

Abstract
The prognosis of patients with malignant glioma is poor in spite of multimodal treatment approaches consisting of neurosurgery, radiochemotherapy and maintenance chemotherapy. Among innovative treatment strategies like targeted therapy, antiangiogenesis and gene therapy approaches, immunotherapy emerges as a meaningful and feasible treatment approach for inducing long‐term survival in at least a subpopulation of these patients. Setting up immunotherapy for an inherent immunosuppressive tumor located in an immune‐privileged environment requires integration of a lot of scientific input and knowledge of both tumor immunology and neuro‐oncology. The field of immunotherapy is moving into the direction of active specific immunotherapy using autologous dendritic cells (DCs) as vehicle for immunization. In the translational research program of the authors, the whole cascade from bench to bed to bench of active specific immunotherapy for malignant glioma is covered, including proof of principle experiments to demonstrate immunogenicity of patient‐derived mature DCs loaded with autologous tumor lysate, preclinical in vivo experiments in a murine orthotopic glioma model, early phase I/II clinical trials for relapsing patients, a phase II trial for patients with newly diagnosed glioblastoma (GBM) for whom immunotherapy is integrated in the current multimodal treatment, and laboratory analyses of patient samples. The strategies and results of this program are discussed in the light of the internationally available scientific literature in this fast‐moving field of basic science and translational clinical research.
Keywords: clinical trials, dendritic cells, glioblastoma, high‐grade glioma, immunotherapy, translational medicine
INTRODUCTION
High‐grade gliomas (HGGs) consist of World Health Organization (WHO) grade III and WHO grade IV neoplasms: anaplastic/malignant gliomas respectively glioblastomas (GBMs). While GBMs are the most frequent and most malignant of these tumors, they still belong to the category of orphan diseases, with a yearly incidence of about three to four per 100 000 adults (42) and two per million children (120). The treatment for these patients consists primarily of surgery in order to debulk the tumoral mass for symptomatic relief and to obtain tissue for histological diagnosis, as well as radiochemotherapy to induce optimal local tumor control. In spite of improved surgical and radiotherapeutic techniques, and the addition of temozolomide to the multimodal treatment strategy, the prognosis of patients with GBM remains poor: the median overall survival (OS) is about 15 months, with 88% of patients dead within 3 years 117, 118. Indeed, even after maximal treatment with surgery, radiotherapy and chemotherapy, relapse is universal and is believed to be due to the extensive spread of tumor cells into surrounding regions of the brain 21, 68. At the time of relapse, the prognosis is particularly poor, with reports of 100% mortality within 1.5 years (13). As such, there is obviously a need for more effective new therapies. One of the particular challenges with classical chemotherapeutic strategies is overcoming the blood‐brain barrier. Therefore, much of today's preclinical research is focused on alternate approaches, such as more selective therapies, which specifically target intracellular signaling pathways or surface molecules 44, 84, antiangiogenesis strategies (46) and especially immunotherapy. Acknowledging that different aspects of immunotherapy for malignant gliomas have been extensively reviewed in the last couple of years 2, 3, 17, 27, 28, 38, 41, 66, 79, 94, 96, 97, 102, 116, 133, 140, 141, 144, 149, the aim of the present paper is to present our own translational research program efforts as a disease‐specific paradigm for active specific immunotherapy, rather than contributing one more encyclopedic review of dendritic cell (DC) therapy overall.
PRINCIPLES OF DC THERAPY IN GLIOMA
Active specific immunotherapy
Immunotherapy of HGGs covers a broad field comprising several approaches. These include passive immunotherapy with monoclonal antibodies (mAb) (132), passive immune stimulation with cytokines (restorative) 11, 39, 86, 145, 148, treatment with stimulated T cells (adoptive) 100, 106, 113 and active specific immunotherapy. In the latter approach, DCs are most commonly used to stimulate the immune system, given their role as the most powerful antigen‐presenting cells (APCs) known to date. DCs are a subset of white blood cells, critical to most aspects of adaptive immunity due to their central role as specialized APCs in the initiation phase of T cell responses 54, 83. Typically, DCs reside as immature cells in almost every organ and tissue at the interface of potential pathogen entry sites. In this state, they continuously sample antigens. This sampling, however, results in effective antigen presentation only when DCs are also triggered by danger signals derived from pathogens, tissue damage or signs of inflammation. As danger‐triggered DCs start to mature and get activated, they up‐regulate chemokine receptors, which guide them to draining lymph nodes. There, the mature DCs are capable of inducing primary T cell responses due to their high levels of major histocompatibility complex (MHC), adhesion and costimulatory molecule expression. Only mature DCs show the typical dendritic morphology with multiple cytoplasmic extensions (7). Unlike other APCs like macrophages, monocytes and B cells, DCs are able to present and cross‐present the antigenic peptides in the context of both MHC Class II and Class I molecules, respectively 109, 110. In this way, they can prime not only CD4+ T helper cells, but also CD8+ cytotoxic T cells (23). Both effector cell types are believed to be necessary to induce an effective cell‐mediated immune response 34, 72, 121. Direct recognition of tumor cells by unprimed T cells, in contrast, induces T cell anergy rather than immunity as the costimulatory molecules are missing on the tumor cells 126, 127.
DCs are not only sentinels in the adaptive immune response, but have also been shown to be strong activators of natural killer (NK) cells and NK T cells (31), thus linking the innate and adaptive immune responses. In this way, both tumor cells with and without expression of MHC Class I molecules can theoretically be killed (8). All these particular characteristics make DCs the perfect adjuvants in active specific immunotherapeutic strategies, in which one aims to induce a specific immune response in vivo 6, 45, 59, 119, 136. Gliomas have been shown to express several tumor‐associated antigens (TAAs), including epidermal growth factor receptor isoform III (85), glycoprotein 240 (69), tenascin (128), survivin (139), squamous cell carcinoma‐associated reactive antigen for cytotoxic T cells 1 (57), the alpha‐2 chain of interleukin 13 receptor (IL‐13Rα2 chain) (93), melanoma‐associated antigens like tyrosinase, tyrosinase‐related protein 1 and 2, gp100, melanoma antigen (MAGE)‐1 and MAGE‐3 18, 75, 76, 115 and Immunoselected Melanoma‐2 (AIM‐2) (77), all extensively reviewed by Dunn et al (35). Moreover, GBM patients exhibit circulating tumor‐specific CD8+ T cells (122). Until now, however, a universally expressed glioma TAA with a critical downstream cell survival‐related function has not been identified. Therefore, just targeting the known TAAs using individual peptides would inherently lead to immune escape because of the positive clonal selection of antigen‐loss variants (48). In other words, those tumor cell clones that do not express the particular targeted TAA (anymore) will escape immune rejection and thus have an important proliferation advantage as compared with the cell clones that still express the targeted TAA. That heterogeneity in TAA expression in gliomas represents the main reason why most investigators favor the use of whole‐tumor cell antigens as a source of TAAs to load the DCs. For this, acid‐eluted membrane peptides 73, 74, 147, apoptotic bodies after gamma‐irradiation (130), tumor lysates 25, 30, 111, 134, 135, 142, 143, 146 and fusion of DC with tumor cells 64, 65 have all been used to load DCs. Of note, Khan et al (63) isolated proteins out of paraffin‐embedded tissue to load the DCs. To our knowledge, no tumoral RNA has been used to load DCs for administration to patients thus far. In a comparative analysis, the loading with apoptotic bodies or total tumoral RNA seemed to be superior to loading with lysate or fusion of HGG cells with DCs (95). However, loading with apoptotic bodies of HGGs has also been shown to increase the risk of inducing tolerogenic DCs via the cyclooxygenase‐2 (COX2) pathway (4). Loading DCs with specific antigens that play key roles in tumor biology has also been attempted 19, 20, 138. Because glioma cancer stem cells might represent the therapy‐resistant fraction of HGGs, cancer stem cell‐redirected immunotherapy has been explored 98, 99. In our translational research program, we developed patient‐derived autologous mature DCs loaded with lysate of the autologous HGG (DCm‐HGG‐L) for in vitro investigation and for clinical applications. In our murine model, we used DCm loaded with lysate of the GL261 murine glioma tumor cells or with total RNA of GL261 cells (DCm‐GL261‐RNA). The GL261 glioma cell line is derived from Methylcholantrene‐induced glioma tumor cells in C57BL/6J mice.
Experimental rodent models for malignant glioma
Hurdles associated with current animal models
Over the last 4 decades, hundreds of reports of heterotopic and orthotopic rat and mouse GBM models have been published. It is extremely important to be aware of some fundamental differences between those models, for example, between models exploiting spontaneous tumor formation in genetically engineered mice versus engrafted tumor models that are established by implantation of primary tumor cells or tumor cell lines. Although spontaneous tumor models are mimicking the clinical situation of gliomagenesis much better than the engrafted models, the main drawbacks are the poor reproducibility, low tumor penetrance, prolonged latency for tumor formation and the need for advanced in vivo imaging techniques. On the other hand, because engrafted models lack the stepwise genetic changes occurring during tumor progression, many of them are well circumscribed, lack the human counterpart of microvascular proliferation and rarely recapitulate the tumor‐of‐origin phenotype. Nevertheless, based on greater reproducibility, engrafted models are better suited for evaluating preclinical therapies such as DC immunotherapy, as long as the models are studied in immunocompetent animals 16, 22, 43.
Data from the literature
Below, we briefly summarize the most relevant data on the murine GL261 glioma model, as this model is also used in our translational research program. Ni et al underscored the immunogenicity of the GL261 tumor cells by treating mice with intracranial glioma with tumor extract‐pulsed cloned DCs. Cured animals showed increased delayed‐type hypersensitivity (DTH) responses to GL261 cells, with long‐term tumor protection (89). Aoki et al showed that pulsing DCs with a complex of tumor extract with cationic liposomes induces an antitumor immune response against intracerebral glioma in which CD8+ T cells are involved (5). Protective immunity against intracranial glioma growth obtained through immunization with either lysate‐ or RNA‐loaded DCs was reported by Insug et al, while adding recombinant IL‐12 to the vaccine regimen further improved its efficacy (58). A survival benefit of combining vaccination with lysate‐pulsed DCs and interferon (IFN)‐β gene therapy was demonstrated by Saito et al (112). Similarly, the group of Okada et al revealed that the sequential intratumoral delivery of an IFN‐α encoding adenoviral vector and DCs induced long‐term survival and specific cytotoxic T lymphocyte (CTL) activity (123). Furthermore, the same group showed that intratumoral administration of DCs, genetically engineered to secrete IFN‐α, enhances the efficacy of peripheral vaccines with cytokine gene‐transduced tumor cells (70). Kjaergaard et al observed complete tumor regression of established intracranial tumors with infiltration of both CD4+ and CD8+ T cells using vaccines created through electrofusion of DCs and irradiated tumor cells (67). Survivin, a member of the apoptosis inhibition family of proteins, has been the GL261 TAA of choice for Ciesielski et al In particular, the authors exploited the xenogeneic differences between human and murine survivin sequences to develop a more immunogenic tumor vaccine 19, 20. The efficacy of systemic immunotherapy with DCs loaded with GL261 antigens was confirmed by Pellegatta et al Additionally, these authors introduced the concept of cancer stem cells in this model and reported that DC targeting of such stem cells within the GL261 tumor cell pool provides a higher level of protection against GL261 glioma 98, 99. Recently, Grauer et al illustrated the pronounced impact of FoxP3+ regulatory T cells (Tregs) in this model and suggested that Treg elimination is a prerequisite for successful eradication of established glioma using tumor lysate‐pulsed DCs 50, 51.
The concept of DC treatment in the orthotopic GL261 mouse model
We implemented the concept of active glioma immunotherapy with RNA‐loaded DCs in the GL261 murine model step‐by‐step. The rationale for loading DCs with total RNA from tumoral origin is to be able to provide DC immunotherapy for those glioma patients where insufficient tumor tissue is available to produce an optimal amount of DCm‐HGG‐L, for example, in patients with a small tumor mass of a relapsed tumor located in a previously irradiated area. Moreover, RNA molecules are translated into proteins by the DCs themselves and those proteins are subsequently processed in the endogenous pathway of antigen processing, resulting in optimal priming of CD8+ T cells. Researchers have dedicated a lot of effort to the improvement of RNA transfection methods. Three techniques have been investigated: passive transfection, lipid‐mediated transfection and electroporation 47, 53. Using the latter technique, immature DCs are transfected with 15 µg of total tumoral RNA per million DCs through exponential decay electroporation (300 V and 150 µF) with a GenePulser electroporator (Bio‐Rad, Nazareth, Belgium)81, 82. By now, electroporation has proven to be the method of choice for the introduction of exogenous RNA molecules into DCs. It does not require additional reagents and is compatible with clinical use (37). With this technology, improving the intrinsic characteristics of DCs such as cytokine, chemokine and costimulator expression through transfection with additional mRNA species becomes possible and seems very promising 1, 12.
Initially, we focused on testing of our cellular product (murine DCs electroporated with total RNA from GL261 tumor cells, DCm‐GL261‐RNA) in an experimental in vitro design. The procedure of electroporation caused only minor changes in the DC phenotype (Figure 1a), and did not alter the stimulatory capacity of DCs in a mixed lymphocyte reaction (Figure 1b), while the expression of enhanced green fluorescent protein at the protein level could be measured (Figure 1c). In vitro, DCm‐GL261‐RNA were able to stimulate T cells to become specific effector T cells with cytotoxic activity against the GL261 target cells (Figure 2). We noticed a nonspecific T cell cytotoxic activity when stimulated with mock‐electroporated DCm or DCm electroporated with total RNA from Lewis lung cancer (DCm‐LCC‐RNA). Nevertheless, specificity of the induced immune response could be demonstrated in vivo (81). The data obtained from these in vitro stimulation experiments confirmed that DCs loaded with tumor antigens in the form of RNA molecules are capable of inducing a T cell‐mediated antitumor immune response. These results are consistent with published preclinical data from our group 81, 82 and others (10).
Figure 1.
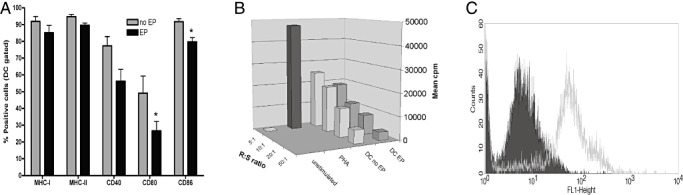
Electroporation of DCs with RNA‐molecules is feasible without loss of cell function in vitro. A. Immature DCs were left untouched (no EP) or electroporated (EP) and immediately afterwards matured with 1 µg/mL (Escherichia coli) lipopolysaccharide for 24 hours. Mature DCs were analyzed by flow cytometry for surface marker expression. EP significantly lowered the expression of CD80 and CD86 on DCs (overall analysis of variance P < 0.001, *P < 0.05). B. The capacity to stimulate allogeneic cells was assessed in a mixed lymphocyte reaction in 96 well format with total 2 × 105 splenocytes from naive BALB/C mice as responder (R) cells and DCs from C57BL/6 mice as stimulator (S) cells. Unstimulated cells and phytohemagglutinin (PHA) were used as negative and positive controls, respectively. Responder to stimulator ratios (R : S) ranged from 5:1 to 50:1. After 4 days of culture, thymidine incorporation was measured by pulsing cultures for 18 hours with 1 µCi [3H] thymidine per well. Mean counts per minute (cpm) are depicted. C. One million DCs were electroporated with 1 µg mRNA encoding enhanced green fluorescent protein (eGFP) and eGFP expression (grey curve in FL1‐Height histogram) was assessed by flow cytometry 24 hours after electroporation. The filled curve represents nonelectroporated DCs.
Figure 2.
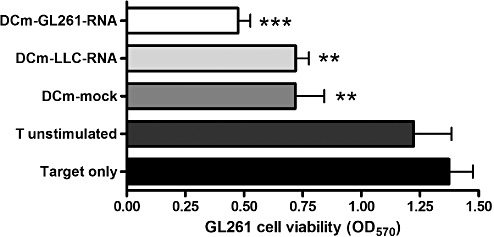
RNA‐loaded DCs induce a T cell‐mediated antitumor immune response in vitro. T cells pooled from spleen and lymph nodes of a naive C57BL/6 mouse were stimulated ex vivo (two stimulation rounds with DC : T cell ratio of 1:10 with addition of 20 U/mL of IL‐2) with either mock‐loaded DCm (DCm‐mock), DCm electroporated with total RNA extracted from Lewis lung carcinoma cells (DCm‐LLC‐RNA) or DCm electroporated with total RNA extracted from GL261 glioma cells (DCm‐GL261‐RNA). Unstimulated T cells were used as the control. Stimulated T cells were then used as effector cells and coincubated with GL261 tumor cells in an effector to target ratio of 10:1. Tumor cell viability was assessed by measuring the metabolization of 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide to formazan (measured at 570 nm) by living cells after 2 days of co‐culture. Pooled results (n = 3) are depicted as optical density values at 570 nm (OD570) and compared to GL261 target cells without effector cells. Overall analysis of variance P < 0.001; **P < 0.01, ***P < 0.001.
Since in vitro systems merely partially mimic the complex tumor microenvironment and cell interactions in vivo, we proceeded to validate our findings in the in vivo experimental mouse glioma model 81, 82. In this model, we hope to unravel new mechanisms governing glioma immunology that can eventually be exploited in translational oncology practice (Figure 3a). We opted for prophylactic immunization because others reported the GL261 model to be highly aggressive, necessitating additional intervention for curative settings 40, 78. Moreover, this model reflects the clinical therapeutic setting, at least locally in the brain, in which DC vaccination is given at a stage of minimal residual disease after (sub)total resection and not at the time of bulky disease. We adapted the model in order to monitor tumor growth consecutively with bioluminescence imaging (BLI) (82). We have shown that there is a strong correlation of the BLI signal with ex vivo measured tumor volumes. Moreover, implementing optical imaging in our experimental glioma model resulted in a more detailed characterization of GL261 glioma behavior in vivo, demonstrating midline crossing of tumor cells, along with the observation of an initial tumor cell adaptation phase before consistent tumor progression.
Figure 3.
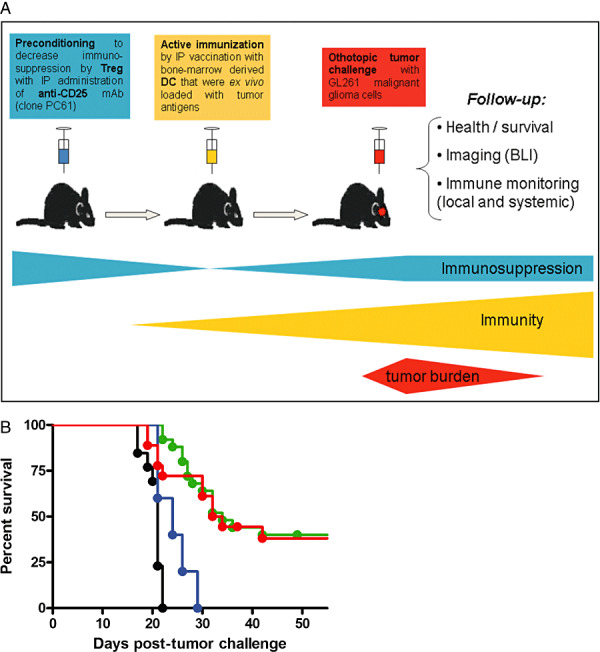
Immunotherapy in the GL261 experimental orthotopic glioma mouse model. A. Overview of the prophylactic DC vaccination model. Adult C57BL/6 mice are orthotopically challenged with 5 × 105 syngeneic GL261 malignant glioma cells injected 1.5 mm posterior and 1 mm lateral from the bregma at a depth of 3 mm under stereotactic guidance. Read‐out consists of monitoring general health parameters (weight and neurological symptoms), overall survival analysis, in vivo bioluminescence (BLI) requiring GL261 cells expressing luciferase in white mutant C57BL/6.Cg.Tyrc‐2J/J mice, and detailed immune monitoring both systemically (in blood, spleen and tumor‐draining cervical lymph nodes) and locally within the brain. Immunotherapy in our preventive treatment setting consists of either active immunization, decreasing regulatory T cell (Treg)‐mediated immune suppression, or combined treatment. Active immunization is performed through intraperitoneal (IP) vaccination on day 14 and day 7 before tumor challenge with one million mature bone‐marrow derived CD11c+ DCs that were loaded ex vivo with tumor antigens (in the form of total RNA). In some experiments, animals were preconditioned with a single intraperitoneal injection (250 µg) of the depleting anti‐CD25 monoclonal antibody (clone PC61). This in vivo depletion of CD25‐expressing cells was performed 1 week before the first vaccination with DC (or 3 weeks before tumor challenge if no DC vaccination was administered). B. Pooled survival data represented as Kaplan–Meier graph. Overall log rank P < 0.001. Survival curves of animals vaccinated with DCm‐GL261‐RNA (green curve, n = 27), DCm‐GL261‐L (DC loaded with GL261 lysate) (red curve, n = 19), DCm‐splenocyte‐RNA (blue curve, n = 5) and untreated animals (black curve, n = 13) are depicted.
Vaccination with DCm‐GL261‐RNA prolonged survival and could cure nearly half of the treated animals subjected to subsequent tumor challenge (81). As shown in Figure 3b, we could not detect a difference in outcome in this model when the mice were treated with DCm‐GL261‐RNA vs. DCm‐GL261‐L (median survival of 36 and 34 days, respectively, compared to 21 days for untreated mice). Mapping of weight loss and tumor‐induced neurologic deficit clearly paralleled the survival data. Finally, histological analysis revealed that DCm‐GL261‐RNA vaccination resulted in infiltration of lymphocytes and nonlymphoid cells, such as macrophages, particularly at the interface of the tumor mass with the normal brain parenchyma (81). These findings are in agreement with the data previously reported by Insug et al (58).
Which cells are players in anti‐GL261 mouse glioma immunotherapy?
Why vaccination only results in tumor protection for half of the treated mice is still not completely understood. Longitudinal imaging of tumor‐bearing mice with BLI revealed that within the animals that eventually succumbed, distinct patterns of nonresponding and partially responding mice can be discriminated (82). We hypothesize that there is a very delicate balance between antitumor immune responses on the one hand (either endogenous or induced/boosted by the DC vaccine) and natural and tumor‐induced immune suppression on the other. In responding mice, it is apparent that treatment with DCs shifts this balance away from tolerance and toward immunity, resulting in protection against tumor growth. In contrast, in mice refractory to DC vaccination, DCs might actually induce or expand immune‐suppressive Tregs and/or immune‐suppressive and tumor‐promoting myeloid‐derived suppressor cells (MDSCs) (103).
Our data provided a rationale to investigate in‐depth the mode of action of DC treatment and to implement strategies that are aimed at lowering the detrimental immune suppression within the tumor microenvironment. In order to study the individual role of separate cell populations, we used depleting mAb to selectively remove the targeted cell population (Figure 3a). When CD8+ T cells were removed, tumor‐challenged mice had worse outcomes as compared with the control‐challenged mice, suggesting that the injection of GL261 cells as such induces a minor endogenous CD8+ T cell response in the immunocompetent hosts. When CD8‐depleted mice were subsequently treated with DCm‐GL261‐RNA, the survival was still prolonged as compared with CD8+‐depleted or control‐untreated animals, but none of the mice ultimately survived. These data suggest that besides a major role of CD8+ T cells in inducing the antitumoral immune effect leading to long‐term survival, other immune cells stimulated via DC vaccination play a role as well. When Tregs were depleted via anti‐CD25 mAb, all mice survived the subsequent tumor challenge, without or with DC treatment. Although the injection of anti‐CD25 mAb only transiently depleted circulating CD4+CD25+FoxP3+ Tregs, we found an increased amount and specific antitumoral activity of brain‐infiltrating lymphocytes in these Treg‐depleted animals, even stronger than in the animals that were treated with DCm‐GL261‐RNA. Whereas an infiltration of MDSCs was found in the control tumor‐challenged mice, similar to published data from others 49, 125, we found an increased infiltration of macrophages with M1 phenotype in the Treg‐depleted animals, releasing or unmasking an endogenous tumor rejecting immunosurveillance. Finally, upon rechallenge of long‐term surviving mice after Treg depletion and/or DC vaccination, only those mice that were treated with DC vaccination depicted a prolonged antitumor protective immune response. Upon rechallenge, the CD25‐depleted surviving animals that were not vaccinated showed mortality rates similar to untreated mice. All together, our data suggest that there is a complex interaction between different cell populations: tumor cells, effector T cells, memory T cells, Tregs and macrophages with different functional phenotypes. Most likely all these cells interact with each other, and the net result of complex interactions determines the outcome of the mice upon vaccination.
Human DCm‐HGG‐L
In preparation for potential clinical use, we studied the loading of human DCs with tumor lysates in vitro, as well as the potency of DCm‐HGG‐L 26, 29. DCs were differentiated out of monocytes, obtained after two adherence cycles of peripheral blood mononuclear cells. Differentiation was induced during 6 days with medium change and addition of IL‐4 and Granulocyte/Macrophage Colony‐Stimulating Factor (GM‐CSF) at day 1, with 30% medium and 100% cytokine refreshments at days 3 and 5. Loading with lysate was performed at day 6, after which, maturation stimuli were added to the cultures for one day: IL‐1β, tumor necrosis factor (TNF)‐α and prostaglandin E2 (PGE2). The phenotypic and functional characteristics of these so‐called early mature DCs have been described 26, 29. After two cycles of stimulation, the first stimulation in the presence of IL‐6 and IL‐12 and the second in the presence of IL‐4 and IL‐7, DCm‐HGG‐L were able to induce T cell proliferation. Upon stimulation, a shift towards Th1 cytokine production was observed. Finally and most importantly, DCm‐HGG‐L induced tumoricidal and tumor‐suppressive activity by the T cells, which was antigen specific, MHC specific and MHC restricted, pointing to CD8+ cytotoxic T cell activity. These cells were prepared as cellular product to be injected intradermally in the shoulder region of patients.
It is important to note here that multiple steps in the production process, as well as the administration of the DC vaccine, are amenable to significant improvement. In fact, no comparative clinical data are currently available pertaining to the optimal cellular source of DCs, cytokine cocktail to induce DC differentiation, source of tumor material, maturation cocktail, amount of cells to be injected, volume in which the cells should be suspended, route of injection, site of injection, schedule for vaccination, method for boosting the immune response, and combinations with other antitumoral and immunomodulatory treatment strategies. Still, progress has been made over the last few years, and currently, most groups use DCs that were matured ex vivo, injecting these DC vaccines intradermally or directly into lymph nodes.
DESIGN OF THERAPEUTIC STUDIES
Selection of patients
Upon literature search, three case reports and thirteen phase I, phase I/II or phase II trials have been published on DC vaccination of glioma patients since 2000 (Table 1). The median patient number in these reports was only 12, ranging from 1 to 56. The inclusion criteria, immunotherapeutic designs and interpretations varied significantly among and even within the reports, being based entirely on single‐center approaches and hypotheses. Hence, no firm conclusions can be derived regarding the optimal immunotherapeutic strategy, nor would it make sense to perform a meta‐analysis on these data. Already in our first reports 25, 111, we stressed the importance of a minimal residual disease setting for adjuvant postoperative DC vaccination in HGG patients. In the largest series published to date (30), we confirmed the importance of a gross total resection prior to DC vaccination for GBM patients in a multivariate analysis. General awareness and agreement on this important issue is mounting and even culminating in the assumption that therapeutic tumor vaccinations should be directed to early disease states. Most of the reports describe patients treated with immunotherapy at a stage of minimal residual disease, after gross‐total or near‐total resection. Interestingly, patients have been under maintenance corticosteroid treatment just prior and/or during immunotherapy in some trials 142, 143, 146, which one would expect to have a negative effect on the generation of an effective immune response.
Table 1.
Overview of reported clinical trials. Abbreviations: CTL = cytotoxic T lymphocyte; DC = dendritic cell; DTH = delayed‐type hypersensitivity; GBM = glioblastoma multiforme; IFNγ = interferon‐γ; IL‐4 = interleukin‐4; JAM = just another method; MHC = major histocompatibility complex; mo = month; NK = natural killer; no. = number; OS = overall survival; pat = patient; PBMC = peripheral blood mononuclear cell; PFS = progression‐free survival; qPCR = quantitative polymerase chain reaction; rhIL‐12 = recombinant human interleukin‐12; RTE = naive recent thymic emigrant T cell; TTP = time to progression; TTS = time to survival; w = week.
| Author | Year | No. of patients (type of trial) | Antigen source | Administration | Treatment schedule | Immune response | Clinical response |
|---|---|---|---|---|---|---|---|
| Liau et al (73) | 2000 | 1 (case report) | Allogeneic MHC class I‐matched GBM peptides | Intradermal | 3 vaccinations | T‐cell proliferation | None |
| Yu et al (147) | 2001 | 9 (phase I) | Tumor‐specific MHC‐I associated peptides | Subcutaneous | 3 vaccinations at 2‐week intervals | Systemic CTL cytotoxicity against tumor (n = 4) (JAM assay) | Prolonged survival compared to control group |
| Kikuchi et al (64) | 2001 | 8 (phase I) | DC fusion with autologous glioma cells | Intradermal | 3–7 vaccinations at 3‐week intervals | Increase in NK cells in PMBCs (n = 4); increased IFNγ in supernatant (n = 6) | Mixed response (n = 1); steroids during vaccination (n = 5); 2 minor responses |
| Yamanaka et al (142) | 2003 | 10 (phase I‐II) | Autologous tumor lysate | Intradermal and/or intratumor (Ommaya) | 1–10 vaccinations at 3‐week intervals | Increase in NK cells in PBMCs (n = 5); positive DTH reaction to tumor lysate (n = 3); increased T‐cell mediated antitumor activity (n = 2); Elispot IFNγ | Minor response (n = 2) |
| Wheeler et al (135) | 2003 | 17 (phase I) | Autologous tumor lysate | Not reported | 3 vaccinations at 2‐ week intervals | Not reported (study on CD8+ RTE) | Not reported |
| 17 (phase II) | 3 vaccinations at 2‐week intervals; fourth vaccination 6 weeks after third (n = 10) | ||||||
| Yu et al (146) | 2004 | 14 (phase I‐II) | Autologous tumor lysate | Subcutaneous | 3 vaccinations at 2‐week intervals | IFNγ release in PBMCs (n = 6); expansion of CD8+ antigen specific T cell clones (n = 4); systemic T cell cytotoxicity against tumor (n = 1) | Significant increase in median survival |
| De Vleeschouwer et al (25) | 2004 | 1 (case report) | Autologous tumor lysate | Intradermal | 6 vaccinations; w1, w3 and further every 4 weeks | Positive DTH reaction to tumor lysate | Long‐lasting tumor‐free survival (>7 years after vaccination) |
| Rutkowski et al (111) | 2004 | 12 (phase I) | Autologous tumor lysate | Intradermal | 2–7 vaccinations; w1, w3 and further every 4 weeks | Positive DTH reaction to tumor lysate (n = 6) | Partial response (n = 1); tumor‐free survival (n = 2; 5 years after vaccination) |
| Kikuchi et al (65) | 2004 | 15 (phase I‐II) | DC fusion with autologous glioma cells | Intradermal | Vaccination + rhIL‐12 | Positive DTH reaction to tumor lysate (n = 15); increased cytotoxic activity (n = 2); increased intracellular IFNγ in CD8+ T cells (n = 1) | Partial response (n = 4); mixed response (n = 1) |
| Liau et al (74) | 2005 | 12 (multicohort dose‐escalation study) | Acid‐eluted tumor associated peptides (autologous) | Intradermal | ≤3 vaccinations | CTL response (n = 6) | Median TTP 19.9 mos—OS 18 to >58 mos—median OS 35.8 mos |
| Yamanaka et al (143) | 2005 | 24 (phase I‐II) | Autologous tumor lysate | Intradermal or intradermal + intratumor (Ommaya) | 1–10 vaccinations at 3‐week intervals | Positive DTH reaction to tumor lysate (n = 8); positive IFNγ Elispot (n = 7) | Partial response (n = 1); minor response (n = 3); significantly increased median survival |
| Okada et al (92) | 2007 | 2 (A) / 5 (B) | IL‐4 transfected fibroblasts + apoptotic glioma cells (A) / autologous tumor lysate (B) | Intradermal | 2 vaccinations with 2‐week interval | A: increased T cell reactivity; B: no response | A: clinical and radiological response in both patients; B: no response |
| Wheeler et al (134) | 2008 | 34 (phase II) | Autologous tumor lysate | Subcutaneous | 3 vaccinations at 2‐week intervals; fourth vaccination 6 weeks after third | Post‐vaccine antigen‐directed IFNγ response in PBMCs (qPCR‐based assay) (n = 17); DTH‐test resulted in cutaneous GBM in 1 pat (DTH was subsequently discontinued) | Significant positive correlation between post‐vaccine response magnitude on one hand and TTS and TTP spanning chemotherapy on the other hand |
| Walker et al (130) | 2008 | 13 (phase I) | Single‐cell suspension of autologous tumor cells | Intradermal | Priming phase consisting of 6 vaccinations at 2‐week intervals; further vaccinations at 6‐week intervals | Increased T cell infiltration in post‐vaccination tumor specimens compared to pre‐vaccination specimens (n = 3) | 9‐month survival (9/13); 12‐month survival (6/13); 18‐month or longer survival (3/13) |
| De Vleeschouwer et al (30) | 2008 | 56 (phase I‐II) | Autologous tumor lysate | Intradermal | Cohort comparison | Positive DTH reaction to tumor lysate (9/21 at time of diagnosis and 9/17 after 2 vaccinations) | PFS 3 mos; OS 9.6 mos; 2‐year OS 14.8%; total resection is a predictor for better PFS; younger age and total resection are predictors for better OS in univariable analysis; tendency towards improved PFS when faster DC vaccination schedule with tumor lysate boosting was applied |
In order to avoid this confounding variable, our trial has been restricted to patients with relapsed HGG, who could obtain a total or near‐total resection and be rapidly weaned from corticosteroids within 1 to 2 weeks after surgery. There are multiple arguments supporting the need for these restrictions. First, because there are major immune‐suppressive mechanisms at play within the tumor microenvironment (33) and even systemically (36), only a gross‐total resection results in a clinically effective recovery of normal immune system function (107). Second, we have not been able to produce good‐quality DCs out of monocytes isolated at the time of corticosteroid treatment. Third, we have previously faced an overwhelming peritumoral inflammatory reaction in one patient with bulky residual tumor (111). In this patient, the vaccine‐induced inflammatory immune reaction occurred with a progressively increasing delay after the vaccine injection. This is compatible with the hypothesis that the growing tumor and, as such, the increasingly induced immune suppression counteracts the vaccine‐induced immune reaction. This observation pointed to a tumor‐specific reaction induced by the vaccine, but maybe also to the gradually shifting balance in favor of tumor‐induced immune suppression.
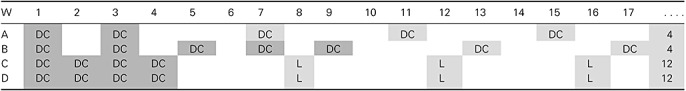
In our initial experiments, we detected an effective antitumoral reaction only 4 months after the start of vaccination (25). This observation and the finding of an increasing tumor‐mediated immune suppression (111) brought us to design an accelerated and intensified schedule of induction vaccinations to establish immune control before tumor regrowth occurs and induces tumor‐associated immune suppression (Table 2). Two DCm‐HGG‐L vaccines were administered within 3 weeks in cohort A; five DCm‐HGG‐L vaccines were administered within 9 weeks in cohort B, whereas four DCm‐HGG‐L vaccines were administered within 4 weeks in the cohorts C and D.
Table 2.
Vaccination schedule in the different cohorts of the cohort comparison trial HGG‐IMMUNO‐2003 for patients with relapsed HGG. Abbreviations: W = Week; DC = injections with DCm‐HGG‐L; L = injections with HGG‐L without DC; Dark gray area = the time schedule of the induction vaccinations; Gray area = the start of the maintenance boost vaccinations; . . . . = Boost vaccines were given each 4 weeks for cohort A and B. After three 4‐weekly lysate injections, further boosts were given each 12 weeks for cohort C and D.
Selection of outcome measures
Already before the consensus on study design and clinical end points in DC vaccination was published by the Cancer Vaccine Clinical Trial Working group (56), we designed a cohort comparison trial in which small cohorts of at least 20 patients at a time were treated, with small modifications in DC‐based vaccination strategies implemented in a stepwise fashion from one cohort to the next. As such, each cohort served as a control for the next cohort, enabling the investigators to detect or sometimes validate stepwise improvements in the vaccine. The available reports in the literature on DC‐based vaccination in patients with malignant glioma result in an overall clinical response rate of 11.4% based on the Macdonald criteria (80), ranging from 0% to 33%, taking into account that in 21% of all reported patients, clinical response was not within the scope of the study (28). Overall, this is still in accordance with the 5%–10% reported clinical responses upon DC‐based vaccination in other malignancies, such as melanoma (71). Immunological responses, assessed by a wide variety of immunomonitoring tools, have been demonstrated in 50% of reported cases, ranging from 20% to 100%. Only recently, Wheeler et al (134) found stronger evidence between immunological responses and survival in vaccinated glioma patients. Although immunological responses are mandatory to map the way and to provide proof of the principle for this therapeutic strategy, “proof of efficacy” can only be established in large well‐designed comparative clinical trials, ideally in controlled randomized trials.
Especially in HGG, an old paradigm states that pretreatment prognostic variables might have more impact on outcome than any (new) potentially active therapy or treatment strategy. To that end, recursive partitioning analysis (RPA) as originally described and validated for newly diagnosed malignant gliomas by the Radiation Therapy Oncology Group (RTOG) in 1993, provides us with an excellent model of prognostic classes of pretreatment and treatment‐related patient's variables (24). Using this RPA classification, the impact of new treatments on OS in HGG can be examined in clinically similar patient groups and compared with large databases of conventionally treated patients. To date, there is consensus that clinical responses, as defined by the Macdonald criteria, do not apply for biological treatment modalities such as tumor vaccination. The paradigm of a therapeutic vaccine leading to (detectable) immunological responses and hence to (detectable) clinical response with increased OS is at least incomplete. Even immunomonitoring tools directed toward the affected organ, that is, the brain, will have to take into account that the presence of tumor‐infiltrating lymphocytes and other immune effectors do not unequivocally correlate with a good or a bad outcome (35). Proof of efficacy of immunotherapy asks for impact on OS in patients with HGG, of whom the different prognostic risk factors are clearly categorized.
Nature of DCs
All clinical reports have used monocyte‐derived DCs for their trials. The methodology for DC preparation is now fairly well established, and gives a sufficient yield of DCm for injections into patients (124). Most of the older trials have used immature DCs 73, 74, 134, 135, 142, 146, 147, while others have used maturation stimuli like TNF‐α64, 65, penicillin‐killed Streptococcus pyogenes (OK‐432) (143), monocyte‐conditioned medium (130), IFN‐γ and TNF‐α in combination with IL‐4‐secreting fibroblasts (92). Only one phase I study focused on the dose of DCs and could not find any dose‐limiting toxicity (74).
In order to guide the use of DCm‐HGG‐L stepwise into clinical practice with the goal of improving clinical outcome, we set up a cohort comparison trial (HGG‐IMMUNO‐2003) for end‐stage patients with relapsed HGG, in which each cohort is designed based on recent knowledge and experience and aimed to provide results that can be compared with earlier cohorts of patients. A cohort comparison approach has been published with the aim to define improved chemotherapeutic control of HGG in childhood (137). Within our approach, we varied some parameters, including not only the vaccination schedule but also the maturation stimuli, while other principle parameters were preserved. All patients received freshly cultured monocyte‐derived DCs loaded with autologous HGG lysate (DCm‐HGG‐L). The monocytes were obtained after plastic adherence and were differentiated with IL‐4 and GM‐CSF. In cohorts A, B and C, maturation during 24 hours was induced with TNF‐α, IL‐1β and PGE2, a cocktail, which is essentially based on the so‐called Jonuleit cocktail (60). However, we opted to omit IL‐6 from the cocktail, as it is currently known to play a major role in the induction of a Th17 phenotype T cell response (131). As a next step, PGE2 was omitted in cohort D based on recent discussions on the activity of PGE2 at the time of maturation. For instance, PGE2 had already been linked to the induction of a DC2 type years ago (62). Because of its importance mainly for the induction of the mobility of DCs (114), it was kept in the classical maturation cocktail. In more recent studies, however, PGE2 has been shown to induce indoleamine 2,3‐dioxygenase (IDO) activity in human DCs, thereby creating a tolerating DC phenotype (9). Moreover, PGE2 up‐regulates CD25 on DCs, the latter believed to be a marker of strong DC maturation, while on the other hand representing a marker that is shed and thereby consumes the IL‐2 needed for autocrine T cell activation. CD25 expression was analyzed on 226 samples of mature DCs prepared in our hands, and the median CD25 expression was 15% (range: 0% to 76%). Because incompletely maturated DCs themselves play a role in tolerance induction (87), we wanted to apply a recently described method to induce in vivo DC maturation after preparation of the injection site with imiquimod 88, 108. Imiquimod binds to Toll‐like receptor 7 and induces strong DC maturation and activation. Its role in the generation of immune responses in a preclinical in vivo model of HGG has been described (104). In cohort D, we combined ex vivo maturation with TNF‐α and IL‐1β with in vivo maturation with imiquimod.
We subsequently moved to a third major change in our strategy, besides changing the vaccination schedule and maturation cocktail, by introducing boost vaccines with lysate only, starting with cohort C. The rationale for this was a publication from Jouanneau et al (61), clearly demonstrating in the murine GL261 model that optimal survival was reached when priming of the immune system was performed with DCm loaded with tumor antigens, whereas for boosting purposes, only lysate was used. Although not elaborated extensively, one might hypothesize based on these results and our own results in the murine model (81) that profound and/or protracted DC vaccination might result in a higher proportion of Tregs and MDSCs that ultimately down‐regulate the global immune reactivity, while HGG‐L injection more indirectly maintains the ongoing immune response and the induction of memory T cell responses. Hence, from cohort C on, boost vaccines were given using crude tumor lysates. In cohort D, the lysates were injected intradermally at sites pretreated with imiquimod cream.
Injection site
At the time of the design of most clinical trials so far, no knowledge was available if and which lymph nodes would be the optimal destination of injected DCs. Later on, however, data became available that priming of T cells by DCs within the cervical lymph nodes induced an integrin homing pattern toward intracerebral locations (15). Other preclinical in vivo brain tumor models clearly showed an enrichment of tumor‐specific Tregs in the blood and cervical lymph nodes (40). Although intradermal injections in the cervical lymph node region are feasible, we have thus far opted to inject the DCm‐HGG‐L and HGG‐L in the ventral region of shoulder and upper arm, with the idea of targeting the axillary lymph nodes for T cell stimulation.
As pointed out by Tuyaerts et al (124), the question of whether DCm‐HGG‐L should be injected intradermally, intravenously or in the lymph nodes is not yet resolved. This becomes particularly important as only a small amount of the intradermally injected DCs ultimately reaches the T cell area of lymph nodes (129). However, taking into account the documented failure rate of even experienced radiologists injecting DCs under ultrasound guidance, and the desire to spread DC vaccination technology to more centers in order to perform large‐scale clinical trials, we pragmatically favored the intradermal route for DC injection, similar to most researchers in the field. One particular issue that should become of major interest is the local injection of DCm‐HGG‐L directly into the tumor region. In their study, Yamanaka et al (143) showed some benefit from intratumoral plus intradermal injections of DCs as compared with only intradermal injections, although one should note that this interesting observation is not derived from a prospective randomized approach. Nevertheless, a local change from an immune‐suppressive into a more immune stimulatory microenvironment might be induced by intratumoral administration of DCs.
Monitoring
Immune responses following vaccination have been monitored in most trials. These analyses have included positive DTH skin reaction, T cell reactivity and NK cell enrichment in peripheral blood, as well as measuring T cell infiltration in tumoral tissue taken after vaccination. The reported immune monitoring remains very global in these clinical trials, mainly due to the lack of specific antigens to be targeted.
We have also identified a potential role of imaging to monitor immune reactions in the tissue surrounding the resection cavity or residual tumor lesion (25). Although data are still preliminary, advanced magnetic resonance imaging (MRI) eventually combined with positron emission tomography (PET) may soon provide better tools to monitor the effects of immunotherapy of HGG.
RESULTS
Because all clinical trials reported thus far comprise case reports, phase I, phase I/II or phase II trials, the questions answered in these trials relate to feasibility, toxicity and early efficacy, immunologically or oncologically (Table 1). In all trials, it seemed to be feasible to administer DC‐based vaccines. Moreover, all reports conclude that the toxicity is minimal except for the case reported by our group where we observed an overwhelming inflammatory reaction around a residual tumor (111). Of major importance, none of the reports describe autoimmune phenomena induced by DC vaccination.
Data from the literature
Besides feasibility, toxicity and clinical outcome, all published studies on DC‐based immunotherapy have aimed to assess immune responses. Several immune assays have been developed for this purpose, but so far, results have been difficult to interpret. Moreover, immunological responses are not detected in all patients. Seven studies 25, 30, 65, 111, 134, 142, 143 report positive DTH reactions to tumor lysate in only a subset of treated patients. Liau et al (73) described a patient, in which a strong T cell proliferative response occurred against the allogeneic tumor peptide preparation that was used to pulse the DCs for vaccination. Similarly, an increase in systemic CTL activity was seen in a subset of treated patients in five studies 65, 74, 142, 146, 147. Other immune monitoring assays that have been used are expression of CD8+ antigen‐specific T cell clones (146), increase of intracellular IFN‐γ in CD8+ T cells (65) and an increase in NK cells 64, 142. Also IFN‐γ release/response by peripheral blood mononuclear cells (PBMCs) has been assessed in five studies. Kikuchi et al (64) measured IFN‐γ in supernatans of a culture of PBMCs with autologous glioma Enzyme‐Linked Immuno Sorbent Assay. IFN‐γ‐Elispot assays were used by Yamanaka et al 142, 143, while Wheeler et al (134) and Yu et al (146) used quantitative polymerase chain reaction assessment of IFN‐γ responsiveness. T cell infiltrates in tumor tissue taken after vaccination was documented by several groups 73, 74, 130, 142, 146, 147. A correlation between immunologically measurable effects and clinical outcome was reported in several papers 74, 134, 135. In the study by Wheeler et al (134), a significant progressive (logarithmic) association was found between time to survival after inclusion and postvaccine IFN‐γ enhancement. This allowed them to propose that single‐vaccine response metrics can quantitatively reflect inhibition of tumor progression by T cells in GBM patients.
Based on review of the literature, at least three groups in the United States, one group in Europe, one in Japan, one in Asia and one in Australia are actively performing clinical trials. All these trials are small, and include a heterogeneous group of patients with regard to histology and/or time of vaccination. Some studies included patients with all subtypes of HGGs 64, 92, 130, 134, 142, 146, 147, while others focused solely on patients with GBM 25, 30, 73, 74, 111, 135. Some studied patients with relapsed HGG 64, 73, 142, while others included patients with a first diagnosis of HGG, either exclusively (147) or combined with patients treated at recurrence 74, 92, 130, 134, 135, 146. Only the European group reported on children with relapsed GBM 25, 30, 111. Moreover, as pointed out before, the vaccination protocol varied enormously among research groups. Due to this heterogeneity in study design and execution, sound comparative analyses on clinical outcome are difficult.
Nevertheless, in most trials, some clinical effect is observed and some important clues for future clinical studies are provided, especially from trials where there are comparisons with historical controls or between internal subgroups. For example, Yu et al (147) reported that seven patients with newly diagnosed GBM who received three biweekly intradermal vaccinations with peptide‐pulsed DCs had median survival times of 455 days as compared with 257 for 42 nonvaccinated but otherwise comparably treated patients in the same institute. Although compared with historical control patients and although no statistics are provided, the prolongation of the median survival is an interesting observation in this phase I clinical trial. The same group reported on vaccinations at the time of relapse in eight GBM patients, and made a similar comparison with 26 historical control patients (146). Both patient groups underwent second craniotomy for relapsed GBM. Whereas the median survival was 30 weeks in the control group, subsequent vaccination after new resection resulted in a significant change of median survival to 133 weeks, with some patients surviving for more than 250 weeks. In a recent paper, the group demonstrated that vaccine responders exhibited more favorable clinical outcomes relative to nonresponders. Moreover, the vaccine‐induced responses elicited therapeutic benefits primarily by sensitizing tumors to subsequent chemotherapy (134). In the phase I study reported by Liau et al (74), the patients with stable tumors or no residual disease at time of DC vaccination compared favorably even with historical/concurrent data as the best prognostic subgroup of GBM patients treated during the same time period. In contrast, Okada et al (92) did not show any benefit in progression‐free survival (PFS) in patients treated with DCs loaded with autologous tumor lysate, in combination with IL‐4 producing fibroblasts (92). In the study on recurrent HGG patients reported by Yamanaka et al (142), radiologic responses were observed in the group of five patients who received intratumoral plus intradermal vaccinations, while no response was detected in the five patients who were only injected intradermally.
Clinical experiences on a large group of patients with recurrent GBM
In our clinical practice, we have implemented immunotherapy with DCm‐HGG‐L for the treatment of patients with relapsed HGG, as a stepwise process using a cohort‐comparison study concept (HGG‐IMMUNO‐2003). Children above the age of 3 years, adults and elderly patients up to 70 years with relapsed HGG can be included into the trial, if as much as possible and at least 1.5 cm3 viable tumor material is available in sterile, dry and frozen (−80°C or liquid N2) condition, if (sub)total resection is documented after operation, if the patients can be tapered off of corticosteroids within 1 week after operation and if pathology is confirmed. By following this strategy, we have been able to compare each cohort within the trial with the other cohorts. Thus far, two major issues in the complexity of DC immunotherapy have been addressed: the vaccination schedule and the maturation cocktail.
We have analyzed a subgroup of the first 75 adults with relapsed GBM who were treated according to the four cohorts described earlier. As shown in Figure 4, there was no difference in median age or in extent of resection among the patients in the four cohorts. The median PFS and OS times of the total group were 2.6 and 8.62 months, respectively, with a 2‐year survival of 13.6% [standard error of the mean (SEM) = 4.6]. Complete resection (n = 39) vs. partial or subtotal resection (n = 36) improved the median PFS from 1.87 to 3.51 months (log rank test: P = 0.004), and the OS from 6.97 to 10.08 months (log rank test: P = 0.0168). We reported earlier that the extent of resection, but not the Karnofsky index nor the age, was an independent risk factor in a multivariate analysis (30). Interestingly, we observed a stepwise improvement in the PFS curves from cohort A to cohort C by shortening the interval between the induction DC vaccines, with further improvements noted by changing the maturation cocktail in cohort D (Figure 4). The median PFS for the four cohorts was: 1.94, 1.67, 3.23 and 2.72 months, respectively; the PFS at 2 years was 4% in cohort C and 15.3% in cohort D. Although the median OS did not differ significantly between the four cohorts, we noticed a 2‐year survival of 16.7% (SEM = 7.6) in cohort C and of 27.8% (SEM = 9.9) in cohort D.
Figure 4.
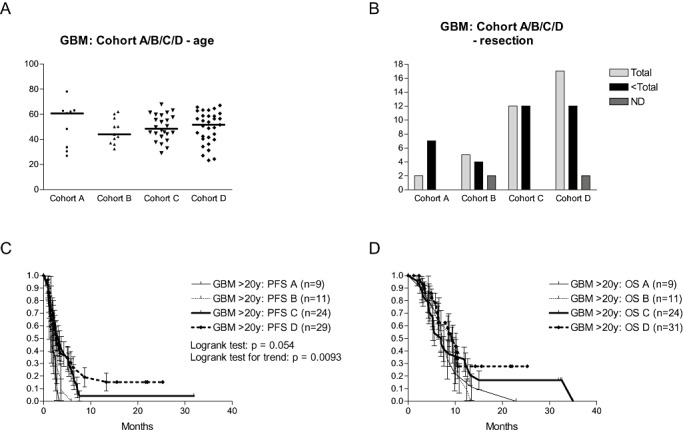
HGG‐IMMUNO‐2003 protocol: DC vaccination for adults with relapsed GBM. Patients with relapsed GBM were reoperated (removal of as much tumor tissue as feasible) and received subsequent immunotherapy. Patients were divided among four consecutive cohorts. A. The age (expressed in years on the y‐axis) for the four cohorts is shown, and median age is indicated. B. The number of patients with total resection or less than total resection of relapsed GBM is shown for each cohort (ND = not documented). C. Progression‐free survival (PFS) for the four cohorts of patients. D. Overall survival (OS) for the four cohorts of patients.
Considering the whole subgroup of 88 children and adults treated for relapsed GBM with new surgery and immunotherapy, the younger long‐term surviving patients published earlier (30) illustrate a level 1c medical evidence of clinical efficacy (http://www.cebm.net/levels_of_evidence.asp). This high level of evidence points to the change from 100% mortality of relapsed GBM after resection alone (90) toward a situation where at least some patients survive the disease after subsequent immunotherapy (the intervention). The patient published in 2004 (25) is the longest disease‐free survivor in a series of 88 patients with relapsed GBM (children and adults), with a follow‐up of 86 months. The significant shift in PFS of adults treated with immunotherapy in the consecutive cohorts of patients further illustrate level 2b efficacy. The latter efficacy level points to significance in individual cohort studies without randomization.
Further analysis is currently being performed on a subgroup of the first 117 adult patients with (all types of) relapsed HGG according to allocation in the different RPA classes I to VI as described by the RTOG (manuscript in preparation). The RPA classes reflect the integrated importance of age, pathology, mental status, Karnofsky performance score, extent of resection and intensity of radiation therapy. The latter factor is irrelevant for the patients with relapsed HGG as radiotherapy could not be implemented anymore. With a median follow‐up of 20.8 months, 80% of patients were allocated to classes III to V and a highly significant difference in OS was noted according to RPA class. With median survival times for classes I to VI of more than 36 months (to date), 14.2, 14.0, 10.6, 6 and 4 months, respectively, as well as 2‐year survival rates (after start of vaccination) of 100%, 30.8%, 25%, 11.5%, 0% and 0% for these classes, these results are comparable with survival data of newly diagnosed HGG patients in the original RTOG database, even though no additional radiotherapy was performed in the relapsed setting. Moreover, we could demonstrate that the use of imiquimod resulted in a significant survival benefit in patients in class III and as such, this might be the first report on the significant and relevant clinical advantage of the use of Toll‐like receptors (TLR) agonists in tumor vaccination strategies.
MRI of patients who obtained DC vaccination for relapsed GBM
Radiologic follow‐up is routinely performed with serial MRI scans in GBM patients. In some cases, no contrast enhancing mass is seen. However, others display contrast enhancement along with variable perilesional edema and mass effects. The differentiation between a vaccine‐induced inflammatory immune reaction and early tumor relapse remains challenging as the radiological characteristics of both entities are similar. As such, only further follow‐up with repeat imaging and clinical correlation makes it possible to distinguish these two possibilities. Furthermore, a PET scan with the radiolabeled amino acid methionine may help, as this technique provides metabolic data for the lesion in question (25).
Four different MRI patterns at follow‐up are illustrated in Figure 5. All patients presented with relapsed GBM and received vaccination therapy after neurosurgical resection. Case 1 is a 36‐year‐old man with a recurrent tumor in the left frontal lobe. MRI at 6 weeks after the first vaccine revealed perioperative contrast enhancement, which retrospectively most likely represented surgical changes, given the resolution of the enhancement on consecutive images. The patient still has no further clinical or radiographic evidence of recurrence at 3 years. Case 2 is a 16‐year‐old woman with no clear changes on MRI until 4 to 6 months after the start of immune therapy, when a large area around the resection cavity revealed contrast enhancement, albeit with no significant mass effect or edema. Subsequent imaging showed resolution of the contrast enhancement with no evidence of recurrence on the last MRI, taken 6 years later. The patient remains clinically stable at 86 months after treatment. Case 3 is a 59‐year‐old man with stable clinical and radiological disease for over 3 years, although at 6 weeks following administration of the first vaccine, MRI showed contrast enhancement in the area around the resection cavity. Over time, the surgical cavity collapsed, while the contrast enhancement remained relatively unchanged. Case 4 is a 60‐year‐old man with rapid clinical deterioration 6 to 7 months after the first vaccine. Parallel to the clinical evolution, contrast enhancement was observed in the area around the resection cavity, 6 months after administration of the first vaccine. Consecutive MRI revealed a significant increase in contrast enhancement with progressive midline shift, indicative of mass effect.
Figure 5.
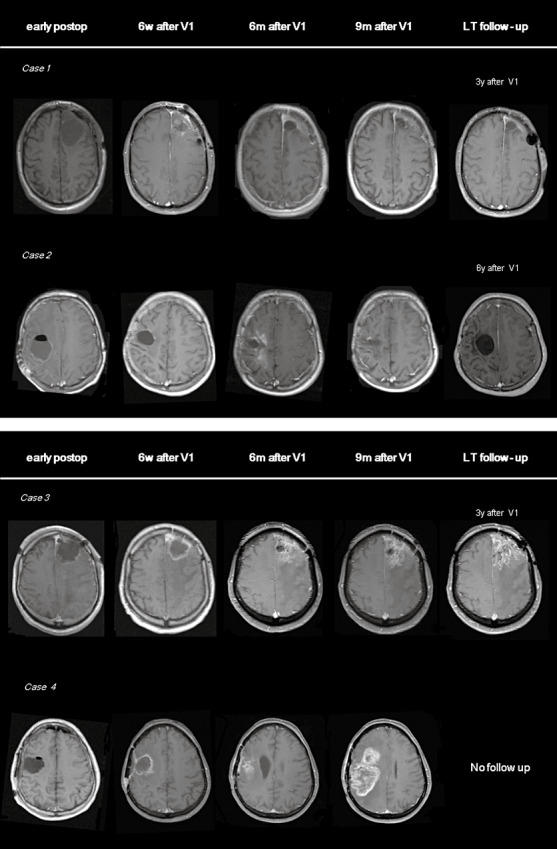
MRI findings during and after DC vaccination for relapsed GBM. Transverse T1‐weighted MR images after contrast administration, obtained 1 day after debulking surgery for relapsed GBM (early postoperative period), 6 weeks after the first vaccine (6 w after V1), 6 months after the first vaccine (6 m after V1), 9 months after the first vaccine (9 m after V1) and after long‐term follow‐up (LT follow‐up), 3 to 6 years after the first vaccine according to the case (3–6 years after V1). Case 1 is a patient with a resection in the left frontal lobe. MR imaging showed discrete contrast enhancement around the resection cavity 6 weeks after administration of the first vaccine. No contrast enhancement was documented in follow‐up imaging. Case 2 is a patient after surgery in the right frontal lobe. Transient contrast enhancement was seen at 6 and 9 months after the start of the vaccination therapy. Follow‐up MR scans showed subsequent resolution of the contrast enhancement. Case 3 shows follow‐up MR images in a patient with surgery in the left frontal lobe. Over consecutive time points, the resection cavity collapsed and a comparable area of contrast enhancement has remained stable for 3 years. Case 4 is a patient with a resection in the right frontoparietal region. Subsequent MRI shows an increasing region of contrast enhancement, suggestive of tumor relapse/progression.
As current “routine” MRI does not enable adequate documentation of the time course and nature of the vaccine‐induced inflammatory immune responses, further research is being performed to characterize these responses in both patients and animal models, using advanced magnetic resonance (MR) techniques like MR spectroscopy 32, 52, 91, MR diffusion‐weighted 91, 105, perfusion‐weighted 55, 91, 105 and diffusion tensor imaging (101). Hopefully, such research will provide clues for early prediction of treatment response and thereby allow for earlier changes in treatment approach as necessary.
The integration of immunotherapy in the primary multimodal treatment strategy
Based on previous experience, a “cohort C” approach has been integrated into the primary treatment regimen for patients with newly diagnosed GBM at our medical center. After radiochemotherapy, four weekly DCm‐HGG‐L are injected, and lysate boosts are administered at day 8 of the 28 day cycles 1, 2, 3 and 6 of temozolomide maintenance therapy. In total, 66 adults with a primary diagnosis of GBM are available for further analysis, with a median age of 55 years. The data are still preliminary due to short follow‐up of recently included patients. Nonetheless, the median PFS and OS times are currently 10.3 and 17.9 months, respectively, the 6‐month PFS is 75%, and the 2‐year OS is 31.6%. The incidence of side effects has been minimal, in that the registered quality of life has not been affected by therapy and most patients have been treated in an ambulatory setting.
CONCLUSIONS
Immunotherapy for patients with HGG is a novel therapeutic approach that opens new opportunities for enhanced survival without major toxicity. This is of particular importance, taking into account that HGGs cause relatively high community burdens, not only with many years of life lost due to cancer (14), but also because of the major morbidity from the tumor and subsequent treatments. In the assessment of immunotherapeutic results for patients with HGG, the improvement of PFS and, particularly, the significant increase of OS with satisfactory quality of life are, of course, by far the most important variables, much more than immunologic surrogate markers for response.
The studies in preclinical animal models for human gliomas, especially the murine models, are interesting in order to obtain more insight into the basic biology underlying disease evolution and immunotherapeutic mechanisms. However, timely translation of new concepts into clinical practice should be the primary objective.
With several groups entering into clinical practice in this field, new data are now being generated. However, it is of paramount importance not to discredit this promising approach based on data from poorly designed trials with inappropriate end points. Thus far, the observations with regard to both immunological responses and clinical responses are promising and beneficial effects are reproducible. Of particular interest is the fact that no induction of autoimmunity or other major toxicities have been observed to date. However, larger and more homogeneous patient groups with more restricted inclusion criteria should be studied for more rapid progress in this field. Moreover, confirmatory studies with an appropriate randomization versus a control patient group, preferably even stratified for molecular tumor signature, should be implemented in the near future. For this, collaborative efforts between well‐organized and experienced vaccination centers should be established.
ACKNOWLEDGMENTS
This work has been supported by the Olivia Hendrickx Research Fund (http://www.olivia.be). Support was also obtained from Electrabel Netmanagement Vlaanderen, CAF Belgium, Baxter, the Herman Memorial Research Fund (http://www.hmrf.be), the James E. Kearney Memorial Fund and gifts from private families and service clubs. Additionally, grants were obtained from “Stichting tegen Kanker,” IWT (TBM project), the Stem Cell Institute Leuven, the Emmanuel van der Schueren Fund, the International Union against Cancer, the Klinisch Onderzoeksfonds UZ Leuven, and the Fund for Scientific Research—Flanders (FWO‐V). We are very grateful for the technical assistance from Katja Vandenbrande, Goedele Stegen and Vallentina Schaiko. We thank the neuro‐oncology team in the hospital for fruitful patient discussion, and the staff of the Laboratory of Experimental Immunology for basic scientific discussions.
Stefaan Van Gool is senior clinical investigator at the Fund for Scientific Research Flanders (FWO‐V). Wim Maes is supported by the Olivia Hendrickx Research Fund. Hilko Ardon and Tina Verschuere are research fellows supported by the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT). Sofie Van Cauter is research fellow in the Department of Radiology. Steven De Vleeschouwer is supported by the Klinisch Onderzoeksfonds UZ Leuven.
REFERENCES
- 1. Aarntzen EH, Figdor CG, Adema GJ, Punt CJ, De Vries IJ (2008) Dendritic cell vaccination and immune monitoring. Cancer Immunol Immunother 57:1559–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akasaki Y, Black KL, Yu JS (2005) Dendritic cell‐based immunotherapy for malignant gliomas. Expert Rev Neurother 5:497–508. [DOI] [PubMed] [Google Scholar]
- 3. Akasaki Y, Black KL, Yu JS (2005) T cell immunity in patients with malignant glioma: recent progress in dendritic cell‐based immunotherapeutic approaches. Front Biosci 10:2908–2921. [DOI] [PubMed] [Google Scholar]
- 4. Akasaki Y, Liu G, Chung NH, Ehtesham M, Black KL, Yu JS (2004) Induction of a CD4+ T regulatory type 1 response by cyclooxygenase‐2‐overexpressing glioma. J Immunol 173:4352–4359. [DOI] [PubMed] [Google Scholar]
- 5. Aoki H, Mizuno M, Natsume A, Tsugawa T, Tsujimura K, Takahashi T, Yoshida J (2001) Dendritic cells pulsed with tumor extract‐cationic liposome complex increase the induction of cytotoxic T lymphocytes in mouse brain tumor. Cancer Immunol Immunother 50:463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banchereau J, Palucka AK (2005) Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol 5:296–306. [DOI] [PubMed] [Google Scholar]
- 7. Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392:245–252. [DOI] [PubMed] [Google Scholar]
- 8. Basse PH, Whiteside TL, Chambers W, Herberman RB (2001) Therapeutic activity of NK cells against tumors. Int Rev Immunol 20:439–501. [DOI] [PubMed] [Google Scholar]
- 9. Bergwelt‐Baildon MS, Popov A, Saric T, Chemnitz J, Classen S, Stoffel MS et al (2006) CD25 and indoleamine 2,3‐dioxygenase are up‐regulated by prostaglandin E2 and expressed by tumor‐associated dendritic cells in vivo: additional mechanisms of T‐cell inhibition. Blood 108:228–237. [DOI] [PubMed] [Google Scholar]
- 10. Boczkowski D, Nair SK, Snyder D, Gilboa E (1996) Dendritic cells pulsed with RNA are potent antigen‐presenting cells in vitro and in vivo . J Exp Med 184:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boethius J, Blomgren H, Collins VP, Greitz T, Strander H (1983) The effect of systemic human interferon‐alpha administration to patients with glioblastoma multiforme. Acta Neurochir (Wien) 68:239–251. [DOI] [PubMed] [Google Scholar]
- 12. Bonehill A, Van Nuffel AM, Corthals J, Tuyaerts S, Heirman C, Francois V et al (2009) Single‐step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin Cancer Res 15:3366–3375. [DOI] [PubMed] [Google Scholar]
- 13. Brada M, Hoang‐Xuan K, Rampling R, Dietrich PY, Dirix LY, Macdonald D et al (2001) Multicenter phase II trial of temozolomide in patients with glioblastoma multiforme at first relapse. Ann Oncol 12:259–266. [DOI] [PubMed] [Google Scholar]
- 14. Burnet NG, Jefferies SJ, Benson RJ, Hunt DP, Treasure FP (2005) Years of life lost (YLL) from cancer is an important measure of population burden—and should be considered when allocating research funds. Br J Cancer 92:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Calzascia T, Masson F, Berardino‐Besson W, Contassot E, Wilmotte R, Aurrand‐Lions M et al (2005) Homing phenotypes of tumor‐specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity 22:175–184. [DOI] [PubMed] [Google Scholar]
- 16. Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE et al (2007) Intracranial glioblastoma models in preclinical neuro‐oncology: neuropathological characterization and tumor progression. J Neurooncol 85:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carpentier AF, Meng Y (2006) Recent advances in immunotherapy for human glioma. Curr Opin Oncol 18:631–636. [DOI] [PubMed] [Google Scholar]
- 18. Chi DD, Merchant RE, Rand R, Conrad AJ, Garrison D, Turner R et al (1997) Molecular detection of tumor‐associated antigens shared by human cutaneous melanomas and gliomas. Am J Pathol 150:2143–2152. [PMC free article] [PubMed] [Google Scholar]
- 19. Ciesielski MJ, Apfel L, Barone TA, Castro CA, Weiss TC, Fenstermaker RA (2006) Antitumor effects of a xenogeneic survivin bone marrow derived dendritic cell vaccine against murine GL261 gliomas. Cancer Immunol Immunother 55:1491–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ciesielski MJ, Kozbor D, Castanaro CA, Barone TA, Fenstermaker RA (2008) Therapeutic effect of a T helper cell supported CTL response induced by a survivin peptide vaccine against murine cerebral glioma. Cancer Immunol Immunother 57:1827–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Claes A, Idema AJ, Wesseling P (2007) Diffuse glioma growth: a guerilla war. Acta Neuropathol 114:443–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Claes A, Schuuring J, Boots‐Sprenger S, Hendriks‐Cornelissen S, Dekkers M, Van Der Kogel AJ et al (2008) Phenotypic and genotypic characterization of orthotopic human glioma models and its relevance for the study of anti‐glioma therapy. Brain Pathol 18:423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clarke SR (2000) The critical role of CD40/CD40L in the CD4‐dependent generation of CD8+ T cell immunity. J Leukoc Biol 67:607–614. [DOI] [PubMed] [Google Scholar]
- 24. Curran WJ Jr., Scott CB, Horton J, Nelson JS, Weinstein AS, Fischbach AJ et al (1993) Recursive partitioning analysis of prognostic factors in three radiation therapy oncology group malignant glioma trials. J Natl Cancer Inst 85:704–710. [DOI] [PubMed] [Google Scholar]
- 25. De Vleeschouwer S, Van Calenbergh F, Demaerel P, Flamen P, Rutkowski S, Kaempgen E et al (2004) Transient local response and persistent tumor control of recurrent malignant glioma treated with combination therapy including dendritic cell therapy. J Neurosurg (Pediatrics) 100:492–497. [DOI] [PubMed] [Google Scholar]
- 26. De Vleeschouwer S, Arredouani M, Ade M, Cadot P, Vermassen E, Ceuppens JL, Van Gool SW (2005) Uptake and presentation of malignant glioma tumor cell lysates by monocyte‐derived dendritic cells. Cancer Immunol Immunother 54:372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Vleeschouwer S, Van Gool SW, Van Calenbergh F (2005) Immunotherapy for malignant gliomas: emphasis on strategies of active specific immunotherapy using autologous dendritic cells. Childs Nerv Syst 21:7–18. [DOI] [PubMed] [Google Scholar]
- 28. De Vleeschouwer S, Rapp M, Sorg RV, Steiger HJ, Stummer W, Van Gool S, Sabel M (2006) Dendritic cell vaccination in patients with malignant gliomas: current status and future directions. Neurosurgery 59:988–999. [DOI] [PubMed] [Google Scholar]
- 29. De Vleeschouwer S, Spencer Lopes I, Ceuppens JL, Van Gool SW (2007) Persistent IL‐10 production is required for glioma growth suppressive activity by Th1‐directed effector cells after stimulation with tumor lysate‐loaded dendritic cells. J Neurooncol 84:131–140. [DOI] [PubMed] [Google Scholar]
- 30. De Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, Goffin J et al (2008) Postoperative adjuvant dendritic cell‐based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res 14:3098–3104. [DOI] [PubMed] [Google Scholar]
- 31. Dhodapkar KM, Cirignano B, Chamian F, Zagzag D, Miller DC, Finlay JL, Steinman RM (2004) Invariant natural killer T cells are preserved in patients with glioma and exhibit antitumor lytic activity following dendritic cell‐mediated expansion. Int J Cancer 109:893–899. [DOI] [PubMed] [Google Scholar]
- 32. Di CA, Scarabino T, Trojsi F, Popolizio T, Catapano D, Giannatempo GM et al (2008) Proton MR spectroscopy of cerebral gliomas at 3 T: spatial heterogeneity, and tumour grade and extent. Eur Radiol 18:1727–1735. [DOI] [PubMed] [Google Scholar]
- 33. Dix AR, Brooks WH, Roszman TL, Morford LA (1999) Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol 100:216–232. [DOI] [PubMed] [Google Scholar]
- 34. Dredge K, Marriott JB, Todryk SM, Dalgleish AG (2002) Adjuvants and the promotion of Th1‐type cytokines in tumour immunotherapy. Cancer Immunol Immunother 51:521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dunn GP, Dunn IF, Curry WT (2007) Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human glioma. Cancer Immun 7:12. [PMC free article] [PubMed] [Google Scholar]
- 36. Elliott LH, Brooks WH, Roszman TL (1987) Activation of immunoregulatory lymphocytes obtained from patients with malignant gliomas. J Neurosurg 67:231–236. [DOI] [PubMed] [Google Scholar]
- 37. Erdmann M, Dorrie J, Schaft N, Strasser E, Hendelmeier M, Kaempgen E et al (2007) Effective clinical‐scale production of dendritic cell vaccines by monocyte elutriation directly in medium, subsequent culture in bags and final antigen loading using peptides or RNA transfection. J Immunother 30:663–674. [DOI] [PubMed] [Google Scholar]
- 38. Everson RG, Graner MW, Gromeier M, Vredenburgh JJ, Desjardins A, Reardon DA et al (2008) Immunotherapy against angiogenesis‐associated targets: evidence and implications for the treatment of malignant glioma. Expert Rev Anticancer Ther 8:717–732. [DOI] [PubMed] [Google Scholar]
- 39. Farkkila M, Jaaskelainen J, Kallio MJ, Blomstedt G, Raininko R, Virkkunen P et al (1994) Randomised, controlled study of intratumoral recombinant gamma‐interferon treatment in newly diagnosed glioblastoma. Br J Cancer 70:138–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA et al (2006) Systemic anti‐CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res 12:4294–4305. [DOI] [PubMed] [Google Scholar]
- 41. Fenstermaker RA, Ciesielski MJ (2004) Immunotherapeutic strategies for malignant glioma. Cancer Control 11:181–191. [DOI] [PubMed] [Google Scholar]
- 42. Fleury A, Menegoz F, Grosclaude P, Daures JP, Henry Amar M, Raverdy N et al (1997) Descriptive epidemiology of cerebral gliomas in France. Cancer 79:1195–1202. [DOI] [PubMed] [Google Scholar]
- 43. Fomchenko EI, Holland EC (2006) Mouse models of brain tumors and their applications in preclinical trials. Clin Cancer Res 12:5288–5297. [DOI] [PubMed] [Google Scholar]
- 44. Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A et al (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21:2683–2710. [DOI] [PubMed] [Google Scholar]
- 45. Gabrilovich DI (2002) Dendritic cell vaccines for cancer treatment. Curr Opin Mol Ther 4:452–458. [PubMed] [Google Scholar]
- 46. Gerstner ER, Duda DG, Di Tomaso E, Sorensen G, Jain RK, Batchelor TT (2007) Antiangiogenic agents for the treatment of glioblastoma. Expert Opin Investig Drugs 16:1895–1908. [DOI] [PubMed] [Google Scholar]
- 47. Gilboa E, Vieweg J (2004) Cancer immunotherapy with mRNA‐transfected dendritic cells. Immunol Rev 199:251–263. [DOI] [PubMed] [Google Scholar]
- 48. Gilboa E, Nair SK, Lyerly HK (1998) Immunotherapy of cancer with dendritic‐cell‐based vaccines. Cancer Immunol Immunother 46:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Graf MR, Sauer JT, Merchant RE (2005) Tumor infiltration by myeloid suppressor cells in response to T cell activation in rat gliomas. J Neurooncol 73:29–36. [DOI] [PubMed] [Google Scholar]
- 50. Grauer OM, Nierkens S, Bennink E, Toonen LW, Boon L, Wesseling P et al (2007) CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo . Int J Cancer 121:95–105. [DOI] [PubMed] [Google Scholar]
- 51. Grauer OM, Sutmuller RP, Van Maren W, Jacobs JF, Bennink E, Toonen LW et al (2008) Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate‐pulsed dendritic cells in a murine glioma model. Int J Cancer 122:1794–1802. [DOI] [PubMed] [Google Scholar]
- 52. Gruber S, Stadlbauer A, Mlynarik V, Gatterbauer B, Roessler K, Moser E (2005) Proton magnetic resonance spectroscopic imaging in brain tumor diagnosis. Neurosurg Clin N Am 16:101–114, vi. [DOI] [PubMed] [Google Scholar]
- 53. Grunebach F, Muller MR, Nencioni A, Brossart P (2003) Delivery of tumor‐derived RNA for the induction of cytotoxic T‐lymphocytes. Gene Ther 10:367–374. [DOI] [PubMed] [Google Scholar]
- 54. Heath WR, Carbone FR (2001) Cross‐presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol 19:47–64. [DOI] [PubMed] [Google Scholar]
- 55. Hirai T, Murakami R, Nakamura H, Kitajima M, Fukuoka H, Sasao A et al (2008) Prognostic value of perfusion MR imaging of high‐grade astrocytomas: long‐term follow‐up study. AJNR Am J Neuroradiol 29:1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hoos A, Parmiani G, Hege K, Sznol M, Loibner H, Eggermont A et al (2007) A clinical development paradigm for cancer vaccines and related biologics. J Immunother 30:1–15. [DOI] [PubMed] [Google Scholar]
- 57. Imaizumi T, Kuramoto T, Matsunaga K, Shichijo S, Yutani S, Shigemori M et al (1999) Expression of the tumor‐rejection antigen SART1 in brain tumors. Int J Cancer 83:760–764. [DOI] [PubMed] [Google Scholar]
- 58. Insug O, Ku G, Ertl HC, Blaszczyk‐Thurin M (2002) A dendritic cell vaccine induces protective immunity to intracranial growth of glioma. Anticancer Res 22:613–321. [PubMed] [Google Scholar]
- 59. Jefford M, Maraskovsky E, Cebon J, Davis ID (2001) The use of dendritic cells in cancer therapy. Lancet Oncol 2:343–353. [DOI] [PubMed] [Google Scholar]
- 60. Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E et al (1997) Proinflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal‐calf serum‐free conditions. Eur J Immunol 27:3135–3142. [DOI] [PubMed] [Google Scholar]
- 61. Jouanneau E, Poujol D, Gulia S, Le Mercier I, Blay JY, Belin MF, Puisieux I (2005) Dendritic cells are essential for priming but inefficient for boosting antitumour immune response in an orthotopic murine glioma model. Cancer Immunol Immunother 55:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kalinski P, Hilkens CMU, Wierenga EA, Kapsenberg ML (1999) T‐cell priming by type‐1 and type‐2 polarized dendritic cells: the concept of a third signal. Immunol Today 20:561–567. [DOI] [PubMed] [Google Scholar]
- 63. Khan JA, Yaqin S (2006) Dendritic cell therapy with improved outcome in glioma multiforme—a case report. J Zhejiang Univ Sci B 7:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T (2001) Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother 50:337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kikuchi T, Akasaki Y, Abe T, Fukuda T, Saotome H, Ryan JL et al (2004) Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J Immunother 27:452–459. [DOI] [PubMed] [Google Scholar]
- 66. Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S (2000) Therapeutic efficacy of OX‐40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res 60:5514–5521. [PubMed] [Google Scholar]
- 67. Kjaergaard J, Wang LX, Kuriyama H, Shu S, Plautz GE (2005) Active immunotherapy for advanced intracranial murine tumors by using dendritic cell‐tumor cell fusion vaccines. J Neurosurg 103:156–164. [DOI] [PubMed] [Google Scholar]
- 68. Kleihues P, Soylemezoglu F, Schauble B, Scheithauer BW, Burger PC (1995) Histopathology, classification, and grading of gliomas. Glia 15:211–221. [DOI] [PubMed] [Google Scholar]
- 69. Kurpad SN, Zhao XG, Wikstrand CJ, Batra SK, McLendon RE, Bigner DD (1995) Tumor antigens in astrocytic gliomas. Glia 15:244–256. [DOI] [PubMed] [Google Scholar]
- 70. Kuwashima N, Nishimura F, Eguchi J, Sato H, Hatano M, Tsugawa T et al (2005) Delivery of dendritic cells engineered to secrete IFN‐alpha into central nervous system tumors enhances the efficacy of peripheral tumor cell vaccines: dependence on apoptotic pathways. J Immunol 175:2730–2740. [DOI] [PubMed] [Google Scholar]
- 71. Lesterhuis WJ, Aarntzen EH, De Vries IJ, Schuurhuis DH, Figdor CG, Adema GJ, Punt CJ (2008) Dendritic cell vaccines in melanoma: from promise to proof? Crit Rev Oncol Hematol 66:118–134. [DOI] [PubMed] [Google Scholar]
- 72. Levitsky HI, Lazenby A, Hayashi RJ, Pardoll DM (1994) In vivo priming of two distinct antitumor effector populations: the role of MHC class I expression. J Exp Med 179:1215–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liau LM, Black KL, Martin NA, Sykes SN, Bronstein JM, Jouben‐Steele L et al (2000) Treatment of a glioblastoma patient by vaccination with autologous dendritic cells pulsed with allogeneic Major Histocompatibility Complex Class I‐matched tumor peptides: case report. Neurosurgical Focus 9:e8. [DOI] [PubMed] [Google Scholar]
- 74. Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ et al (2005) Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T‐cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res 11:5515–5525. [DOI] [PubMed] [Google Scholar]
- 75. Liu G, Khong HT, Wheeler CJ, Yu JS, Black KL, Ying H (2003) Molecular and functional analysis of tyrosinase‐related protein (TRP)‐2 as a cytotoxic T lymphocyte target in patients with malignant glioma. J Immunother 26:301–312. [DOI] [PubMed] [Google Scholar]
- 76. Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS (2004) HER‐2, gp100, and MAGE‐1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res 64:4980–4986. [DOI] [PubMed] [Google Scholar]
- 77. Liu G, Yu JS, Zeng G, Yin D, Xie D, Black KL, Ying H (2004) AIM‐2: a novel tumor antigen is expressed and presented by human glioma cells. J Immunother 27:220–226. [DOI] [PubMed] [Google Scholar]
- 78. Lumniczky K, Desaknai S, Mangel L, Szende B, Hamada H, Hidvegi EJ, Safrany G (2002) Local tumor irradiation augments the antitumor effect of cytokine‐producing autologous cancer cell vaccines in a murine glioma model. Cancer Gene Ther 9:44–52. [DOI] [PubMed] [Google Scholar]
- 79. Luptrawan A, Liu G, Yu JS (2008) Dendritic cell immunotherapy for malignant gliomas. Rev Recent Clin Trials 3:10–21. [DOI] [PubMed] [Google Scholar]
- 80. Macdonald DR, Cascino TL, Schold SC Jr., Cairncross JG (1990) Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 8:1277–1280. [DOI] [PubMed] [Google Scholar]
- 81. Maes W, Galicia Rosas G, Verbinnen B, Boon L, De Vleeschouwer S, Ceuppens JL, Van Gool SW (2008) DC vaccination with anti‐CD25 treatment leads to long‐term immunity against experimental glioma. Neurooncol 2009 Mar 31. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Maes W, Deroose C, Reumers V, Krylyshkina O, Gijsbers R, Ceuppens J et al (2009) In vivo bioluminescence imaging in an experimental mouse model for dendritic cell based immunotherapy against malignant glioma. J Neurooncol 91:127–139. [DOI] [PubMed] [Google Scholar]
- 83. Maldonado‐Lopez R, Moser M (2001) Dendritic cell subsets and the regulation of Th1/Th2 responses. Semin Immunol 13:275–282. [DOI] [PubMed] [Google Scholar]
- 84. Mason WP (2008) Emerging drugs for malignant glioma. Expert Opin Emerg Drugs 13:81–94. [DOI] [PubMed] [Google Scholar]
- 85. McLendon RE, Wikstrand CJ, Matthews MR, Al‐Baradei R, Bigner SH, Bigner DD (2000) Glioma‐associated antigen expression in oligodendroglial neoplasms. Tenascin and epidermal growth factor receptor. J Histochem Cytochem 48:1103–1110. [DOI] [PubMed] [Google Scholar]
- 86. Merchant RE, McVicar DW, Merchant LH, Young HF (1992) Treatment of recurrent malignant glioma by repeated intracerebral injections of human recombinant interleukin‐2 alone or in combination with systemic interferon‐alpha. Results of a phase I clinical trial. J Neurooncol 12:75–83. [DOI] [PubMed] [Google Scholar]
- 87. Moser M (2003) Dendritic cells in immunity and tolerance—do they display opposite functions? Immunity 19:5–8. [DOI] [PubMed] [Google Scholar]
- 88. Nair S, McLaughlin C, Weizer A, Su Z, Boczkowski D, Dannull J et al (2003) Injection of immature dendritic cells into adjuvant‐treated skin obviates the need for ex vivo maturation. J Immunol 171:6275–6282. [DOI] [PubMed] [Google Scholar]
- 89. Ni HT, Spellman SR, Jean WC, Hall WA, Low WC (2001) Immunization with dendritic cells pulsed with tumor extract increases survival of mice bearing intracranial gliomas. J Neurooncol 51:1–9. [DOI] [PubMed] [Google Scholar]
- 90. Nieder C, Grosu AL, Molls M (2000) A comparison of treatment results for recurrent malignant gliomas. Cancer Treat Rev 26:397–409. [DOI] [PubMed] [Google Scholar]
- 91. Oh J, Henry RG, Pirzkall A, Lu Y, Li X, Catalaa I et al (2004) Survival analysis in patients with glioblastoma multiforme: predictive value of choline‐to‐N‐acetylaspartate index, apparent diffusion coefficient, and relative cerebral blood volume. J Magn Reson Imaging 19:546–554. [DOI] [PubMed] [Google Scholar]
- 92. Okada H, Lieberman FS, Walter KA, Lunsford LD, Kondziolka DS, Bejjani GK et al (2007) Autologous glioma cell vaccine admixed with interleukin‐4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J Transl Med 5:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Okano F, Storkus WJ, Chambers WH, Pollack IF, Okada H (2002) Identification of a novel HLA‐A*0201‐restricted, cytotoxic T lymphocyte epitope in a human glioma‐associated antigen, interleukin 13 receptor alpha2 chain. Clin Cancer Res 8:2851–2855. [PubMed] [Google Scholar]
- 94. Parajuli P, Sloan AE (2004) Dendritic cell‐based immunotherapy of malignant gliomas. Cancer Invest 22:405–416. [DOI] [PubMed] [Google Scholar]
- 95. Parajuli P, Mathupala S, Sloan AE (2004) Systematic comparison of dendritic cell‐based immunotherapeutic strategies for malignant gliomas: in vitro induction of cytolytic and natural killer‐like T cells. Neurosurgery 55:1194–1204. [DOI] [PubMed] [Google Scholar]
- 96. Parajuli P, Mathupala S, Mittal S, Sloan AE (2007) Dendritic cell‐based active specific immunotherapy for malignant glioma. Expert Opin Biol Ther 7:439–448. [DOI] [PubMed] [Google Scholar]
- 97. Parney IF, Hao C, Petruk KC (2000) Glioma immunology and immunotherapy. Neurosurgery 46:778–791. [DOI] [PubMed] [Google Scholar]
- 98. Pellegatta S, Poliani PL, Corno D, Grisoli M, Cusimano M, Ubiali F et al (2006) Dendritic cells pulsed with glioma lysates induce immunity against syngeneic intracranial gliomas and increase survival of tumor‐bearing mice. Neurol Res 28:527–531. [DOI] [PubMed] [Google Scholar]
- 99. Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, Suarez‐Merino B et al (2006) Neurospheres enriched in cancer stem‐like cells are highly effective in eliciting a dendritic cell‐mediated immune response against malignant gliomas. Cancer Res 66:10247–10252. [DOI] [PubMed] [Google Scholar]
- 100. Plautz GE, Yang ZY, Wu BY, Gao X, Huang L, Nabel GJ (1993) Immunotherapy of malignancy by in vivo gene transfer into tumors. Proc Natl Acad Sci USA 90:4645–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Price SJ, Jena R, Burnet NG, Carpenter TA, Pickard JD, Gillard JH (2007) Predicting patterns of glioma recurrence using diffusion tensor imaging. Eur Radiol 17:1675–1684. [DOI] [PubMed] [Google Scholar]
- 102. Prins RM, Liau LM (2004) Cellular immunity and immunotherapy of brain tumors. Front Biosci 9:3124–3136. [DOI] [PubMed] [Google Scholar]
- 103. Prins RM, Scott GP, Merchant RE, Graf MR (2002) Irradiated tumor cell vaccine for treatment of an established glioma. II. Expansion of myeloid suppressor cells that promote tumor progression. Cancer Immunol Immunother 51:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Prins RM, Craft N, Bruhn KW, Khan‐Farooqi H, Koya RC, Stripecke R et al (2006) The TLR‐7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen‐specific T cell priming: relation to central nervous system antitumor immunity. J Immunol 176:157–164. [DOI] [PubMed] [Google Scholar]
- 105. Provenzale JM, Mukundan S, Barboriak DP (2006) Diffusion‐weighted and perfusion MR imaging for brain tumor characterization and assessment of treatment response. Radiology 239:632–649. [DOI] [PubMed] [Google Scholar]
- 106. Quattrocchi KB, Miller CH, Cush S, Bernard SA, Dull ST, Smith M et al (1999) Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. J Neurooncol 45:141–157. [DOI] [PubMed] [Google Scholar]
- 107. Rapp M, Ozcan Z, Steiger HJ, Wernet P, Sabel MC, Sorg RV (2006) Cellular immunity of patients with malignant glioma: prerequisites for dendritic cell vaccination immunotherapy. J Neurosurg 105:41–50. [DOI] [PubMed] [Google Scholar]
- 108. Rechtsteiner G, Warger T, Osterloh P, Schild H, Radsak MP (2005) Cutting edge: priming of CTL by transcutaneous peptide immunization with imiquimod. J Immunol 174:2476–2480. [DOI] [PubMed] [Google Scholar]
- 109. Rock KL, Clark K (1996) Analysis of the role of MHC class II presentation in the stimulation of cytotoxic T lymphocytes by antigens targeted into the exogenous antigen‐MHC class I presentation pathway. J Immunol 156:3721–3726. [PubMed] [Google Scholar]
- 110. Rock KL, Gamble S, Rothstein L (1990) Presentation of exogenous antigen with class I major histocompatibility complex molecules. Science 249:918–921. [DOI] [PubMed] [Google Scholar]
- 111. Rutkowski S, De Vleeschouwer S, Kaempgen E, Wolff JEA, Kuhl J, Demaerel P et al (2004) Surgery and adjuvant dendritic cell‐based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br J Cancer 91:1656–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Saito R, Mizuno M, Nakahara N, Tsuno T, Kumabe T, Yoshimoto T, Yoshida J (2004) Vaccination with tumor cell lysate‐pulsed dendritic cells augments the effect of IFN‐beta gene therapy for malignant glioma in an experimental mouse intracranial glioma. Int J Cancer 111:777–782. [DOI] [PubMed] [Google Scholar]
- 113. Saris SC, Spiess P, Lieberman DM, Lin S, Walbridge S, Oldfield EH (1992) Treatment of murine primary brain tumors with systemic interleukin‐2 and tumor‐infiltrating lymphocytes. J Neurosurg 76:513–519. [DOI] [PubMed] [Google Scholar]
- 114. Scandella E, Men Y, Gillessen S, Forster R, Groettrup M (2002) Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte‐derived dendritic cells. Blood 100:1354–1361. [DOI] [PubMed] [Google Scholar]
- 115. Scarcella DL, Chow CW, Gonzales MF, Economou C, Brasseur F, Ashley DM (1999) Expression of MAGE and GAGE in high‐grade brain tumors: a potential target for specific immunotherapy and diagnostic markers. Clin Cancer Res 5:335–341. [PubMed] [Google Scholar]
- 116. Sikorski CW, Lesniak MS (2005) Immunotherapy for malignant glioma: current approaches and future directions. Neurol Res 27:703–716. [DOI] [PubMed] [Google Scholar]
- 117. Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoom MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Eng J Med 352:987–996. [DOI] [PubMed] [Google Scholar]
- 118. Stupp R, Hegi ME, Mason WP, Van Den Bent MJ, Taphoorn MJ, Janzer RC et al (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol 10:459–466. [DOI] [PubMed] [Google Scholar]
- 119. Svane IM, Soot ML, Buus S, Johnsen HE (2003) Clinical application of dendritic cells in cancer vaccination therapy. APMIS 111:818–834. [DOI] [PubMed] [Google Scholar]
- 120. Tamber MS, Rutka JT (2003) Pediatric supratentorial high‐grade gliomas. Neurosurg Focus 14:e1. [DOI] [PubMed] [Google Scholar]
- 121. Tanaka H, Shimizu K, Hayashi T, Shu S (2002) Therapeutic immune response induced by electrofusion of dendritic and tumor cells. Cell Immunol 220:1–12. [DOI] [PubMed] [Google Scholar]
- 122. Tang J, Flomenberg P, Harshyne L, Kenyon L, Andrews DW (2005) Glioblastoma patients exhibit circulating tumor‐specific CD8+ T cells. Clin Cancer Res 11:5292–5299. [DOI] [PubMed] [Google Scholar]
- 123. Tsugawa T, Kuwashima N, Sato H, Fellows‐Mayle WK, Dusak JE, Okada K et al (2004) Sequential delivery of interferon‐alpha gene and DCs to intracranial gliomas promotes an effective antitumor response. Gene Ther 11:1551–1558. [DOI] [PubMed] [Google Scholar]
- 124. Tuyaerts S, Aerts JL, Corthals J, Neyns B, Heirman C, Breckpot K et al (2007) Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol Immunother 56:1513–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K et al (2008) Tumor‐infiltrating myeloid‐derived suppressor cells are pleiotropic‐inflamed monocytes/macrophages that bear M1‐ and M2‐type characteristics. J Leukoc Biol 83:1136–1144. [DOI] [PubMed] [Google Scholar]
- 126. Van Gool SW, Van den Hove L, Ceuppens JL (2000) Activation of the immune system in cancer patients. Med Pediatr Oncol 34:1–9. [DOI] [PubMed] [Google Scholar]
- 127. Van Gool SW, Vandenberghe P, De Boer M, Ceuppens JL (1996) CD80, CD86 and CD40 provide accessory signals in a multiple step T cell activation model. Immunol Rev 153:47–83. [DOI] [PubMed] [Google Scholar]
- 128. Ventimiglia JB, Wikstrand CJ, Ostrowski LE, Bourdon MA, Lightner VA, Bigner DD (1992) Tenascin expression in human glioma cell lines and normal tissues. J Neuroimmunol 36:41–55. [DOI] [PubMed] [Google Scholar]
- 129. Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, Van Rossum MM et al (2009) Limited amounts of dendritic cells migrate into the T‐cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res 15:2531–2540. [DOI] [PubMed] [Google Scholar]
- 130. Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C (2008) Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: potential interaction with adjuvant chemotherapy. J Clin Neurosci 15:114–121. [DOI] [PubMed] [Google Scholar]
- 131. Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM (2006) Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity 24:677–688. [DOI] [PubMed] [Google Scholar]
- 132. Wersall P, Ohlsson I, Biberfeld P, Collins VP, Von Krusenstjerna S, Larsson S et al (1997) Intratumoral infusion of the monoclonal antibody, mAb 425, against the epidermal‐growth‐factor receptor in patients with advanced malignant glioma. Cancer Immunol Immunother 44:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wheeler CJ, Black KL (2005) Dendritic cell vaccines and immunity in glioma patients. Front Biosci 10:2861–2881. [DOI] [PubMed] [Google Scholar]
- 134. Wheeler CJ, Black KL, Liu G, Mazer M, Zhang XX, Pepkowitz S et al (2008) Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res 68:5955–5964. [DOI] [PubMed] [Google Scholar]
- 135. Wheeler CJ, Black KL, Liu G, Ying H, Yu JS, Zhang W, Lee PK (2003) Thymic CD8(+) T cell production strongly influences tumor antigen recognition and age‐dependent glioma mortality. J Immunol 171:4927–4933. [DOI] [PubMed] [Google Scholar]
- 136. Whiteside TL, Odoux C (2004) Dendritic cell biology and cancer therapy. Cancer Immunol Immunother 53:240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Wolff JE, Classen CF, Wagner S, Kortmann RD, Palla SL, Pietsch T et al (2008) Subpopulations of malignant gliomas in pediatric patients: analysis of the HIT‐GBM database. J Neurooncol 87:155–164. [DOI] [PubMed] [Google Scholar]
- 138. Wu AH, Xiao J, Anker L, Hall WA, Gregerson DS, Cavenee WK et al (2005) Identification of EGFRvIII‐derived CTL epitopes restricted by HLA A0201 for dendritic cell based immunotherapy of gliomas. J Neurooncol 76:23–30. [DOI] [PubMed] [Google Scholar]
- 139. Yamada Y, Kiroiwa T, Nakagawa T, Kajimoto Y, Dohi T, Azuma H et al (2003) Transcriptional expression of survivin and its splice variants in brain tumors in humans. J Neurosurg 99:338–345. [DOI] [PubMed] [Google Scholar]
- 140. Yamanaka R (2006) Novel immunotherapeutic approaches to glioma. Curr Opin Mol Ther 8:46–51. [PubMed] [Google Scholar]
- 141. Yamanaka R (2008) Cell‐ and peptide‐based immunotherapeutic approaches for glioma. Trends Mol Med 14:228–235. [DOI] [PubMed] [Google Scholar]
- 142. Yamanaka R, Abe T, Yajima N, Tsuchiya N, Homma J, Kobayashi T et al (2003) Vaccination of recurrent glioma patients with tumour lysate‐pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer 89:1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T et al (2005) Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res 11:4160–4167. [DOI] [PubMed] [Google Scholar]
- 144. Yamanaka R, Yajima N, Abe T, Tsuchiya N, Homma J, Narita N et al (2003) Dendritic cell‐based glioma immunotherapy. Int J Oncol 23:5–15. [PubMed] [Google Scholar]
- 145. Yoshida J, Wakabayashi T, Mizuno M, Sugita K, Yoshida T, Hori S et al (1992) Clinical effect of intra‐arterial tumor necrosis factor‐alpha for malignant glioma. J Neurosurg 77:78–83. [DOI] [PubMed] [Google Scholar]
- 146. Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ (2004) Vaccination with tumor lysate‐pulsed dendritic cells elicits antigen‐specific, cytotoxic T‐cells in patients with malignant glioma. Cancer Res 64:4973–4979. [DOI] [PubMed] [Google Scholar]
- 147. Yu JS, Wheeler CJ, Zeltzer PM, Ying H, Finger DN, Lee PK et al (2001) Vaccination of malignant glioma patients with peptide‐pulsed dendritic cells elicits systemic cytotoxicity and intracranial T‐cell infiltration. Cancer Res 61:842–847. [PubMed] [Google Scholar]
- 148. Yung WK, Prados M, Levin VA, Fetell MR, Bennett J, Mahaley MS et al (1991) Intravenous recombinant interferon beta in patients with recurrent malignant gliomas: a phase I/II study. J Clin Oncol 9:1945–1949. [DOI] [PubMed] [Google Scholar]
- 149. Zeltzer PM, Moilanen B, Yu JS, Black KL (1999) Immunotherapy of malignant brain tumors in children and adults: from theoretical principles to clinical application. Childs Nerv Syst 15:514–528. [DOI] [PubMed] [Google Scholar]