

Abstract
The number of patients with intractable epilepsy undergoing surgical management in China is increasing rapidly. We retrospectively reviewed 435 consecutive cases of intractable epilepsy receiving surgical resection from 2005 to 2008 in our hospital, looking specifically at the neuropathological findings. The three most common causes of intractable epilepsy were focal cortical dysplasia (FCD; 52.9%), scar lesions (22.8%) and brain tumors (11.7%). Hippocampal sclerosis was identified in 74 cases (17.0%), although most of these were accompanied by dual pathology with FCD (especially Palmini type IB), scar lesions or tumors. Among FCD cases, Palmini type I lesions are the most frequently observed abnormality, with a preferred location in the temporal lobe (60.1%) and often accompanied by dual pathology. In contrast, Palmini type II FCD lesions occurred predominantly in the frontal regions and with a lower age of onset. Most tumors were mixed neuronal–glial tumors, mainly ganglioglioma (19 cases) and dysembryoplastic neuroepithelial tumor (10 cases), with a trend toward a temporal location and usually accompanied by cortical dysplasia in the peritumor area. Our data on the neuropathology of intractable epilepsy in China show that glioneuronal lesions are the most prominent cause of intractable epilepsy, and this is consistent with reports from other countries.
Keywords: focal cortical dysplasia, glioneuronal lesion, intractable epilepsy, malformation of cortical development, mixed neuronal–glial tumors
INTRODUCTION
Epilepsy is a common neurological disorder affecting 0.5%–1% of the population worldwide (2). Malformations of cortical development (MCD) and mixed neuronal–glial tumors are the most crucial cause of intractable epilepsy, especially in children and adolescents. Resective epilepsy surgery can be beneficial for intractable epilepsy because of MCD, particularly focal cortical dysplasia (FCD), and unilateral hemispheric malformations (18).
Recently, researchers proposed using the term glioneuronal lesions to include FCD and glioneuronal tumor. There have been several large studies of consecutive cases of pharmacologically refractory epilepsy receiving surgical treatment from the USA, Germany, Japan, France and India 5, 18, 23, 27, 29, 31. More recently, findings from several large case series of FCD or temporal lobe epilepsy have been published 6, 12, 13, 14, 19, 30. According to the China Association Against Epilepsy (CAAE, 2005), there are approximately 9 million patients with epilepsy in China. Among them, about 3 million patients are resistant to antiepileptic drugs (AEDs) and are considered to have “intractable epilepsy.” Surgery is an option for some of these patients, and the number of patients accepting surgical management in China is increasing rapidly. No large studies have provided neuropathological descriptions, so we retrospectively reviewed the neuropathological findings in consecutive cases of intractable epilepsy receiving surgical resection in the epilepsy center of our hospital.
METHODS
Case collection
All cases of intractable epilepsy who received surgical treatment in the Department of Neurosurgery of the Beijing Institute of Functional Neurosurgery, Xuanwu Hospital, Capital Medical University, between 2005 and 2008 were collected. We retrospectively analyzed their clinical data (gender, ages at onset and surgery, neurological examination findings and seizure types), magnetic resonance imaging (MRI) examinations and pathological findings.
Histopathology
We had used a standard approach to processing surgical specimens from patients with intractable epilepsy since 2005. For all specimens, coronal sections were performed. Selected blocks were processed for embedding in paraffin wax; 4 µm or 8 µm‐thick sections (according to the staining required) were cut and stained with hematoxylin and eosin (H&E) and Luxol fast blue. Selected sections were also processed for immunohistostaining using the polymer horse radish peroxidase (HRP) detection system [Polink‐1HRP Broad Spectrum DAB Detection Kit, Golden Bridge International (GBI), Mukilteo, WA, USA]. For primary antibodies, we used mouse monoclonal antibodies against neuronal nuclear antigen (NeuN; Chemicon, Temecula, CA, USA; 1:4000), neurofilament protein (NF; Zymed, San Francisco, CA, USA; 1:100), microtubule‐associated protein‐2 (MAP‐2; Zymed; 1:200), nestin (R&D, Minneapolis, MN, USA; 1:200), vimentin (Zymed; 1:200), epithelial membrane antigen (EMA; Zymed; 1:50), parvalbumin (PV; Sigma, St Louis, MO, USA; 1:1000), CD34 (Zymed; 1:50) and Ki‐67 (MIB‐1; Labvision, Fremont, CA, USA; 1:50), as well as rabbit polyclonal antibodies against glial fibrillary acidic protein (GFAP; Dako, Glostrup, Denmark; 1:1000), Olig2 (Immuno‐Biological Laboratories, Takasaki, Japan; 1:200) and synaptophysin (Biogenics, San Ramon, CA, USA; 1:50).
For PV immunohistochemistry, we chose 20 cases with FCD (4 of FCD IA, 7 of FCD IB, 5 of FCD IIA and 4 of FCD IIB) and, as controls, 4 cases of brain tumors (peritumor region) without history of epilepsy. We calculated the average number of PV‐positive neurons per highest power field (×400) in the most intensely stained background. The focal and peripheral regions of FCD type II were calculated, respectively. The rank sum test was used to compare data, with P < 0.05 indicating that the differences were statistically significant.
Histopathological diagnoses were made using Palmini et al's terminology and classification of cortical dysplasias (26) or the World Health Organization (WHO) Classification of Tumors of the Central Nervous System (20) by at least two neuropathologists (LD‐H and CL at the time of initial resection, and PY‐S in a retrospective fashion).
Statistical analysis
We analyzed the cases statistically. Differences in age at onset of the different FCD subgroups were tested by Student's t‐test or by one‐way analysis of variance (ANOVA). Comparison of the regional distribution between FCD subgroups was assessed by the χ2 test. Calculations were performed using commercial software SPSS® 11.5 for Windows (SPSS, Chicago, IL, USA). The level of statistical significance was set at P < 0.05.
RESULTS
In total, 435 cases [male (M) : female (F) = 294:141] of intractable epilepsy received surgical treatment between 2005 and 2008 (Table S1). The age of onset was from 0 years to 57 years (average 11.5 years), and the disease duration ranged from 0.1 years to 32 years (average 11.1 years). The most common presentation was complex partial seizure. In only a few cases did neurological examinations show mild defects such as hemiparesis and slightly lower intelligence.
MCD
Neuropathologically, malformations were the most common finding and were seen in more than half of the cases (249 cases, M : F = 166:83), with mild MCD (14 cases) and FCD (216 cases) being the predominant histopathological findings (230/249, 92.4%) (Table S1). In cases of FCD, there were 173 cases of type I and 43 cases of type II. Most cases with FCD had an onset age under 20 years (188 cases, 87%). Of interest, patients with FCD type II often had their onset at an earlier age than those with FCD type I (7.8 vs. 11.2, Student's t‐test, P = 0.011).
Neuroimaging was available for 132 cases. About 26/107 (24.3%) of cases of FCD type I and 13/25 (52%) of cases of FCD type II showed abnormal lesions in the cortices and/or subcortical white matter, indicating cortical dysplasia. These abnormalities can be detected as a thickened cortex, blurred cortex–white matter junction and abnormal signals in the white matter on T2‐weighted images (9). More than half the cases of FCD type I had hippocampus atrophy (50 cases) or signal abnormalities (18 cases). However, there were 22 cases of FCD type I and 8 cases of type II with no abnormalities on neuroimaging.
Histopathologically, FCD type IB was the most common subtype of FCD. In its regional distribution, FCD type I tended to be seen in the temporal region (104/173, 60.1%). In contrast, FCD type II tended to be seen in the extratemporal region (41/43, 95.3%), especially the frontal lobe (26/41, 63.4%) (Table S2), and this finding was statistically significant (χ2 test, P = 0.000).
The histopathological findings of FCD mainly include cortical architectural abnormalities and cytoarchitectural abnormalities, as Palmini et al described. In details, the former consisted of increased numbers of neurons in the layer I/white matter, cortical laminar disorganization, a microcolumnar arrangement of cortical neurons and neuronal clustering (Figure 1A–D). The latter consisted of giant neurons, immature neurons, dysmorphic neurons and balloon cells (Figure 1E–H). In FCD type IB, in addition to cortical architectural abnormalities, giant or immature, but not dysmorphic, neurons were detected. FCD type II included more pronounced architectural and cytoarchitectural disturbances such as dysmorphic neurons (in FCD type IIA) and additional balloon cells (in FCD type IIB).
Figure 1.
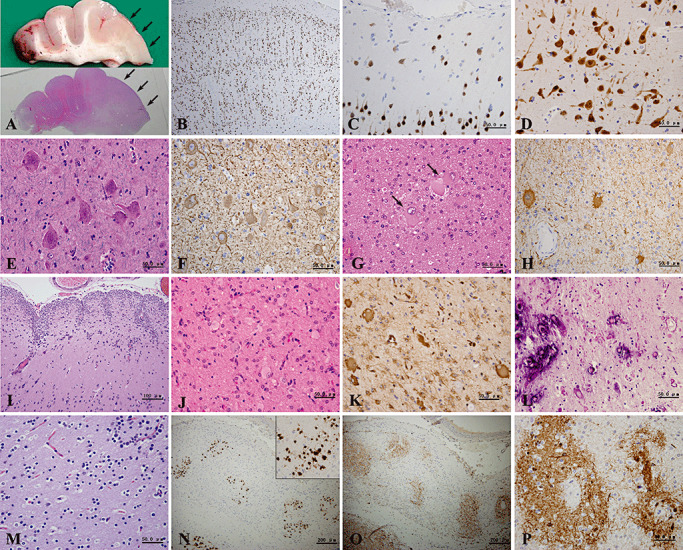
(A–I) Histopathological findings of focal cortical dysplasia (FCD). Macroscopically, thickened cortex and blurred cortex–white matter junction are observed in FCD type II (A, arrows). Microscopically, microcolumnar arrangement of cortical neurons (B), increased numbers of neurons in layer I (C) and cortical laminar disorganization (D) are detected by neuronal nuclear antigen (NeuN) immunostaining. Dysmorphic neurons reveal abnormal size and cytoskeletal structure (E) and rich cytoplasmic neurofilament (F). FCD type IIB is defined by the presence of balloon cells (G), which can be immunolabeled with glial fibrillary acidic protein (GFAP) antibody (H). Numerous corpora amylacea in the subpial molecular layer and abnormal gyration are also noted (I). (J–L) Histopathological findings of tuberous sclerosis. Magnified views from the cortical tuber show proliferation of many giant cells (J,K), appearance of Rosenthal fibers and calcifications (L). (M–P) Histopathological features of the island distribution of residual neurons seen in the scar lesions. (M) With hematoxylin and eosin (H&E) staining, these neurons have small round nuclei and perinuclear haloes and resemble oligodendroglia. (N) Most of them are immunolabeled by NeuN. (O) There is background staining and many axons detected by neurofilament protein (NF) in the island. (P) On microtubule‐associated protein‐2 (MAP‐2) immunostaining, the background and several neurons in the island are prominently labeled. (A,G,I,J,L,M) H&E; (E) H&E/Luxol fast blue; (B,C,D,N) NeuN immunostaining; (F,O) NF immunostaining; (H) GFAP immunostaining; (K) vimentin immunostaining; (P) MAP‐2 immunostaining.
Moreover, we also often noted gliosis and the appearance of corpora amylacea in the subpial molecular layer, dilatation of perivascular spaces in the white matter and abnormal gyration (Figure 1I).
Immunohistochemistry revealed that NeuN was an excellent marker of neuronal cells, including giant, immature and dysmorphic neurons, and normal pyramidal cells (Figure 1B–D). NF and MAP‐2 antibodies could also detect neuronal cells. Giant and dysmorphic neurons were stained particularly strongly with NF (Figure 1F), while immature neurons were stained strongly with MAP‐2. Most balloon cells immunostained with GFAP and/or vimentin antibodies (Figure 1H). Moreover, a few balloon cells showed nestin immunopositivity also.
On PV immunohistochemistry, compared with the relatively normal cortices from patients without epilepsy, FCD cortices showed dramatically decreased numbers of PV‐positive interneurons and PV background staining, especially in the foci of FCD type II (Figure 2).
Figure 2.
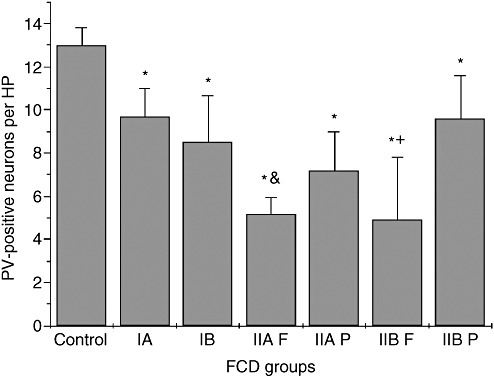
Bar graphs showing the mean (number ± SD) number of parvalbumin (PV)‐positive neurons in the controls and focal cortical dysplasia (FCD) subtypes. *, There are fewer PV‐positive neurons in all FCD subtypes than in controls (P < 0.05). &, There are fewer PV‐positive neurons in FCD IIA foci compared with FCD IIA peripheral regions (P = 0.036). +, There are fewer PV‐positive neurons in FCD IIB foci compared with FCD IIB peripheral regions (P = 0.043). Abbreviations: HP = high power field; IA = FCD IA regions; IB = FCD IB regions; IIA F = FCD IIA foci regions; IIA P = FCD IIA peripheral regions; IIB F = FCD IIB foci regions; IIB P = FCD IIB peripheral regions.
There were seven cases of tuberous sclerosis (Figure 1J–L), four cases of nodular neuronal heterotopia, three cases of polymicrogyria and one case of porencephaly. In addition, there were 14 cases of nodular neuronal heterotopia accompanied by FCD (13 cases, especially type IB) or polymicrogyria (1 case).
Tumors
Fifty‐one cases (11.7%, M : F = 34:17) were diagnosed as having tumors, with an average age of onset of 13.2 years (range 0 years–44 years) and a disease duration of 8.9 years (range 0.1 years–34 years) (Table S3). However, on MRI examination, there are only 11 cases of suspected tumorous lesions, without distinct mass effects or gadolinium‐diethylenetriamine penta‐acetic acid (DTPA) enhancement. The remaining cases were reported as presenting abnormal signals in the cortices (14 cases) or hippocampus (4 cases), softening (5 cases), hippocampus atrophy (3 cases), vascular malformations (2 cases), no detectable abnormalities (2 cases), brain atrophy (1 case), a multicystic lesion (1 case), arachnoid cyst (1 case) and dysplasia (1 case).
With regard to regional distribution, the tumors tended to be seen in the temporal region (37/51, 72.5%), and seven cases (13.7%) were accompanied by hippocampal sclerosis (HS). Histopathologically, mixed neuronal–glial tumors were the major common type, with 19 cases of ganglioglioma (GG) (37.3% overall, WHO grade I or II) and 10 cases of dysembryoplastic neuroepithelial tumor (DNT) (19.6% overall, WHO grade I). On the one hand, GG (Figure 3A,B) was the most common tumor associated with epilepsy and was seen in the temporal lobe particularly (16/19, 84.2%). On the other hand, DNT (Figure 3C,D) was mainly localized in the temporal lobe (6/10, 60%), with three cases in the frontal lobe (Table S3). Most samples that had sufficient peritumor cortices for examination had a dual lesion of FCD.
Figure 3.
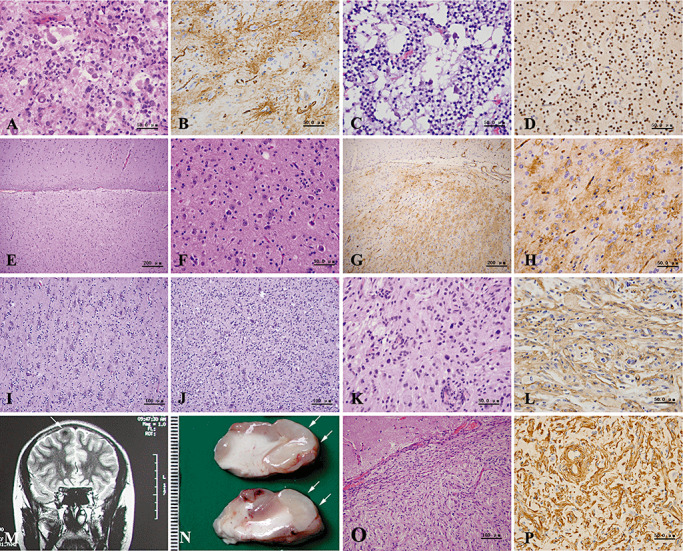
(A–L) Histopathological findings of mixed neuronal–glial tumors. Gangliogliomas are composed of large ganglion‐like cells and neoplastic glial cells (A), and show a bushy pattern of CD34 positivity (B). Dysembryoplastic neuroepithelial tumors with a microcystic morphology contain isolated, mature large neurons (C) and oligodendroglia‐like cells, which are immunolabeled with Olig2 antibody (D). (E–H) Mixed neuronal–glial tumors with preliminary features of ganglioglioma. With low magnification, obvious cortical architectural disturbances can be seen (E). Note a normal appearance of cortex seen in the upper part. High magnification shows randomly scattered neurons and glial cells of somewhat high cellularity (F). CD34 immunostainig reveals diffuse and solitary patterns (G,H). (I,J) Mixed neuronal–glial tumors with preliminary features of dysembryoplastic neuroepithelial tumors. The area with normal neurons surrounded by proliferating oligodendrocytes (I) is adjacent to the area with an admixture of neuronal and proliferating oligodendrocytic elements (J). (K,L) Pleomorphic xanthoastrocytoma shows nuclear pleomorphism and xanthomatous change (K), as well as diffuse CD34 immunostaining (L). (M–P) Neuroimaging and histopathological findings of meningioangiomatosis. T2‐weighted magnetic resonance imaging (MRI) image displays an abnormal signal in the cortex of the right frontal lobe (M, arrow). Macroscopically, partial gyri appear tumid and pale (N, arrows). Microscopically, there were proliferating spindle cells and vessels invading into the superficial cortex with rich collagen deposition (O,P). (A,C,E,F,I,J,K,O) hematoxylin and eosin (H&E); (B,G,H,L) CD34 immunostaining; (D) Olig2 immunostaining; (P) vimentin immunostaining.
In addition, there were 13 cases (25.5%) of mixed neuronal–glial tumors of uncertain subtype. Of interest, these cases contained an admixture of neuronal and glial elements but were distinct from typical cases of either GG or DNT. These tumors had a preference for the temporal lobes, especially the hippocampal formation. We named these tumors “mixed neuronal–glial tumors with transitional features,” depending on histopathological features. In details, some tumors had transitional features between glioneuronal hamartia and GG, showing disorganized mature‐appearing neurons and glial cells with slightly proliferated capillary vessels (preliminary histological features of GG, Figure 3E–H). The other tumors had preliminary histological features of DNT (Figure 3I,J). The peritumor region often showed obvious cortical architectural abnormalities and glioneuronal nodules.
Epilepsy‐associated glial tumors were also seen, including four cases of pleomorphic xanthoastrocytoma (PXA; WHO grade II) (Figure 3K,L) and one case of angiocentric glioma (AG; WHO grade I). In addition, we encountered a case of a 14‐year‐old female with meningioangiomatosis (Figure 3M–P), a type of hamartomatous lesion (Table S3).
On immunohistochemical study, a remarkable presence of CD34 in all cases of GG (Figure 3B) and PXA (Figure 3L), with three patterns of “diffuse,”“solitary” and “bushy,” was observed (4). Ten of 13 cases with “mixed glioneuronal tumors with transitional features” expressed CD34 (Figure 3G,H). However, such CD34 immunostaining was seldom detected in other tumorous and FCD lesions.
Scar lesions
There were 99 cases (23%, M : F = 74:25) of scar lesions with an average age of onset of 10.5 years (range 0 years–46 years) and a disease duration of 11.2 years (range 0.3 years–36 years). With regard to regional distribution, surgical specimens were resected from multiple lobes in 43 cases, the frontal lobe in 20 cases, the temporal lobe in 10 cases, the occipital lobe in 5 cases and the parietal lobe in 4 cases. The other 17 cases received hemispherectomy.
Histopathologically, destruction of the cortices with severe neuron loss, fibrous gliosis and cyst formation was seen. In some cases, an island distribution of residual neurons, which had an oligodendroglia‐like appearance, was observed, especially in cases caused by ischemia/hypoxia (Figure 1M–P). In 36 cases (36.4%), hemosiderin deposits or hemosiderin‐laden macrophages were detected. Among them, 15 cases had a considerable history of trauma.
Dual pathology
HS was identified in 74 cases (17.0%). Most of these were accompanied by FCD (53 cases), scar lesions (10 cases) or tumor (7 cases). There were only four cases of pure HS.
Among the cases of FCD accompanied by HS, most cases were type IB (40 cases), accounting for 75.5% of FCD cases. In this condition, FCD lesions were detected in the neocortex of lobectomy specimen, and HS was noted in the separated hippocampi specimen, respectively. Apart from the obvious neuronal loss and gliosis seen in the CA1, CA3 and CA4 regions, we often observed abnormally enlarged neurons (dysmorphic neuron‐like) in the area of the CA4 region. Moreover, dispersion of granular cells in the dentate gyrus was also noted.
Others
Inflammatory lesions included viral encephalitis (three cases), neurocysticercosis (three cases), Rasmussen's encephalitis (two cases) and tuberculous meningitis (one case), as well as nonspecific chronic inflammations.
Vascular malformations included six cases of cavernous angioma and four cases of arteriovenous malformation.
Benign cystic lesions (six cases) included two cases of epidermoid cyst.
In nine cases where the specimens were small, only nonspecific findings, including gliosis and accumulation of corpora amylacea, were observed.
DISCUSSION
In this study, we report the largest series of patients with intractable epilepsy receiving surgical resection in China. Our specialized epilepsy center, with its multidisciplinary team of highly trained physicians, has been developing surgical treatments for epilepsy since 2001. As our results have shown, the three most common causes of refractory epilepsy were FCD (52.9%), scar lesions (22.8%) and brain tumors (11.7%). Reviewing previous studies, we note that the top three neuropathological findings are medial temporal sclerosis (MTS)/HS (18.3%–48.2%), tumor (17.6%–31.2%) and cortical dysplasia (13.2%–23.5%) 18, 23, 27, 29, 31.
Of note, in our series, 22.8% of cases were histopathologically diagnosed as scar lesions, resulting from hypoxia/ischemia or trauma. A study from the Cleveland Clinic Foundation with 133 consecutive cases of extratemporal‐based intractable epilepsy reported that 18% showed evidence of remote ischemic damage or infarct being confined to the frontal lobe (7). Our results also show an extratemporal preference for scar lesion, but cases commonly involved multiple lobes. As recent studies indicate, patients with ulegyria often have a history of perinatal asphyxia and present with pharmacoresistant seizures 16, 25. Taken together, we speculate that perinatal hypoxic‐ischemic brain injury remains one of the most prevalent complications in the newborn, with a relatively high frequency in China.
Interestingly, we found residual neurons in an island distribution, which is often seen in scar lesions. With routine H&E staining, these neurons resembled oligodendroglia. However, they were all strongly immunopositive for NeuN, and some immunostained with MAP‐2 and NF, with the appearance of processes. They were immunonegative for Olig2, GFAP, vimentin, nestin, CD56 and CD34 antibodies, and none of these cells were positive for Ki‐67. We think these neurons originated from neuroblasts that have not migrated, organized and matured normally. In 1995, Mischel et al proposed that MCD lesions reflect events occurring within discrete time windows in development and proposed a stratification of these lesions into those occurring with early, intermediate and late gestational events (24).We therefore speculate that these island‐distributed neurons, resembling nodular glioneuronal heterotopia, may have formed during late gestational time events.
MCD is increasingly recognized as an important cause of developmental delay, epilepsy and other neurological disorders, and this has followed the development of MRI examinations and the recent increase in the surgical treatment of neocortical epilepsy 1, 6, 19, 21. The incidence of cortical dysplasia in epilepsy surgery series varies from 12% to 40%. FCD is one of the most common neuropathological findings in resection specimens from pediatric patients with refractory epilepsy. It is reported in nearly 50% of the pediatric patients from centers specialized for pediatric epilepsy surgery 15, 28. In our series, FCD accounted for 60% of the total cases, with mean onset age of 10.5 years, and this was similar to the incidences reported previously.
MRI examinations improve the detection of abnormalities of cortical development (17). Consequently, selection of cases undergoing surgical treatment has been broadened, in particular, to include patients who will be histopathologically diagnosed with FCD. In our series, distinguishable MRI abnormalities were encountered in 52% of FCD type II cases, but only 24% of FCD type I cases. This agrees with previous studies showing that the use of MRI to diagnose FCD type I is highly challenging, in contrast to FCD type II 10, 13, 14, 22, 30. However, more recently, Krsek et al reported a high proportion of MRI‐positive FCD type I cases, using MRI as an adjunct to their protocol for detecting malformations (15).
As regards our histopathological findings, FCD type I was the more frequently observed abnormality, with a preferred location in the temporal lobe (104/173, 59.5%) and often accompanied by dual pathology. In contrast, FCD type II occurred predominantly in the frontal and central regions, and had a lower age at onset. These findings correspond with previously reported studies 6, 10, 14, 15, 19, 24. However, FCD type IB was the most common subtype of FCD in our series, a finding at variance with that reported by other groups. The reason may be that all cases with dual pathology were divided into the FCD group, but not the HS group. Furthermore, our immunohistochemistry revealed decreased numbers of PV‐positive interneurons and PV background staining in all FCD subtypes, suggesting the absence of the inhibitory amino acid neurotransmitter system components in FCD cortices. This appearance was also reported by Garbelli et al (8) and Zamecnik et al (32).
NeuN is a useful antibody for revealing cortical lamination and organization, as well as for detecting cytoarchitectural disturbances such as dysmorphic neurons. Because of their immature features, immature neurons preferably stain with MAP‐2 antibody rather than with NF antibody 10, 12, 26, whereas balloon cells have both glial and neuronal immunohistochemical features (staining with nestin), which accords with the idea that there is some degree of heterogeneity among balloon cells with partial commitment toward glia or neuron differentiation 1, 26, 27.
Most tumors in our patients with intractable epilepsy were benign neoplasms localized in the temporal lobe, where the most frequent type was mixed neuronal–glial tumors (42/51). These neuronal–glial tumors have several features: epilepsy is the exclusive symptom, there is no obvious mass effect with neuroimaging, and the histopathology is usually accompanied by cortical dysplasia in the peritumor area. Moreover, we observed some tumors to have transitional features between glioneuronal hamartia 9, 31 and mixed neuronal–glial tumor. These tumors resembled the second major subtype of glioneuronal hamartoma reported by Blümcke et al exhibiting properties of a tumor‐like lesion with a close admixture of dysplastic neuronal cells and glial elements (3). Consequently, we speculate that cortical malformations, transitional types of glioneuronal tumors and glioneuronal tumors (GG and DNT) form a morphological continuity. The morphological continuity, CD34 immunopositive reactions and the common region of the temporal lobe strengthen our opinion that glioneuronal hamartia and mixed neuronal–glial tumors have similar pathomechanisms and represent a characteristic spectrum of disease of glioneuronal lesions with a different growth pattern 2, 3, 28. In 1998, Hirose and Scheithauer reported a case of mixed DNT and GG with a detailed histological, immunochemical and ultrastructural report that suggested that a close histogenetic relationship existed between DNT and GG (11). CD34 is now known to be a useful marker of neuron stem cells. However, in our examinations, CD34 was exclusively detected in GG, PXA and some glioneuronal tumors with transitional features. CD34 immunoreactivity was seldom seen in FCD cases and DNT. This provides support for the idea of different origins for GG (and PXA) and DNT (4). The underlying molecular pathogenesis requires further studies.
In conclusion, glioneuronal lesions appear to be the most common disease associated with intractable epilepsy. As Palmini et al and Mathern suggested, cellular–molecular studies of the mechanisms of epileptogenicity and clinical–imaging–electrical–pathological correlations are needed to further understand these disorders 22, 26.
Supporting information
Table S1. Summary of neuropathological diagnosis of our series of intractable epilepsy.
Table S2. Clinical data of malformation of cortical development in our series.
Table S3. Clinical data of tumors in our series.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
We thank Yang Hong, Zhao Li‐Hong and Wei Li‐Feng for their technical assistance. This study was supported by the Beijing Nova program (No. 2006B63) and the Capital Medical Development Research Fund (No.2007‐2070).
REFERENCES
- 1. Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB (2005) A developmental and genetic classification for malformations of cortical development. Neurology 65:1873–1887. [DOI] [PubMed] [Google Scholar]
- 2. Becker AJ, Blümcke I, Urbach H, Hans V, Majores M (2006) Molecular neuropathology of epilepsy‐associated glioneuronal malformations. J Neuropathol Exp Neurol 65:99–108. [DOI] [PubMed] [Google Scholar]
- 3. Blümcke I, Lobach M, Wolf H, Wiestler OD (1999) Evidence for developmental precursor lesions in epilepsy‐associated glioneuronal tumors. Microsc Res Tech 46:53–58. [DOI] [PubMed] [Google Scholar]
- 4. Deb P, Sharma MC, Tripathi M, Chandra PS, Gupta A, Sarkar C (2005) Expression of CD34 as a novel marker for glioneuronal lesions associated with chronic intractable epilepsy. Neuropathol Appl Neurobiol 32:461–468. [DOI] [PubMed] [Google Scholar]
- 5. Farrell MA, DeRosa MJ, Curran JG, Secor DL, Cornford ME, Comair YG et al (1992) Neuropathologic findings in cortical resections (including hemispherectomies) performed for the treatment of intractable childhood epilepsy. Acta Neuropathol 83:246–259. [DOI] [PubMed] [Google Scholar]
- 6. Fauser S, Huppertz HJ, Bast T, Strobl K, Pantazis G, Altenmueller DM et al (2006) Clinical characteristics in focal cortical dysplasia: a retrospective evaluation in a series of 120 patients. Brain 129:1907–1916. [DOI] [PubMed] [Google Scholar]
- 7. Frater JL, Prayson RA, Morris HH III, Bingaman WE (2000) Surgical pathologic findings of extratemporal‐based intractable epilepsy: a study of 133 consecutive resections. Arch Pathol Lab Med 124:545–549. [DOI] [PubMed] [Google Scholar]
- 8. Garbelli R, Meroni A, Magnaghi G, Beolchi MS, Ferrario A, Tassi L et al (2006) Architectural (Type IA) focal cortical dysplasia and parvalbumin immunostaining in temporal lobe epilepsy. Epilepsia 47:1074–1078. [DOI] [PubMed] [Google Scholar]
- 9. Gómez‐Ansón B, Thom M, Moran N, Stevens J, Scaravilli F (2000) Imaging and radiological‐pathological correlation in histologically proven cases of focal cortical dysplasia and other glial and neuronoglial malformative lesions in adults. Neuroradiology 42:157–167. [DOI] [PubMed] [Google Scholar]
- 10. Hildebrandt M, Pieper T, Winkler P, Kolodziejczyk D, Holthausen H, Blümcke I (2005) Neuropathological spectrum of cortical dysplasia in children with severe focal epilepsies. Acta Neuropathol 110:1–11. [DOI] [PubMed] [Google Scholar]
- 11. Hirose T, Scheithauer BW (1998) Mixed dysembryoplastic neuroepithelial tumor and ganglioglioma. Acta Neuropathol 95:649–654. [DOI] [PubMed] [Google Scholar]
- 12. Kakita A, Kameyama S, Hayashi S, Masuda H, Takahashi H (2005) Pathologic features of dysplasia and accompanying alterations observed in surgical specimens from patients with intractable epilepsy. J Child Neurol 20:341–350. [DOI] [PubMed] [Google Scholar]
- 13. Kim DW, Lee SK, Chu K, Park KI, Lee SY, Lee CH et al (2009) Predictors of surgical outcome and pathologic considerations in focal cortical dysplasia. Neurology 72:211–216. [DOI] [PubMed] [Google Scholar]
- 14. Krsek P, Maton B, Korman B, Pacheco‐Jacome E, Jayakar P, Dunoyer C et al (2008) Different features of histopathological subtypes of pediatric focal cortical dysplasia. Ann Neurol 63:758–769. [DOI] [PubMed] [Google Scholar]
- 15. Krsek P, Pieper T, Karlmeier A, Hildebrandt M, Kolodziejczyk D, Winkler P et al (2009) Different presurgical characteristics and seizure outcomes in children with focal cortical dysplasia type I or II. Epilepsia 50:125–137. [DOI] [PubMed] [Google Scholar]
- 16. Kuchukhidze G, Unterberger I, Dobesberger J, Embacher N, Walser G, Haberlandt E et al (2008) Electroclinical and imaging findings in ulegyria and epilepsy: a study on 25 patients. J Neurol Neurosurg Psychiatry 79:547–552. [DOI] [PubMed] [Google Scholar]
- 17. Kuzniecky RI, Barkovich AJ (2001) Malformations of cortical development and epilepsy. Brain Dev 23:2–11. [DOI] [PubMed] [Google Scholar]
- 18. Lahl R, Villagran R, Teixeira W (2003) Morphological findings on compartments from our own study material. In: Neuropathology of Focal Epilepsies: An Atlas, Chapter 5. Lahl R, Villagran R, Teixeira W (eds), pp. 13–15. John Libby: London, Paris, Rome, Sydney. [Google Scholar]
- 19. Lerner JT, Salamon N, Hauptman JS, Velasco TR, Hemb M, Wu JY et al (2009) Assessment and surgical outcomes for mild type I and severe type II cortical dysplasia: a critical review and the UCLA experience. Epilepsia 50:1310–1335. [DOI] [PubMed] [Google Scholar]
- 20. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007) WHO Classification of Tumours of the Central Nervous System. IARC: Lyon, France. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lüders H, Schuele SU (2006) Epilepsy surgery in patients with malformations of cortical development. Curr Opin Neurol 19:169–174. [DOI] [PubMed] [Google Scholar]
- 22. Mathern GW (2009) Challenges in the surgical treatment of epilepsy patients with cortical dysplasia. Epilepsia 50(Suppl. 9):45–50. [DOI] [PubMed] [Google Scholar]
- 23. Mihara T, Matsuda K, Tottori T, Otsubo T, Baba K, Nishibayashi H et al (2004) Long‐term seizure outcome following resective surgery at National Epilepsy Center in Shizuoka, Japan. Psychiatry Clin Neurosci 58:S22–S25. [DOI] [PubMed] [Google Scholar]
- 24. Mischel PS, Nguyen LP, Vinters HV (1995) Cerebral cortical dysplasia associated with pediatric epilepsy. Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol 54:137–153. [DOI] [PubMed] [Google Scholar]
- 25. Nikas I, Dermentzoglou V, Theofanopoulou M, Theodoropoulos V (2008) Parasagittal lesions and ulegyria in hypoxic‐ischemic encephalopathy: neuroimaging findings and review of the pathogenesis. J Child Neurol 23:51–58. [DOI] [PubMed] [Google Scholar]
- 26. Palmini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary‐Schaefer N et al (2004) Terminology and classification of the cortical dysplasias. Neurology 62:S2–S8. [DOI] [PubMed] [Google Scholar]
- 27. Pasquier B, Péoc'H M, Fabre‐Bocquentin B, Bensaadi L, Pasquier D, Hoffmann D et al (2002) Surgical pathology of drug‐resistant partial epilepsy. A 10‐year‐experience with a series of 327 consecutive resections. Epileptic Disord 4:99–119. [PubMed] [Google Scholar]
- 28. Rickert CH (2006) Cortical dysplasia: neuropathological aspects. Childs Nerv Syst 22:821–826. [DOI] [PubMed] [Google Scholar]
- 29. Sarkar C, Sharma MC, Deb P, Singh VP, Chandra PS, Gupta A et al (2006) Neuropathological spectrum of lesions associated with intractable epilepsies: a 10‐year experience with a series of 153 resections. Neurol India 54:144–150. [PubMed] [Google Scholar]
- 30. Tassi L, Colombo N, Garbelli R, Francione S, Lo Russo G, Mai R et al (2002) Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain 125:1719–1732. [DOI] [PubMed] [Google Scholar]
- 31. Wolf HK, Wiestler OD (1993) Surgical pathology of chronic epileptic seizure disorders. Brain Pathol 3:371–380. [DOI] [PubMed] [Google Scholar]
- 32. Zamecnik J, Krsek P, Druga R, Marusic P, Benes V, Tichy M, Komarek V (2006) Densities of parvalbumin‐immunoreactive neurons in non‐malformed hippocampal sclerosis‐temporal neocortex and in cortical dysplasias. Brain Res Bull 68:474–481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of neuropathological diagnosis of our series of intractable epilepsy.
Table S2. Clinical data of malformation of cortical development in our series.
Table S3. Clinical data of tumors in our series.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item