

Abstract
Plant cytochrome P450s play key roles in the diversification and functional modification of plant natural products. Although over 200,000 plant P450 gene sequences have been recorded, only seven crystalized P450 genes severely hampered the functional characterization, gene mining and engineering of important P450s. Here, we combined Rosetta homologous modeling and MD-based refinement to construct a high-resolution P450 structure prediction process (PCPCM), which was applied to 181 plant P450s with identified functions. Furthermore, we constructed a ligand docking process (PCPLD) that can be applied for plant P450s virtual screening. 10 examples of virtual screening indicated the process can reduce about 80% screening space for next experimental verification. Finally, we constructed a plant P450 database (PCPD: http://p450.biodesign.ac.cn/), which includes the sequences, structures and functions of the 181 plant P450s, and a web service based on PCPCM and PCPLD. Our study not only developed methods for the P450-specific structure analysis, but also introduced a universal approach that can assist the mining and functional analysis of P450 enzymes.
Keywords: Plant P450 database, Homologous modeling, Ligand docking, Database, Web service
Introduction
Cytochrome P450 enzymes have been identified in all kingdoms of life, which can catalyze more than 20 types of reactions, including hydroxylation, epoxidation, cyclization, and so on [1]. Due to their functional diversity, they can generate terpenoids [2], flavonoids [3], alkaloids [4] and fatty acid compounds [5], etc. P450 genes play a vital role in the biosynthesis of plant natural products. With the rapid development of DNA sequencing technology, about 200,000 plant P450 gene sequences have been identified and collected in public databases [6]. However, a small proportion of the plant P450s have been functionally characterized, due to the high cost required for screening a P450 enzyme with a specific catalytic function. Some studies have exploited the potentials of comparative genomic or transcriptomic analysis to reduce the screening scope [[7], [8], [9]]. However, to our knowledge, no universal method can obviously assist the mining and functional analysis of plant P450 enzymes.
The tertiary structure of enzymes and their corresponding ligand-binding motifs can help us analyze their reaction types and function preferences [10,11]. However, the crystal structures of plant P450s are difficult to resolve due to the membrane localization, which leads to the structure easily broken and degraded during purification and crystal growth [12]. Currently, there are only seven plant P450 protein crystals in the Protein Data Bank (PDB) database, which limits the further structural analysis of plant P450s [[13], [14], [15], [16], [17], [18]]. In this study, we developed a template-based structure prediction process and a ligand docking process specifically for plant P450 enzymes, designated as PCPCM (plant cytochrome P450 comparative modelling) and PCPLD (plant cytochrome P450 ligand docking), respectively. Using PCPCM and PCPLD, we constructed the structures of the plant P450 enzymes with known functions and accurately docked the enzymes with the corresponding ligands. With the cross-docking experiments, we successfully validated the potential of PCPLD in P450 virtual screening. Finally, we built a plant P450 database (PCPD) web platform (http://p450.biodesign.ac.cn/) to share the sequences, structures, and functions of the plant P450s, and the web-based tools of PCPCM and PCPLD.
Result
Plant P450s with known functions and crystal structures
Collecting all functionally characterized P450s not only helps to predict the catalytic function of other P450s based on sequence similarity but also facilitates the design of new pathways to biosynthesize natural and non-natural products by synthetic biology [[19], [20], [21]]. Totally 181 plant P450s with known functions were collected from the KEGG database, the BRENDA database, and published literatures (Fig. 1A and Table S1). Among them, 118 cases catalyze hydroxylation reactions, 11 cases catalyze oxidation reactions, 11 cases catalyze epoxidation reactions, and 3 cases catalyze cyclization reactions (Fig. 1A). P450s catalyze the conversions of 133 substrates among 181 plant P450s, including 71 terpenoids, 10 flavonoids, 15 alkaloids, and so on (Fig. 1A). Based on the phylogenetic tree and sequence similarity, the 181 plant P450s were classified into 46 gene families and 113 subfamilies (Fig. 1A) [22]. The P450s in the same subfamily (sequence identity >60%) often have a similar catalytic function, the P450s in different subfamilies or families often display entirely different catalytic functions [23,24]. But, except for true orthologs, this classification can never accurately predict the function of P450s. Therefore, besides sequence similarity, other analytic features, such as structure information, are also considered for the function prediction of so massive P450s.
Fig. 1.

Data of plant cytochrome P450 enzymes with known functions. (A) The phylogenetic tree of 181 plant P450s with known functions. The outermost ring indicated the types of substrates, and the internal ring indicated the types of catalytic reactions. (B) Structural alignment of five plant P450 protein crystals in PDB. The pairwise amino acid identities were shown on the structures.
Among seven collected plant P450 protein crystals in the PDB database (ID: 3DSK, 3DAN, 5YLW, 6A15, 6J95, 6L8H, and 6VBY), 3DSK and 3DAN come from the CYP74A subfamily, 6J95 and 6L8H come from the CYP97 family, other three P450s (i.e., 5YLW, 6A15, 6VBY) come from CYP76, CYP90, and CYP73 family, respectively. To avoid the function biases of similar sequences and structures from the same families, this study only compared and analyzed five protein crystals from different families (3DSK (lower resolution than 3DAN), 5YLW, 6A15, 6J95 (presence of the substrate in the structure) and 6VBY) (Fig. 1B). Although the sequence identities among these five P450s are lower than 30%, all of them have very conserved secondary structures and super-secondary domains in the tertiary structures (Fig. 1B and Table S2) [[25], [26], [27]]. Structure analysis of all P450 crystal structures from the PDB database also suggested a similar structural arrangement (Fig. S1). The conserved structural arrangement provides us an opportunity to predict P450 structures by homology modeling, even the query P450s have relatively low sequence similarities with their templates.
PCPCM: plant cytochrome P450 comparative modelling
By integrating the widely used homology modeling method RosettaCM with the MD-based structure refinement method for structure optimization [[28], [29], [30], [31], [32]], we built a structure prediction process specific for plant P450 (PCPCM). PCPCM predicts the plant P450 structure with the following five steps (Fig. 2A): 1) The transmembrane region of the target P450 protein sequence was clipped according to the transmembrane topology prediction; 2) The homologous template was selected by the blast search for a local P450 structure library which included all P450 crystal structures; 3) The initial model of the target P450 structure was constructed by homology modeling method; 4) The heme prosthetic group was added into the initial model by structure alignment; 5) The complex structure was optimized by molecular dynamics (MD) simulation, and the structure with the lowest potential energy was selected as the final structure for the target P450 gene.
Fig. 2.

PCPCM workflow and model evaluations. (A) The workflow of PCPCM. The workflow mainly includes: preprocessing of the input sequence (TMHMM step) and blast search (BLAST+ step); homology modeling to predict initial model (RosettaCM step); addition of active heme prosthetic group (Add HEM step); molecular dynamics optimization of the composite structure (Amber step). (B) Comparison of five structure prediction methods for P450 structural modeling. For each crystal structure, five methods were ranked based on the protein structure quality measures. Ranking first represents the method that had the best performance for the corresponding crystal structure and quality measure. Each color represents a modeling method, the Y-axis represents the number of five rankings of each method. (C) Comparison of RMSD values of five structure prediction methods. The RMSD values were calculated by comparing the crystal structures with the modeling structures from five methods using LGA (http://proteinmodel.org/AS2TS/LGA/lga.html).
In order to evaluate the prediction accuracy of PCPCM. PCPCM and the other four structure prediction methods (Swiss-Model [33], I-TASSER [34], MODELLER [35], OROIN [36]) are used to blindly predict the structures of the five crystallized plant P450 proteins, respectively. To obtain a more accurate assessment of these methods, the real crystal structure had been removed from the corresponding template library in the modeling process (Materials and Methods). Six protein structure quality measures (namely WHATCHECK [37], Verify 3D [38], ERRAT [39], Prove [40], PROCHECK [41], and QMEAN [42]) were used to evaluate the predicted models (Fig. 2B, Fig. S2, and Table S3). For 30 (5 × 6) evaluations from 5 structures and 6 metrics, PCPCM ranked first in 24 evaluations and ranked second in 6 evaluations. Furthermore, the Root Mean Squared Deviation (RMSD) values were obtained by comparing the predicted models with the crystal structures (Fig. 2C). PCPCM obtained very low RMSD values (<2 Å) in all five comparisons, which indicated that our process has a higher prediction accuracy compared to other methods. In addition, due to the consideration of the heme prosthetic groups and the further MD optimization for the structures, PCPCM showed high performance in the stacking of active centers and heme-binding regions, which was especially important for substrate recognition and catalysis (Fig. S3).
PCPLD: plant cytochrome P450 ligand docking
The P450s recognize substrates with high regio- and stereo-selectivity, and the P450 with specific function often recognizes the specific substrate [43,44]. Based on the P450 structure prediction process PCPCM, we furthermore constructed a ligand docking process (PCPLD) that can be applied for plant P450s virtual screening (Fig. 3A). PCPLD mainly includes the following four steps: 1) The parameterization (distance, dihedral angle, and so on) of ligands and the obtainments of the cytochrome P450 Compound I (CpdI) (CpdI is the high-energy intermediate state of heme in the catalytic reaction cycle) complex and heme-ligated cysteine [45]; 2) Docking the ligand to the active centers of plant P450s; 3) Screening the docking poses with energy function and clustering the docking poses based on the backbone atoms with Calibur software [46]. The top 10 poses in each cluster were filtered by distance value (between CpdI oxygen and ligand atoms) to obtain suitable candidates; 4) A Score value for each candidate structure was calculated by integrating the Rosetta total energy and the ligand binding free energy, and the docking pose with the highest Score was selected as the final complex [29,47]. To evaluate the docking accuracy of PCPLD, we docked the corresponding ligands to three crystal structures with native ligands. As expected, all docked ligands aligned well like the ligand arrangements in the crystal structures (Fig. 3B). We also analyzed the relationship between the positional difference of docked ligand with native ligand and the docking score, and a significant negative correlation (R2 = 0.82) between them was observed (Fig. S5), indicating that the calculated docking score can well reflect the docking accuracy.
Fig. 3.

Docking workflow and evaluations of PCPLD. (A) The Docking workflow of PCPLD. (B) Plant P450 crystal structures (gray) and predicted structures (purple) docking with corresponding ligands, plant P450 crystal structure (PDB ID: 3DSK, 6A15, and 6J95), and corresponding ligands (vanillic acid, cholesterol, and retina). Since there are no corresponding ligands and analogs in the crystal structure of 5YLW and 6B9Y, there is no docking. (C) 10 ligands and 181 plant P450s were selected for cross-docking, and the results were shown in the figures. The red marked short-term represented the correct docking. The 10 ligands are abbreviated as Te (terpenoids), Fa (fatty acids), Aa (amino acids), Fl (flavonoids), and Al (alkaloids).
Furthermore, we investigated that whether PCPLD has the potential for the virtual screening of plant P450s. Firstly, we constructed a structure screening library including the 181 predicted plant P450 structures with known functions; Secondly, we selected 10 representative ligands (including 4 terpenoids, 3 flavonoids, 1 alkaloid, 1 fatty acid, and 1 amino acid) (Table 1) which could be catalyzed in the structure screening library; Thirdly, a cross-docking between 10 selected ligands and 181 Plant P450s was conducted, and 1810 (10 × 181) docking scores were calculated and sorted (Materials and Methods). For all selected ligands, we found the native plant P450 often has the higher docking score towards the native ligands, and the docking scores of all 10 native plant P450s are distributed in top 20% (Fig. 3C). Our results indicated that the PCPLD has the potential for the virtual screening of plant P450s, which is of reference significance for mining some important P450s.
Table 1.
10 P450 catalytic reactions for the test of P450s virtual screening.
| Number | Enzymes | Catalytic function | Reactions | Sources |
|---|---|---|---|---|
| 1 | CYP51H10 | 12,13β-epoxy-β-amyrin 16β-hydroxylase | 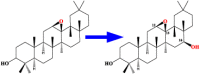 |
Avena strigose [48] |
| 2 | CYP706X | Apigenin 6-hydroxylase | 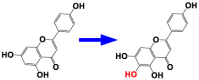 |
Erigeron breviscapus [7] |
| 3 | CYP93C | Isoflavone synthase | 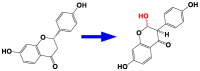 |
Glycine max [49] |
| 4 | CYP90B2 | Cholesterol 22-hydroxylase | 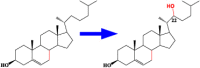 |
Dioscorea zingiberensis [50] |
| 5 | CYP76AH3 | Ferruginol 2,11- hydroxylase | 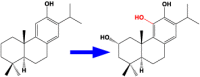 |
Salvia miltiorrhiza [51] |
| 6 | CYP75A | Dihydrokaempferol 3′,5′-hydroxylase | 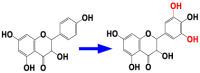 |
Vitis vinifera [52] |
| 7 | CYP76AH1 | Miltiradiene 12-hydroxylase | 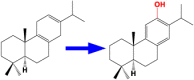 |
Salvia miltiorrhiza [53] |
| 8 | CYP79D1 | l-Valine N-monooxygenase | 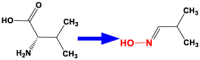 |
Manihot esculenta [54] |
| 9 | CYP80B1 | (S)–N-methylcoclaurine 3′-hydroxylase | 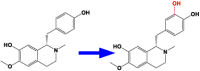 |
Eschscholzia californica [55] |
| 10 | CYP94A5 | fatty acid omega-hydroxylase | 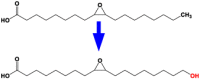 |
Nicotiana tabacum [56] |
PCPD: plant P450 enzyme database
At last, we constructed a database of the 181 plant P450s (PCPD), which included the sequences, functions, and PDB structures predicted by PCPCM and PCPLD (Fig. 4, http://p450.biodesign.ac.cn/). Besides, a web server for PCPCM and PCPLD was provided for structure prediction and ligand docking of plant P450 enzymes. The usage information is shown below briefly. Firstly, users need to provide their own username and email address to facilitate the checking of data results; Secondly, selecting one tool between PCPCM and PCPLD. For PCPCM, a FASTA formatted sequence file of target plant P450 is required. For PCPLD, a PDB formatted structure file of target plant P450 and a corresponding SDF formatted ligand file are both required.
Fig. 4.

The main function interface of PCPD. (A) The data browsing and downloading interface of the website. It mainly includes: browsing of plant P450 family, catalytic and function and reaction, as well as downloading of sequence and structure. (B) The application interface of PCPCM and PCPLD. Users can choose the needed method and submit input file according to the User Guide.
The submitted task will run on the server with several days. Once the task was completed, the output result files will be sent to the user's email. PCPCM's result files include: prediction information of sequence transmembrane region, the P450 secondary structure prediction information, homologous template selection and comparison information, and the final structure model file. The PCPLD result file is a PDB formatted complex with docked ligand.
Discussion
Over the last few years, there has been remarkable progress in the gene number and function characterization for plant P450s with the rapid development of sequencing technology, omics analysis, and synthetic biology [57]. Many databases, such as Cytochrome P450 Homepage (https://drnelson.uthsc.edu/cytochromeP450.html), Cytochrome P450 Engineering Database (https://cyped.biocatnet.de) and Plant GDB (http://www.plantgdb.org/site/acknowledgments.php), had been constructed to collect the sequence and function information of plant P450s. However, the structure information is often ignored, due to the huge difficulties in protein purification and crystal growth [58], and only seven crystalized P450 proteins were collected in the PDB database. With the development of computational technologies for protein structure prediction, many methods such as RosettaCM, Swiss-Model, I-TASSER, etc. have been widely used in protein structure prediction [59,60]. In this study, the PCPCM, integrating RosettaCM and MD simulation, showed better performance in plant P450s homology modeling than all other methods. However, the performance of PCPCM is not always the best for the construction of the P450 ligand-binding pockets (Table S4). The missing of highly similar templates in active site pockets compared to backbone regions may account for the poor performances (The backbone region refers to helices F, I, C and β1–5, between helices F and F′, between helices K and β1-4, and between β3-2 and β3-3 in P450s) [16,61].
On the basis of PCPCM, we further developed PCPLD with the potential for P450s virtual screening. However, the request for tremendous computational resources (both one structure prediction and one docking need about 1 day) severely restricted the application for large-scale structure prediction and virtual screening. Due to these limitations, the PCPCM and PCPLD were only applied to 181 plant P450s with identified functions. Besides, our PCPLD prediction process only generated a preliminary rough confirmation of the active sites due to the very variable active pocket in the P450 structure. Therefore, our docking results can only be used as a reference for the prediction of substrates. With the development of deep learning algorithms, the Google team has used co-evolution information combined with deep neural networks to develop AlphaFold to generate spatial constraints and reduce spatial search [62]. David Baker's team integrates deep learning algorithms and Rosetta modeling software to develop trRosetta to reduce the consumption of resources and time for prediction [63]. In the future, we will integrate the methods such as co-evolution, deep learning, and deep neural networks to accelerate the speed of plant P450 structure prediction and ligand docking, realize the structure prediction and functional screen of million P450 genes.
Materials and Methods
Data collection of plant P450 enzymes to identify functions
We manually search for the keyword “plant cytochrome P450” from public databases (KEGG, NCBI, UniProt, EMBL-EBI, etc.) and plant-related journals (Plant Physiology, The Plant Journal, Nature plants, etc.) to find plant P450s with identified functions. The sequence information, mutation modification, catalytic reaction, participation in metabolic pathways, and other related information were also searched. At last, 181 plant P450s with known functions were summarized in Table S1 and our PCPD database. All these data will be updated every six months.
PCPCM process
Preprocessing of the input file: The user submits the amino acid sequence (target sequence) of plant P450, which is required to be the standard fasta format (i.e., Target.fasta). Since the N-terminal part of the plant P450 enzyme is a trans-membrane region, in order to improve the accuracy of sequence alignment and the feasibility of later molecular dynamics optimization, TMHMM Server v.2.0 (the most popular transmembrane area prediction software) was first used to predict and clip Target.fasta transmembrane region sequence to obtain Target_RTM.fasta [64,65]. NCBI-blast-2.8.1 was then used to retrieve the homologous structure sequence of Target_RTM.fasta in the P450 structure library which include all 101 PDB crystal structures filtered with 90% identity, and up to five crystal structures with the highest identity were set as the template structures for homology modeling (template1.pdb, template2.pdb, template3.pdb …). The template library will be updated every six months.
Generation of initial conformation: RosettaCM was used to build a three-dimensional structure model for plant P450s, and Clustal Omega was used to compare three template and target sequences [66]. Totally 500 models for the target were produced, and the model with the lowest total_score was chosen as the candidate. And then the all-atom refinement of the candidate structure was implemented using Rosetta relax application. 100 models were produced to fully sample the local conformers. Finally, the model with the lowest total_score was selected as the result. The REF2015 energy function was used in RosettaCM and Rosetta relax applications [29,47].
Adding the prosthetic group HEM: In the plant P450 enzyme structure, the CYS (heme-ligated cysteine) near the C-terminus and the prosthetic group HEM are coordinated to form the active reaction center. The presence of HEM will change the conformation around the active center. For the optimization of the spatial position of the HEM, we collected all relative information of heme and ligated CYS from all P450 crystal structures, and constructed the PDB_CST database (dihedral angle, angle, and distance data between heme, CYS, and CYS adjacent amino acids). According to the types of CYS and adjacent amino acids in the model, search for matching template data in the PDB_CST database, so as to place heme in the model structure. Therefore, the spatial position of the HEM in the unknown structure can be optimized through the existing HEM and the surrounding spatial position relationship. UCSF Chimera's StructSeqAlign was used to align the candidate structure with the template, so as to determine the general position of the HEM in the candidate structure according to the position of the HEM in the template [67]. Then, by analyzing the relative positional relationship between HEM and CYS in all P450 enzymes, the spatial position of HEM is optimized (Fig. S4), and a plant P450 enzyme complex structure with catalytic activity is obtained.
Molecular Dynamics Protocol: By using AMBER16, the plant P450 structure model was optimized by molecular dynamics simulation with 15ns. Firstly, pdb4amber was used to preprocess the protein structure (hydrogen deletion and residue fixes). The heme molecule file and parameter file (HEM.mol2 and HEM.frcmod) were directly obtained from a previous study (Shahrokh et al.) [68]. The force field parameter files and corresponding library files for the ligands were generated with the parmchk and tleap modules from amber package. Secondly, ff14SB [69], GAFF [70], and TIP3P [71] force fields were used to load protein and ligand files in tleap. The whole system constructed is solvated into an explicit octahedral TIP3P water box (box radius is 12.0 Å), and the system was neutralized by the addition of explicit counterions (Na+ and Cl−). Thirdly, we performed a 1000-step steepest descent for the complex system followed by a 1000-cycle conjugate gradient, and the system was minimized with a 50 kcal/mol/Å2 constraint on protein atoms [72]. After 2500 steps of steepest descent followed by a 2500-cycle conjugate gradient, the system was minimized without restraint for a short period of time, reducing the conflict between the protein and the water box. Fourthly, the system was heated from 0 K to 300 K under constant pressure, and the total time is 50ps, and the step size is 2 fs [73]. The backbone atoms of the protein are constrained by 30.0 kcal/mol/Å2. Fifthly, we performed a constant volume MD of 50ps, and constrained the protein atoms with 20 kcal/mol/Å2 to adjust the density of the system. Then, we applied a constant pressure of 500 ps and constrained the protein atoms to 10 kcal/mol/Å2. Sixthly, under constant temperature and pressure conditions (ntb = 2, ntp = 1), through six consecutive stages, the constraints on the protein backbone were gradually reduced and removed, and MD without constraints was run for 10 ns. In the process of operation, SHAKE constraints were used for hydrogen atoms (ntc = 2, ntf = 2), and the temperature was controlled by a time step of 2fs and Langevin dynamics (ntt = 3, gamma_ln = 2.0) [74]. The generated trajectory file was analyzed using Amber16's cpptraj program, and the optimized optimal structure model was obtained by calculating temperature, density, total energy, and RMSD.
PCPLD process
Ligand Docking: In our docking process, we use CpdI (Active oxidant) instead of heme as a cofactor for plant P450 enzymes [75,76]. We pretreatment the protein structure, CpdI (coordinated with CYS), and ligand before docking using the RosettaLigand program. The RosettaLigand application was used to dock the ligand molecule to the active center of the protein structure and a monte carlo minimization process was performed to generate a total of 10,000 docking decoys.
Scoring functions for docking: Firstly, the top 100 docking poses with the highest score (Total_score and interface_delta_X) were selected; Secondly, the selected poses were clustered with Calibur software and the top 10 poses in each cluster were then filtered by distance (between CpdI oxygen and ligand atoms) to obtain suitable candidates (3.5~5 Å). Based on filtering results, two scoring items of RosettaLigand docking results: total score (Total_score) and ligand binding free energy (interface_delta_X) are analyzed. The empirical formula is: Score = K1·Vtotal + K2·Vx, where Vtotal and Vx are the average values of the Total_score and interface_delata_X in all candidates, K1 and K2 are set to −0.1 and −0.9, respectively. Finally, one Score value was calculated based on the formula above and each docking experiment corresponds to one Score value.
Application of other homology modeling methods: In this article, for the model to predict the real crystal structure, the Swiss-Model and OROIN methods of the webserver, and the localized scripts of the I-TASSER and MODELLER methods are used. Among them, the localization script command used by I-TASSER is: perl/PATH/I-TASSER5.0/I-TASSERmod/runI-TASSER.pl -pkgdir/PATH/I-TASSER5.0 -libdir/PATH/I-TASSER5 0.0/ITLIB -ntemp 50 -LBS true -EC true -GO true -seqname Target.fasta -datadir/PATH/I-TASSER5.0/Target_Dir/-java_home/usr/bin/java -light true -hours 10 -outdir/PATH/I-TASSER5.0/Target_Dir/-homoflag = benchmark.
We use the parameter -homoflag = benchmark to exclude templates that are homologous in the database.
The localization process of MODELLER is: First, finding the structure related to the target sequence (search.py); second, preparing all the structure templates and target sequence (compare.py); then, aligning the template structure with the target sequence (production alignment file): Finally, performing homology modeling (get-model.py). The scripts used here are all provided by MODELLER.
Availability and implementation
The important parameter and configuration files of PCPD are available at https://github.com/JiangLab2020/PCPD.
CRediT authorship contribution statement
Hui Wang: authors discussed the results and commented on the manuscript, wrote the manuscript, constructed PCPCM and PCPLD processes. Qian Wang: authors discussed the results and commented on the manuscript, constructed PCPCM and PCPLD processes. Yuqian Liu: constructed the web-based plant P450 database, authors discussed the results and commented on the manuscript. Xiaoping Liao: authors discussed the results and commented on the manuscript, constructed the web-based plant P450 database. Huanyu Chu: authors discussed the results and commented on the manuscript. Hong Chang: authors discussed the results and commented on the manuscript. Yang Cao: authors discussed the results and commented on the manuscript. Zhigang Li: authors discussed the results and commented on the manuscript. Tongcun Zhang: authors discussed the results and commented on the manuscript. Jian Cheng: wrote the manuscript, authors discussed the results and commented on the manuscript. Huifeng Jiang: designed the study, authors discussed the results and commented on the manuscript.
Declaration of competing interest
The authors declare no competing financial interests.
Acknowledgments
This work was supported by grants from the National Key R&D Program of China (2020YFC1316400 and No. 2019YFA0905700), the National Natural Science Foundation of China (NSFC; Grant Nr. 31670100); as well as grants by the NSFC (81560621); and the National Science Fund for Excellent Young Scholars (31922047).
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.synbio.2021.04.004.
Contributor Information
Jian Cheng, Email: cheng_j@tib.cas.cn.
Huifeng Jiang, Email: jiang_hf@tib.cas.cn, http://p450.biodesign.ac.cn/.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Jung S.T., Lauchli R., Arnold F.H. Cytochrome P450: taming a wild type enzyme. Curr Opin Biotechnol. 2011;22:809–817. doi: 10.1016/j.copbio.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seki H., Tamura K., Muranaka T. P450s and UGTs: key players in the structural diversity of triterpenoid saponins. Plant Cell Physiol. 2015;56:1463–1471. doi: 10.1093/pcp/pcv062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao Q., Cui M.Y., Levsh O., Yang D., Liu J., Li J., Hill L., Yang L., Hu Y., Weng J.K. Two CYP82D enzymes function as flavone hydroxylases in the biosynthesis of root-specific 4'-deoxyflavones in scutellaria baicalensis. Mol Plant. 2018;11:135–148. doi: 10.1016/j.molp.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ikezawa N., Iwasa K., Sato F. Molecular cloning and characterization of CYP80G2, a cytochrome P450 that catalyzes an intramolecular C-C phenol coupling of (S)-reticuline in magnoflorine biosynthesis, from cultured Coptis japonica cells. J Biol Chem. 2008;283:8810–8821. doi: 10.1074/jbc.M705082200. [DOI] [PubMed] [Google Scholar]
- 5.Kandel S., Sauveplane V., Olry A., Diss L., Benveniste I., Pinot F. Cytochrome P450-dependent fatty acid hydroxylases in plants. Phytochemistry Rev. 2006;5:359–372. [Google Scholar]
- 6.Nelson D.R. Cytochrome P450 diversity in the tree of life. Biochimica et biophysica acta. Proteins Proteom. 2018;1866:141–154. doi: 10.1016/j.bbapap.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X., Cheng J., Zhang G., Ding W., Duan L., Yang J., Kui L., Cheng X., Ruan J., Fan W. Engineering yeast for the production of breviscapine by genomic analysis and synthetic biology approaches. Nat Commun. 2018;9:448. doi: 10.1038/s41467-018-02883-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christ B., Xu C., Xu M., Li F.S., Wada N., Mitchell A.J., Han X.L., Wen M.L., Fujita M., Weng J.K. Repeated evolution of cytochrome P450-mediated spiroketal steroid biosynthesis in plants. Nat Commun. 2019;10:3206. doi: 10.1038/s41467-019-11286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng J., Chen J., Liu X., Li X., Zhang W., Dai Z. The origin and evolution of the diosgenin biosynthetic pathway in yam. Plant Commun. 2020;2(1):2590–3462. doi: 10.1016/j.xplc.2020.100079. 100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobson M.P., Kalyanaraman C., Zhao S., Tian B. Leveraging structure for enzyme function prediction: methods, opportunities, and challenges. Trends Biochem Sci. 2014;39:363–371. doi: 10.1016/j.tibs.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang D.E., Morris J.S., Rowley M., Torres M.A., Maksimovich V.A., Facchini P.J., Ng K.K.S. Structure-function studies of tetrahydroprotoberberine N-methyltransferase reveal the molecular basis of stereoselective substrate recognition. J Biol Chem. 2019;294:14482–14498. doi: 10.1074/jbc.RA119.009214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim Y.H., Kwon T., Yang H.J., Kim W., Youn H., Lee J.Y., Youn B. Gene engineering, purification, crystallization and preliminary X-ray diffraction of cytochrome P450 p-coumarate-3-hydroxylase (C3H), the Arabidopsis membrane protein. Protein Expr Purif. 2011;79:149–155. doi: 10.1016/j.pep.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 13.Fujiyama K., Hino T., Kanadani M., Watanabe B., Jae Lee H., Mizutani M., Nagano S. Structural insights into a key step of brassinosteroid biosynthesis and its inhibition. Nat Plants. 2019;5:589–594. doi: 10.1038/s41477-019-0436-6. [DOI] [PubMed] [Google Scholar]
- 14.Niu G., Wang J. Structural basis for plant lutein biosynthesis from α-carotene. Proc Natl Acad Sci Unit States Am. 2020;117:14150–14157. doi: 10.1073/pnas.2001806117. G.Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee D.S., Nioche P., Hamberg M., Raman C.S. Structural insights into the evolutionary paths of oxylipin biosynthetic enzymes. Nature. 2008;455:363–368. doi: 10.1038/nature07307. [DOI] [PubMed] [Google Scholar]
- 16.Li L., Chang Z., Pan Z., Fu Z.Q., Wang X. Modes of heme binding and substrate access for cytochrome P450 CYP74A revealed by crystal structures of allene oxide synthase. Proc Natl Acad Sci USA. 2008;105:13883–13888. doi: 10.1073/pnas.0804099105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu M., Wang M., Guo J., Shi C., Deng J., Huang L., Huang L., Chang Z. Crystal structure of CYP76AH1 in 4-PI-bound state from Salvia miltiorrhiza. Biochem Biophys Res Commun. 2019;511:813–819. doi: 10.1016/j.bbrc.2019.02.103. [DOI] [PubMed] [Google Scholar]
- 18.Zhang B., Lewis K.M., Abril A., Davydov D.R., Vermerris W., Sattler S.E., Kang C. Structure and function of the cytochrome P450 monooxygenase cinnamate 4-hydroxylase from sorghum bicolor. Plant Physiol. 2020;183:957–973. doi: 10.1104/pp.20.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamberger B., Bak S. Plant P450s as versatile drivers for evolution of species-specific chemical diversity. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368:20120426. doi: 10.1098/rstb.2012.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee D., Redfern O., Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007;8:995–1005. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- 21.Kufs J.E., Hoefgen S., Rautschek J., Bissell A.U., Graf C., Fiedler J., Braga D., Regestein L., Rosenbaum M.A., Thiele J. Rational design of flavonoid production routes using combinatorial and precursor-directed biosynthesis. ACS Synth Biol. 2020;9:1823–1832. doi: 10.1021/acssynbio.0c00172. [DOI] [PubMed] [Google Scholar]
- 22.Nelson D.R., Koymans L., Kamataki T., Stegeman J.J., Feyereisen R., Waxman D.J., Waterman M.R., Gotoh O., Coon M.J., Estabrook R.W. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Vasav A.P., Barvkar V.T. Phylogenomic analysis of cytochrome P450 multigene family and their differential expression analysis in Solanum lycopersicum L. suggested tissue specific promoters. BMC Genom. 2019;20:1–13. doi: 10.1186/s12864-019-5483-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei K., Chen H. Global identification, structural analysis and expression characterization of cytochrome P450 monooxygenase superfamily in rice. BMC Genom. 2018;19:1–18. doi: 10.1186/s12864-017-4425-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasemann C.A., Boddupalli S.S. Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure. 1995;3:41–62. doi: 10.1016/s0969-2126(01)00134-4. K.R.G. [DOI] [PubMed] [Google Scholar]
- 26.Oezguen N. Analysis of cytochrome P450 conserved sequence motifs between helices E and H: prediction of critical motifs and residues in enzyme functions. J Drug Metabol Toxicol. 2011;2:1000110. doi: 10.4172/2157-7609.1000110. K.S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otyepka M., Anzenbacher P. Is there a relationship between the substrate preferences and structural flexibility of cytochromes P450? Curr Drug Metabol. 2012;13:130–142. doi: 10.2174/138920012798918372. B.K. [DOI] [PubMed] [Google Scholar]
- 28.Song Y., DiMaio F., Wang R.Y., Kim D., Miles C., Brunette T., Thompson J., Baker D. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alford R.F., Leaver-Fay A., Jeliazkov J.R., O'Meara M.J., DiMaio F.P., Park H., Shapovalov M.V., Renfrew P.D., Mulligan V.K., Kappel K. The Rosetta all-atom energy function for macromolecular modeling and design. J Chem Theor Comput. 2017;13:3031–3048. doi: 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feig M., Mirjalili V. Protein structure refinement via molecular-dynamics simulations: what works and what does not? Proteins. 2016;84(Suppl 1):282–292. doi: 10.1002/prot.24871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang G., Su Z. CYPSI: a structure-based interface for cytochrome P450s and ligands in Arabidopsis thaliana. BMC Bioinf. 2012;13:1–10. doi: 10.1186/1471-2105-13-332. Z.Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D., Geng L., Zhao Y.J., Yang Y., Huang Y., Zhang Y., Shen H.B. Artificial intelligence-based multi-objective optimization protocol for protein structure refinement. Bioinformatics. 2020;36:437–448. doi: 10.1093/bioinformatics/btz544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webb B., Sali A. Comparative protein structure modeling using MODELLER. Curr Protocols Bioinfo. 2014;47 doi: 10.1002/0471250953.bi0506s47. 5.6.1-5.6.32. [DOI] [PubMed] [Google Scholar]
- 36.Ghouzam Y., Postic G., Guerin P.E., de Brevern A.G., Gelly J.C. ORION: a web server for protein fold recognition and structure prediction using evolutionary hybrid profiles. Sci Rep. 2016;6:28268. doi: 10.1038/srep28268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.J V.G. What IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 38.Lüthy R., Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. B.J.U. [DOI] [PubMed] [Google Scholar]
- 39.Colovos C. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. Y.T.O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pontius J R.J., Wodak S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J Mol Biol. 1996;264:121–136. doi: 10.1006/jmbi.1996.0628. [DOI] [PubMed] [Google Scholar]
- 41.Laskowski R.A., Moss D.S. Procheck - a program to check the stereochemical quality of protein structures. Thornton J M. 1993;26:283–291. M.M.W. [Google Scholar]
- 42.Studer G., Rempfer C., Waterhouse A.M., Gumienny R., Haas J., Schwede T. QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatics. 2020;36:1765–1771. doi: 10.1093/bioinformatics/btz828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J., DellaPenna D. Defining the primary route for lutein synthesis in plants: the role of Arabidopsis carotenoid beta-ring hydroxylase CYP97A3. Proc Natl Acad Sci USA. 2006;103:3474–3479. doi: 10.1073/pnas.0511207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matthias WüST R.B.C. Hydroxylation of specifically deuterated limonene enantiomers by cytochrome p450 limonene-6-hydroxylase reveals the mechanism of multiple product formation. Biochemistry. 2002;41:1820–1827. doi: 10.1021/bi011717h. [DOI] [PubMed] [Google Scholar]
- 45.Shahrokh K., Orendt A., Yost G.S., Cheatham T.E., 3rd Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J Comput Chem. 2012;33:119–133. doi: 10.1002/jcc.21922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S.C., Ng Y.K. Calibur: a tool for clustering large numbers of protein decoys. BMC Bioinf. 2010;11:25. doi: 10.1186/1471-2105-11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowman G.R., Pande V.S. Simulated tempering yields insight into the low-resolution Rosetta scoring functions. Proteins. 2009;74:777–788. doi: 10.1002/prot.22210. [DOI] [PubMed] [Google Scholar]
- 48.Reed J., Stephenson M.J., Miettinen K., Brouwer B., Leveau A., Brett P., Goss R.J.M., Goossens A., O'Connell M.A., Osbourn A. A translational synthetic biology platform for rapid access to gram-scale quantities of novel drug-like molecules. Metab Eng. 2017;42:185–193. doi: 10.1016/j.ymben.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sawada Y., Akashi T. Key amino acid residues required for aryl migration catalysed by the cytochrome P450 2‐hydroxyisoflavanone synthase. Plant J. 2002;31:555–564. doi: 10.1046/j.1365-313x.2002.01378.x. K.K. [DOI] [PubMed] [Google Scholar]
- 50.Li J., Liang Q., Li C., Liu M., Zhang Y. Comparative transcriptome analysis identifies putative genes involved in dioscin biosynthesis in Dioscorea zingiberensis. Molecules. 2018;23:454. doi: 10.3390/molecules23020454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu H., Song J., Luo H., Zhang Y., Li Q., Zhu Y., Xu J., Li Y., Song C., Wang B. Analysis of the genome sequence of the medicinal plant salvia miltiorrhiza. Mol Plant. 2016;9:949–952. doi: 10.1016/j.molp.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Renault H., Bassard J.E., Hamberger B., Werck-Reichhart D. Cytochrome P450-mediated metabolic engineering: current progress and future challenges. Curr Opin Plant Biol. 2014;19:27–34. doi: 10.1016/j.pbi.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 53.Guo J., Ma X., Cai Y., Ma Y., Zhan Z., Zhou Y.J., Liu W., Guan M., Yang J., Cui G. Cytochrome P450 promiscuity leads to a bifurcating biosynthetic pathway for tanshinones. New Phytol. 2016;210:525–534. doi: 10.1111/nph.13790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jorgensen K., Morant A.V., Morant M., Jensen N.B., Olsen C.E., Kannangara R., Motawia M.S., Moller B.L., Bak S. Biosynthesis of the cyanogenic glucosides linamarin and lotaustralin in cassava: isolation, biochemical characterization, and expression pattern of CYP71E7, the oxime-metabolizing cytochrome P450 enzyme. Plant Physiol. 2011;155:282–292. doi: 10.1104/pp.110.164053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alcantara J., Bird D.A., Franceschi V.R., Facchini P.J. Sanguinarine biosynthesis is associated with the endoplasmic reticulum in cultured opium poppy cells after elicitor treatment. Plant Physiol. 2005;138:173–183. doi: 10.1104/pp.105.059287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinot R. CYP94A5, a new cytochrome P450 from Nicotiana tabacum is able to catalyze the oxidation of fatty acids to the ω‐alcohol and to the corresponding diacid. Eur J Biochem. 2001;268:3083–3090. doi: 10.1046/j.1432-1327.2001.02207.x. L.B.M.S.R.K.I.B.J.P.S.L.S.F.D.F. [DOI] [PubMed] [Google Scholar]
- 57.Liu X., Zhu X., Wang H., Liu T., Cheng J., Jiang H. Discovery and modification of cytochrome P450 for plant natural products biosynthesis. Synth Syst Biotechnol. 2020;5:187–199. doi: 10.1016/j.synbio.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schoch G.A., Attias R., Belghazi M., Dansette P.M., Werck-Reichhart D. Engineering of a water-soluble plant cytochrome P450, CYP73A1, and NMR-based orientation of natural and alternate substrates in the active site. Plant Physiol. 2003;133:1198–1208. doi: 10.1104/pp.103.020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nikolaev D.M., Shtyrov A.A., Panov M.S., Jamal A., Chakchir O.B., Kochemirovsky V.A., Olivucci M., Ryazantsev M.N. A comparative study of modern homology modeling algorithms for rhodopsin structure prediction. ACS Omega. 2018;3:7555–7566. doi: 10.1021/acsomega.8b00721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schaarschmidt J., Monastyrskyy B., Kryshtafovych A., Bonvin A. Assessment of contact predictions in CASP12: Co-evolution and deep learning coming of age. Proteins. 2018;86(Suppl 1):51–66. doi: 10.1002/prot.25407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.≤1-s2.0-S0969212601001344-main.pdf>...
- 62.Senior A.W., Evans R., Jumper J., Kirkpatrick J., Sifre L., Green T., Qin C., Zidek A., Nelson A.W.R., Bridgland A. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. doi: 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- 63.Yang J., Park H. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci Unit States Am. 2020;117:1496–1503. doi: 10.1073/pnas.1914677117. A.I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krogh A L.B., Von Heijne G. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 65.Möller S., Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17:646–653. doi: 10.1093/bioinformatics/17.7.646. C.M.D.R. [DOI] [PubMed] [Google Scholar]
- 66.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 68.Shahrokh K., Orendt A., Yost G.S., Iii T. Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J Comput Chem. 2011;33:119–133. doi: 10.1002/jcc.21922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maier J.A., Kasavajhala K. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theor Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. M.C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang J, Caldwell J.W. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. W.R.M. [DOI] [PubMed] [Google Scholar]
- 71.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 72.Dubey K.D., Wang B., Shaik S. Molecular dynamics and QM/MM calculations predict the substrate-induced gating of cytochrome P450 BM3 and the regio- and stereoselectivity of fatty acid hydroxylation. J Am Chem Soc. 2016;138:837–845. doi: 10.1021/jacs.5b08737. [DOI] [PubMed] [Google Scholar]
- 73.Dubey K.D., Wang B., Vajpai M., Shaik S. MD simulations and QM/MM calculations show that single-site mutations of cytochrome P450BM3 alter the active site's complexity and the chemoselectivity of oxidation without changing the active species. Chem Sci. 2017;8:5335–5344. doi: 10.1039/c7sc01932g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mustafa G., Nandekar P.P., Mukherjee G., Bruce N.J., Wade R.C. The effect of force-field parameters on cytochrome P450-membrane interactions: structure and dynamics. Sci Rep. 2020;10:7284. doi: 10.1038/s41598-020-64129-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hammerer L., Winkler C.K., Kroutil W. Regioselective biocatalytic hydroxylation of fatty acids by cytochrome P450s. Catal Lett. 2017;148:787–812. [Google Scholar]
- 76.Matthews S., Belcher J.D., Tee K.L., Girvan H.M., McLean K.J., Rigby S.E., Levy C.W., Leys D., Parker D.A., Blankley R.T. Catalytic determinants of alkene production by the cytochrome P450 peroxygenase OleTJE. J Biol Chem. 2017;292:5128–5143. doi: 10.1074/jbc.M116.762336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.