

Abstract
Transthyretin (TTR) binds amyloid‐β (Aβ) and prevents Aβ fibril formation in vitro. It was reported that the lack of neurodegeneration in a transgenic mouse model of Alzheimer's disease (AD) (Tg2576 mouse) was associated with increased TTR level in the hippocampus, and that chronic infusion of anti‐TTR antibody into the hippocampus of Tg2576 mice led to increased local Aβ deposits, tau hyperphosphorylation and apoptosis. TTR is, therefore, speculated to prevent Aβ pathology in AD. However, a role for TTR in Aβ deposition is not yet known. To investigate the relationship between TTR and Aβ deposition, we generated a mouse line carrying a null mutation at the endogenous TTR locus and the human mutant amyloid precursor protein cDNA responsible for familial AD (Tg2576/TTR −/− mouse) by crossing Tg2576 mice with TTR‐deficient mice. We asked whether Aβ deposition was accelerated in Tg2576/TTR −/− mice relative to the heterozygous mutant Tg2576 (Tg2576/TTR +/−) mice. Contrary to our expectations, the degree of total and vascular Aβ burdens in the aged Tg2576/TTR −/− mice was significantly reduced relative to the age‐matched Tg2576/TTR +/− mice. Our experiments present, for the first time, compelling evidence that TTR does not suppress but rather accelerates vascular Aβ deposition in the mouse model of AD.
Keywords: Alzheimer's disease, amyloid‐β, apoptosis, tau phosphorylation, Tg2576 mouse, Transthyretin
INTRODUCTION
Insoluble amyloid‐β (Aβ) peptides, the main components of brain amyloid plaques, are thought to be the causative agent of Alzheimer's disease (AD) (11). However, Aβ is normally present in a soluble form in plasma and in the cerebrospinal fluid (CSF) 39, 40, suggesting that some other factors may modulate the aggregation of Aβ fibrils. The hypothesis that transthyretin (TTR) might play some role in the pathogenesis of AD originated from the observation that TTR in the CSF binds Aβ, and prevents Aβ fibril formation in vitro 36, 37. It was further observed that the levels of both TTR and its oxidized forms in the CSF were lower in patients with AD compared with the age‐matched controls 2, 38. The importance of TTR in inhibition of Aβ fibril formation and toxicity in vivo was also suggested in two model systems: transgenic Caenorhabditis elegans and a transgenic mouse model of AD, Tg2576. Link reported that co‐expression of Aβ peptide and TTR in transgenic C. elegans led to a reduction in Aβ deposits (22). Tg2576 line has high level of plasma Aβ peptides 14, 18, and develops brain Aβ deposits similar to that seen in patients with AD 15, 35 and behavioral deficits 13, 53. However, it lacks neurofibrillary tangles (NFT) 27, 48, 49 and neuronal loss (15), which are unique characteristics of patients with AD (5). Stein and Johnson reported that the lack of neurodegeneration was associated with increased level of TTR in the hippocampus of Tg2576 (43). They also reported that chronic infusion of an antibody against TTR into the hippocampus of Tg2576 mice led to increased Aβ deposits, tau hyperphosphorylation, neuronal loss and apoptosis in the CA1 neuronal field (42). Carro et al reported that reduced Aβ burden after insulin‐like growth factor I‐treatment of Tg2576 was paralleled by increased brain levels of TTR (6). Giunta et al reported the inhibition of Aβ aggregation and toxicity and Aβ‐induced apoptotic changes by TTR in cultured cells (10).
All these findings support for the importance of TTR in prevention of Aβ aggregation and toxicity. However, a role for TTR in Aβ deposition is not yet known. To investigate the relationship between TTR and Aβ deposition, we generated a mouse line carrying a null mutation at the endogenous TTR locus and the human mutant amyloid precursor protein (APP) cDNA responsible for familial AD (Tg2576/TTR −/− mouse), by crossing Tg2576 mice with TTR‐deficient mice generated through gene targeting (9). We asked whether Aβ deposition was accelerated in Tg2576/TTR −/− mice relative to the heterozygous mutant Tg2576 (Tg2576/TTR +/−) mice.
METHODS
Animals
Transgenic mice producing human variant APP and lacking endogenous mouse TTR were generated as follows. A male Tg2576 mouse (13) carrying the human mutant APP cDNA with the double mutation K670N and M671L responsible for Swedish familial AD backcrossed to C57BL/6 for 2 generations was mated with TTR −/− female mice backcrossed to C57BL/6 for eight generations (9). The TTR +/− F1 male mice carrying the mutant APP cDNA were mated with TTR −/− female mice. Heterozygous (TTR +/−) F2 male mice carrying the mutant APP cDNA (Tg2576/TTR +/−) were mated with TTR −/− F2 female mice. The TTR +/− and TTR −/− F3 progenies carrying the mutant APP cDNA (Tg2576/TTR +/− and Tg2576/TTR −/−) were used in the present study. The F3 transgenic mice were maintained in cages housing three to six mice each, on separate racks in the same room, kept under a 12‐h light cycle. Regular rodent's chow (Oriental Yeast, Tokyo, Japan) and tap water were freely available.
Transgenic mice were killed by cervical dislocation after anesthesia with diethyl ether. The brains were dissected; the right hemi‐brains were immediately frozen in liquid nitrogen and stored at −80°C while the left hemi‐brains were fixed in 4% buffered paraformaldehyde, and embedded in paraffin. Genotype analysis for each animal was carried out by polymerase chain reaction on DNA, purified from tails, as described 9, 14. The presence and absence of TTR in the serum of Tg2576/TTR +/−, and Tg2576/TTR −/− mice, respectively, were confirmed by western blotting analysis as described (51).
All animal experiments were approved by University of Yamanashi Animal Care and Use Committee.
Immunohistochemistry
For brain Aβ detection, the paraffin‐embedded left hemi‐brain sections (5 µm) were pretreated with 99% formic acid for 3 minutes and immersed in 5% periodic acid for 10 minutes to block endogenous peroxidase. They were then incubated with blocking buffer [5% normal goat serum (Gibco, Carlsbad, CA, USA) in 10‐mM phosphate buffer pH 7.4 and 100‐mM NaCl with 0.05% Tween‐20 (Bio‐Rad, Richmond, CA, USA) containing Block Ace (Dainipponseiyaku, Suita, Japan)] for 1 h, with primary antibody [Ab9204 recognizing normal L‐aspartate at position 1 (34), 0.1 µg/ml] overnight, and with biotinylated anti‐rabbit immunoglobulin G (IgG) antibody (1:200) (Vector Laboratories, Burlingame, CA, USA) for 1 h. Immunoreactivity was visualized with the use of Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA, USA), and 3,3′‐diaminobenzidine, tetrahydrocloride (DAB). Tissue sections were counterstained with hematoxylin.
For phosphorylated tau detection, the paraffin sections were pretreated with periodic acid, as described above and then irradiated in 10‐mM citric acid buffer pH 6.0 for 15 minutes with microwave oven. After blocking, as described above, the sections were stained with the use of primary antibody AT8, recognizing phosphorylated tau at Ser202/Thr205 (1:500) (Innogenetics, Gent, Belgium) or anti‐phosphorylated tau, recognizing phosphorylated tau at Thr231 (Thr231; 1:1000) (Calbiochem, Darmstadt, Germany), and Vectastain ABC Elite kit and counterstained by hematoxylin.
Fragmented DNA of apoptotic cells in the brain was detected by terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling (TUNEL) method with the use of DeadEnd Colorimetric TUNEL System (Promega, Madison, WI, USA) and DAB according to the manufacturer's instructions.
Quantification of Aβ burden by image analysis
For quantification of Aβ burden, immuno‐labeling was examined in the entire cerebral cortex and hippocampal areas of Tg2576/TTR +/− and Tg2576/TTR −/− mice. The amyloid burden was calculated by dividing total area of Aβ deposits by total area of region analyzed (in pixels). Images were captured and analyzed with the use of ImagePro®ver6 software (Media Cybernetics, Silver Spring, MD, USA). Four coronal sections from each of the mice were examined. The burden was expressed as mean ± standard error of the mean.
Protein extraction
Frozen right hemi‐brains were sequentially extracted using two‐step extraction method, as described previously (18). Initially, the frozen brain samples were homogenized in 2% sodium dodecylsulfate (SDS) (150 mg/ml wet weight) with protease inhibitors (complete protease inhibitor cocktail, one tablet in 50‐ml solution; Boehringer Mannheim, Mannheim, Germany) followed by centrifugation at 100,000 g for 1 h at 4°C. The supernatant was then removed (termed SDS fraction), and the resultant pellet was sonicated [(35 s at level 10; XL‐2000 Microson Ultrasonic Cell Disruptor (Misonix Inc., Farmingdale, NY, USA)] in 70% formic acid in water. After sonication, the samples were centrifuged, as described above, and the supernatant was removed (termed FA fraction). Total protein concentration measurement for SDS fraction was carried out with the use of BCA Kit (Pierce, Rockford, IL, USA).
Western blotting analysis
The SDS fractions of brain extracts (30 µg of protein) were electrophoresed on 4–12% gradient Bis‐Tris gels (NuPage, Invitrogen, Carlsbad, CA, USA) and transferred to polyvinylidene difluoride membranes (Tefco, Tokyo, Japan). Membranes were labeled with the use of primary antibody, Saeko (1:1000), recognizing C terminal 30 amino acids of both human and mouse APP (18) overnight at 4°C, incubated with horseradish peroxidase‐linked anti‐rabbit IgG antibody (Amersham Biosciences, Buckingham, UK) (1:2000) for 1 h, and the immunoreactivity was visualized with the use of Supersignal (Pierce, Rockford, IL, USA). Images were captured by Fuji Bas‐1000 imaging analyzer (Fujifilm, Tokyo, Japan), and the intensity of the bands was quantified with the use of Scion Image (Scion Corp., Frederick, MD, USA).
Sandwich enzyme‐linked immunosorbent assay
Amyloid‐β 40 and Aβ42 in the brain extracts (SDS and FA fractions) were measured by sandwich enzyme‐linked immunosorbent assay (ELISA), as described previously 18, 24, 25. Microplates (Immunoplate I, Nunc, Rockilde, Denmark) were pre‐coated with anti‐Aβ monoclonal antibody BNT77 (IgA isotype specific for Aβ11‐16) that recognizes both Aβ40 and Aβ42, then incubated for 24 h at 4°C with 100 µl/well of samples. The microplates were further incubated for 24 h at 4°C with either horseradish‐peroxidase‐conjugated BA27 (anti‐Aβ1‐40, specific for Aβ40) or BC‐05 (anti‐Aβ35‐43, specific for Aβ42 and Aβ43). Color was developed with 3,3′,5,5′‐tetramethylbenzidine and evaluated at 450 nm on a microplate Reader (Molecular Devices, Menlo Park, CA, USA). The SDS fractions were diluted 400 times in EC buffer [20‐mM phosphate buffer, pH 7.0, 400‐mM NaCl, 2‐mM EDTA, 0.4% Block Ace (Dainipponseiyaku, Suita, Japan), 0.2% bovine serum albumin, 0.05% CHAPS and 0.05% sodium azide] containing 0.005% SDS. The FA fraction was neutralized by a 1:50 dilution into 1‐M Tris‐HCl, pH 8.0 and then further diluted 20 times in EC buffer. The program Softmax (Molecular Devices, Menlo Park, CA, USA) was used to calculate Aβ concentration (in picomolar) by comparing the sample absorbance with the absorbance of known concentrations of synthetic Aβ42 or Aβ40 standards (Sigma, St Louis, MO, USA) assayed identically on the same plate. Using the wet weight of brain in the original homogenate, the final values of Aβ in brain were expressed as picomoles per gram wet weight.
Statistical analysis
The difference in the Aβ burden between Tg2576/TTR +/− and Tg2576/TTR −/− mice was examined with ANOVA followed by the Student's unpaired t‐test with GraphPad Prism, Version 4.0 (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered significant.
RESULTS
There is no significant difference in the brain levels of full‐length APP between Tg2576/TTR+/− and Tg2576/TTR−/− mice
Amyloid‐β peptides are derived from APP. To determine whether or not TTR affected the level of full‐length APP, the groups of two Tg2576/TTR +/− and Tg2576/TTR −/− littermates were killed at 16, 18 and 20 months of age, and relative levels of full‐length APP in the SDS fractions prepared from the brain were determined by western blotting with the use of Saeko, as described under Methods. Significant differences were never detected in the levels of full‐length APP among any of the Tg2576/TTR +/− and Tg2576/TTR −/− mice examined (Figure 1). Thus, TTR does not affect the level of full‐length APP in the brain of Tg2576 mice.
Figure 1.

Western blotting analysis of full‐length amyloid precursor protein (APP). The arrow on the left indicates the location of full‐length APP.
Transthyretin deficiency does not increase but decreases the degree of total and vascular Aβ burdens in the brain of Tg2576 mice
Total Aβ burden
To evaluate whether or not TTR affected Aβ deposition, we compared the onset, progression and distribution of amyloid deposition between the brain of Tg2576/TTR +/− and Tg2576/TTR −/− mice, measuring the area occupied by Aβ deposits around the vascular wall of the meninx and cerebral parenchyma (termed cerebral amyloid angiopathy; CAA) and inside the brain parenchyma (termed Aβ plaque), as described under Methods. A time‐course analysis of the total Aβ deposition in the brain was performed by assessing mice of ages 7–20 months. The number and age of mice examined were shown in Table 1. Aβ deposits were not detected in any of the six 7–11‐month‐old Tg2576/TTR +/− and Tg2576/TTR −/− mice examined. A small amount of Aβ deposits was first observed at 12 months of age in both the mice (data not shown). With advancing age, total Aβ burden increased (Figure 2A), and Aβ deposits were observed in the cerebral cortex, neocortex and hippocampus (Figure 3A), but not in the cerebellum (data not shown) in both the mice. Although there was a trend to reduction of total Aβ burden in 12–17‐month‐old Tg2576/TTR −/− mice relative to the age‐matched Tg2576/TTR +/− mice, there was no statistically significant difference in the onset, progression and distribution of total Aβ deposition in the entire cerebral cortex between Tg2576/TTR +/− and Tg2576/TTR −/− mice (Figure 2A). The size of Aβ deposits in Tg2576/TTR −/− mice was also much the same as that in the age‐matched Tg2576/TTR +/− mice. In 18–20‐month‐old Tg2576/TTR −/− mice, however, total Aβ burden was significantly reduced relative to the age‐matched Tg2576/TTR +/− mice (P < 0.05) (Figure 2A). Thus, contrary to our expectations, total Aβ burden is not increased, but rather decreased by eliminating TTR in Tg2576 mice.
Table 1.
The number and age of mice examined by immunohistochemistry. Abbreviation: n = number of mice.
| Age (months) | Tg2576/TTR +/− (n) | Tg2576/TTR −/− (n) |
|---|---|---|
| 7 | 2 | 2 |
| 8 | 2 | 2 |
| 11 | 2 | 2 |
| 12 | 2 | 2 |
| 13 | 5 | 5 |
| 14 | 6 | 6 |
| 15 | 6 | 6 |
| 16 | 6 | 6 |
| 17 | 3 | 3 |
| 18 | 6 | 6 |
| 20 | 2 | 2 |
| Total | 42 | 42 |
Figure 2.

The Aβ burden in the brain of Tg2576/TTR +/− and Tg2576/TTR −/− mice. The total Aβ burden (vascular amyloid and plaques) (A) vascular Aβ burden (B) and Aβ plaque burden (C) in the entire cerebral cortex were calculated by dividing total area of Aβ deposits by total area of analyzed cortex. The hippocampal total Aβ burden (D) was calculated by dividing area of total Aβ deposits (vascular amyloid and plaques) by area of analyzed hippocampus. All data are expressed as mean ± standard error of the mean. Numbers in parentheses denote numbers of mice examined. *P < 0.05. TTR = transthyretin.
Figure 3.
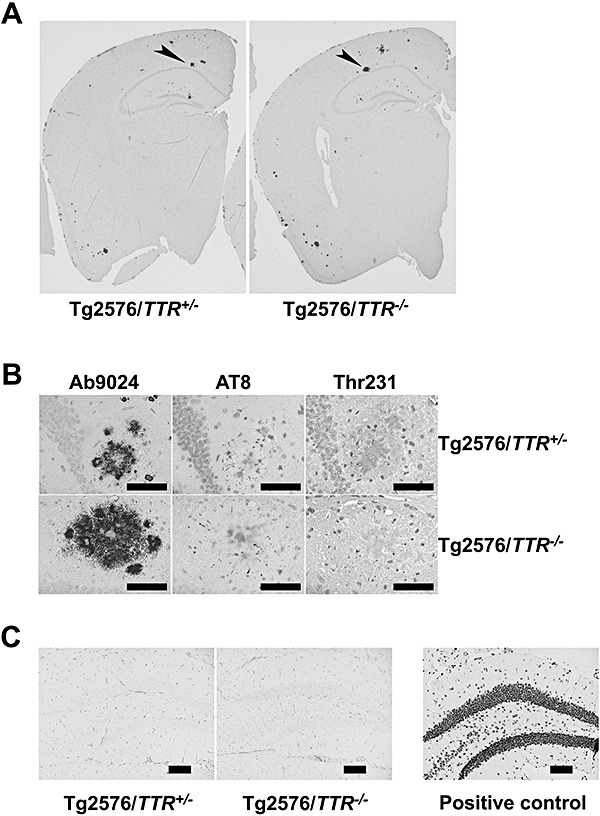
Immunohistochemistry of Tg2576/TTR +/− and Tg2576/TTR −/− brains. Immuno‐labeling of left hemi‐brain sections of 18‐month‐old Tg2576/TTR +/− and Tg2576/TTR −/− mice with Ab9204. A. The higher magnification of the hippocampal Aβ plaque with giant cores indicated by an arrowhead in A (B, left panels). Serial sections (5 µm) were labeled with AT8, and anti‐phosphorylated tau (Thr231). AT8 and Thr‐231 labeled punctate dystrophic neurites in and around Aβ plaques (B, middle and right panels, respectively). Scale bar; 50 µm. The hippocampal dentate gyrus areas of 18‐month‐old Tg2576/TTR +/− and Tg2576/TTR −/− mice stained with transferase‐mediated dUTP nick end labeling. C. No apoptotic cells were found in the hippocampus. A DNaseI‐treated sample was stained in parallel with the samples as a positive control. Scale bar; 100 µm. TTR = transthyretin.
Vascular Aβ burden
It had been reported that Tg2576 mice developed abundant vascular amyloid while aging, especially in leptomeningeal vessels (31). In order to determine whether the onset and degree of particular form of Aβ deposition were affected by TTR, we separately assessed vascular amyloid and plaque burdens in the brain of Tg2576/TTR +/− and Tg2576/TTR −/− mice, as described under Methods.
A time‐course analysis of vascular Aβ burden was performed by assessing the mice of ages 7–20 months. A few vascular Aβ deposits were first observed at 12 months of age in both Tg2576/TTR +/− and Tg2576/TTR −/− mice. With advancing age, total vascular Aβ burden increased in both the mice (Figure 2B). Vascular Aβ deposits were detected only in the wall of leptomeningeal vessels of 12–16‐month‐old Tg2576/TTR +/− and Tg2576/TTR −/− mice, while in the 17–20‐month‐old Tg2576/TTR +/− and Tg2576/TTR −/− mice, the deposits were detected in the vascular wall of cerebral parenchyma as well as the wall of leptomeningeal vessels (data not shown). There was no significant difference in the onset, progression and distribution of vascular Aβ deposition in the entire cerebral cortex between Tg2576/TTR +/− and Tg2576/TTR −/− mice up to 17 months of age. However, a significant reduction in vascular Aβ burden by 47.1% was found in 18–20‐month‐old Tg2576/TTR −/− mice relative to the age‐matched Tg2576/TTR +/− mice (P < 0.05) (Figure 2B). These findings suggested that TTR does not decrease but rather increases the degree of vascular Aβ burden in Tg2576 mice.
Amyloid‐β plaque burden
Aβ plaques were first detected in both Tg2576/TTR +/− and Tg2576/TTR −/− mice at 12 months of age, and both the size and number of the plaques increased with advancing age (Figure 2C). Although there was a trend to reduction of total Aβ plaque burden in 12– 20‐month‐old Tg2576/TTR −/− mice relative to the age‐matched Tg2576/TTR +/− mice, there was no statistically significant difference in the onset, degree and distribution of Aβ plaque deposition between Tg2576/TTR +/− and Tg2576/TTR −/− mice (Figure 2C). These findings suggested that TTR does not decrease Aβ plaque burden in the brain of Tg2576 mice.
Transthyretin deficiency does not affect Aβ deposition in the hippocampus of Tg2576 mice
The hippocampus is highly susceptible area to Aβ deposition in both humans (5) and Tg2576 mice (15). To investigate the effect of TTR deficiency on Aβ deposition in the hippocampus, we measured the total Aβ burden in the hippocampus of Tg2576/TTR +/− and Tg2576/TTR −/− mice. The Aβ deposits were first detected in the hippocampus of both the mice at 13 months of age, and showed an age‐related increase (Figure 2D). Although the total Aβ burden in Tg2576/TTR +/− mice was consistently greater than that in Tg2576/TTR −/− mice, the difference was not statistically significant. Thus, the TTR deficiency does not affect Aβ deposition in the hippocampus of Tg2576 mice.
Transthyretin deficiency does not increase but decreases the level of Aβ40 in the brain of Tg2576 mice
Different forms of Aβ, biochemically distinguishable by their solubility properties, are present in varying amounts during the lifetime of Tg2576 mice. Detergent‐soluble Aβ (SDS fraction) is present throughout life; however, detergent‐insoluble Aβ (FA fraction) is absent up to age 6 months (18). It had been reported in AD that the predominant Aβ peptide present in CAA is Aβ40; however, in brain parenchymal plaques, it is Aβ42 1, 7, 17, 29, 44. To evaluate whether or not TTR affects the level of different forms of Aβ, we quantified the Aβ40 and Aβ42 in SDS and FA fractions of brain homogenates from Tg2576/TTR +/− and Tg2576/TTR −/− mice by sandwich ELISA, as described under Methods. The number and age of 13–20‐month‐old Tg2576/TTR +/− and Tg2576/TTR −/− mice examined were shown in Table 2. Aβ40 and Aβ42 levels in SDS and FA fractions increased with age in both the mice. There was no significant difference in the levels of Aβ40 and Aβ42 in both the fractions between Tg2576/TTR +/− and Tg2576/TTR −/− mice up to 17 months of age. In 18–20‐month‐old Tg2576/TTR −/− mice, however, the levels of Aβ40 in both SDS and FA fractions were significantly reduced by 35.2% and by 41.6%, respectively, relative to the age‐matched Tg2576/TTR +/− mice (P < 0.05) (Figure 4A,B). The level of Aβ42 in SDS fraction was also significantly reduced by 57.8% in 18–20‐month‐old Tg2576/TTR −/− mice relative to the age‐matched Tg2576/TTR +/− mice (P < 0.01) (Figure 4C). On the other hand, there was no significant difference in the levels of Aβ42 in FA fraction between Tg2576/TTR +/− and Tg2576/TTR −/− mice (Figure 4D). The mean level of Aβ42 in FA fraction is much higher than that in SDS fraction. Thus, there was no significant difference in the sum of Aβ42 levels in both the fractions between aged Tg2576/TTR +/− and Tg2576/TTR −/− mice. Thus, TTR deficiency does not increase but rather decreases the level of Aβ40 in the brain of aged Tg2576 mice, a result, which is in good agreement with the immunohistochemistry data, suggesting that TTR increases the vascular Aβ burdens in the brain of aged mice (Figure 2).
Table 2.
The number and age of mice examined by sandwich enzyme‐linked immunosorbent assay. Abbreviation: n = number of mice.
| Age (months) | Tg2576/TTR +/− (n) | Tg2576/TTR −/− (n) |
|---|---|---|
| 13 | 2 | 2 |
| 14 | 3 | 3 |
| 15 | 2 | 2 |
| 16 | 3 | 3 |
| 17 | 2 | 2 |
| 18 | 5 | 5 |
| 20 | 2 | 2 |
| Total | 19 | 19 |
Figure 4.
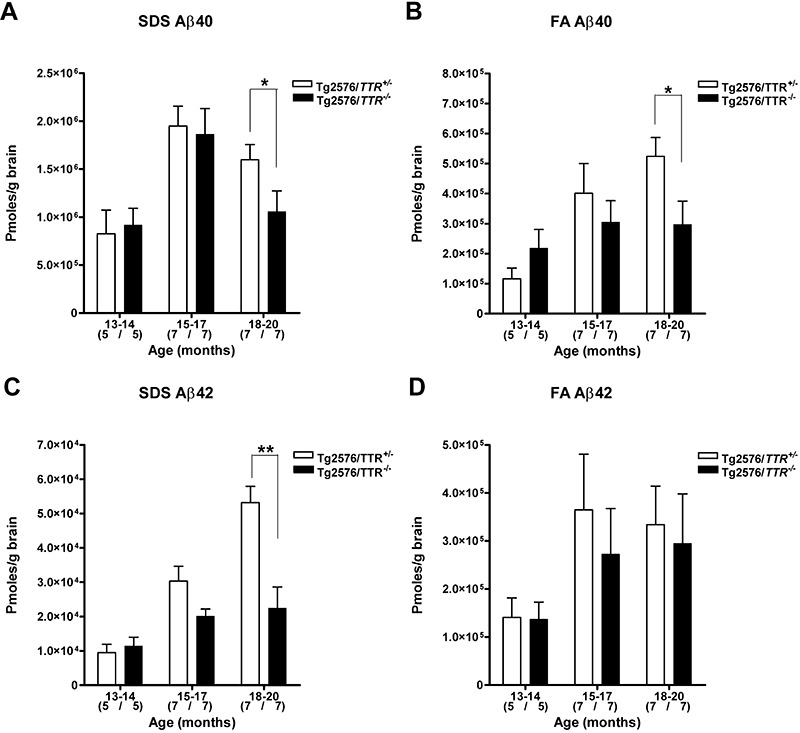
The Aβ level in the brain of Tg2576/TTR +/− and Tg2576/TTR −/− mice. The Aβ40 (A,B) and Aβ42 (C,D) in Tg2576/TTR +/− and Tg2576/TTR −/− brains were quantified by sandwich enzyme‐linked immunosorbent assay. The samples were sequentially extracted in 2% sodium dodecylsulfate (SDS) (A,C) and 70% FA (B,D). All data are expressed as mean ± standard error of the mean. Numbers in parentheses denote numbers of mice examined. *P < 0.05, **P < 0.01. TTR = transthyretin.
Transthyretin deficiency does not affect the distribution and degree of tau phosphorylation in the brain of Tg2576 mice
In contrast to human AD, Tg2576 mice lack NFT, and develop the phosphorylated tau‐immunoreactive aberrant structures that are exclusively associated with congophilic Aβ plaques 27, 48, 49. Stein et al reported that chronic infusion of an antibody against TTR into the hippocampus of Tg2576 led to an increase of tau phosphorylation within the CA1 neuronal field (42). To investigate whether or not TTR deficiency affected the distribution and degree of tau phosphorylation, the brain slices of 16–20‐month‐old Tg2576/TTR +/− and Tg2576/TTR −/− mice were stained with either AT8 or Thr231 antibody, as described under Methods. Both the antibodies reacted only with the punctate dystrophic neurites (DNs) within the Aβ plaques in hippocampus and cerebral cortex in both the mice (Figure 3B). The abundance of the DNs immunopositive with the antibodies in Tg2576/TTR −/− mice was much the same as that in Tg2576/TTR +/− mice (Figure 3B). No NFT was detected in any of the mice examined. Thus, TTR deficiency does not affect tau phosphorylation in the brain of Tg2576 mice.
No apoptotic cells are detected in the hippocampus of Tg2576/TTR+/− and Tg2576/TTR−/− mice
Tg2576 mice do not develop severe neuronal loss observed in AD (15). Stein and Johnson suggested that high level of TTR in the hippocampus of Tg2576 mice might protect the mice from severe neuronal loss (43). Furthermore, the same group reported that chronic infusion of an antibody against TTR into the hippocampus of Tg2576 mice led to an increase of neuronal loss and apoptosis within the CA1 neuronal field (42). To determine whether or not TTR deficiency induces apoptosis in the hippocampus of Tg2576 mice, the brain sections from 18–20‐month‐old Tg2576/TTR +/− and Tg2576/TTR −/− mice were subjected to TUNEL immunohistochemistry, as described under Methods. Apoptotic cells were never detected in the hippocampus or other parts of brain of any of the mice examined (Figure 3C). These results indicate that TTR deficiency does not induce apoptosis in the brain of Tg2576 mice.
DISCUSSION
To investigate the role of TTR in the Aβ deposition in vivo, we generated a mouse line carrying a null mutation at the endogenous TTR locus and the human mutant APP cDNA with the Swedish mutation (Tg2576/TTR −/− mouse) by crossing Tg2576 mice with TTR‐deficient mice generated through gene targeting. We then asked whether Aβ deposition was accelerated in Tg2576/TTR −/− mice relative to the heterozygous mutant Tg2576 (Tg2576/TTR +/−) mice. Contrary to our expectations, the degree of total Aβ deposition, tau phosphorylation and apoptosis in the brain was not increased by eliminating TTR in Tg2576 mice. Moreover, the degree of vascular Aβ burden in the aged Tg2576/TTR −/− mice was significantly reduced relative to the age‐matched Tg2576/TTR +/− mice. Our experiments present, for the first time, compelling evidence that TTR does not suppress but rather accelerates vascular Aβ deposition in the mouse model of AD.
We confirmed that there was no significant difference in the onset, progression and distribution of total Aβ deposition between Tg2576/TTR +/− and Tg2576/TTR −/− mice up to age 17 months by immunohistochemistry (Figure 2A). However, total Aβ burden in 18–20‐month‐old Tg2576/TTR −/− mice was significantly reduced relative to the age‐matched Tg2576/TTR +/− mice (P < 0.05) (Figure 2A). The result suggested that TTR does not suppress but rather accelerates Aβ deposition in the brain of Tg2576 mice. Although both Tg2576/TTR +/− and Tg2576/TTR −/− mice are smaller than non‐transgenic littermates, both of them display no obvious phenotypic abnormalities, and their fertility is normal up to age 10 months. This observation is consistent with the immunohistochemistry data.
We then separately assessed vascular amyloid and plaque burdens in the brain of Tg2576/TTR +/− and Tg2576/TTR −/− mice. Although Aβ plaque burden was much the same between 7–20‐month‐old Tg2576/TTR +/− and Tg2576/TTR −/− mice (Figure 2C), vascular amyloid burden in the aged (18–20‐month‐old) Tg2576/TTR −/− mice was significantly reduced relative to the age‐matched Tg2576/TTR +/− mice (P < 0.05) (Figure 2B). The quantification of Aβ40 and Aβ42 in the brain homogenates from Tg2576/TTR +/− and Tg2576/TTR −/− mice by sandwich ELISA demonstrated that TTR deficiency does not increase, but rather decreases the level of Aβ40 in the aged Tg2576 mice (Figure 4). Because the predominant Aβ peptide present in vascular amyloid deposits is reportedly Aβ40 1, 7, 44, the result is also in good agreement with our immunohistochemistry data (Figure 2), suggesting that TTR increases the vascular Aβ burden in the brain of aged Tg2576 mice.
The reason why vascular amyloid burden is increased by TTR is not clear. Amyloid deposits of all types, including Aβ deposits, contain glycosaminoglycans (GAGs) and serum amyloid P component (SAP). A role for GAGs in amyloidosis is inferred from the observation that small molecules that interfere with GAG/amyloid interactions reduce murine experimental amyloid A (AA) amyloid progression (19). An amyloid‐binding protein SAP protects amyloid fibrils from proteolysis in vitro (46), and the induction of AA amyloidosis is significantly retarded in the SAP‐deficient mice relative to wild‐type mice 4, 47. On the other hand, recent evidence indicates that Aβ is mainly cleared out of the brain to blood via transport through the blood‐brain barrier, and via the interstitial fluid (ISF) bulk flow along periarterial drainage pathways into the CSF, and from there into the blood 26, 33, 52, 56. It is the CSF and perhaps the ISF and not the brain parenchyma (41) that is enriched in TTR. Thus we think it likely that when Aβ drains from the brain parenchyma along periarterial drainage pathways, it may come into contact with TTR which may protect Aβ deposits from proteolysis like GAG and SAP, thereby, slightly increases vascular amyloid burden over the ages.
Schwarzman et al reported that TTR in the CSF binds Aβ, and prevents Aβ fibril formation in vitro. They, however, also reported that apoE prevents Aβ fibril formation too 36, 37. It has been well established that apoE promotes assembly of Aβ fibril 23, 32. Thus, TTR may promote the fibrillization of Aβ too. Moreover, Holtzman et al found that a transgenic mouse model of AD on an apoE−/− background had significantly reduced Aβ deposition relative to the same mouse model expressing wild‐type murine apoE (apoE+/+), human apoE3 (apoE3+/−) or human apoE4 (apoE4+/−) (12). Therefore, TTR null Tg2576 (Tg2576/TTR −/−) mice may represent mice that are unable to form Aβ fibrils, and the Aβ detected in the brain of the mice could be due in part to apoE.
Stein et al reported that chronic infusion of anti‐TTR antibody into the hippocampus of Tg2576 mice increased Aβ burden, and led to tau hyperphosphorylation, neuronal loss and apoptosis in the CA1 neuronal field (42). These observations suggest the importance of TTR in inhibition of Aβ fibril formation and toxicity. However, contrary to these reports, our experiments suggested that TTR does not suppress but rather enhances Aβ deposition in Tg2576 mice. The reason for the discrepancy between data of other authors and our data is not clear. TTR is complexed with retinol‐binding protein (RBP) and thyroid hormone in vivo. In the in vitro Aβ aggregation assay, however, recombinant TTR alone, not complexed with RBP or thyroid hormone, was used to examine its ability to inhibit Aβ fibril formation 36, 37. Association of TTR with RBP and thyroid hormone may affect its binding capacity with Aβin vivo. Tau phosphorylation and apoptosis were induced by Aβ in hippocampal cultures (42). Thus, as suggested by Stein et al, high intrahippocampal concentration of Aβ, induced temporarily by the disruption of TTR binding of Aβ by the antibody, might have caused localized neurodegeneration in the CA1 field. The neurodegeneration reportedly detected in the antibody‐infused limited area of hippocampus of Tg2576 mice (42), however, was not detected in the entire brain of TTR‐deficient Tg2576 mice.
Stein and Johnson reported that the lack of neurodegeneration was associated with increased level of TTR synthesized in the hippocampus of Tg2576 mice (43). However, Lazarov et al reported that the individual levels of TTR mRNA in the hippocampi of a transgenic mouse model of AD, which co‐expresses familial AD‐linked mutant APP, and presenilin 1 (PS1) cDNAs were considerably variable (20). Furthermore, it had been reported that choroid plexus is the sole site of TTR synthesis within the brain; in this regard, Sousa et al recently confirmed that TTR is not produced in the brain parenchyma of either wild‐type or Tg2576 mice, using laser dissection microscopy (41). The finding suggests that contamination by choroid plexus might lead to misinterpretation of the role of TTR in Aβ deposition in the brain.
In the present study, we compared the onset and progression of Aβ pathology in TTR null Tg2576 (Tg2576/TTR −/−) mice with those in heterozygous mutant Tg2576 mice (Tg2576TTR +/−). Thus, one factor which causes the discrepancy between our data and other authors' data obtained by examining only Tg2576 mice homozygous for the wild‐type TTR gene (Tg2576/TTR +/+) for comparison might be the difference in the levels of TTR. However, the onset and progression of Aβ pathology in our Tg2576/TTR +/− mice are rather delayed than accelerated relative to those in Tg2576/TTR +/+ mice previously reported by other authors 6, 18, 45, 55. Thus, the possibility that homozygous levels of TTR would be required to prevent Aβ pathology appears to be remote.
On the other hand, Nunes et al reported that peptidylglycine α‐amidating monooxygenase, the rate‐limiting enzyme in neuropeptide maturation, is over‐expressed in the peripheral, and central nervous systems of TTR −/− mice that, consequently, display increased neuropeptide Y (NPY) levels relative to wild‐type mice (28). NPY is known to be a substrate of neprilysin, which is an Aβ‐degrading protease (50). Another Aβ‐degrading enzyme, insulin‐degrading enzyme (IDE) is also known as insulin and amylin protease 30, 50. Hyperinsulinaemia is known to increase the risk of developing AD. Thus, it is suggested that hyperinsulinaemia may elevate Aβ level through insulin's competition with Aβ for IDE (30). Analogous to the competition, the increase in NPY levels in TTR −/− mice might competitively reduce neprilysin clearance of Aβ. Thus, Tg2576/TTR −/− mice might display enhanced Aβ deposition relative to Tg2576/TTR +/− mice through NPY's competition with Aβ for neprilysin. Contrary to the expectation, Tg2576/TTR −/− mice display rather suppressed Aβ deposition relative to Tg2576/TTR +/− mice (2, 4). The results say that TTR does not suppress but rather accelerates Aβ deposition in Tg2576 mice.
Contrary to our findings, Choi et al recently reported that in a different transgenic mouse model of AD heterozygous for the disrupted TTR gene (TTR +/−), brain Aβ deposition is significantly accelerated relative to the age‐matched model homozygous for the wild‐type TTR gene (TTR +/+) (8). Their observation, which suggests that TTR suppresses Aβ deposition, contradicts ours. It is important to note that there are several critical differences in the experimental designs which might have caused the contradiction between their data and ours: (i) the AD model mouse we examined (Tg2576) is distinct from that Choi et al examined. They used ceAPPswe/PS1ΔE9 mice that harbor not only the human mutant APP cDNA with the double mutation K670N and M671L linked to a Swedish familial AD but also the human mutant PS1 cDNA with the exon 9 deletion linked to a familial AD 16, 21. In contrast to Tg2576 mice, their control singly transgenic mice that express the human mutant APP cDNA alone are free of brain Aβ deposits up to age 14 months and co‐expression of human mutant APP, and PS1 accelerates the amyloid deposition 3, 21. Furthermore, comparative analysis of cortical gene expression patterns between Tg2576 mice homozygous for the PS1 knock‐in mutation (Tg2576/PS1264L/264L) and control Tg2576 mice heterozygous for the PS1 mutation (Tg2576/PS1P264L/+) by DNA micro‐array analysis revealed that the patterns are distinct, although there were some common regulated genes (54). All these observations suggest that the molecular pathogenesis of Aβ deposition in the two mouse models is different; (ii) the level of human variant APP in the brain of Tg2576 mice is more than fourfold higher than that of endogenous brain APP (14). On the other hand, although the level of human variant APP in the brain of ceAPPswe/PS1ΔE9 mice is not described (8), variant PS1ΔE9 reportedly elevates Aβ42/Aβ40 ratio (3). Thus the contradiction between their results and ours might be caused by the significant difference in the levels of Aβ42 and/or Aβ40 between Tg2576 and ceAPPswe/PS1ΔE9 mice; and (iii) Choi et al compared the degree of Aβ deposition between the brains of ceAPPswe/PS1ΔE9 mice heterozygous for the disrupted TTR gene (TTR +/−) and the mice homozygous for the wild‐type TTR gene (TTR +/+) (8), in contrast to the TTR null (TTR −/−) and TTR +/− Tg2576 mice we examined for comparison. Thus in their study, in contrast to our study, the individual differences in the levels of brain TTR among the TTR +/− and control TTR +/+ mice should critically affect the results, and hence, the elucidation of the relationship between TTR and Aβ deposition. They described that the levels of immunoreactive TTR in the extracts from the brains of ceAPPswe/PS1ΔE9/TTR +/− mice are clearly lower relative to the age‐matched ceAPPswe/PS1ΔE9/ TTR+/+ mice at all ages examined (8). However, the report is lacking in important details about the levels of TTR in the individual animals that would make the data more compelling. For example, given only the pictorial data with sample number of 1, it is not clear that the differences in the brain levels of TTR between ceAPPswe/PS1ΔE9/TTR +/+ and ceAPPswe/PS1ΔE9/TTR +/− mice are really significant. On the other hand, it is possible that TTR, as a peripheral Aβ binding protein, may have the ability to act as a peripheral Aβ‘sink’; whereby, it pulls Aβ from the brain into the periphery, hence decreasing the amount of Aβ in the brain 26, 33, 52, 56. Thus, if we had examined Tg2576TTR +/+ mice, we, too, might have detected a decrease in Aβ deposition as Choi et al did.
All the above differences may cause the contradiction between their data and ours. Moreover, they described that the levels of brain TTR were significantly lower in human AD patients compared with age‐matched controls and negatively correlated with the abundance of amyloid plaques. However, the references they cited didn't refer to the brain TTR levels but to the CSF TTR levels in the patients (8) and, to our knowledge, the comparison of the brain TTR levels between AD patients and control disease‐free individuals has not yet been reported.
In conclusion, our results indicated, for the first time, TTR does not suppress but accelerates vascular Aβ burden in the brain of Tg2576 mice. However, the mechanism(s) by which TTR affects the Aβ deposition in vivo are not yet elucidated. Taken together with the Choi et al's contradictory finding (8), our finding suggests that the role of TTR in the pathogenesis of AD remains to be understood.
ACKNOWLEDGMENTS
The authors would like to thank Drs. K. H. Ashe and Takaomi C. Saido for provision of Tg2576 mice and Ab9204 antibody, respectively. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology, Japan: Grants‐in‐aids for Scientific Research (18390098; to SM); and by grants to the Amyloidosis Research Committee for the Research on Intractable Diseases from the Ministry of Health, Labour and Welfare, Japan (to MS & SM).
REFERENCES
- 1. Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM (1998) Progression of cerebral amyloid angiopathy: accumulation of amyloid‐beta40 in affected vessels. J Neuropathol Exp Neurol 57:353–359. [DOI] [PubMed] [Google Scholar]
- 2. Biroccio A, Del Boccio P, Panella M, Bernardini S, Di Ilio C, Gambi D et al (2006) Differential post‐translational modifications of transthyretin in Alzheimer's disease: a study of the cerebral spinal fluid. Proteomics 6:2305–2313. [DOI] [PubMed] [Google Scholar]
- 3. Borchelt DR, Ratovitski T, Van Lare J, Lee MK, Gonzales V, Jenkins NA et al (1997) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19:939–945. [DOI] [PubMed] [Google Scholar]
- 4. Botto M, Hawkins PN, Bickerstaff MC, Herbert J, Bygrave AE, McBride A et al (1997) Amyloid deposition is delayed in mice with targeted deletion of the serum amyloid P component gene. Nat Med 3:855–859. [DOI] [PubMed] [Google Scholar]
- 5. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol (Berl) 82:239–259. [DOI] [PubMed] [Google Scholar]
- 6. Carro E, Trejo JL, Gomez‐Isla T, LeRoith D, Torres‐Aleman I (2002) Serum insulin‐like growth factor I regulates brain amyloid‐beta levels. Nat Med 8:1390–1397. [DOI] [PubMed] [Google Scholar]
- 7. Castano EM, Prelli F, Soto C, Beavis R, Matsubara E, Shoji M, Frangione B (1996) The length of amyloid‐beta in hereditary cerebral hemorrhage with amyloidosis, Dutch type. Implications for the role of amyloid‐beta 1‐42 in Alzheimer's disease. J Biol Chem 271:32185–32191. [DOI] [PubMed] [Google Scholar]
- 8. Choi SH, Leight SN, Lee VM, Li T, Wong PC, Johnson JA, Saraiva MJ, Sisodia SS (2007) Accelerated Abeta deposition in APPswe/PS1deltaE9 mice with hemizygous deletions of TTR (transthyretin). J Neurosci 27:7006–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Episkopou V, Maeda S, Nishiguchi S, Shimada K, Gaitanaris GA, Gottesman ME, Robertson EJ (1993) Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc Natl Acad Sci USA 90:2375–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Giunta S, Valli MB, Galeazzi R, Fattoretti P, Corder EH, Galeazzi L (2005) Transthyretin inhibition of amyloid beta aggregation and toxicity. Clin Biochem 38:1112–1119. [DOI] [PubMed] [Google Scholar]
- 11. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 12. Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ et al (2000) Apolipoprotein E isoform‐dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 97:2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S et al (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102. [DOI] [PubMed] [Google Scholar]
- 14. Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W et al (1995) Age‐related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 15:1203–1218. [DOI] [PubMed] [Google Scholar]
- 15. Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT (1997) APPSw transgenic mice develop age‐related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 56:965–973. [DOI] [PubMed] [Google Scholar]
- 16. Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR (2001) Co‐expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng 17:157–165. [DOI] [PubMed] [Google Scholar]
- 17. Joachim CL, Duffy LK, Morris JH, Selkoe DJ (1988) Protein chemical and immunocytochemical studies of meningovascular beta‐amyloid protein in Alzheimer's disease and normal aging. Brain Res 474:100–111. [DOI] [PubMed] [Google Scholar]
- 18. Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age‐dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 21:372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kisilevsky R, Lemieux LJ, Fraser PE, Kong X, Hultin PG, Szarek WA (1995) Arresting amyloidosis in vivo using small‐molecule anionic sulphonates or sulphates: implications for Alzheimer's disease. Nat Med 1:143–148. [DOI] [PubMed] [Google Scholar]
- 20. Lazarov O, Lee M, Peterson DA, Sisodia SS (2002) Evidence that synaptically released beta‐amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci 22:9785–9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lazarov O, Robinson J, Tang YP, Hairston IS, Korade‐Mirnics Z, Lee VM et al (2005) Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 120:701–713. [DOI] [PubMed] [Google Scholar]
- 22. Link CD (1995) Expression of human beta‐amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci USA 92:9368–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma J, Yee A, Brewer HB, Das S, Potter H (1994) Amyloid‐associated proteins α1‐antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β‐protein into filaments. Nature 372:92–94. [DOI] [PubMed] [Google Scholar]
- 24. Matsubara E, Bryant‐Thomas T, Pacheco Quinto J, Henry TL, Poeggeler B, Herbert D et al (2003) Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer's disease. J Neurochem 85:1101–1108. [DOI] [PubMed] [Google Scholar]
- 25. Matsubara E, Ghiso J, Frangione B, Amari M, Tomidokoro Y, Ikeda Y et al (1999) Lipoprotein‐free amyloidogenic peptides in plasma are elevated in patients with sporadic Alzheimer's disease and Down's syndrome. Ann Neurol 45:537–541. [PubMed] [Google Scholar]
- 26. Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V et al (2003) Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to beta‐amyloid. J Neurosci 23:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Noda‐Saita K, Terai K, Iwai A, Tsukamoto M, Shitaka Y, Kawabata S et al (2004) Exclusive association and simultaneous appearance of congophilic plaques and AT8‐positive dystrophic neurites in Tg2576 mice suggest a mechanism of senile plaque formation and progression of neuritic dystrophy in Alzheimer's disease. Acta Neuropathol (Berl) 108:435–442. [DOI] [PubMed] [Google Scholar]
- 28. Nunes AF, Saraiva MJ, Sousa MM (2006) Transthyretin knockouts are a new mouse model for increased neuropeptide Y. Faseb J 20:166–168. [DOI] [PubMed] [Google Scholar]
- 29. Prelli F, Castano E, Glenner GG, Frangione B (1988) Differences between vascular and plaque core amyloid in Alzheimer's disease. J Neurochem 51:648–651. [DOI] [PubMed] [Google Scholar]
- 30. Qiu WQ, Folstein MF (2006) Insulin, insulin‐degrading enzyme and amyloid‐beta peptide in Alzheimer's disease: review and hypothesis. Neurobiol Aging 27:190–198. [DOI] [PubMed] [Google Scholar]
- 31. Rensink AA, De Waal RM, Kremer B, Verbeek MM (2003) Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev 43:207–223. [DOI] [PubMed] [Google Scholar]
- 32. Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, Wisniewski T (2006) Blocking the apolipoprotein E/amyloid‐beta interaction as a potential therapeutic approach for Alzheimer's disease. Proc Natl Acad Sci USA 103:18787–18792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R et al (2007) Clearance of amyloid‐β by circulating lipoprotein receptors. Nat Med 13:1029–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S (1995) Dominant and differential deposition of distinct beta‐amyloid peptide species, a beta N3(pE), in senile plaques. Neuron 14:457–466. [DOI] [PubMed] [Google Scholar]
- 35. Sasaki A, Shoji M, Harigaya Y, Kawarabayashi T, Ikeda M, Naito M et al (2002) Amyloid cored plaques in Tg2576 transgenic mice are characterized by giant plaques, slightly activated microglia, and the lack of paired helical filament‐typed, dystrophic neurites. Virchows Arch 441:358–367. [DOI] [PubMed] [Google Scholar]
- 36. Schwarzman AL, Goldgaber D (1996) Interaction of transthyretin with amyloid beta‐protein: binding and inhibition of amyloid formation. Ciba Found Symp 199:146–160.discussion 60–64. [DOI] [PubMed] [Google Scholar]
- 37. Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ et al (1994) Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci USA 91:8368–8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Serot JM, Christmann D, Dubost T, Couturier M (1997) Cerebrospinal fluid transthyretin: aging and late onset Alzheimer's disease. J Neurol Neurosurg Psychiatry 63:506–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seubert P, Vigo‐Pelfrey C, Esch F, Lee M, Dovey H, Davis D et al (1992) Isolation and quantification of soluble Alzheimer's beta‐peptide from biological fluids. Nature 359:325–327. [DOI] [PubMed] [Google Scholar]
- 40. Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM et al (1992) Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258:126–129. [DOI] [PubMed] [Google Scholar]
- 41. Sousa JC, Cardoso I, Marques F, Saraiva MJ, Palha JA (2007) Transthyretin and Alzheimer's disease: where in the brain? Neurobiol Aging 28:713–718. [DOI] [PubMed] [Google Scholar]
- 42. Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA (2004) Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J Neurosci 24:7707–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stein TD, Johnson JA (2002) Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci 22:7380–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suzuki N, Iwatsubo T, Odaka A, Ishibashi Y, Kitada C, Ihara Y (1994) High tissue content of soluble beta 1‐40 is linked to cerebral amyloid angiopathy. Am J Pathol 145:452–460. [PMC free article] [PubMed] [Google Scholar]
- 45. Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M et al (2000) Age‐related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol 157:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tennent GA, Lovat LB, Pepys MB (1995) Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc Natl Acad Sci USA 92:4299–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Togashi S, Lim SK, Kawano H, Ito S, Ishihara T, Okada Y et al (1997) Serum amyloid P component enhances induction of murine amyloidosis. Lab Invest 77:525–531. [PubMed] [Google Scholar]
- 48. Tomidokoro Y, Harigaya Y, Matsubara E, Ikeda M, Kawarabayashi T, Shirao T et al (2001) Brain Abeta amyloidosis in APPsw mice induces accumulation of presenilin‐1 and tau. J Pathol 194:500–506. [DOI] [PubMed] [Google Scholar]
- 49. Tomidokoro Y, Ishiguro K, Harigaya Y, Matsubara E, Ikeda M, Park JM et al (2001) Abeta amyloidosis induces the initial stage of tau accumulation in APP (SW) mice. Neurosci Lett 299:169–172. [DOI] [PubMed] [Google Scholar]
- 50. Wang DS, Dickson DW, Malter JS (2006) beta‐Amyloid degradation and Alzheimer's disease. J Biomed Biotechnol 2006:58406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wei L, Kawano H, Fu X, Cui D, Ito S, Yamamura K et al (2004) Deposition of transthyretin amyloid is not accelerated by the same amyloid in vivo. Amyloid 11:113–120. [DOI] [PubMed] [Google Scholar]
- 52. Weller RO, Cohen NR, Nicoll JA (2004) Cerebrovascular disease and the pathophysiology of Alzheimer's disease. Implications for therapy. Panminerva Med 46:239–251. [PubMed] [Google Scholar]
- 53. Westerman MA, Cooper‐Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH et al (2002) The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci 22:1858–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu ZL, Ciallella JR, Flood DG, O'Kane TM, Bozyczko‐Coyne D, Savage MJ (2006) Comparative analysis of cortical gene expression in mouse models of Alzheimer's disease. Neurobiol Aging 27:377–386. [DOI] [PubMed] [Google Scholar]
- 55. Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS et al (2005) Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280:5892–5901. [DOI] [PubMed] [Google Scholar]
- 56. Zlokovic BV (2004) Clearing amyloid through the blood‐brain barrier. J Neurochem 89:807–811. [DOI] [PubMed] [Google Scholar]