUNEL assay to investigate whether early‐ or late‐HIF‐1α expression regulated apoptosis following ischemic stress. As shown in Figure 6B, focal ischemia was associated with a significant increase in TUNEL‐positive cells and caspase‐3 expression (Figure S4A). 2ME2 treatment at 0.5 h after an ischemic insult markedly reduced the number of apoptotic cells in vivo (F 4,15 = 60.11, P < 0.0001). Newman–Keuls post hoc comparison revealed differences between the MCAO and the MCAO plus 2ME2 (0.5 h) group (P < 0.05). Conversely, 2ME2 administered 8 h after the insult increased apoptosis compared with that seen in the MCAO group (P < 0.05). Intracerebroventricular injections of 2ME2 alone had no effect on the population or apoptosis of NeuN‐positive neurons (2, 6). No significant difference was observed between the sham‐control and 2ME2 group (P > 0.05). The similar result was also observed in primary neurons (F 4,15 = 40.56, P < 0.0001; Figure 6C). Furthermore, primary neurons were transfected with siRNA targeting HIF‐1α to specifically downregulate HIF‐1α expression. Figure 6D shows the number of apoptotic cells after OGD. Transfection with siRNA targeting HIF‐1α 30 h before OGD markedly decreased the number of apoptotic cells compared with that observed in the OGD group (F 7,24 = 50.09, P < 0.0001). However, transfection 12 h after OGD did not have any effect. Newman–Keuls post hoc comparison revealed significant differences between the naive‐control group and the OGD, OGD plus HIF‐1α siRNA (−30 h), OGD plus HIF‐1α siRNA (12 h), OGD plus luciferase siRNA (−30 h) and OGD plus luciferase siRNA (12 h) groups (P < 0.05). No significant difference was observed between the naive‐control group and the HIF‐1α siRNA or luciferase siRNA groups (P > 0.05). In summary, these results indicate that inhibition of early‐phase HIF‐1α expression decreases brain injury following ischemic stress, which is exacerbated when the late phase of HIF‐1α expression is attenuated.
Figure 6.
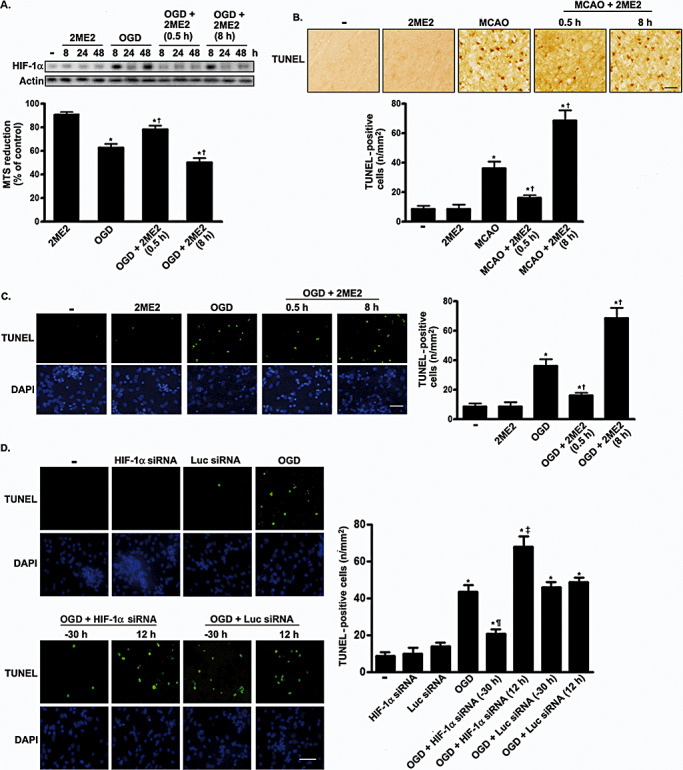
Representative images of TUNEL staining in cortex and primary neurons at 4 days after 2ME2 treatment or transfection with HIF‐1α siRNA. A. The viability of the cultured cortical neurons was assessed by performing the MTS assay. The HIF‐1α expression level was used as an internal control for the MTS assay. Experiments were performed in triplicate. Values are expressed as mean (SD) (n = 3). The percentage of cell viability in treated cells is relative to that in untreated cells. B. and C. Rat or primary neurons were treated with 2ME2 0.5 h or 8 h after ischemic challenge. Four days later, the cells were fixed, and TUNEL staining was performed. B,C. Scale bar, 100 µm. D. siRNA targeting HIF‐1α or luciferase was transfected into primary cortical neurons either 30 h before or 16 h after OGD. TUNEL staining was performed 4 days after OGD treatment. TUNEL‐positive cell counting analysis was determined under each condition. Scale bars, 50 µm. In panels B–D, quantification of TUNEL‐positive cells was presented as mean (SD) (n = 4). Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. the naive‐control and sham‐control group. †P < 0.05 vs. OGD and MCAO group. ¶P < 0.05 vs. OGD plus luciferase siRNA (−30 h) group. ‡P < 0.05 vs. OGD plus luciferase (12 h) group. Abbreviations: TUNEL = terminal deoxyuridine‐triphosphate nick‐end labeling; 2ME2 = 2‐methoxyestradiol; HIF‐1α = hypoxia‐inducible factor‐1‐alpha; siRNA = Small interfering RNA; MTS = methanethiosulfonate; SD = standard deviation; OGD = oxygen–glucose deprivation; MCAO = middle cerebral artery occlusion; DAPI = 4′,6‐diamidino‐2‐phenylindole; Luc = luciferase.
The pro‐survival or pro‐death effect of HIF‐1α was also determined by transfecting primary neurons with either Nix or VEGF shRNA. As shown in Figure 7A, the results of TUNEL staining suggests that the number of apoptotic cells was lower in the Nix shRNA plus OGD group than in the OGD group. In contrast, the knockdown of VEGF with VEGF shRNA increased apoptosis (F 7,32 = 177.6, P < 0.0001). Newman–Keuls post hoc comparison revealed differences between the naive‐control group and the OGD, OGD plus VEGF siRNA, OGD plus Nix siRNA and OGD plus luciferase siRNA groups (P < 0.05). Cell viability in the naive‐control group was similar to that in the Nix siRNA, VEGF siRNA and luciferase siRNA groups (P > 0.05). Finally, we investigated the effects of Nix and VEGF on neuronal viability following OGD. On the basis of the expression of Nix and VEGF, which had been knocked down (Figure 7B), we investigated cell viability with or without OGD. OGD for 24 h reduced cell viability (F 7,16 = 41.52, P < 0.0001; Figure 7B). Newman–Keuls post hoc comparison revealed differences between the naive‐control group and the OGD, OGD plus VEGF siRNA and OGD plus luciferase siRNA groups (P < 0.05). Cell viability was similar in the naive‐control group and Nix siRNA, VEGF siRNA, luciferase siRNA and OGD plus Nix siRNA groups (P > 0.05). Thus, Nix plays an important role in cell death caused by HIF‐1α, whereas VEGF mediates cell survival.
Figure 7.
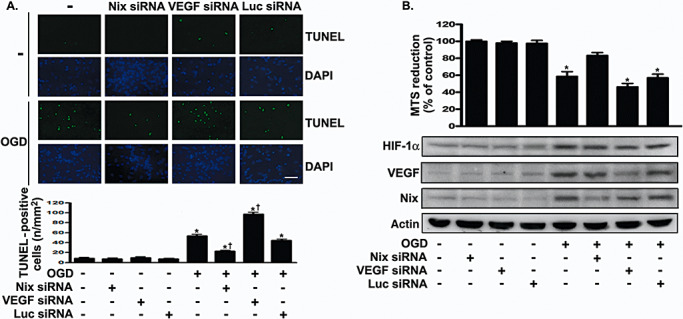
Effects of siRNA targeting Nix or VEGF on cellular apoptosis after ischemic challenge. A. siRNA targeting Nix, VEGF or luciferase was transfected into primary cortical neurons 30 h before OGD. TUNEL staining was performed 96 h after OGD challenge. TUNEL‐positive cell counting analysis was determined under each condition. Quantification of TUNEL‐positive cells was presented as mean (SD) (n = 4). Scale bars, 50 µm. B. Nix or VEGF in OGD‐treated or untreated primary neurons was knocked down by using the appropriate siRNA. The viability of the cultured cortical neurons was assessed by performing the MTS assay. The Nix and VEGF expression levels were used as an internal control for the MTS assay. Experiments were carried out independently in triplicate. The values that were normalized to the protein content are provided as the means (SD) (n = 3) and have been expressed as percentage viability relative to the untreated cells. Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. the naive‐control group. †P < 0.05 vs. OGD plus luciferase siRNA group. Abbreviations: siRNA = small interfering RNA; Nix = Nip‐like protein X; VEGF = vascular endothelial growth factor; OGD = oxygen–glucose deprivation; TUNEL = terminal deoxyuridine‐triphosphate nick‐end labeling; MTS = methanethiosulfonate; SD = standard deviation.
DISCUSSION
The role of the transcription factor HIF‐1α in neuronal survival after an ischemic stroke is controversial. Here, we showed that MCAO triggered a robust, biphasic (early and late phase) HIF‐1α accumulation that was mainly restricted to the neurons of the ischemic cortex and striatum. Early‐phase HIF‐1α expression facilitated apoptosis, but late‐phase expression increased cell survival. These results offer a novel insight into the role of HIF‐1α in ischemia.
In hypoxia, HIF‐1α accumulates because of increased protein stability and by binding with HIF‐1β to form a heterodimer. The heterodimer helps regulate transcription of hypoxia‐inducible genes involved in angiogenesis, glucose transport and metabolism, erythropoiesis, inflammation, apoptosis and cellular stress 17, 36. HIF‐1α is also activated by environmental, physiological and pathological stimuli, for example, iron chelators (28), hormones (1) and bacterial infection at normoxia (24). Two of our findings support the conclusion that HIF‐1α expression is induced at two periods after ischemia onset. First, MCAO induced biphasic HIF‐1α expression at 4–8 h and after 48 h (Figure 1C and Figure S1). Second, OGD also induced biphasic HIF‐1α expression at 4–12 h and after 48 h (Figure 1D). HIF‐1α messenger RNA (mRNA) determined by Reverse transcription polymerase chain reaction (RT‐PCR) was not increased in the early phase, but it did increase in the late phase (data not shown). These data suggest that HIF‐1α accumulation early after MCAO is because of increased protein stability, but that which occurs during the late phase results from augmented transcription. Therefore, we show that different mechanisms modulated HIF‐1α expression in a time‐dependent manner.
We also investigated the contribution of other mechanism to regulation of HIF‐1α expression. Figure S2 suggests that glutamate levels are correlated with HIF‐1α expression. Blockade of the N‐methyl‐D‐aspartic acid (NMDA) receptor with the inhibitor 2‐amino‐5‐phosphonovaleric acid also attenuated the early‐phase rise in HIF‐1α (Figure S2C). These data reveal that glutamate might be related to the induction of HIF‐1α during the early period of ischemia. BDNF has been reported to induce VEGF through HIF‐1α(23). Here, we showed that a BDNF scavenger reversed the late, but not early, phase induction of HIF‐1α (Figure S2F). Thus, ischemia induces an early‐ and late‐phase induction of HIF‐1α expression in neurons, which may be partly mediated by glutamate and BDNF, respectively. However, these findings do not exclude the contribution of other factors.
It is generally believed that HIF‐1α is an inducible protein and acts as a transcriptional factor in the nucleus. However, the results obtained from the immunofluorescence analysis revealed that HIF‐1α was expressed both in the cytoplasm and nucleus after ischemic stroke (Figure S1A). Moreover, Western blotting revealed that HIF‐1α was expressed in normal control cells (Figure 1). A previous study indicated a significant difference between normal and cancer cells with regard to cytoplasm–nuclear trafficking of HIF‐1α(21). The data suggested that HIF‐1α was constitutively expressed in normal [mouse embryonic fibroblasts (NIH3T3) and humanembryonic kidney 293 cells (HEK293)] and cancer cells (U87) under normoxia, and was significantly elevated after CoCl2 or hypoxic treatment. Furthermore, following CoCl2 exposure or hypoxia, HIF‐1α was observed to accumulate in the cytoplasm as well as the nucleus of NIH373 cells, whereas HIF‐1α preferentially accumulated in the nucleus of U87 cells. Lately, it is becoming evident that subcellular distribution and degradation of HIF‐1α are regulated in a cell‐specific manner (37). Although it is generally accepted that subcellular localization of HIF‐1α in mammalian cells depends on the von Hippel–Lindau tumor suppressor protein (12), it has not been fully investigated whether HIF‐1α expression is regulated in the cytoplasm or nucleus. Moreover, the shuttling mechanisms of HIF‐1α between the cytoplasm and nucleus remain poorly understood. Thus, we speculate that HIF‐1α continuously shuttles between the cytoplasm and nucleus, and that HIF‐1α expression is regulated in both compartments, following ischemic stroke. The distribution of HIF‐1α in cortical neurons after ischemic stroke is a new finding, which warrants further investigation.
Several studies suggest that HIF‐1α protects neurons from environmental stress. Prolyl 4‐hydroxylase inhibition and hypoxic preconditioning both induced HIF‐1α expression and protected neurons from damage after focal cerebral ischemia 18, 30. HIF‐1α also rescued neurons from nerve growth factor deprivation (33). In contrast, another study indicates that HIF‐1α is involved in hypoxia‐induced cell death by activating the expression of various pro‐death genes (32). 2ME2 had protective and damaging effects in experimental stroke 9, 38. Here, we provide several lines of evidence that support the finding that HIF‐1α has protective and damaging effects in neurons during ischemia. First, 2ME2 treatment early after MCAO decreased brain injury, but it increased damage when given at a later stage (Figure 2). Second, all the behavior tests indicated that 2ME2 given early after the onset of ischemia improved behavioral deficits but led to worsening of symptoms when given 8 h after MCAO (Figure 3A–3C). Third, we found that early administration of 2ME2 significantly increased survival of rats, whereas treatment at 8 h after MCAO not only removed the treatment effect but also reduced survival (Figure 3D). Fourth, early 2ME2 treatment as well as knockdown of HIF‐1α with HIF‐1α siRNA reduced apoptosis caused by MCAO or OGD (Figure 6B–6D). These results explain the discrepancy why some previous reports show that HIF‐1α protects neurons and other reports show that the protein promotes injury.
HIF‐1α regulates the expression of target genes by binding to the HIF‐1 binding element in response to hypoxia (35). Recent studies using microarrays revealed that HIF‐1αmut overexpression and hypoxia upregulated various genes, including those involved in apoptosis (eg, Nix) and oncogenes (eg, VEGF) 10, 31. In this study, we found that HIF‐1α modulated both Nix and VEGF at different time points after hypoxia/ischemia (Figure 4). Nix induction might be involved in apoptosis occurring during ischemia: 2ME2 or HIF‐1α siRNA that blocked the early phase of HIF‐1α expression attenuated Nix induction (4, 5), and knockdown of Nix with Nix shRNA prevented apoptosis during OGD (Figure 7). Previous studies confirm that Nix is involved in apoptosis (34). We also suggest that little VEGF expression is induced early after ischemia onset, but it increases at later stages after MCAO or OGD (Figure 4B). Accordingly, HIF‐1α might induce Nix in the early period of ischemia if the neurons were markedly stressed by the insult, and the cells could then undergo apoptosis. However, HIF‐1α in the late phase of ischemia could strongly induce VEGF and rescue neurons if the stress was mild or moderate. Repression of Nix expression at the initiation of ischemia may be important to shield neurons from apoptosis and give the cells enough time to produce pro‐survival VEGF.
After ET‐1‐induced MCAO, the ischemic area was localized to the motor and somatosensory cortices (Figure 2A). A previous study showed that ET‐1‐induced MCAO causes infarction of the lateral cortex (5). Two findings in this study support the conclusion that MCAO damaged the lateral cortex dependent of 2ME2 treatment. First, TTC staining indicated that the extent of the brain damage in the 2ME2 plus MCAO group and the MCAO group was limited to the cortex (Figure 2A). Second, the IgG‐positive region was limited to the cortex (Figure 2B). MCAO damaged the motor and somatosensory cortices (Figure 2A–2C), which resulted in motor and sensory dysfunction, respectively (Figure 3A–3C). In the in vivo study, we administered 2ME2 either 0.5 h or 8 h after MCAO to investigate the role of biphasic HIF‐1α in ischemic stroke. Although the differences in the damaged areas were statistically significant among the MCAO, MCAO plus 2ME2 (0.5 h) and MCAO plus 2ME2 (8 h) groups (Figure 2A–2C), the differences in behavioral deficits and survival rates within 4 days after MCAO were not statistically significant in these groups (Figure 3), which may be the result of compensatory brain functions. In addition, the differences in the rate of onset and the extent of behavioral deficits in these groups 4 days after MCAO may be the result of increased brain damage. A previous study has shown that the extent of the penumbra region is associated with functional recovery after stroke (11). Thus, we hypothesize that damage of the penumbra region but not the core region is associated with biphasic HIF‐1α activation.
In conclusion, our results provide evidence that time‐dependent HIF‐1α expression has pro‐death and pro‐survival effects during ischemic injury and that diverse signaling pathways mediate actions opposing those of HIF‐1α in an ischemic stress model. These data will assist the design of more effective therapeutic interventions against stroke.
Supporting information
Figure S1. Immunostaining of HIF‐1α at various time points of recovery from MCAO. A. Double immunostaining of HIF‐1α and NeuN at various time points of recovery from MCAO. Brain sections were obtained at various time points after MCAO and were subsequently probed with anti‐HIF‐1α antibodies and anti‐NeuN antibody. HIF‐1α was detected with Alexa 488 (green); NeuN, with Alexa 568 (red). Top panels show images taken from the cortex of non‐ischemic and ischemic hemispheres. Scale bars, 200 μm. Bottom panels show higher‐magnification views of neurons expressing HIF‐1α after MCAO. Scale bar, 50 μm. Merged images are shown in yellow. B. Quantification of HIF‐1α‐positive neurons in the total granule cell population was presented as mean (SD) (n = 4). Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; **P < 0.01 significantly different from contralateral and ipsilateral hemisphere.
Figure S2. Effects of glutamate and BDNF on HIF‐1α expression in primary neurons. A. Glutamate was measured in the supernatant of control neurons or neurons that had been exposed to 60 minutes of OGD. Values were normalized to the amount of cell protein in each well and presented as mean (SD) (n = 4). Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. the naive‐control group. B. Total lysates of the primary neurons were obtained at various time points following glutamate (100 μM) treatment. HIF‐1α expression was detected by performing Western blotting with anti‐HIF‐1α antibody, using actin as an internal control. C. NMDA receptor antagonist DL‐APV (20 μM) or AMPA receptor antagonist CNQX (20 μM) was used for pretreating the primary neurons for 30 minutes before application of glutamate. D. Total lysates of the primary neurons were obtained at various time points following OGD challenge. BDNF concentration was measured with a BDNF ELISA kit. Graphs illustrate mean (SD) (n = 4). Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. the naive‐control group. E. Total lysates of the primary neurons were obtained at various time points following BDNF (1 ng/mL) treatment. HIF‐1α expression was detected by performing Western blotting with anti‐HIF‐1α antibody, using actin as an internal control. F. The effects of BDNF on time‐dependent HIF‐1α expression. BDNF scavenger TrkB‐IgG (2 μg/mL) was used for pretreating the primary neurons for 30 minutes before exposure to OGD challenge. Total lysates were prepared at various time points of recovery from OGD and immunoblotted with an anti‐HIF‐1α antibody. Treatment with TrkB‐IgG alone had no effect on HIF‐1α expression. In panels B, C and E, F, the HIF‐1α expression levels were quantified in three independent experiments and were expressed as a percentage of the levels in the naive‐controls. Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. the naive‐control group.
Figure S3. Role 2ME2 in brain function. A. and B. Line graphs show temporal profiles of functional change from rats in 2ME2 treatment. Rats received 2ME2 were detected using the swing test (A) and adhesive‐removal patch test (B). C. Kaplan–Meier survival analysis after 2ME2 treatment. In panels A and B, statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests.
Figure S4. Effects of focal ischemia on caspase‐3 expression. A. Total lysates of the primary neurons were obtained at 24 h following OGD challenge, and then the expression level of caspase‐3 was determined by using immunoblotting with an anti‐caspase‐3 antibody. Actin was used as an internal control. B. The primary neurons were fixed, permeabilized with 1% Triton X‐100 and then probed with an anti‐NeuN antibody. NeuN was detected with Alexa 568 (red). The nucleus was stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; blue). Scale bars, 50 μm.
Figure S5. Transfection efficiency of Lipofectamine™ 2000 in primary neurons. A. and B. Alexa Fluor® 555‐labeled Red Fluorescent Oligo was transfected into cortical neurons with vehicle alone (A) or Lipofectamine 2000 (B) as described. Twenty‐four hours after transfection, the Alexa Fluor® 555‐positive cells are determined by fluorescence microscopy. Scale bars, 50 μm.
Supporting info item
ACKNOWLEDGMENTS
This work was supported by the National Cheng‐Kung University project of the Program for Promoting Academic Excellence and Developing World Class Research Centers, together with grants NSC 97‐2320‐B‐006‐016‐MY3 and NSC 97‐2311‐B‐006‐002‐MY3 obtained from the National Science Council, Republic of China.
REFERENCES
- 1. Aleffi S, Petrai I, Bertolani C, Parola M, Colombatto S, Novo E et al (2005) Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology 42:1339–1348. [DOI] [PubMed] [Google Scholar]
- 2. Aminova LR, Chavez JC, Lee J, Ryu H, Kung A, Lamanna JC, Ratan RR (2005) Prosurvival and prodeath effects of hypoxia‐inducible factor‐1alpha stabilization in a murine hippocampal cell line. J Biol Chem 280:3996–4003. [DOI] [PubMed] [Google Scholar]
- 3. Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC (2007) Neuron‐specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci 27:6320–6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17:472–476. [DOI] [PubMed] [Google Scholar]
- 5. Biernaskie J, Chernenko G, Corbett D (2004) Efficacy of rehabilitative experience declines with time after focal ischemic brain injury. J Neurosci 24:1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blouin CC, Page EL, Soucy GM, Richard DE (2004) Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia‐inducible factor 1alpha. Blood 103:1124–1130. [DOI] [PubMed] [Google Scholar]
- 7. Bogaert L, Scheller D, Moonen J, Sarre S, Smolders I, Ebinger G, Michotte Y (2000) Neurochemical changes and laser Doppler flowmetry in the endothelin‐1 rat model for focal cerebral ischemia. Brain Res 887:266–275. [DOI] [PubMed] [Google Scholar]
- 8. Chavez JC, LaManna JC (2002) Activation of hypoxia‐inducible factor‐1 in the rat cerebral cortex after transient global ischemia: potential role of insulin‐like growth factor‐1. J Neurosci 22:8922–8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen W, Jadhav V, Tang J, Zhang JH (2008) HIF‐1alpha inhibition ameliorates neonatal brain injury in a rat pup hypoxic‐ischemic model. Neurobiol Dis 31:433–441. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10. Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM (2006) Concordant regulation of gene expression by hypoxia and 2‐oxoglutarate‐dependent dioxygenase inhibition: the role of HIF‐1alpha, HIF‐2alpha, and other pathways. J Biol Chem 281:15215–15226. [DOI] [PubMed] [Google Scholar]
- 11. Ginsberg MD (1997) The new language of cerebral ischemia. AJNR Am J Neuroradiol 18:1435–1445. [PMC free article] [PubMed] [Google Scholar]
- 12. Groulx I, Lee S (2002) Oxygen‐dependent ubiquitination and degradation of hypoxia‐inducible factor requires nuclear‐cytoplasmic trafficking of the von Hippel‐Lindau tumor suppressor protein. Mol Cell Biol 22:5319–5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gundersen HJ, Jensen EB (1987) The efficiency of systematic sampling in stereology and its prediction. J Microsc 147:229–263. [DOI] [PubMed] [Google Scholar]
- 14. Hagen T, D'Amico G, Quintero M, Palacios‐Callender M, Hollis V, Lam F, Moncada S (2004) Inhibition of mitochondrial respiration by the anticancer agent 2‐methoxyestradiol. Biochem Biophys Res Commun 322:923–929. [DOI] [PubMed] [Google Scholar]
- 15. Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C et al (2005) Brain‐specific knock‐out of hypoxia‐inducible factor‐1alpha reduces rather than increases hypoxic‐ischemic damage. J Neurosci 25:4099–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kinouchi H, Sharp FR, Chan PH, Koistinaho J, Sagar SM, Yoshimoto T (1994) Induction of c‐fos, junB, c‐jun, and hsp70 mRNA in cortex, thalamus, basal ganglia, and hippocampus following middle cerebral artery occlusion. J Cereb Blood Flow Metab 14:808–817. [DOI] [PubMed] [Google Scholar]
- 17. Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW (2004) Hypoxia‐inducible factor (HIF‐1)alpha: its protein stability and biological functions. Exp Mol Med 36:1–12. [DOI] [PubMed] [Google Scholar]
- 18. Liu J, Narasimhan P, Yu F, Chan PH (2005) Neuroprotection by hypoxic preconditioning involves oxidative stress‐mediated expression of hypoxia‐inducible factor and erythropoietin. Stroke 36:1264–1269. [DOI] [PubMed] [Google Scholar]
- 19. Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS et al (2003) 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 3:363–375. [DOI] [PubMed] [Google Scholar]
- 20. Macrae IM, Robinson MJ, Graham DI, Reid JL, McCulloch J (1993) Endothelin‐1‐induced reductions in cerebral blood flow: dose dependency, time course, and neuropathological consequences. J Cereb Blood Flow Metab 13:276–284. [DOI] [PubMed] [Google Scholar]
- 21. Moroz E, Carlin S, Dyomina K, Burke S, Thaler HT, Blasberg R, Serganova I (2009) Real‐time imaging of HIF‐1alpha stabilization and degradation. PLoS One 4:e5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagle DG, Zhou YD (2006) Natural product‐derived small molecule activators of hypoxia‐inducible factor‐1 (HIF‐1). Curr Pharm Des 12:2673–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakamura K, Martin KC, Jackson JK, Beppu K, Woo CW, Thiele CJ (2006) Brain‐derived neurotrophic factor activation of TrkB induces vascular endothelial growth factor expression via hypoxia‐inducible factor‐1alpha in neuroblastoma cells. Cancer Res 66:4249–4255. [DOI] [PubMed] [Google Scholar]
- 24. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL et al (2005) HIF‐1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest 115:1806–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phan TG, Wright PM, Markus R, Howells DW, Davis SM, Donnan GA (2002) Salvaging the ischaemic penumbra: more than just reperfusion? Clin Exp Pharmacol Physiol 29:1–10. [DOI] [PubMed] [Google Scholar]
- 26. Sarnico I, Lanzillotta A, Boroni F, Benarese M, Alghisi M, Schwaninger M et al (2009) NF‐kappaB p50/RelA and c‐Rel‐containing dimers: opposite regulators of neuron vulnerability to ischaemia. J Neurochem 108:475–485. [DOI] [PubMed] [Google Scholar]
- 27. Schmidt‐Kastner R, van Os J, Steinbusch HWM, Schmitz C (2006) Gene regulation by hypoxia and the neurodevelopmental origin of schizophrenia. Schizophr Res 84:253–271. [DOI] [PubMed] [Google Scholar]
- 28. Semenza GL (2000) Expression of hypoxia‐inducible factor 1: mechanisms and consequences. Biochem Pharmacol 59:47–53. [DOI] [PubMed] [Google Scholar]
- 29. Sharkey J, Ritchie IM, Kelly PA (1993) Perivascular microapplication of endothelin‐1: a new model of focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab 13:865–871. [DOI] [PubMed] [Google Scholar]
- 30. Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC et al (2005) Hypoxia‐inducible factor prolyl 4‐hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem 280:41732–41743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang V, Davis DA, Haque M, Huang LE, Yarchoan R (2005) Differential gene up‐regulation by hypoxia‐inducible factor‐1alpha and hypoxia‐inducible factor‐2alpha in HEK293T cells. Cancer Res 65:3299–3306. [DOI] [PubMed] [Google Scholar]
- 32. Wang Y, Pakunlu RI, Tsao W, Pozharov V, Minko T (2004) Bimodal effect of hypoxia in cancer: role of hypoxia inducible factor in apoptosis. Mol Pharm 1:156–165. [DOI] [PubMed] [Google Scholar]
- 33. Xie L, Johnson RS, Freeman RS (2005) Inhibition of NGF deprivation‐induced death by low oxygen involves suppression of BIMEL and activation of HIF‐1. J Cell Biol 168:911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC et al (2002) Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med 8:725–730. [DOI] [PubMed] [Google Scholar]
- 35. Zagorska A, Dulak J (2004) HIF‐1: the knowns and unknowns of hypoxia sensing. Acta Biochim Pol 51:563–585. [PubMed] [Google Scholar]
- 36. Zarember KA, Malech HL (2005) HIF‐1alpha: a master regulator of innate host defenses? J Clin Invest 115:1702–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng X, Ruas JL, Cao R, Salomons FA, Cao Y, Poellinger L, Pereira T (2006) Cell‐type‐specific regulation of degradation of hypoxia‐inducible factor 1 alpha: role of subcellular compartmentalization. Mol Cell Biol 26:4628–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou D, Matchett GA, Jadhav V, Dach N, Zhang JH (2008) The effect of 2‐methoxyestradiol, a HIF‐1 alpha inhibitor, in global cerebral ischemia in rats. Neurol Res 30:268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Immunostaining of HIF‐1α at various time points of recovery from MCAO. A. Double immunostaining of HIF‐1α and NeuN at various time points of recovery from MCAO. Brain sections were obtained at various time points after MCAO and were subsequently probed with anti‐HIF‐1α antibodies and anti‐NeuN antibody. HIF‐1α was detected with Alexa 488 (green); NeuN, with Alexa 568 (red). Top panels show images taken from the cortex of non‐ischemic and ischemic hemispheres. Scale bars, 200 μm. Bottom panels show higher‐magnification views of neurons expressing HIF‐1α after MCAO. Scale bar, 50 μm. Merged images are shown in yellow. B. Quantification of HIF‐1α‐positive neurons in the total granule cell population was presented as mean (SD) (n = 4). Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; **P < 0.01 significantly different from contralateral and ipsilateral hemisphere.
Figure S2. Effects of glutamate and BDNF on HIF‐1α expression in primary neurons. A. Glutamate was measured in the supernatant of control neurons or neurons that had been exposed to 60 minutes of OGD. Values were normalized to the amount of cell protein in each well and presented as mean (SD) (n = 4). Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. the naive‐control group. B. Total lysates of the primary neurons were obtained at various time points following glutamate (100 μM) treatment. HIF‐1α expression was detected by performing Western blotting with anti‐HIF‐1α antibody, using actin as an internal control. C. NMDA receptor antagonist DL‐APV (20 μM) or AMPA receptor antagonist CNQX (20 μM) was used for pretreating the primary neurons for 30 minutes before application of glutamate. D. Total lysates of the primary neurons were obtained at various time points following OGD challenge. BDNF concentration was measured with a BDNF ELISA kit. Graphs illustrate mean (SD) (n = 4). Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. the naive‐control group. E. Total lysates of the primary neurons were obtained at various time points following BDNF (1 ng/mL) treatment. HIF‐1α expression was detected by performing Western blotting with anti‐HIF‐1α antibody, using actin as an internal control. F. The effects of BDNF on time‐dependent HIF‐1α expression. BDNF scavenger TrkB‐IgG (2 μg/mL) was used for pretreating the primary neurons for 30 minutes before exposure to OGD challenge. Total lysates were prepared at various time points of recovery from OGD and immunoblotted with an anti‐HIF‐1α antibody. Treatment with TrkB‐IgG alone had no effect on HIF‐1α expression. In panels B, C and E, F, the HIF‐1α expression levels were quantified in three independent experiments and were expressed as a percentage of the levels in the naive‐controls. Statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests; *P < 0.05 vs. the naive‐control group.
Figure S3. Role 2ME2 in brain function. A. and B. Line graphs show temporal profiles of functional change from rats in 2ME2 treatment. Rats received 2ME2 were detected using the swing test (A) and adhesive‐removal patch test (B). C. Kaplan–Meier survival analysis after 2ME2 treatment. In panels A and B, statistical analysis was carried out using the one‐way ANOVA with appropriate post hoc tests.
Figure S4. Effects of focal ischemia on caspase‐3 expression. A. Total lysates of the primary neurons were obtained at 24 h following OGD challenge, and then the expression level of caspase‐3 was determined by using immunoblotting with an anti‐caspase‐3 antibody. Actin was used as an internal control. B. The primary neurons were fixed, permeabilized with 1% Triton X‐100 and then probed with an anti‐NeuN antibody. NeuN was detected with Alexa 568 (red). The nucleus was stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; blue). Scale bars, 50 μm.
Figure S5. Transfection efficiency of Lipofectamine™ 2000 in primary neurons. A. and B. Alexa Fluor® 555‐labeled Red Fluorescent Oligo was transfected into cortical neurons with vehicle alone (A) or Lipofectamine 2000 (B) as described. Twenty‐four hours after transfection, the Alexa Fluor® 555‐positive cells are determined by fluorescence microscopy. Scale bars, 50 μm.
Supporting info item