

Abstract
Lung adenocarcinoma (LUAD) is the most frequent subtype of lung cancer worldwide. However, the survival rate of LUAD patients remains low. N6-methyladenosine (m6A) and long noncoding RNAs (lncRNAs) play vital roles in the prognostic value and the immunotherapeutic response of LUAD. Thus, discerning lncRNAs associated with m6A in LUAD patients is critical. In this study, m6A-related lncRNAs were analyzed and obtained by coexpression. Univariate, least absolute shrinkage and selection operator (LASSO), and multivariate Cox regression analyses were conducted to construct an m6A-related lncRNA model. Kaplan-Meier analysis, principal-component analysis (PCA), functional enrichment annotation, and nomogram were used to analyze the risk model. Finally, the potential immunotherapeutic signatures and drug sensitivity prediction targeting this model were also discussed. The risk model comprising 12 m6A-related lncRNAs was identified as an independent predictor of prognoses. By regrouping the patients with this model, we can distinguish between them more effectively in terms of the immunotherapeutic response. Finally, candidate compounds aimed at LUAD subtype differentiation were identified. This risk model based on the m6A-based lncRNAs may be promising for the clinical prediction of prognoses and immunotherapeutic responses in LUAD patients.
Keywords: N6-methyladenosine, m6A, long noncoding RNAs, lncRNAs, prognosis, lung adenocarcinoma, LUAD, immunotherapy
Graphical abstract
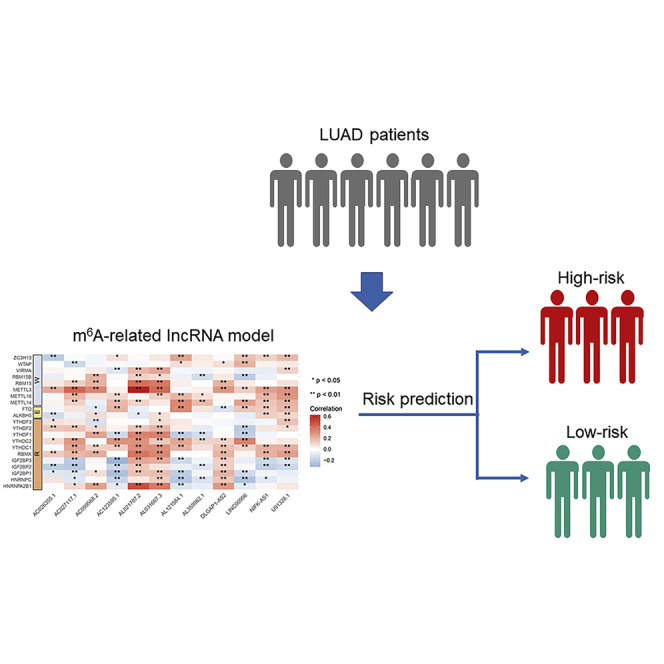
Xu et al. constructed and validated a risk model based on m6A-related lncRNAs in lung adenocarcinoma. This risk model may be promising for the clinical prediction of prognoses and immunotherapeutic responses in lung adenocarcinoma patients.
Introduction
Lung adenocarcinoma (LUAD) is the most frequent subtype of lung cancer worldwide.1, 2, 3 With advances in diagnosis, surgery, radiotherapy, and molecular therapeutics, the clinical outcome of LUAD patients has significantly improved. However, the 5-year survival rate of LUAD patients is still at a low level.4,5 Presently, evidence has shown that the discovery and application of molecular biomarkers can provide prognostic value.6
N6-methyladenosine (m6A), the most abundant RNA modification of eukaryotic cells, is vital in various biological processes and mRNA metabolism, such as RNA processing, transport, and stability.7,8 m6A modification comprises methyltransferase, signal transducers, and demethylase, which are also called writers, readers and erasers, respectively. Additionally, m6A modification is also a reversible RNA epigenetic process.9 Changes in the RNA structure can affect various cell processes; therefore, the effect of m6A-regulated long noncoding RNAs (lncRNAs) may be crucial for the proliferation and migration of cancer cells.10
Recent studies have shown that m6A modification regulates oncogenesis and tumor development. For example, FEZF1-AS1 influenced by m6A modification regulates the ITGA11/miR-516b-5p axis and finally is upregulated in non-small cell lung cancer (NSCLC).11 Additionally, m6A methyltransferase-like 3 (METTL3)-induced lncRNA ABHD11-AS1 is upregulated in NSCLC, and its ectopic expression is closely related to the poor prognosis of patients with NSCLC.12 Recently, several bioinformatics studies have shown that the dysregulation of m6A regulators is involved in LUAD.13,14 The specific role of m6A regulators in lncRNAs remains unclear; therefore, understanding the mechanism of m6A-related lncRNAs in the development of LUAD may be useful for prognostic targets.
In our study, we abstracted the expression profiles of 14,142 lncRNAs and 21 m6A genes from The Cancer Genome Atlas (TCGA) dataset. Next, we identified the m6A-associated lncRNAs using Pearson’s correlation analysis. This model is based on a novel prognostic model of m6A, which was developed to forecast the overall survival (OS) of patients with LUAD. Next, using the publicly available drug sensitivity database, we discovered candidate drugs targeting this m6A-related lncRNA signature. Additionally, we explored the relationship association with immunotherapy responses. Finally, we established a nomogram to predict the OS of patients with LUAD.
Results
Identification of m6A-related lncRNAs in patients with LUAD
The detailed workflow for risk model construction and subsequent analyses is shown in Figure 1. The matrix expression of 21 m6A genes and 14,142 lncRNAs was abstracted from the TCGA database. We defined m6A-related lncRNAs as lncRNAs that were significantly related to greater than or equal to one of the 21 m6A genes (|Pearson R| > 0.3 and p < 0.001). Finally, the m6A-lncRNA coexpression network was visualized using the Sankey diagram in Figure 2A, and 1,149 m6A-related lncRNAs were discerned as m6A-related lncRNAs. The correlation between m6A genes and m6A-related lncRNAs in the TCGA entire set is shown in Figure 2B.
Figure 1.

Flow chart of this study
Figure 2.

Identification of m6A-related lncRNAs in LUAD patients
(A) Sankey relational diagram for 21 m6A genes and m6A-related lncRNAs. (B) Heatmap for the correlations between 21 m6A genes and the 12 prognostic m6A-related lncRNAs.
Construction and validation of a risk model according to m6A-related lncRNAs in LUAD patients
We screened m6A-associated prognostic lncRNAs from 1,149 m6A-associated lncRNAs in the TCGA training set using univariate Cox regression analysis. Thirty-eight m6A-related lncRNAs in the TCGA dataset were significantly correlated with OS (Figure 3A). LASSO-penalized Cox analysis is a common method of multiple regression analysis. The application of this method not only enhances the forecast accuracy and explainability of the statistical model but also makes variable options and regularization simultaneously. This method is extensively applied for the optimal choice of characteristics in high-dimensional data with an inferior correlation and prominent forecasted value to avoid overfitting. Consequently, this method can effectively discern the most available forecast markers and produce a prognostic indicator to predict clinical results. The dashed perpendicular line illustrates the first-rank value of log λ with the minimum segment likelihood bias. Hence, 24 m6A-related lncRNAs were selected for the subsequent multivariate analysis (Figures 3B and 3C). Next, we used multivariate Cox ratio hazard regression analysis to distinguish autocephalous prognostic proteins. Twelve m6A-related lncRNAs were prognostic proteins independently correlated with OS in the training queue and were used to construct a risk model to assess the prognostic risk of patients with LUAD (Figure 3D). LUAD samples were categorized into low- and high-risk groups based on the median value of the prognostic risk grade. The distribution of risk grades between the low-risk and high-risk groups is depicted in Figure 4A, and the survival status and survival time of patients in the two different risk groups are shown in Figure 4B. The relative expression standards of the 12 m6A-related lncRNAs for each patient are shown in Figure 4C. The survival analysis demonstrated that the OS of the low-risk group was longer than that of the high-risk group (p < 0.001) (Figure 4D).
Figure 3.

Risk model for LUAD patients based on m6A-related lncRNAs
(A) Univariate Cox regression analysis revealed that the selected lncRNAs significantly correlated with clinical prognosis. (B) The tuning parameters (log λ) of OS-related proteins were selected to cross-verify the error curve. According to the minimal criterion and 1-se criterion, perpendicular imaginary lines were drawn at the optimal value. (C) The LASSO coefficient profile of 24 OS-related lncRNAs and perpendicular imaginary line were drawn at the value chosen by 10-fold cross-validation. (D) Multivariate Cox regression analysis showed 12 independent prognostic lncRNAs.
Figure 4.

Prognostic value of the risk patterns of the 12 m6A-related lncRNAs in the TCGA training set
(A) Distribution of m6A-related lncRNA model-based risk score. (B) Different patterns of survival status and survival time between the high- and low-risk groups. (C) Clustering analysis heatmap shows the expression standards of the 12 prognostic lncRNAs for each patient. (D) Kaplan-Meier survival curves of the OS of patients in the high- and low-risk groups.
To test the prognostic capability of this established model, we calculated risk scores for every patient in the test set and entire set using the uniform formula. Figure 5 depicts the distribution of risk grades, pattern of survival status and survival time, and expression of the m6A-related lncRNAs in the testing set (Figures 5A–5C) and entire set (Figures 5E–5G). Kaplan-Meier survival analyses performed on the testing set and entire set showed no differences in the outcomes in the TCGA training set: the OS of LUAD patients with higher risk scores was worse than that of patients with lower risk scores (Figures 5D and 5H). To further predict the ability of the prognostic model, the disease-free interval (DFI), progression-free interval (PFI), and disease-specific survival (DSS) were explored to distinguish high- and low-risk LUAD patients. As predicted, DFI, PFI, and DSS were different between the low- and high-risk groups, indicating that the m6A-related lncRNA model level affected the prognosis of LUAD patients (Figures S1A–S1C).
Figure 5.

Prognostic value of the risk model of the 12 m6A-related lncRNAs in the TCGA testing and entire sets
(A) Distribution of m6A-related lncRNA model-based risk score for the testing set. (B) Patterns of the survival time and survival status between the high- and low-risk groups for the testing set. (C) Clustering analysis heatmap shows the display levels of the 12 prognostic lncRNAs for each patient in the testing set. (D) Kaplan-Meier survival curves of the OS of patients in the high- and low-risk groups for the testing set. (E) Distribution of the m6A-related lncRNA model-based risk score for the entire set. (F) Patterns of the survival time and survival status between the high- and low-risk groups for the entire set. (G) Clustering analysis heatmap shows the expression levels of the 12 prognostic lncRNAs for each patient for the entire set. (H) Kaplan-Meier survival curves of OS of patients in the low- and high-risk groups for the entire set.
The discrepancies in OS stratified by the universal clinicopathologic characteristics were analyzed between the low- and high-risk groups in the TCGA entire set. According to the subgroups classified by gender, age, stage, or tumor stage, the OS of the low-risk group continued to be superior to that of the high-risk group (Figure 6).
Figure 6.

Kaplan-Meier curves of OS differences stratified by gender, age, tumor grade, or TNM stage between the high- and low-risk groups in the TCGA entire set
Principal-component analysis (PCA) further verifies the grouping ability of the m6A-related lncRNA model
PCA was conducted to test the difference between the low-risk and high-risk groups based on the entire gene expression profiles, 21 m6A genes, 12 m6A-related lncRNAs, and risk model classified by the expression profiles of the 12 m6A-related lncRNAs (Figures 7A–7D). Figures 7A–7C display that the distributions of the high- and low-risk groups were relatively scattered. However, the results obtained based on our model illustrated that the low- and high-risk groups had different distributions (Figure 7D). These results suggest that the prognostic signature can distinguish between the low- and high-risk groups.
Figure 7.

Principal component analysis between the high- and low-risk groups based on
(A) entire gene expression profiles, (B) 21 m6A genes, (C) 12 m6A-related lncRNAs, and (D) risk model based on the representation profiles of the 12 m6A-related lncRNAs in the TCGA entire set
Estimation of the tumor immune microenvironment and cancer immunotherapy response using the m6A-related lncRNA model
The enrichment level and activity of several immune cells, pathways, or functions in LUAD were further analyzed based on the m6A-related lncRNA model from 504 LUAD samples. The low-risk and high-risk groups showed prominent differences in the expression of immune indicators (Figure 8A). To explore the underlying molecular mechanisms of the m6A-based model, we performed Gene Ontology (GO) enrichment analysis, which revealed the involvement of many immune-related biological processes (Figure 8B). We next investigated the correlations between the m6A-related lncRNA model and immunotherapeutic biomarkers. Unsurprisingly, we discovered that the high-risk group was more likely to respond to immunotherapy than the low-risk group, indicating that this m6A-based classifier index might serve as an indicator for predicting Tumor Immune Dysfunction and Exclusion (TIDE) (Figure 8C). Using the R package maftools, the mutation data were analyzed and summarized. The mutations were stratified based on the variant effect predictor. The top 20 driver genes with the highest alteration frequency between the high- and low-risk subgroups are shown in Figures 8D and 8E. We then calculated TMB scores based on the TGCA somatic mutation data. The TMB in the low-risk group exceeded that in the high-risk group, showing that the m6A-based classifier index had a high correlation with TMB (Figure 8F). TP53 mutations are correlated with a worse survival and can be used as a prognostic marker in lung cancer. Therefore, we tested whether the m6A-related lncRNA model could predict the OS outcome better than TP53 mutation status. Patients with TP53 mutation and wild-type TP53 in the high-risk groups (defined as TP53 mutation/high and TP53 wild/high, respectively) presented a worse OS than patients with TP53 mutation and wild-type TP53 in the low-risk groups (TP53 mutation/low and TP53 wild/low, respectively) (Figure 8G). Interestingly, patients with wild-type TP53 in the high-risk group (TP53 wild/high) had worse survival outcomes than patients with TP53 mutation in the low-risk group (TP53 mutation/low). The survival curve of patients with TP53 mutation in the high-risk group (TP53 mutation/high) was similar to that of patients with wild-type TP53 in the high-risk group (TP53 wild/high), indicating that the TP53 mutation status failed to distinguish the survival rate in the high-risk group. Thus, these findings indicate that the m6A-related lncRNA model may have greater prognostic significance than the TP53 mutation status.
Figure 8.

Estimation of the tumor immune microenvironment and cancer immunotherapy response using the m6A-related lncRNA model in the TCGA entire set
(A) The indicated standards of the immunity index for each patient. (B) GO enrichment analysis. (C) TIDE prediction difference in the high- and low-risk patients. (D and E) Waterfall plot displays mutation information of the genes with high mutation frequencies in the high-risk group (D) and low-risk group (E). (F) TMB difference in the high- and low-risk patients. (G) Kaplan-Meier curve analysis of OS is shown for patients classified according to the TP53 mutation status and m6A-related lncRNA model.
Identification of novel candidate compounds targeting the m6A-related lncRNA model
To identify potential drugs targeting our lncRNA model for treating LUAD patients, we used the pRRophetic algorithm to estimate the therapeutic response based on the half-maximal inhibitory concentration (IC50) available in the Genomics of Drug Sensitivity in Cancer (GDSC) database for each sample. We found that 78 compounds were screened out for significant differences in the estimated IC50 between these two groups, and the high group was more sensitive to all of these compounds. Figure S2 displays the top 20 compounds that might be used for further analysis in patients with LUAD.
Evaluation of the prognostic risk model of m6A-related lncRNAs and clinical features of LUAD
We performed univariate and multivariate Cox regression analyses to evaluate whether the risk model of 12 m6A-related lncRNAs had independent prognostic characteristics for LUAD. The HR of the risk score and 95% confidence interval (CI) were 1.06 and 1.04–1.08 (p < 0.001) in univariate Cox regression analysis, respectively. In multivariate Cox regression analysis, the HR was 1.07 and 95% CI was 1.05–1.09 (p < 0.001) (Figure 9A), indicating that the risk model of the 12 m6A-related lncRNAs was unrelated to clinicopathological parameters, such as gender, age, tumor/node/metastasis (TNM) stage, and tobacco smoking history. The conformance index of the risk score and the area under the ROC curve (AUC) were assessed. This process was conducted to better assess the uniqueness and susceptibility of risk scores in predicting outcomes in patients with LUAD. With increasing time, the concordance index of the risk score was always greater than that of other clinical factors, suggesting that the risk grade could better forecast the prognosis of LUAD (Figure 9B). The AUC of the risk grade was also higher than the AUCs of other clinicopathological characteristics, showing that the prognostic risk model of the 12 m6A-related lncRNAs for LUAD was comparatively dependable (Figure 9C).
Figure 9.

Assessment of the prognostic risk model of the m6A-related lncRNAs and clinical features in LUAD in the TCGA entire set
(A) Univariate and multivariate analyses of the clinical characteristics and risk score with the OS. (B) Concordance indexes of the risk score and clinical characteristics. (C) ROC curves of the clinical characteristics and risk score.
Construction and evaluation of the prognostic nomogram
The nomogram comprising the risk grade and clinical risk characteristics was fabricated to predict the 1-, 2-, and 3-year OS incidences. By comparison with clinical factors, the risk grade of the prognostic model showed predominant predictive ability in the nomogram (Figure 10A). Correlation charts displayed that the observed versus predicted rates of the 1-, 2-, and 3-year OS revealed ideal consistency (Figures 10B–10D).
Figure 10.

Construction and evaluation of a prognostic nomogram
(A) The nomogram predicts the probability of the 1-, 2-, and 3-year OS. (B–D) The calibration plot of the nomogram predicts the probability of the 1-, 2-, and 3-year OS.
Discussion
As the most common subtype of lung cancer,15 many medical researchers have focused on studying the occurrence, development, and treatment of LUAD in recent years.9,16,17 Accumulating studies have shown that different lung cancer subtypes have distinct clinical characteristics and clinical outcomes; thus, an increasing number of studies focusing on identifying signatures with noncoding RNAs have been conducted to predict the survival and immunotherapeutic response in patients with LUAD.18, 19, 20
As the most abundant posttranscriptional modification in eukaryotic mRNAs and lncRNAs, m6A has extensive regulatory usefulness in regulating mRNA transcription, splicing, and translation and influencing the structure and effect of lncRNAs.10 Additionally, lncRNA-related studies have drawn attention in numerous cancer fields.19 m6A regulators can modify specific lncRNAs to maintain malignant n6-methyladenosine and lncRNAs in various tumors.20 Studies have shown that m6A modification of lncRNAs can affect the occurrence and development of tumors, and lncRNAs may target m6A regulators as competitive endogenous RNAs, affecting tumor invasive progression.10 m6A modification can adjust lncRNA function by supplying binding sites to m6A reader proteins. It can also regulate local RNA structure to permit concrete RNA-binding proteins to enter the surrounding m6A residue. Additionally, m6A modification can affect the formation of the RNA-DNA triple helix, in which a lncRNA binds to the series through the Hoogsteen base pair in the main groove of double-stranded DNA.21 Furthermore, m6A may impact the reciprocity site between lncRNAs and specific DNA.22 Both m6A and lncRNAs are important regulators of LUAD tumorigenesis.23 However, studies on the pathological role of m6A and lncRNAs in LUAD progression remain limited, and studies on the biological mechanisms and prognostic biomarkers of LUAD concerning m6A-related lncRNAs are still lacking.24,25 In the present study, we were inspired by the function of m6A and lncRNAs in LUAD; thus, we attempted to construct an independent model based on m6A-related lncRNAs.
In our study, 1,149 m6A-related lncRNAs from the TCGA dataset were identified in this paper to explore the prognostic function of m6A-related lncRNAs. The TCGA dataset confirmed the prognostic value of 24 m6A-related lncRNAs, and 12 of them were applied to construct an m6A-related lncRNA model to predict the OS of patients with LUAD. Among them, DLGAP1-AS2 contributes to glioma cell proliferation, migration, and apoptosis by upregulating the expression of the downstream target YAP1.26 Additionally, as immune-related lncRNAs, AC123595.1 and AC026355.1 can increase the predicted value of LUAD.27 Additionally, other lncRNAs were revealed for the first time. Subsequently, LUAD patients were separated into high- and low-risk groups based on the intermediate risk score, and the high-risk group had apparently poor clinical results. Multivariate Cox regression analysis showed that the m6A-related lncRNA model was an autocephalous risk element of OS. ROC analysis showed that the model was superior to conventional clinical features in the survival prediction for LUAD. We also established a nomogram showing perfect consistency between the observed and predicted rates for the 1-year, 3-year, and 5-year OS. Finally, the observed versus 1-year, 3-year, and 5-year OS forecasting rates displayed excellent consistency. The risk model based on 12 m6A-related lncRNAs that were independently associated with OS was fairly accurate, and this prediction model could identify novel biomarkers for subsequent studies.
The TMB is the total number of somatic coding mutations and is related to the emergence of neoantigens that trigger antitumor immunity.28 Recent studies revealed the TMB as a valid biomarker to predict the response to PD-L1 treatment.29 We found that the TMB in the low-risk group exceeded that the in high-risk group. In addition, an increasing number of studies have used the TIDE prediction score, which is a computational framework developed for immunotherapeutic prediction,30 and its predictive function has been validated successfully. In our study, the prediction of the TIDE algorithm suggests that patients with the high-risk subtype have a superior response to immunotherapy. According to the above results, we infer that this prediction model may provide dependable immune biomarkers for oncotherapy. Additionally, our study provides new insight into the molecular biological mechanism of m6A-related lncRNAs in LUAD.
In the clinic, the pathological stage is the decisive factor in the prognosis of LUAD.31 However, LUAD patients at the same stage always have different clinical outcomes, suggesting that the present periodization systems in providing dependable predictions and reflecting the heterogeneity of LUAD are inaccurate.32 Therefore, latent predictive and therapeutic biomarkers should be explored. The established m6A-related lncRNA model provides a new method for prognostic prediction in LUAD patients. The results also provide insight for future studies on the process and mechanism of m6A modification of lncRNAs. In our study, several methods were used to confirm this novel model, and we might choose the optimal model and utilize it evenly. We assumed that the prediction model was acceptable without the external data validation. We are also aware of some shortcomings and limitations in this study. External validation by other clinical datasets would be beneficial, and the biological mechanism of m6A-related lncRNAs has not been fully elucidated. Thus, we will recollect clinical samples and expand the sample size. Additionally, we will attempt to validate the accuracy of this model via more external experiments to explore the role of lncRNAs and their interaction with m6A-related genes in our following work.
In conclusion, our study provides clues for prognostic prediction in patients with LUAD and may help elucidate the process and mechanism of m6A-regulated lncRNAs. In addition, the prediction model shows sensitivity in identifying LUAD patients who may respond well to immunotherapy.
Materials and methods
Acquisition of information of patients with LUAD
Using VarScan software, we obtained RNA sequence transcriptome data, relevant clinical information, and mutation data of LUAD patients from the TCGA (https://cancergenome.nih.gov/) database. To reduce statistical bias in this analysis, LUAD patients with missing OS values were excluded.
Selection of m6A genes and m6A-related lncRNAs
We obtained the profiles of lncRNAs and m6A genes from the TCGA database. According to previous studies, the expression matrixes of 21 m6A genes were retrieved from the TCGA, including the expression data of writers (METTL3, METTL14, METTL16, VIRMA, RBM15, RBM15B, ZC3H13, and WTAP), readers (IGF2BP1, IGF2BP2, IGF2BP3, YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, HNRNPA2B1, HNRNPC, and RBMX), and erasers (ALKBH5 and FTO).9 We screened m6A-related lncRNAs by Pearson’s correlation analysis, and 1,149 m6A-related lncRNAs were identified. The process used the criteria of |Pearson R| >0.3 and p <0.001.
Establishment and validation of the risk signature
The entire TCGA set was randomized as a training set and the testing set. The training set was utilized to construct an m6A-related lncRNA model, and the entire set and testing set were applied to validate this established model. Table S1 shows the baseline characteristics of these two sets. No significant differences in clinical properties were observed between the two datasets (p > 0.05). Combined with LUAD survival information in TCGA, we screened the prognosis of m6A-related lncRNAs from 1,149 m6A-related lncRNAs in the TCGA dataset (p < 0.05), and univariate Cox regression was used in this study.33 Using the R package glmnet to conduct LASSO Cox regression (using the penalty parameter estimated by 10-fold cross-validation), we found that 24 m6A-related lncRNAs were distinctly related to the OS of LUAD patients from TCGA datasets.34 Multifactor Cox regression was applied to analyze the 24 m6A-related lncRNAs, and a 12-m6A-related lncRNA risk model was ultimately established.3 The following formula was used to calculate the risk score: Risk score = coef (lncRNA1) × expr (lncRNA1) + coef (lncRNA2) × expr (lncRNA2) + …… + coef (lncRNAn) × expr (lncRNAn), where coef indicates the coefficients, coef (lncRNAn) was the coefficient of lncRNAs correlated with survival, and expr (lncRNAn) was the expression of lncRNAs. According to the median risk score, subgroups including low- and high- risk groups were established.35
Functional analysis
We performed GO analysis to identify the differentially expressed genes. This process utilizes the R package clusterProfiler. The analysis threshold was determined by the p value, and p <0.05 indicates that the functional comment is significantly enriched.16,36
Exploration of the model in the immunotherapeutic treatment
We used the R package maftools to evaluate and sum the mutation data. The TMB was measured according to tumor-specific mutated genes.37 We used the TIDE algorithm to predict the likelihood of the immunotherapeutic response.33
PCA and Kaplan-Meier survival analysis
PCA was used for effective dimensionality reduction, model identification, and grouping visualization of high-dimensional data of the entire gene expression profiles, 21 m6A genes, 12 m6A-related lncRNAs, and risk model according to the expression patterns of the 12 m6A-related lncRNAs.38 We used Kaplan-Meier survival analysis to appraise diversities in the OS between the high-risk and low-risk groups. The R packages survMiner and survival were tools to enable this process.3
Exploration of potential compounds targeting m6A-related lncRNA model in clinical treatment
To obtain potential compounds in the clinic for LUAD treatment, we calculated the IC50 of compounds obtained from the GDSC website in the TCGA project of the LUAD dataset. The R package pRRophetic was used to predict the IC50 of compounds obtained from the GDSC website in patients with LUAD.
Independence of the m6A-related lncRNA model
Multivariate and univariate Cox regression analyses were conducted to test whether the prognostic pattern was an independent variable considering other clinical characteristics (gender, age, TNM stage, T stage, N stage, M stage, and tobacco smoking history) in the patients with LUAD.16
Establishing and proving a predictive nomogram
The predictive ability of the nomogram and other predictors (age, gender, risk score, TNM stage, T stage, N stage, M stage, and tobacco smoking history) for the 1-, 3-, and 5-year OS was set up. Correction curves based on the Hosmer-Lemeshow test were applied to illustrate the uniformity between the practical outcome and model prediction outcome.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81672640); the Grant for Key Disciplinary Project of Clinical Medicine under the Guangdong High-level University Development Program; the 2020 Li Ka Shing Foundation Cross-Disciplinary Research Grant (2020LKSFG04A and 2020LKSFG10A); the Dengfeng Project for the construction of high-level hospitals in Guangdong Province–The First Affiliated Hospital of Shantou University Medical College Supporting Funding (2019-70); the Guangdong Basic and Applied Basic Research Foundation (2021A1515010137, 2020A1515011519); and the Medical Science and Technology Research Foundation of Guangdong Province (A2021409, A2020430).
Author contributions
F.X., X.H., and Y.L. designed the study, analyzed data, and wrote the manuscript. F.X., L.L., and Y.C. provided funding acquisition. L.L. and Y.C. supervised the research, analyzed data, and wrote the manuscript. All authors read and approved the final submitted manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.04.003.
Contributor Information
Yongsong Chen, Email: yongsongchen@126.com.
Ling Lin, Email: llinc@163.net.
Supplemental information
References
- 1.Cao M., Li H., Sun D., Chen W. Cancer burden of major cancers in China: A need for sustainable actions. Cancer Commun. (Lond.) 2020;40:205–210. doi: 10.1002/cac2.12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferlay J., Colombet M., Soerjomataram I., Dyba T., Randi G., Bettio M., Gavin A., Visser O., Bray F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer. 2018;103:356–387. doi: 10.1016/j.ejca.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Xu F., He L., Zhan X., Chen J., Xu H., Huang X., Li Y., Zheng X., Lin L., Chen Y. DNA methylation-based lung adenocarcinoma subtypes can predict prognosis, recurrence, and immunotherapeutic implications. Aging (Albany NY) 2020;12:25275–25293. doi: 10.18632/aging.104129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang C., Zhang J., Xu F.P., Wang Y.G., Xie Z., Su J., Dong S., Nie Q., Shao Y., Zhou Q. Genomic Landscape and Immune Microenvironment Features of Preinvasive and Early Invasive Lung Adenocarcinoma. J. Thorac. Oncol. 2019;14:1912–1923. doi: 10.1016/j.jtho.2019.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jurisic V., Vukovic V., Obradovic J., Gulyaeva L.F., Kushlinskii N.E., Djordjević N. EGFR Polymorphism and Survival of NSCLC Patients Treated with TKIs: A Systematic Review and Meta-Analysis. J. Oncol. 2020;2020:1973241. doi: 10.1155/2020/1973241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiao G., Wang B. NK Cell Subtypes as Regulators of Autoimmune Liver Disease. Gastroenterol. Res. Pract. 2016;2016:6903496. doi: 10.1155/2016/6903496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun T., Wu R., Ming L. The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 2019;112:108613. doi: 10.1016/j.biopha.2019.108613. [DOI] [PubMed] [Google Scholar]
- 8.Liu Z.X., Li L.M., Sun H.L., Liu S.M. Link Between m6A Modification and Cancers. Front. Bioeng. Biotechnol. 2018;6:89. doi: 10.3389/fbioe.2018.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tu Z., Wu L., Wang P., Hu Q., Tao C., Li K., Huang K., Zhu X. N6-Methylandenosine-Related lncRNAs Are Potential Biomarkers for Predicting the Overall Survival of Lower-Grade Glioma Patients. Front. Cell Dev. Biol. 2020;8:642. doi: 10.3389/fcell.2020.00642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou K.I., Parisien M., Dai Q., Liu N., Diatchenko L., Sachleben J.R., Pan T. N(6)-Methyladenosine Modification in a Long Noncoding RNA Hairpin Predisposes Its Conformation to Protein Binding. J. Mol. Biol. 2016;428(5 Pt A):822–833. doi: 10.1016/j.jmb.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song H., Li H., Ding X., Li M., Shen H., Li Y., Zhang X., Xing L. Long non-coding RNA FEZF1-AS1 facilitates non-small cell lung cancer progression via the ITGA11/miR-516b-5p axis. Int. J. Oncol. 2020;57:1333–1347. doi: 10.3892/ijo.2020.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue L., Li J., Lin Y., Liu D., Yang Q., Jian J., Peng J. m6 A transferase METTL3-induced lncRNA ABHD11-AS1 promotes the Warburg effect of non-small-cell lung cancer. J. Cell. Physiol. 2021;236:2649–2658. doi: 10.1002/jcp.30023. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., Liu X., Liu L., Li J., Hu Q., Sun R. Expression and Prognostic Significance of m6A-Related Genes in Lung Adenocarcinoma. Med. Sci. Monit. 2020;26:e919644. doi: 10.12659/MSM.919644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y., Gu J., Xu F.K., Zhu Q.L., Chen Y.W., Ge D., Lu C.L. Molecular characterization, biological function, tumor microenvironment association and clinical significance of m6A regulators in lung adenocarcinoma. Brief Bioinform. 2020 doi: 10.1093/bib/bbaa225. Published online October 1, 2020. [DOI] [PubMed] [Google Scholar]
- 15.Bade B.C., Dela Cruz C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020;41:1–24. doi: 10.1016/j.ccm.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Zhao X., Liu X., Cui L. Development of a five-protein signature for predicting the prognosis of head and neck squamous cell carcinoma. Aging (Albany NY) 2020;12:19740–19755. doi: 10.18632/aging.104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong H.X., Wang R., Jin X.Y., Zeng J., Pan J. LncRNA DGCR5 promotes lung adenocarcinoma (LUAD) progression via inhibiting hsa-mir-22-3p. J. Cell. Physiol. 2018;233:4126–4136. doi: 10.1002/jcp.26215. [DOI] [PubMed] [Google Scholar]
- 18.Tian Y., Yu M., Sun L., Liu L., Wang J., Hui K., Nan Q., Nie X., Ren Y., Ren X. Distinct Patterns of mRNA and lncRNA Expression Differences Between Lung Squamous Cell Carcinoma and Adenocarcinoma. J. Comput. Biol. 2020;27:1067–1078. doi: 10.1089/cmb.2019.0164. [DOI] [PubMed] [Google Scholar]
- 19.Song Q., Shang J., Yang Z., Zhang L., Zhang C., Chen J., Wu X. Identification of an immune signature predicting prognosis risk of patients in lung adenocarcinoma. J. Transl. Med. 2019;17:70. doi: 10.1186/s12967-019-1824-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H., Guo L., Chen J. Rationale for Lung Adenocarcinoma Prevention and Drug Development Based on Molecular Biology During Carcinogenesis. OncoTargets Ther. 2020;13:3085–3091. doi: 10.2147/OTT.S248436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghafouri-Fard S., Shoorei H., Branicki W., Taheri M. Non-coding RNA profile in lung cancer. Exp. Mol. Pathol. 2020;114:104411. doi: 10.1016/j.yexmp.2020.104411. [DOI] [PubMed] [Google Scholar]
- 22.Loewen G., Jayawickramarajah J., Zhuo Y., Shan B. Functions of lncRNA HOTAIR in lung cancer. J. Hematol. Oncol. 2014;7:90. doi: 10.1186/s13045-014-0090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu L., Wang H., Wei D., Wang B., Zhang C., Zhu T., Ma Z., Li Z., Wu Y., Yu G. The value of CEP55 gene as a diagnostic biomarker and independent prognostic factor in LUAD and LUSC. PLoS ONE. 2020;15:e0233283. doi: 10.1371/journal.pone.0233283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fazi F., Fatica A. Interplay Between N6-Methyladenosine (m6A) and Non-coding RNAs in Cell Development and Cancer. Front. Cell Dev. Biol. 2019;7:116. doi: 10.3389/fcell.2019.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo Z.B., Lai G.E., Jiang T., Cao C.L., Peng T., Liu F.E. A Competing Endogenous RNA Network Reveals Novel lncRNA, miRNA and mRNA Biomarkers With Diagnostic and Prognostic Value for Early Breast Cancer. Technol. Cancer Res. Treat. 2020;19 doi: 10.1177/1533033820983293. 1533033820983293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao W., Li N., Gu B., Yi G., Su Z., Cheng H. LncRNA DLGAP1-AS2 modulates glioma development by up-regulating YAP1 expression. J. Biochem. 2020;167:411–418. doi: 10.1093/jb/mvz108. [DOI] [PubMed] [Google Scholar]
- 27.Li J.P., Li R., Liu X., Huo C., Liu T.T., Yao J., Qu Y.Q. A Seven Immune-Related lncRNAs Model to Increase the Predicted Value of Lung Adenocarcinoma. Front. Oncol. 2020;10:560779. doi: 10.3389/fonc.2020.560779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allgäuer M., Budczies J., Christopoulos P., Endris V., Lier A., Rempel E., Volckmar A.L., Kirchner M., von Winterfeld M., Leichsenring J. Implementing tumor mutational burden (TMB) analysis in routine diagnostics-a primer for molecular pathologists and clinicians. Transl. Lung Cancer Res. 2018;7:703–715. doi: 10.21037/tlcr.2018.08.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Topalian S.L., Taube J.M., Anders R.A., Pardoll D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jurišić V., Obradovic J., Pavlović S., Djordjevic N. Epidermal Growth Factor Receptor Gene in Non-Small-Cell Lung Cancer: The Importance of Promoter Polymorphism Investigation. Anal. Cell. Pathol. (Amst.) 2018;2018:6192187. doi: 10.1155/2018/6192187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafei H., El-Bahesh E., Finianos A., Nassereddine S., Tabbara I. Immune-based Therapies for Non-small Cell Lung Cancer. Anticancer Res. 2017;37:377–387. doi: 10.21873/anticanres.11330. [DOI] [PubMed] [Google Scholar]
- 33.Xu F., Zhan X., Zheng X., Xu H., Li Y., Huang X., Lin L., Chen Y. A signature of immune-related gene pairs predicts oncologic outcomes and response to immunotherapy in lung adenocarcinoma. Genomics. 2020;112:4675–4683. doi: 10.1016/j.ygeno.2020.08.014. [DOI] [PubMed] [Google Scholar]
- 34.Xu F., Lin H., He P., He L., Chen J., Lin L., Chen Y. A TP53-associated gene signature for prediction of prognosis and therapeutic responses in lung squamous cell carcinoma. OncoImmunology. 2020;9:1731943. doi: 10.1080/2162402X.2020.1731943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong W., Liang L., Gu Y., Qi Z., Qiu H., Yang X., Zeng W., Ma L., Xie J. Immune-Related lncRNA to Construct Novel Signature and Predict the Immune Landscape of Human Hepatocellular Carcinoma. Mol. Ther. Nucleic Acids. 2020;22:937–947. doi: 10.1016/j.omtn.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gong K., Guo G., Beckley N., Zhang Y., Yang X., Sharma M., Habib A.A. Tumor necrosis factor in lung cancer: Complex roles in biology and resistance to treatment. Neoplasia. 2021;23:189–196. doi: 10.1016/j.neo.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Z., Wang M., Liu Q., Liu Y., Zhu K., Chen L., Guo H., Li Y., Shi B. Identification of gene expression profiles and immune cell infiltration signatures between low and high tumor mutation burden groups in bladder cancer. Int. J. Med. Sci. 2020;17:89–96. doi: 10.7150/ijms.39056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X., Li Y., Yu X., Jin F. Identification and validation of stemness-related lncRNA prognostic signature for breast cancer. J. Transl. Med. 2020;18:331. doi: 10.1186/s12967-020-02497-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.