

Abstract
Background
Women may experience differing types of pain and discomfort following birth, including cramping pain (often called after‐birth pain) associated with uterine involution, where the uterus contracts to reduce blood loss and return the uterus to its non‐pregnant size. This is an update of a review first published in 2011.
Objectives
To assess the effectiveness and safety of pharmacological and non‐pharmacological pain relief/analgesia for the relief of after‐birth pains following vaginal birth.
Search methods
For this update, we searched Cochrane Pregnancy and Childbirth’s Trials Register, ClinicalTrials.gov, the WHO International Clinical Trials Registry Platform (ICTRP) (31 October 2019), and reference lists of retrieved studies.
Selection criteria
Randomised controlled trials comparing two different types of analgesia or analgesia versus placebo or analgesia versus no treatment, for the relief of after‐birth pains following vaginal birth. Types of analgesia included pharmacological and non‐pharmacological. Quasi‐randomised trials were not eligible for inclusion.
Data collection and analysis
Two review authors independently assessed trials for inclusion, conducted 'Risk of bias' assessment, extracted data and assessed the certainty of the evidence using the GRADE approach.
Main results
In this update, we include 28 studies (involving 2749 women). The evidence identified in this review comes from middle‐ to high‐income countries. Generally the trials were at low risk of selection bias, performance bias and attrition bias, but some trials were at high risk of bias due to selective reporting and lack of blinding. Our GRADE certainty of evidence assessments ranged from moderate to very low certainty, with downgrading decisions based on study limitations, imprecision, and (for one comparison) indirectness.
Most studies reported our primary outcome of adequate pain relief as reported by the women. No studies reported data relating to neonatal adverse events, duration of hospital stay, or breastfeeding rates. Almost half of the included studies (11/28) excluded breastfeeding women from participating, making the evidence less generalisable to a broader group of women.
Non‐steroidal anti‐inflammatory drugs (NSAIDs) compared to placebo
NSAIDs are probably better than placebo for adequate pain relief as reported by the women (risk ratio (RR) 1.66, 95% confidence interval (CI) 1.45 to 1.91; 11 studies, 946 women; moderate‐certainty evidence). NSAIDs may reduce the need for additional pain relief compared to placebo (RR 0.15, 95% CI 0.07 to 0.33; 4 studies, 375 women; low‐certainty evidence). There may be a similar risk of maternal adverse events (RR 1.05, 95% CI 0.78 to 1.41; 9 studies, 598 women; low‐certainty evidence).
NSAIDs compared to opioids
NSAIDs are probably better than opioids for adequate pain relief as reported by the women (RR 1.33, 95% CI 1.13 to 1.57; 5 studies, 560 women; moderate‐certainty evidence) and may reduce the risk of maternal adverse events (RR 0.62, 95% CI 0.43 to 0.89; 3 studies, 255 women; low‐certainty evidence). NSAIDs may be better than opioids for the need for additional pain relief, but the wide CIs include the possibility that the two classes of drugs are similarly effective or that opioids are better (RR 0.37, 95% CI 0.12 to 1.12; 2 studies, 232 women; low‐certainty evidence).
Opioids compared to placebo
Opioids may be better than placebo for adequate pain relief as reported by the women (RR 1.26, 95% CI 0.99 to 1.61; 5 studies, 299 women; low‐certainty evidence). Opioids may reduce the need for additional pain relief compared to placebo (RR 0.48, 95% CI 0.28 to 0.82; 3 studies, 273 women; low‐certainty evidence). Opioids may increase the risk of maternal adverse events compared with placebo, although the certainty of evidence is low (RR 1.59, 95% CI 0.99 to 2.55; 3 studies, 188 women; low‐certainty evidence).
Paracetamol compared to placebo
Very low‐certainty evidence means we are uncertain if paracetamol is better than placebo for adequate pain relief as reported by the women, the need for additional pain relief, or risk of maternal adverse events (2 studies, 123 women).
Paracetamol compared to NSAIDs
Very low‐certainty evidence means we are uncertain if there are any differences between paracetamol and NSAIDs for adequate pain relief as reported by the women, or the risk of maternal adverse events. No data were reported about the need for additional pain relief comparing paracetamol and NSAIDs (2 studies, 112 women).
NSAIDs compared to herbal analgesia
We are uncertain if there are any differences between NSAIDs and herbal analgesia for adequate pain relief as reported by the women, the need for additional pain relief, or risk of maternal adverse events, because the certainty of evidence is very low (4 studies, 394 women).
Transcutaneous nerve stimulation (TENS) compared to no TENS
Very low‐certainty evidence means we are uncertain if TENS is better than no TENS for adequate pain relief as reported by the women. No other data were reported comparing TENS with no TENS (1 study, 32 women).
Authors' conclusions
NSAIDs may be better than placebo and are probably better than opioids at relieving pain from uterine cramping/involution following vaginal birth. NSAIDs and paracetamol may be as effective as each other, whereas opioids may be more effective than placebo. Due to low‐certainty evidence, we are uncertain about the effectiveness of other forms of pain relief. Future trials should recruit adequate numbers of women and ensure greater generalisability by including breastfeeding women. In addition, further research is required, including a survey of postpartum women to describe appropriately their experience of uterine cramping and involution. We identified nine ongoing studies, which may help to increase the level of certainty of the evidence around pain relief due to uterine cramping in future updates of this review.
Plain language summary
Relief of pain caused by uterine cramping or involution after giving birth
This updated review investigates the effectiveness and safety of drug and non‐drug pain relief in women experiencing after‐birth pains following vaginal birth. Giving an agent for pain relief was compared to an inactive placebo, to no treatment, or to a different type of agent in randomised controlled trials.
What is the issue?
Women may experience cramping pain and discomfort following the birth of their baby, as the uterus contracts and returns to its normal pre‐pregnancy size. These pains usually last for two to three days after the birth. Women who have previously had a baby are more likely to experience after‐birth pains. Breastfeeding stimulates the uterus to contract and increases the severity of the pains.
Types of pain relief used to treat the pain include paracetamol, non‐steroidal anti‐inflammatory drugs (NSAIDs) ibuprofen and naproxen, opioids including codeine, and non‐medicine methods such as herbal preparations and transcutaneous electrical nerve stimulation (TENS).
Why is this important?
Management of pain after birth is important, as the pain can affect a mother carrying out her normal activities as well as bonding with and caring for her baby. After‐pains can interfere with establishing breastfeeding.
What evidence did we find?
We searched for evidence from randomised controlled trials (October 2019) and identified 28 studies (2749 mothers) who were in hospital after uncomplicated single births. Most of the evidence is low‐certainty because the studies did not include sufficient numbers of women. Many of the studies excluded breastfeeding women.This makes the evidence less relevant to a broader group of women. No studies reported evidence on adverse events in the newborn infants.
NSAIDs are probably better than placebo (a dummy treatment) in giving adequate pain relief as reported by the women (11 studies, 946 women; moderate‐certainty evidence), and they may reduce the need for additional pain relief (4 studies, 375 women; low‐certainty evidence). There may be little difference between NSAIDs and placebo in the risk of adverse events in the mother (9 studies, 598 women; low‐certainty evidence).
NSAIDs are probably better than opioids in providing adequate pain relief as reported by the women (5 studies, 560 women; moderate‐certainty evidence) and may reduce the risk of adverse events in the mother (3 studies, 255 women; low‐certainty evidence). NSAIDs may slightly reduce the need for additional pain relief compared with opioids (2 studies, 232 women; low‐certainty evidence).
Opioids may be better than placebo for adequate pain relief as reported by the women (5 studies, 299 women; low‐certainty evidence) and for the need for additional pain relief (3 studies, 273 women; low‐certainty evidence). Opioids may increase the risk of adverse events in the mother compared with placebo (3 studies, 188 women; low‐certainty evidence).
Very low‐certainty evidence means we are uncertain if paracetamol is better than placebo for adequate pain relief as reported by the women, the need for additional analgesia, or risk of maternal adverse events (2 studies, 123 women).
Very low‐certainty evidence means we are uncertain if there are any differences between paracetamol and NSAIDs for adequate pain relief as reported by the women, or the risk of maternal adverse events (2 studies, 112 women).
Very low‐certainty evidence means we are uncertain if NSAIDs are better than herbal pain relief for adequate pain relief as reported by the women (4 studies, 394 women), the need for additional pain relief (1 study, 90 women) or risk of maternal adverse events (1 study, 108 women).
Very low‐certainty evidence means we are uncertain if there is any difference between TENS and no TENS for adequate pain relief as reported by the women (1 study, 32 women).
What does this mean?
NSAIDs may be better than placebo and are probably better than opioids at relieving after‐birth pains following vaginal birth. The quality of the evidence was poor and we are uncertain about the effectiveness of other forms of pain relief. Future trials should recruit adequate numbers of women and ensure greater relevance by including breastfeeding women. Further research could also include a survey of women after delivery to capture their experience of after‐birth pains following vaginal birth.
Summary of findings
Background
Description of the condition
Women may experience differing types of pain and discomfort following the birth of their baby. This may include incisional pain after a caesarean section, perineal pain following perineal trauma or episiotomy during vaginal birth, nipple pain from breastfeeding and cramping pain (often called after‐birth pain) associated with involution of the uterus. Following birth, the uterus returns to its normal size through involution, a process of intermittent uterine contractions. These involutionary contractions may be painful and are commonly felt for two or three days after birth (Paliulyte 2017).
The incidence and severity of after‐birth pains is not widely reported. However, multiparous women usually experience more pain as the lost tone of the uterus of the multiparous woman contracts and relaxes alternately (Blackburn 2013). This is also true of a uterus that is greatly distended by a multiple pregnancy or polyhydramnios (Pessel 2019). It has been further hypothesised that childbirth can induce central neural changes that increase predisposition for pain during the postpartum period, suggesting multiparous women's perception of uterine cramp pain is increased through a process of central sensitisation of nociceptive neurons (Marieb 2019). Endogenous oxytocin released during breastfeeding stimulates the uterus to contract and increases the severity of after‐birth pains felt by the mother (Wambach 2021). Thus after‐birth pains may hinder successful breastfeeding, reducing the mother's ability to care for her new baby and may impair the establishment of good‐quality mother‐baby interactions. In contrast, the uterus of the primiparous woman remains contracted after birth (Rankin 2017), hence these women do not commonly experience after‐birth pains. It has been documented that some women consider their after‐birth pains to be a major burden requiring powerful analgesia (Baddock 2019).
A number of randomised trials comparing the safety and effectiveness of various pharmacological and non‐pharmacological forms of pain relief have been published. After‐birth pains and perineal tissue injury after vaginal birth are established clinical pain conditions that call for the investigation of the efficacy of new pain relief treatments including non‐pharmacological and pharmacological analgesia (Bloomfield 1998; Mall 2019).
Description of the intervention
Analgesia is any agent used to relieve pain. The Oxford Advanced Learner's Dictionary defines the term analgesia as "the loss of the ability to feel pain while still conscious" (Oxford 2020). Analgesia includes pharmacological and non‐pharmacological interventions aiming to relieve pain. Pharmacological analgesia can be further classified: simple analgesics (including paracetamol and non‐steroidal anti‐inflammatory drugs (NSAIDs) like aspirin and naproxen), opioid analgesics (including codeine and morphine) (MIMS 2020), and herbal analgesics. Herbal remedies are usually not required to be registered and are derived from a plant or plant part or an extract or mixture of these (Merriam‐Webster 2020). There are many different herbal preparations thought to have anti‐inflammatory properties which have been used for centuries for this purpose (Maroon 2010). There is interest in alternative anti‐inflammatory options, given the side‐effect profile of many NSAIDs (Oguntibeju 2018).
Non‐pharmacological analgesia may include massage, heat packs, cold packs, hypnotherapy, hydrotherapy, acupuncture and transcutaneous electrical nerve stimulation (TENS) (Coutaux 2017; Gallo 2018).
How the intervention might work
Analgesia can stop or decrease pain or the perception of pain in several ways. Systemic analgesic drugs can be categorised into different classes:
Simple analgesics like paracetamol inhibit central nervous system prostaglandin synthesis (Ritter 2019);
NSAIDs, including aspirin and naproxen, have an anti‐inflammatory action (Ritter 2019);
Narcotic analgesics including codeine and morphine reduce perception of pain by inhibiting pain‐transmission neurons and reducing the psychological response to pain (Ritter 2019);
Herbal preparations used as analgesics are believed to inhibit inflammatory pathways, similarly to NSAIDs (Maroon 2010);
TENS is thought to inhibit nociception through somatosensory electrical input (Peng 2019).
Why it is important to do this review
Women may experience pain after birth from several sources, including uterine involution and perineal trauma. Management of pain after birth is important and can impact on a woman's return to normal activities and caring for her baby.
There is little in the literature to guide women and clinicians in the management of pain from uterine cramping/involution. The aim of this review is to systematically assess what is known about the effectiveness and safety of analgesia for relief of pain from uterine cramping/involution.
This review is an update of a review first published in 2011 and will contribute to what is known about the management of postpartum pain.
Objectives
To assess the effectiveness and safety of pharmacological and non‐pharmacological pain relief/analgesia for the relief of after‐birth pains following vaginal birth.
Methods
Criteria for considering studies for this review
Types of studies
We assessed all identified published and unpublished randomised controlled trials (RCTs), comparing two different types of analgesia or analgesia versus placebo or analgesia versus no treatment, for the relief of after‐birth pains following vaginal birth. We have included studies that met the inclusion criteria, including those which were reported in abstract form only. We excluded abstracts reporting interventions for postpartum pain that did not separately report on pain from uterine involution. We also excluded studies reporting interventions specifically for the prevention of pain due to uterine cramping/involution. We further excluded studies where pain due to uterine cramps was not reported separately from other pain. We have not included quasi‐randomised studies in this review. Cluster‐randomised trials were eligible for inclusion.
Types of participants
Women who have given birth vaginally, requiring analgesia for after‐birth pains.
Types of interventions
Randomised controlled trials comparing any type of analgesia (excluding pharmacological analgesics that are no longer available or that are not approved for use in this population) for after‐birth pains following vaginal birth versus:
any other type of analgesia;
placebo;
no treatment.
Analgesic intervention may be administered once as a single dose or with the dosage repeated at therapeutic intervals.
Types of outcome measures
Primary outcomes
Adequate pain relief as reported by the woman, or by determination of > 50% relief of pain (either as stated by the woman or calculated using a formula)*
Secondary outcomes
Need for additional pain relief
Pain relief, however measured by the authors
Number of women with adverse events, including nausea, vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness, psychological impact
Number of infants with adverse events, including vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness
Duration of hospital stay
Any breastfeeding at hospital discharge
Any breastfeeding at six weeks postpartum
Maternal views (using a validated questionnaire)
Maternal postpartum depression
*Assessment of 50% pain relief via summed pain intensity difference (SPID) scores (1.23 x SPID%max ‐ 2.3 = proportion with 50%) (Cooper 1997; Moore 1997a; Moore 1997b).
Search methods for identification of studies
The following Methods section is based on a standard template used by Cochrane Pregnancy and Childbirth.
Electronic searches
For this update, we searched Cochrane Pregnancy and Childbirth’s Trials Register by contacting their Information Specialist (31 October 2019).
The Register is a database containing over 25,000 reports of controlled trials in the field of pregnancy and childbirth. It represents over 30 years of searching. For full current search methods used to populate Pregnancy and Childbirth’s Trials Register, including the detailed search strategies for CENTRAL, MEDLINE, Embase and CINAHL; the list of handsearched journals and conference proceedings, and the list of journals reviewed via the current awareness service, please follow this link.
Briefly, Cochrane Pregnancy and Childbirth’s Trials Register is maintained by their Information Specialist and contains trials identified from:
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
weekly searches of MEDLINE (Ovid);
weekly searches of Embase (Ovid);
monthly searches of CINAHL (EBSCO);
handsearches of 30 journals and the proceedings of major conferences;
weekly current awareness alerts for a further 44 journals plus monthly BioMed Central email alerts.
Search results are screened by two people and the full text of all relevant trial reports identified through the searching activities described above is reviewed. Based on the intervention described, each trial report is assigned a number that corresponds to a specific Pregnancy and Childbirth review topic (or topics), and is then added to the Register. The Information Specialist searches the Register for each review using this topic number rather than keywords. This results in a more specific search set that has been fully accounted for in the relevant review sections (Included studies; Excluded studies; Ongoing studies).
In addition, we searched ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform (ICTRP) for unpublished, planned and ongoing trial reports (31 October 2019), using the search methods detailed in Appendix 1.
Searching other resources
We tried to contact the original trial authors for clarification or additional data (this is identified in the tables under included or excluded studies), and searched the reference lists of trials and review articles.
We did not apply any language restrictions.
Data collection and analysis
For methods used in the previous version of this review, seeDeussen 2011.
The following Methods section is based on a standard template used by Cochrane Pregnancy and Childbirth.
For this update, we used the following methods for assessing the 56 reports that we identified as a result of the updated search.
We defined the number of participants achieving adequate pain relief as one of the following:
The number of women reporting 'good' or 'excellent' pain relief when asked about their level of pain relief four to six hours after receiving their allocated treatment (we extracted the information as dichotomous data);
The number of women who reported 50% pain relief, or greater;
The number of women who achieved 50% pain relief, or greater, as calculated by using derived pain relief scores (TOTPAR (total pain relief) or SPID) over four to six hours.
It is common to use categorical or visual analogue scales for pain intensity and to calculate the results for each participant over periods of four or six hours, as SPID or TOTPAR (Moore 1996). From these categorical scales, it was possible to convert results into dichotomous data (the proportion of participants achieving at least 50%, or greater, max TOTPAR) using standard formulae (Moore 1996; Moore 1997b). Converting data in this way allowed us to use these data in a meta‐analysis (Moore 1997a; Moore 1997b). We used the following equations to estimate the proportions of participants achieving at least 50% of maximum TOTPAR.
Proportion with more than 50% maxTOTPAR = (1.33 x mean %maxTOTPAR – 11.5)
With %maxTOTPAR = mean TOTPAR x 100/(maximum score x number of hours) Cooper 1997; Moore 1997b).
Proportion with more than 50% maxTOTPAR = (1.36 x mean %maxSPID – 2.3)
With %maxSPID = mean SPID x 100/(maximum score x number of hours) (Cooper 1997; Moore 1997a).
The number of participants achieving at least 50% maxTOTPAR was then calculated by multiplying the proportions of participants with at least 50% maxTOTPAR by the total number of participants in the treatment groups. The number of participants with at least 50% maxTOTPAR was then used to calculate the relative benefit and number needed to treat for an additional beneficial outcome.
Where studies used more than one method of calculating adequate pain relief, our preference for analyses and reporting purposes, in order of decreasing preference, was: i) the proportion with at least 50% maxTOTPAR calculated using SPID; ii) the proportion with at least 50% maxTOTPAR calculated using TOTPAR; and iii) the number of participants reporting 'good' or 'excellent' pain relief/number of participants reporting at least 50% pain relief. We also assessed the number of participants who re‐medicated in the period of four to eight hours, as well as the median time to re‐medication, if data were available.
Selection of studies
Two review authors independently assessed for inclusion all the potential studies identified as a result of the search strategy. We resolved any disagreement through discussion or, if required, we consulted a third review author.
Data extraction and management
We designed a form to extract data. For eligible studies, two review authors extracted the data using the agreed form. We resolved discrepancies through discussion or, if required, we consulted a third review author. We entered the data into Review Manager 5 software (RevMan 2014) and checked them for accuracy.
When information about any of the above was unclear, we planned to contact authors of the original reports to provide further details.
We contacted a number of authors of the original reports to provide us with further details. However, the response rate was low, and is identified in the tables of included and excluded studies (seeCharacteristics of included studies and Characteristics of excluded studies).
Assessment of risk of bias in included studies
Two review authors independently assessed risks of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2020a). Any disagreement was resolved by discussion or by involving a third assessor.
(1) Random sequence generation (checking for possible selection bias)
We describe for each included study the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups.
We assessed the method as:
low risk of bias (any truly random process, e.g. random‐number table; computer random‐number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital or clinic record number);
unclear risk of bias.
(2) Allocation concealment (checking for possible selection bias)
We describe for each included study the method used to conceal allocation to interventions prior to assignment and assessed whether intervention allocation could have been foreseen in advance of or during recruitment, or changed after assignment.
We assessed the methods as:
low risk of bias (e.g. telephone or central randomisation; consecutively‐numbered sealed opaque envelopes);
high risk of bias (open random allocation; unsealed or non‐opaque envelopes; alternation; date of birth);
unclear risk of bias.
(3.1) Blinding of participants and personnel (checking for possible performance bias)
We describe for each included study the methods used, if any, to blind study participants and personnel from knowledge of which intervention a participant received. We considered that studies were at low risk of bias if they were blinded, or if we judged that the lack of blinding unlikely to affect results. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed the methods as:
low, high or unclear risk of bias for participants;
low, high or unclear risk of bias for personnel.
(3.2) Blinding of outcome assessment (checking for possible detection bias)
We describe for each included study the methods used, if any, to blind outcome assessors from knowledge of which intervention a participant received. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed methods used to blind outcome assessment as:
low, high or unclear risk of bias.
(4) Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
We describe for each included study, and for each outcome or class of outcomes, the completeness of data including attrition and exclusions from the analysis. We state whether attrition and exclusions were reported and the numbers included in the analysis at each stage (compared with the total randomised participants), reasons for attrition or exclusion where reported, and whether missing data were balanced across groups or were related to outcomes. Where sufficient information was reported, or could be supplied by the trial authors, we planned to re‐include missing data in the analyses which we undertook.
We assessed methods as:
low risk of bias (e.g. no missing outcome data; missing outcome data balanced across groups);
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups; ‘as treated’ analysis done with substantial departure of intervention received from that assigned at randomisation);
unclear risk of bias.
(5) Selective reporting (checking for reporting bias)
We describe for each included study how we investigated the possibility of selective outcome reporting bias and what we found.
We assessed the methods as:
low risk of bias (where it is clear that all of the study’s prespecified outcomes and all expected outcomes of interest to the review have been reported);
high risk of bias (where not all the study’s prespecified outcomes have been reported; one or more reported primary outcomes were not prespecified; outcomes of interest are reported incompletely and so cannot be used; study fails to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias.
(6) Other bias (checking for bias due to problems not covered by (1) to (5) above)
We describe for each included study any important concerns we had about other possible sources of bias.
Measures of treatment effect
Dichotomous data
For dichotomous data, we present results as the summary risk ratio (RR) with its 95% confidence interval (CI).
Continuous data
We used the mean difference (MD) if outcomes were measured in the same way between trials. We used the standardised mean difference (SMD) to combine trials that measured the same outcome, but used different methods.
Unit of analysis issues
Cluster‐randomised trials
We intended to include cluster‐randomised trials in the analyses along with individually‐randomised trials, although none were identified. If identified in future updates, we will adjust their sample sizes or standard errors using the methods described in the Cochrane Handbook, Section 23.1.4 (Higgins 2020b), using an estimate of the intracluster correlation co‐efficient (ICC) derived from the trial, if possible, from a similar trial or from a study of a similar population. If we use ICCs from other sources, we will report this and conduct sensitivity analyses to investigate the effect of variation in the ICC. If we identify both cluster‐randomised trials and individually‐randomised trials, we plan to synthesise the relevant information. We will consider it reasonable to combine the results from both if there is little heterogeneity between the study designs and we consider the interaction between the effect of intervention and the choice of randomisation unit to be unlikely. We will also acknowledge heterogeneity in the randomisation unit and perform a sensitivity or subgroup analysis to investigate the effects of the randomisation unit.
Cross‐over trials
We identified cross‐over trials as not being appropriate for this intervention.
Dealing with missing data
For included studies, we noted levels of attrition. In future updates, if more eligible studies are included, we will explore the impact of including studies with high levels of missing data in the overall assessment of treatment effect by using sensitivity analysis.
For all outcomes, we conducted analyses, as far as possible, on an intention‐to‐treat basis, i.e. we attempted to include all participants randomised to each group in the analyses. The denominator for each outcome in each trial was the number randomised minus any participants whose outcomes were known to be missing.
Assessment of heterogeneity
We assessed statistical heterogeneity in each meta‐analysis by visual inspection of the forest plot and by using I2 and Chi2 statistics. We interpreted I2 as follows:
0% to 40%: heterogeneity might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity
We were unable to explore substantial heterogeneity by subgroup analysis as the range of analgesia was so wide that subgroup comparison was not possible. We had intended to explore the data with a subgroup analysis for caesarean section, but it was too difficult to differentiate between incisional pain and uterine cramping; hence we excluded these data from the review.
Assessment of reporting biases
Where there were 10 or more studies in the meta‐analysis we investigated reporting biases (such as publication bias) using funnel plots. We assessed funnel plot asymmetry visually, and if asymmetry was suggested by a visual assessment we performed exploratory analyses to investigate it.
Data synthesis
We carried out statistical analysis using the Review Manager 5 software (RevMan 2014). We used a fixed‐effect meta‐analysis for combining data where it was reasonable to assume that studies were estimating the same underlying treatment effect, i.e. where trials were examining the same intervention, and we judged the trials’ populations and methods to be sufficiently similar.
If there was clinical heterogeneity sufficient to expect that the underlying treatment effects differed between trials, or if we detected substantial statistical heterogeneity, we used a random‐effects meta‐analysis to produce an overall summary if we considered an average treatment effect across trials to be clinically meaningful. The random‐effects summary was treated as the average range of possible treatment effects and discussed the clinical implications of treatment effects differing between trials. If the average treatment effect was not clinically meaningful, we did will not combine trials. Where we used random‐effects analyses, we present the results as the average treatment effect with a 95% confidence interval.
Subgroup analysis and investigation of heterogeneity
We intended to explore possible sources of heterogeneity using subgroup analyses. However, this was not possible with the included trials. The range of analgesia, the timing of observations and the types of observations were too varied.
In future updates of this review, as more data become available, we plan to carry out the following subgroup analyses:
nulliparous versus primiparous;
up to six hours after birth versus more than six hours; up to 12 hours after birth versus more than 12 hours; up to 18 hours after birth versus more than 18 hours; up to 24 hours after birth versus more than 24 hours; up to 48 hours after birth versus more than 48 hours; up to 72 hours after birth versus more than 72 hours;
type of anaesthesia during birth (for example, epidural anaesthesia versus no anaesthesia).
We will restrict subgroup analyses to the primary outcomes.
We will assess subgroup differences by interaction tests available within RevMan 5 (RevMan 2014). We will report the results of subgroup analyses quoting the Chi2 statistic and P value, and the interaction test I2 value.
Sensitivity analysis
We intended to conduct sensitivity analyses by comparing the outcomes before and after exclusion of the trials at high risk of bias or unclear risk of bias for sequence generation or allocation concealment; however, the included trials and their outcomes were too varied.
Summary of findings and assessment of the certainty of the evidence
For this update we assessed the certainty of the evidence using the GRADE approach, as outlined in the GRADE handbook, to consider the certainty of the body of evidence relating to the following comparisons.
NSAID versus placebo
NSAID versus opioid
Opioid versus placebo
Paracetamol versus placebo
Paracetamol verses NSAID
NSAID versus herbal analgesia
TENS versus no TENS
We included the following outcomes in the assessment of the certainty of evidence:
Adequate pain relief as reported by the woman;
Need for additional pain relief;
Number of women with adverse events, including nausea, vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness, psychological impact;
Number of infants with adverse events, including vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness;
Duration of hospital stay;
Any breastfeeding at hospital discharge;
Any breastfeeding at six weeks postpartum.
We used GRADEpro Guideline Development Tool to create ’Summary of findings’ tables. We produced a summary of the intervention effect and a measure of certainty for each of the above outcomes using the GRADE approach. The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the certainty of the body of evidence for each outcome. The evidence can be downgraded from 'high certainty' by one level for serious (or by two levels for very serious) limitations.
Results
Description of studies
Results of the search
See Figure 1.
1.
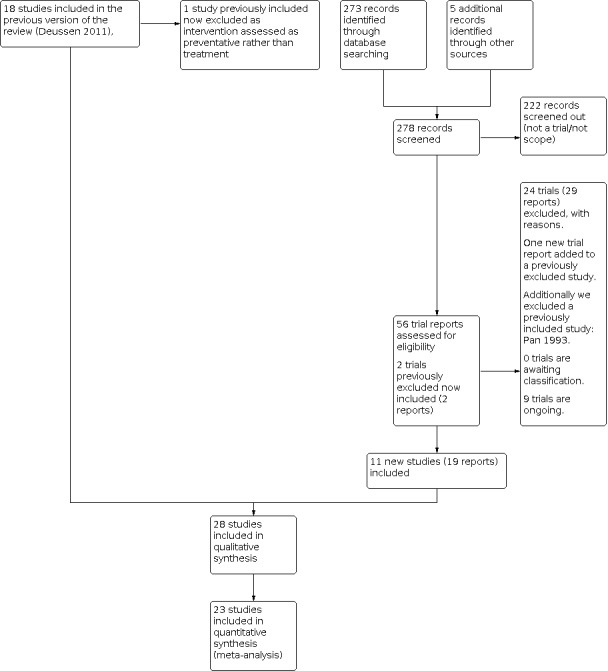
Study flow diagram.
For this update we identified 56 trial reports to assess.
We included nine new trials (17 reports) (Asti 2011; Chananeh 2018; Dastjerdi 2019; De Sousa 2014; Kantor 1984a; Kheiriyat 2016; Ozgoli 2017; Pourmaleky 2013; Simbar 2015). We excluded 24 new studies (29 reports) (Afravi 2019; Bachar 2018; Bahri 2019; Barhan 2019; Bilgin 2016; Blue 2018; Can 2015; Cunha 2011; Katz 2019; Kayman‐Kose 2014; Kenton 2011; Kim 2019; Kumbar 2017; Li 2014; Li 2015; Mirror 2019; Narimatsu 2001; Nazari 2018; Ozgoli 2018; Parsa 2019; Soltani 2017; Tafazoli 2013; Vaziri 2017; Yogev 2015) and added one trial report to a previously excluded study (Sunshine 1983). We considered two trials, previously excluded because they were conference proceedings that did not include enough detail for inclusion, now eligible for inclusion in this update (Bloomfield 1983; Bloomfield 1986c), although still providing no data.
There are nine ongoing studies (IRCT2015050322053N1; IRCT20190217042739N1; NCT04037202; IRCT2016070428240N2; IRCT2016100930238N1; IRCT20171208037792N1; IRCT201707283860N33; IRCT20180428039454N1; NCT03617900), all of which are trial registrations. We have been in contact with one author (IRCT201707283860N33), whose study has been submitted for publication with the findings embargoed until publication.
Included studies
There are 28 studies included with 2749 women (Asti 2011; Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Chananeh 2018; Dastjerdi 2019; De Sousa 2014; Jain 1978; Kantor 1984a; Kheiriyat 2016; Laska 1981 Study 1; Laska 1981 Study 2; Mehlhorn 2005; Okun 1982; Olsen 2007; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Skovlund 1991a; Skovlund 1991b; Tehrani 2015).
Design
All of the included studies are randomised controlled trials. Two of these randomised trials used a sequential trial design (Skovlund 1991a; Skovlund 1991b).
Twelve studies were randomised studies with two arms (Asti 2011; Chananeh 2018; Dastjerdi 2019; De Sousa 2014; Kheiriyat 2016; Olsen 2007; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Skovlund 1991a; Skovlund 1991b; Tehrani 2015). Five studies had three arms (Bettigole 1981; Bloomfield 1977 Study 2; Bloomfield 1986c; Jain 1978; Kantor 1984a). Three studies had four arms (Bloomfield 1977 Study 1; Bloomfield 1986b; Mehlhorn 2005). One study had five arms (Bloomfield 1986a). One report included two studies, one with six arms (Laska 1981 Study 1) and a second with seven arms (Laska 1981 Study 2).
Four studies with five arms (Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Okun 1982) and one study with four arms (Bloomfield 1987) included medications that are no longer in use; therefore only arms with current medications or placebo were included. Three studies reported two arms that could be included (Bloomfield 1978; Bloomfield 1987; Okun 1982) and one study reported three arms that could be included (Bloomfield 1981).
Sample sizes
The samples sizes range from 21 women (Olsen 2007) to 203 women (Bloomfield 1986c).
Setting
All of the studies included in this review enrolled women who were hospital inpatients following the birth of their baby. Thirteen studies enrolled women in the USA (Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Jain 1978; Kantor 1984a; Okun 1982); eight studies enrolled women in Iran (Asti 2011; Chananeh 2018; Dastjerdi 2019; Kheiriyat 2016; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Tehrani 2015); two studies enrolled women in Venzuela (Laska 1981 Study 1; Laska 1981 Study 2); two enrolled women in Norway (Skovlund 1991a; Skovlund 1991b); one study enrolled women in Sweden (Olsen 2007); one in Germany (Mehlhorn 2005), and one in Brazil (De Sousa 2014).
Participants
All of the studies included women with postpartum pain from uterine cramping, which was assessed and reported separately from other sources of pain. Six studies specifically excluded women with perineal pain or trauma (Asti 2011; Dastjerdi 2019; Olsen 2007; Ozgoli 2017; Simbar 2015; Tehrani 2015) or 3rd and 4th degree trauma (Chananeh 2018); five specified that uterine cramp pain should be greater than perineal pain (Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1986a; Bloomfield 1986b). Two studies reported that when pain was assessed uterine pain and perineal pain (if applicable) were assessed and reported separately (Skovlund 1991a; Skovlund 1991b).
Age as an inclusion/exclusion is specified in seven studies (Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1986a; Bloomfield 1986b; Chananeh 2018; Ozgoli 2017), with five specifying 18 years or older and one study specifying 20 to 30 years. Age was reported in the results of 12 studies (Bettigole 1981; Dastjerdi 2019; De Sousa 2014; Jain 1978; Laska 1981 Study 1; Laska 1981 Study 2; Okun 1982; Olsen 2007; Simbar 2015; Skovlund 1991a; Skovlund 1991b; Tehrani 2015), but ranges were not consistently reported.
Only two studies specified singleton pregnancy (Asti 2011; Chananeh 2018). No studies specified inclusion of twin or higher‐order pregnancies.
Twenty‐three of the studies (Asti 2011; Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1987; Chananeh 2018; Dastjerdi 2019; De Sousa 2014; Jain 1978; Kheiriyat 2016; Laska 1981 Study 1; Laska 1981 Study 2; Okun 1982; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Skovlund 1991a; Skovlund 1991b; Tehrani 2015) included women who had normal vaginal or uncomplicated births (assumed to be vaginal). Mode of birth was not specified in five studies (Bloomfield 1983; Bloomfield 1986c; Kantor 1984a; Mehlhorn 2005; Okun 1982) and assumed to be inclusive of women with normal births only.
The intention or ability to breastfeed was specified as an inclusion criterion in two studies (Chananeh 2018; Dastjerdi 2019), pain whilst breastfeeding was specified as an inclusion criterion in one study (De Sousa 2014). Breastfeeding was specified as an exclusion criterion in 11 studies (Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1987; Kantor 1984a; Laska 1981 Study 1; Laska 1981 Study 2; Okun 1982).
Interventions and comparisons
This review includes studies comparing an intervention for pain relief of uterine cramps with a placebo or another form of pain relief.
Pharmacological interventions included: aspirin 650 mg compared with placebo in two studies (Bloomfield 1978; Okun 1982); compared with aspirin 800 mg plus caffeine 64 mg in one study (Jain 1978); compared with placebo, flurbiprofen 50 mg, codeine 60 mg and codeine 120 mg in one study (Bloomfield 1986a); compared with placebo and naproxen 275 mg in one study (Bloomfield 1977 Study 2); compared with placebo and paracetamol 650 mg in one study (Bloomfield 1981); and compared with placebo, ketorolac 5 mg and ketorolac 10 mg in one study (Bloomfield 1986b).
One study (Bloomfield 1986c) with five arms compares aspirin 650 mg, aspirin 1000 mg, paracetamol 650 mg, paracetamol 1000 mg and placebo.
Fenoprofen at different doses is compared with codeine 60 mg and placebo in one three‐arm study (Bettigole 1981), where the fenoprofen dose was 200 mg; one six‐arm study (Laska 1981 Study 1) where the doses of fenoprofen were 50 mg, 100 mg, 200 mg and 300 mg; and in one seven‐arm study (Laska 1981 Study 2) where the doses of fenoprofen were 12.5 mg, 25 mg, 50 mg, 100 mg and 200 mg.
Naproxen 300 mg and 600 mg is compared with codeine 60 mg and placebo in one study (Bloomfield 1977 Study 1). Naproxen 550 mg is compared with placebo in one study (Bloomfield 1987). Naproxen 500 mg is compared with paracetamol 1000 mg in one study (Skovlund 1991a).
Paracetamol 1000 mg is compared with placebo in one study (Skovlund 1991b).
Nalbuphine 15 mg is compared with codeine 60 mg and placebo in one study (Kantor 1984a).
Different doses, 100 mg, 200 mg and 400 mg ibuprofen are compared with aspirin 650mg and placebo in one study with five arms (Bloomfield 1983).
Ibuprofen 400 mg is compared with fennel essence in one study (Asti 2011).
Mefenamic acid 250 mg is compared with a herbal analgesic in seven studies, including melissa officinalis 150 mg (Dastjerdi 2019); ginger 250mg (Pourmaleky 2013); anethum graveolens extract (dill extract) 1.5 mg/kg body weight (Kheiriyat 2016); anise 60 mg (Ozgoli 2017); pimpinella anisum, apium graveolens and crocus sativus (PAC) 500 mg (Simbar 2015); fennelin (fennel extracts) 30 mg (Tehrani 2015). One two‐armed study compared mefenamic acid with mefenamic acid and Nigella Sativa (Chananeh 2018).
One study with four arms (Mehlhorn 2005) compared combinations of TENS (fixed 100‐Hz), metamizole 625 mg, placebo TENS and placebo metamizole.
One study compared high‐intensity TENS (50 mA for one minute) (HI) with low‐intensity TENS (10‐10 5 mA for one minute) (LI) (Olsen 2007).
One study compared TENS (100‐Hz current and 75 msec pulse for 40 mi) with no treatment (De Sousa 2014).
We noted inconsistencies in the doses of oral analgesics administered across studies. For pharmacological preparations, a number of studies administered doses that are above (Bloomfield 1986a) or below (Bloomfield 1981; Dastjerdi 2019; Kheiriyat 2016; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Tehrani 2015) recognised therapeutic doses used currently in clinical practice. For herbal preparations, therapeutic doses are largely unknown and therefore these could not be assessed (Asti 2011; Dastjerdi 2019; Kheiriyat 2016; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Tehrani 2015). While a number of studies were identified that included comparisons of different doses of the same analgesic (Bloomfield 1977 Study 1; Bloomfield 1986a; Bloomfield 1986b; Laska 1981 Study 1; Laska 1981 Study 2), none of these studies were adequately designed or powered to identify the optimal dose.
Outcomes
Adequate pain relief as reported by the woman
Summed pain intensity differences (SPID) scores were used to calculate the number of women with adequate pain relief for the meta‐analysis in 11 trials (Asti 2011; Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1986b; Bloomfield 1987; Laska 1981 Study 1; Laska 1981 Study 2; Okun 1982). One of these studies (Bloomfield 1981) reported SPID scores and the number of women with at least 50% pain relief (or similar), but the number did not agree with the number calculated from the SPID. The reason for the discrepancy was not clear, so for consistency we used the number derived from SPID. We used the estimation of one pain intensity difference (PID) to calculate the number of women with adequate pian relief in four studies (Dastjerdi 2019; De Sousa 2014; Simbar 2015; Tehrani 2015).
One study (Bloomfield 1986a) reported a total pain relief (TOTPAR) score, which we used to calculate number of women reporting adequate pain relief. The number of women with at least 50% pain relief was reported, but did not agree with the number calculated from the TOTPAR. The reason for the discrepancy was not clear, so for consistency we used the number derived from TOTPAR.
Trials varied by the length of time following administration of the intervention when participants' pain was assessed; time intervals from treatment to final assessment included 30 minutes (Kheiriyat 2016; Mehlhorn 2005; Pourmaleky 2013); one hour (Chananeh 2018; Simbar 2015; Tehrani 2015); three hours (Dastjerdi 2019); four hours (Asti 2011; Jain 1978; Skovlund 1991a; Skovlund 1991b); five hours (Laska 1981 Study 1; Laska 1981 Study 2); six hours (Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Kantor 1984a; Ozgoli 2017); seven hours (Bloomfield 1977 Study 2; Bloomfield 1978) and eight hours (Bettigole 1981; Bloomfield 1977 Study 1; Okun 1982. One study assessed pain immediately after treatment (Olsen 2007). One study assessed pain during the breastfeed following treatment (De Sousa 2014).
Need for additional analgesia
The need for additional pain relief was reported by 12 studies (Asti 2011; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1987; De Sousa 2014; Kantor 1984a; Mehlhorn 2005; Skovlund 1991a; Skovlund 1991b) and 11 of these reported data that could be included in meta‐analysis (Mehlhorn 2005 only reported the statistical significance of the difference between groups).
Pain relief, however measured by the authors
One study reported the number of women rating their pain at 1 to 4 points on a 1 ‐ 10 visual analogue scale (VAS) used for assessing pain (Mehlhorn 2005). One study reported the VAS assessing pain (De Sousa 2014).
Pain relief reported by four studies (Jain 1978; Olsen 2007; Skovlund 1991a; Skovlund 1991b) could not be included in meta‐analysis. Jain 1978 reported pain at four hours following the intervention as a percentage of the baseline pain assessed by a VAS. Olsen 2007 reported the median decrease in VAS seven hours after the intervention. Two studies reported the difference in pain intensity at two hours (Skovlund 1991a) and four hours (Skovlund 1991b) following the intervention.
Maternal adverse events
Maternal adverse events were reported by 16 trials (Asti 2011; Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1987; De Sousa 2014; Jain 1978; Okun 1982; Olsen 2007; Simbar 2015; Skovlund 1991a; Skovlund 1991b), although one of these studies (Jain 1978) with very small numbers reported no maternal adverse events and did not contribute data to the meta‐analysis. Side effects reported included nausea, vomiting, diarrhoea, dizziness, constipation, sleepiness, drowsiness, headache, blurred vision, hypertension, hypotension, sweating, tingling, fatigue and 'other'.
Neonatal adverse events
Not reported by any of the included studies.
Duration of hospital stay
Not reported by any of the included studies.
Any breastfeeding at hospital discharge
Not reported by any of the included studies.
Any breastfeeding at six weeks postpartum
Not reported by any of the included studies.
Maternal views
One study only reported maternal views of their treatment, assessed as women's satisfaction (Mehlhorn 2005).
Maternal postnatal depression
Not reported by any of the included studies.
Dates of study
Very few studies reported the timing of recruitment into their studies. We estimate that recruitment occurred prior to the earliest publications in 1977 (Bloomfield 1977 Study 1; Bloomfield 1977 Study 2) and at least until 2016 (Dastjerdi 2019).
The following studies reported the timing of recruitment into their studies: Okun 1982: 2004; Simbar 2015: April 2011 until February 2012.
Dates for some trials with available trial registrations that were retrospective have been included: Dastjerdi 2019: August to November of 2016; and Ozgoli 2017: September to December 2013.
Funding sources
Ten studies did not report the source of their funding (Asti 2011; Bettigole 1981; Bloomfield 1983; Bloomfield 1986c; Kantor 1984a; Laska 1981 Study 1; Laska 1981 Study 2; Mehlhorn 2005; Okun 1982; Olsen 2007). Seven studies were funded by the authors' universities (Chananeh 2018; Dastjerdi 2019; De Sousa 2014; Kheiriyat 2016; Ozgoli 2017; Pourmaleky 2013; Tehrani 2015). Two studies were funded by their national government (Skovlund 1991a; Skovlund 1991b). Nine studies were funded by pharmaceutical companies that manufacturer one or more of the interventional products within the study (Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1987; Jain 1978; Simbar 2015).
Declarations of interest
Three studies declared that they had no conflicts of interest (Simbar 2015; Skovlund 1991a; Skovlund 1991b). The remaining studies made no declarations about conflicts or absence of conflicts (Asti 2011; Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Chananeh 2018; Dastjerdi 2019; De Sousa 2014; Jain 1978; Kantor 1984a; Kheiriyat 2016; Laska 1981 Study 1; Laska 1981 Study 2; Mehlhorn 2005; Okun 1982; Olsen 2007; Ozgoli 2017; Pourmaleky 2013; Tehrani 2015).
Excluded studies
We excluded 59 studies in this update. We excluded 24 studies because they included participants with other sources of postpartum pain, including pain from perineal trauma, and did not distinguish between pain source in the analyses (Azpiroz 1971; Beaver 1980; Benson 1963; Bonica 1957; Bruni 1965; Finch 1957; Goodman 2005; Gruber 1962; Gruber 1963; Gruber 1979; Hartemann 1968; Kantor 1984b; Nunlee 2000; Olson 1984; Ray 1993; Redick 1980; Rubin 1984; Smith 1973; Sunshine 1983; Sunshine 1985; Sunshine 1986; Sunshine 1989; Van Wering 1972; Von Pein 1974).
We excluded three studies because the methods were unclear or not well enough described to include (Gruber 1971a; Gruber 1971b; Laska 1983). Two studies were quasi‐randomised and therefore excluded (Baptisti 1971; Prockop 1960). One study was a case‐control design (Linder 1997).
One study was excluded as the interventional medications are no longer available (Gindhart 1971).
One study was excluded because it was an abstract with insufficient inclusion details and confirmed by the author as not completed (Mehlhorn 2006). Another two studies were registered with the Oxford Perinatal Trials Register but not published (personal communications to the Oxford Register from the first author confirms that the studies were not published and not likely to be published: Bloomfield 1988a; Bloomfield 1988b).
Reasons for exclusion were the intervention was for prevention of pain rather than treatment (Bachar 2018; Bahri 2019; Barhan 2019; Bilgin 2016; Can 2015; Cunha 2011; Katz 2019; Kayman‐Kose 2014; Li 2014; Mirror 2019; Narimatsu 2001; Nazari 2018; Ozgoli 2018; Pan 1993; Soltani 2017); outcomes were not reported separately for uterine cramp pain (including perineal pain) or by mode of birth (including caesarean birth) (Blue 2018; Kenton 2011; Kim 2019; Kumbar 2017; Vaziri 2017); one study was investigating joint pain postpartum ( Li 2015); one study (Yogev 2015) had two arms, a prevention arm where women who had not begun breastfeeding were randomised to the dental device or not to prevent pain and a second arm where all women who had begun feeding were given the device and acted as their own control, with pain measured before and after use. Three studies were excluded because women had access to additional analgesia, either routinely and as needed (Afravi 2019) or as needed (Parsa 2019; Tafazoli 2013), none of these studies included sufficient information to assess whether they could be considered as controlled.
Risk of bias in included studies
We assessed the included studies for risks of bias on the basis of selection bias (allocation concealment and sequence generation), performance bias (blinding), attrition bias (incomplete outcome data), and selective reporting bias (seeMethods above and Figure 2 and Figure 3). Fifteen of the included studies were published between 1977 and 1991, prior to the first published version (1996) of Consolidated Standards of Reporting Trials (CONSORT 2010).
2.

Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies.
3.

Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
Allocation
Random sequence generation
Only four studies were considered to be at low risk of bias for random sequence generation (Dastjerdi 2019; De Sousa 2014; Mehlhorn 2005; Olsen 2007), with the remaining 24 studies assessed as unclear because they did not provide sufficient information to describe adequate random sequence generation (Asti 2011; Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Chananeh 2018; Jain 1978; Kantor 1984a; Kheiriyat 2016; Laska 1981 Study 1; Laska 1981 Study 2; Okun 1982; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Skovlund 1991a; Skovlund 1991b; Tehrani 2015).
Allocation concealment
Two studies were judged as low risk of bias for allocation concealment (Mehlhorn 2005; Olsen 2007). Twenty‐four studies did not provide sufficient information to permit judgement and were therefore assessed at unclear risk of bias (Asti 2011; Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Chananeh 2018; De Sousa 2014; Jain 1978; Kantor 1984a; Kheiriyat 2016; Laska 1981 Study 1; Laska 1981 Study 2; Okun 1982; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Skovlund 1991a; Skovlund 1991b). We rated two studies at high risk of bias: one states that the allocation was not concealed (Dastjerdi 2019), and the second study states that the researcher and pharmacist were aware of the allocation (Tehrani 2015).
Blinding
Blinding of participants and personnel
Sixteen of the included studies described their study as 'double blind' and or that the medications were of identical taste or appearance, or both; we judged these studies to be at low risk of performance bias (Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Dastjerdi 2019; Laska 1981 Study 1; Laska 1981 Study 2; Mehlhorn 2005; Okun 1982; Ozgoli 2017). Eight studies did not report sufficient information to permit assessment and were therefore judged as unclear (Asti 2011; Bettigole 1981; Jain 1978; Kantor 1984a; Pourmaleky 2013; Simbar 2015; Skovlund 1991a; Skovlund 1991b). Two studies were at high risk of bias as women and researchers were unblinded due to the nature of the intervention (De Sousa 2014; Olsen 2007). A third study, at high risk of bias, states that women nor researchers were blinded (Tehrani 2015). A fourth study, judged as high risk (Kheiriyat 2016), the women were blinded but not the researchers.
Blinding of outcome assessment (checking for possible detection bias)
Three studies were judged as being at low risk of bias (Laska 1981 Study 1; Laska 1981 Study 2; Mehlhorn 2005). These low‐risk studies stated that the researchers were blinded or that the study was double‐blinded. Twenty‐four studies did not provide information on blinding of outcome assessors and were therefore judged at unclear risk of detection bias (Asti 2011; Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Chananeh 2018; Dastjerdi 2019; De Sousa 2014; Jain 1978; Kantor 1984a; Okun 1982; Olsen 2007; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Skovlund 1991a; Skovlund 1991b; Tehrani 2015). We rated one study (Kheiriyat 2016) at high risk of detection bias as the researchers were not blinded.
See also Figure 3.
Incomplete outcome data
Eighteen studies were judged at low risk of attrition bias (Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1987; Chananeh 2018; De Sousa 2014; Jain 1978; Kantor 1984a; Mehlhorn 2005; Okun 1982; Olsen 2007; Ozgoli 2017; Skovlund 1991a; Skovlund 1991b; Tehrani 2015). Seven studies were judged as unclear risk, reporting insufficient information to permit judgement (Asti 2011; Bettigole 1981; Bloomfield 1983; Bloomfield 1986c; Kheiriyat 2016; Pourmaleky 2013; Simbar 2015). Three studies were judged at high risk of attrition bias (Dastjerdi 2019; Laska 1981 Study 1; Laska 1981 Study 2) in both of the studies by Laska et al, women who gave birth by caesarean were inadvertently randomised (21% and 12% respectively) but not included in the analyses. Dastjerdi 2019 had 13% attrition following randomisation in both groups, and further states that women who did not experience pain (relief) in the first hour were given mefenamic acid and removed from the study.
Selective reporting
Three of the included studies had prospective trial registrations available (Chananeh 2018; De Sousa 2014; Tehrani 2015), and we judged them to be at low risk for reporting bias. A third study with an unpublished protocol available was judged as low risk (Mehlhorn 2005). We rated 21 studies at unclear risk, as there were no trial registrations or protocols available (Asti 2011; Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Jain 1978; Kantor 1984a; Kheiriyat 2016; Laska 1981 Study 1; Laska 1981 Study 2; Okun 1982; Olsen 2007; Pourmaleky 2013; Skovlund 1991a; Skovlund 1991b). We judged three studies to be at high risk of reporting bias as their studies were registered retrospectively (Dastjerdi 2019; Ozgoli 2017; Simbar 2015).
Other potential sources of bias
We found no other sources of bias in 18 studies and judged them to be at low risk (Asti 2011; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1983; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1986c; Bloomfield 1987; Dastjerdi 2019; De Sousa 2014; Jain 1978; Mehlhorn 2005; Simbar 2015; Tehrani 2015).
Women in two studies may have had other pain relief at varying times before being randomised into the studies, although all women who were randomised had pain and were requesting analgesia (Skovlund 1991a; Skovlund 1991b). In addition, Skovlund 1991a has errors in the labelling of graphs in the report. We considered that these studies were at low risk of other bias.
We rated 9 studies at unclear risk of other bias. Five studies included women with perineal pain, and it is unclear if randomisation was stratified by source of pain; uterine cramp or episiotomy (Bettigole 1981; Kantor 1984a; Laska 1981 Study 1; Laska 1981 Study 2; Okun 1982). Four studies (Chananeh 2018; Kheiriyat 2016; Ozgoli 2017; Pourmaleky 2013) were judged unclear as they were translated, with only the abstracts in English.
Olsen 2007 was the exception, with discrepancies found in the reported number of participants randomised and the number of participants with outcome data, and was therefore rated as at high risk of other bias.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7
Summary of findings 1. NSAID compared to placebo for relief of pain due to uterine cramping/involution after birth.
| NSAID compared to placebo for relief of pain due to uterine cramping/involution after birth | |||||
| Patient or population: women who have given birth vaginally, requiring analgesia for after‐birth pains. Setting: hospital obstetric inpatients (USA, Venezuela, and one trial setting unspecified) Intervention: NSAID Comparison: placebo | |||||
| Outcomes | № of participants (studies) | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with placebo | Risk difference with NSAID | ||||
| Adequate pain relief as reported by the woman (5 to 8 hours) |
946 (11 RCTs) | ⊕⊕⊕⊝ MODERATEa | RR 1.66 (1.45 to 1.91) | Study population | |
| 441 per 1000 | 291 more per 1000 (198 more to 401 more) | ||||
| Need for additional pain relief (5 to 8 hours) |
375 (4 RCTs) | ⊕⊕⊝⊝ LOWa,b | RR 0.15 (0.07 to 0.33) | Study population | |
| 160 per 1000 | 136 fewer per 1000 (149 fewer to 107 fewer) | ||||
| Maternal adverse events (4 to 8 hours) |
598 (9 RCTs) | ⊕⊕⊝⊝ LOWa,c | RR 1.05 (0.78 to 1.41) | Study population | |
| 239 per 1000 | 12 more per 1000 (52 fewer to 98 more) | ||||
| Neonatal adverse events | Not reported | ||||
| Duration of hospital stay | Not reported | ||||
| Any breastfeeding at hospital discharge | Not reported | ||||
| Any breastfeeding at 6 weeks postpartum | Not reported | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
aDowngraded one level due to serious concerns about limitations in study design: risk of bias ‐ unclear random sequence generation, allocation concealment, blinding of outcome assessors and selective reporting. bDowngraded one level due to serious concerns about imprecision: few events. cDowngraded one level due to serious concerns about imprecision: wide 95% confidence interval that is consistent with possible harm and possible benefit.
Summary of findings 2. NSAID compared to opioid for relief of pain due to uterine cramping/involution after birth.
| NSAID compared to opioid for relief of pain due to uterine cramping/involution after birth | |||||
| Patient or population: women who have given birth vaginally, requiring analgesia for after‐birth pains. Setting: hospital obstetric inpatients (USA, Venezuela, and one trial setting unspecified) Intervention: NSAID Comparison: opioid | |||||
| Outcomes | № of participants (studies) | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with opioid | Risk difference with NSAID | ||||
| Adequate pain relief as reported by the woman (5 to 8 hours) |
560 (5 RCTs) | ⊕⊕⊕⊝ MODERATEa | RR 1.33 (1.13 to 1.57) | Study population | |
| 539 per 1000 | 178 more per 1000 (70 more to 307 more) | ||||
| Need for additional pain relief (6 to 8 hours) |
232 (2 RCTs) | ⊕⊕⊝⊝ LOWa,b | RR 0.37 (0.12 to 1.12) | Study population | |
| 61 per 1000 | 39 fewer per 1000 (54 fewer to 7 more) | ||||
| Maternal adverse events (6 to 8 hours) |
255 (3 RCTs) | ⊕⊕⊝⊝ LOWa,c | RR 0.62 (0.43 to 0.89) | Study population | |
| 440 per 1000 | 167 fewer per 1000 (251 fewer to 48 fewer) | ||||
| Neonatal adverse events | Not reported | ||||
| Duration of hospital stay | Not reported | ||||
| Any breastfeeding at hospital discharge | Not reported | ||||
| Any breastfeeding at 6 weeks postpartum | Not reported | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
aDowngraded one level due to serious concerns about limitations in study design: risk of bias ‐ unclear random sequence generation, allocation concealment, blinding of outcome assessors and selective reporting. bDowngraded one level due to serious concerns around imprecision: few participants and wide 95% confidence interval that is consistent with possible harm and possible benefit. cDowngraded one level due to serious concerns around imprecision: few participants.
Summary of findings 3. Opioid compared to placebo for relief of pain due to uterine cramping/involution after birth.
| Opioid compared to placebo for relief of pain due to uterine cramping/involution after birth | |||||
| Patient or population: women who have given birth vaginally, requiring analgesia for after‐birth pains. Setting: hospital obstetric inpatients (USA, Venezuela, and one trial setting unspecified) Intervention: opoid Comparison: placebo | |||||
| Outcomes | № of participants (studies) | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with placebo | Risk difference with opioid | ||||
| Adequate pain relief as reported by the woman (5 to 8 hours) |
299 (5 RCTs) | ⊕⊕⊝⊝ LOWa,b | RR 1.26 (0.99 to 1.61) | Study population | |
| 396 per 1000 | 103 more per 1000 (4 fewer to 241 more) | ||||
| Need for additional pain relief (6 to 8 hours) |
273 (3 RCTs) | ⊕⊕⊝⊝ LOWa,c | RR 0.48 (0.28 to 0.82) | Study population | |
| 223 per 1000 | 116 fewer per 1000 (161 fewer to 40 fewer) | ||||
| Maternal adverse events (6 to 8 hours) |
188 (3 RCTs) | ⊕⊕⊝⊝ LOWa,b | RR 1.59 (0.99 to 2.55) | Study population | |
| 266 per 1000 | 157 more per 1000 (30 fewer to 412 more) | ||||
| Neonatal adverse events | Not reported | ||||
| Duration of hospital stay | Not reported | ||||
| Any breastfeeding at hospital discharge | Not reported | ||||
| Any breastfeeding at 6 weeks postpartum | Not reported | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
aDowngraded one level due to serious concerns about limitations in study design: risk of bias ‐ unclear risk in most domains. bDowngraded one level due to serious concerns around imprecision: few participants and wide 95% confidence interval that is consistent with possible harm and possible benefit. cDowngraded one level due to serious concerns around imprecision: few participants.
Summary of findings 4. Paracetamol compared to placebo for relief of pain due to uterine cramping/involution after birth.
| Paracetamol compared to placebo for relief of pain due to uterine cramping/involution after birth | |||||
| Patient or population: women who have given birth vaginally, requiring analgesia for after‐birth pains. Setting: hospital obstetric inpatients (Norway, and USA) Intervention: paracetamol Comparison: placebo | |||||
| Outcomes | № of participants (studies) | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with placebo | Risk difference with paracetamol | ||||
| Adequate pain relief as reported by the woman (6 hours) |
48 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | RR 1.27 (0.80 to 2.00) | Study population | |
| 538 per 1000 | 145 more per 1000 (108 fewer to 538 more) | ||||
| Need for additional pain relief (up to 4 hours) |
75 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | RR 0.74 (0.21 to 2.54) | Study population | |
| 139 per 1000 | 36 fewer per 1000 (110 fewer to 214 more) | ||||
| Maternal adverse events (up to 4 hours and at 6 hours) |
123 (2 RCTs) | ⊕⊝⊝⊝ VERY LOWa,b | RR 2.27 (0.97 to 5.33) | Study population | |
| 97 per 1000 | 123 more per 1000 (3 fewer to 419 more) | ||||
| Neonatal adverse events | Not reported | ||||
| Duration of hospital stay | Not reported | ||||
| Any breastfeeding at hospital discharge | Not reported | ||||
| Any breastfeeding at 6 weeks postpartum | Not reported | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
aDowngraded one level due to serious concerns about limitations in study design: risk of bias ‐ unclear risk in most domains. bDowngraded two levels due to very serious concerns about imprecision: few participants, few events and wide confidence intervals.
Summary of findings 5. Paracetamol compared to NSAID for relief of pain due to uterine cramping/involution after birth.
| Paracetamol compared to NSAID for relief of pain due to uterine cramping/involution after birth | |||||
| Patient or population: women who have given birth vaginally, requiring analgesia for after‐birth pains. Setting: hospital obstetric inpatients (Norway, and USA) Intervention: paracetamol Comparison: NSAID | |||||
| Outcomes | № of participants (studies) | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with NSAID | Risk difference with paracetamol | ||||
| Adequate pain relief as reported by the woman (6 hours) |
48 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | RR 0.89 (0.62 to 1.26) | Study population | |
| 769 per 1000 | 85 fewer per 1000 (292 fewer to 200 more) | ||||
| Need for additional pain relief | Not reported | ||||
| Maternal adverse events (up to 4 hours and at 6 hours) |
112 (2 RCTs) | ⊕⊝⊝⊝ VERY LOWa,b | RR 0.99 (0.52 to 1.86) | Study population | |
| 241 per 1000 | 2 fewer per 1000 (116 fewer to 207 more) | ||||
| Neonatal adverse events | Not reported | ||||
| Duration of hospital stay | Not reported | ||||
| Any breastfeeding at hospital discharge | Not reported | ||||
| Any breastfeeding at 6 weeks postpartum | Not reported | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
aDowngraded one level due to serious concerns about limitations in study design: risk of bias ‐ unclear risk in most domains. bDowngraded two levels due to very serious concerns about imprecision: few participants and wide confidence intervals.
Summary of findings 6. NSAID compared to herbal analgesia for relief of pain due to uterine cramping/involution after birth.
| NSAID compared to herbal analgesia for relief of pain due to uterine cramping/involution after birth | |||||
| Patient or population: women who have given birth vaginally, requiring analgesia for after‐birth pains. Setting: hospital obstetric inpatients (Iran) Intervention: NSAID Comparison: herbal analgesia | |||||
| Outcomes | № of participants (studies) | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with placebo | Risk difference with NSAID versus herbal analgesia | ||||
| Adequate pain relief as reported by the woman (1 to 4 hours) |
394 (4 RCTs) | ⊕⊝⊝⊝ VERY LOW a,b | RR 0.96 (0.78 to 1.18) | Study population | |
| 462 per 1000 | 18 fewer per 1000 (102 fewer to 83 more) | ||||
| Need for additional pain relief (4 hours) |
90 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b,c | RR 1.00 (0.44 to 2.29) | Study population | |
| 200 per 1000 | 0 fewer per 1000 (112 fewer to 258 more) | ||||
| Maternal adverse events (1 hour) |
108 (1 RCTs) | ⊕⊝⊝⊝ VERY LOWb,c,d | RR 5.00 (0.60 to 41.39) | Study population | |
| 19 per 1000 | 74 more per 1000 (7 fewer to 748 more) | ||||
| Neonatal adverse events | Not reported | ||||
| Duration of hospital stay | Not reported | ||||
| Any breastfeeding at hospital discharge | Not reported | ||||
| Any breastfeeding at 6 weeks postpartum | Not reported | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
aDowngraded two levels for very serious concerns about limitations in study design: risk of bias ‐ some of the studies included have some domains of risk of bias assessed as unclear (selective outcome reporting) or high risk (allocation concealment, blinding, selective outcome reporting). bDowngraded one level due to serious concerns about indirectness: some study interventions were not therapeutic doses. cDowngraded two levels due to very serious concerns about imprecision: few participants, few events and wide confidence intervals. dDowngraded one level due to serious concerns about limitations in study design: risk of bias ‐ unclear risk of selective outcome reporting.
Summary of findings 7. TENS compared to no TENS for relief of pain due to uterine cramping/involution after birth.
| TENS compared to no TENS for relief of pain due to uterine cramping/involution after birth | |||||
| Patient or population: women who have given birth vaginally, requiring analgesia for after‐birth pains. Setting: hospital obstetric inpatients (Brazil and Germany) Intervention: TENS Comparison: no TENS | |||||
| Outcomes | № of participants (studies) | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with no TENS | Risk difference with TENS | ||||
| Adequate pain relief as reported by the woman (during next breast feed) |
32 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | RR 4.00 (0.50 to 31.98) | Study population | |
| 63 per 1000 | 188 more per 1000 (31 fewer to 1,936 more) | ||||
| Need for additional pain relief | Not reported | ||||
| Maternal adverse events | One study (32 women) stated "there were few adverse effects associated with TENS". | ||||
| Neonatal adverse events | Not reported | ||||
| Duration of hospital stay | Not reported | ||||
| Any breastfeeding at hospital discharge | Not reported | ||||
| Any breastfeeding at 6 weeks postpartum | Not reported | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||
aDowngraded one level due to serious concerns about limitations in study design: risk of bias ‐ high risk for performance and detection bias, and outcome assessment bias, unclear risk for allocation concealment. bDowngraded two levels for very serious concerns about imprecision: very few participants and wide CI.
Comparison 1: NSAID versus placebo
Twelve studies (Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1977 Study 2; Bloomfield 1978; Bloomfield 1981; Bloomfield 1986a; Bloomfield 1986b; Bloomfield 1987; Jain 1978; Laska 1981 Study 1; Laska 1981 Study 2; Okun 1982) compared non‐steroidal anti‐inflammatory drugs (NSAIDs) versus placebo, and reported data suitable for meta‐analysis.
Primary outcome
Adequate pain relief as reported by the woman
NSAIDs are probably more effective than placebo for adequate pain relief (risk ratio (RR) 1.66, 95% confidence interval (CI) 1.45 to 1.91; I2 = 0%; 11 studies, 946 women; moderate‐certainty evidence; Analysis 1.1; Table 1).
1.1. Analysis.

Comparison 1: NSAID versus placebo, Outcome 1: Adequate pain relief as reported by the woman.
Secondary outcomes
Need for additional pain relief
NSAIDs may be more effective than placebo for the need for additional analgesia (RR 0.15, 95% CI 0.07 to 0.33; 4 studies, 375 women; low‐certainty evidence; Analysis 1.2; Table 1).
1.2. Analysis.

Comparison 1: NSAID versus placebo, Outcome 2: Need for additional pain relief
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
Comparing NSAIDs and placebo, there may be a similar risk of adverse events in the mother (RR 1.05, 95% CI 0.78 to 1.41; 9 studies, 598 women; low‐certainty evidence; Analysis 1.3; Table 1).
1.3. Analysis.

Comparison 1: NSAID versus placebo, Outcome 3: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 2: NSAID versus opioid
NSAIDs and opioids were compared in five studies (Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1986a; Laska 1981 Study 1; Laska 1981 Study 2).
Primary outcomes
Adequate pain relief
NSAIDS are probably more effective than an opioid for adequate pain relief (RR 1.33, 1.13 to 1.57; I2 = 0%; 5 studies, 560 women; moderate‐certainty evidence; Analysis 2.1; Table 2).
2.1. Analysis.

Comparison 2: NSAID versus opioid, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
NSAIDs may reduce the need for additional pain relief when compared with opioid, although the confidence interval includes the possibility that the two classes of drugs are similar or that opioids are superior (RR 0.37, 95% CI 0.12 to 1.12; I2 = 0%; 2 studies, 232 women; low‐certainty evidence; Analysis 2.2; Table 2).
2.2. Analysis.

Comparison 2: NSAID versus opioid, Outcome 2: Need for additional pain relief
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
NSAIDs may lower the risk of maternal adverse events compared with opioids (RR 0.62, 95% CI 0.43 to 0.89; I2 = 55%; 3 studies, 255 women; low‐certainty evidence; Analysis 2.3Table 2). The statistical heterogeneity (I2 = 55%) is likely to be explained by the different doses of codeine used in the trials, so we judged it appropriate to use a fixed‐effect model rather than random‐effects, since the different effects observed are not down to chance.
2.3. Analysis.

Comparison 2: NSAID versus opioid, Outcome 3: Maternal adverse events
Included studies reported up to 12 adverse events, including nausea, dizziness and drowsiness. Bloomfield 1986a, with five arms, included two arms with codeine, 60 mg and 120 mg. Almost 90% of women receiving 120 mg of codeine reported adverse events, predominantly drowsiness and dizziness, compared with only 30% in th 60 mg arm. Most reported adverse events were for drowsiness and dizziness. Adverse events were similar between groups for the other included studies.
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 3: Opioid versus placebo
An opioid was compared with placebo in six studies (Bettigole 1981; Bloomfield 1977 Study 1; Bloomfield 1986a; Kantor 1984a; Laska 1981 Study 1; Laska 1981 Study 2).
Primary outcomes
Adequate pain relief
Opioids may be more effective for adequate pain relief compared with placebo. However the 95% CI indicates the possibility that there may be little difference between opioids and placebo (RR 1.26, 95% CI 0.99 to 1.61; I2 = 18%; 5 studies, 299 women; low‐certainty evidence; Analysis 3.1; Table 3).
3.1. Analysis.

Comparison 3: Opioid versus placebo, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
Opioids may be better than placebo for the need for additional analgesia (RR 0.48, 95% CI 0.28 to 0.82; I2 = 0%; 3 studies, 273 women, low‐certainty evidence; Analysis 3.2; Table 3).
3.2. Analysis.

Comparison 3: Opioid versus placebo, Outcome 2: Need for additional pain relief
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
Opioids may increase the risk of maternal adverse events compared with placebo ( RR 1.59, 95% CI 0.99 to 2.55; I2 = 67%; 3 studies, 188 women; low‐certainty evidence; Analysis 3.3; Table 3), although the 95% CI indicates the possibility that the true effect may show little or no difference. The statistical heterogeneity (I2 = 67%) is likely to be explained by the different doses of codeine used in the trials, so we judged it appropriate to use a fixed‐effect analysis rather than random‐effects, since the different effects observed are not down to chance. Included studies reported up to 12 adverse events, including nausea, dizziness and drowsiness. Bloomfield 1986a, with five arms, included two arms with codeine, at doses of 60 mg and 120 mg. Almost 90% of women receiving 120 mg of codeine reported adverse events, predominantly drowsiness and dizziness, compared with only 30% in th 60 mg arm.Most reported adverse events were for drowsiness and dizziness. Adverse events were similar between groups for the other included studies.
3.3. Analysis.

Comparison 3: Opioid versus placebo, Outcome 3: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 4: Paracetamol versus placebo
Two studies compared paracetamol with placebo (Bloomfield 1981; Skovlund 1991a).
Primary outcomes
Adequate pain relief
We are uncertain if paracetamol is better than placebo for adequate pain relief because the certainty of evidence is very low (RR 1.27, 95% CI 0.80 to 2.00; 1 study, 48 women; very low‐certainty evidence; Analysis 4.1Table 4).
4.1. Analysis.

Comparison 4: Paracetamol versus placebo, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
We are uncertain if paracetamol is better than placebo for the need for additional pain relief, because the certainty of evidence is low and the 95% CI indicates the possibility of appreciable harm and appreciable benefit (RR 0.74, 95% CI 0.21 to 2.54; 1 study, 75 women; very low‐certainty evidence; Analysis 4.2; Table 4).
4.2. Analysis.

Comparison 4: Paracetamol versus placebo, Outcome 2: Need for additional pain relief
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
We are uncertain if there is any difference in the risk of maternal adverse events comparing paracetamol with placebo (RR 2.27, 95% CI 0.97 to 5.33; I2 = 0%; 2 studies, 123 women; very low‐certainty evidence; Analysis 4.3; Table 4). Women in the paracetamol group of Bloomfield 1981 reported predominantly drowsiness, while Skovlund 1991a only reported the numbers of adverse events.
4.3. Analysis.

Comparison 4: Paracetamol versus placebo, Outcome 3: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 5: Paracetamol versus NSAID
Paracetamol was compare with a NSAID in two studies (Bloomfield 1981; Skovlund 1991b).
Primary outcomes
Adequate pain relief
We are uncertain if there is any difference between NSAID and paracetamol for adequate pain relief (RR 0.89, 95% CI 0.62 to 1.26; 1 study, 48 women; very low‐certainty evidence; Analysis 5.1; Table 5).
5.1. Analysis.

Comparison 5: Paracetamol versus NSAID, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
Not reported.
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
We are uncertain if there is any difference in the risk of maternal adverse events comparing paracetamol with NSAID (RR 0.99, 95% CI 0.52 to 1.86; I2 = 47%; 2 studies, 112 women; very low‐certainty evidence; Analysis 5.2; Table 5). One of the included studies (Skovlund 1991b) reported only the number of adverse events.
5.2. Analysis.

Comparison 5: Paracetamol versus NSAID, Outcome 2: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 6: NSAID versus herbal analgesia
Seven studies compared a NSAID with a herbal analgesic (Asti 2011; Dastjerdi 2019; Kheiriyat 2016; Ozgoli 2017; Pourmaleky 2013; Simbar 2015; Tehrani 2015). However, three studies (Kheiriyat 2016; Ozgoli 2017; Pourmaleky 2013) did not report data for inclusion in this meta‐analysis.
Primary outcomes
Adequate pain relief
We are uncertain if there is any difference between a herbal analgesic compared with NSAID for adequate pain relief as reported by the women (RR 0.96, 95% CI 0.78 to 1.18; I2 = 0%; 4 studies, 394 women; Analysis 6.1; very low‐certainty evidence; Table 5).
6.1. Analysis.

Comparison 6: NSAID versus herbal analgesia, Outcome 1: Adequate pain relief as reported by the woman
Data from trials not included in the analysis
See Table 8.
1. Comparison 6: NSAID versus herbal analgesia ‐ conclusions of studies without data.
| Study | Conclusions |
| Tafazoli 2013 | Cuminum was more effective than placebo and less effective than and mefenamic acid for relief of pain due to uterine cramping/involution (P = 0.001) |
| Kheiriyat 2016 | Mean of postpartum pain decreased after the intervention, but no statistically significant difference was observed between 2 groups (P > 0.05) |
| Ozgoli 2017 | Results revealed that the reduction of the pain was significantly higher in the anise capsule group (P < 0.05). The anise capsule is effective for relief of postpartum after‐pain |
Secondary outcomes
Need for additional pain relief
We are uncertain if there is any difference between NSAID and herbal analgesia for the need for additional analgesia (RR 1.00, 95% CI 0.44 to 2.29; 1 study, 90 women; Analysis 6.2; very low‐certainty evidence; Table 6).
6.2. Analysis.

Comparison 6: NSAID versus herbal analgesia, Outcome 2: Need for additional pain relief
Pain relief, however measured by the authors
There is little evidence of a difference in pain between herbal analgesia when assessed by women using 0 ‐ 10 point VAS (mean difference (MD) 0.21, 95% CI −0.13 to 0.55; 1 study, 108 women; Analysis 6.3).
6.3. Analysis.

Comparison 6: NSAID versus herbal analgesia, Outcome 3: Pain however measured by the authors
Maternal adverse events of the intervention
We are uncertain if there is any difference in the risk of maternal adverse events comparing herbal analgesia with NSAID (RR 5.00, 95% CI 0.60 to 41.39; 1 study, 108 women; Analysis 6.4; very low‐certainty evidence; Table 6). Only one study (Simbar 2015) was included for this outcome, with women in the mefenamic acid group reported nausea, constipation and gastritis compared with one woman experiencing dizziness in the herbal analgesia group.
6.4. Analysis.

Comparison 6: NSAID versus herbal analgesia, Outcome 4: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 7: TENS versus no TENS
TENS versus no TENS was compared in two studies: in Mehlhorn 2005 the comparison was versus placebo, and in De Sousa 2014 TENS was compared with no treatment.
Primary outcomes
Adequate pain relief
We are uncertain if TENS is better than no TENS for adequate pain relief (RR 4.00, 95% CI 0.50 to 31.98; 1 study, 32 women; Analysis 7.1; very low‐certainty evidence; Table 7).
7.1. Analysis.

Comparison 7: TENS versus no TENS, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
Not reported.
Pain relief, however measured by the authors
It is unclear if there is any difference in pain relief between TENS and no TENS when assessed by women as 1 ‐ 4 points on a 1 ‐ 10 point VAS 30 minutes after intervention (RR 1.04, 95% CI 0.29 to 3.73; 1 study, 55 women; Analysis 7.2).
7.2. Analysis.

Comparison 7: TENS versus no TENS, Outcome 2: Pain however measured by the authors decrease in VAS
It is unclear if there is any difference in pain relief between TENS and no TENS when assessed by women using a 0 ‐ 10 point VAS (MD −1.25 Pain 0 ‐ 10, 95% CI −2.70 to 0.20; 1 study, 32 women; Analysis 7.3); the confidence interval indicates the possibility of benefit or harm.
7.3. Analysis.

Comparison 7: TENS versus no TENS, Outcome 3: Pain however measured by the authors
Maternal adverse events of the intervention
Mehlhorn 2005 did not report adverse events, while De Sousa 2014 stated "there were few adverse effects associated with TENS".
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
TENS compared with no TENS may be better for maternal satisfaction with treatment (RR 1.70, 95% CI 1.14 to 2.55; 1 study, 55 women; Analysis 7.4).
7.4. Analysis.

Comparison 7: TENS versus no TENS, Outcome 4: Maternal views of treatment
Maternal postpartum depression
Not reported.
Comparison 8: Aspirin versus naproxen
One study compared aspirin with naproxen (Bloomfield 1977 Study 2).
Primary outcomes
Adequate pain relief
It is unclear if there is any difference between aspirin and naproxen for adequate pain relief (RR 1.04, 95% CI 0.89 to 1.21; 1 study, 60 women; Analysis 8.1).
8.1. Analysis.

Comparison 8: Aspirin versus naproxen, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
Not reported.
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
It is unclear if there is any difference between aspirin and naproxen for the risk of maternal adverse events (RR 0.80, 95% CI 0.24 to 2.69; 1 study, 60 women; Analysis 8.2). Adverse events that were reported by women for naproxen were drowsiness, headache, nausea and hot flushes, whilst women in the aspirin group reported drowsiness and dizziness.
8.2. Analysis.

Comparison 8: Aspirin versus naproxen, Outcome 2: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 9: Aspirin versus flurbiprofen
Aspirin was compared with flurbiprofen in one study (Bloomfield 1986a).
Primary outcomes
Adequate pain relief
It is unclear if there is any difference between flurbiprofen and aspirin for adequate pain relief (RR 0.81, 95% CI 0.63 to 1.05; 1 study, 64 women; Analysis 9.1).
9.1. Analysis.

Comparison 9: Aspirin versus flurbiprofen, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
It is unclear if there is any difference between flurbiprofen and aspirin for the need for additional pain relief (RR 4.43, 95% CI 0.22 to 88.74; 1 study, 64 women; Analysis 9.2).
9.2. Analysis.

Comparison 9: Aspirin versus flurbiprofen, Outcome 2: Need for additional pain relief
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
It is unclear if there is any difference between flurbiprofen and aspirin for the risk of maternal adverse events (RR 1.18, 95% CI 0.46 to 3.01; 1 study, 64 women; Analysis 9.3). Adverse events could include drowsiness, dizziness, fatigue, nervousness and 'other', and were similar between groups.
9.3. Analysis.

Comparison 9: Aspirin versus flurbiprofen, Outcome 3: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 10: Aspirin versus ketorolac
One study compared aspirin with ketorolac 5 mg and ketorolac 10 mg (Bloomfield 1986b).
Primary outcomes
Adequate pain relief
It is unclear if there is any difference between ketorolac compared with aspirin for adequate pain relief (RR 0.95, 95% CI 0.81 to 1.11; 1 study, 90 women; Analysis 10.1).
10.1. Analysis.

Comparison 10: Aspirin versus ketorolac, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
It is unclear if there is any difference between ketorolac compared with aspirin for need for additional pain relief (RR 1.18, 95% CI 0.16 to 8.52; 1 study, 90 women; Analysis 10.1).
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
It is unclear if there is any difference between ketorolac compared with aspirin for the risk of maternal adverse events (RR 1.69, 95% CI 0.86 to 3.31; 1 study, 90 women; Analysis 10.1). Reported adverse events were similar between groups, and included drowsiness, dizziness, headache, nausea, sweating, jitters, nervousness, tiredness and visual disturbance.
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 11: Naproxen: different doses
One study compared naproxen at different doses (300 mg versus 600 mg) (Bloomfield 1977 Study 1).
Primary outcomes
Adequate pain relief
There is little evidence for a difference between naproxen 600 mg compared with naproxen 300 mg for adequate pain relief (RR 0.90, 95% CI 0.72 to 1.13; 1 study, 70 women; Analysis 11.1).
11.1. Analysis.

Comparison 11: Naproxen different doses, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
Not reported.
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
It is unclear if there is any difference in the risk of maternal adverse events between naproxen 300 mg compared with naproxen 600 mg (RR 1.00, 95% CI 0.45 to 2.22; 1 study, 70 women; Analysis 11.2).
11.2. Analysis.

Comparison 11: Naproxen different doses, Outcome 2: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 12: Ketorolac: different doses
Different doses of ketorolac (5 mg and 10 mg) were compared in one study (Bloomfield 1986b).
Primary outcomes
Adequate pain relief
There is little evidence for a difference between ketorolac 5 mg and ketorolac 10 mg for adequate pain relief (RR 0.90, 95% CI 0.77 to 1.05; 1 study, 60 women, Analysis 12.1).
12.1. Analysis.

Comparison 12: Ketorolac different doses, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
It is unclear if there is any difference between ketorolac 5 mg and ketorolac 10 mg for the need for additional analgesia (RR 0.86, 95% CI 0.33 to 2.25; 1 study, 60 women; Analysis 12.2).
12.2. Analysis.

Comparison 12: Ketorolac different doses, Outcome 2: Need for additional pain relief
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
It is unclear if there is any difference between ketorolac 5 mg and ketorolac 10 mg for the risk of maternal adverse events (RR 0.86, 95% CI 0.33 to 2.25; 1 study, 60 women; Analysis 12.3).
12.3. Analysis.

Comparison 12: Ketorolac different doses, Outcome 3: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 13: Codeine versus nalbuphine
One study compared codeine with nalbuphine (Kantor 1984a).
Primary outcomes
Adequate pain relief
Not reported.
Secondary outcomes
Need for additional pain relief
It is unclear if there is any difference between codeine compared with nalbuphine for the need for additional pain relief (RR 0.59, 95% CI 0.21 to 1.64; 1 study, 72 women; Analysis 13.1).
13.1. Analysis.

Comparison 13: Codeine versus nalbuphine, Outcome 1: Need for additional pain relief
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
Not reported.
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 14: Codeine: different doses
Diffferent doses of codeine (60 mg versus 120 mg) were compared in one study (Bloomfield 1986a).
Primary outcomes
Adequate pain relief
It is unclear if there is any difference between codeine 60 mg compared with codeine 120 mg for adequate pain relief (RR 1.07, 95% CI 0.75 to 1.51; 1 study, 63 women; Analysis 14.1).
14.1. Analysis.

Comparison 14: Codeine different doses, Outcome 1: Adequate pain relief as reported by the woman
Secondary outcomes
Need for additional pain relief
It is unclear if there is any difference between codeine 60 mg compared with codeine 120 mg for the need for additional pain relief (RR 0.97, 95% CI 0.06 to 14.82; 1 study, 63 women; Analysis 14.2).
14.2. Analysis.

Comparison 14: Codeine different doses, Outcome 2: Need for additional pain relief
Pain relief, however measured by the authors
Not reported.
Maternal adverse events of the intervention
There was a lower risk of maternal adverse events with codeine 60 mg compared with codeine 120 mg (RR 0.36, 95% CI 0.21 to 0.61; 1 study, 63 women; Analysis 14.3). Almost 90% of women receiving 120 mg of codeine reported adverse events, predominantly drowsiness and dizziness, compared with only 30% in th 60 mg arm.
14.3. Analysis.

Comparison 14: Codeine different doses, Outcome 3: Maternal adverse events
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
Not reported.
Maternal postpartum depression
Not reported.
Comparison 15: Metamizol versus placebo
Metamizol was compared with placebo in one study (Mehlhorn 2005).
Primary outcomes
Adequate pain relief
Not reported.
Secondary outcomes
Need for additional pain relief
Not reported.
Pain relief, however measured by the authors
It is unclear if there is any difference between metamizole compared with placebo for pain relief assessed by women as 1 ‐ 4 points on a 1 ‐ 10 point VAS 30 minutes after intervention (RR 1.06, 95% CI 0.31 to 3.57; 1 study, 61 women; Mehlhorn 2005).
Maternal adverse events of the intervention
Not reported.
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
It is unclear if there is any difference between metamizole compared with placebo for women's satisfaction with treatment (RR 0.97, 95% CI 0.58 to 1.62; 1 study, 61 women; Analysis 15.2).
15.2. Analysis.

Comparison 15: Metamizol versus placebo, Outcome 2: Maternal views of treatment
Maternal postpartum depression
Not reported.
Comparison 16: TENS plus metamizole versus placebo
One study compared TENS plus metamizole versus placebo (Mehlhorn 2005).
Primary outcomes
Adequate pain relief
Not reported.
Secondary outcomes
Need for additional pain relief
Not reported.
Pain relief, however measured by the authors
It is unclear if there is any difference between TENS plus metamizole compared with placebo for pain relief assessed by women as 1 ‐ 4 points on a 1 ‐ 10 point VAS 30 minutes after intervention (RR 2.57, 95% CI 0.92 to 7.13; 1 study, 58 women; Analysis 16.1).
16.1. Analysis.

Comparison 16: TENS plus metamizol versus placebo, Outcome 1: Pain however assessed by the authors
Maternal adverse events of the intervention
Not reported.
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
TENS plus metamizole compared with placebo may be better for women's satisfaction with treatment (RR 1.60, 95% CI 1.06 to 2.41; 1 study, 58 women; Analysis 16.2).
16.2. Analysis.

Comparison 16: TENS plus metamizol versus placebo, Outcome 2: Maternal views of treatment
Maternal postpartum depression
Not reported.
Comparison 17: TENS plus metamizole versus TENS
One study compared TENS plus metamizole with TENS (Mehlhorn 2005).
Primary outcomes
Adequate pain relief
Not reported.
Secondary outcomes
Need for additional pain relief
Not reported.
Pain relief, however measured by the authors
It is unclear if there is any difference between TENS plus metamizole compared with TENS alone for pain relief assessed by women as between 1 ‐ 4 points on a 1 ‐ 10 point VAS 30 minutes after intervention (RR 2.48, 95% CI 0.89 to 6.86; 1 study, 57 women; Analysis 17.1).
17.1. Analysis.

Comparison 17: TENS plus metamizol versus TENS, Outcome 1: Pain however assessed by the authors
Maternal adverse events of the intervention
Not reported.
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
It is unclear if there is any difference between TENS plus metamizole compared with TENS alone for women's satisfaction with treatment (RR 0.94, 95% CI 0.74 to 1.19; 1 study, 57 women; Analysis 17.2).
17.2. Analysis.

Comparison 17: TENS plus metamizol versus TENS, Outcome 2: Maternal views of treatment
Maternal postpartum depression
Not reported.
Comparison 18: TENS plus metamizole versus metamizole
TENS plus metamizole was compared with metamizole alone in one study (Mehlhorn 2005).
Primary outcomes
Adequate pain relief
Not reported.
Secondary outcomes
Need for additional pain relief
Not reported.
Pain relief, however measured by the authors
It is unclear if there is any difference between TENS plus metamizole compared with metamizole alone for pain relief assessed by women as 1 ‐ 4 points on a 1 ‐ 10 point VAS 30 minutes after intervention (RR 2.42, 95% CI 0.95 to 6.16; 1 study, 63 women; Analysis 18.1).
18.1. Analysis.

Comparison 18: TENS plus metamizol versus metamizol, Outcome 1: Pain however assessed by the authors
Maternal adverse events of the intervention
Not reported.
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
TENS plus metamizole compared with metamizole alone may be similar for women's satisfaction (RR 1.38, 95% CI 0.90 to 2.09; 1 study, 69 women; Analysis 18.2).
18.2. Analysis.

Comparison 18: TENS plus metamizol versus metamizol, Outcome 2: Maternal views of treatment
Maternal postpartum depression
Not reported.
Comparison 19: TENS versus metamizole
One study compared TENS with metamizole (Mehlhorn 2005).
Primary outcomes
Adequate pain relief
Not reported.
Secondary outcomes
Need for additional pain relief
Not reported.
Pain relief, however measured by the authors
It is unclear if there is any difference in pain relief with TENS compared with metamizole, assessed by women as 1 ‐ 4 points on a 1 ‐ 10 point VAS 30 minutes after intervention (RR 0.98, 95% CI 0.29 to 3.29; 1 study, 60 women; Analysis 19.1).
19.1. Analysis.

Comparison 19: TENS versus metamizol, Outcome 1: Pain however assessed by the authors
Maternal adverse events of the intervention
Not reported.
Neonatal adverse events of the intervention
Not reported.
Duration of hospital stay
Not reported.
Exclusive breastfeeding at hospital discharge
Not reported.
Exclusive breastfeeding at six weeks postpartum
Not reported.
Maternal views (using a validated questionnaire)
TENS compared with metamizole may be better for women's satisfaction with treatment (RR 1.65, 95% CI 1.11 to 2.45; 1 study, 63 women; Analysis 19.2).
19.2. Analysis.

Comparison 19: TENS versus metamizol, Outcome 2: Maternal views of treatment
Maternal postpartum depression
Not reported.
Comparison 20: TENS high‐intensity (HI) versus TENS low‐intensity (LI).
Olsen 2007 compared high‐intensity (HI) TENS 50 mA with low‐intensity (LI)TENS 10‐15 mA in a small study of 21 women. The authors found HI TENS to be better than LI TENS; however, these results have a high risk of bias. There is a clear baseline imbalance, with numbers in the abstract differing from those given in the CONSORT flowchart. There is no account given for the discrepancy in the numbers, and we therefore decided not to include these data.
Comparison 21: Herbal analgesia versus placebo they
One study report was an abstract only (Tafazoli 2013), comparing cuminum cyminum versus mefenamic acid 250 mg and versus placebo, but they did not report data for inclusion in this meta‐analysis.
Cuminum cyminum was more effective than placebo and less effective than mefenamic acid for relief of pain due to uterine cramping/involution.
Discussion
Summary of main results
We included 28 trials (involving 2749 women) in this updated review. Interventions were varied, and included the following traditional medications: aspirin, fenoprofen, naproxen, ketorolac, flurbiprofen, ibuprofen, mefenamic acid, codeine, nalbuphine, paracetamol and metamizole. The following herbal preparations were included: fennel essence and fennel extracts, zataria multiflora, melissa officinalis, cuminum cyminum, dill essence and dill extracts (anethum graveolens), anise, and pimpinella anisum, apium graveolens and crocus sativus (PAC). TENS for the relief of pain was also included. Twenty‐three trials reported data that we could include in a meta‐analysis.
NSAID versus placebo
Compared with placebo, NSAIDs probably lead to more women experiencing adequate pain relief and may lower the risk of requiring additional analgesia for the relief of uterine cramping/involution pain.
With regard to possible harm, there may be little difference between NSAIDS and placebo for maternal adverse events.
We downgraded the certainty of the evidence due to risk of bias, including publication bias, and due to imprecision because the studies had few participants (Table 1).
NSAID versus opioid
Moderate‐certainty evidence showed that more women probably experience adequate pain relief from receiving NSAIDs than those receiving opioids for the relief of uterine cramping/involution pain. NSAIDs may reduce the need for additional pain relief compared with opioids, but the wide 95% confidence interval (CI) indicates the possibility of an increased risk. NSAIDs may lower the risk of maternal adverse effects compared with opioid. The certainty of the evidence for these outcomes was moderate to low, due to very low numbers of participants and to risks of bias (Table 2).
Opioid versus placebo
Opioids may be better than placebo for adequate pain relief and may reduce the need for additional analgesia. Opioids may increase the risk of maternal adverse events compared with placebo. We downgraded the evidence for risks of bias and for imprecision (few participants) (Table 3)
Paracetamol versus placebo
We are uncertain if there is any difference between paracetamol and placebo for adequate pain relief, for the need for additional analgesia and for the risk of maternal adverse events. We downgraded the certainty of evidence for this comparison, for risks of bias and for imprecision (few participants) (Table 4).
Paracetamol versus NSAID
We are uncertain if there is any difference between paracetamol and NSAIDs for adequate pain relief or for maternal adverse events. We found no evidence relating to the need for additional pain relief following treatment with paracetamol compared with NSAIDs. We downgraded the certainty of the evidence for risks of bias and for imprecision (few participants) (Table 5)
NSAID versus herbal analgesia
We are uncertain if there are any differences between NSAIDs and herbal analgesia for adequate pain relief, for the need for additional analgesia and for the risk of maternal adverse events. We downgraded the evidence for risk of bias as some studies had high risk of bias for allocation concealment, blinding and selective outcome reporting. We downgraded for indirectness, as the adequacy of therapeutic doses of herbal preparations are unknown, and for imprecision as there were few participants (Table 6).
TENS versus no TENS
We are uncertain if there are any differences between TENS and no TENS for adequate pain relief. We downgraded the evidence for risks of bias and for outcome assessment imprecision, as there were very few participants (Table 7).
Safety of analgesia during breastfeeding
A primary concern about interventions for the management of pain among breastfeeding mothers is for infant safety. Potential infant adverse events were not investigated in any of the identified studies, and in many of them women were actually excluded if they were breastfeeding. Information on the safety of analgesics during breastfeeding must therefore be drawn from other studies.
Transfer of high‐dose aspirin (acetylsalicylic acid) results in disproportionately higher salicylic acid levels in breast milk. Long‐term, high‐dose maternal aspirin ingestion probably caused metabolic acidosis in one breastfeeding infant. Reye's syndrome is associated with aspirin administration to infants with viral infections, but the risk of Reye's syndrome from salicylate in breast milk is unknown. For this reason experts advise against the use of aspirin during breastfeeding (Hale 2019).
Ibuprofen is considered among the safest NSAIDs to use for breastfeeding women. The amount of transfer into breast milk has been reported as minimal, due to high protein binding. No infant concerns have arisen due to exposure through breast milk (Hale 2019).
There is little published evidence for the use of fenoprofen during lactation, with a suggestion that some clinicians consider fenoprofen to be acceptable for breastfeeding. Other agents may be preferred (LactMed 2018a).
Flurbiprofen (LactMed 2018b) and ketorolac have been studied in a limited number of women. Levels are difficult to detect in breast milk following recommended dosages and are considered safe for breastfeeding women. Caution has been advised when using systemic ketorolac for a longer period, in particular when breastfeeding a preterm infant, but there is a lack of evidence to substantiate this (Drugs.com 2019a).
Metamizole has been removed from sale in many countries, due to serious adverse events including agranulocytosis, aplastic anaemia and other dyscrasias. It has been studied in a very small number of breastfeeding women and detected in small amounts in their breast milk. Two cyanotic episodes in one infant were noted 30 minutes after the breastfeeding mother consumed 1500 mg. Metamizole is not recommended, as safer alternatives are available (Hale 2019).
Naproxen is considered moderately safe for breastfeeding women in short‐term use. The amount of transfer into breast milk has been reported as minimal. It has a relatively long half‐life of 12‐15 hours. One case has been documented of an infant with prolonged bleeding, haemorrhage and acute anaemia. Long‐term use of naproxen in breastfeeding women may be hazardous (Hale 2019).
Paracetamol (acetaminophen) is an analgesic and antipyretic. It has been well‐researched in breastfeeding women; amounts passed into breast milk are considered too small to be hazardous and in recommended doses it is considered safe. However, judicious use is recommended, as there has been considerable debate in recent years that paracetamol may be linked to an increase in asthma in infants and children, although other studies have refuted this (Hale 2019).
The amount of codeine secreted though breast milk differs according to maternal dose and metabolism. Several cases of neonatal sedation, apnoea, bradycardia, cyanosis and one infant death have been linked to codeine usage (Koren 2006). Hereditary polymorphisms of the drug‐metabolising enzyme cytochrome P450 2D6 (CYP2D6) mean that some individuals lacking this enzyme will find codeine ineffective, but others may be ultra‐rapid metabolisers of codeine to morphine (Dean 2017). This means that some lactating women may pass potentially fatal concentrations of morphine to their infants through breast milk. One infant death has been reported (Lam 2012). Breastfeeding women should be informed of the risks and if they decide to take codeine preparations need to watch for signs of sedation and codeine toxicity in their infants (Hale 2019).
Nalbuphine is a potent synthetic narcotic similar in potency to morphine and excreted into breast milk in very small amounts. Nalbuphine has poor oral absorption, so is therefore unlikely to affect the breastfeeding infant and is considered safe (Drugs.com 2019b).
The effects of herbal analgesia in lactation have not been well studied; it is therefore not possible to determine infant risks.
Overall completeness and applicability of evidence
The evidence identified in this review comes from middle‐ to high‐income countries, and is specifically focused on women with postpartum pain from uterine cramping. The applicability of the evidence may be limited by the fact that the studies generally included only women with singleton pregnancies and uncomplicated births. Furthmore, many studies excluded breastfeeding women. which clearly limits the applicability of the evidence, given that women are generally encouraged to breastfeed.
While most studies reported adequate pain relief and maternal adverse events, our secondary outcomes were not well reported. Uncertainty therefore remains around the effects of pain relief on the risk of neonatal adverse events, duration of hospital stay, breastfeeding and postnatal depression.
We identified nine ongoing studies, which may help to increase the level of certainty of the evidence around pain relief due to uterine cramping in future updates of this review.
Quality of the evidence
Generally the trials were at low risk of selection bias, performance bias and attrition bias, but some trials were at high or unclear risk of bias due to concerns about selective reporting and lack of blinding. Many of the studies were published prior to the requirement to register clinical trials prospectively on a clinical trials registry, and we therefore could not judge whether the prespecified outcomes were reported.
To establish the GRADE certainty of evidence we decided to downgrade for risk of bias because most trials did not describe their methods in sufficient detail for us to be sure that robust randomisation, allocation concealment, blinding and outcome reporting methods were used. We also downgraded for imprecision, because many studies included few participants; studies that do not recruit adequate numbers of women cannot reach a precise estimate of effectiveness due to under‐powered study samples. We also downgraded evidence from one comparison (comparison 6: herbal analgesia) for indirectness, because some study interventions were not therapeutic doses.
Potential biases in the review process
With a comprehensive literature search, unrestricted by language or publication status, we made every effort to identify all the relevant evidence and to contact study authors for clarification or for further data. However, it is possible that we may have missed some evidence.
We further reduced bias in the review process by having two review authors conduct independent data extraction, 'Risk of bias' assessment and GRADE assessments.
Agreements and disagreements with other studies or reviews
The results of this review are in agreement with previous Cochrane Reviews assessing the use of NSAIDs or paracetamol for relief of pain due to perineal trauma. These reviews identified low‐certainty evidence supporting the use of aspirin (Molakatalla 2017), NSAIDs (Wuytack 2016), or paracetamol (Chou 2013) in the treatment of acute postpartum perineal pain.
The results of this review are also in agreement with a previous Cochrane Review assessing the use of NSAIDs for treating primary dysmenorrhoea (Marjoribanks 2015). This review identified low‐certainty evidence that NSAIDs appear to be a very effective treatment for dysmenorrhoea. The authors also concluded that there was insufficient evidence to determine which (if any) individual NSAID is the safest and most effective for the treatment of dysmenorrhoea.
Authors' conclusions
Implications for practice.
Based on low‐ to moderate‐certainty evidence, NSAIDs appear to be the most effective analgesia for treating postpartum women experiencing pain from uterine cramping and involution after vaginal birth.
Paracetamol may be a possible alternative where the use of NSAIDs is not appropriate, but it can also be used in addition to NSAIDs. Opioids may be more effective than placebo, but with more adverse effects. Due to low‐certainty evidence, we are uncertain about the effectiveness of other forms of pain relief for treating postpartum women experiencing pain from uterine cramping and involution after vaginal birth.
Implications for research.
Further research is required, including a survey of postpartum women to describe appropriately their experience of uterine cramping and involution.
We believe there is sufficient information about pharmacological analgesia versus placebo. A well‐controlled study should compare drugs in current use known to be safe in this population.
There is insufficient information about non‐pharmacological analgesia; these should be assessed in well‐designed randomised controlled studies.
Studies should report all outcomes of relevance to women and their babies, and to healthcare providers.
What's new
| Date | Event | Description |
|---|---|---|
| 31 October 2019 | New citation required but conclusions have not changed | Nine new studies included, but conclusions remain unchanged. Two conference proceedings previously excluded are now included, but provide no data (Bloomfield 1983; Bloomfield 1986c). |
| 31 October 2019 | New search has been performed | Search updated and identified 56 new trial reports to assess. |
History
Protocol first published: Issue 3, 2004 Review first published: Issue 5, 2011
Acknowledgements
Thank you to Emily Sheppard from the University of Adelaide for her guidance and support for updating the structure of this review.
The Cochrane Library now has a generic protocol for meta‐analyses of interventional studies for perineal trauma (Chou 2010). The first review using this protocol assessing paracetamol (acetaminophen) has been published (Chou 201). We used this review as guidance.
This project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to Cochrane Pregnancy and Childbirth. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Evidence Synthesis Programme, the NIHR, National Health Service (NHS) or the Department of Health and Social Care.
As part of the pre‐publication editorial process, this review has been commented on by three peers (an editor and two referees who are external to the editorial team), a member of the Pregnancy and Childbirth Group's international panel of consumers and the Group's Statistical Adviser. The authors are grateful to the following peer reviewers for their time and comments: Professor Livia Puljak, MD PhD, Center for Evidence‐Based Medicine and Health Care, Catholic University of Croatia, Croatia; Professor Amita Ray, Department of Obstetrics and Gynaecology, IQ City Medical College, Durgapur, West Bengal, India.
The following people assisted us with translations of papers published in languages other than English: Dr Aidan L Tan: Dept, Organisation: Health Services & Outcomes Research, National Healthcare Group, Singapore. (Li 2014; Li 2015)
Mr Mojtaba Keikha: Department of Epidemiology, Shahroud University of Medical Sciences, Shahroud, Iran. (Chananeh 2018; Pourmaleky 2013; Tafazoli 2013)
Dr Ibrahim Ethem Yaylali: Dentistry, Endodontics, Isparta Military Hospital, Isparta, Turkey. (Bilgin 2016)
Dr Nahid Akbari: Student Research Committee, Iran University Of Medical Sciences, Tehran, Iran.
Selin Biçer: McGill University
Appendices
Appendix 1. Search methods for ICTRP and ClinicalTrials,gov
ICTRP
cramp AND postpartum (including all synonyms)
ClinicalTrials.gov
Advanced search
Interventional Studies | postpartum pain
after pain | Interventional Studies
postpartum | Interventional Studies | Pain
(each line was run separately)
Data and analyses
Comparison 1. NSAID versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Adequate pain relief as reported by the woman. | 11 | 946 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.45, 1.91] |
| 1.1.1 Aspirin 650 mg | 6 | 282 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [1.09, 1.61] |
| 1.1.2 Naproxen 275 mg | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [0.98, 2.31] |
| 1.1.3 Naproxen 300 mg | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.90, 2.36] |
| 1.1.4 Naproxen 550 mg | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.56 [1.43, 4.57] |
| 1.1.5 Naproxen 600 mg | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.54 [1.00, 2.38] |
| 1.1.6 Flurbiprofen 50 mg | 1 | 46 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.93, 2.08] |
| 1.1.7 Ketorolac 5 mg | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.73 [0.92, 3.27] |
| 1.1.8 Ketorolac 10 mg | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.88, 3.16] |
| 1.1.9 Fenoprofen 12.5 mg | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.22 [0.37, 13.48] |
| 1.1.10 Fenoprofen 25 mg | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.78 [0.47, 16.56] |
| 1.1.11 Fenoprofen 50 mg | 2 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.72 [1.03, 13.39] |
| 1.1.12 Fenoprofen 100 mg | 2 | 69 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.86 [1.04, 7.89] |
| 1.1.13 Fenoprofen 200 mg | 3 | 93 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [1.15, 6.23] |
| 1.1.14 Fenoprofen 300 mg | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.41 [0.73, 7.99] |
| 1.2 Need for additional pain relief | 4 | 375 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.07, 0.33] |
| 1.2.1 Aspirin 650 mg | 2 | 85 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.02, 0.63] |
| 1.2.2 Ketorolac 5 mg | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.02, 1.65] |
| 1.2.3 Ketorolac 10 mg | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.02, 1.65] |
| 1.2.4 Naproxen 275 mg | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.01, 2.02] |
| 1.2.5 Naproxen 300 mg | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.01, 1.98] |
| 1.2.6 Naproxen 600 mg | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 2.09] |
| 1.2.7 Naproxen 550 mg | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.05, 0.94] |
| 1.3 Maternal adverse events | 9 | 598 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.78, 1.41] |
| 1.3.1 Aspirin 650 mg | 6 | 238 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.58, 1.47] |
| 1.3.2 Flurbiprofen 50 mg | 1 | 46 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.31, 3.71] |
| 1.3.3 Naproxen 275 mg | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.27, 5.70] |
| 1.3.4 Naproxen 300 mg | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.35, 2.21] |
| 1.3.5 Naproxen 550 mg | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.00 [0.67, 5.94] |
| 1.3.6 Naproxen 600 mg | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.36, 2.36] |
| 1.3.7 Ketorolac 5 mg | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.24, 4.18] |
| 1.3.8 Ketorolac 10 mg | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.29, 4.73] |
| 1.3.9 Fenoprofen 200 mg | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.50, 2.88] |
Comparison 2. NSAID versus opioid.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Adequate pain relief as reported by the woman | 5 | 560 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [1.13, 1.57] |
| 2.1.1 Aspirin 650 mg versus codeine 60mg | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.65, 1.61] |
| 2.1.2 Aspirin 650 mg versus codeine 120 mg | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.69, 1.84] |
| 2.1.3 Fenoprofen 12.5 mg versus codeine 60 mg | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.35, 3.52] |
| 2.1.4 Fenoprofen 25 mg versus codeine 60 mg | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.42, 2.04] |
| 2.1.5 Fenoprofen 50 mg versus codeine 60 mg | 2 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.68, 2.27] |
| 2.1.6 Fenoprofen 100 mg versus codeine 60 mg | 2 | 69 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.77, 2.66] |
| 2.1.7 Fenoprofen 200 mg versus codeine 60 mg | 3 | 92 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.81, 2.47] |
| 2.1.8 Fenoprofen 300 mg versus codeine 60 mg | 1 | 37 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.84 [0.73, 4.65] |
| 2.1.9 Flurbiprofen 50 mg versus codeine 60 mg | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.86, 1.85] |
| 2.1.10 Flurbiprofen 50 mg versus codeine 120 mg | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.86, 1.96] |
| 2.1.11 Naproxen 300 mg versus codeine 60 mg | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.90, 2.36] |
| 2.1.12 Naproxen 600 mg versus codeine 60 mg | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.71 [1.06, 2.77] |
| 2.2 Need for additional pain relief | 2 | 232 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.12, 1.12] |
| 2.2.1 Aspirin 650 mg versus codeine 60 mg | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.06, 13.82] |
| 2.2.2 Aspirin 650 mg versus codeine 120 mg | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [0.12, 60.93] |
| 2.2.3 Flurbiprofen 50 mg versus codeine 60 mg | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 2.2.4 Flurbiprofen 50 mg versus codeine 120 mg | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.02, 8.08] |
| 2.2.5 Naproxen 300 mg versus codeine 60 mg | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.01, 1.98] |
| 2.2.6 Naproxen 600 mg versus codeine 60 mg | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 2.09] |
| 2.3 Maternal adverse events | 3 | 255 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.43, 0.89] |
| 2.3.1 Aspirin 650 mg versus codeine 60 mg | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.24, 2.32] |
| 2.3.2 Aspirin 650 mg versus codeine 120mg | 1 | 33 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.11, 0.65] |
| 2.3.3 Fenoprofen 200 mg versus codeine 60 mg | 1 | 23 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [0.60, 5.61] |
| 2.3.4 Flurbiprofen 50 mg versus codeine 60 mg | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.18, 2.22] |
| 2.3.5 Flurbiprofen 50 mg versus codeine 120 mg | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.08, 0.65] |
| 2.3.6 Naproxen 300 mg versus codeine 60 mg | 1 | 52 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.39, 3.05] |
| 2.3.7 Naproxen 600 mg versus codeine 60 mg | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.41, 3.25] |
Comparison 3. Opioid versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Adequate pain relief as reported by the woman | 5 | 299 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.99, 1.61] |
| 3.1.1 Codeine 60 mg versus placebo | 5 | 252 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [1.01, 1.76] |
| 3.1.2 Codeine 120 mg versus placebo | 1 | 47 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.65, 1.64] |
| 3.2 Need for additional pain relief | 3 | 273 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.28, 0.82] |
| 3.2.1 Codeine 60 mg versus placebo | 3 | 173 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.24, 1.02] |
| 3.2.2 Codeine 120 mg versus placebo | 1 | 47 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.02, 1.52] |
| 3.2.3 Nalbuphine versus placebo | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.25, 1.36] |
| 3.3 Maternal adverse events | 3 | 188 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.59 [0.99, 2.55] |
| 3.3.1 Codeine 60 mg versus placebo | 3 | 141 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.54, 1.67] |
| 3.3.2 Codeine 120 mg versus placebo | 1 | 47 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.65 [1.66, 13.00] |
Comparison 4. Paracetamol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.1 Paracetamol 650 mg versus placebo | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2 Need for additional pain relief | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2.1 Paracetamol 1000 mg versus placebo | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.3 Maternal adverse events | 2 | 123 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.27 [0.97, 5.33] |
| 4.3.1 Paracetamol 650 mg versus placebo | 1 | 48 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.36 [0.95, 5.88] |
| 4.3.2 Paracetamol 1000 mg versus placebo | 1 | 75 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.85 [0.17, 19.50] |
Comparison 5. Paracetamol versus NSAID.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.1.1 Paracetamol 650 mg versus aspirin 650 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.2 Maternal adverse events | 2 | 112 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.52, 1.86] |
| 5.2.1 Paracetamol 650 mg versus aspirin 650 mg | 1 | 48 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.65, 2.64] |
| 5.2.2 Paracetamol 1000 mg versus naproxen 500 mg | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.08, 1.97] |
Comparison 6. NSAID versus herbal analgesia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Adequate pain relief as reported by the woman | 4 | 394 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.78, 1.18] |
| 6.1.1 Mefenamic acid 250 mg versus PAC 500 mg | 1 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.69, 1.35] |
| 6.1.2 Mefenamic acid 250 mg versus Melissa Officinalis 395 mg | 1 | 110 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.37, 1.45] |
| 6.1.3 Mefenamic acid 250 mg versus fennel 300 mg | 1 | 86 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.71, 1.41] |
| 6.1.4 Ibuprofen 400 mg versus fennel essence 20% | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.66, 1.69] |
| 6.2 Need for additional pain relief | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.2.1 Ibuprofen 400 mg versus fennel essence 20% | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.3 Pain however measured by the authors | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.3.1 VAS 0‐10 | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.4 Maternal adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.4.1 Mefenamic acid 250 mg versus PAC 500 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 7. TENS versus no TENS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.1 TENS versus no treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.2 Pain however measured by the authors decrease in VAS | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.2.1 TENS versus placebo | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.3 Pain however measured by the authors | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 7.3.1 TENS versus no treatment | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 7.4 Maternal views of treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.4.1 Maternal satisfaction with treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 8. Aspirin versus naproxen.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1.1 Aspirin 650 mg versus naproxen 275 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.2 Maternal adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.2.1 Aspirin 650 mg versus naproxen 275 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 9. Aspirin versus flurbiprofen.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.1 Aspirin 650 mg versus Flurbiprofen 50 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.2 Need for additional pain relief | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.2.1 Aspirin 650 mg versus Flurbiprofen 50 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.3 Maternal adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.3.1 Aspirin 650 mg versus Flurbiprofen 50 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 10. Aspirin versus ketorolac.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 10.1 Adequate pain relief as reported by the woman | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.81, 1.11] |
| 10.1.1 Aspirin 650 mg versus Ketorolac 5 mg | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.78, 1.28] |
| 10.1.2 Aspirin 650 mg versus Ketorolac 10 mg | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.73, 1.11] |
| 10.2 Need for additional pain relief | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.16, 8.52] |
| 10.2.1 Aspirin 650 mg versus Ketorolac 5 mg | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.00 [0.13, 29.81] |
| 10.2.2 Aspirin 650 mg versus Ketorolac 10 mg | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.03, 14.97] |
| 10.3 Maternal adverse events | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [0.86, 3.31] |
| 10.3.1 Aspirin 650 mg versus Ketorolac 5 mg | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.61, 4.59] |
| 10.3.2 Aspirin 650 mg versus Ketorolac 10 mg | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.71 [0.70, 4.20] |
10.2. Analysis.

Comparison 10: Aspirin versus ketorolac, Outcome 2: Need for additional pain relief
10.3. Analysis.

Comparison 10: Aspirin versus ketorolac, Outcome 3: Maternal adverse events
Comparison 11. Naproxen different doses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 11.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 11.1.1 Naproxen 300 mg versus Naproxen 600 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 11.2 Maternal adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 11.2.1 Naproxen 300 mg versus Naproxen 600 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 12. Ketorolac different doses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 12.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1.1 Ketorolac 5 mg versus Ketorolac 10 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.2 Need for additional pain relief | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.2.1 Ketorolac 5 mg versus Ketorolac 10 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.3 Maternal adverse events | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.33, 2.25] |
| 12.3.1 Ketorolac 5 mg vs Ketorolac 10 mg | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.33, 2.25] |
Comparison 13. Codeine versus nalbuphine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 13.1 Need for additional pain relief | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.1 Codeine 60mg vs Nalbuphine15mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 14. Codeine different doses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 14.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 14.1.1 Codeine 60 mg versus codeine 120 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 14.2 Need for additional pain relief | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 14.2.1 Codeine 60 mg versus codeine 120 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 14.3 Maternal adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 14.3.1 Codeine 60 mg versus codeine 120 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 15. Metamizol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 15.1 Pain however assessed by the authors | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 15.1.1 Reduction in VAS | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 15.2 Maternal views of treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 15.2.1 Maternal satisfaction with treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
15.1. Analysis.

Comparison 15: Metamizol versus placebo, Outcome 1: Pain however assessed by the authors
Comparison 16. TENS plus metamizol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 16.1 Pain however assessed by the authors | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 16.1.1 Reduction in VAS | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 16.2 Maternal views of treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 16.2.1 Maternal satisfaction with treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 17. TENS plus metamizol versus TENS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 17.1 Pain however assessed by the authors | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 17.1.1 Reduction in VAS | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 17.2 Maternal views of treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 17.2.1 Maternal satisfaction with treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 18. TENS plus metamizol versus metamizol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 18.1 Pain however assessed by the authors | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 18.1.1 Reduction in VAS | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 18.2 Maternal views of treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 18.2.1 Maternal satisfaction with treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 19. TENS versus metamizol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 19.1 Pain however assessed by the authors | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 19.1.1 Reduction in VAS | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 19.2 Maternal views of treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 19.2.1 Maternal satisfaction with treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Asti 2011.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: Obstetrics ward, Assli Hospital, Khoramabad – time frame not stated Inclusion criteria: 90 multiparous women with postpartum pain following a singleton cephalic vaginal delivery at term (confirmed with author that pain was due to postpartum uterine cramping) Exclusion criteria: women with episiotomy, perineal laceration, prolonged delivery, caesarean section, severe postpartum bleeding, drug sensitivity, regular use of NSAID and opioids during and after pregnancy |
|
| Interventions | Experimental intervention/comparison: fennel essence 20% (N = 45) Experimental intervention: ibuprofen (400 mg) 1 tablet (plus ranitidine (150 mg) 1 tablet) (N = 45) |
|
| Outcomes | Adequate pain relief as reported by the woman: women were asked before the 1st dose of the intervention to give their pain score by VAS, then hourly for 4 hours after the 1st dose. Severity of pain, rated on a 10 cm VAS from 0 (no pain) to 10 (worst pain ever). VAS scores were collected by 1 of the investigators
|
|
| Notes | Funding: Study was unfunded. No declaration of interests stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. Quote: "The qualified women were randomly allocated to receive either ibuprofen or fennel orally by stratified random sampling technique." |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Each treatment pack contained 1 tablet of ibuprofen 400 mg and 1 tablet of ranitidine 150 mg or 1 tablet of 20% fennel essence |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 45 women randomised to each group, the number of women analysed is not reported. The methods report a total of 90 women randomised. Results state 45 women in each group with similar demographics. There are no withdrawals/exclusions reported |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | Low risk | No other risk of bias identified |
Bettigole 1981.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: Massachusetts, USA, no information about time frame of study Inclusion criteria: postpartum women aged 46 years or less with acute uterine cramp pain, review authors have assumed women had vaginal delivery. Women gave informed consent Exclusion criteria: women who had received analgesics in the preceding 4 hours |
|
| Interventions | Following initial pain assessment women were randomly allocated to 1 of 3 treatment groups:
|
|
| Outcomes | Adequate pain relief as reported by the woman: pain assessed by an observer before the 1st dose and at hourly intervals for 8 hours
|
|
| Notes | Paper does not state how many women were randomised Dates of study: not stated Funding sources: not stated Declarations of interest: no declaration of interests statement |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study medications were "prescribed in random order". |
| Allocation concealment (selection bias) | Unclear risk | Concealment not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study medication was presented as "capsules of identical appearance". Study described as double‐blind, but unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Study described as double‐blind, but unclear who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The number of women who were randomised is not given. Includes only participants for whom complete data were available. Baseline demographics and results include 35 women with pain from uterine cramping |
| Selective reporting (reporting bias) | Unclear risk | No protocol published or trial registration available |
| Other bias | Unclear risk | Unclear if randomisation was stratified by source of pain; uterine cramp or episiotomy although data analysed separately |
Bloomfield 1977 Study 1.
| Study characteristics | ||
| Methods | Randomised controlled trial. | |
| Participants | Setting: single‐centre study at Cincinnati General Hospital ‐ time frame not given Inclusion criteria: women with moderate or severe postpartum uterine cramp pain within 48 hours of an uncomplicated birth Exclusion criteria: women experiencing episiotomy pain greater than their uterine cramp pain; unmarried women less than 18 years of age; women with history of aspirin or codeine allergy; women given analgesics, sedatives or other psychotropic within previous 6 hours; women breastfeeding their babies. Known drug dependence |
|
| Interventions | Following initial pain assessment women were randomly allocated to 1 of 4 treatment groups, stratified by initial pain intensity, moderate or severe, and given a single dose of study medication.
|
|
| Outcomes | Adequate pain relief as reported by the woman: women were interviewed by 1 nurse observer before drug administration and ½ hour post‐drug administration, then hourly for 7 hours
|
|
| Notes | Dates of study: not stated Funding sources: study funded in part by a grant from Syntex Laboratories (pharmaceutical company) Declarations of interest: no declaration of interests statement |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Women were randomised using a "predetermined balanced schedule, which assured that all treatment groups were of equal size. The randomization also provided for stratification of patients when first interviewed on the basis of pain (moderate or severe), with treatment groups equalized within the strata". Unclear exactly how random sequence was generated |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study medication presented as capsules of identical appearance.Trial capsules were identical in appearance and taste, and were packaged in code numbered individual dose vials |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data from all women randomised were included in analyses. There were 35 women allocated to each of 4 groups. There were 8 participants who required additional analgesia, 4 women in the codeine group and 4 women in the placebo group. These women were withdrawn from the study and were not interviewed about pain following withdrawal. Subsequent pain intensity scores were adjusted to their pretreatment score and used in the analysis |
| Selective reporting (reporting bias) | Unclear risk | No protocol published or trial registration available |
| Other bias | Low risk | No other risks of bias identified |
Bloomfield 1977 Study 2.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: single‐centre study at Cincinnati General Hospital ‐ time frame not given Inclusion criteria: women with severe or moderate postpartum uterine cramp pain within 48 hours of an uncomplicated birth Exclusion criteria: women experiencing episiotomy pain greater than their uterine cramp pain; unmarried women less than 18 years of age; women with history of aspirin or codeine allergy; women given analgesics, sedatives or other psychotropic within previous 6 hours; women breastfeeding their babies. Known drug dependence |
|
| Interventions | Following initial pain assessment women were randomly allocated to 1 of 3 treatment groups, stratified by initial pain intensity, moderate or severe, and given a single dose of study medication.
|
|
| Outcomes | Adequate pain relief as reported by the woman: women were interviewed by 1 nurse observer before drug administration and ½ hour post drug administration then hourly for 7 hours.
|
|
| Notes | Dates of study: not stated Funding sources: study funded in part by a grant from Syntex Laborattories (pharmaceutical company) Declarations of interest: no declaration of interests statement |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned using a "predetermined balanced schedule, which assured that all treatment groups were of equal size. The randomization also provided for stratification of patients when first interviewed on the basis of pain (moderate or severe), with treatment groups equalized within the strata". Unclear exactly how random sequence was generated |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study was double‐blind using tablets which were identical in appearance and taste and were packaged in code numbered individual dose vials |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data from all women randomised were included in analyses. There were 30 women randomised to each group. There were 4 participants who required additional analgesia, all from the placebo group. These women were withdrawn from the study and were not interviewed about pain following withdrawal. Subsequent pain intensity scores were adjusted to their pretreatment score and used in the analysis |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | Low risk | No other risks of bias identified |
Bloomfield 1978.
| Study characteristics | ||
| Methods | Randomised controlled trial. | |
| Participants | Setting: single centre study at Cincinnati General Hospital ‐ time frame not given Inclusion criteria: women who had given birth within the previous 48 hours with moderate or severe uterine cramp pain as assessed by the woman Women were 18 years or older Exclusion criteria: women experiencing episiotomy pain greater than their uterine cramp pain; unmarried women less than 18 years of age; women with history of aspirin allergy; women given analgesics, sedatives or other psychotropic within previous 6 hours; women breastfeeding their babies |
|
| Interventions | Following initial pain assessment women were randomly allocated to 1 of 2 treatment groups, stratified by initial pain intensity, moderate or severe, and given a single dose of 1 of 2 study medications when required.
|
|
| Outcomes | Adequate pain relief as assessed by the woman: pain was assessed at ½ hour post‐study medication then hourly for 7 hours. All interviews were conducted by the same trained nurse observer
|
|
| Notes | Additional study arms: this study included an additional 3 arms of fendosal 100 mg, 200 mg and 400 mg. This medication is no longer available, so these arms were not included Dates of study: not stated Funding sources: study funded in part by grant from pharmaceutical company Hoechst‐Roussel Declarations of interest: no declaration of interests statement |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned using a "predetermined balanced schedule, stratified by pretreatment pain intensity". Unclear exactly how random sequence was generated |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was "double blind". "All capsules were identical in taste and appearance." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data from all women randomised were included in analyses. There were 20 women allocated to each group. There were 2 women who required additional analgesia, 1 woman from each group, placebo and aspirin. These women were withdrawn from the study and were not interviewed about pain following withdrawal. Subsequent pain intensity scores were adjusted to their pretreatment score and used in the analysis |
| Selective reporting (reporting bias) | Unclear risk | No protocol published or trial registration available |
| Other bias | Low risk | No other risks of bias identified |
Bloomfield 1981.
| Study characteristics | ||
| Methods | Randomised controlled trial. | |
| Participants | Setting: single‐centre study at Cincinatti General Hospital ‐ time frame not given Inclusion criteria: women with moderate or severe postpartum uterine cramp pain within 48 hours of an uncomplicated birth Exclusion criteria: women given analgesics or other central nervous system drugs within previous 6 hours; women with a known allergy to aspirin or paracetamol (acetaminophen); women breastfeeding their babies |
|
| Interventions | Women were randomly allocated to 1 of 3 treatment groups, stratified by initial pain intensity, moderate or severe, and given a single oral dose (2 capsules) of 1 of the following:
Placebo (N = 26) |
|
| Outcomes | Adequate pain relief as assessed by the woman: women were interviewed by a trained nurse observer at baseline, ½ hour post‐treatment and hourly for 6 hours
|
|
| Notes | Additional study arms: this study included an additional 2 arms of pirprofen 200 mg and 400 mg. This medication is no longer available, so these arms were not included. Study included a comparison of pirprofen. Since this medication not in current used it was not included Dates of study: not stated. Funding sources: study funded in part by grant from pharmaceutical company CIBA‐GEIGY Corporation Declarations of interest: no declaration of interests statement |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Within each of the 2 pain strata there was a separate, balanced randomisation of patients". Women with moderate and women with severe pain intensity were evenly divided between the treatment groups Unclear exactly how random sequence was generated |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Use of "identical coded capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data on all women randomised were reported. There were 26 women allocated to the aspirin and placebo groups and 22 women allocated to the paracetamol group. There are no reported withdrawals and the number of women included at baseline and reported for all outcomes is the same |
| Selective reporting (reporting bias) | Unclear risk | No protocol published or trial registration available |
| Other bias | Low risk | No other risk of bias identified |
Bloomfield 1983.
| Study characteristics | ||
| Methods | RCT Setting: USA |
|
| Participants | 150 women with moderate or severe postpartum uterine cramps Number per group not reported. Inclusion/exclusion criteria not reported. |
|
| Interventions | Group 1: 100 mg ibuprofen Group 2: 200 mg ibuprofen Group 3: 400 mg ibuprofen Group 4: 650 mg aspirin Group 5: placebo |
|
| Outcomes | Summed and peak analgesia | |
| Notes | No useable data. No outcome data presented by intervention group Dates of study: not reported Funding sources: not reported Declarations of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | not reported ‐ "stratified, randomized, double‐blind design" |
| Allocation concealment (selection bias) | Unclear risk | not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | not reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | no information reported on numbers providing data |
| Selective reporting (reporting bias) | Unclear risk | No protocol, outcome data not reported in full by group |
| Other bias | Low risk | Nothing to indicate any other source of bias. |
Bloomfield 1986a.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: single‐centre study at Cincinnati General Hospital ‐ time frame not given Inclusion criteria: hospitalised postpartum women, 18 years or older with an uncomplicated birth and moderate or severe uterine cramps Exclusion criteria: women who experienced episiotomy pain greater than their uterine cramp pain; women with history of hypersensitivity to aspirin or codeine; women given analgesics, sedatives or other psychotropic within previous 6 hours; known drug dependence; women breastfeeding their babies |
|
| Interventions | Women were randomly allocated to 1 of 5 treatment groups, stratified by initial pain intensity, moderate or severe, and given a single oral dose (2 capsules) of 1 of the following.
|
|
| Outcomes | Adequate pain relief as assessed by the woman: pain assessed by 1 of 2 trained nurse observers before the 1st dose and at ½hour or hourly intervals for 6 hours
|
|
| Notes | Quote: "The pain intensity score for each unperformed interview was adjusted to the pretreatment value, and the adjusted scores were analysed" Dates of study: not stated. Funding sources: study funded in part by grant from pharmaceutical company Upjohn Declarations of interest: no declaration of interests statement |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Women were "randomly assigned". "On enrolment. patients underwent a two‐way stratification according to morning or afternoon shifts of the two clinical nurse observers, and according to moderate or severe pretreatment pain intensity" Unclear exactly how random sequence was generated |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind, the participant and the caregiver. Medications were "pre packed, code numbered", and were identical in appearance and taste |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data on all women randomised have been included in the reported outcomes. There were 30 women allocated to the flurbiprofen group, 34 women to the aspirin group, 32 women to each of the codeine 120mg group and placebo, and 321 women to the codeine 60mg group, including appropriately‐estimated data for the 9 women who withdrew. Five women from the placebo group, two women from the aspirin group and one woman each from the two codeine groups were withdrawn so that they could receive rescue analgesia. These women were not interviewed about pain following withdrawn. Subsequent pain intensity scores were adjusted to their pretreatment score and used in the analysis |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | Low risk | No other risk of bias identified |
Bloomfield 1986b.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: single‐centre study at Cincinnati General Hospital ‐ time frame not given Inclusion criteria: hospitalised women with moderate or severe uterine cramp pain within 48 hours of an uncomplicated vaginal birth Exclusion criteria: women who experienced episiotomy pain greater than their uterine cramp pain; unmarried women less than 18 years of age; women with history of hypersensitivity to aspirin or other NSAIDs; women given analgesics, sedatives or other psychotropic within previous 4 hours; known drug dependence; women breastfeeding their babies. |
|
| Interventions | Following initial pain assessment women were randomly allocated to 1 of 4 treatment groups and given appropriate study medication on demand. Randomisation was stratified by initial pain intensity and by 1 of 2 nurse observers
|
|
| Outcomes | Adequate pain relief as assessed by the woman: women were interviewed by 1 of 2 trained nurse observers before drug administration and ½ hour post‐treatment and then hourly for 6 hours.
|
|
| Notes | Dates of study: not stated Funding sources: Syntex Research. Ketorolac marketed by Syntex Inc in 1991 Declarations of interest: none stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Women were assigned using a "predetermined balanced randomisation schedule". "On enrolment, patients underwent two‐way stratification: first according to clinical nurse‐observer (morning or afternoon shift) and second, according to initial pain intensity (moderate or severe). Within each of these 4 strata, patients were allocated to 1 of 4 treatment groups according to a predetermined, balanced, randomization schedule that assured that all groups were of equal size and matched with respect to initial intensity of pain and nurse‐observers." Unclear exactly how random sequence was generated |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Capsules were identical in appearance and taste and were packaged in individual code numbered containers.." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data on all women randomised have been included in the reported outcomes. There were 30 women allocated to each of the groups. Including appropriately estimated data for the 10 women who withdrew. Seven women from the placebo group, and one woman each from the other three groups were withdrawn so that they could receive rescue analgesia. These women were not interviewed about pain following withdrawn. Subsequent pain intensity scores were adjusted to their pretreatment score and used in the analysis |
| Selective reporting (reporting bias) | Unclear risk | No protocol published or trial registration available |
| Other bias | Low risk | No other risk of bias identified |
Bloomfield 1986c.
| Study characteristics | ||
| Methods | RCT Setting: USA |
|
| Participants | 203 women with moderate or severe postpartum uterine cramps Number per group not reported Inclusion/exclusion criteria not reported |
|
| Interventions | Intervention: single oral dose 650 mg aspirin Intervention: single oral dose 1000 mg acetaminophen Comparator: placebo |
|
| Outcomes | Pain relief and pain intensity | |
| Notes | No useable data. No outcome data presented by intervention group Dates of study: not reported Funding sources: not reported Declarations of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | not reported ‐ "stratified, randomized, double‐blind design" |
| Allocation concealment (selection bias) | Unclear risk | not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | not reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | no information reported on numbers providing data |
| Selective reporting (reporting bias) | Unclear risk | No protocol, no outcome data presented by group |
| Other bias | Low risk | Nothing to indicate any other source of bias |
Bloomfield 1987.
| Study characteristics | ||
| Methods | Randomised controlled trial Sample size calculation not stated. |
|
| Participants | Setting: single‐centre study at Cincinnati General Hospital ‐ time frame not given Inclusion criteria: hospitalised women with moderate of severe uterine cramp pain within 48 hours of an uncomplicated vaginal birth Exclusion criteria: women were excluded with known hypersensitivity to aspirin or NSAIDs, if they had been given other analgesia or were breastfeeding their babies |
|
| Interventions | Following initial pain assessment women were randomly allocated to 1 of 2 treatment groups and given appropriate study medication on demand. Randomisation was stratified by initial pain intensity and by 1 of 3 nurse observers
|
|
| Outcomes | Adequate pain relief as assessed by the woman: women were interviewed by 1 of 3 trained nurse observers before drug administration and ½ hour post‐treatment and then hourly for 6 hours
|
|
| Notes | Additional study arms: this study included an additional 2 arms of anirolac 50 mg and 100 mg. This medication is no longer available, so these arms were not included Dates of study: not stated. Funding sources: Sytnex Research ‐ manufacturer of anirolac Declarations of interest: none stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Women were assigned using a "predetermined balanced randomized schedule". "On enrolment, patients underwent two‐way stratification: first according to clinical nurse‐observer (morning or afternoon shift) and second, according to initial pain intensity (moderate or severe). Within each of these 6 strata, patients were allocated to 1 of 6 treatment groups according to a predetermined, balanced, randomization schedule that assured that all groups were of equal size and matched with respect to initial intensity of pain and nurse‐observers." (Only 2 of the 6 strata have been included in this meta‐analysis). Unclear exactly how random sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study medications were "packaged in code‐numbered individual dose containers". "All capsules identical in taste and appearance." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data on all women randomised have been reported. There were 30 women allocated to each of the groups. One women in the placebo group was disqualified as she was inadvertently given a medication contraindicated with study medications. One women in the naproxen group had received analgesia prior to enrolling in the study and was also disqualified. These women were replaced |
| Selective reporting (reporting bias) | Unclear risk | No protocol published or trial registration available |
| Other bias | Low risk | No other risk of bias identified |
Chananeh 2018.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Persian Gulf Shohada Hospital of Bushehr Hospital Participants: 100 multiparous women, aged 15 ‐ 44 years, with natural delivery complaining of moderate‐to‐severe postpartum pain as measured using VAS 2 hours after delivery; women with pain score > 4 enrolled Inclusion criteria: 1.Being Iranian; 2. can read and write; 3. vaginal delivery; 4. spontaneous exit of placenta and membranes; 5. Multiparous; 6. gestational age between 37 ‐ 42 weeks; 7. complained of moderate or severe pain after delivery; 8. feeding is started and will be continued; 9. mother age between 15 ‐ 44 years; 10. deliver a singleton and healthy baby Exclusion criteria: 1. Instrumental delivery or pressure on uterus; 2. 3rd or 4th degree laceration;.3. history of caesarian section or pelvic operation; 4. use of any narcotic drug during labor and delivery or used at least 4 hours ago; 5. used epidural or spinal anaesthesia during labour and delivery; 6. mother has any drug addiction; 7. maternal history of herbal drug allergy; 8. maternal history of chronic disease Exclusion criteria during trial: 1. herbal drug sensitivity in mother during study; 2. mother uses other methods or drugs to relieve pain during study. 3. mother is suffering serious complications after delivery (such as high blood pressure, severe bleeding after childbirth, fever, etc.); 4. breastfeeding discontinued for mother or baby reasons; 5. mothers withdraws consent |
|
| Interventions | 1. Nigella sativa (500 mg capsule) + mefenamic acid (250 mg capsule), women received 4 tablets, 6 hourly for 24 hours; n = 50 2. Placebo + mefenamic acid (250 mg capsule), women received 4 tablets, 6‐hourly for 24 hours; n = 50 | |
| Outcomes | Severity and duration of pain measured using VAS before, and 1 hour after medication administered. Side effects | |
| Notes | Dates of study: 05 May 2017 ‐ final date not available (information from trial registration) Sponsor: Faculty of Nursing and Midwifery, Shahid Behesti Medical Science University Declarations of interest: none stated. Abstract in English, full paper in Perisan. Author emailed 8 February 2020 Translation requested 22 July 2020 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised double‐blind clinical trial Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | No information provided to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All women received 1 capsule of mefenamic acid, women in the intervention group additionally received a capsule of Nigella Sativa, women in the control group received a capsule similar in appearance to the Nigella Sativa but without Nigella Sativa Paper states "mothers and investigators were blinded". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Paper states "mothers and investigators were blinded" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Results reported for all randomised women |
| Selective reporting (reporting bias) | Low risk | Stated outcomes were reported. Trial registration available |
| Other bias | Unclear risk | Abstract only in English, full text required translation |
Dastjerdi 2019.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: Asgariyeh Hospital, Isfahan City, 2016 Trial registration states that recruitment was expected to occur between August and November 2016 Inclusion criteria: 126 women with uncomplicated normal vaginal delivery without epidural or spinal anaesthesia; able to breastfeed, with moderate to severe postpartum pain requiring narcotics. Women with uterine cramp pain (author email 6 January 2020) Exclusion criteria: women with history of previous caesarean section or abdominal surgery; serious maternal complications after the birth (postpartum bleeding, temperature > 39 °C; BP > 140/90); allergy to Melissa or other herbal drugs; pre‐existing chronic disease such as diabetes, hypertension, thyroid disorder. Women with pain from episiotomy (author email 6 January 2020). |
|
| Interventions | Experimental intervention: Melissa Officinalis (1 capsule, containing 150 mg dried extracts of Melissa Officinalis) 2 hours after delivery, then 6‐hourly for 24 hours (N = 63) Control/comparison: Mefenamic acid (1 capsule, 250 mg) 2 hours after delivery, then 6‐hourly for 24 hours (N = 63) | |
| Outcomes | Adequate pain relief as reported by the woman: intensity of uterine involution pain was assessed 1. Before Intervention 2. At 1, 2 and 3 hours after 1st capsule
|
|
| Notes | All 63 women received intervention of Melissa Officinalis; 8 discontinued intervention and excluded from analysis (due to headache (2), stomach ache (3), use of alternative herbal medicine (3)) All 63 women received mefenamic acid intervention; 8 excluded from analysis: 4 declined participation, 4 discontinued intervention (due to stomach ache (2), use of alternative herbal medicine (2)) Funding: Vice Chancellor for Research of Shahid Beheshti University of Medical Sciences(SBUMS) No declaration of interests stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random‐number generation using Excel |
| Allocation concealment (selection bias) | High risk | Allocation not concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Interventions similar in appearance. The M. officinalis and mefenamic acid capsules were in the same packaging. They were quite similar in colour and odour and were placed by the pharmacist within the envelopes encoded 1(mefenamic acid) and 2(M. officinalis), named as “a” and “b” respectively on the envelopes. The researcher and the samples were therefore unaware of the nature of the codes and blinding was achieved |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not explicitly stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Excluded those with adverse events from use of other pain relief. 63 women were randomly allocated to each group, There were 8 women (13%) excluded from mefenamic acid group, 4 women declined to participate and 4 women discontinued the intervention, 2 due to stomach ache and 2 in favour of different treatment. There were 8 women (13%) excluded from the M. Officinalis group, 2 due to headache, 3 due to stomachache and 3 in favour of different treatment. Baseline characteristics and outcomes were reported for 55 women in each group. Rated as high risk because authors state "In the treatment group, if a patient, an hour after taking the capsule, expressed that no pain (?relief) had occurred, mefenamic acid capsule was given for her lack of pain and the sample was excluded from the study." |
| Selective reporting (reporting bias) | High risk | Retrospective registration of trial |
| Other bias | Low risk | No other risks of bias identified |
De Sousa 2014.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: maternity hospital in Sao Paulo, Brazil Trial registration states recruitment was expected to start in July 2010 Inclusion criteria: women after vaginal delivery, multiparity, without complications postpartum, experiencing uterine contraction pain while breastfeeding, pain level greater than ‘1’ by numeric rating scale. No analgesia in 6 hours prior to study entry Exclusion criteria: intolerance to stimulus generated by use of electrical stimulation and allergy to the use of electrode, pacemaker, complications that require medical intervention (e.g. bleeding, infection) |
|
| Interventions | Women had their pain assessed and were randomly allocated to 1 of 2 study groups: Experimental intervention: TENS: women monitored in standardised position during 1 feed with no treatment, TENS then administered during next feed. The TENS device was programmed to generate a 100‐Hz current and 75 msec pulse for 40 min. Any increase in intensity was decided by the participants, after being instructed to keep a strong and tolerable stimulation without muscle contraction (N = 16) Control/comparison: no treatment; women monitored in standardised position during 2 consecutive feeds. (N = 16) |
|
| Outcomes | Adequate pain relief as assessed by the woman: uterine contraction pain was assessed by means of an 11‐point numerical rating scale during pre‐ and post‐intervention breastfeeds, in which 0 means absence of pain and 10 represents extreme pain
|
|
| Notes | The participants were informed that they could ask for pain medication at any time during the study without influencing the care received at the maternity hospital, but that this would result in their necessary exclusion from the study Funding: University Escola de Enfermagem de Ribeirao Preto da Universidade de Sao Paulo No declaration of interests stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐based randomisation |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not possible to blind intervention |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not explicitly stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All outcomes reported for all randomised participants. There were no losses to follow‐up, 32 women randomised (16 per group) and all were included in analysis |
| Selective reporting (reporting bias) | Low risk | Trial registered prospectively and all outcomes have been reported as prespecified |
| Other bias | Low risk | Participants were informed that they could ask for pain medication at any time during the study without influencing the care received at the maternity hospital, and that this would result in their necessary exclusion from the study. According to the flow chart no participants were excluded after randomisation |
Jain 1978.
| Study characteristics | ||
| Methods | Randomised controlled trial Sample size calculation not stated |
|
| Participants | Setting: New Orleans, USA. Time of study not stated Inclusion criteria: postpartum women who had an uncomplicated vaginal birth with moderate or severe uterine cramp pain (self‐rated pain score of 60% or more). The women were aged between 16 and 35 years Exclusion criteria: women dependent on analgesics or tranquillisers or hypersensitive to salicylates or caffeine. Women with gastrointestinal, hepatic or renal disease or history of psychiatric illness |
|
| Interventions | Following initial pain assessment participants were allocated to 1 of 3 treatment groups and given 1 dose of study medication
|
|
| Outcomes |
|
|
| Notes | This study included women with perineal pain and reported the majority of pain assessments including both groups of women Dates of study: not stated Funding sources: blinded drugs supplied by American Home Products. Statistical support from Ives Laboratories and Wyeth Laboratories Declarations of interest: none stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Methods state "patients were separated at random". Insufficient information to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data on all women randomised are reported. There were 7 women allocated to the aspirin 650 mg group, and 8 women to each of the aspirin 800 mg and placebo groups. Baseline pain and all reported pain outcomes were included for the 23 women with uterine pain |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration |
| Other bias | Low risk | No other risk of bias identified |
Kantor 1984a.
| Study characteristics | ||
| Methods | Randomised controlled trial Sample size calculation not stated |
|
| Participants | Setting: single‐centre study, Bellevue Hospital, New York. Time not given Inclusion criteria: postpartum women who complained of moderate or severe uterine cramp pain (review authors have assumed vaginal birth) Exclusion criteria: women breastfeeding; previous severe adverse reactions to narcotics; treated with other analgesia or sedative‐tranquillisers; severe renal, hepatic, cardiac or neurological deficits; history of drug abuse |
|
| Interventions | Women were randomly allocated to 1 of 3 treatment groups and given 1 dose of study medication followed by observations at 30 minutes and hourly for 6 hours
|
|
| Outcomes | Need for additional analgesia: number of women who dropped out or required additional analgesia were recorded | |
| Notes | The formulation of codeine (phosphate or sulfate) is not stated 121 women randomised (N = 39, C = 42, P = 40), 3 post‐randomisation exclusions (1 from each group). Women with episiotomy pain were included and most of the analyses included all women. There were 3 episiotomy women in N, 4 in C and 3 in P Dates of study: not stated Funding sources: none stated Declarations of interest: none stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to make a judgement. Study described as "randomized" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to make a judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to make a judgement |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to make a judgement |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3 of the women randomised were excluded from analyses because of "administrative deviations from protocol". There were 35 women allocated to the nalbuphine group, 37 women to the codeine group and 36 women to the placebo group. Quote: "Subjects were excluded if they had previous severe narcotic adverse events, or had severe renal, hepatic, cardiac, of neurologic deficits. Patients with a history of drug abuse and nursing mothers were excluded." The study does not report which treatment group the excluded women had been allocated to. There was 2% attrition of randomised participants. 27 women required additional analgesia toward the end of the 6‐hour post‐treatment observation, did not complete the 6‐hour period of observation, but all after the 2nd hour and were therefore included in outcome reporting. (14 women in the placebo group, 8 women in the nalbuphine group and 5 women in the codeine group). |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | Unclear risk | Unclear if randomisation was stratified by source of pain; uterine cramp or episiotomy, only 1 outcome reported separately |
Kheiriyat 2016.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: Rahzi Hospital, Ahwaz, Iran. 2015 Inclusion criteria: multiparous women with a normal‐term vaginal delivery and postpartum pain Exclusion criteria: women who were unable to be sedated and other interventions required, dystocia, prolonged labour, history of caesarean section or other abdominal surgery, any history of postpartum haemorrhage; history of underlying disease | |
| Interventions | Women had their pain assessed and were randomly divided into 1 of 2 study groups (below), they received their allocated study medication 6‐hourly up to 4 times if required Experimental intervention: dill essence (Anethum graveolens extract) 1.5 mg/kg body weight, up to 4 doses (N = 54) Control/comparison: 250 mg Mefenamic acid, up to 4 doses (N = 54) |
|
| Outcomes |
|
|
| Notes | Funding: Ahvaz Jundishapur University of Medical Sciences No declaration of interests stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Women were randomly divided, no other detail provided |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Study not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Study not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only, total numbers randomised reported but not the number analysed. There were 54 women allocated to each group |
| Selective reporting (reporting bias) | Unclear risk | No published full text or protocol available. Trial registration available but study report is abstract only |
| Other bias | Unclear risk | Full article not available in English. Translation to be requested |
Laska 1981 Study 1.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: multicentre study; Hospital Maternidad ‐ Concepcion Palacios and University Hospital in Caracas Venezuela. Time not given Inclusion criteria: postpartum women who had a vaginal birth with severe postpartum uterine cramp pain; gave consent and had no complicating illness, and were expected to tolerate the medication well Exclusion criteria: women breastfeeding, with complicating illness and expected not to tolerate the medication well |
|
| Interventions | Following initial pain assessment by a trained nurse observer women were randomly allocated to 1 of 6 treatment groups and given 1 of 6 study preparations:
|
|
| Outcomes | Adequate pain relief as assessed by the woman: pain intensity was assessed at baseline and 1, 2, 3, 4 and 5 hours post‐study medication
|
|
| Notes | Primary objective of this study was to assess the dose‐response of fenoprofen Some women who delivered by caesarean were randomised into the study but excluded from the analyses, 'N' above exclude these women Dates of study: not stated Funding sources: not stated Declarations of interest: none |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Women were "assigned according to a random code" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to make this judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study was double‐blind. Study medications were "identical in appearance". "Neither the patient or the observer knew which medication was being given" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study was double‐blind. Study medications were "identical in appearance". "Neither the patient or the observer knew which medication was being given." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 37 women were randomised into the study who had delivered by caesarean, but were not included in the analysis. There were 28 women allocated to the fenoprofen 50 mg and placebo groups, the remaining 4 groups had 29 women each. Table 1 reports the number of women in each study group, results tables do not provide the number of women included in each study group. It is unclear at which point the women who birthed by caesarean were excluded and if any women withdrew during the first 2 hours of the study. Women who withdrew before the second hour for additional analgesia were withdrawn from the study. For women who withdrew after the second hour to receive additional pain relief, their responses to the last observation were assumed for the duration of the experiment |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | Unclear risk | Women who gave birth by caesarean were randomised but excluded from the analyses. Unclear if randomisation was stratified by source of pain; uterine cramp or episiotomy although data analysed separately |
Laska 1981 Study 2.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: multicentre study; Hospital Maternidad ‐ Concepcion Palacios and University Hospital in Caracas Venezuela. Time not given Inclusion criteria: 188 postpartum women, who had a vaginal birth with severe postpartum uterine cramp pain and gave consent, with no complicating illness and were expected to tolerate the medication well Exclusion criteria: women breastfeeding, with complicating illness and expected not to tolerate the medication well |
|
| Interventions | Following initial pain assessment by a trained nurse observer women were randomly allocated to 1 of 7 treatment groups and given 1 of 7 study preparations.
|
|
| Outcomes | Adequate pain relief as assessed by the woman: pain intensity was assessed at baseline and 1, 2, 3, 4 and 5 hours post‐study medication
|
|
| Notes | Primary objective of this study was to assess the dose‐response of fenoprofen. Dates of study: not stated Funding sources: not stated Declarations of interest: none |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Women were "assigned according to a random code" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to make this judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study was double‐blind. Study medications were "identical in taste and appearance". "Neither the patient or the observer knew which medication was being given" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study was double‐blind. Study medications were "identical in taste and appearance". "Neither the patient or the observer knew which medication was being given" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 23 women were randomised into the study who had delivered by caesarean, but were not included in the analysis. There were 26 women allocated to the fenoprofen 50 mg group, all other groups had 27 women each. Table 1 reports the number of women in each study group; results tables do not provide the number of women included in each study group. It is unclear at which point the women who birthed by caesarean were excluded and if any women withdrew during the first 2 hours of the study. Women who withdrew before the second hour for additional analgesia were withdrawn from the study. For women who withdrew after the second hour to receive additional pain relief, their responses to the last observation were assumed for the duration of the experiment |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | Unclear risk | Women who gave birth by caesarean were randomised but excluded from the analyses. Unclear if randomisation was stratified by source of pain; uterine cramp or episiotomy although data analysed separately. |
Mehlhorn 2005.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: single‐centre study at Frauenklinik Friedrich‐Alexander Universitat Erlangen, Erlangen, Germany. (Time not stated) Inclusion criteria: multiparous women randomised into 4 groups Initial pain score of all women was between VAS 5 ‐ 6 (median) before intervention Exclusion criteria: women excluded if minimal involutionary pain. |
|
| Interventions | 4 treatment groups as follows:
Maximum dose was 4 x 25 Metamizole drops (625 mg) in 24 hours (total of 2500 mg. Pain score obtained with VAS; scaled 1 ‐ 10 where 1 is no pain and 10 is maximum pain |
|
| Outcomes | Pain however measured by the authors: women rated their pain using a visual analogue pain scale ‐ VAS scaled 1 ‐ 10. Number and percentage of women rating their pain as 1 to 4 points on a 1 ‐ 10 point VAS following treatment. Pain assessment documented at 2‐minute, 10‐minute, 20‐minute and 30‐minute intervals | |
| Notes | RM emailed 1st author (December 2009 and January 2010) and corresponded to ascertain information re randomisation, adequate sequence generation, allocation concealment and blinding. All correspondence in German RM emailed 1st author November 2010 to determine time and place of study ‐ no response Abstract in German and English. The 2005 paper translated by Ruth Martis Dates of study: not stated Funding sources: none stated Declarations of interest: none stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Multiparous women were assigned to 1 of 4 groups. Randomised via computer‐generated allocation |
| Allocation concealment (selection bias) | Low risk | Corresponding random number was on TENS devices and trial medication bottles |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded ‐ placebo medication (drops) and placebo TENS were used. The women, administrators and trial co‐ordinator were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blinded ‐ placebo medication (drops) and placebo TENS were used. The women, administrators and trial co‐ordinator were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data from 118 women who participated in the intervention were included and reported for all outcomes |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the Methods were reported. No protocol cited but available from first author on request |
| Other bias | Low risk | None of the included women had used TENS before. Full study not available in English. Review author RM (fluent in German) reviewed article in German |
Okun 1982.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: Los Angelas, USA. Time not stated Inclusion criteria: postpartum women, within 48 hours of delivery, with moderate or severe uterine cramp pain as assessed by the woman. The women ranged in age from 18 years to 42 years Exclusion criteria: women who were breastfeeding or using other analgesia or psychotropic drugs, or both |
|
| Interventions | Following initial pain assessment women were randomly allocated to 1 of 2 groups, stratified by initial pain intensity, moderate, severe or very severe. They were given a dose of 1 of the 2 study preparations in the form of 2 identical capsules
|
|
| Outcomes | Adequate pain relief as assessed by the woman: The same nurse assessed the women's pain intensity at 1, 2, 3, 4, 5, 6 and 7 hours after the initial dose
|
|
| Notes | Study included a comparison of fendosal, since this medication not in current use it has not been included. This study included a second group of participants who had episiotomy pain, not included in this meta‐analysis. One woman in the study was inadvertently given aspirin and 100 mg of fendosal; she was included in both of these study groups. The authors do not report whether she was included for uterine pain or episiotomy pain. Since our review does not include the fendosal arms, this woman is not counted twice in data included in this review No data for meta‐analyses Dates of study: not stated Funding sources: not stated Declarations of interest: none |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Assignment to treatment was randomized between groups" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to make this judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was "double‐blind". Study medications "were administered as identical‐looking capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Study described as "double‐blind", but unclear who was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data on all women randomised were reported for all outcomes. There were 32 women allocated to the aspirin group and 31 women to the placebo group |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | Unclear risk | Unclear if randomisation was stratified by source of pain; uterine cramp or episiotomy although data analysed separately |
Olsen 2007.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: single‐centre study at the Sahlgrenska University Hospital, Gothenburg, Sweden during 2004 Inclusion criteria: 22 women following uncomplicated vaginal birth and painful postpartum uterine contractions requiring pain relief Exclusion criteria: women with systemic disorders; abnormal pregnancy; operative delivery and receiving analgesic treatment for other pain; Swedish as 2nd language |
|
| Interventions | Women were randomly allocated to 1 of 2 treatment groups:
In both groups the TENS electrodes were placed over the lower part of the abdomen bilaterally over the uterus |
|
| Outcomes | Adequate pain relief as reported by the woman: women were asked to estimate their pain intensity using a 100 mm VAS ranging from no pain to worst possible pain, before and after treatment
|
|
| Notes | Dates of study: women recruited in 2004 Funding sources: not stated Declarations of interest: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Women were randomised using a "computer generated random table" |
| Allocation concealment (selection bias) | Low risk | Quote: "Groups coded and transferred to pre‐sealed opaque envelopes" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not possible to blind study personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 13 women allocated to the HI TENS group and 8 women to the LI TENS group. The data from 1 of 13 women (8%) in 1 allocated to the HI TENS intervention group was excluded because she withdrew after experiencing discomfort |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | High risk | Baseline imbalance ‐ numbers in abstract differ from those given in results |
Ozgoli 2017.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: Shohada Hospital, Ghochan, Iran. Timing from trial registration ‐ September to December 2013 Inclusion criteria: Iranian; speak the Persian language; normal vaginal delivery between 37 and 42 weeks, ability to breastfeed, with postpartum pain of degree 4 or more on numeric scale (not specified) and requiring narcotics. Women aged from 20 to 30 years. Women with uterine cramp pain (author email 6 January 2020) Exclusion criteria: Instrumental or caesarean birth or previous caesarean birth, epidural or spinal anaesthetic, smoking or drug abuse, chronic disease including hypertension, diabetes, heart disease or infectious disease. Allergy to anise. Women with pain from episiotomy (author email 6 January 2020). | |
| Interventions | Following initial pain assessment, women were randomised into 1 of 2 groups: Experimental intervention: The samples were given an Anise oral capsule containing 60 mg of dried extracts of anise (produced in Shahid Beheshti pharmacy faculty), 1 hour after delivery then every 6 hours for 24 hours. N = unknown Control/comparison: the samples were given a mefenamic acid oral capsule 250 mg, 1 hour after delivery then every 6 hours for 24 hours. N = unknown |
|
| Outcomes |
|
|
| Notes | 96 women randomised, N per group was not reported The 2 groups were matched in the number of parity and intensity of the pain before intervention. Funding: Vice Chancellor for research of Shahid Beheshti University of Medical Sciences(SBUMS) Declaration of interest: none stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Authors state: "Capsules are put in the bar coded packets and given to the patients randomly" |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This research is a double‐blind study in which researcher and patients are not aware about prescribed capsules |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Translation states 96 women recruited and randomised, abstract and translation state 96 women included in the trial |
| Selective reporting (reporting bias) | High risk | No published protocol. Trial registration retrospective |
| Other bias | Unclear risk | Abstract only in English, full text required translation |
Pourmaleky 2013.
| Study characteristics | ||
| Methods | Single‐blind randomised trial | |
| Participants | Multiparous women who had normal vaginal delivery in Mahshahr Hospital, Iran. The pregnant women were randomly divided into 2 groups of 61 cases in zintoma and 61 cases in mefenamic acid groups. Using the VAS, after‐pain was determined during the first 2 hours after delivery and participants received zintoma and mefenamic acid if the pain score ≥ 4 was expressed by participants | |
| Interventions |
|
|
| Outcomes | The intensity of after‐pain, before intervention and 30 minutes after intervention, for each of the 4 doses. English abstract reports no side effects experienced by any of the women | |
| Notes | Translation received 26 July 2020 Funding University of Medical Sciences, Ahvaz, Iran |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Only states study is randomised |
| Allocation concealment (selection bias) | Unclear risk | Allocation states use of ‘pocket’. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided about blinding of participants. Abstract describes this as "single blind", and translation indicates that researchers were blinded: 'In order for researcher not to be aware of the prescribed drug, the drugs were coded and given by 1 of the midwife’s colleagues'. It is unclear if women were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | English abstract states 61 women randomised to each group, translation states 120 in total. Data have been reported on 115 women. Only 4% ‐ 6% of randomised women lost to follow‐up. Unclear how many women included in reporting of outcomes |
| Selective reporting (reporting bias) | Unclear risk | Data have not been included in the translation |
| Other bias | Unclear risk | Abstract only in English, full text required translation |
Simbar 2015.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | Setting: Alzahra Hospital, Tehran, Iran. From April 2011 to February 2012
Inclusion criteria: primi‐ or multiparous women with moderate‐to‐severe after‐pain (score > 4 on VAS and with need for analgesia), normal vaginal delivery Exclusion criteria: women with birth complicated by perineal laceration, prolonged labour, macrosomia, instrumental birth, analgesics in previous 2 weeks |
|
| Interventions | Following initial assessment of pain, women were randomly allocated to 1 of 2 groups:
|
|
| Outcomes | Adequate pain relief as reported by the women
Pain however reported by the authors:
|
|
| Notes | Funding sources: Goldaru Company ‐ manufacturer of Menstrogol Declarations of interests: the authors declare that they have no conflicts of interest |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information related to sequence generation |
| Allocation concealment (selection bias) | Unclear risk | Quote: "participants randomly selected a packet that was coded A or B containing 4 capsules of PAC of MAC". Unclear if codes visible |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Single‐blind, assume participants were blinded but personnel may not have been blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial registration states that study is double‐blind but publication states single‐blind. Therefore unclear if they were excluded if they experienced any side effects or requested to withdraw from the study. Unclear how many participants applied to withdraw |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The trial included 108 women, 54 in each group. Pain severity before and after treatment does not include the number of women. Latency for medication effect reports data from 54 women in each group. Study report states that "participants were excluded if they experienced any side effects or requested to withdraw from the study". Unclear how many participants this applied to |
| Selective reporting (reporting bias) | High risk | Trial registered retrospectively |
| Other bias | Low risk | No other risk of bias identified |
Skovlund 1991a.
| Study characteristics | ||
| Methods | Randomised controlled trial with a sequential trial design | |
| Participants | Location: country Norway but site not clearly identified (maybe Akershus Central hospital, Oslo, Norway) and time of study not stated Included: postpartum women with uterine cramps and possible concomitant episiotomy pain after vaginal birth requesting analgesia Excluded: women allergic to paracetamol |
|
| Interventions | Postpartum women with uterine pain and possible concomitant episiotomy pain after vaginal birth and who were asking for analgesic and consented to participate were randomly allocated to 1 of 2 groups:
The medications were identical in appearance |
|
| Outcomes |
|
|
| Notes | Dates of study: not stated Funding sources: 1st author supported by a grant from the Norwegian Research Council for Science and the Humanities Declarations of interest: declared as none |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study simply described as randomised, but no further details provided |
| Allocation concealment (selection bias) | Unclear risk | No information related to allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind. "Identical appearing tablets." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 woman (placebo group) excluded as she had received analgesia 1 hour prior to inclusion. There were 39 women allocated to the paracetamol group and 36 women to the placebo group. Results included for 1 woman in placebo group who was under study when trial stopped. 2 women withdrew after 2 hours observation, no 4‐hour data included. Appropriately‐imputed data were used for women who withdrew before 2 hours |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | Low risk | Similar numbers of women in both groups (10 of the 39 women in the paracetamol group and 11 of the 38 women in the control group) had pain medication before enrolling in the study; inclusion criteria stated that women had pain and were requesting analgesia. Graphs presenting results appear to be mislabelled |
Skovlund 1991b.
| Study characteristics | ||
| Methods | Randomised controlled trial with a sequential trial design | |
| Participants | Location: country Norway but site not clearly identified (maybe Akershus Central hospital, Oslo, Norway) and time of study not stated Included: postpartum women with uterine cramps and possible concomitant episiotomy pain after vaginal birth requesting analgesia. Data from 56 participants were included in the sequential test but 60 participants were included in the estimation effect on uterine cramping Excluded: Women allergic to paracetamol or naproxen or with peptic disease |
|
| Interventions | Women were randomly allocated to 1 of 2 groups.
The medications were Identical in appearance |
|
| Outcomes |
|
|
| Notes | Sequential design Dates of study: not stated Funding sources: 1st author supported by a grant from the Norwegian Research Council for Science and the Humanities Declarations of interest: declared as none |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study simply described as randomised |
| Allocation concealment (selection bias) | Unclear risk | No information related to allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | States "double dummy technique used to make the study double blind" but unclear who was blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 36 women allocated to the paracetamol group and 28 women to the naproxen group. 3 women excluded as misunderstood administration of medication (2 in paracetamol group, 1 in naproxen group). 1 excluded from paracetamol group as experienced only episiotomy pain Results included for 4 women in paracetamol group who were under study when the stopping boundaries for this sequential design trial were reached. |
| Selective reporting (reporting bias) | Unclear risk | No published protocol or trial registration available |
| Other bias | Low risk | Similar numbers of women in both groups (18 of the 36 paracetamol participants and 9 of the 28 naproxen group) had pain medication before enrolled into the trial, inclusion criteria stated they had pain and were requesting analgesia |
Tehrani 2015.
| Study characteristics | ||
| Methods | Randomised controlled trial | |
| Participants | This trial was conducted on 86 mothers with postpartum pain after vaginal delivery at Baharloo Hospital in Tehran, Iran in 2014 ‐ 2015 Inclusion criteria: normal vaginal delivery; gestational age:37 to 42 weeks; postpartum women with after‐pain intensity score of 4 or more on a 0 ‐ 10 visual analogue score; literate women; woman's with infant weight range about 2500 ‐ 4000 g; women without difficult or prolonged labour; no addiction; no herbal allergy history; no caesarean section and abdominal surgery history; no postpartum haemorrhage history; no underlying disease. Absence of grade 3 and 4 perineal tears Exclusion criteria: if drugs could not sedate the mother and other interventions was necessary; history of ulcers or gastrointestinal bleeding |
|
| Interventions | Postpartum pain was measured 2 hours after childbirth, using VAS. Volunteers with scores higher than 4 were included in the study. Participants were randomly divided into 2 groups (43 cases per group):
|
|
| Outcomes | Pain intensity was measured by VAS before and 1 hour after each round of intervention. Participants used the medicines 4 times a day (with 4 ‐ 6 hour intervals) | |
| Notes | Funding: Tehran University of Medical Science Research Conflicts of interest: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Cards with A or B were placed in similar unmarked envelopes. Women selected an envelope with a card and then were given an envelope marked with corresponding A or B |
| Allocation concealment (selection bias) | High risk | The cards were put inside an envelope and eligible mothers were asked to pick a card. Mothers had no information about the type of medicines, whereas both the researcher and pharmacist were fully aware of the content of envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Researcher and pharmacist were aware of envelope contents |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 45 women allocated to each group. Data on 86 of 90 women (96%) randomised are reported for study outcome |
| Selective reporting (reporting bias) | Low risk | Outcomes reported as stated in trial registration |
| Other bias | Low risk | None identified |
NSAID: non‐steroidal anti‐inflammatory drugs; SPID: summed pain intensity differences; TENS: transcutaneous nerve stimulation; VAS: visual analogue scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Afravi 2019 | Emailed author to clarify the following 16 July 2020, no response It is unclear if this study was part prevention part treatment as not all women who enrolled had pain, and pain is not a trial entry criterion Use of analgesia ‐ it is reported that "all patients routinely received mefenamic acid (500 mg) or acetaminophen (325 mg) (based on its availability) 4 and 10 hours after delivery." and "In the early 10 hours after delivery, based on the postpartum ward routine, the mothers received painkillers and also were allowed to use extra painkillers if they requested. The amount of painkiller was compared between intervention and control groups.." Although the authors have reported "no difference" in analgesic use, they have not provided data. We have considered this trial to be uncontrolled |
| Azpiroz 1971 | Women with any postpartum pain included without differentiation between origin of pain Translated from Spanish to English by Ruth Martis with the assistance of a translation software |
| Bachar 2018 | Preventive intervention rather than treatment |
| Bahri 2019 | Preventive intervention rather than treatment |
| Baptisti 1971 | Inappropriate study design for this review. Insufficient detail on who participated and how pain scores were derived and analysed |
| Barhan 2019 | Preventive intervention rather than treatment |
| Beaver 1980 | Women with episiotomy pain and uterine cramp pain included and analysed together. Separate members for pain subgroups not available |
| Benson 1963 | Includes women with any postpartum pain ‐ no subgroup analyses |
| Bilgin 2016 | Inclusion criteria do not specify women with postpartum pain from uterine cramping. Range of initial pre‐treatment pain assessment scores on visual analogue scale included 0. More than 80% of women reported perineal pain prior to intervention. Unclear if post‐intervention pain assessment only for pain from uterine cramping |
| Bloomfield 1988a | Trial registered with Oxford Perinatal Trials Database ‐ never reported. Confirmed by author |
| Bloomfield 1988b | Trial registered with Oxford Perinatal Trials Database ‐ never reported. Confirmed by author |
| Blue 2018 | Included women who gave birth by caesarean, outcomes not reported separately for mode of birth |
| Bonica 1957 | Results combine perineal pain and uterine cramp pain |
| Bruni 1965 | Source of postpartum pain not specified or separated for analyses |
| Can 2015 | Preventive intervention rather than treatment |
| Cunha 2011 | Preventive intervention rather than treatment |
| Finch 1957 | Type of pain not separated. Not a suitable study design for inclusion |
| Gindhart 1971 | The medications tested are no longer available for use, as it was associated with severe adverse effects |
| Goodman 2005 | Data not separated into source of pain. Confirmation from author |
| Gruber 1962 | Results for pain intensity and change in pain intensity; do not separate uterine cramp pain from incisional (episiotomy) pain. No useable data |
| Gruber 1963 | Results for pain intensity and change in pain intensity do not separate uterine cramp pain from incisional (episiotomy) pain |
| Gruber 1971a | Participants may have participated on more than 1 day and therefore included as 2 participants. No suitable data could be extracted |
| Gruber 1971b | Participants may have participated on more than 1 day and therefore included as 2 participants. No suitable data could be extracted |
| Gruber 1979 | No useful data ‐ conclusions based on data pooled from both sources of pain ‐ uterine cramp and episiotomy. Paper focus is on methods of analyses |
| Hartemann 1968 | Does not differentiate between postpartum pains. Overall only gives "good and bad results" Translated from French to English with the assistance of Philippa Middleton and by Ruth Martis with the assistance of a translation software |
| Kantor 1984b | Results for pain intensity and change in pain intensity do not separate uterine cramp pain from incisional (episiotomy) pain |
| Katz 2019 | Preventive intervention rather than treatment |
| Kayman‐Kose 2014 | Preventive intervention rather than treatment |
| Kenton 2011 | Includes women with other sources of postpartum pain ‐ no subgroup analyses (NCT01271855 2011) |
| Kim 2019 | Includes women with any postpartum pain ‐ no subgroup analyses. Author emailed 17 December 2019; 8 January 2020. No response |
| Kumbar 2017 | Includes women with any postpartum pain ‐ no subgroup analyses |
| Laska 1983 | Not a suitable study design for this review. Some analyses of source of pain but does not separate the various doses of the medications being tested |
| Li 2014 | English abstract, paper written in Chinese. Translated by Aidan Tan, 08 July 2020. The paper states that the intervention is being tested to prevent pain, it is likely that some women had pain and were included for relief. Data not reported separately. Trials of prevention are not included in this review. Author emailed 09 July 2020 ‐ email address not active |
| Li 2015 | English abstract, paper written in Chinese. Translated by Aidan Tan 08 July 2020; this is a study of an intervention for joint pain postpartum |
| Linder 1997 | Women were not randomised. They were selected by a nurse who attempted to match baseline characteristics |
| Mehlhorn 2006 | Abstract only in supplement of journal. Data analysis not completed, as confirmed by email with author. Translated into English by Ruth Martis |
| Mirror 2019 | This study is about prevention, not treatment |
| Narimatsu 2001 | This study is about prevention, not treatment |
| Nazari 2018 | This study is about prevention, not treatment |
| Nunlee 2000 | Results do not separate uterine cramp pain from incisional (episiotomy pain). Attempts to contact author unsuccessful, (Internet, email and mail) |
| Olson 1984 | Results for pain intensity and change in pain intensity do not separate uterine cramp pain from incisional (episiotomy) pain |
| Ozgoli 2018 | Abstract and trial registration give different information on the use of ibuprofen whilst undergoing trial interventions and assessment of pain, therefore not possible to define intervention and not possible to separate the effect of ibuprofen from the effect of the trial intervention. The authors have not provided any further information |
| Pan 1993 | Preventive intervention rather than treatment. This trial was included in 2011 |
| Parsa 2019 | All women in the study had access to other analgesia as required and therefore it is not possible to separate the effect of the other analgesia from the effect of the intervention. This study was translated 04 August 2020 |
| Prockop 1960 | Study method not suitable for inclusion. Randomisation was by ward, analyses by individual. One of the medications tested (ASA compound) is no longer in use |
| Ray 1993 | Does not differentiate between types of pain. Attempts to contact author unsuccessful |
| Redick 1980 | This study was done on analgesia for postpartum women with no description of the source of the pain |
| Rubin 1984 | Study done on postpartum women with episiotomies. No description of type of pain other than this. Not certain uterine cramp pain included |
| Smith 1973 | No definition of postpartum pain. Paper about analgesic‐sedative effect of drug combination. Analyses include post‐surgical men and women |
| Soltani 2017 | Preventive intervention rather than treatment. Author emailed 8 January 2020 |
| Sunshine 1983 | Analyses do not separate pain from uterine cramping and pain from episiotomies |
| Sunshine 1985 | Inlcudes episiotomy, CS and uterine cramps. Episiotomy and CS pain analysed together. Uterine pain was not analysed alone as the numbers were too small |
| Sunshine 1986 | Analyses do not separate type of pain |
| Sunshine 1989 | Ineligible because women with pain from different sources were included, not possible to differentiate pain due to involution |
| Tafazoli 2013 | All women in the study had access to other analgesia as required and therefore it is not possible to separate the effect of the other analgesia from the effect of the intervention. This study was translated 26 July 2020 |
| Van Wering 1972 | Includes any source of postpartum pain; analyses do not separate type of pain |
| Vaziri 2017 | Includes women with perineal pain ‐ no subgroup analyses |
| Von Pein 1974 | Does not describe source of pain in puerperium |
| Yogev 2015 | Trial not eligible. Trial has 2 parts: 1. Prevention arm where women who had not begun breastfeeding were be randomised to the dental device or not to prevent pain. 2. All women who had begun feeding were given the device and acted as their own control – pain measured before and after use |
CS: caesarean section
Characteristics of ongoing studies [ordered by study ID]
IRCT2015050322053N1.
| Study name | Comparison of the effect of chamomila matricaria and mefenamic acid capsules on postpartum pain and haemorrhage |
| Methods | Randomised double‐blind study |
| Participants | 70 multiparous women. Location: Iran Inclusion criteria: age:18 ‐ 35 years, normal vaginal delivery, live birth, gestational age: 37 to 42, multiparity, cephalic presentation, neonatal weight is 2500 ‐ 4000 g, singleton, having moderate or severe after‐pain based on a visual scale, no consumption of benzodiazepine, barbiturates, narcotics, alcohol, aspirin, warfarin and heparin 11, no sensitivity to Chamomila, no history of medical illness, no caesarean section history, no severe bleeding, no rupture of the cervix and uterus, no history of postpartum haemorrhage, no addiction of mother to drugs and alcohol, no high‐risk pregnancy, no rupture of membranes more than 12 hours Exclusion criteria: mother's unwillingness to continue to participate in study, severe bleeding |
| Interventions |
|
| Outcomes |
|
| Starting date | Expected recruitment start date: 12 January 2015 |
| Contact information | rezvanifardm911@mums.ac.ir |
| Notes | Expected recruitment end date: 12 June 2015 |
IRCT2016070428240N2.
| Study name | The effect of acupressure and touch point (SP6) on pain intensity after delivery of 88 qualified mothers gave birth on 22 Bahman Hospital in Gonabad City |
| Methods | Randomised controlled trial |
| Participants | 88 women following 2nd birth by uncomplicated term vaginal delivery, no episiotomy, with moderate or severe postpartum pain (score > 4) |
| Interventions |
|
| Outcomes |
|
| Starting date | Expected ‐ 22 July 2016; expected completion 18 February 2018 |
| Contact information | Fatemeh Yaghoobi Moghadam Bilondi; yaghoobi@shmu.ac.ir |
| Notes | Notes: trial registration only. Emailed 16 July 2020, email not active |
IRCT2016100930238N1.
| Study name | Comparative study of the effect of fennel capsules and Ibuprofen on postpartum after pain in multiparous women admitted in postpartum ward of Sanandaj Beasat hospital |
| Methods | RCT Location: Iran |
| Participants | Target sample size: 70 Inclusion criteria: age 17 ‐ 50; vaginal delivery; term gestational age (40 ‐ 37 weeks); live births; singleton; fetal weight 4000 ‐ 2500 g; lack of specific diseases such as ulcers or gastrointestinal bleeding or known cardiovascular disease; higher pain score of 4 Exclusion criteria: severe bleeding after childbirth; evacuating corpus luteum or manual removal of placenta; use of tools (vacuum, forceps),having perineal tear grade 3 and 4; long and difficult labour; sensitivity to fennel "In case of dissatisfaction with the continued cooperation in the study for any reason and at any stage of the research will be withdrawn." |
| Interventions |
|
| Outcomes |
|
| Starting date | Expected recruitment start date January 2017, expected recruitment end date March 2020 |
| Contact information | Leila Hasheminasab: hasheminasab.l.2014@gmail.com Parya Foroughi: oroghip@gmail.com, p.foroughi@muk.ac.ir |
| Notes |
IRCT201707283860N33.
| Study name | Clinical trial study of the effect of ginger plant capsules on reducing the pain in women with postpartum pain |
| Methods | Randomised trial |
| Participants | Iranian women with after‐birth pain who met the following inclusion criteria: literate; vaginal birth at 38 weeks or more; birth weight > 2500 grams; absence of drug addiction; not allergic to ginger or herbal medicine; did not have the following analgesia for labour: epidural anaesthesia, spinal anaesthesia, entonox and pethidine. Exclusion Criteria: Maternal complications including postpartum haemorrhage; pyrexia; hypertension; or chronic disease. |
| Interventions |
|
| Outcomes |
|
| Starting date | 06 August 2017; completion date 23 October 2017 |
| Contact information | Gity Ozgoli; g.ozgoli@gmail.com |
| Notes | Email correspondence 10 July 2020 ‐ Emailed author, this paper has been submitted to the Journal of Research in Medical Sciences. The author emailed the journal about sharing results and the journal has embargoed until publication. Expected to be published mid to late September 2020 |
IRCT20171208037792N1.
| Study name | The effect of foot reflexology on reduction of postpartum after‐pain |
| Methods | Randomised controlled trial |
| Participants | 68 eligible postpartum women (34 in the reflexology group and 34 in the control group) referring to the Department of Obstetrics of Razi Hospital of Ahvaz Inclusion criteria: Spontaneous vaginal delivery; Singleton pregnancy; Cephalic presentation; Mother's complaint of moderate or severe postpartum pain (a pain score of 3.1 or above based on VAS); Age between 18 ‐ 35; 2nd to 4th parity Exclusion criteria: Hard and prolonged delivery; Mother's substance abuse; Postpartum haemorrhage; Underlying illnesses (blood pressure, diabetes, kidney problem, etc.); Any problem with the soles such as corns, burns, cuts, fungal infection, varicose veins, warts or any numbness in the foot; Still births |
| Interventions |
|
| Outcomes | Severity of after‐pains and duration of after‐pains |
| Starting date | Proposed start date 04 April 2018 proposed completion date 05 June 2018. Study retrospectively registered 14 July 2018 |
| Contact information | Galiya Bakhtiyari Niya |
| Notes | Emailed 16 July 2020 |
IRCT20180428039454N1.
| Study name | Effect of parpin ala (portulaca Oleracea) capsule on the postpartum pain and haemorrhage volume |
| Methods | RCT Location: Iran Participants, investigators, data analysers all blinded |
| Participants | Target sample size: 106 Inclusion criteria: 15 ‐ 35 years old, vaginal delivery, nullipara, to take an oral drug, no history of medical and psychological disease, no sensitivity to herbal medicine, no drug addiction, no need to have surgery after delivery Exclusion criteria: obesity and body mass index > 35, caesarean section, multiple births, polyhydramnios, pre‐eclampsia, haemoglobin < 9 |
| Interventions |
|
| Outcomes |
|
| Starting date | Expected recruitment start date: March 2018 Expected recruitment end date: May 2018 |
| Contact information | Samira Shiralinezhad: shiralinezhad.s@tak.iums.ac.ir |
| Notes | Eamiled 16 July 2020; email was not active |
IRCT20190217042739N1.
| Study name | Comparing the effect of Salvia hydrangea and mefenamic acid on postpartum pain |
| Methods | Randomised double blind study Participants, care providers, investigators, outcome assessors, and data analysers are all blinded |
| Participants | 100 women Inclusion criteria: vaginal delivery, moderate or severe pain after delivery, no maternal history of herbal drugs allergy, mother's age between 18 ‐ 35 years, deliver a singleton and healthy baby. No entry criteria: maternal history of herbal drugs allergy, 3 or 4 degree laceration, maternal history of chronic disease, history of caesarean section or pelvic operation |
| Interventions |
|
| Outcomes |
|
| Starting date | Start date March 2019 Recruitment reported as complete July 2019 |
| Contact information | kheyri.rezvan@sbmu.ac.ir |
| Notes | Emailed 16 July 2020; no response |
NCT03617900.
| Study name | Efficacy of ginger abstract (compare between the ginger preparation of ancient concept of Thai practitioner, standard drug and placebo) by using pain score to evaluate after pain of three groups of first normal postpartum women |
| Methods | Randomised trial |
| Participants | Setting: Naphatsaran Roekruangrit, Thammasat University, Thailand Inclusion criteria: women aged 20 ‐ 34 years, healthy, showing no symptoms of disorder, no history of pre‐eclampsia, liver disease, kidney disease or gastrointestinal bleeding during pregnancy or after participating in a research project, consents Exclusion criteria: postpartum haemorrhage, unable to travel conveniently, allergic to modern medicine or herbal remedies, gallstones, regular medications, smoking or consuming alcohol in pregnancy |
| Interventions | Drug: ginger extract is contains 100 mg/capsules. Use 2 capsules 3 times/day Drug: placebo oral capsule lactose monohydrate 400 mg/capsules Drug: paracetamol 500 mg paracetamol. use in need
|
| Outcomes |
|
| Starting date | 29 August 2018. Enrolment completed 25 May 2019 |
| Contact information | Preecha Wanichsetakul, M.D. No email provided in registration |
| Notes | The primary objective of this study is to compare the efficacy of ginger extract on pain relief at the following anatomical locations, uterus, episiotomy and breast. It is unclear if the authors intend to report pain relief separately for each source of pain. Funding: Thammasat University |
NCT04037202.
| Study name | Effect of foot massage on postpartum comfort and pain level of the mothers who had vaginal birth |
| Methods | Randomised controlled trial |
| Participants | 66 primiparous women aged 18 t0 35 with a normal vaginal delivery within the previous 24 hours, including women with episiotomy, no complications in the infant. |
| Interventions |
|
| Outcomes |
For control group, all assessments recorded at equivalent time points |
| Starting date | Began 03 July 2017; completed 01September 2017 |
| Contact information | Rabia Genc, Ege University. No email contact within registration |
| Notes | Trial registration only. Awaiting publication to check type of postpartum pain being assessed: perineal/uterine cramp pain |
PCS: postpartum comfort scale VAS: visual analogue scale
Differences between protocol and review
We changed the protocol to exclude studies reporting after‐birth pain in women following caesarean birth.
We changed the protocol to exclude meta‐analyses on drugs no longer in use or contraindicated in lactation.
We have updated the methods to reflect the latest Cochrane Handbook and Cochrane Pregnancy and Childbirth's methodological guidelines.
Sheree Agett is no longer an author on this review.
We did not carry out the additional searching (Embase, CINAHL and MIDIRS) specified in the protocol.
Title changed from Analgesia for the relief of pain due to uterine cramping/involution after birth to Relief of pain due to uterine cramping/involution after birth.
Differences between 2011 and 2020
Additional fields added for risk of bias (RoB).
Studies included in 2011 reassessed for RoB, including new fields and applying stricter assessment of the 'selective outcome reporting'.
Inclusion criteria updated to exclude studies of the prevention of pain due to uterus cramping/involution.
Assessment of heterogeneity changed to reflect guidance in new version of Cochrane Handbook.
Contributions of authors
For this update of the review, Andrea Deussen (AD), Pat Ashwood (PA), Ruth Martis (RM) and Luke Grzeskowiak (LG) reviewed the inclusion and exclusion criteria and outcomes. AD, PA and LG screened all of the studies identified in the new search and reviewed all of the studies from the previous version against the new criteria. AD, PA, RM and LG completed data extraction and 'Risk of bias' assessments in pairs for all studies from the previous version of the review and newly‐identified studies. This was done in pairs and a third author resolved disagreement. AD entered data into Review Manager 5 and these were checked by the remaining authors. AD, LG and Fiona Stewart (FS) completed the GRADE assessment. AD, RM, LG and FS drafted the manuscript, with all authors reviewing and agreeing on the final version.
Sources of support
Internal sources
-
The University of Adelaide ‐ Australian Research Centre for Health of Women and Babies (ARCH), Australia
Release time for attending a 1 week dedicated work‐in for data analyses.
-
The University of Adelaide, School of Medicine, Other
Provided facilities,infrastructure and research resources.
External sources
-
Australasian Cochrane Centre, Monash University, Melbourne, Australia
One week on‐site guidance and support for progressing with review.
Declarations of interest
Andrea R Deussen: none known
Pat Ashwood: none known
Ruth Martis: none known
Luke Grzeskowiak: none known
Fiona Stewart: none known
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Asti 2011 {published data only}
- Asti P, Delfan B, Masudi M, Ebrahimzadeh F. Ibuprofen versus fennel for the relief of postpartum pain; a randomized controlled trial. Journal of Family and Reproductive Health 2011;5(2):65-7. [Google Scholar]
Bettigole 1981 {published data only}
- Bettigole JB. A double-blind comparison of placebo, codeine, and fenoprofen in patients with postpartum pain. Current Therapeutic Research 1981;29:778-84. [Google Scholar]
Bloomfield 1977 Study 1 {published data only}
- Bloomfield SS, Barden TP, Mitchell J. Aspirin and codeine in two postpartum pain models. Clinical Pharmacology and Therapeutics 1976;20(4):499-503. [DOI] [PubMed] [Google Scholar]
- Bloomfield SS, Barden TP, Mitchell J. Naproxen, aspirin, and codeine in postpartum uterine pain. Clinical Pharmacology and Therapeutics 1977;21(4):414-21. [DOI] [PubMed] [Google Scholar]
Bloomfield 1977 Study 2 {published data only}
- Bloomfield SS, Barden TP, Mitchell J. Aspirin and codeine in two postpartum pain models. Clinical Pharmacology and Therapeutics 1976;20(4):499-503. [DOI] [PubMed] [Google Scholar]
- Bloomfield SS, Barden TP, Mitchell J. Naproxen, aspirin, and codeine in postpartum uterine pain. Clinical Pharmacology and Therapeutics 1977;21(4):414-21. [DOI] [PubMed] [Google Scholar]
Bloomfield 1978 {published data only}
- Bloomfield SS, Barden TP, Mitchell J. Fendosal and aspirin in postpartum uterine pain. Clinical Pharmacology and Therapeutics 1978;23(4):390-6. [DOI] [PubMed] [Google Scholar]
Bloomfield 1981 {published data only}
- Bloomfield SS, Barden TP, Mitchell J. A comparison of pirprofen, aspirin, acetaminophen and placebo in postpartum uterine cramp pain. Current Therapeutic Research 1981;30:S139-S145. [Google Scholar]
Bloomfield 1983 {published data only}
- Bloomfield SS, Mitchell J, Bichlmeir LP, Barden TP. Low dose ibuprofen and aspirin analgesia for postpartum uterine cramps. American Society for Clinical Pharmacology and Therapeutics 1983;33:194. [DOI] [PubMed] [Google Scholar]
Bloomfield 1986a {published data only}
- Bloomfield SS, Barden TP, Mitchell J, Bichlmeier G. Flurbiprofen, aspirin, and codeine in postpartum uterine pain. Clinical Pharmacology and Therapeutics 1981;29:234-5. [DOI] [PubMed] [Google Scholar]
- Bloomfield SS, Barden TP, Mitchell J, Bichlmeir G. A comparison of flurbiprofen, aspirin and placebo in postpartum uterine pain. Current Therapeutic Research 1981;30:670-9. [Google Scholar]
- Bloomfield SS, Cissell GB, Mitchell J, Barden TP. Codeine and aspirin analgesia in postpartum uterine cramps: qualitative aspects of quantitative assessments. Clinical Pharmacology and Therapeutics 1983;34(4):488-95. [DOI] [PubMed] [Google Scholar]
- Bloomfield SS, Mitchell J, Cissell G, Barden TP. Flurbiprofen, aspirin, codeine, and placebo for postpartum uterine pain. American Journal of Medicine 1986;80(3A):65-70. [DOI] [PubMed] [Google Scholar]
Bloomfield 1986b {published data only}
- Bloomfield SS, Mitchell J, Cissell G, Barden TP. RS-37619 and aspirin analgesia for postpartum uterine cramps. Clinical Pharmacology and Therapeutics 1984;35(2):228. [DOI] [PubMed] [Google Scholar]
- Bloomfield SS, Mitchell J, Cissell GB, Barden TP, Yee JP. Ketorolac versus aspirin for postpartum uterine pain. Pharmacotherapy 1986;6(5):247-52. [DOI] [PubMed] [Google Scholar]
- Bloomfield SS, Mitchell J, Cissell, G, Peters N, Nelson ED, Barden TP. Ketorolac vs aspirin with or without codeine for postpartum or postoperative pain. In: Proceedings of 10th International Congress of Pharmacology; 1987 Aug; Sydney, Australia. 1987:P590.
Bloomfield 1986c {published data only}
- Bloomfield SS, Kantor TG, Hopper M, Mitchell J. Aspirin (S) and acetaminophen (C), regular vs extra-strength, for postpartum uterine cramps. Clinical Pharmacology and Therapeutics 1986;38:181. [Google Scholar]
Bloomfield 1987 {published data only}
- Bloomfield SS, Cissell GB, Nancy NM, Mitchell J, Nelson ED, Bardon TP. Anirolac vs Naproxen for postpartum uterine pain. Clinical Pharmacology and Therapeutics 1987;42(1):89-95. [DOI] [PubMed] [Google Scholar]
- Bloomfield SS, Nelson ED, Mitchell J, Cissel GB, Peters N, Barden TP. Anirolac and naproxen analgesia for postpartum uterine cramp pain. Clinical Pharmacology and Therapeutics 1986;39(2):181. [DOI] [PubMed] [Google Scholar]
Chananeh 2018 {published data only}
- Chananeh M, Ataei PJ, Dolatian M, Mojab F, Nasiri M. Effects of the combination of nigella sativa and mefenamic acid and mefenamic acid alone on the severity of postpartum pain in multiparous women: a double-blind clinical trial. Iranian Journal of Obstetrics, Gynecology and Infertility 2018;21(4):62-71. [Google Scholar]
- IRCT2017041033356N1. Comparison between Nigella Sativa capsule and mefenamic acid capsule on after pain in multiparous women.. www.irct.ir/trial/25749 (first received 8 August 2017).
- Keikha M. Translation for study by Chananeh 2018. Email to: A Deussen 24 July 2020.
Dastjerdi 2019 {published data only}
- Dastjerdi M, Darooneh T, Nasiri M, Moatar F, Esmaeili S, Ozgoli G. Investigating the effect of Melissa Officinalis on after-pains: a randomized single-blind clinical trial. Journal of Caring Sciences 2019;8(3):129-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgoli G, IRCT201608023860N25. A comparative study on the effects of herbal capsule of Melisa and Mefenamic acid on postpartum after pain. en.irct.ir/trial/3991 (first received 28 December 2016).
- Ozgoli G. Email correspondence. Email to: A Deussen 06 January 2020.
De Sousa 2014 {published data only}
- De Sousa L, ACTRN12610000644066. Effectiveness of transcutaneous electrical nerve stimulation for painful postpartum uterine contraction during breastfeeding. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12610000644066 (first received 5 August 2010).
- De Sousa L, Gomes-Sponholz FA, Nakano AM. Transcutaneous electrical nerve stimulation for the relief of post-partum uterine contraction pain during breast-feeding: a randomized clinical trial. Journal of Obstetrics and Gynaecology Research 2014;40(5):1317-23. [DOI] [PubMed] [Google Scholar]
Jain 1978 {published data only}
- Jain AK, McMahon FG, Ryan JR, Unger D, Richard V. Aspirin and aspirin-caffeine in postpartum pain relief. Clinical Pharmacology and Therapeutics 1978;24(1):69-75. [DOI] [PubMed] [Google Scholar]
- Jain AK, McMahon FG, Ryan JR, Unger D, Richard W. A comparison of aspirin-caffeine versus aspirin: results of two double-blind placebo controlled studies in postpartum pain. Clinical Pharnacology and Therapeutics 1978;23(1):116. [DOI] [PubMed] [Google Scholar]
Kantor 1984a {published data only}
- Kantor T, Hopper M. Oral nalbuphine in postpartum pain. Clinical Pharmacology and Therapeutics 1984;35(1):46-9. [DOI] [PubMed] [Google Scholar]
Kheiriyat 2016 {published data only}
- Kheiriyat F, Najafabadi MT, Mousavi P, Haghighizadeh H, Namjuyan F. Effect of dill essence and mefenamic acid on postpartum pain. Iranian Journal of Obstetrics, Gynecology and Infertility 2016;19(4):8-16. [Google Scholar]
- Najafabadi MT, IRCT2015070623102N1. The effect of anethum graveolens extract on after pain. en.irct.ir/trial/19790 (first received 27 July 2015).
Laska 1981 Study 1 {published data only}
- Laska EM, Sunshine A. Fenoprofen and codeine analgesia. Clinical Pharmacology and Therapeutics 1981;29(5):606-16. [DOI] [PubMed] [Google Scholar]
- Offen WW, Gruber CM. Dose response to fenoprofen calcium using placebo and codeine as controls. Journal of Medicine 1985;16(4):439-52. [PubMed] [Google Scholar]
- Sunshine A, Laska E, Zighelboim I, Desenne J. A comparison of the analgesic responses of fenoprofen, codeine, and placebo in postpartum and postoperative pain. Current Therapeutic Research 1981;29(5):771-7. [Google Scholar]
Laska 1981 Study 2 {published data only}
- Laska EM, Sunshine A. Fenoprofen and codeine analgesia. Clinical Pharmacology and Therapeutics 1981;29(5):606-16. [DOI] [PubMed] [Google Scholar]
- Offen WW, Gruber CM. Dose response to fenoprofen calcium using placebo and codeine as controls. Journal of Medicine 1985;16(4):439-52. [PubMed] [Google Scholar]
- Sunshine A, Laska E, Zighelboim I, Desenne J. A comparison of the analgesic responses of fenoprofen, codeine, and placebo in postpartum and postoperative pain. Current Therapeutic Research 1981;29(5):771-7. [Google Scholar]
Mehlhorn 2005 {published data only}
- Mehlhorn G, Beckmann MW, Schild RL, Binder H. Analgesia of afterpains with transcutaneous nerve stimulation (TENS) vs. metamizole. A prospective, randomized placebo controlled double blind study [Analgesie von schmerzhaften Nachwehen mittels transkutaner elektrischer Nervenstimulation (TENS) vs. Metamizol]. Geburtshilfe und Frauenheilkunde 2005;65:266-71. [Google Scholar]
- Melhorn G. Email correspondence. Email to: A Deussen 12 January 2020.
Okun 1982 {published data only}
- Okun R. Evaluation of the analgesic effect of fendosal in patients with postpartum uterine cramp or episiotomy pain. Current Therapeutic Research, Clinical and Experimental 1982;32(1):65-73. [Google Scholar]
Olsen 2007 {published data only}
- Olsen MF, Elden H, Janson ED, Litjer H, Stener-Victorin E. A comparison of high- versus low-intensity, high-frequency transcutaneous electric nerve stimulation for painful postpartum uterine contractions. Acta Obstetrica et Gynecologica Scandinavica 2007;86(3):310-4. [DOI] [PubMed] [Google Scholar]
Ozgoli 2017 {published data only}
- Akbari N. Translation for Ozgoli 2017. Email to: A Deussen 02 August 2020.
- Ozgoli G, IRCT201512203860N21. A comparative study on the effects of herbal capsule of anise and mefenamic acid on postpartum after pain. en.irct.ir/trial/3987 (first received 7 February 2016).
- Ozgoli G, Khodadadie A, Sheikhan Z, Taleb S, Jambarsang S, Mojab F. Comparison of efficacy between herbal capsule of anise and mefenamic acid on after-pain [درد زايماني انيسون و مفناميك اسيد بر پس مقايسه تأثير كپسول گياهي]. Journal of Medicinal Plants 2017;16(62):38-49. [Google Scholar]
- Ozgoli G. Email correspondence. Email to: A Deussen 06 January 2020.
Pourmaleky 2013 {published data only}
- Keikha M. Translation for Pourmaleky 2013. Email to: A Deussen 24 July 2020.
- Poormaleky S, IRCT2013052613475N1. The comparison effect of ginger and mefenamic acid on after delivery pain in multiparous women. en.irct.ir/trial/13337 (first received 20 July 2013).
- Pourmaleky S, Najar S, Montazery S, Hossein Haghighizadeh M. Comparison between the effects of zintoma (ginger) and mefenamic acid on after pain during postpartum in multiparous women [مقایسه تأثیر کپسول زینتوما (زنجبیل) و مفنامیک اسید بر پس درد زایمانی زنان چندزا]. Iranian Journal of Obstetrics, Gynecology and Infertility 2013;16(79):18-25. [Google Scholar]
Simbar 2015 {published data only}
- Simbar M, IRCT201201228801N1. A comparative study on the effects of Menstrogol® and mefenamic acid on postpartum pain. en.irct.ir/trial/9284 (first received 21 April 2012).
- Simbar M, Shadipour M, Salamzadeh J, Ramezani-Tehrani F, Nasiri N. The combination of "pimpinella anisum, apium graveolens and crocus sativus (PAC)" is more effective than "mefenamic acid" on postpartum after-pain. Journal of Herbal Medicine 2015;5(1):20-5. [Google Scholar]
Skovlund 1991a {published data only}
- Skovlund E, Fyllingen G, Landre H, Nesheim BL. Comparison of postpartum pain treatments using a sequential trial design. I. Paracetamol versus placebo. European Journal of Clinical Pharmacology 1991;40(4):343-7. [DOI] [PubMed] [Google Scholar]
Skovlund 1991b {published data only}
- Skovlund E, Fyllingen G, Landre H, Nesheim BL. Comparison of postpartum pain treatments using a sequential trial design. II. Naproxen verus paracetamol. European Journal of Clinical Pharmacology 1991;40:539-42. [DOI] [PubMed] [Google Scholar]
Tehrani 2015 {published data only}
- Shahnaz GT, IRCT2014020515338N2. The comparison of fennel and mefenamic acid effects on post partum after pain. en.irct.ir/trial/14612 (first received 20 May 2014).
- Tehrani SG, Mirmohammadali M, Moghadam AS, Mehran A, Zadeh MT, Baleghi M. The comparison of fennel and mefenamic acid effects on postpartum after pain. Journal of Babol University of Medical Sciences 2015;17(8):7-13. [Google Scholar]
References to studies excluded from this review
Afravi 2019 {published data only}
- Afravi S, Abbaspoor Z, Montazeri S, Cheraghian B. The effect of Hugo point pressure on postpartum pain in multiparous women. Family Medicine and Primary Care Review 2019;21(1):7-11. [Google Scholar]
Azpiroz 1971 {published data only}
- Azpiroz P, Garcia G. Clinical trial of a new analgesic CI-473 Parke Davis, in puerperal pain. [Ensayo clinico de un nuevo analgesico, CI-473 Parke Davis, en los entuertos puerperales]. Toko-Ginecologia Practica 1971;30:135-59. [Google Scholar]
Bachar 2018 {published data only}
- Bachar G, NCT04087317. Comparison between 2 pain analgesic protocols following vaginal delivery. clinicaltrials.gov/ct2/show/NCT04087317 (first received 12 August 2018).
Bahri 2019 {published data only}
- Bahri N, IRCT20181214041962N1. The effect of foot reflexology in the fourth stage of labor on after pain and postpartum hemorrhage. en.irct.ir/trial/36099 (first received 4 February 2019).
Baptisti 1971 {published data only}
- Baptisti A, Gruber CM, Santos EL. The effectiveness and side-effect liability of propoxyphene hydrochloride and propoxyphene napsylate in patients with postpartum uterine cramping. Toxicology and Applied Pharmacology 1971;19(3):519-27. [DOI] [PubMed] [Google Scholar]
Barhan 2019 {published data only}
- Barhan S, NCT03903172. Post-partum non-pharmacologic pain management. clinicaltrials.gov/ct2/show/NCT03903172 (first received 4 April 2019).
Beaver 1980 {published data only}
- Beaver WT, McMillan D. Methodological considerations in the evaluation of analgesic combinations: acetaminophen (paracetamol) and hydrocodone in postpartum women. British Journal of Clinical Pharmacology 1980;10(Suppl 2):215S-223S. [PMC free article] [PubMed] [Google Scholar]
Benson 1963 {published data only}
- Benson RC. Double blind evaluation of analgesic agents in the postpartum patient. Western Journal of Surgery 1963;71:167-9. [PubMed] [Google Scholar]
Bilgin 2016 {published data only}
- Bilgin Z, Komurcu N. Effect of uterine massage in the perception of women's postpartum pain intensity. Zeynep Kamil tip Bulteni 2016;47(2):39-44.
- Bilgin Z. Email Correspondence. Email to: A Deussen 13 July 2020.
- Selin B. Translation. Email to: A Deussen 06 August 2020.
- Yaylali E. Email Correspondence re translation. Email to: A Deussen 26 July 2020.
Bloomfield 1988a {published data only (unpublished sought but not used)}
- Bloomfield SS. The comparative efficacy of Voltaren (diclofenac), naproxen sodium and placebo in the treatment of postpartum uterine cramp pain. Personal communication 1988.
Bloomfield 1988b {published data only (unpublished sought but not used)}
- Bloomfield SS. A double-blind placebo-controlled parallel evaluation of the comparative analgesic efficacy between intramuscularly administered lysine acetylsalicylate (LAS) and oral aspirin (ASA) in postpartum uterine cramp pain. Personal Communication 1988.
Blue 2018 {published data only}
- Blue NR, Drake-Lavelle S, Weinberg D, Holbrook BD, Katukuri VR, Leeman L, et al. Effect of ibuprofen versus acetaminophen on postpartum hypertension in preeclampsia with severe features: a double-masked, randomized controlled trial. American Journal of Obstetrics and Gynecology 2018;218(1 Suppl):S604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bonica 1957 {published data only}
- Bonica JJ, Hadfield D, Bennett B. The management of postpartum pain with dihydrohydroxycodeinone (percodan) evaluation with codeine and placebo. Western Journal of Surgery 1957;65(2):84-8. [PubMed] [Google Scholar]
Bruni 1965 {published data only}
- Bruni JR, Holt RE. Controlled double-blind evaluation of three analgesic medications for postpartum discomfort. Obstetrics and Gynecology 1065;25:76-81. [PubMed] [Google Scholar]
Can 2015 {published data only}
- Can HO, Saruhan A. Evaluation of the effects of ice massage applied to large intestine 4 (hegu) on postpartum pain during the active phase of labor. Iranian Journal of Nursing and Midwifery Research 2015;20(1):129-38. [PMC free article] [PubMed] [Google Scholar]
Cunha 2011 {published data only}
- Cunha A, Almeida A, Almeida L, Cruz A, Vilhena I, Costa C, et al. Pospartum analgesia - What can we do? Regional Anesthesia and Pain Medicine 2011;36(5 Suppl 2):E239. [Google Scholar]
Finch 1957 {published data only}
- Finch JS, DeKornfeld TJ. Clonixin: a clinical evaluation of a new oral analgesic. Journal of Clinical Pharmacology 1971;11(5):371-7. [PubMed] [Google Scholar]
Gindhart 1971 {published data only}
- Gindhart JD. A rationale for studying analgesia. A double blind study in postpartum patients. Current Therapeutic Research 1971;13(4):240-50. [PubMed] [Google Scholar]
Goodman 2005 {published data only}
- Goodman SR, Drachenberg AM, Johnson S, Kim-Lo SH, Smiley RM. Decreased use of oral pain medication after vaginal delivery with a single dose of epidural morphine (abstract). Anesthesiology 2001;95(Suppl):A1029. [Google Scholar]
- Goodman SR, Drachenberg AM, Johnson SA, Kim-Loo SH, Smiley RM. Decreased postpartum use of oral pain medication after a single dose of epidural morphine. Anesthesiology 2001;94(1A):A83. [DOI] [PubMed] [Google Scholar]
- Goodman SR, Drachenberg AM, Johnson SA, Negron MA, Kim-Lo SH, Smiley RM. Decreased postpartum use of oral pain medication after a single dose of epidural morphine. Regional Anesthesia and Pain Medicine 2005;30(2):134-9. [DOI] [PubMed] [Google Scholar]
Gruber 1962 {published data only}
- Gruber CM, Baptisti A, Chernish SM. Comparitive evaluation of analgesic agents in postpartum patients: oral dextropropoxyphene, codeine and meperidine. Anesthesia and Analgesia 1962;1(5):538-44. [PubMed] [Google Scholar]
Gruber 1963 {published data only}
- Gruber CM, Baptisti A. Estimating the acceptability of morphine and noracymethadol in postpartum patients. Clinical Pharmacology and Therapeutics 1963;4(2):172-81. [DOI] [PubMed] [Google Scholar]
Gruber 1971a {published data only}
- Gruber CM, Wolen RL, Baptisti A. Analgesia scores as timed responses following oral administration of propoxyphene to postpartum patients. Toxicology and Applied Pharmacology 1971;19(3):504-11. [DOI] [PubMed] [Google Scholar]
Gruber 1971b {published data only}
- Gruber CM, Baptisti A, Kiplinger GF. Relief of postpartum uterine cramping with propoxyphene and aspirin. Toxicology and Applied Pharmacology 1971;19(3):546-53. [DOI] [PubMed] [Google Scholar]
Gruber 1979 {published data only}
- Gruber CM, Bauer RO, Bettigole JB, Lash AF, McDonald JS. A multicenter study for analgesia involving fenoprofen, propoxyphene (alone or in combination) with placebo and aspirin controls in postpartum pain. Journal of Medicine 1979;10(1-2):65-98. [PubMed] [Google Scholar]
Hartemann 1968 {published data only}
- Hartemann J, Landes P, Bertrand P. Results of treatment of post-partum pain with C.B. 3697 [Resultats du traitement des tranches du post-partum par le C.B. 3697]. Bulletin de la Societé Nationale de Gynecologie et d'Obstetrique de France 1968;20(2):176-8. [PubMed] [Google Scholar]
Kantor 1984b {published data only}
- Kantor T, Cavalier MB, Hopper RN, Roepke MS. A double-blind comparison of ketoprofen codeine, and placebo in patients with moderate to severe postpartum pain. Journal of Clinical Pharmacology 1984;24(5-6):228-34. [DOI] [PubMed] [Google Scholar]
- Kantor TG, Hopper M. Oral ketoprofen as an analgesic on a post-partum model. Clinical Pharmacology and Therapeutics 1983;33:196. [Google Scholar]
Katz 2019 {published data only}
- Katz D, NCT04017442. Neuraxial preservative free morphine for normal Spontaneous vaginal delivery: a prospective double blind randomized control trial. clinicaltrials.gov/ct2/show/NCT04017442 (first received 12 July 2019).
Kayman‐Kose 2014 {published data only}
- Kayman-Kose S, Arioz DT, Toktas H, Koken G, Kanat-Pektas M, Kose M, et al. Transcutaneous electrical nerve stimulation (TENS) for pain control after vaginal delivery and cesarean section. Journal of Maternal-Fetal and Neonatal Medicine 2014;27(15):1572-5. [DOI] [PubMed] [Google Scholar]
Kenton 2011 {published data only}
- NCT01271855. Cramps trial: controlled randomized trial assessing maternal post-partum pain with suppositories. clinicaltrials.gov/ct2/show/results/NCT01271855 (first received 7 January 2011).
Kim 2019 {published data only}
- Kim M, Moss D, Crawford P. Battlefield acupuncture for post-partum pain: randomized controlled trial. Explore 2019;15(6):409-14. [DOI] [PubMed] [Google Scholar]
- O'Callaghan M, NCT02526186. Battlefield auricular acupuncture for control of post-partum pain. clinicaltrials.gov/ct2/show/NCT02526186 (first received 18 August 2015).
Kumbar 2017 {published data only}
- Kumbar J, CTRI/2017/11/010702. A clinical study to evaluate the effect of Sootika Paricharya explained by Charakacharya in postnatal mothers. www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=19882 (first received 29 November 2017).
Laska 1983 {published data only}
- Laska EM, Sunshine A, Zighelboim I, Roure C, Marrero I, Wanderling J, et al. Effect of caffeine on acetaminophen analgesia. Clinical Pharmacology and Therapeutics 1983;33(4):498-509. [DOI] [PubMed] [Google Scholar]
Li 2014 {published data only}
- Li LP, Zhuang AW, Bao YH, Chu JM, Dou XQ. Clinical research on acupoint catgut implantation in the prevention and treatment of postpartum pain of uterine contraction with qi and blood deficiency. Chinese Acupuncture & Moxibustion 2014;34(1):34-6. [PubMed] [Google Scholar]
- Tan A. Translation for Li 2014. Email to: A Deussen 08 July 2020.
Li 2015 {published data only}
- Li Z, Chen YC, Su LQ. Clinical research of Tongjing Nazi method acupuncture for postpartum pain. Chinese Community Doctors [zhong guo she qu yi shi] 2015;31(27):93-4.
- Tan A. Translation for Li 2015. Email to: A Deussen 08 July 2020.
Linder 1997 {published data only}
- Linder N, German B, Bessant D, Sirota L, Zylber-Katz E, Martin O, et al. The pharmacological effect on term neonates of analgesic drugs ingested through maternal milk. Canadian Journal of Clinical Pharmacology 1997;4:112-4. [Google Scholar]
Mehlhorn 2006 {published data only}
- Mehlhorn G, Esche C, Fashing PA, Lux MP, Goecke T, Binder H, et al. Analgesia for afterpains by means of transcutaneous electrical nerve stimulation (TENS) vs, placebo TENS. A prospective randomised placebo controlled trial [Analgesie von schmerzhaften Nachwehen mittels transkutane elektrische Nervenstimmulation (TENS) vs. Placebo-TENS. Eine randomisierte placebokontrollierte Doppelblindstudie]. Geburtshilfe and Frauenheilkunde 2006;66 Suppl:S108. [Google Scholar]
Mirror 2019 {published data only}
- Golmakani N, IRCT20181007041266N1. Investigation effect of purslane seed capsule on postpartum hemorrhage and after pain in multiparous mothers. en.irct.ir/trial/35047 (first received 28 December 2018).
- Mirror RA, Golmakani V, Rakhshandeh H, Mazlum SR, Moral F. Effect of purslane seed capsule in prevention of postpartum after pain in multiparous mothers: a randomized clinical trial. Iranian Journal of Obstetrics, Gynecology and Infertility 2019;22(7):47-57. [Google Scholar]
Narimatsu 2001 {published data only}
- Narimatsu A, Ito A. [Usefulness of kyukichoketsuin during puerperium]. Rinsho Lyaku (Journal of Clinical Therapeutics & Medicine) 2001;17:1329-35. [Google Scholar]
Nazari 2018 {published data only}
- Nazari L, IRCT20160722029027N5. Comparison of the effect of TENS and music therapy on pain relief in labor and postpartum: A randomized clinical trial. en.irct.ir/trial/29281 (first received 4 May 2018).
Nunlee 2000 {published data only}
- Nunlee WY, Perry PM, Lawal AH, Ivankovich AD. Does a single dose of epidural morphine provide extended analgesia after vaginal delivery. Anesthesiology 2000;93(3A):A1060. [Google Scholar]
Olson 1984 {published data only}
- Olson N, Sunshine A, Roure C, Colon A, Laska EM, Santiago H, et al. Analgesic efficacy of suprofen, codeine and placebo. Pain 1984;2 Suppl:238. [Google Scholar]
Ozgoli 2018 {published data only}
- Ozgoli G, IRCT20100503003860N35. Effect of acupressure point Liv3 on postpartum pain reduction. en.irct.ir/trial/3995 (first received 9 May 2018).
- Ozgoli G. Email correspondence. Email to: A Deussen 06 January 2020.
Pan 1993 {published data only}
- Pan PH, Moore C, Blass N. Low dose epidural morphine provides better analgesia than oral percocet for post vaginal delivery. Regional Anesthesia 1993;18(2S):15. [Google Scholar]
Parsa 2019 {published data only}
- Akbari N. Translation for Parsa 2019. Email to: A Deussen 02 August 2020.
- Ozgoli G, IRCT2016071128876N1. The effect of zataria multiflora boiss capsule on postpartum pain. en.irct.ir/trial/23333 (first received 3 January 2017).
- Ozgoli. Email correspondence. Email to: A Deussen 06 January 2020 and 17 February 2020.
- Parsa L, Ozgoli G, Mojab F, Nasiri M, Moramezi F, Ghezi M. Comparison of effects of zataria multiflora capsule and Ibuprofen on postpartum pain [و ایبوپروفن بر پسدرد زایمان مقایسه تأثیر کپسول آویشن شیرازی]. Iranian Journal of Obstetrics, Gynaecology and Infertiltiy 2019;22, I(3):32-40. [Google Scholar]
Prockop 1960 {published data only}
- Prockop LD, Eckenhoff JE, McElroy RC. Evaluation of dextropropoxyphene, codeine, and acetylsalicylic compound. Obstetrics & Gynecology 1960;16:113-8. [PubMed] [Google Scholar]
Ray 1993 {published data only}
- Ray S, Swami A, Kadim M, Morgan B. Efficacy of diclofenac in a single prophylactic dose in post partum pain. International Journal of Obstetric Anesthesia 1993;2:58. [Google Scholar]
Redick 1980 {published data only}
- Redick LF, Bromage PR. Postpartum epidural narcotic analgesia. Anesthesiology 1980;53:S297. [Google Scholar]
Rubin 1984 {published data only}
- Rubin A, Winter L. A double-blind randomized study of an aspirin/caffeine combination versus acetaminophen/aspirin combination versus acetaminophen versus placebo in patients with moderate to severe post-partum pain. Journal of International Medical Research 1984;12:338-44. [DOI] [PubMed] [Google Scholar]
Smith 1973 {published data only}
- Smith GM, Coletta CG, McBride S, McPeek B. Use of subjective responses to evaluate efficacy of mild analgesic-sedative combinations. Clinical Pharmacology and Therapeutics 1973;15(2):118-29. [DOI] [PubMed] [Google Scholar]
Soltani 2017 {published data only}
- Soltani M, Azhari S, Vakilzadeh AK, Tara F, Mazloum SR. The effect of acupressure on uterine tone and pain after delivery. Iranian Journal of Obstetrics, Gynecology and Infertility 2017;20(9):91-100. [Google Scholar]
Sunshine 1983 {published data only}
- Sunshine A, Zighelboim I, De Sarrazin C, De Castro A, Olson NZ, Laska E. A study of the analgesic efficacy of nalbuphine hydrochloride in patients with postpartum pain. Current Therapeutic Research 1983;33(1):108-14. [Google Scholar]
- Sunshine A, Zighelboim I, Laska E. Oral analgesic study of nalbuphine hydrochloride, codeine, and placebo in postpartum pain. Clinical Pharmacology and Therapeutics 1982;31(2):274. [Google Scholar]
Sunshine 1985 {published data only}
- Sunshine A, Zighelboim I, Olson NZ, De Sarrazin C, Laska E. A comparative oral analgesic study of indoprofen, aspirin, and placebo in postpartum pain. Journal of Clinical Pharmacology 1985;25(5):374-80. [DOI] [PubMed] [Google Scholar]
Sunshine 1986 {published data only}
- Sunshine A, Olson NZ, Siegel C, Laska E. Oral analgesic study of ketoprofen, aspirin and placebo in post-partum pain. Clinical Pharmacology and Therapeutics 1983;33:154. [Google Scholar]
- Sunshine A, Zighelboim I, Laska E, Siegel C, Olson NZ, De Castro A. A double-blind, parallel comparison of ketoprofen, aspirin, and placebo in patients with post-partum pain. Journal of Clinical Pharmacology 1986;26(8):706-11. [DOI] [PubMed] [Google Scholar]
Sunshine 1989 {published data only}
- Sunshine A, Laska E, Siegel C, Zighelboim I, De Castro A, Sorrentino J, et al. Analgesic adjuvancy of caffeine with ibuprofen in three different postpartum pain populations. Clinical Pharmacology and Therapeutics 1989;45:174. [Google Scholar]
Tafazoli 2013 {published data only}
- Keikha. Translation for Tafazoli 2013. Email to: A Deussen 24 July 2020.
- Khadem M, Tafazoli M, Asili J, Esmaila H. A comparison of the effect of cuminum cyminum vs. mefenamic acid on afterpains in multiparous at 17 Shahrivar Hospital in Mashad in 2006-2007. International Journal of Gynecology & Obstetrics 2009;107(Suppl 2):S679. [Google Scholar]
- Tafazoli M, Ahmadabadi MK, Asili J, Esmaili H. Comparison the effects of cuminum and mefenamic acid on after pains in multiparous women. Iranian Journal of Obstetrics, Gynecology and Infertility 2013;16(75):1-11. [Google Scholar]
Van Wering 1972 {published data only}
- Van Wering RF, Bleker OP. Oral analgesia in post-partum pain: a comparison of ibuprofen ('Brufen') and dextropropoxyphene. Current Medical Research and Opinion 1972;1(1):49-52. [DOI] [PubMed] [Google Scholar]
Vaziri 2017 {published data only}
- Najib Z, IRCT2014060910327N7. The effect of lavender aromatherapy on maternal somatic pain, fatigue and mood in the early hours after delivery. en.irct.ir/trial/10829 (first received 1 July 2014).
- Vaziri F, Shiravani M, Najib FS, Pourahmad S, Salehi A, Yazdanpanahi Z. Effect of lavender oil aroma in the early hours of postpartum period on maternal pains, fatigue, and mood: a randomized clinical trial. International Journal of Preventive Medicine 2017;8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Von Pein 1974 {published data only}
- Von Pein W. Double blind study using benzydamine in the puerperium [Doppelblindstudie mit Benzydamine im Wochenbett]. Gynakologische Rundschau 1974;14:327-8. [PubMed] [Google Scholar]
Yogev 2015 {published data only}
- Yogev Y, NCT02399774. Dental support device during breastfeeding as a mean for pain control. clinicaltrials.gov/ct2/show/record/NCT02399774 (first received 26 March 2015).
References to ongoing studies
IRCT2015050322053N1 {published data only}
- IRCT2015050322053N1. Comparison of the effect of chamomila matricaria and mefnamic acid capsules on post partum pain and hemorrhage. en.irct.ir/trial/19115 (first received 25 June 2015).
IRCT2016070428240N2 {published data only}
- IRCT2016070428240N2. The effect of acupressure and touch point (SP6) on pain intensity after delivery 88 qualified mothers give birth on 22 Bahman hospital in Gonabad city. en.irct.ir/trial/22945 (first received 22 August 2016).
IRCT2016100930238N1 {published data only}
- IRCT2016100930238N1. Comparative study of the effect of Fennel capsules and Ibuprofen on postpartum after pain in multiparous women admitted in postpartum ward of Sanandaj Beasat hospital. en.irct.ir/trial/24093 (first received 10 February 2017).
IRCT201707283860N33 {published data only}
- IRCT201707283860N33. Clinical trial study of the effect of ginger plant capsules on reducing the pain in women with postpartum pain. en.irct.ir/trial/3997 (first received 17 November 2017).
- Ozgoli G. Email correspondence. Email to: A Deussen 06 January 2020 and 05 July 2020.
IRCT20171208037792N1 {published data only}
- IRCT20171208037792N1. The effect of foot reflexology on reduction post partum after pain. en.irct.ir/trial/28558 (first received 14 July 2018).
IRCT20180428039454N1 {published data only}
- IRCT20180428039454N1. Effect of portulaca oleracea capsule on the postpartum pain and hemorrhage. en.irct.ir/trial/30837 (first received 31 May 2018).
IRCT20190217042739N1 {published data only}
- IRCT20190217042739N1. Comparing the effect of salvia hydrangea and mefenamic acid on postpartum pain. en.irct.ir/trial/37627 (first received 7 April 2019).
NCT03617900 {published data only}
- NCT03617900. Efficacy of ginger extract (compare between the ginger preparation of ancient concept of Thai traditional practitioner, standard drug and placebo) by using pain score to evaluate after pain of three groups of first normal postpartum women. clinicaltrials.gov/ct2/show/NCT03617900 (first received 7 August 2018).
NCT04037202 {published data only}
- NCT04037202. Effect of foot massage on postpartum comfort and pain level of the mothers who had vaginal birth: a randomised controlled trial. clinicaltrials.gov/ct2/show/NCT04037202 (first received 30 July 2019).
Additional references
Baddock 2019
- Baddock, S. Chapter 27: Overview of physiological changes during the postnatal period. In: Pairman S, Tracy S, Dahlen H, Dixon L, editors(s). Midwifery: Preparation for Practice. 4th edition. Chatswood, NSW, Australia: Elsevier, 2019. [Google Scholar]
Blackburn 2013
- Blackburn ST. Postpartum period and lactation physiology. In: Maternal, Fetal & Neonatal Physiology: A Clinical Perspective. 4th ed edition. Maryland Heights, MO: Elsevier Health Science, 2013:142-60. [Google Scholar]
Bloomfield 1998
- Bloomfield SS, Mitchell J, Cissell G, Barden TP. Analgesic sensitivity of two post-partum models. Pain 1986;27(2):171-9. [DOI] [PubMed] [Google Scholar]
Chou 2010
- Chou D, Abalos E, Gyte GML, Gülmezoglu AM. Drugs for perineal pain in the early postpartum period: generic protocol. Cochrane Database of Systematic Reviews 2009, Issue 3. Art. No: CD007734. [DOI: 10.1002/14651858.CD007734.pub2] [DOI] [Google Scholar]
Chou 2013
- Chou D, Abalos E, Gyte GML, Gülmezoglu AM. Paracetamol/acetaminophen (single administration) for perineal pain in the early postpartum period. Cochrane Database of Systematic Reviews 2013, Issue 1. Art. No: CD008407. [DOI: 10.1002/14651858.CD008407.pub2] [DOI] [PubMed] [Google Scholar]
CONSORT 2010
- Schulz KF, Altman DG, Moher D, for the CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cooper 1997
- Cooper SA. Commentary: Single‐dose analgesic studies: the upside and downside of assay sensitivity. In: Max M, Portenov R, Laska E, editors(s). Advances in Pain Research and Therapy. Vol. 18. New York: Raven Press, 1997:117-24. [Google Scholar]
Coutaux 2017
- Coutaux A. Non-pharmacological treatments for pain relief: TENS and acupuncture. Joint Bone Spine 2017;84(6):657-61. [DOI: ] [DOI] [PubMed] [Google Scholar]
Dean 2017
- Dean L. Codeine therapy and CYP2D6 genotype. In: Pratt VM, McLeod HL, Rubinstein WS, et al, editors(s). Medical Genetics Summaries. National Center for Biotechnology Information, 2017. [https://www.ncbi.nlm.nih.gov/books/NBK100662/] [PubMed] [Google Scholar]
Drugs.com 2019a
- Drugscom 2019. Ketorolac levels and effects while breastfeeding. www.drugs.com/breastfeeding/ketorolac.html (updated 2019).
Drugs.com 2019b
- Drugscom. Nalbuphine use while breastfeeding. www.drugs.com/breastfeeding/nalbuphine.html (updated 2019).
Gallo 2018
- Gallo RB, Santana LS, Marcolin AC, Duarte G, Quintana SM. Sequential application of non-pharmacological interventions reduces the severity of labour pain, delays use of pharmacological analgesia, and improves some obstetric outcomes: a randomised trial. Journal of Physiotherapy 2018;64(1):33-40. [DOI] [PubMed] [Google Scholar]
Hale 2019
- Hale TW. Hale's Medications and Mother's Milk. 18th edition. New York, USA: Springer Publishing Company, 2019. [Google Scholar]
Higgins 2020a
- Higgins JP, Savović J, Page MJ, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). The Cochrane Collaboration, 2020. Available from www.training.cochrane.org/handbook.
Higgins 2020b
- Higgins JP, Eldridge S, Li T (editors). Chapter 23: Including variants on randomized trials. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). The Cochrane Collaboration, 2020. Available from www.training.cochrane.org/handbook.
Koren 2006
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006;368(6536):704. [DOI] [PubMed] [Google Scholar]
LactMed 2018a
- LactMed. Drugs and Lactation Database - Fenoprofen. Bethesda, MD: National Library of Medicine, (updated 2018). [https://www.ncbi.nlm.nih.gov/books/NBK500666/?report=reader#_NBK500666_pubdet] [Google Scholar]
LactMed 2018b
- LactMed. Drugs and Lactation Database - Flurbiprofen. Bethesda, MD: National Library of Medicine, (updated 2018). [https://www.ncbi.nlm.nih.gov/books/NBK500754/] [Google Scholar]
Lam 2012
- Lam J, Matlow JN, Ross CJ, Hayden MR, Carleton BC, Madadi P. Postpartum maternal codeine therapy and the risk of adverse neonatal outcomes: the devil is in the details. Therapeutic Drug Monitoring 2012;34(4):378-80. [DOI: 10.1097/FTD.0b013e31825da19f] [DOI] [PubMed] [Google Scholar]
Mall 2019
- Mall M, Pravati T, Pratibha K. Effect of selected nursing interventions in the reduction of after-pains and involution of uterus among post-natal mothers in selected hospitals. Journal of Biomedical Sciences 2019;6(3):10. [DOI: 10.3126/jbs.v6i3.26842] [DOI] [Google Scholar]
Marieb 2019
- Marieb EN, Hoehn K. Human Anatomy and Physiology. 11th edition. Harlow, Essex, England: Benjamin Cummnings Pearsons, 2019. [Google Scholar]
Marjoribanks 2015
- Marjoribanks J, Ayeleke RO, Farquhar C, Proctor M. Nonsteroidal anti-inflammatory drugs for dysmenorrhoea. Cochrane Database of Systematic Reviews 2015, Issue 7. Art. No: CD001751. [DOI: 10.1002/14651858.CD001751.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Maroon 2010
- Maroon JC, Bost JC, Maroon A. Natural anti-inflammatory agents for pain relief. Surgical Neurology International 2010;1:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Merriam‐Webster 2020
- Merriam-Webster. Dictionary [Herbal medicine]. www.merriam-webster.com/dictionary/herbal%20medicine (Accessed 16 February 2020).
MIMS 2020
- MIMS. MIMS New Zealand. Auckland, New Zealand: MIMS New Zealand Limited, 2020. [Google Scholar]
Molakatalla 2017
- Molakatalla S, Shepherd E, Grivell RM. Aspirin (single dose) for perineal pain in the early postpartum period. Cochrane Database of Systematic Reviews 2017, Issue 2. Art. No: CD012129. [DOI: 10.1002/14651858.CD012129.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 1996
- Moore A, McQuay H, Gavaghan D. Deriving dichotomous outcome measures from continuous data in randomised controlled trials of analgesics. Pain 1996;66(2-3):229-37. [DOI] [PubMed] [Google Scholar]
Moore 1997a
- Moore A, Moore O, McQuary H, Gavaghan D. Deriving dichotomous outcome measures from continuous data in randomised controlled trials of analgesia: use of pain intensity and visual analogue scales. Pain 69;3:311-5. [DOI] [PubMed] [Google Scholar]
Moore 1997b
- Moore A, McQuay H, Gavaghan D. Deriving dichotomous outcome measures from continuous data in randomised controlled trials of analgesia: verification from independent data. Pain 1997;69(1-2):127-30. [DOI] [PubMed] [Google Scholar]
Oguntibeju 2018
- Oguntibeju OO. Medicinal plants with anti-inflammatory activities from selected countries and regions of Africa. Journal of Inflammation Research 2018;11:307-17. [DOI: 10.2147/JIR.S167789] [DOI] [PMC free article] [PubMed] [Google Scholar]
Oxford 2020
- Oxford Advanced Learner's Dictionary. Oxford University Press 2020.
Paliulyte 2017
- Paliulyte V, Drasutiene GS, Ramasauskaite D, Bartkevicien D, Zakareviciene J, Kurmanavicius J. Physiological uterine involution in primiparous and multiparous women: Ultrasound study. Obstetrics and Gynecology International 2017;2017:Article ID 6739345. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Peng 2019
- Peng WW, Tang ZY, Zhang FR, Li H, Kong YZ, Iannetti GD, et al. Neurobiological mechanisms of TENS-induced analgesia. NeuroImage 2019;195:396-408. [DOI: 10.1016/j.neuroimage.2019.03.077] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pessel 2019
- Pessel C, Tsai MC. Chapter 10: The normal puerperium. In: DeCherney AH, Nathan L, Laufer N, Roman, AS, editors(s). Current Diagnosis & Treatment: Obstetrics & Gynecology. 12th edition. USA: McGraw-Hill Education, 2019. [Google Scholar]
Rankin 2017
- Rankin S. Physiology in Childbearing: with Anatomy and Related Biosciences. 4th edition. Edinburgh, Scotland: Elsevier, 2017. [Google Scholar]
RevMan 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Ritter 2019
- Ritter J, Flower R, Henderson G, Loke YK, MacEwan D, Rang H. Rang & Dale's Pharmacology. 9th edition. Elsevier, 2019. [Google Scholar]
Wambach 2021
- Wambach K, Spencer B. Breastfeeding and Human Lactation. 6th edition. Burlington, MA: Jones & Bartlett Learning, 2021. [Google Scholar]
Wuytack 2016
- Wuytack F, Smith V, Cleary BJ. Oral non-steroidal anti-inflammatory drugs (single dose) for perineal pain in the early postpartum period. Cochrane Database of Systematic Reviews 2016, Issue 7. Art. No: CD011352. [DOI: 10.1002/14651858.CD011352.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Deussen 2004
- Deussen AR, Ashwood P, Agett S. Analgesia for relief of pain due to uterine cramping/involution after birth. Cochrane Database of Systematic Reviews 2004, Issue 3. Art. No: CD004908. [DOI: 10.1002/14651858.CD004908] [DOI] [PubMed] [Google Scholar]
Deussen 2011
- Deussen AR, Ashwood P, Martis R. Analgesia for relief of pain due to uterine cramping/involution after birth. Cochrane Database of Systematic Reviews 2011, Issue 5. Art. No: CD004908. [DOI: 10.1002/14651858.CD004908.pub2] [DOI] [PubMed] [Google Scholar]