

Abstract
Background
This is an updated version of the Cochrane Review previously published in 2018.
Epilepsy is a common neurological disorder characterised by recurrent seizures. Most people with epilepsy have a good prognosis and their seizures are well controlled by a single antiepileptic drug, but up to 30% develop drug‐resistant epilepsy, especially people with focal seizures. In this review, we summarised the evidence from randomised controlled trials (RCTs) of gabapentin, when used as an add‐on treatment for drug‐resistant focal epilepsy.
Objectives
To evaluate the efficacy and tolerability of gabapentin when used as an add‐on treatment for people with drug‐resistant focal epilepsy.
Search methods
For the latest update, we searched the Cochrane Register of Studies (CRS Web) and MEDLINE (Ovid) on 11 August 2020. CRS Web includes randomised or quasi‐randomised, controlled trials from PubMed, Embase, ClinicalTrials.gov, the World Health Organization International Clinical Trials Registry Platform (ICTRP), the Cochrane Central Register of Controlled Trials (CENTRAL), and the Specialised Registers of Cochrane Review Groups including Epilepsy. We imposed no language restrictions.
Selection criteria
Randomised, placebo‐controlled, double‐blind, add‐on trials of gabapentin in people with drug‐resistant focal epilepsy. We also included trials using an active drug control group or comparing different doses of gabapentin.
Data collection and analysis
Two review authors independently selected trials for inclusion and extracted the relevant data. We assessed the following outcomes: seizure frequency, seizure freedom, treatment withdrawal (any reason) and adverse effects. Primary analyses were intention‐to‐treat. We also undertook sensitivity best‐case and worst‐case analyses. We estimated summary risk ratios (RR) for each outcome and evaluated dose‐response in regression models.
Main results
We identified no new studies for this update, therefore, the results and conclusions are unchanged.
In the previous update of this review, we combined data from six trials in meta‐analyses of 1206 randomised participants. The overall risk ratio (RR) for reduction in seizure frequency of 50% or more compared to placebo was 1.89 (95% confidence interval (CI) 1.40 to 2.55; 6 studies, 1206 participants; moderate‐certainty evidence). Dose regression analysis (for trials in adults) showed increasing efficacy with increasing dose, with 25.3% (95% CI 19.3 to 32.3) of people responding to gabapentin 1800 mg compared to 9.7% on placebo, a 15.5% increase in response rate (95% CI 8.5 to 22.5). The RR for treatment withdrawal compared to placebo was 1.05 (95% CI 0.74 to 1.49; 6 trials, 1206 participants; moderate‐certainty evidence). Adverse effects were significantly associated with gabapentin compared to placebo. RRs were as follows: ataxia 2.01 (99% CI 0.98 to 4.11; 3 studies, 787 participants; low‐certainty evidence), dizziness 2.43 (99% CI 1.44 to 4.12; 6 studies, 1206 participants; moderate‐certainty evidence), fatigue 1.95 (99% CI 0.99 to 3.82; 5 studies, 1161 participants; low‐certainty evidence) and somnolence 1.93 (99% CI 1.22 to 3.06; 6 studies, 1206 participants; moderate‐certainty evidence). There was no evidence of a difference for the adverse effects of headache (RR 0.79, 99% CI 0.46 to 1.35; 6 studies, 1206 participants; moderate‐certainty evidence) or nausea (RR 0.95, 99% CI 0.52 to 1.73; 4 trials, 1034 participants; moderate‐certainty evidence). Overall, the studies were at low to unclear risk of bias due to information on each risk of bias domain not being available. We judged the overall certainty of the evidence (using the GRADE approach) as low to moderate due to potential attrition bias resulting from missing outcome data and imprecise results with wide CIs.
Authors' conclusions
Gabapentin has efficacy as an add‐on treatment in people with drug‐resistant focal epilepsy, and seems to be fairly well‐tolerated. However, the trials reviewed were of relatively short duration and provide no evidence for the long‐term efficacy of gabapentin beyond a three‐month period. The results cannot be extrapolated to monotherapy or to people with other epilepsy types. Further trials are needed to assess the long‐term effects of gabapentin, and to compare gabapentin with other add‐on drugs.
Plain language summary
Gabapentin as an add‐on for drug‐resistant focal epilepsy
Background
Epilepsy is a disorder where recurrent seizures (fits) are caused by abnormal electrical discharges from the brain. Evidence from randomised controlled trials (well‐designed clinical trials in which people are allocated at random to test a specific drug, treatment or other intervention) are often used to examine how effective and safe antiepileptic medicines are in people who experience such seizures. This review included 12 studies and data from 2607 people with focal seizures (seizures that occur in just one part of the brain).
Study characteristics
Data from six studies were combined in the analysis. All participants (including adults and children) were previously taking at least one antiepileptic medicine and all were continuing to have seizures. Either gabapentin (an antiepileptic medicine) or a placebo (a tablet that contains no medicine) was added to the medicine regimen.
Key results
The results showed that gabapentin effectively reduced seizures when used as an additional treatment. Compared to a placebo, gabapentin was almost twice as likely to reduce seizures by 50% or more. The most common side effects associated with gabapentin were ataxia (poor co‐ordination and unsteady gait), dizziness, fatigue and drowsiness.
Quality of the evidence
Overall, the quality of evidence was low to moderate as information was not reported for all participants in some of the trials and some of the results were imprecise. Research is needed into the effects of the long‐term use of gabapentin, and to compare gabapentin with other add‐on medicines.
The evidence is current to 11 August 2020.
Summary of findings
Summary of findings 1. Gabapentin versus placebo for people with drug‐resistant focal epilepsy.
| Gabapentin versus placebo for people with drug‐resistant focal epilepsy | ||||||
|
Patient or population: people with drug‐resistant focal epilepsy Settings: outpatient Intervention: gabapentin Comparator: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Gabapentin | |||||
|
Reduction in seizure frequency of ≥ 50%: primary analysis
Number of seizures reported in seizure diary Follow‐up: 12–14 weeks |
12 per 100 | 23 per 100 (17 to 31) | RR 1.89 (95% CI 1.40 to 2.55) | 1206 (6 studies) | ⊕⊕⊕⊝ Moderatea | RR > 1 indicated outcome was more likely in gabapentin group. |
|
Treatment withdrawal
Number of participants withdrawn for any reason Follow‐up: 12–14 weeks |
10 per 100 | 11 per 100 (8 to 15) | RR 1.05 (95% CI 0.74 to 1.49) | 1206 (6 studies) | ⊕⊕⊕⊝ Moderatea | RR > 1 indicated outcome was more likely in gabapentin group. |
|
Adverse effects: ataxia
Number of participants experiencing ataxia Follow‐up: 12–14 weeks |
5 per 100 | 10 per 100 (5 to 20) | RR 2.01 (99% CI 0.98 to 4.11) | 787 (3 studies) | ⊕⊕⊝⊝ Lowb,c | RR > 1 indicated outcome was more likely in gabapentin group. |
|
Adverse effects: dizziness
Number of participants experiencing dizziness Follow‐up: 12–14 weeks |
6 per 100 | 14 per 100 (8 to 23) | RR 2.43 (99% CI 1.44 to 4.12) | 1206 (6 studies) | ⊕⊕⊕⊝ Moderatea | RR > 1 indicated outcome was more likely in gabapentin group. |
|
Adverse effects: fatigue
Number of participants experiencing fatigue Follow‐up: 12–14 weeks |
4 per 100 | 7 per 100 (3 to 13) | RR 1.95 (99% CI 0.99 to 3.82) | 1161 (5 studies) | ⊕⊕⊝⊝ Lowa,d | RR > 1 indicated outcome was more likely in gabapentin group. |
|
Adverse effects: nausea
Number of participants experiencing nausea Follow‐up: 12–14 weeks |
7 per 100 | 7 per 100 (4 to 12) | RR 0.95 (99% CI 0.52 to 1.73) | 1034 (4 studies) | ⊕⊕⊕⊝ Moderatea | RR > 1 indicated outcome was more likely in gabapentin group. |
|
Adverse effects: somnolence
Number of participants experiencing somnolence Follow‐up: 12–14 weeks |
7 per 100 | 14 per 100 (9 to 23) | RR 1.93 (99% CI 1.22 to 3.06) | 1206 (6 studies) | ⊕⊕⊕⊝ Moderatea | RR > 1 indicated outcome was more likely in gabapentin group. |
|
Adverse effects: headache
Number of participants experiencing headache Follow‐up: 12–14 weeks |
8 per 100 | 6 per 100 (3 to 10) | RR 0.79 (99% CI 0.46 to 1.35) | 1206 (6 studies) | ⊕⊕⊕⊝ Moderatea | RR < 1 indicated outcome was more likely in control group. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDowngraded once for risk of bias: four studies had missing data and did not carry out intention‐to‐treat analysis. Best‐case and worst‐case scenario analysis demonstrated that missing data may have impacted on the size of the true treatment effect. bDowngraded once for risk of bias: three studies had missing data and did not carry out intention‐to‐treat analysis. cDowngraded once for imprecision: one study with small number of effects and wide confidence intervals; concern regarding the confidence in overall effect. dDowngraded once for imprecision: two studies with small‐study effects and wide confidence intervals; concern regarding the confidence in overall effect.
Background
This is an updated version of the Cochrane Review previously published in 2018 (Panebianco 2018).
The purpose of this updated Cochrane Review is to summarise the current understanding of the role of gabapentin as an add‐on treatment in focal epilepsy resistant to at least one other antiepileptic drug (AED).
Description of the condition
Epilepsy is a common neurological disorder characterised by recurrent seizures. Most people given a diagnosis of epilepsy have a good prognosis and their seizures will be controlled by treatment with a single AED (Reynolds 1981). However, up to 30% will continue to have seizures despite treatment with adequate doses of AEDs, often requiring treatment with a combination (Cockerell 1995). These people represent a significant therapeutic problem taking into account that up to 2% to 3% of the population will experience epilepsy at some time in their lives (Hauser 1993). There is no internationally accepted definition of drug resistance, so, for the purpose of this review, we considered people drug‐resistant if they had focal‐onset seizures (simple focal or complex focal or secondary generalised tonic‐clonic seizures, or a combination of these) and failed to respond to at least one monotherapy AED.
Description of the intervention
Although more than 12 new AEDs have entered the market since 1993, up to 30% of people remain resistant to current treatments. Thus, a concerted effort continues to identify and develop new therapies that will help these people (Barker‐Haliski 2014). Pharmacological treatment remains the first choice for controlling epilepsy (Loscher 2002), although recent decades have seen advances in vagal stimulation (Panebianco 2015), and surgery (West 2019). Current first‐line treatment for focal epilepsy includes: lamotrigine, sodium valproate, carbamazepine, oxcarbazepine and levetiracetam. When first‐line medications fail to achieve seizure freedom, add‐on therapy is required.
How the intervention might work
Gabapentin was licensed for add‐on use in the UK in 1993. The mechanism of action is uncertain (McClean 1995). Gabapentin is a structural analogue of the neurotransmitter gamma‐aminobutyric acid (GABA). However, it does cross the blood‐brain barrier and its activities are believed not to be GABA‐related. Gabapentin has a high volume of distribution, is not significantly protein‐bound or metabolised, and does not induce or inhibit hepatic enzymes; thus, it has minimal‐to‐no known interactions with other AEDs.
Why it is important to do this review
In this review, we summarised evidence from randomised controlled trials (RCTs) on the efficacy and tolerability of gabapentin for people with drug‐resistant focal epilepsy in order to aid clinical decision‐making when considering gabapentin as an add‐on treatment within this population.
Objectives
To evaluate the efficacy and tolerability of gabapentin when used as an add‐on treatment for people with drug‐resistant focal epilepsy.
Methods
Criteria for considering studies for this review
Types of studies
To be included in our review, studies had to meet the following criteria:
RCTs that used an adequate method of concealment of randomisation (e.g. allocation of sequentially sealed packages of medication, sealed opaque envelopes, telephone randomisation);
double‐blind trials, in which both participant and clinician treating or assessing outcome were blinded to treatment allocated;
placebo‐controlled or alternative AED or range of gabapentin doses used as controls;
parallel‐group or cross‐over studies.
Types of participants
People of any age with drug‐resistant focal epilepsy (i.e. experiencing simple focal, complex focal or secondary generalised tonic‐clonic seizures).
Types of interventions
The active treatment group received gabapentin in addition to conventional AED.
The control group received matched placebo, different dose of gabapentin or alternative AED in addition to conventional AED.
Types of outcome measures
Primary outcomes
Reduction in seizure frequency of 50% or more
We chose the proportion of people with a 50% or greater reduction in seizure frequency in the treatment period compared to the prerandomisation baseline period as the primary outcome. This is commonly reported in this type of study and can be calculated for studies that do not report it from baseline seizure data.
Seizure freedom
The proportion of people with complete cessation of seizures during the treatment period.
Secondary outcomes
Treatment withdrawal
We used the proportion of people having treatment withdrawn during the treatment period as a measure of global effectiveness. Treatment is likely to be withdrawn due to adverse effects, lack of efficacy, or a combination of both, and this is an outcome to which the person makes a direct contribution. In trials of short duration, it is likely that adverse effects will be the most common reason for withdrawal.
Adverse effects
-
The proportion of people experiencing the following five common and important adverse effects:
ataxia;
dizziness;
fatigue;
nausea;
somnolence.
The proportion of people experiencing the five most common adverse effects if different from a. to e. above.
Search methods for identification of studies
Electronic searches
Searches were run for the original review in 1998 and subsequent searches were run in 2000, 2003, 2005, 2007, 2010, 2011, 2012, 2013, 2015, 2016 and March 2018. For the latest update, we searched the following databases on 11 August 2020:
Cochrane Register of Studies (CRS Web), using the strategy shown in Appendix 1;
MEDLINE (Ovid, 1946 to 11 August 2020) using the strategy shown in Appendix 2.
CRS Web includes randomised or quasi‐randomised, controlled trials from the Specialized Registers of Cochrane Review Groups including Epilepsy, the Cochrane Central Register of Controlled Trials (CENTRAL), PubMed, Embase, ClinicalTrials.gov and the World Health Organization International Clinical Trials Registry Platform (ICTRP). There were no language restrictions.
Searching other resources
We reviewed the reference lists of included studies to search for additional reports of relevant studies.
Data collection and analysis
Selection of studies
Two review authors (MP and SAB) independently assessed trials for inclusion. Any doubts were resolved by discussion.
Data extraction and management
Two review authors (MP and SAB) independently extracted the following information for each trial using a data extraction sheet. Any discrepancies between the extractions of the two review authors were resolved by discussion.
Methodological/trial design
Method of randomisation and allocation concealment.
Method of double‐blinding.
Whether any participants had been excluded from reported analyses.
Duration of baseline period.
Duration of treatment period.
Dose(s) of gabapentin tested.
Participant/demographic information
Total number of participants allocated to each treatment group.
Age/sex.
Number with focal/secondary generalised seizures.
Seizure types.
Seizure frequency during the baseline period.
Number of background drugs.
Parke Davis sponsored most trials (Pfizer Inc. funded one trial); we asked them to confirm the following information:
method of randomisation;
total number randomised to each group;
number of participants in each group achieving a 50% or greater reduction in seizure frequency per treatment group;
number of participants having treatment withdrawn postrandomisation per treatment group;
-
for those excluded:
the reason for exclusion;
whether any of those excluded completed the treatment phase;
whether any of those excluded had a 50% or greater reduction in seizure frequency during the treatment phase.
Outcomes
We recorded the number of participants experiencing each outcome (see Types of outcome measures) per randomised group.
We contacted authors of trials for any missing information.
Assessment of risk of bias in included studies
Two review authors (SAB and JW) independently assessed the risk of bias for each trial using the Cochrane 'Risk of bias' tool as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We rated included studies as high risk, low risk or unclear risk on six domains applicable to RCTs: randomisation method, allocation concealment, blinding methods, incomplete outcome data, selective outcome reporting and other sources of bias.
We outlined studies failing to meet the criteria to be included in the meta‐analysis in narrative form; statistics for those included in the meta‐analysis are outlined below.
Measures of treatment effect
We presented the outcomes as risk ratios (RR) with 95% or 99% confidence intervals (CI).
Unit of analysis issues
We assessed cross‐over studies to determine if they presented suitable data to allow for inclusion in meta‐analysis using either 'paired' results adjusted for the cross‐over design, or first period results.
One cross‐over trial did not provide suitable data for inclusion in the meta‐analysis, but was discussed in narrative form (Leach 1997).
Dealing with missing data
We sought any missing data from study authors. We carried out intention‐to‐treat (ITT), best‐case and worst‐case analysis to account for any missing data (see Data synthesis).
Assessment of heterogeneity
We assessed clinical heterogeneity by comparing the distribution of important participant factors among trials (e.g. age, seizure type, duration of epilepsy) and trial factors (e.g. methods of randomisation and blinding, missing data). We examined statistical heterogeneity using a Chi2 test (P > 0.10) and the I2 statistic (Higgins 2020).
Assessment of reporting biases
Protocol versus full study
We requested all protocols from study authors to enable a comparison of outcomes of interest. We intended to investigate any suspected outcome reporting bias using the ORBIT matrix system (Kirkham 2010).
Funnel plot
Reporting biases arise when dissemination of research findings is influenced by the nature and direction of results (Higgins 2020). We intended to use funnel plots for investigating reporting biases when 10 or more studies were included in a meta‐analysis, with awareness that they have limited power to detect small‐study effects and we planned to seek statistical advice on their interpretation. For this review, we did not produce funnel plots for outcomes as fewer than 10 studies were included in meta‐analyses.
Data synthesis
We employed a fixed‐effect meta‐analysis to synthesise the data. Comparisons we carried out included:
intervention group versus controls on seizure reduction;
intervention group versus controls on seizure freedom;
intervention group versus controls on treatment withdrawal;
intervention group versus controls on adverse effects.
We performed separate comparisons for different types of control group (i.e. placebo or active control group) and study characteristics (i.e. cross‐over designed trials) to ensure appropriate combination of data.
The preferred estimate was the Mantel‐Haenszel RR. For the outcomes reduction in seizure frequency of 50% of more and treatment withdrawal, we used 95% CIs. For individual adverse effects, we used 99% CIs to make an allowance for multiple testing by using wider CIs. This is not a strict formal adjustment, as the number of individual adverse effects is not known in advance.
Our analyses included all participants in the treatment group to which they had been allocated. For the primary efficacy outcome (reduction in seizure frequency of 50% or more), we undertook three analyses.
-
Primary (ITT) analysis: participants not completing follow‐up or with inadequate seizure data were assumed to be non‐responders. To test the effect of this assumption, we undertook the following sensitivity analyses:
worst‐case analysis: participants not completing follow‐up or with inadequate seizure data were assumed to be non‐responders in the gabapentin group and responders in the placebo group;
best‐case analysis: participants not completing follow‐up or with inadequate seizure data were assumed to be responders in the gabapentin group and non‐responders in the placebo group.
Dose regression analysis
We examined dose‐response relationships using logistic regression (for the five adult trials) and calculated probabilities for the following for differing doses: the percentage of participants having a 50% response; and the difference in the percentage of participants responding to each dose compared to placebo. A binary variable was defined with value 0 if the response was less than 50% and value 1 otherwise. We examined dose‐response relationships using logistic regression, in the framework of generalised linear models, using the package GLIM, with this binary variable as the outcome variable (McCullagh 1989). Trial effects (i.e. adjustment for trial‐specific differences) were not included in the regression models as it was generally not possible to do so as some doses are confounded with trials; in other words, the dose was evaluated in only a single trial. As none of the tests for heterogeneity reached a significance level of less than 30%, it seemed reasonable to proceed without trial effects.
Subgroup analysis and investigation of heterogeneity
We intended to investigate heterogeneity using subgroup analysis of important participant factors among trials (e.g. age, seizure type, duration of epilepsy if deemed appropriate. As there was no important heterogeneity in the meta‐analyses in this review, we conducted no subgroup analysis.
Sensitivity analysis
We intended to carry out sensitivity analysis if there were peculiarities between study quality, characteristics of participants, interventions and outcomes.
Summary of findings and assessment of the certainty of the evidence
We used the GRADE approach, as outlined in the GRADE Handbook (Schünemann 2013), to interpret findings, and GRADEpro GDT software (which imports data from Review Manager 5 software (GRADEpro GDT; Review Manager 2014)), to create a 'Summary of findings' table for the primary outcome (reduction in seizure frequency of 50% or more) and secondary outcomes (treatment withdrawal and adverse effects).
We created a 'Summary of findings' table for the most important comparison (gabapentin versus placebo). We did not create 'Summary of findings' tables for other comparisons with only a single study contributing to the comparison.
Results
Description of studies
Results of the search
Updated searches conducted in August 2020 revealed 29 records identified from the databases outlined in Electronic searches. After removal of three duplicates, 26 records were screened for inclusion in the review. We excluded six clearly irrelevant records and assessed three full‐text articles for eligibility. We did not identify any new studies for this update (French 2016), therefore the results and conclusions are unchanged (see Figure 1).
1.
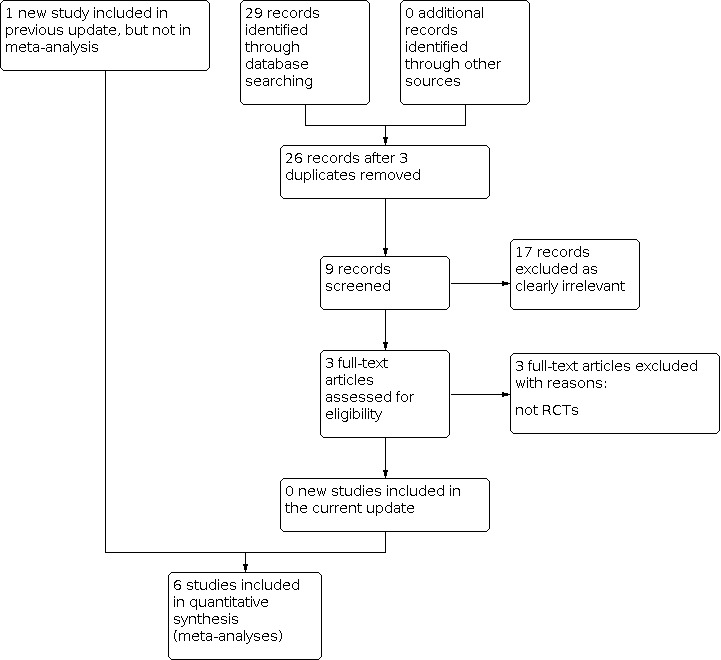
Study flow diagram. Date of search 11 August 2020. RCT: randomised controlled trial.
Included studies
Overall, the review included 12 studies, six of which contributed to the meta‐analyses. The data from the six remaining studies were not combined in meta‐analyses due to the differences in comparisons investigated. Kwan 2000 and Shapiro 2000 are awaiting classification as only abstracts were obtainable, therefore, it was not possible to critique the study design (see Characteristics of studies awaiting classification).
There were seven trials that compared gabapentin to placebo (Anhut 1994; Appleton 1999; Leach 1997; Sivenius 1991; UK Gabapentin 1990; US Gabapentin 1993; Yamauchi 2006), two trials that examined two different doses of gabapentin (Fisher 2001; Tomovic 1999), one trial that compared gabapentin to vigabatrin (Lindberger 2000), one trial that compared gabapentin to lamotrigine (Sethi 2002), and one trial that compared gabapentin to pregabalin (French 2016). All participants had drug‐resistant focal epilepsy and were taking at least one monotherapy AED. Pre‐existing AED regimens remained unchanged throughout the study period. All outcome measures included seizure reduction and adverse effects.
One parallel trial had a 12‐week prerandomisation baseline period and a 12‐week treatment period of gabapentin 900 mg/day (111 participants) or gabapentin 1200 mg/day (52 participants) or placebo (109 participants) (Anhut 1994). Study medication was administered three times daily (TDS). Included participants had a minimum of six focal seizures within the baseline period and were aged 12 years or over. Women of childbearing potential on adequate contraception and participants with additional seizure types were also included in this study.
One multicentre parallel trial had three phases: six weeks of baseline period, nine weeks of double‐blind dose escalation phase and 12 weeks of double‐blind maintenance phase (French 2016). There were two arms: 242 participants randomised to gabapentin (300 mg/day, 600 mg/day, 1200 mg/day, 1500 mg/day and 1800 mg/day) and 242 participants randomised to active control (pregabalin 150 mg/day, 300 mg/day, 450 mg/day and 600 mg/day), but 482 participants (241 in gabapentin group and 241 in pregabalin group) received intended treatment. Participants were adults aged 18 to 80 years. During the nine‐week dose‐escalation phase, the minimum maintenance phase dose was gabapentin 1200 mg/day and pregabalin 300 mg/day TDS. During the 21‐week double‐blind phase, the median doses were gabapentin 1500 mg/day and pregabalin 450 mg/day.
The baseline period in one parallel trial was six weeks with a treatment period of 12 weeks (Appleton 1999). Gabapentin 600 mg/day to 1200 mg/day was administered TDS and was dependent on the weight of the participant. One hundred and twenty‐eight participants received placebo and 119 participants received gabapentin. Participants were children aged less than 12 years and with a minimum of four seizures during the baseline period.
One cross‐over trial was a placebo‐controlled study that did not have a prerandomisation baseline period; however, all participants reported at least four seizures per month for the previous three months (Leach 1997). There were four treatment arms (gabapentin 1200 mg/day, 1800 mg/day and 2400 mg/day, and placebo each administered on a TDS basis). All participants received all doses/placebo in a cross‐over design with a four‐week washout period between each treatment period. The study recruited 27 participants and analysed 23 participants.
One parallel trial had a baseline period of three months in which adults with focal epilepsy experienced four or more seizures a month (Sivenius 1991). Participants received either gabapentin 900 mg/day (16 participants), gabapentin 1200 mg/day (nine participants) or placebo (18 participants). Treatment medication was administered for three months.
One parallel trial had a three‐month baseline period where participants had at least one focal seizure per week (UK Gabapentin 1990). This study had a two‐week initiation phase of gabapentin 600 mg/day or placebo administered TDS, after which 61 participants began a 12‐week treatment period of gabapentin 1200 mg/day TDS and 66 participants received placebo.
One parallel trial recruited 306 adults, randomising 53 participants to gabapentin 600 mg/day, 101 participants to gabapentin 1200 mg/day, 54 participants to gabapentin 1800 mg/day and 98 participants to placebo, all administered TDS for 12 weeks (US Gabapentin 1993). The study implemented an initiation period of two to three days of either gabapentin 300 mg/day or 600 mg/day up to the required dose. The baseline period was three months and included people who had a minimum of four focal seizures per month.
One trial examining gabapentin versus placebo had a baseline period of 12 weeks and included people who had a minimum of eight focal seizures during baseline (Yamauchi 2006). Adults were randomised into one of three treatment arms: gabapentin 1200 mg/day (86 participants), gabapentin 1800 mg/day (41 participants) and placebo (82 participants), taken TDS over 12 weeks.
Two RCTs were gabapentin dose trials that had no placebo group (Fisher 2001; Tomovic 1999). Fisher 2001 compared slow initiation (gabapentin 300 mg on day one, gabapentin 600 mg on day two and then gabapentin 900 mg/day for five days) and rapid initiation of gabapentin (placebo for the first two days followed by gabapentin 900 mg/day for five days). Three hundred and sixty participants were in the slow initiation group and 360 participants were in the rapid initiation dose. There was no baseline period; participants were required to have been taking at least one AED for one month prior to the study and were considered to have inadequate seizure control as defined by the authors. Participants were aged 12 years or older. The trial period was seven days. Tomovic 1999 compared gabapentin 900 mg/day versus gabapentin 1200 mg/day administered TDS over 12 weeks. There were nine participants in each group. There was no formal baseline period; participants were considered to have unsatisfactorily controlled seizures while taking at least one first‐line AED for three months prior to the study, as defined by the authors.
One trial compared gabapentin to lamotrigine; it had an eight‐week baseline period (Lindberger 2000). All participants had tried no more than two AED monotherapy regimens and were on one AED at the time of study (this had to exclude phenytoin). The study required a minimum seizure frequency of four seizures during an eight‐week baseline period and two or more seizures during the last month. One hundred and two participants (aged 12 to 75 years) received either gabapentin or vigabatrin add‐on treatment. There was a flexible dosing regimen over the subsequent 24 weeks: gabapentin variable dose 1800 mg/day minimum, then 2400 mg/day and then a maximum of 3600 mg/day, increased every eight weeks as tolerated. The vigabatrin initial dose was 1000 mg/day, then 2000 mg/day, then 4000 mg/day, increased in the same manner (as tolerated by adverse effects) and increased if complete seizure freedom was not attained. The total trial period was 24 weeks; however, outcome measures were taken at eight weeks (awaiting clarification from author).
One trial compared gabapentin to lamotrigine in participants resistant to the maximum tolerated dose of carbamazepine monotherapy, with a seizure duration of two years or less (Sethi 2002). Twenty‐seven participants received gabapentin and 25 participants received lamotrigine and were aged 10 to 60 years. Baseline seizure frequency was at least four seizures despite treatment (unclear over what time frame). The baseline period was time of enrolment. The trial period was 12 weeks. Treatment was gabapentin 300 mg on day one, 300 mg twice daily on day two and thereafter an increment of 300 mg/day until seizures were controlled or toxic effects appeared. Lamotrigine was started at 50 mg/day for two weeks, then 50 mg twice daily for two weeks, then increased by 50 mg to 100 mg every two weeks until seizures were controlled or there were toxic effects.
For further information on each trial, see the Characteristics of included studies table.
Excluded studies
In this update, we excluded three additional studies as they were not RCTs (Btaiche 1995; Ramsay 1994; Schmidt 2001). There were 11 excluded studies overall.
The details of these studies are given in the Characteristics of excluded studies table.
Risk of bias in included studies
See Figure 2 and Figure 3 for a summary of the 'Risk of bias' in each included study. We rated included studies as having low, high or unclear risk of bias for six domains applicable to RCTs: randomisation method, allocation concealment, blinding methods, incomplete outcome data, selective outcome reporting and other sources of bias.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Six studies did not describe the method of allocation concealment (authors were contacted but we were unable to clarify) and therefore we rated these at unclear in terms of bias (Appleton 1999; Leach 1997; Lindberger 2000; Sethi 2002; Tomovic 1999; Yamauchi 2006). The other six studies achieved randomisation by generating random lists using random permuted blocks and by computer‐generated randomisation; and concealed allocation by dispensing sequentially numbered packages to each participant allocated treatment; we rated these studies at low risk of bias (Anhut 1994; Fisher 2001; French 2016; Sivenius 1991; UK Gabapentin 1990; US Gabapentin 1993).
Blinding
In five studies, the means of blinding was unclear (Appleton 1999; French 2016; Leach 1997; Sethi 2002; Tomovic 1999 ); there were no specific details regarding who was blinded (i.e. participants, study personnel or outcome assessors). The remaining seven studies achieved blinding by providing packaging and tablets that were identical in appearance for the gabapentin and placebo groups; and were at low risk of bias for this domain (Anhut 1994; Fisher 2001; Lindberger 2000; Sivenius 1991; UK Gabapentin 1990; US Gabapentin 1993; Yamauchi 2006).
Incomplete outcome data
We rated three studies at low risk of bias for attrition bias due to the ITT analyses undertaken by the study authors (Appleton 1999; French 2016; Lindberger 2000). Seven studies excluded participants from the study and analysis without providing reasons for this; and therefore we rated these at high in terms of bias (Anhut 1994; Fisher 2001; Leach 1997; Tomovic 1999; UK Gabapentin 1990; US Gabapentin 1993; Yamauchi 2006). Two studies analysed results on an 'as treated' basis, but did report attrition; and were at unclear risk of bias for this domain (Sethi 2002; Sivenius 1991).
Selective reporting
We rated all included studies at low risk of reporting bias as there was no suspicion of selective outcome reporting bias: all expected outcomes were reported in each of the publications.
Other potential sources of bias
Parke Davis, the manufacturers of gabapentin, sponsored nine trials (Anhut 1994; Appleton 1999; Fisher 2001; Leach 1997; Lindberger 2000; Sivenius 1991; UK Gabapentin 1990; US Gabapentin 1993; Yamauchi 2006). Pfizer Inc., the manufacturer of gabapentin and pregabalin, funded one trial (French 2016). Therefore, we rated these 10 trials at high risk of funding bias. Sethi 2002 and Tomovic 1999 were at low risk for this domain. There was no evidence of further bias in any of the included studies.
Effects of interventions
See: Table 1
Gabapentin versus placebo
Seven trials compared gabapentin to placebo (Anhut 1994; Appleton 1999; Leach 1997; Sivenius 1991; UK Gabapentin 1990; US Gabapentin 1993; Yamauchi 2006).
Reduction in seizure frequency of 50% or more
Given that all participants had drug‐resistant focal epilepsy, it seemed reasonable to combine results from the paediatric and adult studies for an overall estimate (irrespective of dose). Data from the paediatric study could not be included in dose regression models, as participants were not randomised to a specific daily dose (Appleton 1999). Seven trials provided data for this outcome (Anhut 1994; Appleton 1999; Leach 1997; Sivenius 1991; UK Gabapentin 1990; US Gabapentin 1993; Yamauchi 2006). See Figure 4 for forest plots. One trial was not suitable for inclusion in the meta‐analysis due to the cross‐over design but was discussed in narrative form below (Leach 1997).
4.

Forest plot of comparison: 1 Gabapentin versus placebo, outcome: 1.1 Reduction in seizure frequency 50% or greater.
Intention‐to‐treat analysis
An analysis pooling data from six studies showed no evidence of heterogeneity (Chi2 = 3.90, P = 0.56, I2 = 0%). The overall RR for reduction in seizure frequency of 50% or more was 1.89 (95% CI 1.40 to 2.55; 1206 participants; moderate‐certainty evidence; Analysis 1.1).
1.1. Analysis.

Comparison 1: Gabapentin versus placebo, Outcome 1: Reduction in seizure frequency ≥ 50%
Best‐case and worst‐case scenarios
Chi2 tests for heterogeneity for a response to gabapentin indicated no significant heterogeneity (best‐case: P = 0.08; worst‐case: P = 0.95). The overall RRs for 50% responders across all studies were 2.52 (95% CI 1.89 to 3.37) for best‐case scenarios and 1.35 (95% CI 1.04 to 1.76) for worst‐case scenarios (Analysis 1.1).
For all three analyses, the results suggested a significant treatment effect. However, there was a considerable difference between estimates.
Dose‐response regression
Intention‐to‐treat analysis
A linear dose‐response model gave a good summary (for the five adult trials) of the log odds of 50% response rate. After adjusting for dose, there was no difference in estimated dose‐response between studies. The log odds of response increase by 0.19 (standard error of the mean (SEM) 0.045) for a 300 mg increase in daily gabapentin dose. This is about a 20% increase in the odds of response with a 300 mg increase in gabapentin dose. The reduction in deviance due to dose was 19.1 on one degree of freedom, and the residual deviance was 10.9 on 13 degrees of freedom. The trial in children was not included, as the doses were prescribed to achieve particular levels of milligrams per kilogram per day.
The results were summarised in Table 2; Table 3; Table 4:
1. Estimated percentage responders and percentage difference compared to placebo per gabapentin dose, intention‐to‐treat.
| Dose | % Responders (95% CI) | % Difference (95% CI) compared to placebo |
| 0 mg (placebo) | 9.7 (7.2 to 12.9) | — |
| 600 mg | 13.6 (11.4 to 16.1) | 3.9 (1.6 to 6.2) |
| 900 mg | 16.0 (13.7 to 18.6) | 6.3 (2.8 to 9.8) |
| 1200 mg | 18.7 (15.8 to 22.1) | 9.0 (4.4 to 13.7) |
| 1800 mg | 25.3 (19.3 to 32.3) | 15.5 (8.5 to 22.5) |
CI: confidence interval.
2. Estimated percentage responders and percentage difference compared to placebo per gabapentin dose, best‐case.
| Dose | Responders % (95% CI) | % Difference (95% CI) compared to placebo |
| 0 mg (placebo) | 10.9 (8.1 to 14.5) | — |
| 600 mg | 17.2 (14.6 to 20.2) | 6.3 (3.9 to 8.8) |
| 900 mg | 21.4 (18.5 to 24.6) | 10.5 (6.8 to 14.2) |
| 1200 mg | 26.2 (22.4 to 30.4) | 15.3 (10.3 to 20.0) |
| 1800 mg | 37.6 (30.0 to 46.0) | 26.7 (19.3 to 34.2) |
CI: confidence interval.
3. Estimated percentage responders and percentage difference compared to placebo per gabapentin dose, worst‐case.
| Dose | Responders % (95% CI) | % Difference (95% CI) compared to placebo |
| 0 mg (placebo) | 13.8 (10.4 to 18.2) | — |
| 600 mg | 16.4 (13.8 to 19.2) | 2.5 (–0.3 to 5.3) |
| 900 mg | 17.8 (15.3 to 20.5) | 3.9 (–0.3 to 8.1) |
| 1200 mg | 19.2 (16.2 to 22.7) | 5.4 (–0.2 to 11.0) |
| 1800 mg | 22.5 (17.1 to 29.0) | 8.6 (0.3 to 17.0) |
CI: confidence interval.
the estimated percentage of participants responding to each gabapentin dose and the percentage difference in participants responding to each gabapentin dose compared to placebo with 95% CI (ITT) (Table 2);
the estimated percentage of participants responding to each gabapentin dose and the percentage difference in participants responding to each gabapentin dose compared to placebo with 95% CI (best‐case) (Table 3);
the estimated percentage of participants responding to each gabapentin dose and the percentage difference in participants responding to each gabapentin dose compared to placebo with 95% CI (worst‐case) (Table 4).
In the best‐case analysis, there was about a 30% increase in the odds of response with a 300 mg increase in gabapentin dose and in the worst‐case analysis, there was about a 10% increase in the odds of response with a 300 mg increase in gabapentin dose.
All three analyses (ITT, best‐case and worst‐case) showed a significant increase in therapeutic effect with increasing dose. However, there was a striking difference in the proportion of responders estimated.
Seizure freedom
Only two trials comparing gabapentin to placebo reported seizure freedom data (Appleton 1999; Yamauchi 2006).
Yamauchi 2006 reported no participants attaining seizure freedom, whereas Appleton 1999 reported 3/119 participants receiving gabapentin as seizure‐free compared to 1/128 participants receiving placebo. Due to the very small numbers of participants achieving seizure freedom in the two trials, the data were not combined in meta‐analyses.
Treatment withdrawal
A Chi2 test of heterogeneity suggested no significant statistical heterogeneity (Chi2 = 4.13, df = 4, P = 0.53, I2 = 0%). The overall RR for withdrawal for any reason was 1.05 (95% CI 0.74 to 1.49; 6 trials, 1206 participants; moderate‐certainty evidence; Analysis 1.2), hence there was insufficient evidence to conclude that people were more likely to withdraw from gabapentin than placebo, but there could have been a substantial withdrawal rate.
1.2. Analysis.

Comparison 1: Gabapentin versus placebo, Outcome 2: Treatment withdrawal
Adverse effects
In addition to reports of ataxia, dizziness, fatigue, nausea and somnolence, headache was among the six most common adverse effects included in our analysis. There were significant differences between gabapentin and placebo for the following adverse effects (see Analysis 1.3): ataxia (RR 2.01, 99% CI 0.98 to 4.11; 3 RCTs; 787 participants; low‐certainty evidence); dizziness (RR 2.43, 99% CI 1.44 to 4.12; 6 RCTs; 1206 participants; moderate‐certainty evidence); fatigue (RR 1.95, 99% CI 0.99 to 3.82; 5 RCTs; 1161 participants; low‐certainty evidence) and somnolence (RR 1.93, 99% CI 1.22 to 3.06; 6 RCTs, 1206 participants; moderate‐certainty evidence). There was no evidence of a difference for headache (RR 0.79, 99% CI 0.46 to 1.35; 6 RCTs, 1206 participants; moderate‐certainty evidence) or nausea (RR 0.95, 99% CI 0.52 to 1.73; 4 RCTs, 1034 participants; moderate‐certainty evidence).
1.3. Analysis.

Comparison 1: Gabapentin versus placebo, Outcome 3: Adverse effects
Cross‐over trial, not included in meta‐analysis
One trial was not suitable for inclusion in the meta‐analysis due to the cross‐over design and due to a lack of a prerandomisation baseline period and was discussed in narrative form (Leach 1997).
Reduction in seizure frequency of 50% or more and seizure freedom
The cross‐over trial evaluated 23/27 participants (although there were six withdrawals, two participants withdrew sufficiently late in the study to provide analysable data). Two participants achieved total seizure control throughout the active treatment phase and none in the placebo phase. For simple focal seizures, two participants showed 'in excess' of 50% reduction in seizure frequency. The median monthly frequency for simple focal seizures was not significantly reduced in the treatment group (P = 0.80). The study reported complex focal seizures with secondary generalisation separately; 5/17 participants had in excess of 50% reduction in seizure frequency (non‐significant).
Treatment withdrawal
Six participants withdrew, five due to adverse effects (four while receiving placebo, one while receiving gabapentin) and one of whom withdrew consent to participate after the second visit. One participant did not provide complete data for seizure frequency and was withdrawn from the study.
Adverse effects
Nineteen (79%) participants reported 47 adverse effects with gabapentin and 15 participants (63%) reported 30 adverse effects with placebo. There was a statistically significant difference (P = 0.006) with gabapentin 2400 mg/day. The types of adverse effects were not reported.
Dose comparison trials with no placebo group
Two studies compared two dose regimens with no placebo (Fisher 2001; Tomovic 1999). For one study, we sent a data extraction form to a translator (Tomovic 1999). Our understanding is that the study combined outcomes for the two treatment arms, thus a comparison between the two treatment groups could not be made. Another study only measured adverse effects at day two and day seven of a slow initiation regimen and a rapid initiation regimen; therefore, this is presented narratively below (Fisher 2001).
Reduction in seizure frequency of 50% or more
In the Tomovic 1999 study, 13/18 (72.2%) participants experienced 50% or greater reduction in seizures (two of whom achieved a 100% reduction). Three participants had a 26% to 49% reduction in seizure frequency. Two participants had worse seizure control. Fisher 2001 did not measure reduction in seizure frequency of 50% or more.
Seizure freedom
In the Tomovic 1999 study, 2/18 (11.1%) participants were seizure‐free during the treatment period; however, it was not reported in which dose group this was achieved. Fisher 2001 did not measure seizure freedom.
Treatment withdrawals
Tomovic 1999 did not report any treatment withdrawals. Fisher 2001 reported only participants who had full exposure to the study medication during the whole period of assessment (i.e. details of withdrawals were not provided).
Adverse effects
Tomovic 1999 reported adverse effects in three participants, two of whom had dizziness and one had excessive sleepiness (they were excluded from the study, therefore, not included in the total number of participants). They also noted bulimia, tremor, diplopia, headache, nausea and ataxia. Fisher 2001 reported adverse effects on day three and day seven of a slow and rapid initiation regimen of gabapentin. See Table 5 for the proportion of people with adverse effects with percentages. There were no statistically significant differences between the two dose regimens, apart from more dizziness in the rapid initiation group compared to the slow initiation group at day three only. In addition, Tomovic 1999 reported 24‐hour electroencephalogram (EEG) recordings pre‐ and postintervention and revealed a reduction in total epileptiform discharges from 229.87 to 167.13.
4. Adverse effects Fisher 2001.
| Adverse effect | Slow initiation (day 2) | % | Rapid initiation (day 2) | % | Slow initiation (day 7) | % | Rapid initiation (day 7) | % |
| Fatigue | 9/280 | 3.2 | 12/294 | 4.1 | 19/274 | 6.9 | 22/294 | 7.5 |
| Dizziness | 18/280 | 6.4 | 31/294 | 10.5 | 45/276 | 16.3 | 59/293 | 19.1 |
| Somnolence | 13/280 | 4.6 | 16/294 | 5.4 | 27/275 | 9.8 | 31/293 | 10.6 |
| Ataxia | 2/280 | 0.7 | 4/294 | 1.4 | 9/275 | 3.3 | 9/294 | 3.1 |
Gabapentin versus vigabatrin
One study compared gabapentin versus vigabatrin (Lindberger 2000).
Reduction in seizure frequency of 50% or more and seizure freedom
The study noted a reduction in seizure frequency of 50% or more and seizure freedom in 27/50 (54%) participants in the gabapentin group and 34/52 (56%) participants in the vigabatrin group (on an ITT basis); the 95% CIs were wide and this was not deemed statistically significant. The proportion seizure‐free without adverse effects was 13/50 (26%) participants in the gabapentin group and 18/52 (35%) participants in the vigabatrin group. This was not statistically significant. The study measured an extra variable of 'improvement rate' (proportion of participants with 50% or greater seizure reduction without adverse effects), which was 24/50 (48%) participants in the gabapentin group and 29/52 (56%) participants in the vigabatrin group. Thirteen out of 50 participants were seizure‐free in the gabapentin group compared to 18/52 participants in the vigabatrin group.
Treatment withdrawals
There were 14 withdrawals from the study as a result of adverse effects, seven in each group. In the gabapentin group, they were status epilepticus, psychiatric problems, epigastric pain, diplopia, vertigo and dizziness (three participants); in the vigabatrin group, they were depression, generalised seizure, rash, numbness and dizziness (three participants).
Adverse effects
In the gabapentin group, three participants experienced serious adverse effects which were status epilepticus, pyelonephritis and psychiatric problems. In the vigabatrin group, four participants had serious adverse effects, which were agitation, depression, weight gain, mononucleosis and a secondary generalised seizure. Thirty‐eight (76%) participants in the gabapentin group and 45 (86.5%) participants in the vigabatrin group experienced adverse effects of any type. The five most common adverse effects were similar in both groups (tiredness, dizziness, respiratory infection, headache and diarrhoea). Specific proportions of individual adverse effects were not provided.
Gabapentin versus lamotrigine
One trial compared gabapentin versus lamotrigine (Sethi 2002).
Reduction in seizure frequency of 50% or more and seizure freedom
There was a 50% or greater reduction in seizure frequency by 77.7% of participants in the gabapentin group and 92% of participants in the lamotrigine group (ITT analysis). There was complete seizure control in 8/27 (29.6%) participants in the gabapentin group; this was not specified in the lamotrigine group.
Treatment withdrawals
Sethi 2002 did not report any treatment withdrawals.
Adverse effects
Twenty‐two out of 27 (81.5%) participants in the gabapentin group and 18/25 (72%) participants in the lamotrigine group reported adverse effects. The most common adverse effects were neurotoxic: dizziness (gabapentin: 22.2%; lamotrigine: 28%), diplopia (gabapentin: 11.11%; lamotrigine: 24%), weakness (gabapentin: 14.8%; lamotrigine: 24%), headache (gabapentin: 25.9%; lamotrigine: 20%), drowsiness (gabapentin: 14.8%; lamotrigine: 12%), tiredness (gabapentin: 14.8%; lamotrigine: 4%), amnesia (gabapentin: 11.11%; lamotrigine: 12%), tingling sensation (gabapentin: 11.11%; lamotrigine: 0%) and anorexia (gabapentin: 11.11%; lamotrigine: 8%).
There were no serious adverse effects in the gabapentin group. In the lamotrigine group, there were two serious adverse effects: Steven‐Johnson's syndrome and anxiety neurosis (corresponding with an increase in seizure frequency). There was an increase in the number of seizures in one participant receiving gabapentin 2400 mg/day. In the gabapentin group, there was a change of seizure type from focal seizures to myoclonic jerks or atypical seizures in five participants during treatment. In the lamotrigine group, seizure type changed to atypical absence (two participants) and pseudoseizures (two participants).
Additionally, the benefit of gabapentin was more pronounced in participants with simple focal seizures with secondary generalisation than in participants with simple and complex focal seizures without secondary generalisation, whereas all subtypes of epilepsy responded similarly in the lamotrigine group.
Gabapentin versus pregabalin
One study compared gabapentin versus pregabalin (French 2016).
Reduction in seizure frequency of 50% or more and seizure freedom
There was a reduction in seizure frequency of 50% or more in 140/240 (58.3%) participants in the gabapentin group and 134/238 (56.3%) participants in the pregabalin groups (on an ITT basis); the 95% CIs were wide and this was not deemed statistically significant. The proportion of seizure‐free participants was 62/182 (34.1%) in the gabapentin group and 58/189 (30.7%) in the pregabalin group; these were not statistically significant. The study measured an extra variable of 'improvement rate' (proportion of participants with 75% or greater seizure reduction) and was 82/240 (34.2%) participants in the gabapentin group and 80/238 (33.6%) participants in the pregabalin group.
Treatment withdrawals
There were 123 withdrawals for any reason from the study, and 31 due to adverse effects (16 in the gabapentin group and 15 in the pregabalin group). In the gabapentin group, the adverse effects were status epilepticus, psychiatric problems, epigastric pain, diplopia, vertigo and dizziness (three participants); in the vigabatrin group, they were depression, generalised seizure, rash, numbness and dizziness (three participants).
Adverse effects
In the gabapentin group, 129/241 (53.5%) participants reported adverse effects and, in the pregabalin group, 142/241 (58.9%) participants reported adverse effects. Both groups had six (2.5%) participants with serious adverse effects. The five most common adverse effects were similar in both groups (somnolence, dizziness, headache, increased weight and dry mouth).
Discussion
Summary of main results
The results of the overall efficacy analysis showed that gabapentin reduced seizure frequency when used as an add‐on AED in people with drug‐resistant focal epilepsy. Compared to placebo, gabapentin was almost twice as likely to reduce seizures by 50% or more; however, there was considerable discrepancy between the results of the ITT and best‐case and worst‐case analyses, hence the ITT analyses need to be interpreted with caution (see Implications for research). The dose‐response regression analysis indicated increasing efficacy with increasing dose. Results suggested that the therapeutic effect of gabapentin 600 mg/day, although statistically significant, was small and 900 mg/day would seem a better initial dose. In addition, there was no apparent 'plateau' of therapeutic effects at the doses tested and it may well be that optimal doses of gabapentin have not been tested.
This was also reflected to a much greater extent by the studies described in narrative form. The Tomovic 1999 study comparing gabapentin doses reported 72.2% of the 18 participants evaluated as having a 50% or greater reduction in seizure frequency outcome (compared to 16% to 22% taken from the studies comparing gabapentin to placebo combined in meta‐analysis) even though the demographics and treatment doses were comparable. Similarly, the active control trials reported a 54% response rate in Lindberger 2000 (compared to vigabatrin), 58% response rate in French 2016 (compared to pregabalin) and 77% response rate in Sethi 2002 (compared to lamotrigine). This could potentially be due to two key differences in methodology: the definition of 'drug‐resistant' focal epilepsy and to the dosing regimens. Lindberger 2000 defined drug resistance as failure to respond to no more than two AED monotherapy regimens and gabapentin was always added to monotherapy. Sethi 2002 only recruited people resistant to carbamazepine monotherapy and 88% of participants in the Tomovic 1999 study were taking one other AED only. As the remaining studies used people with drug‐resistant focal seizures who were established on one or two AEDs and stable doses (apart from Appleton 1999 who allowed three AEDs), the populations are likely to have a less drug‐resistant epilepsy. In addition, Lindberger 2000 and Sethi 2002 used a flexible dosing regimen, allowing doses of gabapentin to be increased as tolerated. This high flexibility made dose adjustments possible in response to a lack of seizure control, with doses of gabapentin 3600 mg/day allowed. This may be reflective of the increased efficacy of gabapentin at higher doses yet this flexible dosing method did result in complexity when interpreting the results as the final doses achieved to maintain seizure control have not been specified. At the other end of the spectrum in the Leach 1997 study, despite allowing doses of gabapentin 2400 mg, only 2/23 (8.7%) participants achieved 50% or greater focal seizure control; this may be reflective of the small sample size and the cross‐over design. All participants received all doses (1200 mg, 1800 mg and 2400 mg) with a washout period of four weeks between doses; this dosing pattern may have influenced the efficacy of gabapentin, which may have resulted in period and carry‐over effects.
Results for the outcome 'Treatment withdrawal' suggested that gabapentin was well tolerated, as there was no significant difference between gabapentin and placebo. However, the efficacy results suggested that optimal doses of gabapentin may not have been tested and it may well be that higher doses of gabapentin were less well tolerated. With respect to adverse effects, dizziness, fatigue and somnolence were significantly more likely to occur in the gabapentin‐treated group. There were insufficient data available for this review to delineate the precise adverse effect profile of this drug.
Overall completeness and applicability of evidence
The studies reviewed were all short duration and no conclusions could be drawn regarding the long‐term efficacy of gabapentin. One trial recruited children only (Appleton 1999), and the estimate for seizure reduction was low in that study. Caution is required when extrapolating the results of this trial to adults.
In terms of seizure subtypes, Sethi 2002 reported gabapentin's more pronounced effects on simple focal seizures and secondary generalised as opposed to complex focal seizures. This is contrary to the US Gabapentin 1993 study, which observed gabapentin to be more efficacious in complex focal seizures. This review focused on the use of gabapentin in drug‐resistant focal epilepsy and the results could not be generalised to add‐on treatment in people with generalised epilepsy. Likewise, no inference can be made about the efficacy and tolerability of gabapentin when used as monotherapy.
The results of this review indicated that gabapentin was an effective add‐on treatment. We found three head‐to‐head trials with no study finding a significant difference between gabapentin and the alternative AED (pregabalin (French 2016), vigabatrin (Lindberger 2000), and lamotrigine (Sethi 2002)). As clinicians are faced with an ever increasing number of AEDs to choose from, more head‐to‐head trials are required to provide the evidence that is needed to enable clinicians to make an evidence‐based choice between AEDs.
It remains difficult to predict the differences between a rapid and slow initiation of gabapentin, as the Fisher 2001 study only observed the effects of rapid initiation on the first day of starting the maximum dose and four days later. However, they did contact participants for the subsequent two weeks to report any serious outcomes. These were not documented in the report; therefore, it is difficult to extrapolate data beyond this period.
Quality of the evidence
Seven out of the 12 studies used adequate methods of concealment of randomisation. All trials were double‐blind; however, often little information was reported as to how personnel/outcome assessors were blinded. For the studies included in the meta‐analysis, apart from Yamauchi 2006, published reports referred to their analyses as being ITT, with 222/1688 participants recruited excluded from analyses. Reported analyses would perhaps be better called 'exploratory,' as participants who had treatment withdrawn during the treatment period and did not meet the original trial inclusion criteria were excluded from the reported analyses, despite completing the treatment period with adequate seizure data.
Additional data, supplied by Parke Davis, revealed that 38 participants did not complete the treatment phase and nine had inadequate seizure data recorded, hence the percentage reduction in seizure frequency could not be calculated for 47 of these participants. Yamauchi 2006 stated that 19 participants were not included in the study and provided reasons for this. The French 2016 study stated that 123 participants withdrew for any reason. Similarly, there was a high risk of attrition bias in the remaining studies, which we discussed in narrative form (Fisher 2001; Leach 1997; Tomovic 1999), apart from Lindberger 2000 for which analysis was completely ITT. Sethi 2002 did not give any information related to dropouts and, therefore, the risk was uncertain. Selective outcome reporting bias was unclear in Leach 1997, Lindberger 2000, Sethi 2002, and Tomovic 1999, as they mentioned 'seizure activity recorded' without details of the methodology.
Overall, the certainty of the evidence for the outcomes was low to moderate due to potential attrition bias resulting from missing outcome data and imprecise results with wide CIs. Research is needed into the effects of the long‐term use of gabapentin and to compare gabapentin with other add‐on drugs.
Potential biases in the review process
There were discrepancies between study designs that may be reflected in the variability of the results.
Parke Davis sponsored most of trials included in this review, apart from Sethi 2002 and Tomovic 1999. Pfizer Inc., the manufacturer of gabapentin and pregabalin, funded one trial (French 2016).
Agreements and disagreements with other studies or reviews
We found no reviews or published information on the use of gabapentin as add‐on therapy for drug‐resistant focal epilepsy.
Authors' conclusions
Implications for practice.
In people with drug‐resistant focal epilepsy, gabapentin has efficacy as an add‐on treatment. Moderate‐certainty evidence for the outcomes from this review suggests that a dose of 1800 mg/day will reduce seizure frequency by at least 50% in 25.3% of people (95% confidence interval 19.3% to 32.3%). Although our results suggest that gabapentin 600 mg has a statistically significant effect on seizure frequency, that effect is small and 900 mg/day would seem a more reasonable initial dose. Regression analyses show no plateau of therapeutic effect and it is likely that optimal doses need to be tested in a more standardised manner and final doses provided so that such results can be included in meta‐analyses in the future. Doses of up to 2400 mg/day are currently recommended in the British National Formulary.
Low‐ to moderate‐certainty evidence suggests that dizziness, fatigue and somnolence occurs significantly more often with gabapentin than placebo, although gabapentin is generally well tolerated.
Implications for research.
The conduct of future 'add‐on' trials
The striking differences between the intention‐to‐treat, worst‐case and best‐case analyses for 50% responder rates has important implications for the conduct of further 'add‐on' studies. For the intention‐to‐treat analysis in this review, all participants lost to follow‐up or excluded from analyses due to inadequate seizure recording were assumed to be 'non‐responders.' The best‐case and worst‐case analyses, although representing the extreme, test the effect of making that assumption. When large discrepancies are found, as in this case, the accuracy of individual trials and hence this review is challenged.
The main problem is that participants having trial treatment withdrawn are no longer followed up. This provides a dataset that allows an explanatory 'on treatment analysis,' but precludes a robust intention‐to‐treat analysis. To minimise this problem, every attempt must be made to follow participants up, even if trial treatment has been withdrawn. This provides the maximum dataset from which analyses other than intention‐to‐treat may be undertaken.
Further evaluation of gabapentin as an antiepileptic drug
To further evaluate the place of gabapentin in the armamentarium of available antiepileptic drugs, further studies are required that address the following:
the efficacy and tolerability of add‐on doses of gabapentin higher than 1800 mg/day in people with drug‐resistant focal epilepsy, in clearly specified doses and a clarification as to maximum doses achieved when flexible regimens are employed;
the long‐term efficacy and tolerability of add‐on gabapentin beyond three months;
how gabapentin compares with other add‐on treatments in drug‐resistant focal epilepsy;
the role of gabapentin in childhood epilepsies;
how gabapentin compares with other standard antiepileptic drugs, such as sodium valproate in generalised epilepsy, as monotherapy.
What's new
| Date | Event | Description |
|---|---|---|
| 11 August 2020 | New search has been performed | Searches updated 11 August 2020; no new studies were identified. |
| 11 August 2020 | New citation required but conclusions have not changed | Conclusions remain unchanged. |
History
Protocol first published: Issue 1, 1999 Review first published: Issue 1, 1999
| Date | Event | Description |
|---|---|---|
| 20 March 2018 | New search has been performed | Searches updated 20 March 2018; one new study has been included. The term 'partial' has been replaced by 'focal', in accordance with the most recent classification of epilepsies of the International League Against Epilepsy (Scheffer 2017). |
| 20 March 2018 | New citation required but conclusions have not changed | Conclusions are unchanged. |
| 14 May 2013 | New search has been performed | Searches updated 14 May 2013. |
| 14 May 2013 | New citation required but conclusions have not changed | Six new studies included. Conclusions remain unchanged. |
| 10 September 2008 | Amended | Converted to new review format. |
| 1 July 2007 | New search has been performed | We re‐ran our searches on 1 July 2007. One potential new study has been identified ‐ this has been added to the 'Studies awaiting classification' section and will be assessed for inclusion at a later date. |
Acknowledgements
We would like to acknowledge Zakaria Kadir and David W Chadwick for contributing to the original review, as well as Miloš Stojadinović for providing the translation of the Serbian paper.
We would like to acknowledge Alexandra Cunliffe and Jennifer Weston for their contributions to previous updates of this review.
Appendices
Appendix 1. Cochrane Register of Studies search strategy
1. MESH DESCRIPTOR Gabapentin EXPLODE ALL AND CENTRAL:TARGET
2. (gabapentin or neurontin):AB,KW,KY,MC,MH,TI AND CENTRAL:TARGET
3. #1 OR #2
4. MESH DESCRIPTOR Epilepsies, Partial EXPLODE ALL AND CENTRAL:TARGET
5. ((partial or focal) and (seizure* or epilep*)):AB,KW,KY,MC,MH,TI AND CENTRAL:TARGET
6. #4 OR #5 AND CENTRAL:TARGET
7. #3 AND #6
8. (monotherap* NOT (adjunct* OR "add‐on" OR "add on" OR adjuvant* OR combination* OR polytherap*)):TI AND CENTRAL:TARGET
9. #7 NOT #8
10. >03/11/2016:CRSINCENTRAL AND CENTRAL:TARGET
11. #9 AND #10
Appendix 2. MEDLINE (Ovid) search strategy
This strategy includes a modification of the Cochrane Highly Sensitive Search Strategy for identifying randomised trials (Lefebvre 2020).
1. exp Gabapentin/
2. (gabapentin or neurontin).tw.
3. 1 or 2
4. exp Epilepsies, Partial/
5. ((partial or focal) and (seizure$ or epilep$)).tw.
6. 4 or 5
7. exp controlled clinical trial/ or (randomi?ed or placebo or randomly).ab.
8. clinical trials as topic.sh.
9. trial.ti.
10. 7 or 8 or 9
11. exp animals/ not humans.sh.
12. 10 not 11
13. 3 and 6 and 12
14. (monotherap$ not (adjunct$ or "add‐on" or "add on" or adjuvant$ or combination$ or polytherap$)).ti.
15. 13 not 14
16. remove duplicates from 15
17. limit 16 to ed=20161103‐20200811
18. 16 not (1$ or 2$).ed.
19. 18 and (2016$ or 2017$ or 2018$ or 2019$ or 2020$).dt.
20. 17 or 19
Data and analyses
Comparison 1. Gabapentin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Reduction in seizure frequency ≥ 50% | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1.1 Primary analysis | 6 | 1206 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.89 [1.40, 2.55] |
| 1.1.2 Sensitivity (best‐case) | 6 | 1206 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.52 [1.89, 3.37] |
| 1.1.3 Sensitivity (worst‐case) | 6 | 1206 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [1.04, 1.76] |
| 1.2 Treatment withdrawal | 6 | 1206 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.74, 1.49] |
| 1.3 Adverse effects | 6 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.3.1 Ataxia | 3 | 787 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.01 [0.98, 4.11] |
| 1.3.2 Dizziness | 6 | 1206 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.43 [1.44, 4.12] |
| 1.3.3 Fatigue | 5 | 1161 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.95 [0.99, 3.82] |
| 1.3.4 Headache | 6 | 1206 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.79 [0.46, 1.35] |
| 1.3.5 Nausea | 4 | 1034 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.95 [0.52, 1.73] |
| 1.3.6 Somnolence | 6 | 1206 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.93 [1.22, 3.06] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Anhut 1994.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study (conducted at 24 centres in Europe, Canada, South Africa and Australia) 3 treatment arms: 1 placebo and 2 gabapentin Prospective prerandomisation baseline period: 12 weeks Treatment period: 12 weeks |
|
| Participants | Adults with drug‐resistant focal epilepsy Total randomised: 272 109 to placebo; 111 to gabapentin 900 mg; 52 to gabapentin 1200 mg 56% men Age range: 12–67 years Other AEDs: ≤ 2 Median baseline seizure frequency per 28 days: 10.2 (range 0.5–634.3) |
|
| Interventions | Gabapentin 900 mg/day Gabapentin 1200 mg/day Placebo All tablets and packaging were identical in appearance. |
|
| Outcomes | Proportion with a 50% reduction in seizure frequency Response ratio (= (T – B)/(T + B) where T = number of seizures during the treatment period and B = number of seizures in the baseline period) Adverse effects |
|
| Notes | 27 participants excluded from published analyses: 10 from placebo group; 15 from gabapentin 900 mg group; 2 from gabapentin 1200 mg group Additional unpublished data allowed the inclusion of participants excluded despite completing the treatment phase with adequate seizure data. The following participants contributed to the best‐case and worst‐case sensitivity analyses in this review: placebo: 7; gabapentin 900 mg: 9; gabapentin 1200 mg: 2. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random permuted blocks to generate sequence for randomisation. |
| Allocation concealment (selection bias) | Low risk | Allocated sequentially, sealed, numbered packages. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Tablets and packaging identical in appearance. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 'As treated' analysis. Disproportionate numbers excluded across groups: 13 in placebo: 17 in gabapentin 900 mg: 2 in gabapentin 1200 mg, some excluded despite completing treatment phase. Exclusions not included in published analyses. |
| Selective reporting (reporting bias) | Low risk | Seizure diary for all groups, same outcomes. Published reports include all prespecified expected outcomes. |
| Other bias | High risk | Trial sponsored by Parke Davis. Study appeared free of other sources of bias. |
Appleton 1999.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study (conducted at 54 centres in Europe, South Africa and the US) Prospective prerandomisation baseline period: 6 weeks Treatment period: 12 weeks |
|
| Participants | Children with drug‐resistant focal seizures (15–16% had generalised seizures also) Total randomised 247 128 to placebo; 119 to gabapentin 54% boys Age range: 3–12 years Other AEDs: ≤ 3 Baseline seizure frequency per 28 days: median 26.7 (range 1.3–2893) |
|
| Interventions | Gabapentin 600–1800 mg/day (equivalent to 23.2–35.3 mg/kg/day) Placebo |
|
| Outcomes | Proportion with a 50% reduction in seizure frequency Response ratio Adverse effects |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not specified. |
| Allocation concealment (selection bias) | Unclear risk | No details provided. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Study mentioned double‐blinding; no details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All evaluated on an ITT basis. |
| Selective reporting (reporting bias) | Low risk | Published reports included all prespecified, expected outcomes. The parent/guardian and physician global assessment of participant seizure frequency and well‐being. |
| Other bias | High risk | Trial sponsored by Parke Davis. Study appeared free of other sources of bias. |
Fisher 2001.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group multicentre study in the US No formal baseline period Treatment period: 7 days |
|
| Participants | Aged ≥ 12 years (range 12–82 years) 720 participants randomised. Initially 360 per dose initiation regimen. Finally, 280 slow initiation regimen and 294 rapid initiation, after withdrawals and exclusions for not fulfilling preprotocol criteria. All participants with a recent history of focal seizures, with or without secondary generalisation with either inadequate seizure control on 1 or 2 anticonvulsants or had been judged to be unable to tolerate therapeutic dosages of these drugs (reaching maximum tolerated dose of ≥ 1 anticonvulsant). 280 slow initiation regimen, 294 rapid initiation regimen Slow initiation; 44.6% male, rapid initiation: 44.2% male |
|
| Interventions | Slow initiation: 300 mg day 1, 600 mg day 2, then 900 mg/day Rapid initiation: 2‐day placebo lead‐in followed by 900 mg/day Total evaluated treatment period: 7 days |
|
| Outcomes | Fatigue Dizziness Somnolence Ataxia |
|
| Notes | Study did not have a baseline period and only measured adverse outcomes over a 7‐day period (day 3 (equivalent to 3rd day of active study medication for slow initiation group and first day for rapid initiation group) and day 7)), therefore unable to include in meta‐analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation schedule that assigned each participant number to either the slow group or the rapid group in a 1‐to‐1 manner. |
| Allocation concealment (selection bias) | Low risk | Number‐specific blister packs. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Matching placebo, all participants had a 2‐day lead‐in phase that was unknown to investigator and participant. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol analysis stated to include participants who met the criteria for evaluation (not ITT analysis). 781 enrolled, only 574 analysed for 3 reasons: inadequate methods, inadequate reasons and reasons for withdrawal. |
| Selective reporting (reporting bias) | Low risk | Appeared all expected and prespecified outcomes were reported. |
| Other bias | High risk | Trial sponsored by Parke Davis. Study appeared free of other sources of bias. |
French 2016.
| Study characteristics | ||
| Methods | Randomised, flexible‐dose, double‐blind, parallel‐group study Multicentre study: 56 centres (Eastern and Western Europe, Asia South and Central America) 2 active treatment arms: 1 gabapentin and 1 pregabalin 3 main phases: 6 weeks of baseline (screening), 9 weeks of double‐blind dose escalation (titration) and 12 weeks of double‐blind maintenance phase (21‐week treatment phase) |
|
| Participants | Adults with drug‐resistant focal epilepsy Inclusion criteria: aged 18–80 years; diagnosis of epilepsy with focal‐onset seizures, inadequately controlled with ≤ 2 to ≥ 5 prior AEDs, receiving 1 or 2 standard AEDs (other than gabapentin or pregabalin), with a minimum of 4 focal‐onset seizures during the 6‐week baseline phase with no 28‐day focal‐onset seizure‐free period 561 participants screened and 484 randomised: 242 to gabapentin and 242 to pregabalin Number of people who received intended treatment: 241 gabapentin and 241 pregabalin Number of people who completed the maintenance phase of the study: 172 gabapentin (69 discontinued treatment) and 187 pregabalin (54 discontinued treatment). Age (mean): gabapentin 35.3 (SD 12.9) years; pregabalin 34.9 (SD 13.0) years Age at epilepsy diagnosis (mean): gabapentin 19.9 years; pregabalin 19.8 years Time since diagnosis of epilepsy: gabapentin 15.8 years; pregabalin 15.6 years Sex of participants: gabapentin 130 men and 111 women; pregabalin 127 men and 114 women |
|
| Interventions | Intervention (241 participants): gabapentin (300 mg/day, 600 mg/day, 1200 mg/day, 1500 mg/day, 1800 mg/day) Active control (241 participants): pregabalin (150 mg/day, 300 mg/day, 450 mg/day, 600 mg/day) During the 9‐week dose‐escalation phase, the minimum maintenance phase dose was gabapentin 1200 mg/day and pregabalin 300 mg/day TDS. During the 21‐week double‐blind phase, the median doses were gabapentin 1500 mg/day and pregabalin 450 mg/day. |
|
| Outcomes | Seizure frequency: ≥ 50% reduction of seizures; ≥ 75% reduction of seizures Seizure freedom (for final 28 days) Withdrawals: any reasons and due to adverse effects Adverse effects (more common): somnolence, dizziness, headache, increased bodyweight and dry mouth |
|
| Notes | Clinical Trials.gov ID NCT00537940 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation method used. |
| Allocation concealment (selection bias) | Low risk | Randomised participants to either gabapentin or pregabalin (1:1). |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No details provided regarding blinding of study personnel, participants and outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 242 participants allocated to gabapentin; 241 received treatment; 172 completed maintenance phase; 69 participants withdrew. 242 participants allocated to pregabalin allocation; 241 received treatment; 187 completed maintenance phase; 54 participants withdrew. Reasons for exclusion were reported. |
| Selective reporting (reporting bias) | Low risk | Protocol unavailable to check a priori outcomes, but appeared all expected and prespecified outcomes were reported. |
| Other bias | High risk | Study funded by Pfizer Inc., the manufacturer of gabapentin and pregabalin. Study appeared free of other sources of bias. |
Leach 1997.
| Study characteristics | ||
| Methods | Double‐blind, random order, cross‐over, placebo‐controlled study in the UK Single centre (Western Infirmary in Glasgow, UK) 12 weeks' treatment or placebo No baseline period; however, all participants reported ≥ 4 seizures/month for 3 months and AED doses remained unchanged for ≥ 3 months prior to study. |
|
| Participants | Adults with focal seizures drug‐resistant to 1 or 2 AEDs Total randomised: 27 participants; 23 analysed after withdrawals Aged 16–67 years, mean 28.4 years 37% men prior to withdrawals |
|
| Interventions | 3 sequential doses of gabapentin 400 mg, 600 mg and 800 mg TDS, each dose increase after 4 weeks) Placebo |
|
| Outcomes | Seizure frequency Seizure freedom Adverse effects (scored, individual adverse effects not mentioned) Neuropsychological tests (psychomotor, memory, cognition, dysphoria, temper, fatigue, worry, tiredness) |
|
| Notes | No baseline period, therefore, not included in meta‐analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details of randomisation provided. |
| Allocation concealment (selection bias) | Unclear risk | No details provided. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind and matched placebo but no further details provided. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 25% of participants excluded and not analysed on an ITT basis. |
| Selective reporting (reporting bias) | Low risk | Included all prespecified expected outcomes. |
| Other bias | High risk | Trial sponsored by Parke Davis. Study appeared free of other sources of bias. |
Lindberger 2000.
| Study characteristics | ||
| Methods | Randomised, double‐blind, dose titration study comparing gabapentin with vigabatrin Multicentre study: 34 centres in Nordic countries (Denmark, Finland, Iceland, Norway and Sweden) 8‐week baseline period, 24‐week treatment period, evaluation period at 8 weeks compared with baseline To allow flexibility, this was a dose adjustment regimen, with increases in doses of drug based on participant tolerance and seizure control, increased if required at 4‐week periods, with a maximum treatment period at each dose of 8 weeks. |
|
| Participants | People with focal epilepsy who had tried ≤ 2 AED monotherapy regimens 102 participants randomised, then 35 (gabapentin group) and 44 (vigabatrin group) postexclusions for not fulfilling criteria |
|
| Interventions | Gabapentin: variable dose 1800 mg/day minimum, then 2400 mg then maximum 3600 mg/day, increased every 8 weeks as tolerated Vigabatrin: initial 1000 mg then 2000 mg then 4000 mg increased in the same manner as gabapentin |
|
| Outcomes | Primary outcome: improvement rate: proportion of participants with 50% seizure reduction without adverse effects Seizure reduction rate: proportion of participants with 50% seizure reduction irrespective of adverse effects Responder rate: proportion of seizure‐free participants without adverse effects Secondary outcomes: quality of life measures, adverse effects, perimetry |
|
| Notes | Results provided did not indicate the doses the participants had achieved of each drug. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified. |
| Allocation concealment (selection bias) | Unclear risk | No details provided. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐dummy technique, participants received active drug and corresponding placebo. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Analysed on an ITT basis. |
| Selective reporting (reporting bias) | Low risk | Seizure activity reported. |
| Other bias | High risk | Trial sponsored by Parke Davis. Study appeared free of other sources of bias. |
Sethi 2002.
| Study characteristics | ||
| Methods | Randomised control trial in India Head‐to‐head trial; 2 treatment groups gabapentin and lamotrigine Treatment period: 12 weeks No formal baseline period (however, all had ≥ 4 seizures, unclear over what time, despite treatment with maximum dose carbamazepine monotherapy) |
|
| Participants | 52 children and adults with drug‐resistant focal seizures 48% male 27 gabapentin (19 male and 8 female), 25 lamotrigine (6 male and 19 female) Aged 10–60 years |
|
| Interventions | Gabapentin: 300 mg day 1, 300 mg twice daily day 2, there after an increment of 300 mg daily until ≥ 50% reduction in seizures or toxic effects Lamotrigine: 50 mg/day for 2 weeks, increased to 50 mg twice daily, subsequently an increase of 50–100 mg daily until above criteria met |
|
| Outcomes | Efficacy: seizure frequency, pattern of seizures, seizure‐free interval. Including % change of seizure frequency from baseline, responder rate (reduction in seizure frequency of ≥ 50%), response ratio Safety: biochemical investigations and adverse effects |
|
| Notes | As no clear baseline period, excluded from meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details provided. |
| Allocation concealment (selection bias) | Unclear risk | No details provided. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No details provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No participants seemed to be excluded from the results, but 1 participant in gabapentin group did not seem to be accounted for. |
| Selective reporting (reporting bias) | Low risk | Included all prespecified expected outcomes. |
| Other bias | Low risk | Study appeared free of other sources of bias. |
Sivenius 1991.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study in Finland 3 treatment arms: 1 placebo and 2 gabapentin Prospective prerandomisation baseline period: 12 weeks Treatment period: 12 weeks |
|
| Participants | Adults with drug‐resistant focal epilepsy Total randomised: 45 participants 18 placebo; 18 gabapentin 900 mg/day; 9 gabapentin 1200 mg/day 47% men Aged 16–59 years Other AEDs: ≤ 2 Median baseline seizure frequency per 12‐week baseline period: 36 placebo; 26 gabapentin 900 mg/day; 23 gabapentin 1200 mg/day |
|
| Interventions | Gabapentin 900 mg/day Gabapentin 1200 mg/day Placebo All treatments and packaging were identical in appearance. |
|
| Outcomes | Median change in seizure frequency % change in seizure frequency Adverse effects |
|
| Notes | 2 people in the gabapentin 900 mg group were excluded from analysis, both excluded 2 weeks' postrandomisation. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Allocated sequentially, sealed, numbered packages. |
| Allocation concealment (selection bias) | Low risk | Random permuted blocks. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Tablets and packaging identical in appearance. Identical analysis of results. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No reasons reported for exclusion of 2 participants in gabapentin 900 mg group. |
| Selective reporting (reporting bias) | Low risk | Quote: "… seizure frequency was recorded." Unclear how, otherwise included all prespecified expected outcomes. |
| Other bias | High risk | Trial sponsored by Parke Davis. Study appeared free of other sources of bias. |
Tomovic 1999.
| Study characteristics | ||
| Methods | Randomised, parallel‐group study in Serbia | |
| Participants | 9 women and 9 men with drug‐resistant focal epilepsy Mean age: 24.7 years, range 17–47 years All had been treated with 1 or 2 first‐line AEDs during 3 months before introducing gabapentin with unsatisfactorily controlled seizures Seizure frequency prior to treatment was unclear. |
|
| Interventions | Gabapentin 900 mg Gabapentin 1200 mg |
|
| Outcomes | Seizure frequency Seizure freedom Haematological and biochemical analyses (4th and 12th week) and 24‐hour EEG before therapy and on final week (week 12). Frequency of epileptiform discharges noted Reduction in seizure activity: 26–49%; 50–99%; 100%; worse state Adverse effects |
|
| Notes | 3 people did not complete the study and were not included in demographics, analysis, etc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation unclear. |
| Allocation concealment (selection bias) | Unclear risk | How participants allocated to each group unclear. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Unclear if tablets and packaging identical in appearance. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Unclear why 3 participants dropped out and excluded from analysis. |
| Selective reporting (reporting bias) | Low risk | Seizure frequency recorded; unclear how seizure activity measured. Otherwise standardised tests for both groups. |
| Other bias | Low risk | Study appeared free of other sources of bias. |
UK Gabapentin 1990.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study (cross‐continent) Prospective prerandomisation baseline period: 12 weeks Treatment period: 14 weeks |
|
| Participants | Adults with drug‐resistant focal epilepsy Total randomised: 127 Placebo: 66 participants; gabapentin 1200 mg/day: 61 participants 39% men Age range: 14–73 years Other AEDs: ≤ 2 Median baseline seizure frequency per 28 days: gabapentin 13 (range 3–368); placebo 13 (range 1–216) |
|
| Interventions | Gabapentin 1200 mg/day Placebo All treatments and packaging identical in appearance |
|
| Outcomes | Proportion with a 50% reduction in seizure frequency Response ratio Adverse effects |
|
| Notes | 14 participants excluded from published analyses: 5 from placebo group; 9 from gabapentin 1200 mg/day group Additional unpublished data allowed the inclusion of participants excluded despite completing the treatment phase with adequate seizure data. The following participants contributed to the best‐case and worst‐case sensitivity analyses in this review: placebo 2; gabapentin 1200 mg 8. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Used random permuted blocks to generate sequence for randomisation. |
| Allocation concealment (selection bias) | Low risk | Allocated sequentially, sealed, numbered packages. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Used tablets and packaging identical in appearance. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rate reported, 14 participants who withdrew were not included in published analyses. Report withdrawals and gave reasons for withdrawal. |
| Selective reporting (reporting bias) | Low risk | Included all prespecified, expected outcomes. |
| Other bias | High risk | Trial sponsored by Parke Davis. Study appeared free of other sources of bias. |
US Gabapentin 1993.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study in the US 4 treatment arms: 1 placebo and 3 gabapentin Prospective prerandomisation baseline period: 12 weeks Treatment period: 12 weeks |
|
| Participants | Adults with drug‐resistant focal epilepsy Total randomised: 306 participants Placebo: 98 participants; gabapentin 600 mg/day: 53; gabapentin 1200 mg/day: 101; gabapentin 1800 mg/day: 54 66% men Aged 16–70 years Other AEDs: ≤ 2 Median baseline seizure frequency per 28 days: 10.8 (range 2.0–1092.7) |
|
| Interventions | Gabapentin 600 mg/day Gabapentin 1200 mg/day Gabapentin 1800 mg/day Placebo All treatments and packages were identical in appearance. |
|
| Outcomes | Proportion with a 50% reduction in seizure frequency Response ratio Adverse effects |
|
| Notes | 18 participants excluded from published analyses: placebo: 3; gabapentin 600 mg/day: 4; gabapentin 1200 mg/day: 10; gabapentin 1800 mg/day: 1 Additional unpublished data allowed the inclusion of participants excluded despite completing the treatment phase with adequate seizure data. The following participants contribute to the best‐case and worst‐case sensitivity analyses in this review: placebo: 2; gabapentin 600 mg/day: 4; gabapentin 1200 mg/day: 10; gabapentin 1800 mg/day: 1 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Used random permuted blocks to generate sequence for randomisation. |
| Allocation concealment (selection bias) | Low risk | Allocated sequentially, sealed, numbered packages. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Used tablets and packaging identical in appearance. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rate reported, 18 participants not included in published analyses and no reasons given. |
| Selective reporting (reporting bias) | Low risk | Included all prespecified expected outcomes. |
| Other bias | High risk | Trial sponsored by Parke Davis. Study appeared free of other sources of bias. |
Yamauchi 2006.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group multicentre study in Japan from March 2000 to October 2002 Prospective prerandomisation baseline period: 12 weeks Treatment period: 12 weeks Dose‐reduction period lasting 8 days, 4 weeks instituted, followed by a 4‐week postdosing observation period 3 treatment arms, 1 placebo and 2 treatment |
|
| Participants | Adults aged ≥ 16 years Most aged 18–44 years, mean age between 3 groups 31–33 years Other AEDs: > 1 Total randomised 209 participants; all with drug‐resistant focal epilepsy 82 participants to placebo (42 men, mean age: 31.8 (SD 11.3) years, 25 secondary generalised seizures, mean duration epilepsy: 19.5 years, mean seizure frequency per 28 days: 19.9, 1 other AED: 19.5%, 2 other AEDs: 80.5%) 86 participants to gabapentin 1200 mg (37 men, mean age 31.3 (SD 10.6) years, 26.3 secondary generalised seizures, mean duration epilepsy: 19.8 years, mean seizure frequency per 28 days: 31.6, 1 other AED: 14%, 2 other AEDs: 86%) 41 participants to gabapentin 1800 mg (22 men, mean age 32.7 (SD 13.7) years, 13 secondary generalised seizures, mean duration epilepsy: 21.2 years, mean seizure frequency per 28 days: 24.2, 1 other AED: 4.9%, 2 other AEDs: 95.1%) 19 excluded; after exclusion placebo: 75, gabapentin 1200 mg: 80, gabapentin 1800 mg: 35 Baseline seizure frequency per 12 weeks: 8 |
|
| Interventions | Gabapentin 1200 mg/day Gabapentin 1800 mg/day Placebo All treatments were identical in appearance (200 mg tablets). |
|
| Outcomes | Improvement in seizure frequency: completely (–100%), markedly improved (–99.9% to –75.0%), moderately improved (–74.9% to –50%), slightly improved (–49.9% to –25%), no change (–24.9% to 0%), aggravated (> +0.1%) Response ratio (= (T – B)/(T + B) where T = number of seizures during the treatment period, and B = number of seizures in the baseline period) Seizure intensity/duration: better, no change and worse Adverse effects Serious treatment‐related adverse effects |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Few/no details regarding randomisation given. Most variables between groups controlled (age, sex, frequency, number of other AED, previous treatments etc.). |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Tablets identical in appearance, all outcomes blinded, monitored and followed up in the same way. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ITT analysis not employed; reasons for exclusions stated; however, 19 participants not included. |
| Selective reporting (reporting bias) | Low risk | Seizure diary for all groups, same outcomes. Published reports include all prespecified expected outcomes. |
| Other bias | High risk | Trial sponsored by Parke Davis. Study appeared free of other sources of bias. |
AED: antiepileptic drug; EEG: electroencephalogram; ITT: intention‐to‐treat; SD: standard deviation; TDS: three times daily.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arya 2013 | Not an RCT |
| Bodalia 2013 | Not an RCT |
| Btaiche 1995 | Not an RCT |
| Crawford 1987 | No seizure data recorded |
| Jacoby 2015 | Ineligible population (new‐onset epilepsy) |
| Korean Gabapentin Study Group 2000 | No gabapentin in add‐on (sodium valproate in add‐on) |
| Nonoda 2014 | Ineligible population (no drug‐resistant epilepsy) |
| Ohtsuka 2014 | Not an RCT |
| Ramsay 1994 | Not an RCT |
| Schmidt 2001 | Not an RCT |
| Semah 2014 | No gabapentin in add‐on |
RCT: randomised controlled trial.
Characteristics of studies awaiting classification [ordered by study ID]
Kwan 2000.
| Methods | Double‐blind, randomised, placebo‐controlled, cross‐over study in China |
| Participants | 43 adults with drug‐resistant focal seizures |
| Interventions | Gabapentin 600 mg/day for 1 week and 1200 mg/day for 12 weeks with matching placebo controls |
| Outcomes | There was a statistically significant difference in seizure frequency from the baseline to the treatment phase between participants receiving placebo and gabapentin 1200 mg, in whom seizure frequency decreased 57%. Gabapentin 900 mg appeared to be ineffective. There was a close relationship between serum gabapentin concentrations and gabapentin dose based on seizure frequency. Serum gabapentin concentrations > 2 μg/mL resulted in a lower frequency of seizures. |
| Notes | No other data available for analysis; all data taken from abstract; author unable to provide further information. |
Shapiro 2000.
| Methods | Randomised, placebo‐controlled trial in US |
| Participants | 76 young children with focal epilepsy |
| Interventions | Syrup formulation of gabapentin 40 mg/kg/day TDS or placebo |
| Outcomes | Main outcome was seizure reduction. Concluded that gabapentin was safe and well tolerated, and reduced the rate of focal seizures; however, this finding did not reach significance. |
| Notes | All information taken from abstract, unable to contact study authors. |
TDS: three times daily.
Differences between protocol and review
The term 'partial' has been replaced by 'focal', in accordance with the most recent classification of epilepsies of the International League Against Epilepsy (Scheffer 2017).
Contributions of authors
MP independently assessed trials for inclusion in the present update. SAB contributed to previous updates of this review. JLH completed the dose‐regression analysis. JLH developed the original protocol. AGM developed the original protocol..
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK
This review update was funded by the National Institute for Health Research (NIHR) [Clinically effective treatments for central nervous system disorders in the NHS, with a focus on epilepsy and Movement Disorders] (SRPG project 16/114/26). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Declarations of interest
MP: none known. SAB: none known. JLH: none known. AGM is part funded by the National Institute for Health Research Applied Research Collaboration North West Coast (NIHR ARC NWC). A consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to University of Liverpool.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Anhut 1994 {published and unpublished data}
- Anhut H, Ashman P, Feuerstein TJ, Sauermann W, Saunders M, Schmidt B. Gabapentin (Neurontin) as add-on therapy in patients with partial seizures: a double-blind, placebo-controlled study. Epilepsia 1994;35(4):795-801. [PMID: ] [DOI] [PubMed] [Google Scholar]
Appleton 1999 {published data only}
- Appleton R, Fichtner K, LaMoreaux L, Alexander J, Halsall G, Murray G, et al, and the Gabapentin Paediatric Study Group. Gabapentin as add-on therapy in children with refractory partial seizures: a 12 week, multicentre, double blind placebo controlled study. Epilepsia 1999;40(8):1147-54. [PMID: ] [DOI] [PubMed] [Google Scholar]
Fisher 2001 {published data only}
- Fisher RS, Sachdeo RC, Pellock J, Penovich PE, Magnus L, Bernstein P. Rapid Initiation of gabapentin: a randomised control trial. Neurology 2001;56(6):743-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
French 2016 {published data only}
- French J, Glue P, Friedman D, Almas M, Yardi N, Knapp L, et al. Adjunctive pregabalin vs gabapentin for focal seizures: interpretation of comparative outcomes. Neurology 2016;87(12):1242-9. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leach 1997 {published data only}
- Leach JP, Girvan J, Paul A, Brodie MJ. Gabapentin and cognition: a double blind, dose ranging, placebo controlled study in refractory epilepsy. Journal of Neurology, Neurosurgery, and Psychiatry 1997;62(4):372-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lindberger 2000 {published data only}
- Lindberger M, Alenius M, Frisen L, Johannessen SI, Larsson S, Malmgren K, et al, on behalf of the GREAT Study Investigators Group. Gabapentin versus vigabatrin as first add-on for patients with partial seizures that failed to respond to monotherapy: a randomised, double-blind, dose titration study. Epilepsia 2000;41(10):1289-95. [PMID: ] [DOI] [PubMed] [Google Scholar]
Sethi 2002 {published data only}
- Sethi A, Chandra D, Puir V, Mallika V. Gabapentin and lamotrigine in Indian patients of partial epilepsy refractory to carbamazepine. Neurology India 2002;50(3):359-63. [PMID: ] [PubMed] [Google Scholar]
Sivenius 1991 {published and unpublished data}
- Sivenius J, Kalviainen R, Ylinen A, Riekkinen P. Double blind study of gabapentin in the treatment of partial seizures. Epilepsia 1991;32(4):539-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
Tomovic 1999 {published data only}
- Tomovic M, Ilic T, Mihajlovic M, Jovicic A. Gabapentin as adjuvant therapy in the treatment of refractory partial epilepsy. Vojnosanitetski Pregled 1999;56(2):151-6. [PMID: ] [PubMed] [Google Scholar]
UK Gabapentin 1990 {published and unpublished data}
- UK Gabapentin Study Group. Gabapentin in partial epilepsy. Lancet 1990;335(8698):1114-7. [PMID: ] [PubMed] [Google Scholar]
US Gabapentin 1993 {published and unpublished data}
- US Gabapentin Study Group No 5. Gabapentin as add-on therapy in refractory partial epilepsy: a double-blind, placebo-controlled, parallel-group study. Neurology 1993;43(11):2292-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Yamauchi 2006 {published data only}
- Yamauchi T, Kaneko S, Vagi K, Sase S. Treatment of partial seizures with gabapentin: double-blind, placebo-controlled, parallel-group study. Psychiatry and Clinical Neurosciences 2006;60(4):507-15. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Arya 2013 {published data only}
- Arya R, Glauser TA. Pharmacotherapy of focal epilepsy in children: a systematic review of approved agents. CNS Drugs 2013;27(4):273-86. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bodalia 2013 {published data only}
- Bodalia PN, Grosso AM, Sofat R, Macallister RJ, Smeeth L, Dhillon S, et al. Comparative efficacy and tolerability of anti-epileptic drugs for refractory focal epilepsy: systematic review and network meta-analysis reveals the need for long term comparator trials. British Journal of Clinical Pharmacology 2013;76(5):649-67. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Btaiche 1995 {published data only}
- Btaiche IF, Woster PS. Gabapentin and lamotrigine: novel antiepileptic drugs. American Journal of Health-system Pharmacy 1995;52(1):61-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Crawford 1987 {published data only}
- Crawford P, Ghadiali E, Lane R, Blumhardt L, Chadwick DW. Gabapentin as an antiepileptic drug in man. Journal of Neurology, Neurosurgery, and Psychiatry 1987;50(6):682-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jacoby 2015 {published data only}
- Jacoby A, Sudell M, Tudur Smith C, Crossley J, Marson AG, Baker GA. Quality-of-life outcomes of initiating treatment with standard and newer antiepileptic drugs in adults with new-onset epilepsy: findings from the SANAD trial. Epilepsia 2015;56(3):460-72. [PMID: ] [DOI] [PubMed] [Google Scholar]
Korean Gabapentin Study Group 2000 {published data only}
- Korean Gabapentin Study Group. Comparative clinical trial of gabapentin and sodium valproate as add-on therapy in refractory partial epilepsies: open randomized multicenter trial. Journal of Korean Epilepsy Society 2000;4(1):19-26. [Google Scholar]
Nonoda 2014 {published data only}
- Nonoda Y, Iwasaki T, Ishii M. The efficacy of gabapentin in children of partial seizures and the blood levels. Brain & Development 2014;36(3):194-202. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ohtsuka 2014 {published data only}
- Ohtsuka Y. New antiepileptic drugs: characteristics and clinical applications. Japanese Journal of Clinical Medicine 2014;72(5):931-8. [PMID: ] [PubMed] [Google Scholar]
Ramsay 1994 {published data only}
- Ramsay RE. Clinical efficacy and safety of gabapentin. Neurology 1994;44(6 Suppl 5):S23-30;discussion S31-2. [PMID: ] [PubMed] [Google Scholar]
Schmidt 2001 {published data only}
- Schmidt D, Elger CE, Brandl U, Schmitz B, Steinhoff BJ, Bauer G, et al. Recommendations for the treatment of partial epilepsies with gabapentin A – a consensus. Nervenheilkunde 2001;20(10):580-5. [Google Scholar]
Semah 2014 {published data only}
- Semah F, Thomas P, Coulbaut S, Derambure P. Early add-on treatment vs alternative monotherapy in patients with partial epilepsy. Epileptic Disorders 2014;16(2):165-74. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Kwan 2000 {published data only}
- Kwan SY, Yiu CH, Su MS. A double blind study of gabapentin in partial seizures. Epilepsia 2000;41(Suppl Florence):69. [Google Scholar]
Shapiro 2000 {published data only}
- Shapiro DY, Nordli D, Glauser TA, Knapp LE, Greiner M, Purcell TJ, et al. Gabapentin as add-on therapy for refractory partial seizures in children 1–36 months of age: a novel, short-term, placebo-controlled trial. Epilepsia 2000;41(Suppl 7):106, Abstract no: 2.065. [Google Scholar]
Additional references
Barker‐Haliski 2014
- Barker-Haliski M, Sills GJ, White HS. What are the arguments for and against rational therapy for epilepsy? Advances in Experimental Medicine and Biology 2014;813:295-308. [PMID: ] [DOI] [PubMed] [Google Scholar]
British National Formulary
- British National Formulary. www.bnf.org/.
Cockerell 1995
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the National General Practice Study of Epilepsy. Lancet 1995;346(8968):140-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Version accessed 2015. Hamilton (ON): McMaster University (developed by Evidence Prime). Available at gradepro.org.
Hauser 1993
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993;34(3):453-68. [PMID: ] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1.
Higgins 2020
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lefebvre 2020
- Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf M-I, et al. Technical Supplement to Chapter 4: Searching for and selecting studies. In: Higgins JP, Thomas J, Chandler J, Cumpston MS, Li T, Page MJ, et al (eds). Cochrane Handbook for Systematic Reviews of Interventions Version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Loscher 2002
- Loscher W. Current status and future directions in the pharmacotherapy of epilepsy. Trends in Pharmacological Sciences 2002;23(3):113-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
McClean 1995
- McClean MJ. Gabapentin. Epilepsia 1995;36(Suppl 2):S73-S86. [PMID: ] [DOI] [PubMed] [Google Scholar]
McCullagh 1989
- McCullagh P, Nelder JA. Generalized Linear Models. 2nd edition. London: Chapman and Hall, 1989. [Google Scholar]
Panebianco 2015
- Panebianco M, Rigby A, Weston J, Marson AG. Vagus nerve stimulation for partial seizures. Cochrane Database of Systematic Reviews 2015, Issue 4. Art. No: CD002896. [DOI: 10.1002/14651858.CD002896.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Reynolds 1981
- Reynolds EH, Shorvon SD, Galbraith AW, Chadwick D, Dellaportas L, Vydelingum L. Phenytoin monotherapy for epilepsy: a long term prospective study, assisted by serum level monitoring, in previously untreated patients. Epilepsia 1981;22(4):475-88. [PMID: ] [DOI] [PubMed] [Google Scholar]
Scheffer 2017
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512-21. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). GRADE Working Group, 2013. Available from gdt.guidelinedevelopment.org/app/handbook/handbook.html (accessed prior to 18 November 2020).
West 2019
- West S, Nolan SJ, Cotton J, Gandhi S, Weston J, Sudan A, et al. Surgery for epilepsy. Cochrane Database of Systematic Reviews 2019, Issue 6. Art. No: CD010541. [DOI: 10.1002/14651858.CD010541.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Al‐Bachari 2013
- Al-Bachari S, Pulman J, Hutton JL, Marson AG. Gabapentin add-on for drug-resistant partial epilepsy. Cochrane Database of Systematic Reviews 2013, Issue 7. Art. No: CD001415. [DOI: 10.1002/14651858.CD001415.pub2] [DOI] [PubMed] [Google Scholar]
Marson 1999
- Marson AG, Kadir ZZ, Hutton JL, Chadwick DW. Gabapentin add-on for drug-resistant partial epilepsy. Cochrane Database of Systematic Reviews 1999, Issue 1. Art. No: CD001415. [DOI: 10.1002/14651858.CD001415] [DOI] [PubMed] [Google Scholar]
Panebianco 2018
- Panebianco M, Al-Bachari S, Weston J, Hutton JL, Marson A. Gabapentin add-on treatment for drug-resistant focal epilepsy. Cochrane Database of Systematic Reviews 2018, Issue 10. Art. No: CD001415. [DOI: 10.1002/14651858.CD001415.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]