

Abstract
Proteomics is increasingly employed in both neurological and oncological research to provide insight into the molecular basis of disease but rarely has a coherent, novel pathophysiological insight emerged. Gliomas account for >50% of adult primary intracranial tumors, with malignant gliomas (anaplastic astrocytomas and glioblastoma multiforme) being the most common. In glioma, the application of proteomic technology has identified altered protein expression but without consistency of these alterations or their biological significance being established. A systematic review of multiple independent proteomic analyses of glioma has demonstrated alterations of 99 different proteins. Importantly 10 of the 99 proteins found differentially expressed in glioma [PHB, Hsp20, serum albumin, epidermal growth factor receptor (EGFR), EA‐15, RhoGDI, APOA1, GFAP, HSP70, PDIA3] were identified in multiple publications. An assessment of protein–protein interactions between these proteins compiled using novel web‐based technology, revealed a robust and cohesive network for glioblastoma. The protein network discovered (containing TP53 and RB1 at its core) compliments recent findings in genomic studies of malignant glioma. The novel perspective provided by network analysis indicates that the potential of this technology to explore crucial aspects of glioma pathophysiology can now be realized but only if the conceptual and technical limitations highlighted in this review are addressed.
Keywords: functional networks, glioblastoma, glioma pathophysiology, proteomics
INTRODUCTION
In 1990 the Human Genome Project formally began its epic 13‐year task to elucidate the entire human genome, which we now know harbors approximately 20 400 genes. The impact has been enormous, with new avenues being opened into the diagnosis of disease, gene therapy, and earlier detection of genetic predispositions. Now, nearly 20 years on, the Human Proteome Project [HPP; coordinated by The Human Proteome Organization (HUPO)] is instigating a plan to identify one protein (excluding post‐translational modifications, isoforms and splice variants) for each of the estimated human genes. This is of particular importance as genes are life's “blueprint,” proteins represent the “bricks and mortar” from which it is built (70) and modifications or mutations at the level of the genome manifest as aberrations of the proteome.
The utility of proteomics (the large‐scale study of multiple proteins) in cancer biology and central nervous system (CNS) disorders is currently one of the most dynamic research areas in medicine 4, 41, 73, 89. The pursuit for disease biomarkers, insights into biological pathways and translating proteomic findings into novel therapies has led to a proliferation of journals addressing aspects of these topics. The generation of large volumes of proteomic data, characterized by long lists of proteins, has also resulted in analogous studies on criteria for quality control and standards required for proteomic studies 20, 46. The need for caution in translating the plethora of proteomic data into functional significance has also been addressed. A sobering paper by Petrak and colleagues (2008) (58) demonstrated that, by analyzing the findings from 169 proteomic studies published in the journal Proteomics during the period 2004–2006, there were many commonly identified proteins found differentially expressed irrespective of the nature of the background disease. The ubiquity of these proteins was considered to arise from cellular stress responses and the limitations of two‐dimensional gel electrophoresis (2DGE). Moreover, two other key areas of caution are the successful application of mass spectrometry (MS) and statistical analysis to ensure that proteins are correctly identified. HUPO have highlighted that most errors in MS based analyses arise not because the present technology is unable to identify specific proteins but because software programmes often misidentify them (70).
The application of proteomics specifically to gliomas is appealing since there has been very limited progress in treatment of malignant glioma in the last 25 years (2). Proteomic studies in gliomas remain limited in number and are characterized by lists of proteins found to be either up‐ or downregulated in tissue specimens compared to normal brain 19, 28. The utility of proteomics in glioma has recently been the subject of two reviews 10, 82 but neither of these articles explored in any detail the specific proteins found to be different in gliomas. The importance of attempting to derive some coherence from the glioma proteomic literature is perhaps even more pressing given the recent publication from The Cancer Genome Atlas Research Network (76). This comprehensive study outlined several pivotal, aberrant genetic pathways found in glioblastoma (GBM) and may provide some direction and focus for proteomic studies in human GBMs. Proteomic data needs to be presented in a way that either confirms or refutes the genetic data. Experiences from other types of malignant tumor 24, 44, 87 suggest that it is highly unlikely that a single GBM biomarker will be identified. However, functional analysis of proteomic data may provide insights into differentially regulated biological pathways that underpin aspects of the pathophysiology of this highly malignant tumor (13). In glioma (and in many other neurological diseases and cancer), a glut of proteomic data have been generated but there has been no unitary approach to establish whether key proteins and/or signaling pathways have been identified. In an attempt to evaluate the current utility of glioma proteomic data, we have performed a systematic review of glioma proteomics to date across multiple publications. Indeed, on cursory evaluation of the data there appears a complete lack of coherence as the validity, reproducibility and comparability of these studies is bedevilled by methodological, analytical and statistical differences. Whether such disparate data can then translate into diagnostic, predictive or prognostic biomarkers and novel therapies is a key question.
In this article we demonstrate a novel and fresh perspective, synthesized from the published glioma proteomic literature. We have performed a systematic review of all the proteins identified in glioma proteomic studies, highlighting where there is commonality between the datasets. We address the robustness and reproducibility of the data and whether any substantive conclusions can be drawn between and from this published series. In particular, we have determined if any coherence can be drawn from the noticeable disparate data published to date, to highlight some putative biological pathways. We have listed all the differentially expressed proteins described so far in human gliomas and have constructed a hierarchy of the most frequently described proteins. We have examined if there is any differential clustering of these proteins to certain parts of the 2D gel from which the proteins were identified to determine if the listed proteins are merely a reflection of 2DGE limitations (58). Finally, we have used sophisticated bioinformatics software to perform functional analyses to determine putative common biological pathways that incorporate these proteins. This analysis will determine whether such an approach adds value to a simple proteomic list. Additionally the findings are compared with biological pathways highlighted as abnormal in glioma, recently described in a genomic study published by The Cancer Genome Atlas Research Network (76). We also discuss how proteomic methodology may be best applied to glioma research in the future.
METHODS
Systematic review of human glioma proteomic literature
Using the search terms “glioma,” “glioblastoma” and “proteomics” we identified 10 peer‐reviewed articles reporting differential protein expression via a range of proteomic technologies (see Search Strategy and Selection Criteria). From each publication the sample size (ie, the number of tumors, “n”), sample origin, tumor pathology [utilizing the World Health Organization (WHO) system], age range of subjects, method of tissue analysis, use of “control” tissue, differential expressed proteins, function (where documented) and protein accession number were extracted. A comprehensive spreadsheet was constructed containing all of the aforementioned data (see 1, 2).
Table 1.
Overview of the glioma proteomic literature. Abbreviations: 2DGE = two‐dimensional gel electrophoresis; GBM = glioblastoma; AA = anaplastic astrocytoma; AO = anaplastic oligodendroglioma; oligo = oligodendroglioma; DNET = dysembryoplastic neuroepithelial tumor; MALDI‐TOF = matrix‐assisted laser desorption ionization—time of flight; nano LC = nanoliquid chromatography; SELDI‐TOF = surface enhanced laser desorption ionization—time of flight.
| Author/reference | n | Pathology who grade | Age range | Sample origin | Method | Control |
|---|---|---|---|---|---|---|
| Chakravarti et al (5) | 94 | 56 GBMs | Not specified | US | Western blot analysis | Lysates from 16 week old fetuses |
| 13 AAIII | ||||||
| 25 low‐grade gliomas | ||||||
| Hiratsuka et al (28) | 5 | 2 GBM | Not specified | JAPAN | 2DGE and MALDI‐TOF | Same patients from which the tumors were taken. |
| 2 Grade III | ||||||
| 1 Grade I | ||||||
| Hobbs et al (29) | 4 | 4 GBM | Not specified | US | SELDI‐TOF‐MS | None |
| Schwartz et al (67) | 18 | 4 GBM | 3 days–80 years (includes control and tumor samples) | US | MALDI‐MS | Temporal lobe specimens from epilepsy surgery. |
| 2 Oligo Grade II | ||||||
| 2 AO Grade III | ||||||
| 2 Embryonal carcinoma | ||||||
| 1 Pheochromcytoma | ||||||
| 1 DNET | ||||||
| 1 Gemistocytic astrocytoma, grade II | ||||||
| Iwadate et al (35) | 85 | 52 GBM | Not specified | JAPAN | 2DGE and MALDI‐TOF | Tissue from patients undergoing surgery or for epilepsy. |
| 13 AA Grade III | ||||||
| 10 Astrocytomas (Grades I and II) | ||||||
| 10 normal brain | ||||||
| Furuta et al (19) | 13 | 6 GBM (Primary)s | Not specified | US | 2DGE | None |
| 7 progressive GBMs | ||||||
| Odreman et al (54) | 20 | 10 Grade II | Not specified | ITALY | 2DGE, LC‐ESI‐MS and Western blot analysis. | “peri‐lesional tissue” from all patients |
| 10 GBM | ||||||
| Schwartz et al (68) | 127 | 29 Grade II glioma | Not specified | US | MALDI‐TOF | 19 patients undergoing surgery for “non‐neoplastic disease”. |
| 22 Grade III glioma | ||||||
| 57 GBM | ||||||
| Chumbalker et al (11) | 27 | 2 Grade I | 3–65 years (includes control and tumor samples) | INDIA | 2DGE and MALDI‐TOF | Tissue from patients undergoing surgery for epilepsy. |
| 1 Grade II | ||||||
| 14 Grade III | ||||||
| 10 GBM | ||||||
| Mustafa et al (52) | 20 | 10 GBM | Control: 24 weeks–76 years | HOLLAND | Nano‐LC prior to MALDI‐TOF/TOF | samples from different patients with a variety of CNS conditions. |
| 10 controls | ||||||
| Tumor: 30–57 years |
This table summarizes details of the clinical samples and methodology used in each of the publications identified from the literature that use proteomics to study glioma, in accordance with our “Search Strategy and Selection Criteria” (samples size “n”; sample origin, tumor pathology utilising the World Health Organization grading system, age range of subjects, method of tissue analysis and origin of “control” tissue).
Table 2.
List of proteins highlighted in our systematic review of the glioma proteomic literature.
| Protein name | Accn No. | Upregulated | Downregulated | Reference |
|---|---|---|---|---|
| Prohibitin (PHB) | P35232 | Gliomas (5), GBM (28) | Grade III (11) | 5, 11, 28 |
| Alpha B crystallin (CRYAB/HSP20) | P02511 | Grades I and II (28), GBM (19) | Grade III (67) | 19, 28, 67 |
| Serum albumin | NP_004468 | Gliomas (5), Grade III and GBM (11) | 5, 11 | |
| Epidermal growth factor receptor (EGFR) ** | P00533 | GBMs (54), GBMs (primary) (68) | 54, 68 | |
| Phosphoprotein enriched in astrocytes (EA‐15) | Q15121 | Grades II and III (5) | Gliomas (29) | 5, 29 |
| RhoGDP dissociator inhibitor (RhoGDI) | P52565 | Grade III (11, 11), GBM (11, 11) | (11, 11) | |
| Apolipoprotein A1 (APOA1) | P02647 | Gliomas (5), GBM (67) | 5, 67 | |
| Glial fibrillary acidic protein (GFAP) | P14136 | Grade II (67), Grade III (11) | 11, 67 | |
| Heat shock 70 kDa protein 5 (HSP 70/HSPA5) | P11021 | GBM (28) | Grade III (11) | 11, 28 |
| Protein disulfide isomerase 3 (PDIA3) | P30101 | Grade I (28), Grade II (67) | 28, 67 | |
| ADAMTS‐19 | 19171152 | GBM (secondary) | (68) | |
| Beta‐actin | P02570 | GBMs | (67) | |
| Cadherin related tumor supressor | Q14517 | GBMs (secondary) | (68) | |
| Fibronectin | P07589 | Gliomas | (52) | |
| hTRT** | CAE75638 | GBMs | (54) | |
| Calcyclin | P06703 | GBMs | (29) | |
| CREB1 | P16220 | GBMs | (28) | |
| CDK4** | CAG4703 | GBMs | (54) | |
| CDKN1A | P38936 | Grades I and II | (28) | |
| Cyclin E1** | NP_001229 | Gliomas | (54) | |
| E2F‐1 | NP_005216 | In 55% gliomas | (54) | |
| HNRPA3 | P51991 | GBMs (secondary) | (68) | |
| Transcription factor BTF3 | Q13890 | GBMs | (67) | |
| WNT II protein precursor | 20532422 | GBMs (secondary) | (68) | |
| SIRT2 | NP_085096 | Gliomas | (5) | |
| Copine I | NP_003906 | Gliomas | (5) | |
| CRMP‐4 | NP_001378 | Gliomas | (5) | |
| Dynein light chain 2 | Q96FJ2 | LTS group | (29) | |
| Ezrin | P15311 | Grade III and GBMs | (28) | |
| NAPG | NP_003817 | Grade III | (11) | |
| Neurocalcin delta | NP_114430 | Gliomas | (5) | |
| Protein kinase C gamma | P05129 | Grade III and GBMs | (28) | |
| Tropomodulin 2 | NP_055363 | Grade III | (11) | |
| Tropomycin | NP_003281 | GBMs | (11) | |
| Tropomycin 4 | NP_003281 | Grade III | GBMs | (11) |
| Tubulin specific chaperone A | O75347 | GBM | (29) | |
| beta tubulin | gi|223486 | GBMs | (11) | |
| Profilin 2 | NP_444252 | Gliomas | (5) | |
| Vimentin | NP_003371 | GBM | (11) | |
| Tenascin X precursor | P22105 | GBMs (primary) | (68) | |
| Alpha‐internexin | Q16352 | GBM | (67) | |
| Adenosine deaminase | 1001165a | Grade III | (11) | |
| ATP synthase | P06576 | GBMs | (11) | |
| ATPase | NP_001686 | Grade III | (11) | |
| Beta synuclein | NP_003076 | Grade III | (11) | |
| Calpactin 1 light chain | P08206 | GBM | (29) | |
| cAMP depended protein kinase | P17612 | Grade II | (67) | |
| Catechol‐O‐methytransferase | NP_000745 | Gliomas | (5) | |
| Cathepsin D precursor | P07339 | Grade III and GBMs | (28) | |
| Dihydropteridine reductase | P09417 | Grade II | (67) | |
| DUOX2 | NP_054799 | GBMs (secondary) | (68) | |
| Enolase 1 | AHH27725 | GBMs (primary) | (68) | |
| ERK kinase 1 | Q02750 | Grade III and GBMs | (28) | |
| Fatty acid binding protein 5 | Q01469 | Grade III | (29) | |
| Fatty acid binding protein 7 | NP_001437 | Gliomas | (5) | |
| Glutamate dehydrogenase 1 | P00367 | Grades I and II | (28) | |
| Glutathione S transferase | P09488 | Grade III and GBMs | (28) | |
| Glutathione S transferase P | P09211 | Grades I and II | (28) | |
| human mitochondrial aldehyde dehydrogenase | gi|6137677 | GBMs | (11) | |
| Nucleolar GTP binding protein 1 | Q9BZE4 | Grade III and GBMs | (28) | |
| Peroxiredoxin 1 | Q06830 | GBMs | (67) | |
| Peroxiredoxin 6 | P30041 | GBMs | (67) | |
| Phosphoglycerate mutase 1 | P18669 | Grade III and GBMs | (28) | |
| Phosphopyruvate hydratase | P06733 | Grades I and II | (28) | |
| Phosphoserine phosphatase | NP_004568 | Gliomas | (5) | |
| Plasminogen activator inhibitor 1 | P05121 | Grade III and GBMs | (28) | |
| RAB3A | P20336 | Grade III and GBMs | (28) | |
| Rac 1 | P15154 | GBMs | (28) | |
| Rho A | P06749 | GBMs | (28) | |
| Tyrosine tryptophan mono‐oxidase | NP_036611 | Grade III | (11) | |
| Ubiquitin carboxy‐terminal hydrolase L1 | gi|4185720 | GBMs | (11) | |
| Ubiquitin protein | P09936 | Grade II | (67) | |
| Annexin A5 | P08758 | GBMs | (67) | |
| Annexin II | P07355 | Grade III and GBMs | (28) | |
| Annexin IV | P09525 | Grade III and GBMs | (28) | |
| Annexin V | gi|999926 | Grade III | (11) | |
| T complex protein I | P48643 | Grade II | (67) | |
| Colligin 2 | P50454 | Gliomas | (52) | |
| HSP 27 | P04792 | Grades III and IV | (28) | |
| HSP 60 | P10809 | Grade III | (11) | |
| UCH‐L1 | P09936 | Gliomas | (5) | |
| Oncogene DJ1 | NP00116849 | Grade III | (11) | |
| p14** | GBMs | (54) | ||
| p16 | GBMs | (54) | ||
| p53 | P04637 | GBMs | (54) | |
| Centrosome associated protein 350 | 18378735 | GBMs (primary) | (68) | |
| ERCC6 | 15834617 | GBMs (secondary) | (68) | |
| Eukaryotic initiation factor 4A | P04765 | Grade III and GBMs | (28) | |
| PTEN | NP_000305 | GBMs | (54) | |
| Tumor suppressor pRB | P06400 | GBMs | (54) | |
| Fribrinogen fragment D | 2781208 | GBMs | (67) | |
| alpha‐antitrypsin | P01009 | GBMs | (11) | |
| Ferritin | AAH16715 | Grade III | (11) | |
| Hemopxin | NP_000604 | Gliomas | (5) | |
| Synaptosomal associated protein 25 | NP_003072 | Grade III | (11) | |
| Transthyretin | NP000362 | Gliomas | (5) | |
| Sorcin | NP_003121 | GBMs | (11) | |
| Unamed protein | 16552261 | GBMs (primary) | (68) |
This all the proteins (99 in total) that were reported in the literature as altered in expression level in glioma. This data was extracted from the publications summarized in Table 1. It is important to be aware of two main limitations of the data: (1) the certainty of protein identification varies across studies (some studies report the probability of a definitive protein match and others do not); (2) the validation of identified proteins varies across studies (perhaps due to availability of adequate antibodies for validation of identified proteins by other methodologies such as immunocytochemistry, yet validation is essential to confirm true differences in expression). The proteins presented in this table are: (i) identified by name and accession number (Accn No.); (ii) shown as up‐regulated or down‐regulated in expression level in tumor compared to control tissue; and (iii) shown as to which grade of glioma their altered expression level is reported in. In addition, the data collated is cross‐referenced to the article(s) from which the data was extracted. The 10 proteins found altered in two or more publications (PHB, CRYAB, serum albumin, EGFR, EA‐15, RhoGDI, APOA1, GFAP, HSP70, PDIA3) are shown in bold at the top of the table, and proteins marked ** are proteins that have been shown in previous studies to predict poor patient survival.
Critical appraisal of differentially expressed proteins in glioma: their location on 2D gels
In proteomic analysis, the same proteins are repeatedly identified as altered in a range of biological conditions (58). This reflects in part their functional roles (specifically the involvement of a single protein in numerous diverse cellular processes) and technical issues involving the separation and identification of proteins with particular molecular weights and isoelectric charge (pI). For example, in our own laboratory, 100% of the proteins significantly altered in a study of apoptosis induced by staurosporine, were of medium/high molecular weight (32.9–94.3 kDa) and of low pI (4.7–6.4) (71).
The extent to which proteins of particular molecular weights or charge disproportionately contribute to proteomic analysis of glioma, based on 2DGE, was examined. To determine if the proteins found differentially expressed in our systematic review were positioned randomly on a 2D gel or clustered to one part of the gel (which might indicate technological bias), an in silico gel was constructed that was divided into four quadrants. The x‐axis was assigned a linear scale as the distribution of proteins in IPG strips in the first dimension of 2DGE is predominantly linear. In contrast a logarithmic scale was chosen for the y‐axis because this most closely resembles the pattern of vertical protein migration in gels during the second dimension of 2DGE. The quadrants were defined as: I (30–100 kDa and 3–6.5 pH), II (30–100 kDa and 6.5–10 pH), III (10–30 kDa and 6.5–10 pH) and IV (10–30 kDa and 3–6.5 pH).
Critical appraisal of differentially expressed proteins in glioma: protein classification and protein–protein interactions
To ascertain whether the proteins found differentially expressed in glioma (listed in Table 2) were involved in common biological functions, protein accession numbers were entered into Protein Analysis Through Evolutionary Relationships (PANTHER) Classification System (http://www.pantherdb.org). PANTHER categorized all the proteins found altered in glioma into prespecified protein functional classes.
To gain further insight into whether functional protein–protein interactions existed amongst the list of proteins found altered in glioma, proteins were investigated using Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Mountain View, CA, USA, http://www.ingenuity.com). IPA is web‐based software that comprises a huge knowledge database [Ingenuity Pathways Knowledge Base (IPKB)] of biological and chemical information extracted from the literature. The accession numbers of all proteins found differentially regulated in GBM (WHO grade IV glioma) compared with control brain, were entered into the application and after critical assessment of the protein list, a non‐prejudicial protein interaction network (showing direct interactions and associations only) was generated. The network was scored and ranked according to the inclusion of as many of the proteins that were inputted as possible.
Methodology: search strategy and selection criteria
A comprehensive review of the published literature was performed by searching PubMed, using the search items “proteomics,” “gliomas” and “glioblastomas,” to identify all peer‐reviewed articles reporting differential protein expression via a range of proteomic technologies (eg, MALDI‐TOF) up to October 2008. Papers were also identified from the authors' own files, and references from relevant articles. Only papers published in English were reviewed. All studies that were selected pertained to in vivo samples taken from human gliomas. Eleven studies were identified. One was discarded because of an eminent senior scientist disassociating himself from the publication (38).
RESULTS
What proteomic data have been generated in human glioma and where does this data originate from?
General comment
A total of 99 differentially expressed proteins were identified from 10 peer‐reviewed articles 5, 11, 19, 28, 29, 35, 52, 54, 67, 68. These selected papers vary considerably in terms of sample size, tumor type, age range and control tissue used (see Table 1). The number of tumor specimens analyzed across the series of publications ranges from 4 to 127. In addition, a broad age range of samples was used in several studies (for example, Schwartz et al (67) used tissue from 3‐day‐ to 80‐year‐old patients), whereas others do not stipulate an age range.
Spectrum of proteomic methodology
Proteomic methodology also varies between papers from simple Western blot analysis to 2DGE ± matrix assisted laser desorption ionization—time of flight (MALDI‐TOF) MS, surface enhanced laser desorption ionization—time of flight (SELDI‐TOF) MS or nanoliquid chromatography (Nano LC) with tandem mass spectroscopy. In addition the choice of protein stain, for example Coomassie stain (11), Silver stain 19, 35 or Sypro Ruby (28)[three stains with very different dynamic ranges; see (81)] varies together with the inclusion criteria of the size of proteins studied (ie, 20–120 kDa).
Differences in control tissue
There is also no consistency with control tissue used, since three studies use specimens from epilepsy patients 11, 35, 67, whereas others utilize “normal” peri‐lesional tissue from tumor patients 28, 54 or “normal” tissue from patients with a variety of other CNS conditions 52, 68. These differences give rise to very different experimental comparisons as epileptic tissue may be gliotic and peritumoral tissue may be infiltrated by tumor cells (such as GBM, oligodendroglioma or astrocytoma tumor cells).
Differences in comparisons of glioma proteomic data
Some of the studies exclusively investigate differential protein expression in GBMs versus control (52), whereas others pool different grades of tumor and compare these pooled mixed gliomas versus controls 5, 11, 28, 35, 67 or compare “low‐grade” gliomas to that of “high‐grade” tumors (54). Moreover Furuta et al (19) compare primary GBMs (GBMs that arise de novo) with secondary GBM (GBMs that progress from lower grade gliomas). These differences in experimental design almost certainly contribute to the disparate lists of proteins generated in glioma proteomics.
Differences in the purification of samples
Methodological variation in tissue sample preparation also exists. The majority of the proteomic studies use whole cell lysates generated from gross dissection of the control and tumor specimens 5, 11, 28, 29, 35, 54. However, Furuta et al (19) strived for purer populations of tumor cells by employing selective tissue microdissection prior to 2DGE. Selective tissue microdissection was based on a 10‐µm‐thick tissue section (adjacent to the section used for microdissection) stained for hematoxylin and eosin to identify areas not compromised by inflammation, necrosis or stromal or endothelial proliferation. Mustafa et al (52) also employed a purification step and specifically microdissected glioma blood vessels for investigation by MS. The proteome identified by Mustafa et al (52) was therefore confined to glioma blood vessels and not representative of all proteins altered in glioma tumors.
Differences in statistical methodology
Statistical analysis in glioma proteomic studies to date, range from almost nothing to complex bioinformatic paradigms (13). Moreover, the complex statistical paradigms that are used are diverse and include methodology such as hierarchical cluster analysis 35, 67, symbolic discriminate analysis (SDA) and weighted flexible compound covariate method (WFCCM) (68).
Proteins identified as differentially expressed in gliomas
Despite differences in experimental design, methodology and analysis all of the proteins reported as differentially regulated in glioma tumors were collated (see Table 2). Out of these 99 proteins, 10 proteins [PHB, CRYAB, serum albumin, epidermal growth factor receptor (EGFR), EA‐15, RhoGDI, APOA1, GFAP, HSP70 and PDIA3] were found altered in multiple proteomic studies of glioma (see Table 2). The most commonly identified proteins were Prohibitin (PHB) and Alpha B crystalline (CRYAB), which were found differentially expressed in three distinct studies. Prohibitin is a highly conserved, multifunctional protein, which is found localized to mitochondria and nuclei 47, 48. Overexpression of prohibitin has been reported in human bladder tumors (3), prostate cancer (77) and thyroid carcinomas (17), suggesting that this protein may be widely involved in tumorigenesis. Prohibitin has also been shown to control Ras‐Raf signaling, a major signaling pathway involved in cell growth and malignant transformation 60, 61. In gliomas, prohibitin has been reported to be both up‐ and downregulated [11, 28, 35; see Table 2], a finding that in the first instance seems unclear. However, it is thought that prohibitin may exert different roles in tumorigenesis, having either a permissive action on tumor growth or a tumor suppressor role, depending on the cellular context and/or stage of tumorigenesis 47, 48.
CRYAB is a constitutively expressed molecular chaperone and member of the evolutionary conserved heat shock protein superfamily. CRYAB has also been found regulated in several different types of cancer 9, 30, 55, 56 and may reflect a general cellular stress response to tumorigenesis.
Additional comments about the top 10 proteins identified in glioma
The up‐regulation of serum albumin and apolipoprotein A1 in gliomas is thought to reflect the ability of both these proteins to pass into the interstitium of malignant gliomas because either the blood brain barrier has broken down and/or the tumor capillaries have no BBB (69). It is also reassuring to find both EGFR and GFAP among the top 10 proteins identified in glioma. It has been widely recognized that EGFR is amplified and can be mutated in malignant gliomas (42) and the upregulation of GFAP is considered a fundamental and diagnostic ICC feature of glioma (15). In contrast, the remaining proteins from the list of top 10 have a range of different functional roles and it is not known whether or how they play a role in gliomagenesis. It is important to note that RhoGDI, has been frequently reported as regulated in proteomic studies regardless of the biological process and species studied (58).
Comments about the entire list of proteins found differentially expressed in gliomas (the 99 proteins)
In total, eight out of the total 99 differentially expressed proteins identified in glioma (Table 2) have been reported by Petrak et al (2008) (58) as proteins commonly identified in proteomic studies regardless of experiment, tissue or species (these proteins are: RhoGDI, vimentin, ATP synthase, cathepsin D precursor, enolase 1, peroxiredoxin 1, annexin 4 and HSP27). Aside from these nonspecific findings, however, several important, key putative players in glioma pathophysiology are listed. In particular tumor suppressor proteins p53, phosphatase and tensin homolog (PTEN) and p14 have been identified. Furthermore ferritin, previously reported as regulated in malignant gliomas (31), has been identified.
A critique of the proteins of interest in glioma with respect to 2DGE
To determine whether the differentially expressed proteins (the 99 proteins listed in Table 2) reflected any kind of preferential detection from a certain area of the 2D gel, we located the position of each candidate protein on a virtual gel. Seven proteins, with molecular weights above 100 kDa were excluded from the plot. Ninety‐three proteins were plotted along a logarithmic scale of 10kDa to 100 kDa and against a linear scale of 3 to 10 pH units. Proteins were found distributed (see Figure 1) in the numbered gel quadrants as follows: 39 proteins in quadrant I [42%: 95% confidence intervals (CIs) 32–52%], 19 proteins in quadrant II (20%: 95% CI 13–30%), 14 proteins in quadrant III (15%; 95% CI 9–23%) and 21 proteins in quadrant IV (23%: 95% CI 15–32%). The percentage distribution and 95% CIs of differentially expressed proteins, is not dissimilar to the percentage distribution of the total number of proteins detected on a 2D gel for a malignant glioma. A representative and randomly selected GBM gel (generated from our laboratory) had an overall protein distribution in the four quadrants as follows: 246 proteins in quadrant I (34%: 95% CIs 30–36%), 223 proteins in quadrant II (30%: 95% CI 27–33%), 135 proteins in quadrant III (18%: 95% CI 16–21%) and 131 proteins in quadrant IV (18%; 95% CI 15–21%) (Figure 1).
Figure 1.
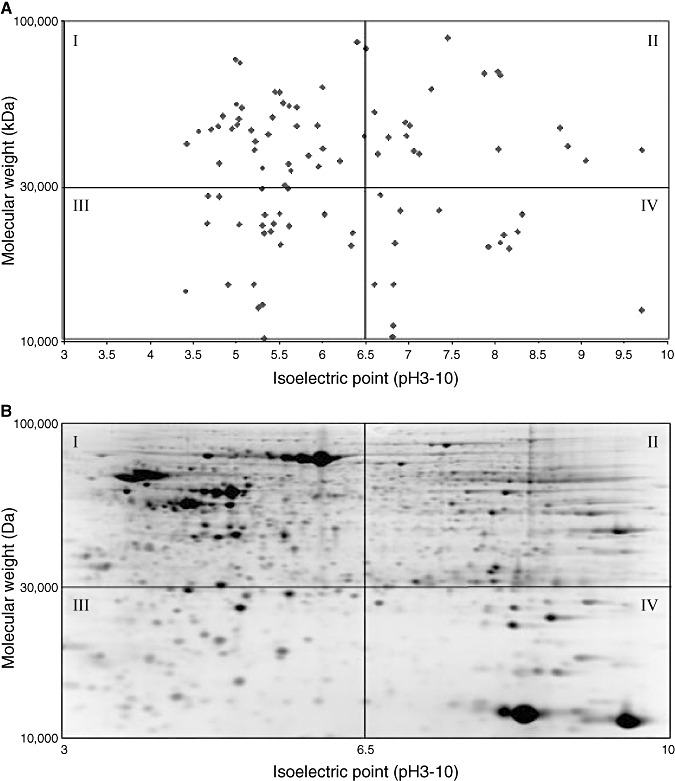
A. Location on two‐dimensional (2D) gels of proteins putatively altered in glioma. B. The distribution of altered proteins in A mirrors the total identifiable protein distribution on a representative 2D gel of human brain tissue. The 99 proteins found altered in glioma proteomic studies (excluding seven proteins with molecular weights above 100 kDa), plotted on a virtual gel. The y‐axis of the virtual gel A has a logarithmic scale of 10 to 100 kDa and the x‐axis has a linear scale of 3–10 pH units to mimic the pattern of protein migration during 2D gel electrophoresis. The proteins altered in glioma are distributed across the gel in a similar fashion (% of proteins per gel quadrant) to total number of proteins in glioma. A representative 2D gel image of a human glioma specimen showing distribution of all proteins (visualized as spots on the gel) is shown in “B”.
Making sense of the individual protein findings: functional insight?
To investigate whether a common biological function characterized the list of 99 proteins found altered in glioma (Table 2), we performed PANTHER analysis on the 99 proteins collated (see the section “Critical appraisal of differentially expressed proteins in glioma: protein classification and protein‐protein interactions”). PANTHER categorized all the proteins (according to prespecified functional groups) into 23 different functional groups (for example, cell structure and motility, cell cycle, intracellular protein trafficking, cell proliferation and differentiation, and signal transduction). Five proteins constituted the most proteins found in any of these functional groups. However, no clear or specific biological process was highlighted (Figure 2).
Figure 2.
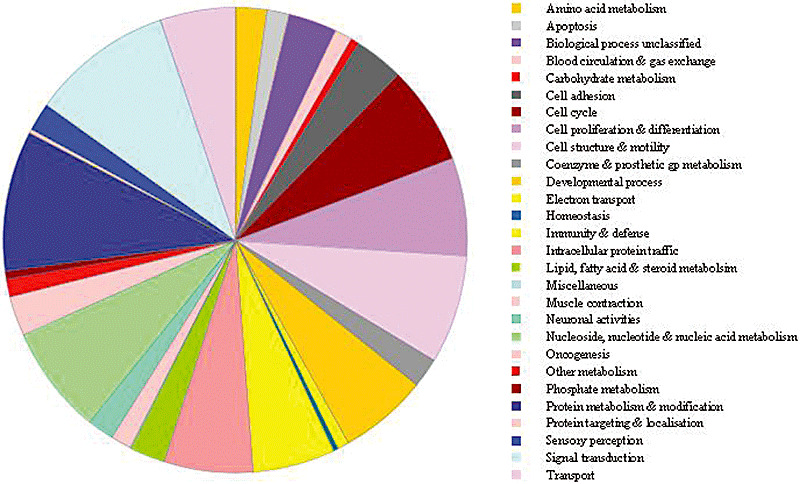
Biological functions of the 99 proteins putatively altered in glioma according to Protein Analysis Through Evolutionary Relationships (PANTHER) classification. A pie chart of the 23 functional groups assigned by PANTHER to categorize the proteins putatively altered in glioma. No clear or specific biological processes are highlighted.
In a further attempt to make some functional sense of the proteomic data generated in glioma, we investigated protein–protein interactions between the differentially regulated 99 proteins using network analysis [Ingenuity Pathway Analysis; (IPA)]. When all these 99 proteins implicated to play a role in glioma pathophysiology were entered into IPA, 8 different protein–protein interaction networks were generated, containing a multitude of indirect protein–protein interactions. No functional coherence was gained from this preliminary IPA analysis. Such a lack of coherence was perhaps not surprising since low‐grade tumors and high‐grade tumors display distinct behavior and are likely to generate quite different protein–protein interaction responses.
However, when the 58 proteins found specifically altered in GBM (WHO grade 4 glioma) were entered into IPA a highly significant primary network was generated (score 57, refer to http://www.ingenuity.com for score details). This network (Figure 3) contains 28 of the proteins that were entered and shows a multitude of direct interactions or associations between all of these molecules. TP53 and RB1 are at the core of this proteomic network which is reassuring given that both genes have been known for some years to be fundamental to gliomagenesis 12, 65. Two other genes frequently described as abnormal in classical molecular studies of GBM are PTEN and EGFR 43, 79 and both these proteins are also central to the functional network generated. Consequently the proteomic network complements recent findings by The Cancer Genome Atlas Research Network (76) who found that GBMs harbor frequent genetic alterations in core components of the RB, TP53 and RTK pathways. In conclusion, the resultant network appears extremely robust and cohesive in terms of “key” players/signaling pathways integral to GBM pathophysiology.
Figure 3.
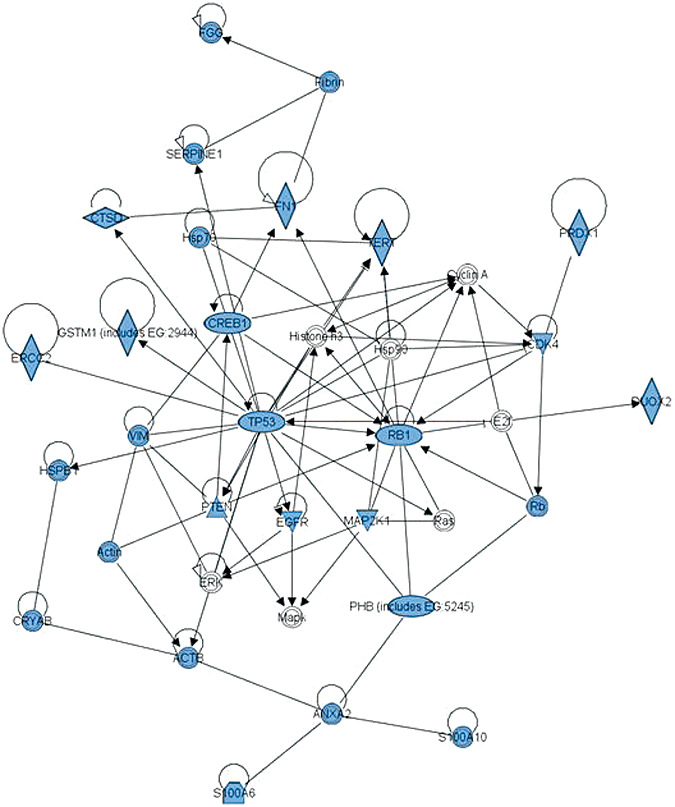
Half of proteins specifically altered in glioblastoma form a coherent network centred on TP53/RB1/PTEN/EGFR. Of the 99 proteins altered in glioma, 58 proteins were reported as altered in glioblastoma. A coherent functional network can be created from 28 of these 58 proteins using Ingenuity Pathway Analysis (IPA). Visual representation of the functional network containing the 28 proteins (in blue) is presented. Each node (blue shape) represents a protein and its association with other proteins, is represented by a line (edge). Nodes have different shapes to represent different molecule types (horizontal diamonds = peptidases, vertical diamonds = enzymes, and circles = “other”; see Ingenuity Systems for detailed node information). Solid lines represent direct interactions between proteins. Direct interactions are defined as those where two proteins make direct physical contact with each other with no intermediate step. Direct interactions may include chemical modifications, for example phosphorylation, but only if there is evidence that the protein can cause the chemical modification directly. The evidence for interactions is obtained from putatively peer‐reviewed publications in “high quality” journals. It should be noted with caution that the evidence (accessible online from IPA) varies markedly in quantity and pertinence for each interaction. This protein–protein interaction network is highly connected with a multitude of direct interactions between proteins altered in glioblastoma.
DISCUSSION
Genetic analysis has dominated glioma research for nearly two decades. Genetics has provided important molecular information (related to therapy response and prognosis) that has further classified tumors within the same histological subtype but has not translated into improved patient outcomes (2). Primary and secondary GBM are indistinguishable histologically but distinct subgroups have been defined genetically (EGFR amplification and loss of PTEN in primary GBM; and overexpression of PDGF, EGF2 and mutations of p53 in secondary GBM). It is now being increasingly recognized that proteomics (the study of multiple proteins simultaneously) is a critical and powerful approach to complement and extend genetic studies, since proteins are the “workhorses” of the cell. The importance of studying protein expression levels and protein dynamics has further been emphasized by multiple discrepancies reported in the literature between the expression levels of genes and proteins in differential analyses of gliomas (L. Salford, pers. comm.).
Proteomics has been adopted in numerous clinical fields of research and has generated enormous amounts of data 63, 73. There has been general criticism however, that proteomic data has not yet yielded significantly novel insights into basic disease mechanisms and translation into either biomarkers or clinical benefit. This is perhaps due to the limitations of some high‐throughput proteomic methodologies, a lack of interpretative tools and immaturity of proteomic analysis compared to genetic analysis (13). Brain tumor proteomics in particular is qualitatively and quantitatively well behind studies of breast, ovarian, colorectal and many other cancers 16, 33. At present, the glioma in vivo proteomic literature is where many studies in systemic cancers were about 5–7 years ago and consequently suffers from the same shortcomings (32).
Differences in analytical methodology between all the glioma proteomic studies raise multiple questions concerning reproducibility, validity and/or comparability of the data. Publications to date have employed a variety of methods with respect to statistical analysis, different protein‐staining techniques and have set a variety of different inclusion criteria for determining which proteins are “key” in glioma pathophysiology. Questions are also raised with regard to the appropriateness of the “control” tissue or group used in these studies. Such methodological considerations may explain why proteins such as PTEN, p53, pRB, tenascin, ferritin, cathepsin, GFAP and EGFR, all of which have been considered fundamental in glioma pathophysiology 27, 45, 79, 90, have been so infrequently described in modern proteomic studies.
The failure of modern high‐throughput proteomic studies to identify other differentially regulated proteins that have been recognized in neuropathological studies using immunocytochemistry or Western blotting is another cause for concern. Some candidate proteins identified in gliomas using classical techniques but not modern proteomics are listed in Table 3. The failure to identify these proteins may reflect their molecular weight, low abundance and/or subcellular location. For example, a protein abundance needs to be >100 ng/g (protein/wet weight tissue) for easy detection on 2D gels, it is a challenge to accurately identify proteins smaller than 30 kDa using MALDI‐TOF MS, and identifying proteins in the membrane bound subcellular fraction is difficult. Even more disconcerting from the translational viewpoint is that several proteins not recognized in high‐throughput proteomic studies have either translated into novel therapies such as EGF, TGF and VGF receptor antagonists or provide prognostic information about the response of GBMs to therapy with temozolomide 25, 45.
Table 3.
List of some proteins found to be biologically important in the pathophysiology of malignant glioma either in vivo or in vitro using classic protein research techniques such as Western blotting and/or immunohistochemistry but not identified using high‐throughput modern proteomic techniques.
| Protein | ∼MW (kDa) | Reference |
|---|---|---|
| Growth factors | ||
| Epidermal growth factor | 6 | Helseth et al (26) |
| Epidermal growth factor receptor VIII | 145 | Wikstrand et al (83) |
| Platelet‐derived growth factor | 31 | Nister et al (53) |
| Vascular endothelial growth factor | 45 | Cheng et al (8) |
| Hepatocyte growth factor | 82 | Moriyama et al (51) |
| Insulin‐like GF | 250 | Gammeltoft et al (21) |
| PI3 kinase | 110 | Chakravarti et al (7) |
| Apoptosis proteins | ||
| MDM2 | 55 | Reifenberger et al (62) |
| Bax | 21 | Krajewski et al (39) |
| Bcl2 | 53 | Krajewski et al (39) |
| Survivin | 16 | Chakravarti et al (6) |
| DNA repair | ||
| MGMT (06methyl guanine transferase) | 22 | Hegi et al (25) |
| Poly ADP ribose polymerase | 116 | Wharton et al (80) |
| Other | ||
| Aquaporin 4 | 32 | Saadoun et al (64) |
One commonality and perhaps pitfall of the glioma high‐throughput proteomic studies reviewed is the use of whole cell lysates. Proteomics of whole cell lysates selects for high abundance proteins with molecular weights between 30 and 100 kDa. Although between 600 and 1000 proteins may be identified, analysis of subcellular fractions may provide insights that are more relevant and focused to glioma pathophysiology. Since dysregulation of cell proliferation and normal apoptosis are two fundamental processes sustaining malignant glioma, evaluation of proteins in the nuclear matrix fraction, chromatin binding fraction and intermediate filaments may be useful 1, 37. Using nanoliquid chromatography‐mass spectroscopy (NLC‐MS MS) up to 2000 proteins can be identified in each of these subnuclear fractions; some of which are unique to one compartment whereas many are common to all three compartments. Energy metabolism is another fundamentally dysregulated process in malignant glioma (66). Proteomic evaluation of the mitochondrial fraction may thus provide mechanistic insights into aberrations of energy metabolism and apoptosis. Protein alterations associated with mitochondrial dysfunction are increasingly becoming a focus of cancer research in general 18, 22. Proteomic analysis of the glioma secretome and exosome may also provide insights into how gliomas attenuate immunological responses and signal to adjacent tumor cells. Moreover, further insights into all these areas may be facilitated by the recent development of mouse glioma models (34). Proteomic studies of these tumors may provide clues into specific mechanistic pathways that may have parallels in humans. Already proteomic studies of melanoma in mice have provided insights into protein pathways in humans (88).
The elucidation of genetic alterations in GBM has greatly advanced our understanding of the molecular basis of this brain tumor. Amplification mutations of EGFR (present in 45% of patients) and PDGFRA (present in 13% of patients) are thought to provide an autocrine drive to tumor growth (76). Disruption of cell cycle regulation (involving p53, CDKN2A, TP53 and RB) is a frequent genetic alteration in GBMs, as it is in tumors of the breast, colon and pancreas 72, 85. Disorder signaling (involving NF1, AKT and particularly PI3K and PTEN) are also frequently observed in GBM. While the genetic advances have underpinned mechanistic models of tumor genesis, proliferation, differentiation and growth, rarely does genetics dictate the clinical course of the disease [the increased survival of patients with mutated isocitrate dehydrogenase 1 and of TERT overexpression in high‐grade tumor are notable exceptions 57, 78]. The extent to which genetic alterations in GBM reflect alterations in protein levels is often poorly defined. Crucially, our network analysis of alterations in GBM (although originating from an amalgamation of proteomic data from different sources) has generated a highly interactive cluster of proteins centred round TP53 (deleted in 35% of patients) and RB1 (deleted in 78% of patients), consistent with key pathways implicated in genetic studies (76). The network offers confidence in the proteomic data generated in glioma. Future network analysis may also prove to be a useful tool for identifying novel proteins, beneath the detection limits of technology, which functionally interact with proteins found altered in major proteomic studies.
A major limitation of contemporary network analyses, however, to understand the molecular mechanisms of tumor progression, is their static nature. Biological systems are highly dynamic with proteins able to translocate between organelles and subcompartments conditional on environment, stimuli and the presence/absence of other proteins such as scaffolding molecules (86). The development and application of more advanced network algorithms, encompassing the concept of dynamic interactions [such as Seeded Bayesian networks (14)] will undoubtedly transform our understanding of pathophysiological mechanisms and provide an excellent platform for the generation of novel hypotheses and experimental verification.
Newer proteomic technologies with higher throughput, quicker identification, and the capability of detecting smaller and lower abundance proteins, will also be invaluable for the future of proteomics in glioma. Technologies such as LC‐MS/MS, stable isotope labeling with amino acids in cell culture (SILAC) and isotope‐coded affinity tag (ICAT) 40, 74 (new, fast, sensitive and accurate technologies) are increasingly being favoured over labor‐intensive 2DGE studies and are revolutionizing progress in cancer proteomics. Antibody tissue arrays and reverse‐phase protein lysate arrays are also proving to be popular, providing systematic approaches for investigating the regulation of major signaling pathways 36, 84. Moreover, a number of phosphoproteomic strategies have been developed for investigating the complexity and dynamics of protein signaling (49). Such phosphoproteomic studies are beginning to yield novel insight into established signaling pathways, such as the EGFR pathway, in response to stimuli and time (50). Additionally technological advances are now facilitating the study of glycosylation of proteins, which is the commonest post‐translational modification of proteins (75).
The utility of proteomics in glioma research has clearly emerged from this critical analysis and systematic review. Proteomics allied to genetic analysis can now be applied to address important clinical questions. Parallel proteomic analysis in glioma may provide important insight into why genetic abnormalities too rarely impact on clinical decisions and prognosis. The interplay between proteins and genes may elucidate the role of stem cells in glioma etiology (59), the molecular mechanisms which make glioma stem cells resistant to therapies, the signals involved in tumor progression (from WHO II gliomas to higher grade gliomas), and the intracellular events associated with chemoradiotherapy in recurrent malignant gliomas. Focusing on the synaptic proteome may also provide insights into how peritumoral brain is functionally altered by the proximity of a glioma (23) and why lower grade gliomas cause seizures whereas higher grade tumors cause focal neurological deficits that are responsive to glucocorticoids (81). The potential of proteomics is now being realized in glioma research. The continuing advances in proteomic technology, interpretative network analyses and in particular, correlation with genetic data will advance our knowledge of glioma pathophysiology and ultimately improve the treatment of this terrible disease.
CONFLICTS OF INTEREST
We have no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported in part by grants and support from the CSO (CZB/4/486); The Melville Trust; Brain Tumour Research Fund and MRC (G0802238).
REFERENCES
- 1. Albrethsen J, Knol JC, Jimenez CR (2009) Unravelling the nuclear matrix proteome. J Proteome 72:71–81. [DOI] [PubMed] [Google Scholar]
- 2. Anderson E, Grant R, Lewis SC, Whittle IR (2008) Randomized phase III controlled trials of therapy in malignant glioma: where are we after 40 years? Br J Neurosurg 22:339–349. [DOI] [PubMed] [Google Scholar]
- 3. Asamoto M, Cohen SM (1994) Prohibitin gene is overexpressed but not mutated in rat bladder carcinomas and cell lines. Cancer Lett 83:201–207. [DOI] [PubMed] [Google Scholar]
- 4. Banks R, Selsby P (2003) Clinical proteomics – insights into pathologies and benefits for patients. Lancet 362:415–416. [DOI] [PubMed] [Google Scholar]
- 5. Chakravarti A, Delaney MA, Noll E, Black PM, Loeffler JS, Muzikansky A et al (2001) Prognostic and pathologic significance of quantitative protein expression profiling in human gliomas. Clin Cancer Res 7:2387–2395. [PubMed] [Google Scholar]
- 6. Chakravarti A, Noll E, Black PM, Finkelstein DF, Finkelstein DM, Dyson NJ et al (2002) Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J Clin Oncol 20:1063–1068. [DOI] [PubMed] [Google Scholar]
- 7. Chakravarti A, Zhai G, Suzuki Y, Sarkesh S, Black PM, Muzikansky A et al (2004) The prognostic significance of phosphatidylinositol 3‐kinase pathway activation in human gliomas. J Clin Oncol 22:1926–1933. [DOI] [PubMed] [Google Scholar]
- 8. Cheng SY, Huang HJ, Nagane M, Ji XD, Wang D, Shih CC et al (1996) Suppression of glioblastoma angiogenicity and tumorigenicity by inhibition of endogenous expression of vascular endothelial growth factor. Proc Natl Acad Sci USA 93:8502–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chin D, Boyle GM, Williams RM, Ferguson K, Pandeya N, Campbell CM et al (2005) Alpha B‐crystallin, a new independent marker for poor prognosis in head and neck cancer. Laryngoscope 115:1239–1242. [DOI] [PubMed] [Google Scholar]
- 10. Chumbalkar V, Sawaya R, Bogler O (2008) Proteomics: the new frontier also for brain tumor research. Curr Probl Cancer 32:143–154. [DOI] [PubMed] [Google Scholar]
- 11. Chumbalkar VC, Subhashini C, Dhople VM, Sundaram CS, Jagannadham MV, Kumar KN et al (2005) Differential protein expression in human gliomas and molecular insights. Proteomics 5:1167–1177. [DOI] [PubMed] [Google Scholar]
- 12. Collins VP (1998) Gliomas. Cancer Surv 32:37–51. [PubMed] [Google Scholar]
- 13. Deighton RF, Short DM, McGregor RJ, Gow AJ, Whittle IR, McCulloch J (2009) The utility of functional interaction and cluster analysis in CNS proteomics. J Neurosci Methods 180:321–329. [DOI] [PubMed] [Google Scholar]
- 14. Djebbari A, Quackenbush J (2008) Seeded Bayesian Networks: constructing genetic networks from microarray data. BMC Syst Biol 2:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duffy PE, Graf L, Rapport MM (1977) Identification of glial fibrillary acidic protein by the immunoperoxidase method in human brain tumors. J Neuropathol Exp Neurol 36:645–652. [DOI] [PubMed] [Google Scholar]
- 16. Fasa VM, Hanash SM (2009) In‐depth proteomics to define the cell surface and secretome of ovarian cancer cells and processes of protein shedding. Cancer Res 69:728–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frazoni A, Dima M, D'Agnostino M, Puppin C, Fabbro D, Loreto CD et al (2009) Prohibitin is overexpressed in papillary thyroid carcinomas bearing the BRAF (V600E) mutation. Thyroid 19:247–255. [DOI] [PubMed] [Google Scholar]
- 18. Frezza C, Gottlieb E (2009) Mitochondria in cancer: not just innocent bystanders. Semin Cancer Biol 19:4–11. [DOI] [PubMed] [Google Scholar]
- 19. Furuta M, Weil RJ, Vortmeyer AO, Huang S, Lei J, Huang TN et al (2004) Protein patterns and proteins that identify subtypes of glioblastoma multiforme. Oncogene 23:6806–6814. [DOI] [PubMed] [Google Scholar]
- 20. Fuxius S, Eravci M, Broedel O, Weist S, Mansmann U, Eravci S et al (2008) Technical strategies to reduce the amount of “false significant” results in quantitative proteomics. Proteomics 8:1780–1784. [DOI] [PubMed] [Google Scholar]
- 21. Gammeltoft S, Ballotti R, Kowalski A, Westermark B, Van Obberghen E (1988) Expression of two types of receptor for insulin‐like growth factors in human malignant glioma. Cancer Res 48:1233–1237. [PubMed] [Google Scholar]
- 22. Grandemange S, Herzig S, Martinou JC (2009) Mitochondrial dynamics and cancer. Semin Cancer Biol 19:50–56. [DOI] [PubMed] [Google Scholar]
- 23. Grant SG (2006) The synapse proteome and phosphoproteome: a new paradigm for synapse biology. Biochem Soc Trans 34:59–63. [DOI] [PubMed] [Google Scholar]
- 24. Griffith OL, Chiu CG, Gown AM, Jones SJ, Wiseman SM (2008) Biomarker panel diagnosis of thyroid cancer: a critical review. Expert Rev Anticancer Ther 8:1399–1413. [DOI] [PubMed] [Google Scholar]
- 25. Hegi ME, Diserens AC, Gorlia T, Hamou MF, De Tribolet N, Weller M et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. [DOI] [PubMed] [Google Scholar]
- 26. Helseth E, Dalen A, Unsgaard G, Vik R (1988) Type beta transforming growth factor and epidermal growth factor suppress the plasminogen activator activity in a human glioblastoma cell line. J Neurooncol 6:277–283. [DOI] [PubMed] [Google Scholar]
- 27. Higuchi M, Ohnishi T, Arita N, Hiraga S, Hayakawa T (1993) Expression of tenascin in human gliomas: its relation to histological malignancy,tumor dedifferentiation and angiogenesis. Acta Neuropathol 85:481–487. [DOI] [PubMed] [Google Scholar]
- 28. Hiratsuka M, Inoue T, Toda T, Kimura N, Shirayoshi Y, Kamitani H et al (2003) Proteomics‐based identification of differentially expressed genes in human gliomas: down‐regulation of SIRT2 gene. Biochem Biophys Res Commun 309:558–566. [DOI] [PubMed] [Google Scholar]
- 29. Hobbs SK, Shi G, Homer R, Harsh G, Atlas SW, Bednarski MD (2003) Magnetic Resonance Image‐guided proteomics of human glioblastoma multiforme. J Magn Reson Imaging 18:530–536. [DOI] [PubMed] [Google Scholar]
- 30. Holcakova J, Hernychova L, Bouchal P, Brozkova K, Zaloudik J, Valik D et al (2008) Identification of alphaB‐crystallin, a biomarker of renal cell carcinoma by SELDI‐TOF MS. Int J Biol Markers 23:48–53. [DOI] [PubMed] [Google Scholar]
- 31. Holtkamp N, Afanasieva A, Elstner A, Van Landeghen FK, Konneker M, Kuhn SA et al (2005) Brain slice invasion model reveals genes differentially regulated in glioma invasion. Biochem Biophys Res Commun 336:1227–1233. [DOI] [PubMed] [Google Scholar]
- 32. Hondermarck H, Vercoutter‐Edouart AS, Revillion F, Lemoine J, El‐Yazidi‐Belkoura I, Nurcombe V et al (2001) Proteomics of breast cancer for marker discovery and signal pathway profiling. Proteomics 1:1216–1232. [DOI] [PubMed] [Google Scholar]
- 33. Hondermarck H, Tastet C, El Yazidi‐Belkoura I, Toillon RA, Le Bourhis X (2008) Proteomics of breast cancer: the quest for markers and therapeutic targets. J Proteome Res 7:1403–1411. [DOI] [PubMed] [Google Scholar]
- 34. Huse JT, Holland EC (2009) Genetically engineered mouse models of brain cancer and the promise ofpreclinical testing. Brain Pathol 19:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iwadate Y, Sakaida T, Hiwasa T, Nagai Y, Ishikura H, Takiguchi M et al (2004) Molecular classification and survival prediction in human gliomas based on proteome analysis. Cancer Res 64:2496–2501. [DOI] [PubMed] [Google Scholar]
- 36. Jiang R, Mircean C, Shmulevich I, Cogdell D, Jia Y, Tabus I et al (2006) Pathway alterations during glioma progression revealed by reverse phase protein lysate arrays. Proteomics 6:2964–2971. [DOI] [PubMed] [Google Scholar]
- 37. Jimenez CR (2009) Sorting and Zooming: subcellular proteomics is booming. J Proteomics 72:1–3. [DOI] [PubMed] [Google Scholar]
- 38. Khalil AA (2007) Biomarker discovery: a proteomic approach for brain cancer profiling. Cancer Sci 98:201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krajewski S, Krajewska M, Ehrmann J, Sikorska M, Lach B, Chatten J et al (1997) Immunohistochemical analysis of Bcl‐2, Bcl‐X, Mcl‐1, and Bax in tumors of central and peripheral nervous system origin. Am J Pathol 150:805–814. [PMC free article] [PubMed] [Google Scholar]
- 40. Kruse U, Bantscheff M, Drewes G, Hopf C (2008) Chemical and pathway proteomics: powerful tools for oncology drug discovery and personalised health care. Mol Cell Proteomics 7:1887–1901. [DOI] [PubMed] [Google Scholar]
- 41. Latterich M, Abramovitz M, Leyland‐Jones B (2008) Proteomics: new technologies and clinical applications. Eur J Cancer 44:2737–2741. [DOI] [PubMed] [Google Scholar]
- 42. Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H et al (1985) Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313:144–147. [DOI] [PubMed] [Google Scholar]
- 43. Liu W, James CD, Frederick L, Alderete BE, Jenkins RB (1997) PTEN/MMAC1 mutations and EGFR amplification in glioblastomas. Cancer Res 57:5254–5257. [PubMed] [Google Scholar]
- 44. Malinowski DP (2007) Multiple biomarkers in molecular oncology. I. Molecular diagnostics applications in cervical cancer detection. Expert Rev Mol Diagn 7:117–131. [DOI] [PubMed] [Google Scholar]
- 45. Mellinghoff IK, Wang MY, Vivanco I, Haas‐Kogan DA, Zhu S, Dia EQ et al (2005) Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 353:2012–2024. [DOI] [PubMed] [Google Scholar]
- 46. Mischak H, Apweiler R, Banks RE, Conaway M, Coon J, Dominiczak A et al (2007) Clinical proteomics: a need to define the field and to begin to set adequate standards. Proteomics Clin Appl 1:148–156. [DOI] [PubMed] [Google Scholar]
- 47. Mishra S, Murphy LC, Nyomba BLG, Murphy LJ (2005) Prohibitin: a potential target for new therapeutics. Trends Mol Med 11:192–197. [DOI] [PubMed] [Google Scholar]
- 48. Mishra S, Murphy LC, Murphy LJ (2006) The Prohibitins: emerging roles in diverse functions. J Cell Mol Med 10:353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morandell S, Stasyk T, Grosstessner‐Haink K, Roitinger E, Mechtler K, Bonn GK et al (2006) Phosphoproteomic stategies for the functional analysis of signal transduction. Proteomics 6:4047–4056. [DOI] [PubMed] [Google Scholar]
- 50. Morandell S, Stasyk T, Skvortsov S, Ascher S, Huber LA (2008) Quantitative proteomics and phosphoproteomics reveal novel insights into complexity and dynamics of the EGFR signalling network. Proteomics 8:4383–4401. [DOI] [PubMed] [Google Scholar]
- 51. Moriyama T, Kataoka H, Tsubouchi H, Koono M (1995) Concomitant expression of hepatocyte growth factor (HGF), HGF activator and c‐met genes in human glioma cells in vitro. FEBS Lett 372:78–82. [DOI] [PubMed] [Google Scholar]
- 52. Mustafa DAN, Burgers PC, Dekker LJ, Charif H, Titulaer MK, Sillevis Smitt PAE et al (2007) Identification of glioma neovascularization‐related proteins by using MALDI‐FTMS and Nano‐LC fractionation to microdissected tumor vessels. Mol Cell Proteomics 6–7:1147–1157. [DOI] [PubMed] [Google Scholar]
- 53. Nistér M, Heldin CH, Wasteson A, Westermark B (1984) A glioma‐derived analog to platelet‐derived growth factor: demonstration of receptor competing activity and immunological crossreactivity. Proc Natl Acad Sci USA 81:926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Odreman F, Vindigni M, Gonzales ML, Niccolini B, Candiano G, Zanotti B et al (2005) Proteomic studies on low‐ and high‐ grade human brain astrocytomas. J Proteome Res 4:698–708. [DOI] [PubMed] [Google Scholar]
- 55. Ou K, Yu K, Kesuma D, Hooi M, Huang N, Chen W et al (2008) Novel breast cancer biomarkers identified by integrative proteomic and gene expression mapping. J Proteome Res 7:1518–1528. [DOI] [PubMed] [Google Scholar]
- 56. Parcellier A, Schmitt E, Brunet M, Hammann A, Solary E, Garrido C (2005) Small heat shock proteins HSP27 and alphaB‐crystallin: cytoprotective and oncogenic functions. Antioxid Redox Signal 7:404–413. [DOI] [PubMed] [Google Scholar]
- 57. Parsons DW, Jones S, Zhang X, Lin JC‐H, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Petrak J, Ivanek R, Toman O, Cmejla R et al (2008) Déjà vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics 8:1744–1749. [DOI] [PubMed] [Google Scholar]
- 59. Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R et al (2009) Glioma stem cell lines expanded in adherent culture have tumor‐specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4:568–580. [DOI] [PubMed] [Google Scholar]
- 60. Rajalingham K, Rudel T (2005) Ras‐Raf signalling needs prohibitin. Cell Cycle 4:1503–1505. [DOI] [PubMed] [Google Scholar]
- 61. Rajalingham K, Wunder C, Brinkmann V, Churin Y, Hekman M, Sievers C et al (2005) Prohibitin is required for Ras‐induced Raf‐MEK‐ERK activation and epithelial cell migration. Nature Cell Biology 7:837–843. [DOI] [PubMed] [Google Scholar]
- 62. Reifenberger G, Liu L, Ichimura K, Schmidt EE, Collins VP (1993) Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res 53:2736–2739. [PubMed] [Google Scholar]
- 63. Reymond MA, Schlegel W (2007) Proteomics in cancer. Adv Clin Chem 44:103–142. [DOI] [PubMed] [Google Scholar]
- 64. Saadoun S, Papadopoulos MC, Davies DC, Krishna S, Bell BA (2002) Aquaporin‐4 expression is increased in oedematous human brain tumours. J Neurol Neurosurg Psychiatry 72:262–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sanson M, Thillet J, Hoang‐Xuan K (2004) Molecular changes in gliomas. Curr Opin Oncol 16:607–613. [DOI] [PubMed] [Google Scholar]
- 66. Santandreu FM, Brell M, Gene AH, Guevara R, Oliver J, Couce ME (2008) Differences in mitochondrial function and anti‐oxidant systams between regions of human glioma. Cell Physiol Biochem 22:757–768. [DOI] [PubMed] [Google Scholar]
- 67. Schwartz SA, Weil RJ, Johnson MD, Toms SA, Caprioli RM (2004) Protein profiling in brain tumours using mass spectrometry: feasibility of a new technique for the analysis of protein expression. Clin Cancer Res 10:981–987. [DOI] [PubMed] [Google Scholar]
- 68. Schwartz SA, Weil RJ, Thompson RC, Shyr Y, Moore JH, Toms SA et al (2005) Proteomic‐based prognosis of brain tumor patients using direct‐tissue matrix‐assisted laser desorption ionization mass spectrometry. Cancer Res 65:7674–7681. [DOI] [PubMed] [Google Scholar]
- 69. Seitz RJ, Wechsler W (1987) Immunohistochemical demonstration of serum proteins in human cerebral gliomas. Acta Neuropathol (Berl) 73:145–152. [DOI] [PubMed] [Google Scholar]
- 70. Service RF (2008) NewsFocus: proteomics ponders prime time. Science 321:1758–1761. [DOI] [PubMed] [Google Scholar]
- 71. Short DM, Heron ID, Birse‐Archbold JLA, Kerr LE, Sharkey J, McCulloch J (2007) Apoptosis induced by staurosporine alters chaperone and endoplasmic reticulum proteins: identification by quantitative proteomics. Proteomics 7:3085–3096. [DOI] [PubMed] [Google Scholar]
- 72. Soni D, King JAJ, Kaye AH, Hovens CM (2005) Genetics of glioblastoma multiforme: mitogenic signalling and cell cycle converge. J Clin Neurosci 12:1–5. [DOI] [PubMed] [Google Scholar]
- 73. Sowell RA, Owen JB, Butterfield DA (2009) Proteomics in animal models of Alzheimer's and Parkinson's diseases. Ageing Res Rev 8:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sun S, Lee NP, Poon RT, Fan ST, He QY, Lau GK, Luk JM (2007) Oncoproteomics of hepatocellular carcinoma: from cancer markers' discovery to functional pathways. Liver Int 27:1021–1038. [DOI] [PubMed] [Google Scholar]
- 75. Taylor AD, Hancock WS, Hincapie M, Taniguchi N, Hanash SM (2009) Towards an integrated proteomic and glycomic approach to finding cancer biomarkers. Genome Med 1:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. The Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterisation defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ummanni R, Junker H, Zimmermann U, Venz S, Teller S, Giebel J et al (2008) Prohibitin identified by proteomic analysis of prostate biopsies distinguishes hyperplasia and cancer. Cancer Lett 266:171–185. [DOI] [PubMed] [Google Scholar]
- 78. Wager M, Menei P, Guilhot J, Levillain P, Michalak S, Bataille B et al (2008) Prognostic molecular markers with no impact on decision‐making: the paradox of gliomas based on a prospective study. Br J Cancer 98:1830–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, Parsons R (1997) Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res 57:4183–4186. [PubMed] [Google Scholar]
- 80. Wharton SB, McNelis U, Bell HS, Whittle IR (2000) Expression of poly(ADP‐ribose) polymerase and distribution of poly(ADP‐ribosyl)ation in glioblastoma and in a glioma multicellular tumour spheroid model. Neuropathol Appl Neurobiol 26:528–535. [DOI] [PubMed] [Google Scholar]
- 81. Whittle IR (1992) The origins and management of peritumoural brain dysfunction. Neurosurg Quart 2:173–198. [Google Scholar]
- 82. Whittle IR, Short DM, Deighton RF, Kerr LE, Smith C, McCulloch J (2007) Proteomic analysis of glioma. Br J Neurosurg 21:576–582. [DOI] [PubMed] [Google Scholar]
- 83. Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD (1997) Cell surface localization and density of the tumor‐associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res 57:4130–4140. [PubMed] [Google Scholar]
- 84. Wong SC, Chan CM, Ma BB, Lam MY, Choi GC, Au TC et al (2009) Advanced proteomic technologies for cancer biomarker discovery. Expert Rev Proteomics 6:123–134. [DOI] [PubMed] [Google Scholar]
- 85. Wrensch M, Jenkins RB, Chang JS, Yeh R‐F, Xiao Y, Decker PA et al (2009) Variants in the CDKN2B and RTEL1 regions are associated with high‐grade glioma susceptibility. Nature Genet 41:905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yu W, Sanders BG, Kline K (2003) RRR‐alpha‐tocopheryl succinate‐induced apoptosis of human breast cells involves Bax translocation to mitochondria. Cancer Res 63:2483–2491. [PubMed] [Google Scholar]
- 87. Yurkovestky ZR, Linkov FY, E Malehon D, Lokshin AE (2006) Multiple biomarker panels for early detection of ovarian cancer. Future Oncol 2:733–741. [DOI] [PubMed] [Google Scholar]
- 88. Zanivan S, Gnad F, Wickstrom SA, Geiger T, Macek B, Cox J et al (2008) Solid tumor proteome and phosphoproteome analysis by high resolution mass spectrometry. J Proteome Res 7:5314–5326. [DOI] [PubMed] [Google Scholar]
- 89. Zhang J, Keene CD, Pan C, Montine KS, Montine TJ (2008) Proteomics of human neurodegenerative diseases. J Neuropathol Exp Neurol 67:923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ et al (2008) p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455:1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]