

Abstract
Background
Patients treated with mechanical ventilation in intensive care units (ICUs) have a high risk of developing respiratory tract infections (RTIs). Ventilator‐associated pneumonia (VAP) has been estimated to affect 5% to 40% of patients treated with mechanical ventilation for at least 48 hours. The attributable mortality rate of VAP has been estimated at about 9%. Selective digestive decontamination (SDD), which consists of the topical application of non‐absorbable antimicrobial agents to the oropharynx and gastroenteric tract during the whole period of mechanical ventilation, is often used to reduce the risk of VAP. A related treatment is selective oropharyngeal decontamination (SOD), in which topical antibiotics are applied to the oropharynx only. This is an update of a review first published in 1997 and updated in 2002, 2004, and 2009.
Objectives
To assess the effect of topical antibiotic regimens (SDD and SOD), given alone or in combination with systemic antibiotics, to prevent mortality and respiratory infections in patients receiving mechanical ventilation for at least 48 hours in ICUs.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), which contains the Cochrane Acute Respiratory Infections (ARI) Group's Specialised Register, PubMed, and Embase on 5 February 2020. We also searched the WHO ICTRP and ClinicalTrials.gov for ongoing and unpublished studies on 5 February 2020. All searches included non‐English language literature. We handsearched references of topic‐related systematic reviews and the included studies.
Selection criteria
Randomised controlled trials (RCTs) and cluster‐RCTs assessing the efficacy and safety of topical prophylactic antibiotic regimens in adults receiving intensive care and mechanical ventilation. The included studies compared topical plus systemic antibiotics versus placebo or no treatment; topical antibiotics versus no treatment; and topical plus systemic antibiotics versus systemic antibiotics.
Data collection and analysis
We used standard methodological procedures expected by Cochrane.
Main results
We included a total of 41 trials involving 11,004 participants (five new studies were added in this update). The minimum duration of mechanical ventilation ranged from 2 (19 studies) to 6 days (one study). Thirteen studies reported the mean length of ICU stay, ranging from 11 to 33 days. The percentage of immunocompromised patients ranged from 0% (10 studies) to 22% (1 study).
The reporting quality of the majority of included studies was very poor, so we judged more than 40% of the studies as at unclear risk of selection bias. We judged all studies to be at low risk of performance bias, though 47.6% were open‐label, because hospitals usually have standardised infection control programmes, and possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting. Regarding detection bias, we judged all included studies as at low risk for the outcome mortality. For the outcome RTIs, we judged all double‐blind studies as at low risk of detection bias. We judged five open‐label studies as at high risk of detection bias, as the diagnosis of RTI was not based on microbiological exams; we judged the remaining open‐label studies as at low risk of detection bias, as a standardised set of diagnostic criteria, including results of microbiological exams, were used.
Topical plus systemic antibiotic prophylaxis reduces overall mortality compared with placebo or no treatment (risk ratio (RR) 0.84, 95% confidence interval (CI) 0.73 to 0.96; 18 studies; 5290 participants; high‐certainty evidence). Based on an illustrative risk of 303 deaths in 1000 people this equates to 48 (95% CI 15 to 79) fewer deaths with topical plus systemic antibiotic prophylaxis. Topical plus systemic antibiotic prophylaxis probably reduces RTIs (RR 0.43, 95% CI 0.35 to 0.53; 17 studies; 2951 participants; moderate‐certainty evidence). Based on an illustrative risk of 417 RTIs in 1000 people this equates to 238 (95% CI 196 to 271) fewer RTIs with topical plus systemic antibiotic prophylaxis.
Topical antibiotic prophylaxis probably reduces overall mortality compared with no topical antibiotic prophylaxis (RR 0.96, 95% CI 0.87 to 1.05; 22 studies, 4213 participants; moderate‐certainty evidence). Based on an illustrative risk of 290 deaths in 1000 people this equates to 19 (95% CI 37 fewer to 15 more) fewer deaths with topical antibiotic prophylaxis. Topical antibiotic prophylaxis may reduce RTIs (RR 0.57, 95% CI 0.44 to 0.74; 19 studies, 2698 participants; low‐certainty evidence). Based on an illustrative risk of 318 RTIs in 1000 people this equates to 137 (95% CI 83 to 178) fewer RTIs with topical antibiotic prophylaxis.
Sixteen studies reported adverse events and dropouts due to adverse events, which were poorly reported with sparse data. The certainty of the evidence ranged from low to very low.
Authors' conclusions
Treatments based on topical prophylaxis probably reduce respiratory infections, but not mortality, in adult patients receiving mechanical ventilation for at least 48 hours, whereas a combination of topical and systemic prophylactic antibiotics reduces both overall mortality and RTIs. However, we cannot rule out that the systemic component of the combined treatment provides a relevant contribution in the observed reduction of mortality. No conclusion can be drawn about adverse events as they were poorly reported with sparse data.
Plain language summary
Topical antibiotics to help reduce death and respiratory infections in people in intensive care receiving mechanical ventilation
Review question
We aimed to assess the effect of two topical antibiotic regimens (selective digestive decontamination (SDD) and selective oropharyngeal decontamination (SOD)) in preventing deaths and respiratory infections in patients receiving mechanical ventilation for at least 48 hours in intensive care units (ICUs). In SDD, non‐absorbable antibiotics are applied to the oropharynx (back third of the tongue, the soft palate, the side and back walls of the throat and tonsils), oesophagus, stomach, and intestine. SOD involves the application of non‐absorbable antibiotics to the oropharynx only. These regimens may be given alone or in combination with systemic antibiotics.
Background
Infections acquired in ICUs are important complications of treatment with ventilation (invasive mechanical breathing support) in patients with very severe diseases who require such treatment. Some of these people will die because of these infections. One method that has been evaluated to reduce these complications is to use antibiotics as a preventative measure.
Search date
This review is current to 5 February 2020.
Study characteristics:
We included 41 trials involving a total of 11,004 patients mechanically ventilated in ICUs to find out whether giving topical antibiotics, alone or in combination with systemic antibiotics, prevents respiratory tract infections and reduces death. Antibiotics were administered either topically (e.g. antibiotics were applied directly to the oropharynx or to the stomach via a nasogastric tube) or systemically (e.g. intravenously (directly into the patient's vein)).
Study funding sources
Twenty‐two studies (52.4%) did not report the funding source; 6 studies (14.3%) were supported by public institutional grants; and 13 studies (30.1%) were totally or partially funded by pharmaceutical companies.
Key results
In patients receiving the combination of topical plus systemic antibiotics, there were fewer deaths (data from 18 studies with 5290 patients) and probably fewer patients with respiratory tract infections (data from 17 studies with 2951 patients) compared to those who received no treatment or placebo, although we cannot exclude the possibility that the systemic component of the treatments contributed to the reduction in deaths. Assuming an illustrative risk of 303 deaths and of 417 cases of respiratory tract infections in 1000 people under mechanical ventilation, we expect 48 fewer death in patients who receive a combination of topical plus systemic antibiotics and 238 fewer cases of respiratory tract infections. When patients who received topical antibiotics only were compared with patients who received no treatment, or when patients who received topical plus systemic antibiotics were compared with patients who received systemic antibiotics alone, the number of deaths was probably similar (data from 22 studies with 4213 patients), although there may be fewer patients with respiratory tract infections in patients who received topical prophylaxis (data from 19 studies; 2698 patients). Adverse events were poorly reported, with limited data.
Certainty of the evidence
We judged the certainty of the evidence as high to moderate for deaths and respiratory tract infections and low to very low for adverse events.
Summary of findings
Background
Description of the condition
Patients admitted to intensive care units (ICUs) are prone to acquire nosocomial infections, which may increase morbidity and mortality (Adrie 2017; Melsen 2013).
Amongst respiratory tract infections (RTIs), ventilator‐associated pneumonia (VAP) has been estimated to affect 5% to 40% of patients treated with mechanical ventilation for at least 48 hours. However, this estimate is highly variable depending on the country, the type of ICU, and the criteria used to define the VAP (American Thoracic Society 2005; Reignier 2016; Seguin 2014).
Data from the US report estimates of incidence of VAP in the order of 1 to 2 cases per 1000 days of ventilation (Dudeck 2013), whilst the European study EU‐VAP/CAP has shown an incidence of 18.3 episodes of VAP per 1000 days of ventilation (Koulenti 2017). Higher incidences were also found in low‐ to middle‐income countries compared to high‐income countries (18.5 versus 9.0 per 1000 days of ventilation) (Bonell 2019). However, it should be noted that these differences are explained, at least partially, by the use of different definitions, and due to differences in microbiological sampling methods (Ego 2015). Incidence rates also vary according to the population studied, that is higher incidences have been reported in cancer patients, Stoclin 2020, and trauma patients (Cook 2010).
Based on aggregated results from 58 randomised studies on VAP prevention, the attributable mortality rate of VAP was reported as 9% (Melsen 2011). A competing risk survival analysis in a cohort of 4479 ICU patients in France reported that intensive care mortality attributable to VAP was approximately 1% on day 30 and 1.5% on day 60 (Bekaert 2011). Data from an individual patient meta‐analysis including 24 studies (6284 patients) showed that mortality was higher in surgical patients and in those with medium‐severity illness at ICU admission, when compared to traumatised and medical patients, or when compared to those with particularly high‐ or low‐severity illness at admission (Melsen 2013).
ICU‐acquired infections are also responsible for increased expenditure in an already costly healthcare setting. Interventions aimed at preventing these complications are therefore encouraged (Kollef 2012; Laupland 2006).
Description of the intervention
Selective digestive decontamination (SDD) and selective oropharyngeal decontamination (SOD) are prophylactic antibiotic interventions used to eradicate colonisation of aerobic gram‐negative bacteria, Staphylococcus aureus, and yeasts, whilst leaving the anaerobic flora intact.
SDD consists of the topical application of non‐absorbable antimicrobial agents to the oropharynx and gastroenteric tract during the whole ICU stay, often in combination with a short initial course of intravenous antibiotics (usually intravenous second‐generation cephalosporin during the first four days of ICU stay).
SOD comprises oropharyngeal application of bactericidal non‐absorbable antibiotics.
Both interventions consist of enteral application of non‐absorbable antimicrobial agents, most often amphotericin B, tobramycin or gentamycin, and colistin, aiming to eradicate yeasts, S aureus, and aerobic gram‐negative bacteria.
Since the main goal of antibiotic prophylaxis is to prevent infections acquired in intensive care, the protocol was usually applied immediately after ICU admission and continued until ICU discharge or extubation (de Jonge 2003; de Smet 2009; Oostdijk 2014; Stoutenbeek 1984; Wittekamp 2018). The target population is patients admitted to ICU for at least 48 hours and undergoing invasive mechanical ventilation.
In early studies, antibiotic prophylaxis was assessed in the ICU setting to prevent VAP in trauma patients who received prolonged mechanical ventilation by using a four‐component “classic” SDD regimen (Hurley 2020). Recent studies have broadened the inclusion criteria beyond trauma patients receiving invasive mechanical ventilation, tested different regimens, and used endpoints other than VAP. Variable definitions for some endpoints, such as VAP and bacteraemia, as well as variable study designs including blind or non‐blind placebo‐controlled groups, receiving or not receiving parenteral antibiotic prophylaxis, or even without any control group, have further clouded the picture. Although no relationship between the administration of SDD and antimicrobial resistance was detected (Daneman 2013), the real impact of SDD on the onset of antibiotic resistance remains, to date, not fully defined. This was mainly due to the fact that most of the studies assessed the effects of SDD at the patient level rather than at the ICU level, and with limited follow‐up time (Sánchez‐Ramírez 2018). Distinguishing ICUs with low prevalence of antibiotic resistance from ICUs with moderate to high prevalence of resistance, three cluster‐randomised studies highlighted that in settings with low prevalence of antibiotic resistance, SDD has been consistently associated with improved patient outcome (de Jonge 2003; de Smet 2009; Oostdijk 2014). These benefits were not confirmed in a large international cluster‐randomised study in settings with moderate‐to‐high prevalence of antibiotic resistance, where clinical relevance of SDD on patient outcomes remains to be seen (Wittekamp 2018).
How the intervention might work
The hypothesis behind antibiotic prophylaxis with SDD/SOD is that intestinal flora may represent the origin of potential pathogenic micro‐organisms which, colonising the upper respiratory tract during hospitalisation, can induce an increased risk of VAP and infection‐related ventilator‐associated complication (IVAC) (Magill 2013).
SDD and SOD have been shown to reduce the incidence of RTIs, the colonisation with antibiotic‐resistant gram‐negative bacteria, and the incidence of nosocomial infections, and to improve patient survival (de Jonge 2018; de Smet 2009; Plantinga 2017; Vincent 2011). The prevention effect with antibiotic prophylaxis seems to exceed the VAP prevention effect of various non‐decontamination methods evaluated in the mechanically ventilated patient group (Landelle 2018).
Why it is important to do this review
Antibiotic prophylaxis, especially SDD, has been discussed in intensive care literature for nearly 40 years (Stoutenbeek 1984). The diversity amongst the studies has fuelled controversy (Hurley 2020).
On the one hand, as early as 25 years ago, the summary evidence derived from more than 40 studies showed an apparent potent prevention effect against VAP, bacteraemia, and mortality (Hurley 1995). On the other hand, opinions regarding antibiotic prophylaxis have varied, even in the Netherlands, where many high‐quality studies have been conducted (Bonten 2001), and where SDD is the standard of care (SWAB 2018).
Although high‐quality evidence supports the use of SDD, its application is still a matter of debate, and it is not widely used in clinical practice (Reis 2015), due to the main concern that it may promote the emergence of antibiotic‐resistant strains (Brink 2013; Halaby 2013).
Given the uncertainty on the efficacy of antibiotic prophylaxis, and in light of the worldwide challenge of multidrug resistance, we considered it necessary to revisit this meta‐analysis, with the aim of redefining the role of the antibiotic prophylaxis in ICU patients requiring invasive mechanical ventilation.
Objectives
To assess the effect of topical antibiotic regimens (selective digestive decontamination (SDD) and selective oropharyngeal decontamination (SOD)), given alone or in combination with systemic antibiotics, to prevent mortality and respiratory infections in patients receiving mechanical ventilation for at least 48 hours in intensive care units (ICUs).
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) and cluster‐RCTs on antibiotic prophylaxis for the prevention of respiratory tract infections (RTIs) and death in adults on mechanical ventilation in intensive care unit (ICU) patients. We included both blinded and unblinded studies.
Types of participants
Adult (≥ 18 years) patients admitted to an ICU for at least 48 hours. We excluded studies where the majority of patients (> 50%) did not undergo mechanical ventilation for at least 48 hours. We also excluded studies if they considered only patients with a higher‐than‐usual risk of infection (e.g. liver transplantation, neutropenic patients).
Types of interventions
Experimental intervention
Topical antibiotic prophylaxis applied to nasopharynx (selective oropharyngeal decontamination (SOD)) or to oropharynx and gastric tube (selective decontamination of the digestive tract (SDD)) for the whole period of mechanical ventilation, given alone or in combination with systemic antibiotic prophylaxis.
Control intervention
Placebo, no prophylaxis, or systemic antibiotic prophylaxis alone.
Types of outcome measures
Primary outcomes
Overall mortality. We considered mortality at hospital discharge if this information was provided; otherwise we considered mortality in the ICU.
Respiratory tract infections. We made no restriction on the type of RTI considered (pneumonia and tracheobronchitis, ventilator‐associated pneumonia (VAP), and infection‐related ventilator‐associated complication (IVAC)), nor on the RTI diagnostic criteria used. We considered both primary (diagnosed within 48 hours from admission) and acquired (diagnosed after 48 hours from admission) infections. In case both were reported, we considered data from the acquired group.
Secondary outcomes
Dropouts due to adverse events.
Participants with gastrointestinal adverse events.
Participants with allergic adverse events.
Search methods for identification of studies
Electronic searches
For this update, we searched the following databases up to 5 February 2020. We imposed no language, publication year, or publication status restrictions. We identified published, unpublished, and ongoing studies by searching the following databases from their inception.
Cochrane Central Register of Controlled Trials (CENTRAL), which contains the Cochrane Acute Respiratory Infections (ARI) Group's Specialised Register, 2020 Issue 3 (searched 5 February 2020) (Appendix 1).
MEDLINE PubMed (up to 5 February 2020) (Appendix 2).
Embase Ovid (up to 5 February 2020) (Appendix 3).
We searched the following trials registries on 5 February 2020:
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (clinicaltrials.gov); and
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/).
Details of the previous search strategies are shown in Appendix 4.
Searching other resources
We searched the reference lists of retrieved included studies, systematic reviews, and meta‐analyses in order to identify other potentially eligible studies.
Data collection and analysis
Selection of studies
Two review authors (SP, SM) independently screened the titles and abstracts of all the references identified by the searches and retrieved and investigated all potentially relevant articles as full text to determine eligibility for inclusion in the review. Any disagreements were resolved by discussion or by involving a third review author (LB) if necessary.
Data extraction and management
Using a standardised data extraction form, three review authors (VP, SM and SP) collected the relevant study data, including study design, sample characteristics, description of the experimental and control interventions, outcomes, study funding, and conflicts of interest. Any disagreements were resolved by discussion. We contacted study authors for clarification when necessary.
Assessment of risk of bias in included studies
Two review authors (SM, VP) independently assessed the risk of bias using the criteria recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The recommended approach for assessing risk of bias is a two‐part tool, addressing the specific domains of sequence generation and allocation concealment (selection bias), blinding of participants and providers (performance bias), blinding of outcome assessor (detection bias), incomplete outcome data (attrition bias), and selective outcome reporting (reporting bias). The first part of the tool involves describing what was reported to have occurred in the study, whilst in the second part a judgement is assigned relating to the risk of bias for each domain, that is low, high, or unclear risk. See Appendix 5 for details.
We assessed the risk of detection bias separately for mortality, RTIs, and adverse events.
Measures of treatment effect
We analysed dichotomous outcomes by calculating the risk ratio (RR) and its relative 95% confidence interval (CI).
Unit of analysis issues
In case of multi‐arm studies, we combined all the relevant experimental or control groups into a single group to avoid double‐counting of participants. In case of cluster‐RCTs, we adjusted the raw data for the 'design effect' by using the effective sample size approach, as recommended in the updated version of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2020).
Dealing with missing data
Missing data are not a relevant issue in this setting and for our outcomes, as patients who meet the inclusion criteria are usually followed up until the end of the study, and outcome values are collected by the researchers.
Assessment of heterogeneity
We analysed heterogeneity using the I2 statistic and the Chi2 test. We considered heterogeneity as substantial if the I² was greater than 75%, or the P value was lower than 0.10 for the Chi2 test for heterogeneity (Higgins 2020).
Assessment of reporting biases
We used the visual inspection of funnel plots (plots of the effect estimate from each study against the effect standard error) to evaluate possible publication bias if there were at least 10 studies included in the meta‐analysis.
Data synthesis
We combined the outcomes from the individual trials through meta‐analysis where possible (comparability of intervention and outcomes between trials), using a random‐effects model, because we expected a certain degree of heterogeneity across trials. If the clinical or statistical heterogeneity was too high (i.e. 75% to 100%), we decided against pooling the data.
Subgroup analysis and investigation of heterogeneity
We did not perform subgroup analysis for type of drug because there were no data to assume a difference in effect amongst the considered prophylactic treatments. This obviously does not mean that all topical and systemic regimens are truly equivalent, but simply reflects our pragmatic working assumption.
Sensitivity analysis
To incorporate our assessment of risk of bias in the review process, we first plotted the intervention effect estimates stratified by risk of bias for allocation concealment (selection bias). We planned that if differences in the results were present amongst studies at different risks of selection bias, we would perform sensitivity analysis by excluding the studies at high risk of selection bias.
Summary of findings and assessment of the certainty of the evidence
We created two 'Summary of findings' tables (Table 1; Table 2) using the following outcomes: overall mortality, RTIs, dropouts due to adverse events, gastrointestinal adverse events and allergic adverse events. We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of a body of evidence as it relates to the studies which contribute data to the meta‐analyses for the prespecified outcomes (Atkins 2004). We used the methods and recommendations described in Chapter 14 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2020), employing GRADEpro GDT software (GRADEpro GDT). We justified all decisions to down‐ or upgrade the quality of studies using footnotes, and made comments to aid the reader's understanding of the review where necessary.
Summary of findings 1. Topical plus systemic prophylaxis versus placebo or no treatment in adults receiving mechanical ventilation for at least 48 hours.
| Topical plus systemic prophylaxisversus placebo or no treatment in adults receiving mechanical ventilation for at least 48 hours | |||||
| Patient or population: adults receiving mechanical ventilation for at least 48 hours Setting: ICU in The Netherlands, France, Spain, Germany, USA, UK, Egypt, Ireland, Tunisia, South Africa, Austria, Greece, Switzerland, Belgium, Australia and New Zealand Intervention: topical and systemic antibiotic prophylaxis Comparison: no prophylaxis | |||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | |
| Risk with no prophylaxis | Risk with topical plus systemic | ||||
| Overall mortality | Study population | RR 0.84 (0.73 to 0.96) | 5290 (18 RCTs) | ⊕⊕⊕⊕ HIGH | |
| 303 per 1000 | 255 per 1000 (224 to 288) | ||||
| Respiratory tract infections | Study population | RR 0.43 (0.35 to 0.53) | 2951 (17 RCTs) | ⊕⊕⊕⊝ MODERATEa | |
| 417 per 1000 | 179 per 1000 (146 to 221) | ||||
| Dropouts due to adverse events | Study population | RR 1.06 (0.30 to 3.76) | 1287 (4 RCTs) | ⊕⊕⊝⊝ LOWb | |
| 6 per 1000 | 7 per 1000 (2 to 24) | ||||
| Gastrointestinal adverse events | Study population | RR 1.08 (0.57 to 2.04) | 2637 (6 RCTs) | ⊕⊕⊝⊝ LOWb | |
| 44 per 1000 | 48 per 1000 (25 to 90) | ||||
| Allergic adverse events | Study population | RR 1.49 (0.09 to 25.33) | 2981 (6 RCTs) | ⊕⊕⊝⊝ LOWb | |
| 0 per 1000 | 1 per 1000 (0 to 9) | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; ICU: intensive care unit; RCT: randomised controlled trial; RR: risk ratio | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | |||||
aDowngraded 1 level for suspected publication bias, as indicated by asymmetry in the funnel plot. bDowngraded 2 levels due to very serious imprecision: sparse data.
Summary of findings 2. Topical prophylaxis versus no topical prophylaxis in adults receiving mechanical ventilation for at least 48 hours.
| Topical prophylaxis versus no topical prophylaxis in adults receiving mechanical ventilation for at least 48 hours | |||||
| Patient or population: adults receiving mechanical ventilation for at least 48 hours Setting: ICU in The Netherlands, France, Spain, Germany, USA, UK, Egypt, Ireland, Tunisia, South Africa, Austria, Greece, Switzerland, Belgium, Australia and New Zealand Intervention: topical antibiotic prophylaxis Comparison: no topical prophylaxis | |||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | |
| Risk with control | Risk with topical | ||||
| Overall mortality | Study population | RR 0.96 (0.87 to 1.05) | 4213 (22 RCTs) | ⊕⊕⊕⊝ MODERATEa | |
| 290 per 1000 | 279 per 1000 (253 to 305) | ||||
| Overall mortality ‐ topical plus systemic prophylaxis versus systemic prophylaxis alone | Study population | RR 0.92 (0.72 to 1.18) | 939 (7 RCTs) | ⊕⊕⊝⊝ LOWb,c | |
| 237 per 1000 | 218 per 1000 (171 to 280) | ||||
| Overall mortality ‐ topical prophylaxis versus placebo or no treatment | Study population | RR 0.97 (0.87 to 1.07) | 3274 (15 RCTs) | ⊕⊕⊕⊝ MODERATEd | |
| 305 per 1000 | 296 per 1000 (265 to 326) | ||||
| Respiratory tract infections | Study population | RR 0.57 (0.44 to 0.74) | 2698 (19 RCTs) | ⊕⊕⊝⊝ LOWe,f,g | |
| 318 per 1000 | 181 per 1000 (140 to 235) | ||||
| Respiratory tract infections ‐ topical plus systemic prophylaxis versus systemic prophylaxis alone | Study population | RR 0.82 (0.58 to 1.16) | 850 (6 RCTs) | ⊕⊕⊝⊝ LOWb,c,f | |
| 303 per 1000 | 248 per 1000 (176 to 352) | ||||
| Respiratory tract infections ‐ topical prophylaxis versus no treatment or placebo | Study population | RR 0.50 (0.36 to 0.69) | 1848 (13 RCTs) | ⊕⊕⊝⊝ LOWf,h | |
| 324 per 1000 | 162 per 1000 (117 to 224) | ||||
| Dropouts due to adverse events | Study population | RR 2.20 (0.57 to 8.54) | 1323 (7 RCTs) | ⊕⊝⊝⊝ VERY LOWc,i | |
| 9 per 1000 | 20 per 1000 (5 to 77) | ||||
| Gastrointestinal adverse events | Study population | RR 2.78 (0.26 to 29.50) | 1859 (3 RCTs) | ⊕⊕⊝⊝ LOWi | |
| 22 per 1000 | 62 per 1000 (6 to 656) | ||||
| Allergic adverse events | Study population | RR 2.64 (0.34 to 20.69) | 2357 (5 RCTs) | ⊕⊝⊝⊝ VERY LOWi,j | |
| 1 per 1000 | 2 per 1000 (0 to 17) | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; ICU: intensive care unit; RCT: randomised controlled trial; RR: risk ratio | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | |||||
a68% of studies at unclear risk and 1 study at high risk of selection bias. bOptimal information size not met. cAll studies at unclear risk of selection bias. d53.3% of studies at unclear risk and 1 study at high risk of selection bias. e74% of studies at unclear risk and 1 study at high risk of selection bias. fDowngraded 1 level for suspected publication bias, as indicated by the Funnel plot showing asymmetry. g We decided against downgrading certainty of evidence of this outcome as, although there is heterogeneity among studies, the direction of all the results favours the experimental intervention. h 62% of studies at unclear risk and 1 study at high risk of selection bias. iDowngraded 2 levels due to very serious imprecision: sparse data. j50% of studies at unclear risk of selection bias.
The GRADE system uses the following criteria for assigning grades of evidence.
High: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
Low: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect.
Very low: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect.
Results
Description of studies
Results of the search
We included 36 trials involving 6914 people in the previous version of this review. In this 2020 update, we identified a total of 3201 records after de‐duplication, of which nine studies were considered as potentially relevant. We excluded seven studies and included two studies (Beshey 2014; Chaari 2014). We also decided to include two previously excluded studies (de la Cal 2005; de Smet 2009), and included one study that was missed in the previous version of the review (Koeman 2006). We identified two ongoing studies (IRCT20180110038298N1; NCT02389036). We included a total of 41 studies. See Figure 1.
1.
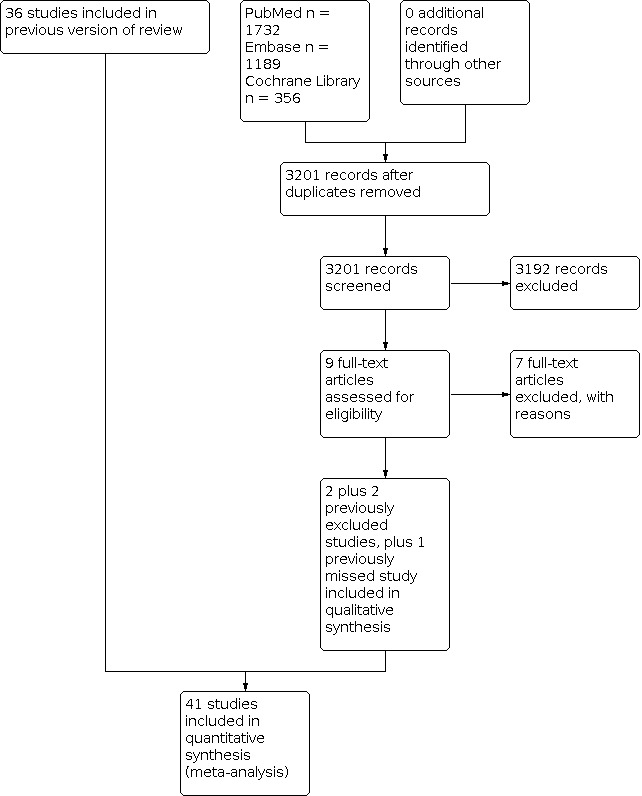
Study flow diagram: 2020 update.
Included studies
We included 41 RCTs in this update. One study was a cluster‐RCT (de Smet 2009). All of the trials were published, 39 as full reports, and two in abstract form (Boland 1991; Finch 1991). See Characteristics of included studies table.
Characteristics of participants
Overall, the trials included 16,329 participants. In our analyses, we considered 11,004 participants as fulfilling the inclusion criteria of a minimum duration of mechanical ventilation of at least 48 hours. The minimum duration of mechanical ventilation was two days in 19 studies, three days in seven studies, four days in three studies, five days in eight studies, and six days in one study. In two studies the minimum duration of mechanical ventilation was not stated.
The number of participants included in the studies ranged from 39 to 4035. The reasons for admission were surgical for 4726 (29%) participants, medical for 5305 (32.5%) participants, and trauma for 2609 (16%) participants.
Length of stay in ICU was reported as median in 18 studies, ranging from 3.5 to 19.5 days, and as mean in 13 studies, ranging from 11.3 to 33 days. Ten studies did not report this information.
In eight studies (Blair 1991; Brun‐Buisson 1989; Cockerill 1992; de Jonge 2003; de la Cal 2005; de Smet 2009; Ulrich 1989; Winter 1992), not all participants were mechanically ventilated. Five studies did not report this information (Beshey 2014; Camus 2005; Cerra 1992; Finch 1991; Laggner 1994).
The percentage of immunocompromised participants ranged from 0% (10 studies) to 22% (1 study).
Characteristics of treatment regimens
Nineteen RCTs compared the combination of topical and systemic antibiotic prophylaxis versus no treatment or placebo (Abele‐Horn 1997; Aerdts 1991; Blair 1991; Boland 1991; Cockerill 1992; de Jonge 2003; de la Cal 2005; de Smet 2009; Finch 1991; Jacobs 1992; Kerver 1988; Krueger 2002; Palomar 1997; Rocha 1992; Sanchez‐Garcia 1998; Stoutenbeek 2007; Ulrich 1989; Verwaest 1997; Winter 1992); 16 RCTs compared topical prophylaxis alone to no treatment or placebo (Bergmans 2001; Beshey 2014; Brun‐Buisson 1989; Camus 2005; Cerra 1992; de Smet 2009; Gastinne 1992; Georges 1994; Koeman 2006; Korinek 1993; Pneumatikos 2002; Pugin 1991; Quinio 1995; Rodriguez‐Roldan 1990; Unertl 1987; Wiener 1995); and seven trials compared the combination of topical and systemic antibiotic prophylaxis versus systemic prophylaxis only (Chaari 2014; Ferrer 1994; Gaussorgues 1991; Hammond 1992; Laggner 1994; Lingnau 1997; Stoutenbeek 1996).
One study had three arms (de Smet 2009): one received the combination of topical and systemic antibiotic prophylaxis, one topical prophylaxis alone, and one arm received no prophylaxis.
Two studies were included in the 'topical SDD plus systemic antibiotic versus systemic antibiotic only' group (Gaussorgues 1991; Laggner 1994), though the use of systemic antibiotics was not explicitly stated in the description of interventions. However, all participants in both arms were treated with systemic antibiotics at admission.
Six studies had more than two arms and were analysed as follows. In two studies (Aerdts 1991; Verwaest 1997), the two control groups were pooled and compared to the treatment group. In Lingnau 1997, the participants in the two treatment arms were summarised and compared with the control arm. In two studies (Koeman 2006; Palomar 1997), one of the two control arms was excluded because the participants received only chlorhexidine and sucralfate, respectively. Another study was a four‐arm factorial design in which we considered only two arms comparing antibiotic prophylaxis versus placebo (Camus 2005). In Chaari 2014, we considered three arms as the experimental group.
Eight studies were conducted in the Netherlands, seven in France, six in Spain, four in Germany, four in the USA, three in the UK, and one each in Egypt, Ireland, Tunisia, South Africa, Austria, Greece, Switzerland, and Belgium. One study was multicentric and was conducted in Europe, Australia, and New Zealand.
Twenty‐two studies (52.4%) did not report the source of funding. Six studies (14.3%) were supported by public institutional grants. Thirteen studies (30.1%) were totally or partially funded by pharmaceutical companies.
Excluded studies
We excluded 33 trials (see Characteristics of excluded studies table). Grounds for exclusion were: only a subgroup of selected patients was included (12 studies); non‐randomised design (two studies); experimental intervention not in the inclusion criteria (two studies); both groups received topical prophylaxis (five studies); control intervention not in the inclusion criteria (four studies); objective of the study was not to assess efficacy and safety of topical prophylaxis on clinical outcomes (one study); unclear description of the interventions being compared (one study); paediatric population (four studies); unpublished study, unable to contact study authors for feedback (one study); and data available only from other published meta‐analyses so it was impossible to retrieve data from the original study (one study).
Risk of bias in included studies
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation
We judged 25 studies (59.5%) as at low risk of bias. No information about the methods of sequence random generation was reported for the other 16 studies which were judged as at unclear risk of bias.
Allocation concealment
We judged 22 studies (52.4%) as at low risk of bias. One study was judged at high risk of bias (Brun‐Buisson 1989). The remaining 18 studies did not report methods of allocation concealment and were therefore judged as at unclear risk of bias.
Blinding
Performance bias
We judged all of the included studies to be at low risk of bias. Twenty (47.6%) studies were open‐label; however, hospitals usually have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. Moreover, all ventilated patients are at high risk for infections independently of antibiotic prophylaxis, so knowledge of it does not change diagnostic workup.
Detection bias
Mortality: We judged all studies to be at low risk of bias.
RTIs: We judged all the double‐blind studies as at low risk of bias. Twenty studies (47.6%) were open‐label. We judged five open‐label studies as at high risk of bias because the diagnosis of RTI was not based on microbiological exams (Blair 1991; Cockerill 1992; Stoutenbeek 2007; Ulrich 1989; Unertl 1987). We judged all the other open‐label studies as at low risk of bias because a standardised set of diagnostic criteria, including results of microbiological exams, was used.
Adverse events (AEs): We judged all studies as at unclear risk of bias because methods for AEs collection were poorly reported, and AEs were heterogeneous.
Incomplete outcome data
We judged four studies as at high risk of bias because they excluded patients from the analyses for reasons that were not stated in the study exclusion criteria.
We judged the remaining 37 studies as at low risk of bias, as there were no dropouts. In 27 of these studies, the number of participants analysed was lower than that of those randomised; the authors of these studies decided to exclude from the analysis those participants who either died too soon after ICU admission, or who were extubated early. We did not judge these studies as at high risk of bias, as antibiotic prophylaxis is usually started on admission, whilst meeting the minimum stay in the ICU inclusion criterion can only be verified after at least 48 hours postrandomisation.
In 14 studies, the number of participants who were randomised and analysed was the same. In these cases, if not otherwise specified, we assumed that either no participants were excluded after randomisation, or that the study authors decided to consider as randomised and analysed only those participants who fulfilled the inclusion criteria after randomisation.
Selective reporting
The protocol was available only for one study (de Smet 2009), which was judged as at low risk of bias as the results for all the outcomes described in the protocol were reported in the final publication. We judged the remaining studies as at unclear risk of bias.
Effects of interventions
We grouped studies in the following comparisons:
topical plus systemic prophylaxis versus no treatment (19 studies); and
topical prophylaxis versus no topical prophylaxis (23 studies).
We further stratified the studies included in the comparison 'topical prophylaxis versus no topical prophylaxis' into two groups:
topical plus systemic prophylaxis versus systemic prophylaxis (7 studies); and
topical prophylaxis versus no treatment (16 studies).
Comparison 1: Topical plus systemic prophylaxis versus placebo or no treatment
See: Table 1
Primary outcomes
1. Overall mortality
We found a significant reduction in overall mortality amongst participants who received topical plus systemic prophylaxis compared to placebo or no treatment (risk ratio (RR) 0.84, 95% confidence interval (CI) 0.73 to 0.96; 18 studies; 5290 participants; high‐certainty evidence; Analysis 1.1).
1.1. Analysis.

Comparison 1: Topical plus systemic prophylaxis versus placebo or no treatment, Outcome 1: Overall mortality
2. Respiratory tract infections
We found a significant reduction in the incidence of respiratory tract infections amongst participants who received topical plus systemic prophylaxis compared to placebo or no treatment (RR 0.43, 95% CI 0.35 to 0.53; 17 studies; 2951 participants; moderate‐certainty evidence; Analysis 1.2).
1.2. Analysis.

Comparison 1: Topical plus systemic prophylaxis versus placebo or no treatment, Outcome 2: Respiratory tract infections
Secondary outcomes
1. Dropouts due to adverse events
We did not find a significant difference in dropouts due to adverse events in participants who received topical plus systemic prophylaxis compared to placebo or no treatment (RR 1.06, 95% CI 0.30 to 3.76; 4 studies; 1287 participants; low‐certainty evidence; Analysis 1.3).
1.3. Analysis.

Comparison 1: Topical plus systemic prophylaxis versus placebo or no treatment, Outcome 3: Dropouts due to adverse events
2. Participants with gastrointestinal or allergic AEs
Gastrointestinal AEs
We did not find a significant difference in participants who received topical plus systemic prophylaxis compared to no treatment (RR 1.08, 95% CI 0.57 to 2.04; 6 studies; 2637 participants; low‐certainty evidence; Analysis 1.4).
1.4. Analysis.

Comparison 1: Topical plus systemic prophylaxis versus placebo or no treatment, Outcome 4: Gastrointestinal adverse events
Allergic AEs
We did not find significant differences between groups (RR 1.49, 95% CI 0.09 to 25.33; 6 studies; 2981 participants; low‐certainty evidence; Analysis 1.5).
1.5. Analysis.

Comparison 1: Topical plus systemic prophylaxis versus placebo or no treatment, Outcome 5: Allergic adverse events
Comparison 2: Topical prophylaxis versus no topical prophylaxis
See: Table 2
Primary outcomes
1. Overall mortality
We did not find a significant difference in overall mortality between participants who received and those who did not receive topical prophylaxis (RR 0.96, 95% CI 0.87 to 1.05; 22 studies, 4213 participants; moderate‐certainty evidence; Analysis 2.1).
2.1. Analysis.

Comparison 2: Topical prophylaxis versus no topical prophylaxis, Outcome 1: Overall mortality
Topical plus systemic prophylaxis versus systemic prophylaxis alone
We did not find a significant difference between groups (RR 0.92, 95% CI 0.72 to 1.18; 7 studies; 939 participants; low‐certainty evidence; Analysis 2.1).
Topical prophylaxis alone versus no treatment
We did not find a significant difference between groups (RR 0.97, 95% CI 0.87 to 1.07; 15 studies; 3274 participants; moderate‐certainty evidence; Analysis 2.1).
2. Respiratory tract infections
We found a significant reduction in the incidence of respiratory tract infections in participants who received topical prophylaxis compared to those who did not receive topical prophylaxis (RR 0.57, 95% CI 0.44 to 0.74; 19 studies; 2698 participants; low‐certainty evidence; Analysis 2.2).
2.2. Analysis.

Comparison 2: Topical prophylaxis versus no topical prophylaxis, Outcome 2: Respiratory tract infections
Topical plus systemic prophylaxis versus systemic prophylaxis alone
We found no significant difference between groups (RR 0.82, 95% CI 0.58 to 1.16; 6 studies; 850 participants; low‐certainty evidence; Analysis 2.2).
Topical prophylaxis alone versus no treatment
We found a significant reduction in the incidence of respiratory tract infections in favour of topical prophylaxis alone (RR 0.50, 95% CI 0.36 to 0.69; 13 studies; 1848 participants; low‐certainty evidence; (Analysis 2.2).
Secondary outcomes
1. Dropouts due to adverse events
We did not find a significant difference in dropouts due to adverse events in participants who received topical prophylaxis compared to those who did not receive topical prophylaxis (RR 2.20, 95% CI 0.57 to 8.54; 7 studies; 1323 participants; very low‐certainty evidence; Analysis 2.3).
2.3. Analysis.

Comparison 2: Topical prophylaxis versus no topical prophylaxis, Outcome 3: Dropouts due to adverse events
2. Participants with gastrointestinal or allergic AEs
Gastrointestinal AEs
We did not find a significant difference in participants who received topical prophylaxis compared to those who did not receive topical prophylaxis (RR 2.78, 95% CI 0.26, to 29.50; 3 studies; 1859 participants; low‐certainty evidence; Analysis 2.4).
2.4. Analysis.

Comparison 2: Topical prophylaxis versus no topical prophylaxis, Outcome 4: Gastrointestinal adverse events
Allergic AEs
We did not find a significant difference between groups (RR 2.64, 95% CI 0.34 to 20.69; 5 studies; 2357 participants; very low‐certainty evidence; Analysis 2.5).
2.5. Analysis.

Comparison 2: Topical prophylaxis versus no topical prophylaxis, Outcome 5: Allergic adverse events
Discussion
Since the introduction of selective digestive decontamination (SDD) as a preventive measure against the development of infections in critically ill patients (Stoutenbeek 1984), its use as an antibiotic prophylaxis has been controversial.
Initial studies aimed at quantifying the effectiveness of SDD in preventing ventilator‐associated pneumonia (VAP) in intensive care units (ICUs) highlighted the difficulty in drawing solid conclusions regarding the effectiveness of the treatment, given the lack of a standardised protocol and the limited numbers of patients included in individual clinical trials (de Jonge 2018; de Smet 2009; Plantinga 2017; Vincent 2011). Concerns were even raised regarding the possible role of the SDD in inducing antimicrobial resistance, as well as the costs associated with its implementation. Furthermore, pneumonia, often considered the target outcome for evaluating efficacy of SDD, can be measured using different clinical, microbiological, and radiological criteria that are often difficult to apply in the ICU setting (Chahoud 2015; Waters 2015). Finally, ICU mortality depends on a number of factors only partially related to VAP.
As those enrolled in SDD trials gradually changed from trauma patients to other patient categories (surgical and medical, with complex medical histories and comorbidity, with prior antibiotic use in the presence of bacteria non‐susceptible to cephalosporin), SDD regimens have varied over time. Endpoints have also changed in some clinical trials from VAP to bacteraemia or ICU mortality (Hurley 2020).
Compared to other published meta‐analyses (Heyland 1994; Hurley 1995; Kollef 1994; Nathens 1999; SDD Group 1993; Vanderbrouk‐Grauls 1991), we decided in our previously published review, D'Amico 2009, to separately analyse trials testing a combination of systemic and topical antibiotics, and those testing topical antibiotics alone. Though there is no consensus on the best way to classify antibiotic prophylaxis regimens, it seemed more appropriate to consider the two groups of trials as distinct approaches to antibiotic prophylaxis. We made this decision a priori, independent of knowing the results.
Summary of main results
We included 41 RCTs with a total of 11,004 participants who were mechanically ventilated for at least 48 hours. Nineteen RCTs compared the combination of topical and systemic antibiotic prophylaxis versus no treatment or placebo; 16 studies compared topical prophylaxis alone to no treatment or placebo; and seven trials compared the combination of topical and systemic antibiotic prophylaxis versus systemic antibiotic only.
Compared to no treatment or placebo, there was a significant reduction in overall mortality (high‐certainty evidence) and a significant reduction of RTIs (moderate‐certainty evidence) in participants receiving topical plus systemic prophylaxis. We also found low‐certainty evidence that participants receiving topical antibiotic prophylaxis achieved a significant reduction of RTI, but moderate‐certainty evidence that mortality did not change, when compared with placebo, no treatment, or systemic prophylaxis alone.
We found low‐certainty evidence of no relevant differences in mortality and RTIs in the subgroup of studies comparing topical plus systemic prophylaxis versus systemic prophylaxis alone. In the subgroup of studies comparing topical prophylaxis alone versus placebo or no treatment, RTIs were significantly reduced (low‐certainty evidence), whilst there was no difference in mortality rate (moderate‐certainty evidence). Although these results could suggest that the systemic component of prophylaxis plays a key role in mortality reduction, caution is advised in interpreting such results on the basis of indirect comparisons. Moreover, as the certainty of the evidence was low, the contribution of adding topical to systemic prophylaxis remains uncertain.
The incidence of adverse events was reported in few studies with inconsistent and uninformative results (low‐ to very low‐certainty evidence).
Overall completeness and applicability of evidence
Overall, the characteristics of participants and ICUs considered in the included studies represent well the actual ICU setting in high‐income countries, although many of the studies were conducted more than 20 years ago. Despite the fact that modalities to prevent the development of infections have changed over time, as well as the characteristics of patients admitted to ICUs, RTIs are still an important cause of mortality.
One limitation of this meta‐analysis is that the patient population, the antibiotic regimens, and the outcome definitions varied across studies. Nevertheless, we believe that it provides the best global picture of the effectiveness of the intervention, despite some recent criticisms on the quality of primary studies and their combination (van Nieuwenhoven 2001), which we feel we have convincingly addressed (Liberati 2001).
Quality of the evidence
Seventy‐three per cent of the included studies were published before the 2001, the year in which the revised CONSORT statement was published and endorsed by many journals. This is the likely explanation for why the reporting of the majority of the studies was poor, which limited our ability to assess risk of bias. We judged more than 40% of the included studies as at unclear risk of selection bias, which led to the downgrading of the certainty of the evidence for the comparison 'topical antibiotic prophylaxis versus no prophylaxis'. In contrast, although about 48% of the studies were open‐label, we judged all studies to be at low risk of performance bias, as hospitals usually have standardised infection control programmes, making subjective decisions on who should be tested for the presence or absence of RTIs unlikely. We judged only five studies as at high risk of detection bias, as they were open‐label and did not use microbiological exams to diagnose RTIs. We judged all studies as at low risk of detection bias for the outcome mortality. We did not find any relevant inconsistency amongst trials. We found asymmetry in the funnel plot indicating possible publication bias for the outcome respiratory tract infection. Finally, we judged evidence for adverse events low or very low due to sparse data
Potential biases in the review process
We performed a comprehensive search without language or publication restrictions. The inspection of funnel plots did not show asymmetry suggestive of possible publication bias. See Figure 4 and Figure 5.
4.

Funnel plot of comparison: 1 Topical plus systemic prophylaxis versus placebo or no treatment, outcome: 1.1 Overall mortality.
5.

Funnel plot of comparison: 2 Topical prophylaxis versus no topical prophylaxis, outcome: 2.1 Overall mortality.
Agreements and disagreements with other studies or reviews
Prior to the publication of the previous version of this systematic review, several non‐Cochrane Reviews and meta‐analyses were published on the effect of SDD on RTIs and mortality (D'Amico 1998; Heyland 1994; Hurley 1995, Kollef 1994; Nathens 1999; Redman 2001; Silvestri 2007; SDD Group 1993; Vanderbrouk‐Grauls 1991). Two systematic reviews were recently published (Price 2014; Righy 2017). All reviews assessing the incidence of RTIs supported the hypothesis that antibiotic prophylaxis is effective, though the magnitude of the treatment effect varied across reviews. The same results were observed in the majority of systematic reviews regarding mortality.
Authors' conclusions
Implications for practice.
Treatments based on topical prophylaxis alone probably reduce respiratory tract infections, but not mortality, in adult patients receiving mechanical ventilation for at least 48 hours, whereas a combination of topical and systemic prophylactic antibiotics reduces both overall mortality and respiratory tract infections. However, it cannot be ruled out that the systemic component of the combined treatment provides a relevant contribution in the observed reduction of mortality. The risk of antimicrobial resistance occurring as a negative consequence of antibiotic use should be explored further using appropriate study designs. No conclusion can be drawn about adverse events as they were poorly reported with sparse data
Implications for research.
The number of randomised controlled trials conducted on antibiotic prophylaxis to date is substantial and provides sufficient evidence to detect a moderate effect of the treatment on mortality. According to this systematic review, the combination of topical and systemic antibiotics should be the standard against which new treatments should be tested. A logical next step for future trials would be the comparison of this protocol against a regimen based on systemic antimicrobials only, as this design was addressed by only seven trials included in this review.
However, it is unlikely that one or more even large conventional trials will mitigate the concerns of those who fear that antimicrobial resistance may occur as a consequence of the widespread use of antibiotics. A more precise definition of the type of drug treatment would also be desirable in order to clarify the possible clinical indications and the risks of resistance. The growing number of publications on this topic has underlined its relevant clinical importance. The lack of appropriately designed studies on microbial resistance leaves much room for future developments, in light of the ever‐increasing complexity of therapies and patients; however, so far there does not seem to be a commercial interest by pharmaceutical companies to support such studies. Similarly, the intensivist community seems rather sceptical about the merits of the intervention and is not willing to embark on new, properly designed and conducted studies. A systematic analysis of the quality and reliability of existing data on antimicrobial resistance might result in a more comprehensive view of the effects of the treatment, especially in particular subgroups of patients.
What's new
| Date | Event | Description |
|---|---|---|
| 5 February 2020 | New search has been performed | Searches conducted 5 February 2020. |
| 5 February 2020 | New citation required but conclusions have not changed | Two review authors have been removed from the byline (Alessandro Liberati and Walter Torri), and three review authors have been added (Silvia Minozzi, Valentina Pecoraro, and Giorgia Montrucchio). We changed the inclusion criteria to include cluster‐randomised controlled trials. We added three outcomes: 'dropouts due to adverse events', 'gastrointestinal adverse events', and 'allergic adverse events'. We included two new trials (Beshey 2014; Chaari 2014), and two previously excluded trials (de la Cal 2005; de Smet 2009). We included one study that was missed in the last update (Koeman 2006). We excluded seven new trials (Camus 2011; Huang 2013; Oostdijk 2014; Oudhuis 2011; Rios 2005; Saidel‐Odes 2012; Wittekamp 2018). We identified two ongoing studies (IRCT20180110038298N1; NCT02389036). |
History
Review first published: Issue 3, 1997
| Date | Event | Description |
|---|---|---|
| 13 March 2009 | New citation required but conclusions have not changed | One study was included in this update (Camus 2005). Two studies whose data were reported in congress proceedings (Lenhart 1994) and were unpublished (Stoutenbeek 2) have been replaced by Krueger 2002 and Stoutenbeek 2007, which are their published versions in peer‐reviewed journals. One study (Jacobs 1995) included in the previous version of this review as a personal contact with the principal investigator, has been excluded due to lack of feedback from the trial author. To date, this study has not been published. |
| 13 March 2009 | New search has been performed | Searches conducted. |
| 30 January 2008 | Amended | Converted to new review format |
| 5 September 2003 | New search has been performed | Searches conducted. Updated review published Issue 4, 2002. |
| 5 December 1999 | New search has been performed | Searches conducted. Review published Issue 3, 1997. |
| 5 December 1995 | New search has been performed | Searches conducted. Updated review published Issue 1, 2004. |
Acknowledgements
We are greatly indebted to Alessandro Liberati, who inspired us to carry on this systematic review and to publish it in the Cochrane Library. Alessandro Liberati and Walter Torri are previous authors of this review.
We thank Tim Kenealy, Viviana Rodriguez Romero, Janet Wale, and Tom Fahey for commenting on the draft of this review update.
Appendices
Appendix 1. Cochrane Library search strategy
#1 (“critical care” OR “intensive care” OR “burn unit” OR “burn units” OR “care unit” OR “care units” OR “recovery room” OR “recovery rooms” OR "critical illness" OR "mechanical ventilation" OR ventilator* OR "artificial respiration" OR respirator*):ti,ab 77406
#2 ("respiratory tract infection" OR "respiratory tract infections" OR pneumon* OR HAP OR VAP OR bronchopneumonia* OR pleuropneumonia* OR pharyngit* OR tracheit*):ti,ab 17417
#3 #1 and #2 8366
#4 'ventilator associated pneumonia':ti,ab 1410
#5 #3 or #4 8383
#6 antibiotic*:ti,ab 26080
#7 #5 and #6 1746
#8 "accession number" near pubmed 664007
#9 "accession number" near embase 538901
#10 #8 or #9 1001070
#11 #7 not #10 with Publication Year from 2012 to 2020, in Trials
Appendix 2. MEDLINE (PubMed) search strategy
#1 Search: "Intensive Care Units"[Mesh]) OR “critical care”[Title/Abstract] OR “intensive care”[Title/Abstract] OR “burn unit”[Title/Abstract] OR “burn units”[Title/Abstract] OR “care unit”[Title/Abstract] OR “care units”[Title/Abstract] OR “recovery room”[Title/Abstract] OR “recovery rooms”[Title/Abstract] OR "Critical Illness"[Mesh]) OR "critical illness"[Title/Abstract]) OR "Ventilators, Mechanical"[Mesh]) OR "mechanical ventilation”[Title/Abstract] OR ventilator*[Title/Abstract] OR "Respiration, Artificial"[Mesh]) OR "artificial respiration"[Title/Abstract] OR respirator*[Title/Abstract]
#2 Search: "Respiratory Tract Infections"[Mesh]) OR "respiratory tract infection"[Title/Abstract] OR "respiratory tract infections"[Title/Abstract] OR "Pneumonia"[Mesh]) OR pneumon*[Title/Abstract] OR HAP[Title/Abstract] OR VAP[Title/Abstract] OR bronchopneumonia*[Title/Abstract] OR pleuropneumonia*[Title/Abstract] OR "Pharyngitis"[Mesh]) OR pharyngit*[Title/Abstract] OR tracheit*[Title/Abstract]
#3 Search: #1 AND #2
#4 Search: "Pneumonia, Ventilator‐Associated"[Mesh]
#5 Search: #3 OR #4
#6 Search: "Antibiotic Prophylaxis"[Mesh] OR "Anti‐Bacterial Agents"[Mesh] OR antibiotic*[Title/Abstract])
#7 Search: #5 AND #6
#8 Search: #7 AND ("random*"[Title/Abstract] OR "placebo"[Title/Abstract] OR "trial*"[Title] OR "Randomized Controlled Trial"[Publication Type] OR "Controlled Clinical Trial"[Publication Type])
#9 Search: ("2012/01/01"[Date ‐ Entry]: "2020/05/21"[Date ‐ Entry]
#10 Search: #8 AND #9
Appendix 3. Embase (Elsevier) search strategy
| No. | Query |
| #23 | #20 AND #21 AND [1‐1‐2012]/sd NOT [21‐5‐2020]/sd |
| #22 | #20 AND #21 |
| #21 | [embase]/lim NOT [medline]/lim |
| #20 | #16 AND #19 |
| #19 | #17 OR #18 |
| #18 | random*:ti,ab OR placebo*:ti,ab OR factorial*:ti,ab OR crossover*:ti,ab OR assign*:ti,ab OR allocat*:ti,ab OR volunteer*:ti,ab OR 'double blind':ti,ab OR 'double blinding':ti,ab OR 'double blinded':ti,ab OR 'single blind':ti,ab OR 'single blinded':ti,ab OR 'single blinding':ti,ab |
| #17 | 'crossover procedure'/exp OR 'single blind procedure'/exp OR 'randomized controlled trial'/exp OR 'controlled clinical trial'/exp |
| #16 | #14 AND #15 |
| #15 | 'antibiotic prophylaxis'/exp OR 'antibiotic agent'/exp OR antibiotic*:ti,ab |
| #14 | #12 OR #13 |
| #13 | 'ventilator associated pneumonia'/exp OR 'ventilator associated pneumonia':ti,ab |
| #12 | #6 AND #11 |
| #11 | #7 OR #8 OR #9 OR #10 |
| #10 | 'artificial ventilation'/exp OR respirator*:ti,ab |
| #9 | 'ventilator'/exp OR ventilator*:ti,ab |
| #8 | 'critical illness'/exp OR 'critically ill':ti,ab OR 'critical illness':ti,ab |
| #7 | 'intensive care unit'/exp OR icu:ti,ab OR 'critical care':ti,ab OR 'intensive care':ti,ab OR 'burn unit':ti,ab OR 'burn units':ti,ab OR 'care unit':ti,ab OR 'care units':ti,ab OR 'recovery room':ti,ab OR 'recovery rooms':ti,ab |
| #6 | #1 OR #2 OR #3 OR #4 OR #5 |
| #5 | 'tracheitis'/exp OR tracheit*:ti,ab |
| #4 | bronchit*:ti,ab OR bronchiolit*:ti,ab OR pharyngit*:ti,ab |
| #3 | pneumon*:ti,ab OR hap:ti,ab OR vap:ti,ab OR bronchopneumonia*:ti,ab OR pleuropneumonia*:ti,ab |
| #2 | 'respiratory tract infection':ti,ab OR 'respiratory tract infections':ti,ab |
| #1 | 'respiratory tract infection'/exp OR 'bronchitis'/exp OR 'pharyngitis'/exp OR 'pneumonia'/exp |
Appendix 4. Details of previous searches
MEDLINE was searched using the following search strategy in conjunction with the Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE: sensitivity‐ and precision‐maximising version (2008 revision); Ovid format (Lefebvre 2011). The same strategy was used to search CENTRAL and adapted to search EMBASE.com. There were no language or publication restrictions.
MEDLINE (Ovid) search strategy
1 exp Respiratory Tract Infections/ 2 respiratory tract infection*.tw. 3 exp Pneumonia/ 4 pneumon*.tw. 5 (HAP or VAP).tw. 6 bronchopneumonia*.tw. 7 pleuropneumonia*.tw. 8 exp Bronchitis/ 9 bronchit*.tw. 10 bronchiolit*.tw. 11 exp Pharyngitis/ 12 pharyngit*.tw. 13 Tracheitis/ 14 tracheit*.tw. 15 or/1‐14 16 exp Intensive Care Units/ 17 icu.tw. 18 exp Critical Care/ 19 critical care.tw. 20 intensive care.tw. 21 burn unit*.tw. 22 care unit*.tw. 23 recovery room*.tw. 24 Critical Illness/ 25 (critic* adj ill*).tw. 26 exp Ventilators, Mechanical/ 27 mechanical ventilat*.tw. 28 ventilator*.tw. 29 Respiration, Artificial/ 30 artificial respiration*.tw. 31 respirator*.tw. 32 or/16‐31 33 15 and 32 34 Pneumonia, Ventilator‐Associated/ 35 33 or 34 36 Antibiotic Prophylaxis/ 37 exp Anti‐Bacterial Agents/ 38 antibiotic*.tw. 39 or/36‐38 40 35 and 39
EMBASE (Elsevier) search strategy
1. 'respiratory tract infection'/exp 2. 'respiratory tract infection':ti,ab OR 'respiratory tract infections':ti,ab 3. 'pneumonia'/exp 4. pneumon*:ti,ab 5. hap:ti,ab OR vap:ti,ab 6. bronchopneumonia*:ti,ab OR pleuropneumonia*:ti,ab 7. 'bronchitis'/exp 8. bronchit*:ti,ab OR bronchiolit*:ti,ab 9. 'pharyngitis'/exp 10. pharyngit*:ti,ab 11. 'tracheitis'/exp 12. tracheit*:ti,ab 13. #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 14. 'intensive care unit'/exp 15. icu:ti,ab OR 'critical care':ti,ab OR 'intensive care':ti,ab OR 'burn unit':ti,ab OR 'burn units':ti,ab OR 'care unit':ti,ab OR 'care units':ti,ab OR 'recovery room':ti,ab OR 'recovery rooms':ti,ab 16. 'critical illness'/exp 17. 'critically ill':ti,ab OR 'critical illness':ti,ab 18. 'ventilator'/exp 19. ventilator*:ti,ab 20. 'artificial ventilation'/exp 21. respirator*:ti,ab 22. #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR # 21 23. #13 AND #22 24. 'ventilator associated pneumonia'/exp 25. 'ventilator associated pneumonia':ti,ab 26. #24 OR #25 27. #23 OR #26 28. 'antibiotic prophylaxis'/exp 29. 'antibiotic agent'/exp 30. antibiotic*:ti,ab 31. #28 OR #29 OR #30 32. #27 AND #31 33. 'randomized controlled trial'/exp 34. 'controlled clinical trial'/exp 35. 'single blind procedure'/exp 36. 'crossover procedure'/exp 37. random*:ti,ab OR placebo*:ti,ab OR factorial*:ti,ab OR crossover*:ti,ab OR assign*:ti,ab OR allocat*:ti,ab OR volunteer*:ti,ab OR 'double blind':ti,ab OR 'double blinding':ti,ab OR 'double blinded':ti,ab OR 'single blind':ti,ab OR 'single blinded':ti,ab OR 'single blinding':ti,ab 38. #33 OR #34 OR #35 OR #36 OR #37 39. #32 AND #38
Appendix 5. Criteria for 'Risk of bias' assessment
| Item | Judgement | Description |
| Random sequence generation (selection bias) | Low risk | The investigators describe a random component in the sequence generation process such as: random number table; computer random number generator; coin tossing; shuffling cards or envelopes; throwing dice; drawing of lots; minimisation. |
| High risk | The investigators describe a non‐random component in the sequence generation process such as: odd or even date of birth; date (or day) of admission; hospital or clinic record number; alternation; judgement of the clinician; results of a laboratory test or a series of tests; availability of the intervention. | |
| Unclear risk | Insufficient information about the sequence generation process to permit judgement of low or high risk | |
| Allocation concealment (selection bias) | Low risk | Investigators enrolling participants could not foresee assignment because one of the following, or an equivalent method, was used to conceal allocation: central allocation (including telephone, web‐based, and pharmacy‐controlled randomisation); sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes. |
| High risk | Investigators enrolling participants could possibly have foreseen assignments because one of the following methods was used: open random allocation schedule (e.g. a list of random numbers); assignment envelopes without appropriate safeguards (e.g. if envelopes were unsealed or nonopaque or not sequentially numbered); alternation or rotation; date of birth; case record number; any other explicitly unconcealed procedure. | |
| Unclear risk | Insufficient information to permit a judgement of low or high risk. This is usually the case if the method of concealment is not described or not described in sufficient detail to permit a definitive judgement. | |
| Blinding of participants and providers (performance bias) | Low risk | No blinding or incomplete blinding, but the review authors judge that the outcome is not likely to be influenced by lack of blinding. Blinding of participants and key study personnel ensured, and it is unlikely that the blinding could have been broken. |
| High risk | No blinding or incomplete blinding, and the outcome is likely to be influenced by lack of blinding. Blinding of key study participants and personnel was attempted, but it is likely that the blinding could have been broken, and the outcome is likely to be influenced by lack of blinding. |
|
| Unclear risk | Insufficient information to permit a judgement of low or high risk | |
| Blinding of outcome assessor (detection bias) | Low risk | No blinding of outcome assessment, but the review authors judge that the outcome measurement is not likely to be influenced by lack of blinding. Blinding of outcome assessment is ensured, and it is unlikely that the blinding could have been broken. |
| High risk | No blinding of outcome assessment, and the outcome measurement is likely to be influenced by lack of blinding. Blinding of outcome assessment, but it is likely that the blinding could have been broken, and the outcome measurement is likely to be influenced by lack of blinding. |
|
| Unclear risk | Insufficient information to permit a judgement of low or high risk | |
| Incomplete outcome data (attrition bias) | Low risk | No missing outcome data. Reasons for missing outcome data are unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias). Missing outcome data are balanced in numbers across intervention groups, with similar reasons for missing data across groups. For dichotomous outcome data, the proportion of missing outcomes compared with observed event risk is not enough to have a clinically relevant impact on the intervention effect estimate. For continuous outcome data, plausible effect size (difference in means or standardised difference in means) amongst missing outcomes is not enough to have a clinically relevant impact on observed effect size. Missing data have been imputed using appropriate methods. All randomised participants are reported/analysed in the group to which they were allocated by randomisation irrespective of non‐compliance and co‐interventions (intention‐to‐treat). |
| High risk | Reason for missing outcome data is likely to be related to true outcome, with either an imbalance in numbers or reasons for missing data across intervention groups. For dichotomous outcome data, the proportion of missing outcomes compared with observed event risk is enough to induce clinically relevant bias in intervention effect estimate. For continuous outcome data, plausible effect size (difference in means or standardised difference in means) amongst missing outcomes is enough to induce clinically relevant bias in observed effect size. ‘As‐treated’ analysis done with substantial departure of the intervention received from that assigned at randomisation. |
|
| Unclear risk | Insufficient information to permit a judgement of low or high risk (e.g. number randomised not stated, no reasons for missing data provided; number of dropouts not reported for each group) | |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. The study protocol is not available, but it is clear that the published reports include all expected outcomes, including those that were prespecified (convincing text of this nature may be uncommon). |
| High risk | Not all of the study’s prespecified primary outcomes have been reported. One or more primary outcomes are reported using measurements, analysis methods, or subsets of the data (e.g. subscales) that were not prespecified. One or more reported primary outcomes were not prespecified (unless clear justification for their reporting is provided, such as an unexpected adverse effect). One or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis. The study report fails to include results for a key outcome that would be expected to have been reported for such a study. |
|
| Unclear risk | Insufficient information to permit a judgement of low or high risk |
Data and analyses
Comparison 1. Topical plus systemic prophylaxis versus placebo or no treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Overall mortality | 18 | 5290 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.73, 0.96] |
| 1.2 Respiratory tract infections | 17 | 2951 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.35, 0.53] |
| 1.3 Dropouts due to adverse events | 4 | 1287 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.30, 3.76] |
| 1.4 Gastrointestinal adverse events | 6 | 2637 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.57, 2.04] |
| 1.5 Allergic adverse events | 6 | 2981 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [0.09, 25.33] |
Comparison 2. Topical prophylaxis versus no topical prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Overall mortality | 22 | 4213 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.87, 1.05] |
| 2.1.1 Topical plus systemic prophylaxis versus systemic prophylaxis alone | 7 | 939 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.72, 1.18] |
| 2.1.2 Topical prophylaxis alone versus no treatment | 15 | 3274 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.87, 1.07] |
| 2.2 Respiratory tract infections | 19 | 2698 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.44, 0.74] |
| 2.2.1 Topical plus systemic prophylaxis versus systemic prophylaxis alone | 6 | 850 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.58, 1.16] |
| 2.2.2 Topical prophylaxis alone versus no treatment | 13 | 1848 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.36, 0.69] |
| 2.3 Dropouts due to adverse events | 7 | 1323 | Risk Ratio (M‐H, Random, 95% CI) | 2.20 [0.57, 8.54] |
| 2.4 Gastrointestinal adverse events | 3 | 1859 | Risk Ratio (M‐H, Random, 95% CI) | 2.78 [0.26, 29.50] |
| 2.5 Allergic adverse events | 5 | 2357 | Risk Ratio (M‐H, Random, 95% CI) | 2.64 [0.34, 20.69] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Abele‐Horn 1997.
| Study characteristics | ||
| Methods | Randomised controlled trial
Blinding: open‐label
Randomisation method: list block randomised assignments maintained by the main investigator
Accrual period: not available Country: Germany |
|
| Participants | Eligibility criteria: intubation within 24 hours of admission, expected ventilation for at least 4 days, first microbial culture within 36 hours of admission
Exclusion criteria: transfer from other hospitals, evidence of infection, prior antibiotic therapy, ARDS, leucopenia, myelosuppression
Number of participants enrolled: 125 Number of participants randomised: not reported Number of patients excluded: 37 because they did not fulfil the inclusion criteria or protocol violation (12 study, 7 control) Number of participants analysed: 88 Percentage of ventilated participants: 100% Length of stay in ICU, mean days (SD): treatment group 18 (7.8), control group 22 (8.8) Diagnosis at admission: surgical unscheduled = 14 (16%); trauma = 29 (84%) Severity score on admission: APACHE II mean = 17; ISS not available Percentage of immunocompromised participants: not available Percentage of participants treated with systemic antibiotic therapy (not stated in protocol) in the first 3 days: not available Stress ulcer prophylaxis applied: sucralfate 1 g x 4 to all participants |
|
| Interventions | Treatment group, randomised = not reported, analysed = 58:
Control group, randomised = not reported, analysed = 30:
Antibiotic prophylaxis was performed only for abdominal, orthopaedic, and neurologic surgery. |
|
| Outcomes | Respiratory infections (acquired pneumonia). Diagnosis was based on Clinical Pulmonary Infection Score as defined by Pugin 1991: new pulmonary infiltrate on X‐ray, increasing amount of tracheal secretions containing > 3 x 104 granulocytes/µL and at least 2 of the following: temperature > 38.5 °C, WBC > 12,000/mm3 or < 4000/mm3, decrease in PaO2 requiring an increase in FiO2. Besides a bacteriological confirmation is required: tracheal aspirates yielding bacteria > 104 CFU/mL and granulocytes > 10/field Mortality: in ICU |
|
| Notes | Data for 37 excluded patients are not available. Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed on the basis of a sequential list of block‐randomised assignments maintained by the principal investigator. |
| Allocation concealment (selection bias) | Low risk | Randomisation was performed on the basis of a sequential list of block‐randomised assignments maintained by the principal investigator. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Objective outcome unlikely to be biased by lack of blinding |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | The chest X‐ray was read by an independent radiologist who was unaware of the participant’s randomisation. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 19 (15%) participants excluded for protocol violation (12 experimental, 7 control). |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Aerdts 1991.
| Study characteristics | ||
| Methods | Randomised study with 3 arms (1 treatment arm versus 2 control arms)
Blinding: open‐label
Randomisation method: sealed envelopes, permuted block method
Accrual period: from May 1986 to September 1987 Country: the Netherlands |
|
| Participants | Eligibility criteria: expected ventilation for at least 5 days, inclusion within 24 hours of admission
Exclusion criteria: age < 16 years, pregnancy, allergy to 1 of the components of the regimen
Number of participants enrolled: 88 Number of participants randomised: 88 Number of patients excluded: 32 because they did not receive mechanical ventilation for at least 5 days or for other reasons Number of participants analysed: 56 Percentage of ventilated participants: 100% Length of stay in ICU, mean days (SD): treatment group 23 (13), control group A 30 (17), control group B 25 (11) Diagnosis at admission: medical = 14 (25%), surgical scheduled = 5 (9%), surgical unscheduled = 11 (20%), trauma = 23 (41%) Severity score on admission: APACHE II mean = 21.8, ISS not available Percentage of immunocompromised participants: 4.6% Percentage of participants treated with systemic antibiotic therapy (not stated in protocol) in the first 3 days: treatment = 35%, CTR = 80% Stress ulcer prophylaxis applied: antiacids until enteral feeding was possible |
|
| Interventions | Treatment group, randomised = 28, analysed = 17:
Infections of unknown origin were treated with cefotaxime + gentamicin and metronidazole if indicated. Control group A, randomised = 30, analysed = 18:
Infections of unknown origin were treated with ampicillin + gentamicin Control group B, randomised = 30, analysed = 21:
Infections of unknown origin were treated with cefotaxime + gentamicin and metronidazole if indicated. |
|
| Outcomes | Respiratory infections (acquired pneumonia and tracheobronchitis). Diagnosis of tracheobronchitis was based on positive culture of the tracheal aspirate and a Gram stain showing many leukocytes as well as the causative organism, associated with 2 of the following: temperature > 38 °C, WBC > 12,000/mm3, purulent tracheal aspirate. Diagnosis of pneumonia was based on a new and persistent pulmonary infiltrate on X‐ray and criteria of tracheobronchitis. Mortality: in ICU |
|
| Notes | Participants enrolled in both control groups were analysed together in the meta‐analyses. Conflict of interest: not reported Funding: partially supported by an educational grant from Roussel by, Iloevelaken |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The severity of acute illness was assessed using the APACHE II score. Participants were stratified for APACHE II score: less than 10, 10 to 19, 20 to 29, and 30 or more. Within these strata, participants were randomly allocated to 1 of the treatment groups by means of the permuted block method. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by the lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | A standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 5 and 3 (9%) participants from the control and experimental groups, respectively, were excluded for reasons other than the inclusion criteria. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Bergmans 2001.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind Randomisation method: randomisation was conducted per hospital Accrual period: from September 1994 to December 1996 Country: the Netherlands |
|
| Participants | Eligibility criteria: intubation within 24 hours of admission and need for mechanical ventilation with an expected duration > 2 days
Exclusion criteria: age < 16 years
Number of participants enrolled: 270 Number of participants randomised: 245 Number of patients excluded: 19 Number of participants analysed: 226 Percentage of ventilated participants: 100% Length of stay in ICU, median (range): treatment group 13 (4 to 54), control group A 15 (4 to 79), control group B 12 (4 to 108) Diagnosis at admission: medical = 78 (35%), surgery = 88 (39%), trauma = 43 (19%), neurology = 14 (6%), other = 3 (1%) Severity score on admission: APACHE II mean = 21.4, ISS not available Percentage of immunocompromised participants: 2% Percentage of participants treated with systematic antibiotic therapy at admission: treatment: 47%, control 42% Stress ulcer prophylaxis applied: treatment = 61%, control = 76% |
|
| Interventions | Treatment group, randomised = 92, analysed = 87. Orabase with 2% gentamicin, 2% colistin, and 2% vancomycin. Orabase was applied in the buccal cavities on a gloved finger every 6 hours. The application of Orabase was started within 24 hours of intubation. Application of treatment was limited to 21 days. Control group A, randomised = 82, analysed = 78: no prophylaxis. This group was studied in ICU in which there were patients receiving topical antimicrobial prophylaxis. Control group B, randomised = 70, analysed = 61: no prophylaxis. This group was studied in ICU in which no topical antimicrobial prophylaxis was used. |
|
| Outcomes | Ventilator‐associated pneumonia: diagnosis of VAP was established on the basis of positive quantitative cultures from BAL (cut‐off point >= 104 CFU/mL) or protected specimen brush (cut‐off point >= 103 CFU/mL), or a positive blood culture unrelated to another source of infection, or a positive culture from pleural fluid in the absence of previous pleural instrumentation. Mortality: in hospital |
|
| Notes | Conflict of interest: not available Funding: supported by Grant 28‐2125‐1 from the Praevention Foundation and a grant from Eli Lilly Netherland bv. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was conducted per hospital and was executed by the Department of Clinical Pharmacy of the University Hospital Maastricht. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. No information about blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 19 excluded because they did not fulfil inclusion criteria. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Beshey 2014.
| Study characteristics | ||
| Methods | Prospective, randomised, placebo‐controlled trial Blinding: open‐label Randomisation method: not available Accrual period: not reported Country: Egypt |
|
| Participants | Eligibility criteria: patients mechanically ventilated for at least 48 hours with an expectation to remain so for at least an additional 72 hours, based on admitting diagnosis, magnitude of haemodynamic instability, respiratory failure, and baseline medical condition and severity of illness according to APACHE II score
Exclusion criteria: pregnancy, receipt of antifungal agents within 7 days before ICU admission, age younger than 18, an expectation that the patient would not survive more than 24 hours, and patients who did not complete the 15‐day period of the study either due to discharge from ICU or death Number of participants enrolled: not reported Number of participants randomised: 75 Number of patients excluded: none Number of participants analysed: 75 Percentage of ventilated participants: not reported Length of stay in ICU: not reported Diagnosis at admission: not reported Severity score at admission: not reported Percentage of immunocompromised participants: not reported Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: not reported Stress ulcer prophylaxis applied: not reported |
|
| Interventions | Treatment group 1, randomised = 25, analysed = 25: participants who received SDD alone without fluconazole
Treatment group 2, randomised = 2, analysed = 25: participants who received fluconazole as a part of their SDD Control group, analysed = 25, randomised = 25: placebo |
|
| Outcomes | Mortality at 15 days and number of positive Candida cultures | |
| Notes | Conflict of interest: none Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Blair 1991.
| Study characteristics | ||
| Methods | Randomised study
Blinding: open‐label
Randomisation method: sealed envelopes
Accrual period: from September 1988 to January 1990 Country: Ireland |
|
| Participants | Eligibility criteria: all admitted patients who do not fulfil the exclusion criteria
Exclusion criteria: patients discharged within 48 hours of ICU admission; admission from cardiac care unit; patients expected to die after 6 hours of ICU admission; patients with discharge anticipated within 48 hours but remaining more than 48 hours; drug overdose; security patients; age < 18 years; patients not randomised within 6 hours of admission; readmission to ICU; burns; miscellaneous Number of participants enrolled: 569 Number of participants randomised: 331 Number of patients excluded: 75 because length of stay was less than 48 hours Number of participants analysed: 256 Percentage of ventilated participants: 93% Length of stay in ICU, median days: 5 Diagnosis at admission: medical = 49 (14%), surgical scheduled = 109 (33%), surgical unscheduled = 43 (13%), trauma = 132 (40%) Severity score on admission: APACHE II mean = 14.4, ISS mean = 24.8 Percentage of immunocompromised participants: 1.8% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment 42%, control 74% Stress ulcer prophylaxis applied: all participants received ranitidine iv plus antiacid therapy if gastric pH was low |
|
| Interventions | Treatment group, randomised = 161, analysed = 126:
Control group, randomised = 170, analysed: 130:
|
|
| Outcomes | Respiratory infections (pneumonia acquired after 48 hours). Diagnosis of infection was based on the fulfilment of Criteria I or Criteria II:
Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: supported in part by Roussel Laboratories and Gelsdich Ltd |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | High risk | No blinding. The diagnosis of RTI was not based on microbiological exams. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 75 excluded because length of stay was less than 48 hours. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Boland 1991.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: computer‐generated randomisation directed by the pharmacy department
Accrual period: from April 1989 to March 1991 Country: the USA |
|
| Participants | Eligibility criteria: all multiple traumatised patients, intubated at the time of admission and likely to stay intubated at least 5 days
Exclusion criteria: patients who did not remain intubated for 5 days Number of participants enrolled: 62 Number of participants randomised: 62 Number of patients excluded: 32 because they did not remain intubated for 5 days Number of participants analysed: 30 Percentage of ventilated participants: 100% Length of stay in ICU, median: 8 days Diagnosis at admission: trauma = 100% Severity score on admission: APACHE II mean = 16.8, ISS not available Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 0%, control = 0% Stress ulcer prophylaxis applied: H2‐blockers or sucralfate (78%) |
|
| Interventions | Treatment group, randomised = 32, analysed = 15:
Control group, randomised = 32, analysed = 15:
|
|
| Outcomes | Respiratory infections (acquired pneumonia and tracheobronchitis). Diagnosis of infection was based on positive sputum culture for bacteria, fever > 38 °C, and leukocytosis (> 10,000 WBC/mm3 of blood). Mortality: in ICU |
|
| Notes | Personal contact with the main investigator provided data for 23 participants who were excluded from the published paper (20 early extubations, 3 early deaths); these data are considered in the analysis. Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation directed by the pharmacy department |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Stated as double‐blind study, no information provided on blinding of outcome assessor. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Stated as double‐blind study, no information provided on blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 32 excluded because they did not remain intubated for 5 days. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Brun‐Buisson 1989.
| Study characteristics | ||
| Methods | Randomised trial
Blinding: open‐label
Randomisation method: odd and even birth year technique
Accrual period: from April 1987 to May 1987 Country: France |
|
| Participants | Eligibility criteria: patients with an admission SAPS > 2 and staying in the ICU more than 48 hours
Exclusion criteria: patients with severe neutropenia routinely receiving oral antibiotic prophylaxis
Number of participants enrolled: 123 Number of participants randomised: 123 Number of patients excluded: 47 because they did not match the inclusion criteria Number of participants analysed: 86 Percentage of ventilated participants: 59% Length of stay in ICU, median days: 3.5 Diagnosis at admission: medical 75%, surgical unscheduled 23%, trauma 2% Severity score on admission: SAPS mean = 10.4, ISS not available Percentage of immunocompromised participants: 12.8% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 41%, control = 53% Stress ulcer prophylaxis applied: none |
|
| Interventions | Treatment group, randomised = 65, analysed = 36:
Control group, randomised = 68, analysed = 50:
|
|
| Outcomes | Respiratory infections (pneumonia acquired in the ICU or within 48 hours from discharge). Diagnosis of infection was based on purulent sputum or tracheal aspirate associated with a new and persistent pulmonary infiltrate on X‐ray and the culture of at least 109 CFU/L from a protected wedged catheter sample of bronchial aspirate, temperature > 38 °C, WBC > 10 x 109. Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | It is reported as "[...] newly admitted patients were allocated, on odd and even birth year to receive either no prophylaxis (group 2) or intestinal decontamination (group 3)". |
| Allocation concealment (selection bias) | High risk | It is reported as "[...] newly admitted patients were allocated, on odd and even birth year to receive either no prophylaxis (group 2) or intestinal decontamination (group 3)". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding, No information provided on blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 47 excluded because they did not match the inclusion criteria. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Camus 2005.
| Study characteristics | ||
| Methods | Randomised trial
Blinding: double‐blind
Randomisation method: performed by computer
Accrual period: from April 1996 to October 1998 Country: France |
|
| Participants | Elegibility criteria: adults intubated for < 48 hours and likely to require mechanical ventilation for > 48 hours
Exclusion criteria: patients with SAPS II > 80 and life expectancy of < 48 hours resulting from brain death of a palliative treatment, a polymorphonuclear count of < 500 cells/mm3, severe diarrhoea, and anyone who had received either a prior decontamination regimen or was already participating in another ongoing clinical trial
Number of participants enrolled: 4444 Number of participants randomised: 516 Number of patients excluded: 1 Number of participants analysed: 515 Of these 515 participants, 130 received polymyxin/tobramycin alone, 130 mupirocin/chlorhexidine alone, 129 both regimens, and 126 placebo. Pecentage of ventilated participants: not reported Length of stay in ICU: not reported Type of admission diagnosis: home/emergency department = 43%, hospital ward = 57% Severity score on admission: median SAPS II = 46 Percentage of immunocompromised participants: 3.9% Percentage of participants treated with systemic antibiotic therapy (not stated in protocol) in the first 3 days: none Stress ulcer prophylaxis applied: not reported |
|
| Interventions | Treatment group, randomised = 130, analysed = 130: solution containing 15 mg/mL polymyxin E and 10 mg/mL tobramycin Control group, randomised = 127, analysed = 126: placebo | |
| Outcomes | Respiratory infections: acquired infections Mortality: in ICU | |
| Notes | The study is a 4‐arm, 2 x 2 factorial design. In this review we considered only the 2 arms comparing SDD regimen versus placebo. Conflict of interest: all the authors state that they have no financial interest to disclose. Funding: supported in part by the Programme Hospitalier de Recherche Clinique (Clinical Research Hospital Program) 94, Direction des Hôpitaux, Paris, France, and by a grant from GlaxoSmithKline. Some study drugs were provided by Astra‐Zeneca. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation by a computer‐generated list |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Stated as double‐blind; no information on blinding of outcome assessor. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Unclear risk | Stated as double‐blind study, no information provided on blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 1 patient was excluded. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Cerra 1992.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: sealed envelopes
Accrual period: from September 1988 to January 1990 Country: the USA |
|
| Participants | Eligibility criteria: admission within 48 hours from surgery, trauma or other acute event, expected stay at least 5 days, hypermetabolisms (VO2 > 140 mL/m2 or urinary nitrogen excretion > 10 g/day) without progressive MOSF (normal transaminases, stable bilirubin and creatinine)
Exclusion criteria: cirrhosis, allergy to used drugs, chemo‐radiotherapy, progressive MOSF, gastrointestinal leak or fistula Number of participants enrolled: 48 Number of participants randomised: 48 Number of patients excluded: 2 because they stayed in the ICU less than 5 days Number of participants analysed: 46 Percentage of ventilated participants: not reported Lenght of stay in ICU: not reported Diagnosis at admission: surgical = 46 (96%), trauma = 2 (4%) Severity score on admission: not reported Percentage of immunocompromised participants: not reported Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: not reported Stress ulcer prophylaxis applied: not reported |
|
| Interventions | Treatment group, randomised = 25, analysed = 25: norfloxacin 500 mg x 3, nystatin 1 million units x 4 applied enterally until discharge Control group, randomised = 23, analysed 21: placebo |
|
| Outcomes | Respiratory infections: not possible to evaluate Mortality |
|
| Notes | Conflict of interest: not reported Funding: this study was supported by Public Health Service grant AI23484 and DK34931 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐derived random number generator |
| Allocation concealment (selection bias) | Low risk | The research pharmacist randomised the participants, then logged in the patient and dispensed the experimental drug or placebo sealed envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind study; no information provided on blinding of outcome assessor. However, outcome is unlikely to be biased by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Stated as double‐blind study, no information provided on blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 excluded because they stayed in the ICU less than 5 days. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Chaari 2014.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: not described
Accrual period: from August 2011 to November 2011 Country: Tunisia |
|
| Participants | Eligibility criteria: age > 15 years, need for MV within the few hours after trauma, and predicted duration of MV > 48 hours
Exclusion criteria: patients who received prior antibiotics for 48 hours, those who are known to be allergic to beta‐lactams, and those with a contraindication of gastric tube were excluded Number of participants enrolled: 61 Number of participants randomised: 44 Number of patients excluded: none Number of participants analysed: 44 Percentage of ventilated participants: 100% Lenght of stay in ICU: not reported Diagnosis at admission: trauma = 44 (100%). Severity score on admission: SAPS II mean 37 (13) Percentage of immunocompromised participants: not reported Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: not reported Stress ulcer prophylaxis applied: omeprazole |
|
| Interventions | Treatment group, randomised = not reported, analysed = 31: systemic antibiotic therapy (cefotaxime with a dose of 1 g 3 times a day for 4 consecutive days) in association with:
The suspension was administered orally (10 mL) for oropharyngeal decontamination and through a gastric tube (20 mL) for gastric decontamination. Control group, randomised = not reported, analysed = 13: systemic antibiotic prophylaxis plus subglottic and gastric placebo |
|
| Outcomes | Ventilator‐associated pneumonia: the diagnosis was considered if the chest X‐ray showed evidence of new and persistent (> 48 hours) pulmonary infiltrates with at least 2 of the following criteria: fever 38 °C or hypothermia, 35 °C, WBC 12,000 or 4000/mm3, purulent respiratory secretion, and/or worsening of respiratory insufficiency Mortality Adverse events |
|
| Notes | Conflict of interest: the authors have no conflicts of interest to declare Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was conducted per hospital and executed by the Department of Clinical Pharmacy of the Habib Bourguiba University Hospital. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | It is reported that: "only the study supervisor and the hospital pharmacist were aware of this inclusion scheme". |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | It is reported that: "only the study supervisor and the hospital pharmacist were aware of this inclusion scheme". |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Cockerill 1992.
| Study characteristics | ||
| Methods | Randomised study: intention‐to‐treat
Blinding: open‐label
Randomisation method: randomisation table at a remote site in the pharmacy
Accrual period: from 1986 to 1989 Country: the USA |
|
| Participants | Eligibility criteria: all patients admitted to the mixed ICU if their condition suggested a prolonged stay (> 3 days), age > 18 years
Exclusion criteria: age < 18 years, pregnancy, allergy to 1 component of the regimen, infections, antibiotic therapy 24 hours before randomisation Number of participants enrolled: 150 Number of participants randomised:150 Number of patients excluded: none Number of participants analysed: 150 Percentage of ventilated participants: 85% Length of stay in ICU, median days: 4.5 Diagnosis at admission: medical = 261 (8%), surgical = 72 (48%), surgical unscheduled = 30 (20%), trauma = 22 (15%) Severity score on admission: APACHE II mean = 19.4, ISS mean = 24.3 Percentage of immunocompromised participants: 4% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 75%, control = 80% Stress ulcer prophylaxis applied: H2‐blockers (80%) |
|
| Interventions | Treatment group, randomised = 75, analysed = 75:
Control group, randomised = 75, analysed = 75:
|
|
| Outcomes | Respiratory infections (only acquired infections)
Mortality: in hospital |
|
| Notes | Conflict of interest: not reported Funding: in part by Hoechst‐ Roussel Pharmaceutical, Inc |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation tables |
| Allocation concealment (selection bias) | Low risk | It is reported as "Blocking within stratification with randomisation to the control or decontamination group was achieved using randomisation tables at a remote site by a neutral observer in the pharmacy". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | High risk | The diagnosis of RTI was not based on microbiological exams. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 10 participants dropped out of the SDD group because of dislike or AEs. All included in the analysis. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
de Jonge 2003.
| Study characteristics | ||
| Methods | Randomised study
Blinding: open‐label
Randomisation method: computer‐generated random number codes kept in sealed envelopes Accrual period: from September 1999 to December 2001 Country: the Netherlands |
|
| Participants | Eligibility criteria: adult patients admitted to ICU with an expected stay of at least 72 hours and an expected duration of mechanical ventilation of at least 48 hours
Exclusion criteria: previous admission to ICU within 3 months, hypersensitivity to study medication, pregnancy, and perceived imminent death Number of participants enrolled: 1090 Number of participants randomised: 1090 Number of patients excluded: 102 because consent was denied or unable to ask consent Number of participants entered in the study: 934 Percentage of ventilated participants: 85% Length of stay in ICU: median days, treatment group 6.8 days, control group 8.5 Diagnosis at admission: elective surgery = 54 (17%), urgent surgery = 50 (21%), medical = 135 (35%) Severity score at admission: APACHE II mean = 18.7, SAPS II in SDD group mean = 17.9, SAPS II in control group mean = 17.1 Percentage of immunocompromised participants: SDD group = 2.4%, control group = 1.7% Information on prescribed antibiotics per 1000 patients available in the main publication. Stress ulcer prophylaxis by protocol: none |
|
| Interventions | Treatment group, randomised = 537, analysed = 466: topical plus systemic treatment
Control group, randomised = 533, analysed = 468:
|
|
| Outcomes | Colonisation, antibiotic resistance Mortality: in ICU and hospital | |
| Notes | Participants were allocated to either an SDD or a control unit to prevent cross‐colonisation between SDD and ICU control participants. Standard care was the same in the 2 units. Conflict of interest: none Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Unless beds were available in 1 unit only, on admission participants were allocated to 1 of the 2 units, according to computer‐generated random number codes kept in sealed envelopes. |
| Allocation concealment (selection bias) | Low risk | It is reported as "Which unit would be the SDD unit and which the control unit was randomly decided before the study. Patients were assigned to treatment groups by nursing staff not involved in the study". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is unlikely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 102 excluded because consent was denied or unable to ask consent. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
de la Cal 2005.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled trial
Blinding: double‐blind
Randomisation method: not described
Accrual period: from May 1997 to January 2000 Country: Spain |
|
| Participants | Eligibility criteria: all patients > 14 years old with burns of > 20% of total body surface area and/or suspected inhalation injury and an interval between injury and burn ICU admission < 3 days were eligible
Exclusion criteria: a stay in the burn ICU < 3 days, withdrawal of treatment within 3 days, immunosuppression, pregnancy, and inhalation injury not requiring mechanical ventilation within the first 3 days Number of participants enrolled: 117 Number of participants randomised: 117 Number of patients excluded: 10 because length of stay was less than 3 days Number of participants entered in the study: 107 Percentage of ventilated participants: 76% Length of stay in ICU, mean days: treatment 30.6 (30.9), control 33.6 (24.5) Diagnosis at admission: not available Severity score at admission: not available Percentage of immunocompromised participants: not available Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: not reported Stress ulcer prophylaxis applied: not available |
|
| Interventions | Treatment group, randomised = 58, analysed = 53:
Control group, randomised = 59; analysed = 54:
|
|
| Outcomes | Mortality Infection |
|
| Notes | Conflict of interest: not reported Funding: this study has been partially supported by 2 grants from Fondo de Investigación Sanitaria: FIS 02/1883 and Respira C 03/11 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | The randomisation result was introduced in a sealed envelope that was kept in the Department of Pharmacy. The hospital pharmacist was the only person to be informed regarding the identity of the study medication. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. The hospital pharmacist was the only person to be informed regarding the identity of the study medication. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. The hospital pharmacist was the only person to be informed regarding the identity of the study medication. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 10 excluded because length of stay was less than 3 days. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
de Smet 2009.
| Study characteristics | ||
| Methods | Cluster‐randomised study
Blinding: open‐label
Randomisation method: computer software
Accrual period: from May 2004 to July 2006 Country: the Netherlands |
|
| Participants | Eligibility criteria: patients admitted to the ICU with an expected duration of mechanical ventilation of more than 48 hours or an anticipated ICU stay of more than 72 hours were eligible
Exclusion criteria: pregnant patients and patients with documented or presumed allergy to any component of the antimicrobial study regimens were excluded Number of participants enrolled: 6565 Number of participants randomised: 5939 Number of patients excluded: none Number of participants analysed: 5939 Percentage of ventilated participants: 91.5% Length of stay in ICU: not reported Diagnosis at admission: surgical = 2762 (47%), medical = 3165 (53%) Severity score on admission: APACHE II score mean: SDD 19.6 (7.8), SOD 19.5 (8.2), standard care 18.6 (7.9) Percentage of immunocompromised participants: 2.5% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: the median number of defined daily doses of systemic antibiotic agents per patient‐day was reported Stress ulcer prophylaxis applied: none |
|
| Interventions | Treatment group:
Control group, randomised = 1990, analysed = 1990:
|
|
| Outcomes | Mortality Adverse events |
|
| Notes | Conflict of interest: Dr Bonten reports receiving advisory board fees from Ipsat Therapies, 3M, and Novartis; consulting fees from Novartis, 3M, and Bayer; and lecture fees from Cepheid and Pfizer. Dr Kluytmans reports receiving consulting fees from 3M, NovaBay, and Wyeth and lecture fees from 3M and Becton Dickinson. Dr Cooper reports receiving a lecture fee from Novartis. Dr Voss reports receiving grants from 3M and Medica. No other potential conflicts of interest were reported. Funding: not reported The study reported results in term of adjusted odds ratios (ORs) estimated using random logistic regression (de Smet 2009); covariates included in the model were scored on the Acute Physiology and Chronic Health Evaluation (APACHE II), intubation status, medical specialty (classified as surgical or other), age, and sex. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer software |
| Allocation concealment (selection bias) | Low risk | Randomisation was performed by a clinical pharmacist who was not involved in patient care in any of the participating units and who was unaware of the identity of each ICU. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs were poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout due to non‐compliance: SOD: 4.3%, SDD 2.5%. All participants included in the analysis. |
| Selective reporting (reporting bias) | Low risk | ISRCTN registry number ISRCTN35176830. All outcomes listed in the protocol were reported in the final publication. |
Ferrer 1994.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: computer‐generated table
Accrual period: from January 1991 to March 1992 Country: Spain |
|
| Participants | Eligibility criteria: all mechanically ventilated patients expected to remain intubated for more than 3 days
Exclusion criteria: patients with HIV‐related diseases or treated with antineoplastic chemotherapy as well as patients who had received transplants, extubation or death within 72 hours Number of participants enrolled: 101 Number of participants randomised: 101 Number of patients excluded: 21 because they were intubated less that 72 hours Number of participants analysed: 80 Percentage of ventilated participants: 100% Length of stay in ICU, median days: 7.5 Diagnosis at admission: medical = 66%, surgical scheduled = 6.9%, surgical unscheduled 6.9%, trauma = 19.8% Severity score on admission: SAPS mean = 12.1, ISS not available Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: not available Stress ulcer prophylaxis applied: sucralfate, except in participants with paralytic ileus or with upper gastrointestinal bleeding, who were treated with ranitidine |
|
| Interventions | Treatment group, randomised = 51, analysed = 39:
Control group, randomised = 50, analysed = 41:
*participants infected on admission received adequate antibiotic treatment instead of cefotaxime |
|
| Outcomes | Respiratory infections (pneumonia acquired after 4 days of mechanical ventilation). Diagnosis of infection was based on clinical criteria plus brush or BAL confirmation. Clinical criteria: new or progressive pulmonary X‐ray infiltrate for at least 48 hours, purulent tracheal secretions, temperature > 38.5 °C, and leukocytosis >= 12 x 109 WBC/L or leukopenia <= 4 x 109 Mortality: in ICU |
|
| Notes | Personal contact with the main investigator provided data regarding 21 participants who were excluded from the published paper (14 early extubations, 6 early deaths, 1 transfer); these data are considered in the analysis. Conflict of interest: not reported Funding: in part by grant Hospital Clinic 1991, a grant from the Beques de Formaciod'Investigadors del Departament d'Ensenyament de la Generalitat de Catalunya 1992, and grant Fiss 92/0104 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation was done using a computer‐generated random number. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. Study authors were blinded in the recovery of the results. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. Study authors were blinded in the recovery of the results. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 21 excluded because they were intubated less that 72 hours. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Finch 1991.
| Study characteristics | ||
| Methods | Randomised study
Blinding: open‐label
Randomisation method: sealed envelopes. Randomisation series made available to the hospital pharmacy only.
Accrual period: from August 1987 to September 1989 Country: the UK |
|
| Participants | Eligibility criteria: all patients whose length of stay was > 60 hours, age > 16 years
Exclusion criteria: none Number of participants enrolled: 49 Number of participants randomised: not reported Number of patients excluded: 5 Number of participants analysed: 44 Percentage of ventilated participants: not reported Length of stay in ICU: not reported Diagnosis at admission: medical = 59%, surgical scheduled = 27%, surgical unscheduled = 10%, trauma = 4% Severity score on admission: SAPS mean = 10.5, ISS not available Percentage of immunocompromised participants: 22% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 58%, control = 68% Stress ulcer prophylaxis applied: not reported |
|
| Interventions | Treatment group A, randomised = 24, analysed = 20:
Control group B, randomised = 25, analysed = 24:
|
|
| Outcomes | Respiratory infections (acquired pneumonia). Diagnosis of pneumonia was based on tracheal aspirate with numerous leukocytes associated with any of the following: a single bacterial species with a growth density > 105 CFU, diagnosis of septicaemia, clinical signs of pulmonary infections (fever, leukocytosis, and appropriate radiological findings). Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Sealed envelopes |
| Allocation concealment (selection bias) | Low risk | Randomisation series made available to the hospital pharmacy by sealed envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is unlikely to be biased due to lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs were poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A total of 49 participants were admitted, of whom 44 (90%) were evaluated; reasons for exclusion not provided. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Gastinne 1992.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, multicentre (15 ICUs) study. Intention‐to‐treat
Blinding: double‐blind
Randomisation method: a randomised list of consecutive treatment assignments, performed separately in each unit
Accrual period: from February 1990 to June 1990 Country: France |
|
| Participants | Eligibility criteria: all patients > 15 years of age who required mechanical ventilation and with intubation performed no more than 48 hours before randomisation
Exclusion criteria: patients with ventilation for less than 24 hours, drug or alcohol overdose, neutropenia (WBC < 500/mm3), SAPS > 24 or GCS < 4, chronic degenerative central nervous system disease or spinal cord injury above level of C4, acute severe enteropathy, pregnancy, participation in another ongoing clinical trial, refusal of consent, patients with conditions in which survival was strongly related to status on admission Number of participants enrolled: 445 Number of participants randomised: 445 Number of patients excluded: none Number of participants analysed: 445 Percentage of ventilated participants: 100% Length of stay in ICU, median: 12 days Diagnosis at admission: medical = 72%, surgical scheduled = 3%, surgical unscheduled = 10%, trauma = 15% Severity score on admission: SAPS mean = 13.5, ISS not available, GCS mean = 11.7 Percentage of immunocompromised participants: 18% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 89%, control = 84% Stress ulcer prophylaxis applied: sucralfate (43% participants), H2‐blockers (13% participants) |
|
| Interventions | Treatment group, randomised = 220, analysed = 220: tobramycin 80 mg, polymyxin E 100 mg, amphotericin B 100 mg applied enterally and as a 2% paste to the oropharynx 4 times a day throughout the period of ventilation Control group, randomised = 225, analysed = 225: placebo |
|
| Outcomes | Respiratory infections (pneumonia diagnosed within 48 hours and acquired): diagnosis of infection was based on: purulent tracheal aspirate, temperature > 38.5 °C, peripheral leukocytosis (> 10,000 WBC/mm3 of blood), and a new and persistent infiltrate on the chest film. Brushing was recommended but not mandatory. Mortality: in hospital |
|
| Notes | Conflict of interest: not reported Funding: supported in part by grants from Lilly France and the Commission de la Rocheeche Clinique de l'Assistance Publique Hopitaux de Paris |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | List of consecutive treatment assignment |
| Allocation concealment (selection bias) | Unclear risk | Performed separately in each centre |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor. However, the outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. No information about blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Gaussorgues 1991.
| Study characteristics | ||
| Methods | Randomised study. Intention‐to‐treat
Blinding: open‐label
Randomisation method: odd‐even numbers
Accrual period: from September 1988 to September 1989 Country: France |
|
| Participants | Eligibility criteria: all patients admitted to the ICU who required mechanical ventilation and inotropic drugs for haemodynamic reasons
Exclusion criteria: neutropenia Number of participants enrolled: 118 Number of participants randomised: 118 Number of patients excluded: none Number of participants analysed: 118 Percentage of ventilated participants: 100% Length of stay in ICU: not reported Diagnosis at admission: medical = 83%, surgical scheduled = 17% (all participants were infected on admission) Severity score on admission: SAPS mean = 17.5 Percentage of immunocompromised participants: not reported Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 100%, control = 100% Stress ulcer prophylaxis applied: sucralfate 4 g to all participants |
|
| Interventions | Treatment group, randomised = 59, analysed = 59:
Control group, randomised = 59, analysed = 59:
|
|
| Outcomes | Respiratory infections: not possible to evaluate Mortality: in ICU |
|
| Notes | All participants were infected on admission.
Data about respiratory infections are not provided. Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Georges 1994.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: open‐label
Randomisation method: sealed envelopes
Accrual period: from June 1990 to April 1992 Country: France |
|
| Participants | Eligibility criteria: polytrauma, expected mechanical ventilation for at least 4 days, age > 18 years
Exclusion criteria: hypersensitivity to the used agents, protocol violation, obesity, ventilation < 4 days, patients on mechanical ventilation 2 days before admission, severe maxillo‐facial lesions Number of participants enrolled: 138 Number of participants randomised: 64 Number of patients excluded: none Number of participants analysed: 64 Length of stay in ICU, mean days: 33 Percentage of ventilated participants: 100%. Length of ventilation, mean: 16 days Diagnosis at admission: trauma 100% Severity score on admission: APACHE II mean = 15, ISS = 41 Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy in the first 3 days: almost 100% Stress ulcer prophylaxis: H2‐blockers |
|
| Interventions | Treatment group, randomised = 31, analysed = 31:
Control group, randomised = 33, analysed = 33:
|
|
| Outcomes | Respiratory infections (acquired pneumonia). Diagnosis of infection was based on: fever > 38.5 °C, leukocytosis > 12,000/mm3, new infiltrates in the chest X‐rays, purulent pulmonary secretions, positive bacteriologic findings (> 103 CFU/mL) obtained through a protected catheter Mortality: in ICU and hospital |
|
| Notes | Antibiotic prophylaxis was free, and almost all participants in both groups were treated with systemic antibiotics. Conflict of interest: not available Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Coin flip |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Hammond 1992.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: computer‐generated random numbers
Accrual period: from January 1989 to December 1990 Country: South Africa |
|
| Participants | Eligibility criteria: expected intubation for longer than 48 hours and stay in ICU for at least 5 days
Exclusion criteria: hypersensitivity to the study drugs, patients with asthma, drug overdose, and patients admitted electively after surgery Number of participants enrolled: 322 Number of participants randomised: 322 Number of patients excluded: 83 (80 short duration of ventilation, 3 protocol violations) Number of participants analysed: 239 Percentage of ventilated participants: 100% Length of stay in ICU, median days: 11 Diagnosis at admission: medical = 55%, surgical scheduled = 3%, surgical unscheduled = 11%, trauma = 31% Severity score on admission: APACHE II mean = 13.9, ISS mean = 28.7 Percentage of immunocompromised participants: 0.8% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 54%, control = 58% Stress ulcer prophylaxis applied: none, H2‐blockers only to high‐risk patients |
|
| Interventions | Treatment group, randomised = 162, analysed = 114:
Control group B, randomised = 160, analysed = 126:
|
|
| Outcomes | Respiratory infections (infections acquired after 48 hours)
Mortality: in hospital |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated numbers |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor. However, the outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. No information about blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs were poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 83 (80 short duration of ventilation, 3 protocol violations) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Jacobs 1992.
| Study characteristics | ||
| Methods | Randomised study
Blinding: open‐label
Randomisation method: sealed envelopes
Accrual period: from July 1989 to August 1990 Country: the UK |
|
| Participants | Eligibility criteria: expected stay in ICU > 3 days
Exclusion criteria: none Number of participants enrolled: 91 Number of participants randomised: 91 Number of patients excluded: 11 length of stay shorter than 3 days, 1 suspected to be an HIV carrier Number of participants analysed: 79 Percentage of ventilated participants: 100% Length of stay in ICU, median days: SDD 9 (3 to 49), control 10 (3 to 52) Diagnosis at admission: medical = 25%, surgical = 57%, trauma = 18% (high percentage of neurologic and neurosurgical patients 52%) Severity score on admission: APACHE II mean = 17.5, ISS not available Percentage of immunocompromised participants: not available Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: next to 100% Stress ulcer prophylaxis applied: policy of maintenance a low gastric pH, H2‐blockers only if peptic ulcer or steroid therapy (33%), sucralfate 4 g to all participants not on enteral feeding |
|
| Interventions | Treatment group, randomised = 45, analysed = 36:
Control group, randomised = 46, analysed = 43:
|
|
| Outcomes | Respiratory infections (acquired pneumonia). Diagnosis of infection was based on alveolar infiltrates on 2 or more chest X‐rays, moderate or copious purulent tracheal aspirate, rectal temperature > 38.4 °C, leucocytosis > 13 x 109/L, a heavy growth of organisms from tracheal aspirate with a high polymorphonuclear leucocytes/epithelial cell ratio. Mortality |
|
| Notes | Almost 100% of participants received systemic antibiotic therapy on admission. Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Closed‐envelope system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 11 excluded because length of stay shorter than 3 days, 1 suspected to be an HIV carrier. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Kerver 1988.
| Study characteristics | ||
| Methods | Randomised study
Blinding: open‐label
Randomisation method: odd/even numbers
Accrual period: from January 1985 to May 1986 Country: the Netherlands |
|
| Participants | Eligibility criteria: all patients admitted to the surgical ICU who required care > 5 days
Exclusion criteria: none Number of participants enrolled: not reported Number of participants randomised: 96 Number of patients excluded: none Number of participants analysed: 96 Percentage of ventilated participants: 100% Length of stay in ICU: not reported Diagnosis at admission: surgical = 60%, trauma = 28%, other = 12% Severity score on admission: APACHE II mean = 14.8, ISS not reported Percentage of immunocompromised participants: not reported Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: about 85% Stress ulcer prophylaxis applied: not reported |
|
| Interventions | Treatment group, randomised = 49, analysed = 49:
Control group, randomised = 47, analysed = 47:
|
|
| Outcomes | Respiratory infections (primary pneumonia and pneumonia acquired after 48 h). Diagnosis of infection was based on X‐ray findings and the presence of 3 of the following criteria on the same day: rectal temperature > 38.5 °C for at least 12 h, WBC count > 10 x 103 or < 4 x 103/µL, at least 3% band forming granulocytes, unexplained decrease in platelet count < 100,000/µL, deterioration of renal function due to acute tubular necrosis, unexplained decrease in systolic blood pressure of > 30 mmHg, progressive respiratory failure. Mortality |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. Outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Koeman 2006.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled trial
Blinding: double‐blind
Randomisation method: computerised randomisation schedule
Accrual period: from February 2001 to March 2003 Country: the Netherlands |
|
| Participants | Eligibility criteria: consecutive adult patients (18 years of age) needing mechanical ventilation for at least 48 hours were included within 24 hours after intubation and start of mechanical ventilation
Exclusion criteria: a preadmission immunocompromised status (defined as leucopenia < 3.109/L, cumulative dose of > 750 mg corticosteroids/year, or HIV), pregnancy, and if the physical condition did not permit oral application of study medication Number of participants enrolled: 385 Number of participants randomised: 385 Number of patients excluded: none Number of participants analysed: 385 Percentage of ventilated participants: 100% Length of stay in ICU, mean days: treatment 1 = 13.77, treatment 2 = 13.27, control = 12.45 Diagnosis at admission: trauma = 38 (10%), postoperative = 69 (18%), medical = 278 (72%) Severity score on admission APACHE II score, mean (SD): treatment 1 = 22.2 (7.02), treatment 2 = 23.7 (7.38), control = 21.8 (7.43) Percentage of immunocompromised participants: not available Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: not available Stress ulcer prophylaxis applied: not available |
|
| Interventions | Treatment group 1, randomised = 127, analysed = 127: chlorhexidine 2% in petroleum jelly (Vaseline) FNA, CHX 2% Treatment group 2, randomised = 128, analysed = 128: chlorhexidine 2% in petroleum jelly (Vaseline) FNA, CHX 2% with colistin 2% in Vaseline FNA, and Vaseline FNA Control group, randomised = 130, analysed = 130: placebo |
|
| Outcomes | Time to ventilator‐associated pneumonia All‐cause ICU mortality |
|
| Notes | Conflict of interest: "None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript." Funding: supported by ZonMw, the Netherlands Organisation for Health Research and Development (project number 2200.0046) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised randomisation schedule |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind study. No information provided about blinding of assessor. However, outcome is unlikely to be biased due to lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind study. No information provided about blinding of assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs were poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The study was discontinued in 6 participants: 5 participants (control group, 1 participant; treatment group 1, 2 participants; treatment group 2, 2 participants) refused to accept the oral paste, and 1 participant in treatment group 2 was prematurely withdrawn from the trial protocol due to tongue oedema on the second day. All participants included in the analysis. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Korinek 1993.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, dual‐centre study
Blinding: double‐blind
Randomisation method: randomisation performed by the hospital pharmacist on each unit separately
Accrual period: from March 1989 to September 1990 Country: France |
|
| Participants | Eligibility criteria: all comatose patients with emergency admission to 2 neurosurgical ICUs and intubated within 24 hours for at least 5 days, age > 16 years
Exclusion criteria: age < 16 years, known immunosuppression, antibiotic treatment during the 2 weeks preceding ICU admission, serious injury of oropharyngeal mucosa or epistaxis, abnormal chest X‐ray on admission, extubation or infection occurring within the first 5 days of neurosurgical care Number of participants enrolled: 191 Number of participants randomised: 191 Number of patients excluded: 68 because length of stay shorter than 5 days Number of participants analysed: 123 Percentage of ventilated participants: 100% Length of stay in ICU, mean days: 26 Diagnosis at admission: surgical scheduled = 11%, surgical unscheduled = 39%, trauma = 50% Severity score on admission: SAPS mean = 10.9, ISS not available Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 0%, control = 0% Stress ulcer prophylaxis applied: sucralfate (32%), antiacids (14%), H2‐blockers (20%) until enteral feeding was started |
|
| Interventions | Treatment group, randomised = 96, analysed = 63:
Control group, randomised = 95, analysed = 60:
The non‐absorbable antibiotics were discontinued if any infection requiring a parenteral antibiotic treatment occurred and at the time of participant's extubation. |
|
| Outcomes | Respiratory infections (pneumonia acquired after 5 days). Diagnosis of infection was based on fever > 38.5 °C, leukocytosis > 12,000 cells/mm3, purulent sputum, new and persistent infiltrates on chest X‐ray, and a culture of > 103 CFU/mL obtained with either brush or plugged telescoping catheter. Mortality: in ICU |
|
| Notes | Setting: neurosurgical ICU Conflict of interest: not reported Funding: the study was supported in part by Lilly‐France |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided. |
| Allocation concealment (selection bias) | Low risk | It is reported as "randomisation was performed on each unit separately by hospital's pharmacist, who delivered daily the placebo or treatment capsules for each patients". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind study. No information provided about blinding of assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 68 excluded because length of stay shorter than 5 days. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Krueger 2002.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, double‐centre study
Randomisation method: computer‐generated randomisation
Accrual period: from April 1989 to March 1991 Country: Germany |
|
| Participants | Eligibility criteria: expected stay in ICU > 2 days and at least 1 risk factor for infection
Exclusion criteria: patients expected to die within 48 hours or randomisation was not achieved within 12 hours after admission to ICU
Number of participants enrolled: 546 Number of participants randomised: 546 Number of patients excluded: 19: withdrawal of consent (5 patients), violation of entry criteria (9 patients), and other reasons (5 patients) Number of participants analysed: 527 Percentage of ventilated participants: not reported Length of stay in ICU: not reported Diagnosis at admission: surgical Severity score on admission: APACHE II mean = 20, ISS not available Percentage of immunocompromised participants: not available Percentage of participants treated with systemic antibiotic therapy (not stated in protocol) in the first 3 days: not available Stress ulcer prophylaxis applied: sucralfate to all participants |
|
| Interventions | Treatment group, randomised = not reported, analysed = 265:
Control group, randomised = not reported, analysed = 262:
|
|
| Outcomes | Respiratory infections (acquired pneumonia) Mortality in ICU |
|
| Notes | Information about accrual period and severity score was reported in a previously published abstract. Conflict of interest: not reported Funding: grants from Bayer Vital GmbH, Leverkusen, and Merck, Darmstadt |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme |
| Allocation concealment (selection bias) | Low risk | Sealed‐envelope technique served for assignment to the treatment or placebo group. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind: "All data were noted on standardized documentation sheets and were exclusively collected by a study nurse and a medical doctor in each centre. None of these persons were involved in patient care or in diagnostic or therapeutic decisions" |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind: "All data were noted on standardized documentation sheets and were exclusively collected by a study nurse and a medical doctor in each centre. None of these persons were involved in patient care or in diagnostic or therapeutic decisions" |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs were poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | It is reported as "Nineteen (3.4%) patients were excluded after enrolment (8 of the prophylaxis group and 11 of the control group, all survived) because of withdrawal of consent (five patients), violation of entry criteria (nine patients), and other reasons (five patients)". |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Laggner 1994.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: computer‐generated randomisation in time blocks. Open
Accrual period: from August 1987 to November 1990 Country: Germany |
|
| Participants | Eligibility criteria: expected ventilation for 5 days, age > 18 years and < 80 years, acute onset of respiratory failure
Exclusion criteria: age < 18 years and > 80 years, bleeding of the nasopharynx and of the upper gastrointestinal tract on admission, stress ulcer prophylaxis with other drug therapy than sucralfate, mechanical ventilation for less than 5 days, patients on enteral nutrition or with known allergy to sucralfate or gentamicin Number of participants enrolled: 88 Number of participants randomised: 88 Number of patients excluded: 21 (18 participants ventilated for less than 5 days, 3 participants received enteral nutrition) Number of participants analysed: 67 Percentage of ventilated participants: 100% Length of stay in ICU, mean days: 28.8 Diagnosis at admission: medical = 88%, surgical scheduled = 9%, surgical unscheduled = 1%, trauma = 2% Severity score on admission: APACHE II mean = 23, ISS not available Percentage of immunocompromised participants: 15% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: 100% Stress ulcer prophylaxis applied: sucralfate |
|
| Interventions | Treatment group, randomised = not reported, analysed = 33:
Control group, randomised = not reported, analysed = 34:
|
|
| Outcomes | Respiratory infections (acquired pneumonia). Diagnosis of infection was based on: appearance of a new infiltrate on the chest film with concomitant tracheal colonisation, fever > 38 °C, and > 15,000 or < 5000 WBC/mm3 of blood. Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind study. No information about blinding of outcome assessor. However, outcome is unlikely to be biased due to lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind study. No information provided about blinding of assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 21 participants developed exclusion criteria during the first 5 days. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Lingnau 1997.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study with 3 arms (1 control arm and 2 treatment arms) Intention‐to‐treat
Blinding: double‐blind
Randomisation method: continuous random numbers assigned to blinded study drugs or vehicle by biometric department
Accrual period: from August 1989 to January 1994 Country: Austria |
|
| Participants | Eligibility criteria: non‐infected trauma patients, age > 18 years, expected ventilation for at least 2 days, expected ICU stay for at least 3 days, ISS > 16 and < 74, inclusion within 24 hours of admission
Exclusion criteria: isolated brain injury, prior antibiotic treatment, history of infection Number of participants enrolled: 961 Number of participants randomised: 357 Number of patients excluded: 47 because they developed exclusion criteria Number of participants analysed: 310 Percentage of ventilated participants: 100% Length of stay in ICU, mean days: 20 Diagnosis at admission: trauma = 100% Severity score on admission: APACHE II mean = 15.6, ISS mean = 35.2 Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy (not stated in protocol) in the first 3 days: treatment 1 = 2%, treatment 2 = 2%, control = 6% Stress ulcer prophylaxis applied: free |
|
| Interventions | Treatment group 1, randomised = not reported, analysed = 80:
Treatment group 2, randomised = not reported, analysed = 82:
Control group, randomised = not reported, analysed = 148:
|
|
| Outcomes | Respiratory infections (acquired pneumonia). Diagnosis of infection was based on concomitant occurrence of purulent sputum, positive cultures of bronchial secretions, and deterioration of lung function. Mortality: in ICU |
|
| Notes | Conflict of interest: not available Funding: this study was funded in part by research grant BAY q 3939/0454 and grant of the Leopold Franzers University of Insbruck, Austria |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was carried out using random numbers in blocks of 20. |
| Allocation concealment (selection bias) | Unclear risk | The pharmacist prepared and dispensed all treatments according to the randomisation list. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. Red dye added by the hospital pharmacist to ensure blind protocol to clinicians and investigators. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. Red dye added by the hospital pharmacist to ensure blind protocol to clinicians and investigators. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 47 excluded because they developed exclusion criteria. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Palomar 1997.
| Study characteristics | ||
| Methods | Randomised, multicentric (10 ICUs) study with 3 arms (1 treatment arm and 2 control arms; 1 control arm was excluded from meta‐analysis because it was the only study arm receiving sucralfate)
Blinding: open‐label
Randomisation method: sealed envelopes
Accrual period: from July 1989 to August 1991 Country: Spain |
|
| Participants | Eligibility criteria: patients requiring mechanical ventilation for more than 4 days, not infected at the time of entry, and not receiving antibiotic therapy
Exclusion criteria: ARDS, leukopenia, pregnancy Number of participants enrolled: 151 Number of participants randomised: 151 Number of patients excluded: 22: early extubation (10), early death (8), protocol violation (3), antibiotic hypersensitivity (1) Number of participants analysed: 129 Percentage of ventilated participants: 100% Length of stay in ICU, median days: 10 Diagnosis at admission: medical = 50 (40%), surgical = 14 (10%), trauma = 65 (50%) Severity score on admission: APACHE II mean = 16.8, ISS not available Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 0%, control = 6% Stress ulcer prophylaxis applied: sucralfate to 1 control group (excluded from meta‐analysis), antiacids or H2‐blockers to the 2 other groups |
|
| Interventions | Treatment group, randomised = 50; analysed = 41:
Control group, randomised = 49, analysed = 42:
Sucralfate group,* randomised = 52, analysed = 46:
*this group was excluded from analysis because it was the only group receiving sucralfate |
|
| Outcomes | Respiratory infections (acquired infections). Diagnosis of pneumonia was based on the CDC criteria of 1980 (clinical or radiologic suspicion with: purulent sputum, organism isolated from blood culture, isolation of pathogen from tracheal aspirate, brush or biopsy). Bacteriologic evaluation was performed with brush or BAL in 50% of participants. Diagnosis of tracheobronchitis was based on the CDC criteria of 1980. Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: grant from Roussel Iberica Laboratories |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random numbers table |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 22 excluded because early extubation (10), early death (8), protocol violation (3), cefotaxime‐related hypertensive reaction (1). |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Pneumatikos 2002.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled
Blinding: outcome assessor
Randomisation method: not stated Accrual period: not available Country: Greece |
|
| Participants | Eligibility criteria: patients with multiple trauma admitted to the ICU who required intubation and had an expected time of mechanical ventilation exceeding 5 days.
Absence of cardiopulmonary disease, negative chest radiography, and a PaO2/FiO2 ratio higher than 300 mmHg Number of participants enrolled: 79 Number of participants randomised: 79 Number of patients excluded: 18 Number of participants analysed: 61 Percentage of ventilated participants: 100% Length of stay in ICU, median days: treatment = 16, control = 23 Diagnosis at admission: trauma = 100% Severity score on admission: APACHE II, treatment = 18.1, control = 19.1 Percentage of immunocompromised participants: not reported Percentage of participants treated with systemic antibiotic therapy: not stated Stress ulcer prophylaxis applied: H2 blockers or sucralfate: treatment = 26%, control = 19% |
|
| Interventions | Treatment group, randomised = 40, analysed = 31:
Control group, randomised = 39, analysed = 30:
|
|
| Outcomes | Ventilator‐associated pneumonia defined as presence of new and persistent pulmonary infiltrates plus 2 of the following criteria: body temperature > 38.3 °C, leukocytosis (> 12,000 leukocytes/mm3) or leukopenia (< 4000 leukocytes/mm3), and purulent tracheal secretions. The diagnosis of VAP was confirmed by quantitative cultures. Mortality: not specified |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | It is reported as "chest physician, a radiologist, and a physician with experience in infectious diseases (blind to the patient’s group assignment)". |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | It is reported as "chest physician, a radiologist, and a physician with experience in infectious diseases (blind to the patient’s group assignment)". |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 18 patients were excluded: 7 extubated, 4 developed pneumonia during the first 48 hours of mechanical ventilation, 1 underwent tracheotomy, and 6 died. It is not reported if these events occurred before or after the first 5 days of mechanical ventilation. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Pugin 1991.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: sealed envelopes
Accrual period: from April 1989 to November 1989 Country: Switzerland |
|
| Participants | Eligibility criteria: all adult patients admitted to the surgical ICU, at high risk of developing pneumonia, and intubated for more than 48 hours
Exclusion criteria: organ transplantation Number of participants enrolled: 79 Number of participants randomised: 79 Number of patients excluded: 27 due to early extubation or early death Number of participants analysed: 52 Percentage of ventilated participants: 100% Length of stay in ICU, mean days: 13.8 Diagnosis at admission: medical = 11%, surgical scheduled = 11%, surgical unscheduled = 22%, trauma = 56% Severity score on admission: APACHE II mean = 15.2, ISS not available Percentage of immunocompromised participants: not available Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 46%, control = 53% Stress ulcer prophylaxis applied: sucralfate (61%), ranitidine (11%) |
|
| Interventions | Treatment group, randomised = 38, analysed = 25: polymyxin B sulphate 150 mg, neomycin sulphate 1 g, vancomycin hydrochloride 1 g applied as a solution to the retropharynx 6 times a day within 24 hours after intubation until extubation or death Control group, randomised = 41, analysed = 27: placebo Withdrawal from the study was possible at any time if the treating physicians estimated that there was any problem related to administration of the drugs or side effects. |
|
| Outcomes | Respiratory infections (pneumonia acquired after 48 hours). Pneumonia was defined as a "clinical pulmonary infections score" (CPIS) ≥ 7 during the course of intubation and that remained elevated (= 7) for at least 3 days (i.e. for 2 consecutive measurements). Mortality: in hospital |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. No information about blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 27 patients excluded due to early extubation or early death. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Quinio 1995.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study. Intention‐to‐treat
Blinding: double‐blind
Randomisation method: randomisation made by the pharmacy service according to computer‐generated random numbers
Accrual period: from January 1991 to January 1993 Country: France |
|
| Participants | Eligibility criteria: trauma patients intubated within 24 h
Exclusion criteria: age < 16 years, antibiotic treatment in the week preceding ICU admission, pregnancy Number of participants enrolled: 148 Number of participants randomised: 148 Number of patients excluded: none Number of participants analysed: 148 Percentage of ventilated participants: 100% Length of stay in ICU, mean days = 20.5 Diagnosis admission: medical = 2%, trauma = 98% Severity score on admission: SAPS mean = 11.2, ISS mean = 31.3, GCS mean = 6.5 Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 35%, control = 26% Stress ulcer prophylaxis applied: sucralfate until enteral feeding was effective, H2‐blockers in high‐risk patients |
|
| Interventions | Treatment group, randomised = 76, analysed = 76: polymyxin E 100 mg, gentamicin 80 mg, amphotericin B 500 mg applied enterally and in the nares and as a 2% paste to the oropharynx 4 times a day until extubation or start of enteral nutrition Control group: randomised = 72, analysed = 72: placebo |
|
| Outcomes | Respiratory infections (pneumonia acquired after 48 h). Diagnosis of infection was based on: purulent tracheal aspirate, fever > 38.5 °C, leukocytosis > 10,000 WBC/mm3, new and persistent infiltrate on chest X‐ray. Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random number |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was performed by the hospital's pharmacists according to a randomised list of consecutive treatment assignments. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor. However, the outcome measurement is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. No information about blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Rocha 1992.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: made by the pharmacy service according to computer‐generated random numbers
Accrual period: from September 1989 to October 1990 Country: Spain |
|
| Participants | Eligibility criteria: expected mechanical ventilation for more than 3 days, stay in ICU more than 5 days
Exclusion criteria: infection or strong suspicion of infection at the start of ventilation, antibiotic treatment in the previous 7 days, neutropenia (WBC < 500/mm3) and fever, pregnancy, history of hypersensitivity to the topical agents Number of participants enrolled: 151 Number of participants randomised: 151 Number of patients excluded: 50 (15 early extubations, 31 early deaths, 2 protocol violation, 2 other) Number of participants analysed: 101 Percentage of ventilated participants: 100% Length of stay in ICU, median days: 8 Diagnosis at admission: medical = 28%, surgical scheduled = 3%, surgical unscheduled = 1%, trauma = 68% Severity score on admission: APACHE II mean = 16.3, ISS not available, GCS mean = 9 Percentage of immunocompromised participants: 0.7% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 0%, control = 0% Stress ulcer prophylaxis applied: H2‐blockers and antiacids |
|
| Interventions | Treatment group, randomised = 74, analysed = 47:
Control group, randomised = 77, analysed = 54:
|
|
| Outcomes | Respiratory infections (acquired pneumonia). Diagnosis of infection was based on purulent pulmonary secretions, new infiltrates in the chest X‐ray, and 1 of the following: fever/hypothermia, leukocytosis/leukopenia, positive physical examination, drop in arterial partial oxygen pressure. Bacteriologic diagnosis, even if performed (with brush in few participants), was not essential. Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random numbers table |
| Allocation concealment (selection bias) | Unclear risk | Made by the pharmacy service |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind study. No information about blinding of outcome assessor. However, outcome is unlikely to be biased due to lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. No information about blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 50 excluded (15 early extubations, 31 early deaths, 2 protocol violation, 2 other). |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Rodriguez‐Roldan 1990.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, dual‐centre study
Blinding: double‐blind
Randomisation method: odd and even numbers
Accrual period: from June 1988 to December 1988 Country: Spain |
|
| Participants | Eligibility criteria: patients intubated and mechanically ventilated for more than 72 hours
Exclusion criteria: patients whose chest X‐rays were difficult to interpret, with suspected inflammatory images during the first 72 hours, patients ventilated for less time Number of participants enrolled: 28 Number of participants randomised: 28 Number of patients excluded: none Number of participants analysed: 28 Percentage of ventilated participants: 100% Length of stay in ICU, median days: 13.5 Diagnosis at admission: medical = 13 (42%), surgical = 5 (16%), trauma = 13 (42%) Severity score on admission: APACHE II mean = 17.1, ISS not available Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 36%, control = 35% Stress ulcer prophylaxis applied: sucralfate or alkaline agents plus ranitidine according to a randomised open protocol |
|
| Interventions | Treatment group: randomised = 13, analysed = 13:
Control group: randomised = 15, analysed = 15:
|
|
| Outcomes | Respiratory infections (pneumonia acquired after 72 hours). Diagnosis of infection was based on the presence of at least 1 in each category of criteria:
Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: this study was supported in part by a grant from the Social Security Health Investigation Foundation of Spain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor for mortality. However, the outcome is unlikely to be biased due to lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind chest radiographs were independently examined by 2 radiologists; independent microbiological surveillance. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Sanchez‐Garcia 1998.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, multicentric (5 ICUs) study
Blinding: double‐blind
Randomisation method: sealed envelopes
Accrual period: not available Country: Spain |
|
| Participants | Eligibility criteria: expected ventilation for longer than 48 hours, age > 16 years
Exclusion criteria: death or extubation before 48 hours, pregnancy, allergy to study antibiotics, organ transplantation, absence or contraindication to nasogastric tube Number of participants enrolled: 271 Number of participants randomised: 271 Number of patients excluded: none Number of participants analysed: 271 Percentage of ventilated participants: 100% Length of stay in ICU, median days: 13 Diagnosis at admission: medical = 191 (70%), surgical scheduled = 5 (3%), surgical unscheduled = 25 (9%), trauma = 45 (18%) Severity score on admission: APACHE II mean = 26.6, ISS not available Percentage of immunocompromised participants: 4.4% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 70%, control = 69% Stress ulcer prophylaxis applied: each group was randomised to receive either sucralfate or H2‐blockers |
|
| Interventions | Treatment group, randomised = 131, analysed = 131:
Control group, randomised = 140, analysed = 140:
|
|
| Outcomes | Respiratory infections (early‐ and late‐acquired pneumonia). Diagnosis of infection was based on new and persistent infiltrate on chest X‐ray and 3 of the following: temperature > 38.5 °C, leukocytosis > 12,000 WBC/mm3 or leukopenia < 3000 WBC/mm3, purulent tracheal aspirate with growth of a potentially pathogenic micro‐organism. Mortality: in ICU |
|
| Notes | Personal contact with the main investigator provided data for 45 patients who were excluded from the published paper (12 early extubations, 12 early deaths, 17 protocol violation, 2 transferring, 2 other); these data are considered in the analysis. Conflict of interest: not reported Funding: supported in part by a grant from Productos Roche S.A., Spain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | It is reported as "Patients were randomised through a computer‐generated random‐number table and were stratified by centre". |
| Allocation concealment (selection bias) | Low risk | Treatment codes were kept in individual sealed envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor for mortality. However, the outcome is unlikely to be biased due to lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. "The microbiologists who examined specimens for the study were blinded to the patient groups" |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs were poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Stoutenbeek 1996.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: closed‐envelope method (the code was known by the pharmacist only)
Accrual period: from November 1984 to August 1986 Country: the Netherlands |
|
| Participants | Eligibility criteria: all patients admitted to the surgical ICU with blunt trauma and an HTI‐ISS > 18, age > 18 years
Exclusion criteria: patients mechanically ventilated for less than 5 days or discharged from ICU within 7 days and with an HTI‐ISS < 18 after 24 hours Number of participants enrolled: 91 Number of participants randomised: 91 Number of patients excluded: 32 (27 early death or early extubations, 5 ISS < 18 after 24 hours) Number of participants entered in the study: 59 Percentage of ventilated participants: 100% Length of stay in ICU, mean days = 15 Diagnosis at admission: trauma = 100% Severity score on admission: APACHE II mean = 10.6, HTI‐ISS mean = 35.1 Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 0%, control = 0% Stress ulcer prophylaxis applied: none, except for participants with history of pre‐existing ulcer or on H2‐blockers |
|
| Interventions | Treatment group, randomised = 49, analysed = 30:
Control group, randomised = 42, analysed = 29:
The blinded medication was discontinued and topical prophylaxis started when a participant developed MOSF not responding to conventional therapy. The code was not broken, and the participant was further evaluated. |
|
| Outcomes | Respiratory infections (tracheobronchitis and pneumonia ‐ early and late infections). Diagnosis of infection was based on clinical criteria: temperature > 38.5 °C, WBC > 12.5 x 109/L or leukopenia < 4 x 109/L, purulent secretions or X‐ray changes and significant growth of bacteria. Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: this study was supported by a grant from Russel Netherland |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Closed‐envelope method. The code was known by the pharmacist only. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor. However, the outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. No information about blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs were poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 27 excluded due to early death or early extubations, 5 ISS < 18 after 24 hours. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Stoutenbeek 2007.
| Study characteristics | ||
| Methods | Randomised, multicentre study. Intention‐to‐treat
Blinding: open‐label
Randomisation method: randomisation lists prepared by the Biometrical Department and supplied with sealed envelopes
Accrual period: from October 1991 to June 1994 Country: Europe, Australia, and New Zealand |
|
| Participants | Eligibility criteria: patients admitted within 24 hours after non‐penetrating blunt trauma with an HTI‐ISS >= 16, necessitating mechanical ventilation, age > 18 years
Exclusion criteria: previous antibiotic use for more than 3 days at study entry, allergy to cefotaxime, referred patients from other hospital or secondary admissions with trauma having occurred > 24 hours before Number of participants enrolled: 405 Number of participants randomised: 405 Number of patients excluded: 4 (2 did not fulfil the inclusion criteria, data from 1 participant not available, 1 participant lost to follow‐up after the 7th day) Number of participants analysed: 401 Percentage of ventilated participants: 100% Length of stay in ICU, median days: SDD = 13, control = 12 days Median duration of mechanical ventilation: SDD = 9 days, control = 8 days Type of admission diagnosis: trauma 100% Severity score on admission: APACHE II median: SDD = 15, control = 14; HTI‐ISS median: SDD = 34, control = 29 Percentage of immunocompromised participants: 0% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 46%, control = 88% Stress ulcer prophylaxis applied: sucralfate, H2‐blockers, omeprazole |
|
| Interventions | Treatment group, randomised = not reported, analysed = 201:
Control group, randomised = not reported, analysed = 200:
|
|
| Outcomes | Primary: mortality from infection or multiple organ failure in ICU or up to 2 weeks after discharge Secondary: incidence of infection, multiple organ failure, and antibiotic usage The maximum observation period was 3 months. Patients dying within 24 hours after injury or dying from craniocerebral trauma were excluded. Diagnosis of pneumonia was based on any of the following: presence of a new and progressive pulmonary infiltrate on chest X‐ray for >= 48 hours, purulent tracheal aspirate, fever > 38.5 °C, leukocytosis > 12,000/mL or leukopenia < 4000/mL. Diagnosis of tracheobronchitis was based on the same criteria except for the radiographic changes. Mortality: in ICU or up to 2 weeks after discharge |
|
| Notes | Conflict of interest: not reported Funding: this study was supported by a grant from Hoechst‐Roussel International |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | It is reported as "A randomisation schedule was prepared separately for each centre in balanced blocks of variable length by Medis GMBH". |
| Allocation concealment (selection bias) | Low risk | It is reported as "Each centre received a set of consecutively numbered sealed envelopes containing the treatment allocation guaranteeing adequacy of concealment of allocation sequence". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | It is reported as "Each death was audited by two independent investigators (R.L. and C.S.) who were blinded to the treatment arm, to confirm the statement of the treating physician about whether death resulted from cranio‐cerebral trauma". |
| Blinding of outcome assessment (detection bias) SRIs | High risk | No blinding. The diagnosis of RTI was not based on microbiological exams. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs were poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 4 participants were excluded from the final analysis after randomisation (2 did not fulfil the inclusion criteria, the data from 1 participant were not available, and 1 participant was lost to follow‐up after the 7th day). |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Ulrich 1989.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: open‐label
Randomisation method: sealed envelopes containing a random code for clusters of 4 patients
Accrual period: from October 1986 to September 1987 Country: the Netherlands |
|
| Participants | Eligibility criteria: patients expected to stay in the ICU more than 5 days and ventilated more than 48 hours Number of participants enrolled: 112 Number of participants randomised: 112 Number of patients excluded: 12 (early death) Number of participants analysed: 100 Percentage of ventilated participants: about 80% Length of stay in ICU, median days = 10 Diagnosis at admission: medical = 34 (34%), surgical scheduled = 19 (19%), surgical unscheduled = 31 (31%), trauma = 16 (16%) Severity score on admission: SAPS mean = 11.7, ISS mean = 36.9 Percentage of immunocompromised participants: 3% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 83%, control = 81% Stress ulcer prophylaxis applied: not available |
|
| Interventions | Treatment group, randomised = 55, analysed = 48:
Control group, randomised = 57, analysed = 52:
|
|
| Outcomes | Respiratory infections (acquired pneumonia). Diagnosis of infection was based on clinical and radiologic signs of pulmonary infiltrations with fever and leukocytosis and a dense growth in cultures of sputum of tracheal aspirate. Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes containing a random code for clusters of 4 patients |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | High risk | No blinding. The diagnosis of RTI was not based on microbiological exams. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 12 patients excluded because they died within 24 hours. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Unertl 1987.
| Study characteristics | ||
| Methods | Randomised study
Blinding: open‐label
Randomisation method: blocked randomisation scheme and sealed‐envelope technique
Accrual period: from May 1984 to January 1985 Country: Germany |
|
| Participants | Eligibility criteria: all patients admitted to the ICU with: intubation within 24 hours after the onset of an acute disease or surgery, expected ventilation > 6 days, interval between intubation and first microbiologic culture < 36 hours
Exclusion criteria: patients with infection, systemic antibiotic treatment, respiratory distress syndrome, leukopenia and myelosuppression on admission, renal failure Number of participants enrolled: 39 Number of participants randomised: 39 Number of patients excluded: none Number of participants analysed: 39 Percentage of ventilated participants: 100% Length of stay in ICU: not reported Diagnosis at admission: medical = 20 (52%), surgical = 6 (15%), trauma = 13 (33%) Severity score on admission: SAPS mean = 12.5, GCS (75% of participants have GCS < 7) Percentage of immunocompromised participants: not available Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: not reported Stress ulcer prophylaxis applied: H2‐blockers to all participants and antiacids if pH < 4 |
|
| Interventions | Treatment group, randomised = 19, analysed = 19:
Control group, randomised = 20, analysed = 20:
|
|
| Outcomes | Respiratory infections (acquired pneumonia). Diagnosis of infection was based on new "definite" infiltrate on chest X‐ray together with increasing amounts of purulent tracheobronchial secretion containing > 3 x 104 granulocytes/µL and at least 2 of the following: new febrile spikes > 38.5 °C, blood leukocyte count > 12,000/µL or < 4000/µL, decrease of PaO2 requiring an increase of the FiO2 of at least 15% to maintain oxygen tension. "Definite" is an infiltrate confirmed by 2 blind independent radiologists and not reversible after chest physiotherapy. Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation scheme |
| Allocation concealment (selection bias) | Low risk | Sealed‐envelope technique |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | High risk | No blinding. The diagnosis of RTI was not based on microbiological exams. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | Methods for collection of AEs were poorly reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Verwaest 1997.
| Study characteristics | ||
| Methods | Randomised study with 3 arms (1 control arm and 2 treatment arms)
Blinding: open‐label
Randomisation method: sealed envelopes with computer‐generated random numbers
Accrual period: from September 1989 to March 1991 Country: Belgium |
|
| Participants | Eligibility criteria: expected ventilation > 48 hours
Exclusion criteria: age < 18 years, pregnancy, recent organ transplantation, serious granulocytopenia (<= 500 WBC/mm3), ventilation < 48 hours, death before 48 hours, missing of essential data in the clinical or bacteriological dossier Number of participants enrolled: 660 Number of participants randomised: 660 Number of patients excluded: 82 (33 early death, 9 early extubation, 40 missing data) Number of participants entered in the study: 578 Percentage of ventilated participants: 100% Length of stay in ICU, mean days = 19.6 Diagnosis at admission: medical = 10%, surgical = 67%, trauma = 23% Severity score on admission: APACHE II mean = 18.1, ISS not available Percentage of immunocompromised participants: not available Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment 1 = 34%, treatment 2 = 31%, control = 34% Stress ulcer prophylaxis applied: sucralfate 2 g x 4 |
|
| Interventions | Treatment group 1, randomised = 220, analysed = 193:
Control group, randomised = 220, analysed = 185:
Treatment group 2, randomised = 220, analysed = 200: this group received non‐absorbable antibiotics administered through a gastric tube as a combination of polymyxin, tobramycin, and amphotericin B. This group was not considered in the analyses.
|
|
| Outcomes | Respiratory infections (pneumonia acquired after 48 h). Diagnosis of infection was based on fever > 38.5 °C, leukocytosis > 10,000 cells/µL, luxuriant growth of potentially pathogenic micro‐organisms in culture of bronchial aspirate, new and persistent infiltrate on chest X‐ray. Mortality: in ICU |
|
| Notes | Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were allocated randomly to 1 of 3 study groups, using a computer‐generated randomisation scheme and the sealed‐envelope technique. |
| Allocation concealment (selection bias) | Low risk | Participants were allocated randomly to 1 of 3 study groups, using a computer‐generated randomisation scheme and the sealed‐envelope technique. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | In 40 participants (6%), balanced between groups, a correct evaluation was impossible because important clinical or bacteriological data were missing. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Wiener 1995.
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study
Blinding: double‐blind
Randomisation method: random number table in blocks of 6 patients
Accrual period: 8 months Country: the USA |
|
| Participants | Eligibility criteria: expected intubation for more than 48 hours, inclusion within 18 hours of intubation, age > 18 years
Exclusion criteria: refusal to consent, allergy to 1 of the components of the regimen, active inflammatory bowel disease Number of participants enrolled: 121 Number of participants randomised: 121 Number of patients excluded: 60 (31 early extubation, 20 early death, 4 transfer to general medical ward, 1 protocol violation) Number of participants entered in the study: 61 Percentage of ventilated participants: 100% Length of stay in ICU, mean days: 11.3 Diagnosis at admission: not available Severity score on admission: APACHE II mean = 27.2, ISS not available Percentage of immunocompromised participants: > 5% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 93%, control = 81% Stress ulcer prophylaxis applied: H2‐blockers to most participants |
|
| Interventions | Treatment group, randomised = not reported, analysed = 30: polymyxin E 100 mg, gentamicin 80 mg, nystatin 2,000,000 U applied enterally 4 times a day and as a 2% paste to the oropharynx until extubation or tracheostomy Control group, randomised = not reported, analysed = 31: placebo |
|
| Outcomes | Respiratory infections (pneumonia acquired after 48 hrs). Diagnosis of infection was based on the presence of the following: persistence of a new or progressive infiltrate on chest‐film, fever > 38.5 °C and/or leukocytosis > 12,000/mm3, growth of > 103 bacteria from a quantitative culture of lower respiratory tract secretions obtained with a blind protected catheter. Mortality: in ICU |
|
| Notes | 60 participants were excluded after randomisation (data not available). Conflict of interest: not reported Funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised in block of 6 using a random number table to receive either the topical antibiotics or placebo. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | Double‐blind. No information about blinding of outcome assessor. However, outcome is unlikely to be biased due to lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | Double‐blind. No information about blinding of outcome assessor. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 60 (31 early extubation, 20 early death, 4 transfer to general medical ward, 1 protocol violation) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
Winter 1992.
| Study characteristics | ||
| Methods | Randomised study
Blinding: open‐label
Randomisation method: sealed envelopes based on a computer‐generated table of random numbers
Accrual period: from July 1988 to May 1990 Country: the UK |
|
| Participants | Eligibility criteria: patients likely to remain in the ICU for at least 48 hours
Exclusion criteria: allergy to the antibiotics used, age > 85 years, pregnancy Number of participants enrolled: 267 Number of participants randomised: 267 Number of patients excluded: none Number of participants entered in the study: 267. Of these, 84 in the historical control group were not considered in the analysis. Percentage of ventilated participants: 92% Length of stay in ICU, median days: 4 Diagnosis at admission: medical = 40%, surgical scheduled = 10%, surgical unscheduled = 37%, trauma = 13% Severity score on admission: APACHE II mean = 15.3, ISS mean = 26.2 Percentage of immunocompromised participants: 2.2% Percentage of participants treated with systemic antibiotic therapy (not stated in the protocol) in the first 3 days: treatment = 47%, control = 64% Stress ulcer prophylaxis applied: all control participants received sucralfate; H2‐blockers were used in participants in both groups with peptic ulcer or pancreatitis |
|
| Interventions | Treatment group, randomised = 91, analysed = 91:
Control group, randomised = 92, analysed = 92:
|
|
| Outcomes | Respiratory infections (pneumonia acquired after 48 h). Diagnosis of infection was based on temperature > 38.5 °C 2 times in 24 hours, WBC < 4 or > 12 x 109/L, positive BAL, 2 of the following: new pulmonary infiltrates on chest X‐ray, purulent sputum, increase of 15% in FiO2 to maintain previous oxygenation. Mortality: in hospital |
|
| Notes | Few participants were excluded after randomisation (data not available). Conflict of interest: not reported Funding: Bristol and Weston Health Authority and Glaxo Research Ltd |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Suitable patients were allocated randomly to contemporaneous control or SDD group with consecutively numbered envelopes prepared by computer‐generated random numbers. |
| Allocation concealment (selection bias) | Low risk | Suitable patients were allocated randomly to contemporaneous control or SDD group with consecutively numbered envelopes prepared by computer‐generated random numbers. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICUs have standardised infection control programmes. Possible subjective decisions on who should be tested for the presence or absence of RTIs are unlikely in an ICU setting because routine care includes daily laboratory tests and scheduled radiologic and microbiologic studies. |
| Blinding of outcome assessment (detection bias) Mortality | Low risk | No blinding. However, outcome is not likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) SRIs | Low risk | No blinding. However, a standardised set of diagnostic criteria, including results of microbiological exams, were used. |
| Blinding of outcome assessment (detection bias) AES | Unclear risk | AEs not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
APACHE: Acute Physiology and Chronic Health Evaluation ARDS: acute respiratory distress syndrome BAL: bronchoalveolar lavage CCU: coronary care unit CDC: Centers for Disease Control and Prevention CFU: colony‐forming unit CHX: chlorhexidine CI: confidence interval CTR: control group FiO2: fractional inspired oxygen FNA: fine needle aspiration GCS: Glasgow Coma Scale HTI‐ISS: Hospital Trauma Index‐Injury Severity Score ICU: Intensive care unit ISS: injury severity score iv: intravenous MOSF: multiple organ system failure MV: mechanical ventilation OR: odds ratio PaO2:arterial oxygen partial pressure RCT: randomised controlled trial RD: risk difference RR: risk ratio RTI: respiratory tract infection SAPS: simplified acute physiology score SD: standard deviation SDD: selective decontamination of the digestive tract VAP: ventilator‐associated pneumonia VO2: oxygen consumption U: unit WBC: white blood count
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arnow 1996 | The study included a selected population of patients undergoing liver transplant. |
| Barret 2001 | The study included only paediatric burns patients. |
| Bion 1994 | The study included a selected population of patients undergoing liver transplant. |
| Bouter 2002 | The study included a selected population undergoing cardiopulmonary bypass. |
| Camus 2011 | Both groups received SDD. |
| Di Filippo 1999 | Experimental intervention not in the inclusion criteria: endonasal mupirocin for only 3 days |
| Flaherty 1990 | The study included a selected population of cardio‐surgical patients. |
| Garbino 2002 | The study tested the effectiveness of fluconazole, as both groups received SDD. |
| Godard 1990 | Non‐randomised study |
| Hellinger 2002 | The study included only liver transplant patients. |
| Huang 2013 | Comparison intervention not in the inclusion criteria: 1) screening and isolation of methicillin‐resistant Staphylococcus aureus (MRSA) carriers; 2) screening, isolation, and decolonisation of MRSA carriers |
| Hunefeld 1989 | Unclear description of the intervention compared |
| Jacobs 1995 | This study, which was included in the previous version of our review as a personal contact with the principal investigator, has now been excluded due to lack of feedback from the trial author. To date, this study has never been published. |
| Lipman 1994 | After contacting the principal investigator, it become apparent that it was not a randomised study. |
| Luiten 1995 | The study included a selected population of patients affected by pancreatitis characterised by a low percentage of ICU admissions. |
| Martinez 1994 | The study compared the effect of 2 different prophylactic regimens without a control group. |
| Martinez‐Pellus 1993 | The study included a selected population of cardio‐surgical patients. |
| Nardi 2001 | The study tested the effectiveness of mupirocin, as both groups received SDD. |
| Oostdijk 2014 | Comparison intervention not in the inclusion criteria (selective oropharyngeal decontamination) |
| Oudhuis 2011 | Comparison intervention not in the inclusion criteria (probiotics) |
| Rayes 2002 | The study included only liver transplant patients. |
| Rios 2005 | Data available only from other published meta‐analyses; not possible to retrieve data from the original study. |
| Rolando 1996 | The study included a selected population of patients with acute hepatic failure. |
| Ruza 1998 | The study included only paediatric burns patients. |
| Saidel‐Odes 2012 | Objective of the study was not to assess efficacy and safety of SDD on clinical outcomes. |
| Schardey 1997 | The study included a selected population of patients undergoing gastric surgery and characterised by a low percentage of ICU admission. |
| Silvestri 2004 | Both groups received topical prophylaxis. |
| Smith 1993 | The study included only paediatric liver transplant patients. |
| Tetteroo 1990 | The study included a selected population of patients undergoing oesophageal resection and characterised by a short length of stay in ICU. |
| Wittekamp 2018 | Comparison intervention not in the inclusion criteria (selective oropharyngeal decontamination, chlorhexidine) |
| Yilmazlar 2009 | Intervention not in the inclusion criteria: SDD for only 3 days |
| Zobel 1991 | The study only included paediatric patients. |
| Zwaveling 2002 | The study only included liver transplant patients. |
ICU: intensive care unit SDD: selective decontamination of the digestive tract
Characteristics of ongoing studies [ordered by study ID]
IRCT20180110038298N1.
| Study name | Effect of oropharyngeal decontamination by topical antibiotics on the ventilator‐associated pneumonia |
| Methods | Randomised, double‐blind, placebo‐controlled trial |
| Participants | 100 patients between 18 and 65 years admitted to the ICU. Performed intubation in the first 24 hours and stay at least 72 hours |
| Interventions | SOD versus no prophylaxis: 2% concentration of polymyxin, nystatin and neomycin, prepared and combined by investigator administered with a syringe on the oral cavity versus saline solution |
| Outcomes | Ventilator‐dependent pneumonia |
| Starting date | 2018 |
| Contact information | |
| Notes | Country: Iran |
NCT02389036.
| Study name | Selective Decontamination of the Digestive Tract in ICU Patients (SuDDICU) |
| Methods | Cluster‐randomised controlled trial |
| Participants | 10,000 patients predicted to remain ventilated beyond the end of the calendar day after the day first ventilated |
| Interventions | SDD oral paste, SDD gastric suspension, intravenous antibiotic versus standard care |
| Outcomes | All‐cause mortality at time of hospital discharge |
| Starting date | 2017 |
| Contact information | jmyburgh@georgeinstitute.org.au |
| Notes | Country: Australia |
ICU: intensive care unit SDD: selective decontamination of the digestive tract SOD: selective oropharyngeal decontamination
Differences between protocol and review
We modified the title to better reflect the topic of the review from 'Antibiotic prophylaxis to reduce respiratory tract infections and mortality in adults receiving intensive care' to 'Topical antibiotic prophylaxis to reduce respiratory tract infections and mortality in adults receiving mechanical ventilation'.
We changed the inclusion criteria to include cluster‐randomised controlled trials. We checked included and excluded trials from previous versions of the review to verify the adherence of the inclusion and exclusion criteria.
We assessed risk of bias of the included studies using the criteria recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
We amended the Methods section to comply with Methodological Expectations of Cochrane Intervention Reviews (MECIR) standards.
We added the outcomes dropouts due to adverse events, gastrointestinal adverse events, and allergic adverse events, as adverse events of a pharmacologic treatment are relevant outcomes.
We also included cluster‐RCTs as they provide useful data to the question addressed in the review.
Contributions of authors
Roberto D'Amico prepared the protocol and review, oversaw the data collection and critical appraisal of studies, and prepared the final version of the manuscript. Luca Brazzi collaborated on the preparation of the protocol. Silvia Pifferi and Silvia Minozzi screened the articles and selected studies for inclusion. Luca Brazzi contributed to screening in the case of doubt or disagreement. Luca Brazzi, Silvia Pifferi, and Giorgia Montrucchio wrote the Background and the Discussion. Silvia Minozzi, Silvia Pifferi, and Valentina Pecoraro contributed to the data extraction. Silvia Minozzi performed the statistical analyses. Valentina Pecoraro and Silvia Minozzi completed the 'Risk of bias' assessment and the assessment of the certainty of the evidence. All review authors contributed to writing and revising the final report.
Sources of support
Internal sources
Italian Cochrane Centre, Italy
External sources
No sources of support supplied
Declarations of interest
Silvia Minozzi: none known. Silvia Pifferi: none known. Luca Brazzi has been member of the advisory Board of the Prodigy trial sponsored by Medtronic. Valentina Pecoraro: none known. Giorgia Montrucchio: none known. Roberto D'Amico: none known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Abele‐Horn 1997 {published data only}
- Abele-Horn M, Dauber A, Bauernfeind A, Russwurm W, Seyfarth-Metzger I, Gleich P, et al. Decrease in nosocomial pneumonia in ventilated patients by selective oropharyngeal decontamination (SOD). Intensive Care Medicine 1997;23(2):187-95. [DOI] [PubMed] [Google Scholar]
Aerdts 1991 {published and unpublished data}
- Aerdts SJ, Dalen R, Clasener HA, Festen J, Lier HJ, Vollaard EJ. Antibiotic prophylaxis of respiratory tract infection in mechanically ventilated patients. Chest 1991;100(3):783-91. [DOI] [PubMed] [Google Scholar]
Bergmans 2001 {published data only}
- Bergmans DC, Bonten MJ, Gailard CA, Paling JC, Geest S, Tiel FH, et al. Prevention of ventilator-associated pneumonia by oral decontamination: a prospective randomized, double blind, placebo controlled study. American Journal of Respiratory and Critical Care Medicine 2001;164(3):382-8. [DOI] [PubMed] [Google Scholar]
Beshey 2014 {published data only}
- Beshey BN, Okasha AS, Eldin ME. Fluconazole and selective digestive decontamination for prevention of Candida infection in high risk critically ill patients. Alexandria Journal of Medicine 2014;50:93-8. [Google Scholar]
Blair 1991 {published and unpublished data}
- Blair P, Rowlands BJ, Lowry K, Webb H, Amstrong P, Smilie J. Selective decontamination of the digestive tract: a stratified, randomized, prospective study in a mixed intensive care unit. Surgery 1991;110(2):303-10. [PubMed] [Google Scholar]
Boland 1991 {published and unpublished data}
- Boland JP, Sadler DL, Stewart W, Wood DJ, Zerick W, Snodgrass KR. Reduction of nosocomial respiratory tract infections in the multiple trauma patients requiring mechanical ventilation by selective parenteral and enteral antisepsis regimen (SPEAR) in the intensive care. In: 17th Congress of Chemotherapy. Berlin, 1991. [ABSTRACT NUMBER: 465]
Brun‐Buisson 1989 {published and unpublished data}
- Brun-Buisson C, Legrand P, Rauss A, Richard C, Montravers F, Besbes M, et al. Intestinal decontamination for control of nosocomial multiresistant Gram-negative bacilli. Annals of Internal Medicine 1989;110(11):873-81. [DOI] [PubMed] [Google Scholar]
Camus 2005 {published data only}
- Camus C, Bellissant E, Sebille W, Perrotin D, Garo B, Legras A, et al. Prevention of acquired infections in intubated patients with combination of two decontaminations regimens. Critical Care Medicine 2005;33(2):307-14. [DOI] [PubMed] [Google Scholar]
Cerra 1992 {published and unpublished data}
- Cerra FB, Maddaus MA, Dunn DL, Wells CL, Konstantinides NN, Lehmann SL, et al. Selective gut decontamination reduces nosocomial infections and length of stay but not mortality or organ failure in surgical intensive care unit patients. Archives of Surgery 1992;127(2):163-9. [DOI] [PubMed] [Google Scholar]
Chaari 2014 {published data only}
- Chaari A, Zribi E, Dammak H, Ghadoun H, Chtara K, Sfar S, et al. Does selective digestive decontamination prevent ventilator-associated pneumonia in trauma patients? American Journal of Therapeutics 2014;21:470-6. [DOI] [PubMed] [Google Scholar]
Cockerill 1992 {published and unpublished data}
- Cockerill FR, Muller SR, Anhalt JP, Marsh HM, Farnell MB, Mucha P, et al. Prevention of infection in critically ill patients by selective decontamination of the digestive tract. Annals of Internal Medicine 1992;117(7):545-53. [DOI] [PubMed] [Google Scholar]
de Jonge 2003 {published data only}
- Jonge E, Schultz JM, Spanjaard L, Bossuyt PM, Vroom MB, Dankert J, et al. Effects of selective decontamination of digestive tract on mortality and acquisition of resistant bacteria in intensive care: a randomised controlled trial. Lancet 2003;362(9389):1011-6. [DOI] [PubMed] [Google Scholar]
de la Cal 2005 {published data only}
- la Cal MA, Cerdà E, Garcia-Hierro P, Saene HK, Gómez-Santos D, Negro E, et al. Survival benefit in critically hill burned patients receiving selective decontamination of the digestive tract. A randomized, placebo-controlled, double-blind trial. Annals of Internal Medicine 1992;117:545-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
de Smet 2009 {published data only}
- Smet AM, Kluytmans JA, Cooper BS, Mascini EM, Benus RF, Werf TS, et al. Decontamination of the digestive tract and oropharynx in ICU patients. New England Journal of Medicine 2009;360:20-31. [DOI] [PubMed] [Google Scholar]
Ferrer 1994 {published and unpublished data}
- Ferrer M, Torres A, Gonzàles J, la Bellacasa JP, El-Ebiary M, Roca M, et al. Utility of selective digestive decontamination in mechanically ventilated patients. Annals of Internal Medicine 1994;120:389-95. [DOI] [PubMed] [Google Scholar]
Finch 1991 {published and unpublished data}
- Finch RG, Tomlinson P, Holliday M, Sole K, Stack C, Rocker G. Selective decontamination of the digestive tract (SDD) in the prevention of secondary sepsis in a medical/surgical intensive care unit. In: 17th Congress of Chemotherapy. Berlin, 1991. [ABSTRACT NUMBER: 471]
Gastinne 1992 {published and unpublished data}
- Gastinne H, Wolff M, Delatour F, Faurisson F, Chevret S. A controlled trial in intensive care units of selective decontamination of the digestive tract with nonabsorbable antibiotics. New England Journal of Medicine 1992;326:594-9. [DOI] [PubMed] [Google Scholar]
Gaussorgues 1991 {published and unpublished data}
- Gaussorgues P, Salord F, Sirodot M, Tigaud S, Cagnin S, Gerard M, et al. Efficacité de la décontamination digestive sur la survenue des bactériémies nosocomiales chez les patients sous ventilation mécanique et recevant des betamimétiques. Reanimation, Soins Intensifs, Medicine d'Urgence 1991;7:169-74. [Google Scholar]
Georges 1994 {published data only}
- Georges B, Mazerolles M, Decun JF, Rouge P, Pomies S, Cougot P, et al. Décontamination digestive sélective résultats d'une ètude chez le polytraumatisé. Reanimation d'Urgence 1994;3:621-7. [Google Scholar]
Hammond 1992 {published and unpublished data}
- Hammond JM, Potgieter PD, Saunders GL, Forder AA. Double blind study of selective decontamination of the digestive tract in intensive care. Lancet 1992;340:5-9. [DOI] [PubMed] [Google Scholar]
Jacobs 1992 {published and unpublished data}
- Jacobs S, Foweraker JE, Roberts SE. Effectiveness of selective decontamination of the digestive tract (SDD) in an ICU with a policy encouraging a low gastric pH. Clinical Intensive Care 1992;3:52-8. [Google Scholar]
Kerver 1988 {published and unpublished data}
- Kerver AJ, Rommes JH, Mevissen-Verhage EA, Hulstaert PF, Vos A, Verhoef J, et al. Prevention of colonization and infection in critically ill patients: a prospective randomized study. Critical Care Medicine 1988;16:1087. [DOI] [PubMed] [Google Scholar]
Koeman 2006 {published data only}
- Koeman M, Ven A, Hak E, Joore H, Kaasjager K, Smet A, et al. Oral decontamination with chlorhexidine reduces the incidence of ventilator-associated pneumonia. American Journal of Respiratory and Critical Care Medicine 2006;173:1348-55. [DOI] [PubMed] [Google Scholar]
Korinek 1993 {published and unpublished data}
- Korinek AM, Laisne MJ, Raskine L, Deroin V, Sanson-Lepors MJ. Selective decontamination of the digestive tract in neurosurgical care units patients: a double blind, randomized, placebo-controlled study. Critical Care Medicine 1993;21:1466-73. [DOI] [PubMed] [Google Scholar]
Krueger 2002 {published data only}
- Krueger WA, Lenhart FP, Neeser G, Ruckdeschel G, Schreckhase H, Eissner HJ, et al. Influence of combined intravenous and topical antibiotic prophylaxis on the incidence of infections, organ dysfunctions and mortality in critically ill surgical patients. American Journal of Respiratory and Critical Care Medicine 2002;166:1029-37. [DOI] [PubMed] [Google Scholar]
- Lenhart FP, Unertl K, Neeser G, Ruckdeschel G, Eckart J, Peter K. Selective decontamination and sucralfate for prevention of acquired infections in intensive care. In: Thirty-fourth Interscience Conference on Antimicrobial Agents and Chemotherapy, October 4-7. Orlando, FL: American Society for Microbiology, Washington, DC, 1994. [ABSTRACT NUMBER: K101]
Laggner 1994 {published and unpublished data}
- Laggner AN, Tryba M, Georgopoulos A, Lenz K, Grimm G, Graninger W, et al. Oropharyngeal decontamination with gentamicin for long-term ventilated patients on stress ulcer prophylaxis with sucralfate? Wiener Klinische Wochenschrift 1994;106:15-9. [PubMed] [Google Scholar]
Lingnau 1997 {published and unpublished data}
- Lingnau W, Berger J, Javorsky F, Lejeune P, Mutz N, Benzer H. Selective intestinal decontamination in multiple trauma patients: prospective, controlled trials. Journal of Trauma 1997;42:687-94. [DOI] [PubMed] [Google Scholar]
Palomar 1997 {published and unpublished data}
- Palomar M, Alvarez-Lerma F, Jorda R, Bermejo B. Prevention of nosocomial infection in mechanically ventilated patients: selective digestive decontamination versus sucralfate. Clinical Intensive Care 1997;8:228-35. [Google Scholar]
Pneumatikos 2002 {published data only}
- Pneumatikos I, Koulouras V, Nathanail C, Goe D, Nakos G. Selective decontamination of subglottic area in mechanically ventilated patients with multiple trauma. Intensive Care Medicine 2002;28:432-7. [DOI] [PubMed] [Google Scholar]
Pugin 1991 {published and unpublished data}
- Pugin J, Auckenthaler R, Lew DP, Suter PM. Oropharyngeal decontamination decreases incidence of ventilator-associated pneumonia. JAMA 1991;265:2704-10. [PubMed] [Google Scholar]
Quinio 1995 {published and unpublished data}
- Quinio B, Albanèse J, Bues-Charbit M, Viviand X, Martin C. Selective decontamination of the digestive tract in multiple trauma patients: prospective, double blind, randomised, placebo-controlled study. Chest 1996;109:765-72. [DOI] [PubMed] [Google Scholar]
Rocha 1992 {published and unpublished data}
- Rocha LA, Martin MJ, Pita S, Paz J, Seco C, Margusino L, et al. Prevention of nosocomial infection in critically ill patients by selective decontamination of digestive tract. Intensive Care Medicine 1992;18:398-404. [DOI] [PubMed] [Google Scholar]
Rodriguez‐Roldan 1990 {published and unpublished data}
- Rodrìguez-Roldàn JM, Altuna-Cuesta A, Lòpez A, Carrillo A, Garcia J, Leòn J, et al. Prevention of nosocomial lung infection in ventilated patients: use of an antimicrobial pharyngeal nonabsorbable paste. Critical Care Medicine 1990;18:1239-42. [DOI] [PubMed] [Google Scholar]
Sanchez‐Garcia 1998 {published and unpublished data}
- Sanchez-Garcia M, Cambronero JA, Lopez J, Cerda E, Rubio J, Aguinaga G, et al. Effectiveness and cost of selective decontamination of the digestive tract (SDD) in critically ill intubated patients. A randomized, double blind, placebo-controlled, multicentric trial. American Journal of Respiratory and Critical Care Medicine 1998;158:908-16. [DOI] [PubMed] [Google Scholar]
Stoutenbeek 1996 {published data only}
- Stoutenbeek CP, Van Saene HK, Miranda DR, Zandstra DF. The effect of selective decontamination of the digestive tract on colonization and infection rate in multiple trauma patients. Intensive Care Medicine 1984;10:185-92. [DOI] [PubMed] [Google Scholar]
Stoutenbeek 2007 {published data only}
- Stoutenbeek CP, Van Saene HK, Little RA, Whitehead A. The effect of selective decontamination on the digestive tract on mortality in multiple trauma patients: a multicentre randomized controlled trial. Intensive Care Medicine 2007;33:261-70. [DOI] [PubMed] [Google Scholar]
Ulrich 1989 {published and unpublished data}
- Ulrich C, Harinck-deWeerd JE, Bakker NC, Jacz K, Doornbos L, Ridder VA. Selective decontamination of the digestive tract with norfloxacin in the prevention of ICU-acquired infections: a prospective randomized study. Intensive Care Medicine 1989;15:424-31. [DOI] [PubMed] [Google Scholar]
Unertl 1987 {published and unpublished data}
- Unertl K, Ruckdeschel G, Selbmann HK, Jensen U, Forst H, Lenhart FP, et al. Prevention of colonization and respiratory infections in long term ventilated patients by local antimicrobial prophylaxis. Intensive Care Medicine 1987;13:106-13. [DOI] [PubMed] [Google Scholar]
Verwaest 1997 {published and unpublished data}
- Verwaest C, Verhaegen J, Ferdinande P, Schets M, Van der Berghe G, Verbist L, et al. Randomized, controlled trial of selective digestive decontamination in 600 mechanically ventilated patients in a multidisciplinary intensive care unit. Critical Care Medicine 1997;25:63-71. [DOI] [PubMed] [Google Scholar]
Wiener 1995 {published data only}
- Wiener J, Itokazu G, Nothan C, Kabins SA, Weinstein RA. A randomized, double-blind, placebo-controlled trial of selective digestive decontamination in a medical-surgical intensive care unit. Clinical Infectious Diseases 1995;20:861-7. [DOI] [PubMed] [Google Scholar]
Winter 1992 {published and unpublished data}
- Winter R, Humphreys H, Pick A, MacGowan AP, Willatts SM, Speller DC. A controlled trial of selective decontamination of the digestive tract in intensive care and its effect on nosocomial infection. Journal of Antimicrobial Chemotherapy 1992;30:73-87. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Arnow 1996 {published data only}
- Arnow PM, Carandanq GC, Irwin ME. Randomized controlled trial of selective bowel decontamination for prevention of infections following liver transplantation. Clinical Infectious Diseases 1996;22:997-1003. [DOI] [PubMed] [Google Scholar]
Barret 2001 {published data only}
- Barret JP, Jeschke MG, Herndon DN. Selective decontamination of the digestive tract in severely burned pediatric patients. Burns 2001;27:439-45. [DOI] [PubMed] [Google Scholar]
Bion 1994 {published data only}
- Bion JF, Badger I, Crosby HA, Hutchings P, Kong KL, Baker J, et al. Selective decontamination of the digestive tract reduces Gram-negative pulmonary colonization but not systemic endotoxemia in patients undergoing elective liver transplantation. Critical Care Medicine 1994;22:40-9. [DOI] [PubMed] [Google Scholar]
Bouter 2002 {published data only}
- Bouter H, Schippers EF, Luelmo SA, Verteegh MI, Ros P, Guiot HF, et al. No effect of preoperative selective gut decontamination on endotoxemia and cytokine activation during cardiopulmonary bypass: a randomized, placebo controlled study. Critical Care Medicine 2002;30:38-43. [DOI] [PubMed] [Google Scholar]
Camus 2011 {published data only}
- Camus C, Bellissant E, Legras A, Renault A, Gacouin A, Lavoué S, et al. Randomized comparison of 2 protocols to prevent acquisition of methicillin-resistant Staphylococcus aureus: results of a 2-center study involving 500 patients. Infection Control and Hospital Epidemiology 2011;32(11):1604-72. [DOI] [PubMed] [Google Scholar]
Di Filippo 1999 {published data only}
- Di Filippo A, Simonetti T. Mupirocina endonasale nella prevenzione delle polmoniti nosocomiali. Minerva Anestesiologica 1999;65:109-13. [PubMed] [Google Scholar]
Flaherty 1990 {published data only}
- Flaherty J, Nathan C, Kabins SA, Weinstein RA. Pilot trial of selective decontamination for prevention of bacterial infection in an intensive care unit. Journal of Infectious Diseases 1990;162:1393-7. [DOI] [PubMed] [Google Scholar]
Garbino 2002 {published data only}
- Garbino J, Lew DP, Romand JA, Hogonnet S, Auckenthaler R, Pittet D. Prevention of severe candida infections in nonneutropenic high risk, critically ill patients: a randomized, double blinded, placebo controlled trial in patients treated by severe digestive decontamination. Intensive Care Medicine 2002;28:1708-17. [DOI] [PubMed] [Google Scholar]
Godard 1990 {published data only}
- Godard J, Guillaume C, Reverdy ME, Bachmann E, Bui-Xuan B, Nageotte A, et al. Intestinal decontamination in a polyvalent ICU. A double-blind study. Intensive Care Medicine 1990;16:307-11. [DOI] [PubMed] [Google Scholar]
Hellinger 2002 {published data only}
- Hellinger WC, Yao JD, Alvarez S, Blair JE, Cawlwy JJ, Paya CV, et al. A randomized, prospective, double blinded evaluation of selective bowel decontamination in liver transplantation. Transplantation 2002;73:1904-9. [DOI] [PubMed] [Google Scholar]
Huang 2013 {published data only}
- Huang SS, Septimus E, Kleinman K, Moody J, Hickok J, Avery TR, et al. Targeted versus universal decolonization to prevent ICU infection. New England Journal of Medicine 2013;368:2255-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hunefeld 1989 {published and unpublished data}
- Hünefeld G. Selective digestive decolonization in long-term ventilated surgical patients [Klinische Studie zur selectiven Darmdekolonisation bei 204 langzeitbeatmeten abdominal und unfallchirurgischen Intensivpatienten]. Anaesthesiologie und Reanimation 1989;14:131-53. [PubMed] [Google Scholar]
Jacobs 1995 {unpublished data only}
- Pifferi S. No title. Correspondence to Jacobs S Fax sent 1995.
Lipman 1994 {published and unpublished data}
- Lipman J, Klugman K, Luyt D, Kraus P, Litmanovitch M, Johnson D, et al. Unique trial design shows SDD to decrease and alter colonization of upper respiratory tract in a multidisciplinary ICU. Critical Care Medicine 1994;22:1. [Google Scholar]
Luiten 1995 {published data only}
- Luiten EJ, Hop WC, Lange JF, Bruining HA. Controlled clinical trial of selective decontamination for the treatment of severe acute pancreatitis. Annals of Surgery 1995;222:57-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Martinez 1994 {published data only}
- Martinez PA, Bru M, Jara P, Ruiz J, Sanmigue lM, Garcia PT. Does cefotaxime prevent early pneumonia in trauma patients receiving pharyngeal decontamination? Medicina Intensiva 1994;18:245-9. [Google Scholar]
Martinez‐Pellus 1993 {published data only}
- Martinez-Pellus AE, Merino P, Bru M, Coneyero R, Seller G, Munoz C, et al. Can selective digestive decontamination avoid the endotoxemia and cytokine activation promoted by cardiopulmonary bypass? Critical Care Medicine 1993;2:1684-91. [DOI] [PubMed] [Google Scholar]
Nardi 2001 {published data only}
- Nardi G, Di Silvestre A, De Monte A, Massarutti D, Proietti A, Grazia Troncon M, et al. Reduction in gram positive pneumonia and antibiotic consumption following the use of a SDD protocol including nasal and oral mupirocin. European Journal of Emergency Medicine 2001;8:203-14. [DOI] [PubMed] [Google Scholar]
Oostdijk 2014 {published data only}
- Oostdijk EA, Kesecioglu J, Schultz MJ, Visser CE, Jonge E, Essen EHR, et al. Effects of decontamination of the oropharynx and intestinal tract on antibiotic resistance in ICUs: a randomized clinical trial. JAMA 2014;312(14):1429-37. [DOI] [PubMed] [Google Scholar]
Oudhuis 2011 {published data only}
- Oudhuis GJ, Bergmans DC, Dormans T, Zwaveling JH, Kessels A, Prins MH, et al. Probiotics versus antibiotic decontamination of the digestive tract: infection and mortality. Intensive Care Medicine 2011;37:110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rayes 2002 {published data only}
- Rayes N, Seehofer D, Hansen S, Boucsein K, Muller AR, Serke S, et al. Early enteral supply of lactobacillus and fiber versus selective bowel decontamination: a controlled trial in liver transplant recipients. Transplantation 2002;74:123-7. [DOI] [PubMed] [Google Scholar]
Rios 2005 {published data only}
- Rios F, Maskin B, Sanez VA. Prevention of ventilator associated pneumonia by oral decontamination: prospective, randomised, double-blind, placebo controlled study. In: American Thoracic Society International Conference 2005 May 20-25. San Diego, United States, 2005:abstracts2view.com/ats05/view.php?nu=ATS5L_3301. [ABSTRACT NUMBER: C95] [POSTER NUMBER: 608]
Rolando 1996 {published and unpublished data}
- Rolando N, Wade J, Stangou A, Gimson AE, Wendon J, Philpott-Howard J, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transplantation and Surgery 1996;2:8-13. [DOI] [PubMed] [Google Scholar]
Ruza 1998 {published data only}
- Ruza F, Alvarado F, Herruzo R, Delgado MA, García S, Dorao P, et al. Prevention of nosocomial infection in a pediatric intensive care unit (PICU) through the use of selective digestive decontamination. European Journal of Epidemiology 1998;14:719-27. [DOI] [PubMed] [Google Scholar]
Saidel‐Odes 2012 {published data only}
- Saidel-Odes L, Polachek H, Peled N, Riesenberg K, Schlaeffer F, Trabelsi Y, et al. A randomized, double-blind, placebo-controlled trial of selective digestive decontamination using oral gentamicin and oral polymyxin E for eradication of carbapenem-resistant Klebsiella pneumoniae carriage. Infection Control and Hospital Epidemiology 2012;33(1):14-9. [DOI] [PubMed] [Google Scholar]
Schardey 1997 {published and unpublished data}
- Schardey HM, Joosten U, Finke U, Stanbach KH, Schauer R, Heiss A, et al. The prevention of anastomotic leakage after total gastrectomy with local decontamination. A prospective double-blind, randomized and placebo-controlled clinical multicenter trial. Annals of Surgery 1997;225:172-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Silvestri 2004 {published data only}
- Silvestri L, Saene HK, Milanese M, Fontana F, Gregori D, Oblach L, et al. Prevention of MRSA pneumonia by oral vancomycin decontamination: a randomised trial. European Respiratory Journal 2004;23:921-6. [DOI] [PubMed] [Google Scholar]
Smith 1993 {published data only}
- Smith SD, Jackson RJ, Hannakan CJ, Wadowsky RM, Tzakis AG, Rowe MI, et al. Selective decontamination in pediatric liver transplant. A randomized prospective study. Transplantation 1993;55:1306-9. [DOI] [PubMed] [Google Scholar]
Tetteroo 1990 {published and unpublished data}
- Tetteroo GW, Wagenvoort JH, Castelei A, Tilanus HW, Ince C, Bruining HA. Selective decontamination to reduce gram-negative colonisation and infections after oesophageal resection. Lancet 1990;335:704-7. [DOI] [PubMed] [Google Scholar]
Wittekamp 2018 {published data only}
- Wittekamp B, Plantinga NL, Cooper BS, Lopez-Contreras J, Coll P, Mancebo J, et al. Decontamination strategies and bloodstream infections with antibiotic-resistant microorganisms in ventilated patients. A randomized clinical trial. JAMA 2018;320(20):2087-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yilmazlar 2009 {published data only}
- Yilmazlar A, Ozyurt G, Kahveci F, Goral G. Selective digestive decontamination can be an infection-prevention regimen for the intoxicated patients. Journal of Pharmacology and Toxicology 2009;4:36-40. [Google Scholar]
Zobel 1991 {published data only}
- Zobel G, Kutting M, Grubbauer HM, Semmelrock HJ, Thiel W. Reduction of colonization and infection rate during pediatric intensive care by selective decontamination of digestive tract. CritIical Care Medicine 1991;19:1242-6. [DOI] [PubMed] [Google Scholar]
Zwaveling 2002 {published data only}
- Zwaveling JH, Maring JK, Klompmaker IJ, Haagsma EB, Bottema JT, Laseur M, et al. Selective decontamination of the digestive tract to prevent postoperative infection: a randomized placebo-controlled trial in liver transplant patients. Critical Care Medicine 2002;30(6):1204-9. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
IRCT20180110038298N1 {published data only}
- IRCT20180110038298N1. Effect of oropharyngeal decontamination by topical antibiotics on the ventilator associated pneumonia. www.who.int/trialsearch/Trial2.aspx?TrialID=IRCT20180110038298N1 23 Febrary 2018.
NCT02389036 {published data only}
- NCT02389036. Selective Decontamination of the Digestive Tract in Intensive Care Unit Patients (SuDDICU). clinicaltrials.gov/ct2/show/NCT02389036 17 March 2015.
Additional references
Adrie 2017
- Adrie C, Garrouste-Orgeas M, Ibn Essaied W, Schwebel C, Darmon M, Mourvillier B, et al. Attributable mortality of ICU-acquired bloodstream infections: impact of the source, causative micro-organism, resistance profile and antimicrobial therapy. Journal of Infection 2017;74:131-41. [DOI] [PubMed] [Google Scholar]
American Thoracic Society 2005
- American Thoracic Society, Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. American Journal of Respiratory and Critical Care Medicine 2005;171:388-416. [DOI] [PubMed] [Google Scholar]
Atkins 2004
- Atkins D, Best D, Briss PA, Eccles M, Falck-Ytter Y, Flottorp S, et al, GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004;328(7454):1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bekaert 2011
- Bekaert M, Timsit JF, Vansteelandt S, Depuydt P, Vesin A, Garrouste-Orgeas M, et al. Attributable mortality of ventilator-associated pneumonia: a reappraisal using causal analysis. American Journal of Respiratory and Critical Care Medicine 2011;184:1133-9. [DOI] [PubMed] [Google Scholar]
Bonell 2019
- Bonell A, Azarrafiy R, Huong VT, Viet TL, Phu VD, Dat VQ, et al. A systematic review and meta-analysis of ventilator-associated pneumonia in adults in Asia: an analysis of national income level on incidence and etiology. Clinical Infectious Diseases 2019;68:511-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bonten 2001
- Bonten MJ, Kullberg BJ, Filius PM, De Stichting Werkgroep Antibioticabeleid. Optimizing antibiotics policy in the Netherlands. VI.SWAB advice: No selective decontamination of intensive care patients on mechanical ventilation. Nederlands Tijdschrift voor Geneeskunde 2001;145:353-7. [PubMed] [Google Scholar]
Brink 2013
- Brink AJ, Coetzee J, Corcoran C, Clay CG, Hari-Makkan D, Jacobson RK, et al. Emergence of OXA-48 and OXA-181 carbapenemases among Enterobacteriaceae in South Africa and evidence of in vivo selection of colistin resistance as a consequence of selective decontamination of the gastrointestinal tract. Journal of Clinical Microbiology 2013;51:369-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chahoud 2015
- Chahoud J, Semaan A, Almoosa KF. Ventilator-associated events prevention, learning lessons from the past: a systematic review. Heart and Lung 2015;44:251-9. [DOI] [PubMed] [Google Scholar]
Cook 2010
- Cook A, Norwood S, Berne J. Ventilator-associated pneumonia is more common and of less consequence in trauma patients compared with other critically ill patients. Journal of Trauma 2010;69:1083-91. [DOI] [PubMed] [Google Scholar]
Daneman 2013
- Daneman N, Sarwar S, Fowler RA, Cuthbertson BH. Effect of selective decontamination on antimicrobial resistance in intensive care units: a systematic review and meta-analysis. Lancet Infectious Diseases 2013;13:328-41. [DOI] [PubMed] [Google Scholar]
de Jonge 2018
- Jonge E, Wilde RB, Juffermans NP, Oostdijk EA, Bernards AT, Essen EH, et al. Carriage of antibiotic-resistant Gram-negative bacteria after discontinuation of selective decontamination of the digestive tract (SDD) or selective oropharyngeal decontamination (SOD). Critical Care 2018;22(1):243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dudeck 2013
- Dudeck MA, Horan TC, Peterson KD, Allen-Bridson K, Morrell G, Anttila A, et al. National healthcare safety network report, data summary for 2011, device-associated module. American Journal of Infection Control 2013;41:286-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ego 2015
- Ego A, Preiser JC, Vincent JL. Impact of diagnostic criteria on the incidence of ventilator-associated pneumonia. Chest 2015;147:347-55. [DOI] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime), Accessed 10 July 2020. Available at gradepro.org.
Halaby 2013
- Halaby T, Al Naiemi N, Kluytmans J, Palen J, Vandenbroucke-Grauls CM. Emergence of colistin resistance in Enterobacteriaceae after the introduction of selective digestive tract decontamination in an intensive care unit. Antimicrobial Agents and Chemotherapy 2013;57:3224-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Heyland 1994
- Heyland DK, Cook DJ, Jaescher R, Griffith L, Lee HN, Guyatt GH. Selective decontamination of the digestive tract. An overview. Chest 1994;105:1221-9. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1.
Higgins 2020
- Higgins JP, Eldridge S, Li T (editors). Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020) Cochrane, 2020. Available from training.cochrane.org/handbook.
Hurley 1995
- Hurley JC. Prophylaxis with enteral antibiotics in ventilated patients: selective decontamination or selective cross-infection? Antimicrobial Agents and Chemotherapy 1995;39:941-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hurley 2020
- Hurley JC. Selective digestive decontamination is neither safe nor efficacious for critically ill patients. Critical Care Medicine 2020;48(5):732-5. [DOI] [PubMed] [Google Scholar]
Kollef 1994
- Kollef MH. The role of selective digestive tract decontamination on mortality and respiratory tract infections. A meta-analysis. Chest 1994;105:1101-8. [DOI] [PubMed] [Google Scholar]
Kollef 2012
- Kollef MH, Hamilton CW, Ernst FR. Economic impact of ventilator-associated pneumonia in a large matched cohort. Infection Control and Hospital Epidemiology 2012;33:250-6. [DOI] [PubMed] [Google Scholar]
Koulenti 2017
- Koulenti D, Tsigou E, Rello J. Nosocomial pneumonia in 27 ICUs in Europe: perspectives from the EU-VAP/CAP study. European Journal of Clinical Microbiology and Infectious Diseases 2017;36:1999-2006. [DOI] [PubMed] [Google Scholar]
Landelle 2018
- Landelle V, Nocquet Boyer M, Abbas M, Genevois E, Abidi N, Naimo S, et al. Impact of a multifaceted prevention program on ventilator‑associated pneumonia including selective oropharyngeal decontamination. Intensive Care Medicine 2018;44:1777-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Laupland 2006
- Laupland KB, Lee H, Gregson DB, Manns BJ. Cost of intensive care unit acquired bloodstream infections. Journal of Hospital Infection 2006;63:124-32. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1.
Liberati 2001
- Liberati A, D'Amico R, Brazzi L, Pifferi S. Influence of methodological quality on study conclusions. JAMA 2001;286:2544-5. [DOI] [PubMed] [Google Scholar]
Magill 2013
- Magill SS, Klompas M, Balk R, Burns SM, Deutschman CS, Diekema D, et al. Developing a new, national approach to surveillance for ventilator-associated events: executive summary. Clinical Infectious Diseases 2013;57(12):1742-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Melsen 2011
- Melsen WG, Rovers MM, Koeman M, Bonten MJ. Estimating the attributable mortality of ventilator-associated pneumonia from randomized prevention studies. Critical Care Medicine 2011;39:2736-42. [DOI] [PubMed] [Google Scholar]
Melsen 2013
- Melsen WG, Rovers MM, Groenwold RH, Bergmans DC, Camus C, Bauer T, et al. Attributable mortality of ventilator-associated pneumonia: a meta-analysis of individual patient data from randomised prevention studies. Lancet Infectious Diseases 2013;13:655-71. [DOI] [PubMed] [Google Scholar]
Nathens 1999
- Nathens AB. Selective decontamination of digestive tract in surgical patients: a systematic review of evidence. Archives of Surgery 1999;134:170-6. [DOI] [PubMed] [Google Scholar]
Plantinga 2017
- Plantinga NL, Smet A, Oostdijk EAN, Jonge E, Camus C, Krueger WA, et al. Selective digestive and oropharyngeal decontamination in medical and surgical ICU patients: individual patient data meta-analysis. Clinical Microbiology and Infection 2017;1(17):30477-9. [DOI] [PubMed] [Google Scholar]
Price 2014
- Price R, MacLennan G, Glen J, on behalf of the SuDDICU collaboration. Selective digestive or oropharyngeal decontamination and topical oropharyngeal chlorhexidine for prevention of death in general intensive care: systematic review and network meta-analysis. BMJ 2014;348:2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Redman 2001
- Redman R, Ludington E, Crocker M, Wittes J, Bellm L, Carlet J, and the VAP Advisory Group. Analysis of respiratory and non-respiratory infections in published trials of selective digestive decontamination. Intensive Care Medicine 2001;27(Suppl):S285. [Google Scholar]
Reignier 2016
- Reignier J, Mercier E, Le Gouge A, Boulain T, Desachy A, Bellec F, et al. Clinical Research in Intensive C, Sepsis G. Effect of not monitoring residual gastric volume on risk of ventilator-associated pneumonia in adults receiving mechanical ventilation and early enteral feeding: a randomized controlled trial. JAMA 2013;309:249-56. [DOI] [PubMed] [Google Scholar]
Reis 2015
- Reis MD, Citerio G, Perner A, Dimopoulos G, Torres A, Hoes A, et al. Use of selective digestive tract decontamination in European intensive cares: the ifs and whys. Minerva Anestesiologica 2015;81:734-42. [PubMed] [Google Scholar]
Righy 2017
- Righy C, Americano do Brasil PE, Vallés J, Bozza FA, Martin-Loeches I. Systemic antibiotics for preventing ventilator‑associated pneumonia in comatose patients: a systematic review and meta-analysis. Annals of Intensive Care 2017;7(1):67. [PMID: DOI: 10.1186/s13613-017-0291-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sánchez‐Ramírez 2018
- Sánchez-Ramírez C, Hípola-Escalada S, Cabrera-Santana M, Hernández-Viera MA, Caipe-Balcázar L, Saavedra P, et al. Long-term use of selective digestive decontamination in an ICU highly endemic for bacterial resistance. Critical Care 2018;22:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Seguin 2014
- Seguin P, Laviolle B, Dahyot-Fizelier C, Dumont R, Veber B, Gergaud S, et al, Study of povidone Iodine to reduce pulmonary Infection in head trauma and cerebral hemorrhage patients (SPIRIT) ICU study group. Effect of oropharyngeal povidone-iodine preventive oral care on ventilator-associated pneumonia in severely brain-injured or cerebral hemorrhage patients: a multicenter, randomized controlled trial. Critical Care Medicine 2014;42:1-8. [DOI] [PubMed] [Google Scholar]
Silvestri 2007
- Silvestri L, Saene HKF, Milanese M, Gregori D, Gullo A. Selective decontamination of the digestive tract reduces bacterial bloodstream infection and mortality in critically ill patients. Systematic review of randomized controlled trials. Journal of Hospital Infection 2007;65:187-203. [DOI] [PubMed] [Google Scholar]
Stoclin 2020
- Stoclin A, Rotolo F, Hicheri Y, Mons M, Chachaty E, Gachot B, et al. Ventilator-associated pneumonia and bloodstream infections in intensive care unit cancer patients: a retrospective 12-year study on 3388 prospectively monitored patients. Supportive Care in Cancer 2020;28:193-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stoutenbeek 1984
- Stoutenbeek CP, Saene HK, Miranda DR, Zandstra DF. The effect of selective decontamination of the digestive tract on colonization and infection rate in multiple trauma patients. Intensive Care Medicine 1984;10(4):185-92. [DOI] [PubMed] [Google Scholar]
SWAB 2018
- Stichting Werkgroep Antibioticabeleid (SWAB). SWAB-Richtlijn: selectieve decontaminatie bij patiënten op de intensive care (herziene versie 2018). http://swab.nl/exec/file/download/90 Accessed 20 September 2019.
Vanderbrouk‐Grauls 1991
- Vanderbrouk-Grauls CM, Vanderbrouke-Grauls JP. Effect of selective decontamination of the digestive tract on respiratory tract infections and mortality in intensive care unit. Lancet 1991;338:859-62. [DOI] [PubMed] [Google Scholar]
van Nieuwenhoven 2001
- Nieuwenhoven CA, Buskens E, Tiel FH, Bonten MJ. Relationship between methodological trial quality and the effects of selective digestive decontamination on pneumonia and mortality in critically ill patients. JAMA 2001;286:335-40. [DOI] [PubMed] [Google Scholar]
Vincent 2011
- Vincent JL, Jacobs F. Effect of selective decontamination on antibiotic resistance. Lancet Infectious Diseases 2011;11(5):337-8. [DOI] [PubMed] [Google Scholar]
Waters 2015
- Waters B, Muscedere J. A 2015 Update on ventilator-associated pneumonia: new insights on its prevention, diagnosis, and treatment. Current Infectious Disease Reports 2015;17:496. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
D'Amico 1998
- D'Amico R, Pifferi S, Leonetti C, Torri V, Tinazzi A, Liberati A, et al. Effectiveness of antibiotic prophylaxis in critically ill adult patients: systematic review of randomised controlled trials. BMJ 1998;316:1275-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
D'Amico 2009
- D'Amico R, Pieri S, Torri V, Brazzi L, Parmelli E, Liberati A. Antibiotic prophylaxis to reduce respiratory tract infections and mortality in adults receiving intensive care. Cochrane Database of Systematic Reviews 2009, Issue 4. Art. No: CD000022. [DOI: 10.1002/14651858.CD000022.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liberati 1997
- Liberati A, D'Amico R, Pifferi S, Leonetti C, Torri V, Brazzi L, et al. Antibiotic prophylaxis in adult patients treated in Intensive Care Units. Cochrane Database of Systematic Reviews 1997, Issue 3. Art. No: CD000022. [DOI: 10.1002/14651858.CD000022.pub2] [DOI] [Google Scholar]
Liberati 2002
- Liberati A, D'Amico R, Pifferi S, Leonetti C, Torri V, Brazzi L, et al. Antibiotics for preventing respiratory tract infections in adults receiving intensive care. Cochrane Database of Systematic Reviews 2002, Issue 4. Art. No: CD000022. [DOI: 10.1002/14651858.CD000022.pub2] [DOI] [PubMed] [Google Scholar]
Liberati 2004
- Liberati A, D'Amico R, Pifferi S, Torri V, Brazzi L. Antibiotic prophylaxis to reduce respiratory tract infections and mortality in adults receiving intensive care. Cochrane Database of Systematic Reviews 2004, Issue 1. Art. No: CD000022. [DOI: 10.1002/14651858.CD000022.pub2] [DOI] [PubMed] [Google Scholar]
SDD Group 1993
- SDD Trialists' Collaborative Group. Meta-analysis of randomised controlled trials of selective decontamination of the digestive tract. BMJ 1993;307:525-32. [DOI] [PMC free article] [PubMed] [Google Scholar]