

Abstract
Little is known about the relationship between soluble amyloid β (Aβ) and age. We have measured soluble and insoluble Aβ by enzyme‐linked immunosorbent assay (ELISA) in post‐mortem frontal cortex in normal brains (16–95 years) and AD. Insoluble Aβ increased with age, and was significantly higher in Alzheimer's disease (AD) than age‐matched controls. However, levels of soluble Aβ declined with age and were significantly greater in younger adults than older adults with or without AD. In AD, insoluble : soluble Aβ ratio was much higher than in age‐matched controls. The high levels of soluble Aβ in young adults included oligomeric species of Aβ1‐42. These observations do not preclude Aβ oligomers as neurotoxic mediators of AD but suggest that if they are, the toxicity may be restricted to certain species (eg, β‐pleated protofibrillar species not detected by our assay) or takes decades to manifest. The dramatically increased insoluble : soluble Aβ in AD points to an altered dynamic equilibrium of Aβ in AD, reflecting both enhanced aggregation and continued overproduction or impaired removal of the soluble peptide in older age, when the concentration of this peptide should be declining.
Keywords: Alzheimer's disease, normal aging, oligomeric Aβ, soluble Aβ
INTRODUCTION
Alzheimer's disease (AD) is characterized pathologically by abnormalities that include amyloid β (Aβ) plaques, composed mainly of Aβ1‐42, and it is the excessive accumulation of Aβ1‐42 within the brain that has been postulated to be responsible for the neurodegeneration observed in AD (14). Aβ production is a normal process that occurs throughout life. It results from sequential cleavage of amyloid precursor protein (APP) by β‐ and γ‐secretases. Aβ exists in a dynamic equilibrium of soluble monomeric, oligomeric, protofibrillar and fibrillar states. Aβ monomers are nontoxic (3), whereas there is increasing evidence demonstrating neurotoxicity of oligomeric species 3, 20, 35, 37. A steady‐state level of Aβ is maintained by balance between its production and removal from the brain. Aβ accumulation reflects a disturbance of this balance: increased Aβ production, decreased removal or a combination of the two.
Age is the most significant non‐genetic risk factor for AD. With age, Aβ levels increase in brains of APP‐transgenic and in non‐transgenic mice 9, 16 primates (41) and humans (8). Levels of formic acid‐extractable (ie, insoluble) Aβ1‐40 and Aβ1‐42 in temporal and frontal cortex from human non‐demented controls were found to be positively associated with age between 18 and 92 years (8). However, little is known about the relationship between soluble nonfibrillar Aβ and age. We have examined total soluble and total insoluble Aβ levels in human post‐mortem brain tissue from normal controls aged 16–95 years and patients with AD. We found total soluble levels to be significantly higher in the younger control brains than in AD or older control brains, with levels being inversely correlated with age in the controls. Additionally, total soluble Aβ showed a negative correlation with insoluble Aβ in controls but a positive correlation in AD. Furthermore, we have demonstrated that the soluble Aβ species found to be negatively associated with age include oligomeric Aβ.
METHODS
Study cohort
Brain tissue was obtained from the South West Dementia Brain Bank (SWDBB), University of Bristol, UK and the MRC Sudden Death Brain and Tissue Bank (SDBTB), University of Edinburgh, UK. The study had approval from Frenchay Local Research Ethics Committee. All cases had been subjected to detailed neuropathological examination, including immunohistochemistry for Aβ and phospho‐tau. The diagnosis of AD was made according to the criteria of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) (25). The AD cohort (n = 84) comprised cases of “definite” AD or “probable” AD. The AD cases ranged from 54 to 98 years in age [mean = 79.5, standard deviation (SD) = 9.0] and comprised 53 females and 31 males. The post‐mortem delays were between 4 and 99 h (mean = 44.3, SD = 24.3). All had had a clinical diagnosis of dementia made during life by experienced clinicians with an interest in dementia and all had had a Mini‐Mental State Examination score of <17 (usually well below this) on several occasions prior to death. Cases with concomitant CNS pathology such as Lewy body disease were excluded from the study. Non‐demented control cases (showing the absence of AD or other neuropathological abnormalities) (n = 68) comprised 21 females and 47 males, with an age range from 16 to 95 years (mean = 60.6, SD = 23.5). These cases had not had a clinical history of cognitive decline or dementia and had no or only sparse neuritic plaques. The post‐mortem delays were between 3 and 115 h (mean = 48.8, SD = 28.1). For some of the analyses, the nondemented cases were stratified according to age‐at‐death into three control groups: <40 years; 40‐60 years; and >60 years (Supporting Information Table S1). The last of these groups was approximately matched in age (mean = 78.5, SD = 7.5) to the AD group (Supporting Information Table S1). Causes of death for all patients included in this study are listed in (Supporting Information Table S2) (controls) and (Supporting Information Table S3) (AD).
Soluble/insoluble Aβ enzyme‐linked immunosorbent assays (ELISAs)
Tissue preparation and ELISA methods used to measure soluble and insoluble Aβ levels in the SWDBB cases (AD and most of the older control) included in this study were reported previously (39). Soluble and guanidine‐HCl extracted (insoluble) fractions were prepared from fresh frozen human brain tissue and measured by sandwich ELISA in which monoclonal anti‐Aβ (4G8 clone, raised against amino acids 18–22; Millipore, Durham, UK) was used for the capture step and biotinylated anti‐human β‐amyloid monoclonal antibody (10H3 clone) (Thermo Fisher Scientific, Northumberland, UK) for the detection step. Frontal neocortex from the SDBTB cases was prepared and assayed following the same protocol.
Oligomeric Aβ1‐42 ELISA
The development and validation of the oligomeric Aβ1‐42 ELISA, and the tissue preparation and measurement of oligomeric Aβ1‐42 levels in the SWDBB subjects included in this study were reported previously (38). A sandwich ELISA was designed in which a rabbit polyclonal pan‐Aβ1‐42 antibody was used for the capture step and monoclonal mouse anti‐oligomeric Aβ antibody (clone 7A1a, New England Rare Reagents, ME, USA) for the detection step. Frontal neocortex (Brodmann area 6) from the SDBTB cases was prepared and assayed following the same protocol.
Western blot and dot blot analyses
Western blot analysis on synthetic Aβ1‐42 peptide (Anaspec, CA, USA) was used to confirm that the anti‐oligomeric Aβ antibody (7A1a) incorporated in the oligomeric Aβ1‐42 ELISA did not detect Aβ1‐42 monomers. Optimal oligomerization conditions had previously been determined and described elsewhere (38); these conditions were used as standard for synthetic Aβ peptide polymerization.
Five microliters of 300 µM synthetic Aβ1‐42 that had either been freshly solubilized or been allowed to oligomerize (overnight incubation at 37°C in phosphate‐buffered saline [PBS]) (32) were diluted to 30 µL in sample buffer (0.5 M tris‐HCl, pH 6.8, 0.25% glycerol, 10% (w/v) sodium dodecyl sulphate (SDS), 0.5% (w/v) bromophenol blue) containing 5% β‐mercaptoethanol. Diluted samples were loaded onto a 4–20% Tris‐HCl pre‐cast gel (Bio‐Rad, Hemel Hempstead, UK) and electrophoresed at 150mV for 1 h. Proteins were subsequently transferred to nitrocellulose membrane overnight at 30 mV. Membranes were blocked for 1 h at room temperature in 10% non‐fat milk in tris‐buffered saline/Tween 20 (TBS‐T; 20 mM tris base, 0.5 M NaCl, 0.05% Tween 20), incubated for 1 h at room temperature in 5% non‐fat milk in TBS‐T with 7A1a antibody (1:250) or 4G8 (1:1000), washed 3 × 10 minutes in TBS‐T and then incubated for 1 h at room temperature with secondary antibody horseradish‐peroxidase (HRP)‐conjugated horse anti‐mouse IgG (1:5000, Vector Laboratories, Burlingame, CA, USA) in 5% non‐fat milk in TBS‐T. The membrane was then washed 3 × 10 minutes in TBS‐T and immunoreactive proteins were detected by enhanced chemiluminescence (GE Healthcare, Piscataway, NJ, USA). In addition, synthetic Aβ that was freshly solubilized, or had been allowed to oligomerize, was sonicated and spun (14,000 g for 15 min) and the supernatant spotted onto a nitrocellulose membrane for 30 minutes for dot blot analysis; the subsequent immunolabelling and detection steps were as described above.
Statistical analysis
Inspection of the ELISA measurements of oligomeric Aβ1‐42 and total insoluble Aβ showed that the distributions were skewed to the right. The data were normalized by logarithmic transformation, and one‐way analysis of variance (ANOVA) with Tukey post hoc testing was used to compare Aβ levels between the three control subgroups (<40 years, 40–60 years and >60 years). Pearson's or Spearman's test was used as appropriate to analyze the correlations between the levels of soluble, oligomeric and insoluble Aβ over the full age range, and to examine the effects of post‐mortem delay. Statistical analyses were performed with the help of Statistical Package for Social Science software (version 12.0.1 for Windows, SPSS Inc., Chicago, IL, USA) and GraphPad Prism 5 (Graphpad Software Inc., La Jolla, CA, USA). Values of P < 0.05 were considered to be statistically significant.
RESULTS
Age‐related decline in soluble Aβ levels in control brains
The level of total soluble Aβ was significantly lower in the >60 years controls than either the <40 years (P < 0.0001) or 40–60 years subgroups (P < 0.001) (Figure 1A). In contrast, total insoluble Aβ level was significantly higher in the older (>60 years) controls than in the <40 years subgroup (P = 0.047) but not the 40–60 years subgroup (Figure 1B). Combining the data from all of the control brains allowed us to examine the correlation between Aβ levels and age from 16 to 95 years. Total soluble Aβ correlated inversely with age (P < 0.0001, r = −0.625) (Figure 1C). In contrast, total insoluble Aβ level increased with age (P = 0.0005, r = 0.437) (Figure 1D).
Figure 1.
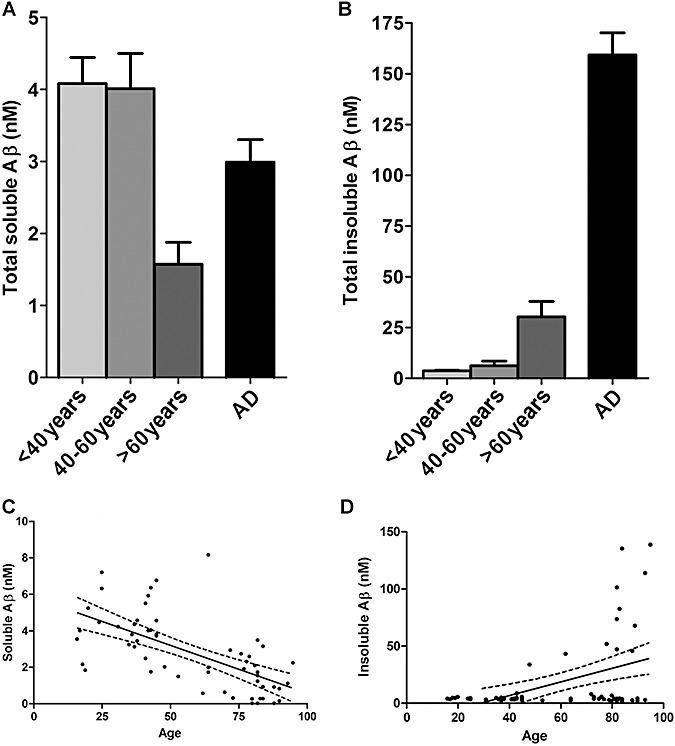
Levels of total soluble amyloid β (Aβ) are highest in younger adults. (A) The level of total soluble Aβ was significantly lower in >60 years controls than in the <40 years (P < 0.0001) or 40–60 years subgroups (P < 0.001). (B) Total insoluble Aβ was significantly higher in >60 years than <40 years (P = 0.047) but not 40–60 years controls. (C) Total soluble Aβ correlated inversely with age (P < 0.0001, r = −0.665). (D) In contrast, total insoluble Aβ levels increased with age (P = 0.0005, r = 0.437). The bars in B and C indicate the mean values ± standard error of the mean. In C and D the individual points represent the mean values for each case. Also shown are the best‐fit linear regression lines (solid) and 95% confidence intervals (interrupted lines).
Total insoluble Aβ level was significantly higher in females than in males (P < 0.01), which is in accordance with our previous findings in older individuals (39). No gender‐dependent significant differences were found for soluble Aβ levels. Within the AD group alone, no correlations were observed between Aβ levels and age.
Relationship between soluble and insoluble forms of Aβ differs in AD and control brains
In control brains the ratio of insoluble : soluble Aβ increased with age (P < 0.0001, r = 0.637) and was higher in >60 years controls than in the <40 years (P = 0.003) or 40–60 years (P = 0.006) subgroups (Figure 2A, B). However, the ratio was much higher still (approximately five‐fold) in AD than in the >60 years controls (P < 0.0001).
Figure 2.
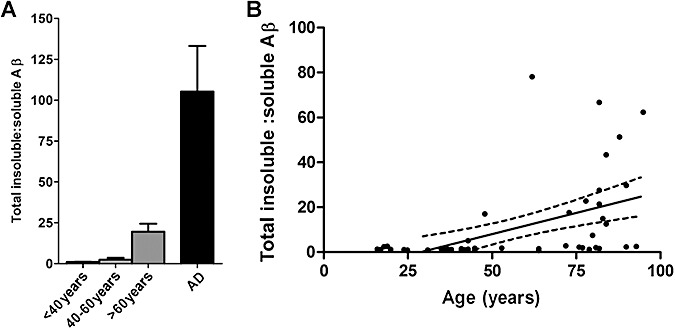
Proportions of soluble and insoluble amyloid β (Aβ) differ in AD and controls. (A) The insoluble : soluble Aβ ratio was significantly higher in the >60 years controls than in the <40 years (P= 0.003) or 40–60 years (P= 0.006) subgroups. The insoluble : soluble ratio was approximately five‐fold higher still in AD brains than the >60 years controls (P < 0.0001). The bars indicate the mean values ± standard error of the mean. (B) In control brains, the ratio of insoluble : soluble Aβ increased with age (P < 0.0001, r = 0.637). The individual points represent the mean values for each case. Also shown are the best‐fit linear regression lines (solid) and 95% confidence intervals (interrupted lines).
No associations were found between total soluble Aβ level and total insoluble Aβ level in the separate control and AD cohorts, or within the study group as a whole (Supporting Information Table S4).
Soluble Aβ levels include oligomeric species
We wished to further examine the composition of the soluble Aβ pool to determine whether this included non‐monomeric Aβ1‐42. Western blot analysis revealed that the smallest Aβ species detected in preparations of either freshly solubilized Aβ1‐42 or peptide that had been allowed to oligomerize overnight had a molecular weight of approximately 8 kDa (Figure 3A). These Aβ species probably represent stable dimers. On dot blot analysis of freshly solubilized or oligomeric Aβ1‐42, 4G8 labelled both forms of Aβ approximately equally but 7A1a labelling of oligomeric Aβ1‐42 was much stronger than that of monomeric Aβ1‐42 (Figure 3B).
Figure 3.

Oligomeric Aβ antibody 7A1a does not detect monomeric Aβ1‐42. (A) The smallest Aβ species detected in preparations of oligomerized synthetic Aβ1‐42 (lane 2) and freshly solubilized peptide (lane 3) had the same molecular weight, of approximately 8 kDa. These Aβ species probably represent stable dimers. The molecular weight markers (kDa) are in lane 1. (B) Freshly solubilized monomeric Aβ1‐42 (sonicated and spun at 14,000 g) and oligomerized Aβ1‐42 probed with pan‐Aβ (4G8) and oligomeric‐specific (7A1a) antibodies reveals that the 7A1a antibody detects negligible levels of monomeric Aβ1‐42.
ELISA measurements revealed that oligomeric Aβ1‐42 level was significantly lower in the >60 years controls than in either of the younger subgroups (<40 years, P < 0.05; 40–60 years, P < 0.01) (Figure 4A) and correlated inversely with age (P < 0.0001, r = −0.515) (Figure 4B). It should be noted that the standards used for the oligomeric Aβ ELISA were supplied by the antibody supplier and differed from those used in the soluble and insoluble Aβ assays; the absolute values are not therefore directly comparable. Oligomeric Aβ1‐42 level correlated directly with soluble Aβ in controls and the combined control and AD cohorts but not AD brains alone (P = 0.009, r = 0.386 in controls; P = 0.016, r = 0.216 in the combined cohorts) (Supporting Information Table S4). In the AD cohort, the level of oligomeric Aβ1‐42 did not correlate with that of insoluble Aβ. In the control and combined cohorts oligomeric Aβ1‐42 level was negatively associated with total insoluble Aβ, although this did not reach significance for the control cohort alone (P = 0.056, r = −0.278 in controls; P = 0.005, r = −0.246 in the combined cohorts) (Supporting Information Table S4).
Figure 4.
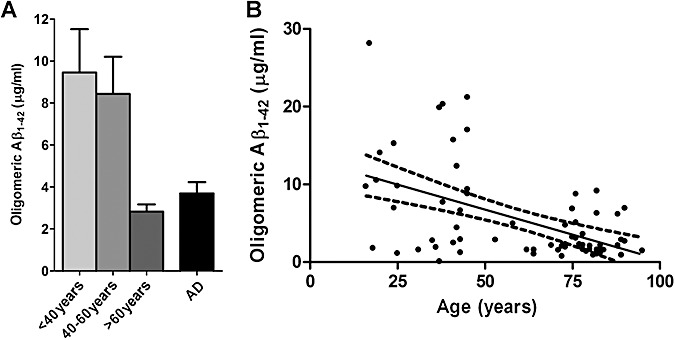
Age‐related decline in level of soluble amyloid β (Aβ) includes oligomeric species. (A) Oligomeric Aβ1‐42 level was significantly lower in the >60 years controls than the other control subgroups (<40 years, P < 0.05; 40–60 years, P < 0.01). The bars indicate the mean values ± standard error of the mean. (B) An inverse correlation was found for oligomeric Aβ1‐42 and age (P = 0.001, r = −0.515). The individual points represent the mean values for each case. Also shown are the best‐fit linear regression lines (solid) and 95% confidence intervals (interrupted lines).
Aβ levels are not affected by post‐mortem delay
No significant correlations were found between levels of soluble (P = 0.637; r = −0.041; n = 136), oligomeric (P = 0.998; r < 0.001; n = 149) or insoluble Aβ (P = 0.051; r = −0.164; n = 142) and post‐mortem interval.
DISCUSSION
Aβ plaques are a neuropathological hallmark of AD, and increase in number with age. In contrast, here we have demonstrated that soluble forms of Aβ are present in highest concentration in younger individuals and decline with normal aging. Furthermore, we have demonstrated that the high Aβ concentrations in young adults are not solely caused by Aβ monomers but that potentially toxic oligomeric Aβ1‐42 species are also present. These unexpected results raise questions about the role of soluble Aβ in neurotoxicity and in normal functioning of the adult brain. We previously reported that case‐to‐case variation in levels of the different forms of Aβ in AD or age‐matched control brains is considerable, and also demonstrated that post‐mortem delay does not contribute to this variability (39).
In contrast to our previous findings on examination of older (>60 years) adults only (39), a relationship between Aβ levels and age was clearly evident across the 16–95 years age range in the present study. Our results demonstrate a shift towards decreased soluble Aβ and increased insoluble fibrillar Aβ with age. The level of soluble Aβ1‐42 was found to decline with age in the cerebrospinal fluid (CSF) (18). The deposition of insoluble Aβ may represent a neuroprotective mechanism for removing toxic oligomers from the interstitial fluid. Alternatively, insoluble Aβ may contribute to the neurodegeneration in AD. On injection of Aβ into rhesus monkeys, the neurotoxicity of the peptide was dependent on its physical form (fibrillar Aβ caused toxic effects whereas soluble Aβ did not) and on the age of the monkeys (toxicity was observed in aged monkeys only) (10). In patients with familial AD or Down's syndrome, soluble Aβ is produced in excess throughout life yet AD‐related cognitive changes and neurofibrillary pathology develop only after the appearance of Aβ plaques 14, 23. The marked increase in the ratio of insoluble : soluble Aβ in AD (much higher than that in age‐matched controls) points to an altered dynamic equilibrium of Aβ in AD. This may reflect enhanced aggregation of Aβ and/or impaired removal of the soluble oligomeric peptide at a stage in life when the level of soluble Aβ would be expected to fall.
The presence of relatively high concentrations of soluble Aβ in younger brains may reflect a physiological role in neural development. Loss of synapses is an early pathological abnormality in AD but is also a developmental process, a means of achieving neural plasticity in the developing and adult nervous system (22). A requirement for Aβ production (or, at least, for β‐ and γ‐secretase activities) during neural development may drive the elevated expression of APP in developing neurons and axons. β‐secretase activity is needed to produce a soluble amino‐terminal fragment of APP (N‐APP), which is a ligand for death receptor 6 (DR6); this fragment of APP initiates an active caspase‐dependent process of axonal pruning and has been reported to have a key role in sculpting the nervous system during development (29). γ‐Secretase activity is required for generation of APP intracellular domain (AICD), which is thought to regulate the transcription of multiple genes (36), including genes important for organization and dynamics of the cytoskeleton (27).
Although a pathological diagnosis of AD requires the presence of Aβ plaques, which consist predominantly of insoluble fibrillar Aβ, Aβ plaque deposition can occur in the absence of cognitive impairment (2). Recent in vivo and in vitro evidence suggests that soluble oligomeric forms of Aβ are the major toxic species in AD 3, 20, 35, 37. Levels of oligomeric Aβ are increased in brain tissue from AD patients 19, 21, 24, 38, 40, and correlate with the degree of cognitive decline 11, 21, 40 and density of neurofibrillary tangles (24). Aβ is produced throughout life, and there is experimental evidence that Aβ oligomers may be present in the brain from an early age and in the absence of AD. In evaluating the neurotoxicity of Aβ*56 in Tg2576(APPSWE) mice, Lesnéet al (20) detected Aβ trimers and hexamers before cognitive decline first became apparent. In humans, Kuo et al (19) demonstrated that both AD and control brains contained a continuous distribution of Aβ species from monomers up to oligomers in excess of 100kDa, with the major contribution coming from low‐n oligomers ranging from dimers to octamers. In addition, high levels of Aβ were measured in the leptomeninges from age‐matched controls as well as AD brains (13); and stable dimers of Aβ42 were identified in the putamen and mamillary bodies from non‐demented subjects aged 24–87 years (28).
Although Aβ oligomers may be produced from early adulthood if not earlier, toxicity may be dependent on the particular types of oligomer that are present. Several lines of evidence support the suggestion that the various oligomeric combinations of Aβ differ in their toxicity, although precisely which species of oligomeric Aβ are most damaging remain unclear. Data from Lesnéet al (20) pointed to Aβ*56 as a major mediator of neurotoxicity, Aβ trimers and hexamers being present in Tg2576 mice before memory impairment could be detected. In Swiss Webster mice, Aβ trimers fully inhibited long‐term potentiation (LTP) whereas dimeric and tetrameric species caused inhibition that was incomplete (37). Soluble Aβ aggregates from AD cases were reported to be more toxic than soluble Aβ aggregates from age‐matched controls (30). More recently, Aβ dimers that had been extracted from AD brains were found to be particularly synaptotoxic (35). Biochemical and immunohistochemical analyses of the composition of Aβ species present in the J20 transgenic AD mouse model revealed the presence of multiple Aβ forms, including aggregated Aβ species, throughout the life of these mice, highlighting the difficulty in identifying the neurotoxic species (34).
It has also been argued that the toxicity of Aβ oligomers is age‐dependent (1). In a Caenorhabditis elegans model, aging impaired detoxification of small aggregates of Aβ1‐42; this was attributed to reduced activity of DAF‐16 and HSF‐1, two components of the insulin/insulin growth factor‐1‐like signaling pathway. There is also evidence that Aβ aggregation can follow either a toxic (“on‐pathway”) or a non‐toxic (“off‐pathway”) route, through competitive reactions that tend with age towards toxic aggregate production 7, 26. It may be relevant to present findings that cognitive decline in healthy educated adults may start as early as the third or fourth decade (33). However, more work is required to characterize the composition of soluble Aβ species during the first few decades of life and to determine whether, if oligomers are indeed produced throughout life, those oligomers present in younger adults differ in toxicity from those present in later life and AD.
A recent study found a strong correlation between soluble oligomeric Aβ1‐42 and detergent‐insoluble Aβ in 54 autopsy brains, in a population‐based series that included both demented and non‐demented elderly (42); from the data presented it is not clear whether or not the relationship between oligomeric and insoluble Aβ differed between AD and control brains. In our series direct correlation between soluble oligomeric Aβ1‐42 and detergent‐insoluble Aβ was restricted to AD with a trend towards an inverse correlation in controls. However, technical differences limit the extent to which data in that and the present study can usefully be compared. Woltjer et al (42) measured the concentration of oligomeric Aβ by means of a Luminex assay in which antibody A11 (17) was used. This antibody was originally reported to recognize micellar but not low molecular weight Aβ (less than approximately 40 kDa). We have previously shown that 7A1a recognises both low and higher molecular weight oligomers of Aβ(38), and provided further evidence here that this antibody does not detect monomeric Aβ. 7Ala was designed to detect α‐helical Aβ conformation and therefore would not detect β‐pleated sheet fibrillar or protofibrillar forms of Aβ.
There are inherent difficulties in obtaining brain tissue from young control subjects. The control cases in the present study included younger individuals with histories of depression and hypertension, and three patients who died after head injury (Supporting Information Table S2). It is possible that those individuals may have been at risk of later development of AD. Although evidence is inconclusive, depression, hypertension and traumatic head injury have all been linked to an increased risk of later AD 4, 6, 12 and our results should be interpreted with a degree of caution. Most studies of the possible relationship between depression or hypertension and AD have been in the elderly. Within an elderly population, depression was reported to be a prodromal manifestation of AD (12). However, other studies found no such association 5, 15. There is epidemiological evidence of a modest association between AD, and depression more than 25 years beforehand (12). Hypertension is a risk factor for vascular disease and dementia, including AD, yet antihypertensive treatment trials have yielded contradictory results (6). Aβ plaques may form within hours of traumatic brain injury, although some evidence suggests that this is probably age‐dependent (31). The level of soluble Aβ1‐42 was elevated in temporal cortex resected from patients (of average age 46.2 years) with severe traumatic brain injury (4); however, the increase in soluble Aβ42 was found only in a small number of patients (n = 6) whose brain tissue contained plaques, whereas no such increase was found in 13 patients whose tissue was plaque‐free.
In conclusion, we have demonstrated that soluble Aβ species are not AD‐specific but are produced throughout life, the levels being highest in younger adults. The precise role of soluble Aβ in the development of AD still needs clarification, as does its possible contribution to normal brain development.
Supporting information
Table S1. Summary of demographic and neuropathological characteristics of study subjects.
Table S2. Causes of death in controls.
Table S3. Cause of death in AD patients.
Table S4. Summary of relationships between Aβ species.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
This work was supported by Alzheimer's Research Trust. We are grateful to the MRC Sudden Death Brain and Tissue Bank, University of Edinburgh, UK, for providing tissue.
REFERENCES
- 1. Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A (2006) Opposing activities protect against age‐onset proteotoxicity. Science 313:1604–1610. [DOI] [PubMed] [Google Scholar]
- 2. Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M et al (1988) Clinico‐pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease. Neurology 38:1682–1687. [DOI] [PubMed] [Google Scholar]
- 3. Dahlgren KN, Manelli AM, Stine WB Jr, Baker LK, Krafft GA, LaDu MJ (2002) Oligomeric and fibrillar species of amyloid‐β peptides differentially affect neuronal viability. J Biol Chem 277:32046–32053. [DOI] [PubMed] [Google Scholar]
- 4. DeKosky ST, Abrahamson EE, Ciallella JR, Paljug WR, Wisniewski SR, Clark RS et al (2007) Association of increased cortical soluble aβ42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol 64:541–544. [DOI] [PubMed] [Google Scholar]
- 5. Dufouil C, Fuhrer R, Dartigues JF, Alperovitch A (1996) Longitudinal analysis of the association between depressive symptomatology and cognitive deterioration. Am J Epidemiol 144:634–641. [DOI] [PubMed] [Google Scholar]
- 6. Duron E, Hanon O (2008) Hypertension, cognitive decline and dementia. Arch Cardiovasc Dis 101:181–189. [DOI] [PubMed] [Google Scholar]
- 7. Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R et al (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off‐pathway oligomers. Nat Struct Mol Biol 15:558–566. [DOI] [PubMed] [Google Scholar]
- 8. Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC (2004) β‐secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol 164:719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C et al (1995) Alzheimer‐type neuropathology in transgenic mice overexpressing V717F β‐amyloid precursor protein. Nature 373:523–527. [DOI] [PubMed] [Google Scholar]
- 10. Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA (1998) Aging renders the brain vulnerable to amyloid β‐protein neurotoxicity. Nat Med 4:827–831. [DOI] [PubMed] [Google Scholar]
- 11. Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE et al (2003) Alzheimer's disease‐affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA 100:10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Green RC, Cupples LA, Kurz A, Auerbach S, Go R, Sadovnick D et al (2003) Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol 60:753–759. [DOI] [PubMed] [Google Scholar]
- 13. Hamano T, Yoshimura M, Yamazaki T, Shinkai Y, Yanagisawa K, Kuriyama M et al (1997) Amyloid β‐protein (Aβ) accumulation in the leptomeninges during aging and in Alzheimer's disease. J Neuropathol Exp Neurol 56:922–932. [DOI] [PubMed] [Google Scholar]
- 14. Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 12:383–388. [DOI] [PubMed] [Google Scholar]
- 15. Henderson AS, Korten AE, Jacomb PA, Mackinnon AJ, Jorm AF, Christensen H et al (1997) The course of depression in the elderly: a longitudinal community‐based study in Australia. Psychol Med 27:119–129. [DOI] [PubMed] [Google Scholar]
- 16. Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S et al (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274:99–102. [DOI] [PubMed] [Google Scholar]
- 17. Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW et al (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489. [DOI] [PubMed] [Google Scholar]
- 18. Kester MI, Blankenstein MA, Bouwman FH, Van Elk EJ, Scheltens P, Van Der Flier WM (2009) CSF biomarkers in Alzheimer's disease and controls: associations with APOE genotype are modified by age. J Alzheimers Dis 16:601–607. [DOI] [PubMed] [Google Scholar]
- 19. Kuo YM, Emmerling MR, Vigo‐Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH et al (1996) Water‐soluble Aβ (N‐40, N‐42) oligomers in normal and Alzheimer disease brains. J Biol Chem 271:4077–4081. [DOI] [PubMed] [Google Scholar]
- 20. Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A et al (2006) A specific amyloid‐β protein assembly in the brain impairs memory. Nature 440:352–357. [DOI] [PubMed] [Google Scholar]
- 21. Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L et al (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155:853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luo L, O'Leary DD (2005) Axon retraction and degeneration in development and disease. Annu Rev Neurosci 28:127–156. [DOI] [PubMed] [Google Scholar]
- 23. Mann DM, Younis N, Stoddart RW, Jones D (1992) The time course of pathological events concerned with plaque formation in Down's syndrome with particular reference to the involvement of microglial cells. Neurodegeneration 1:201–215. [Google Scholar]
- 24. McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K et al (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46:860–866. [DOI] [PubMed] [Google Scholar]
- 25. Morris JC, Heyman A, Mohs RC, Hughes JP, Van Belle G, Fillenbaum G et al (1989) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology 39:1159–1165. [DOI] [PubMed] [Google Scholar]
- 26. Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6:11–22. [DOI] [PubMed] [Google Scholar]
- 27. Müller T, Concannon CG, Ward MW, Walsh CM, Tirniceriu AL, Tribl F et al (2007) Modulation of gene expression and cytoskeletal dynamics by the amyloid precursor protein intracellular domain (AICD). Mol Biol Cell 18:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakabayashi J, Yoshimura M, Morishima‐Kawashima M, Funato H, Miyakawa T, Yamazaki T et al (1998) Amyloid β‐protein (Aβ) accumulation in the putamen and mamillary body during aging and in Alzheimer's disease. J Neuropathol Exp Neurol 57:343–352. [DOI] [PubMed] [Google Scholar]
- 29. Nikolaev A, McLaughlin T, O'Leary DD, Tessier‐Lavigne M (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30. Piccini A, Russo C, Gliozzi A, Relini A, Vitali A, Borghi R et al (2005) β‐amyloid is different in normal aging and in Alzheimer disease. J Biol Chem 280:34186–34192. [DOI] [PubMed] [Google Scholar]
- 31. Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI (1994) B amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry 57:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ryu JK, Franciosi S, Sattayaprasert P, Kim SU, McLarnon JG (2004) Minocycline inhibits neuronal death and glial activation induced by β‐amyloid peptide in rat hippocampus. Glia 48:85–90. [DOI] [PubMed] [Google Scholar]
- 33. Salthouse TA (2009) When does age‐related cognitive decline begin? Neurobiol Aging 30:507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shankar GM, Leissring MA, Adame A, Sun X, Spooner E, Masliah E et al (2009) Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Aβ assembly forms throughout life. Neurobiol Dis 36:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shankar GM, Li S, Mehta TH, Garcia‐Munoz A, Shepardson NE, Smith I et al (2008) Amyloid‐β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14:837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Slomnicki LP, Lesniak W (2008) A putative role of the Amyloid Precursor Protein Intracellular Domain (AICD) in transcription. Acta Neurobiol Exp 68:219–228. [DOI] [PubMed] [Google Scholar]
- 37. Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ (2006) Effects of secreted oligomers of amyloid β‐protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol 572:477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Helmond Z, Heesom K, Love S (2009) Characterisation of two antibodies to oligomeric Aβ and their use in ELISAs on human brain tissue homogenates. J Neurosci Methods 176:206–212. [DOI] [PubMed] [Google Scholar]
- 39. Van Helmond Z, Miners JS, Kehoe PG, Love S (2009) Oligomeric Aβ in Alzheimer's disease: relationship to plaque and tangle pathology, APOE genotype and cerebral amyloid angiopathy. Brain Pathol doi: 10.1111/j.1750‐3639.2009.00321.x [E‐pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang J, Dickson DW, Trojanowski JQ, Lee VM (1999) The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol 158:328–337. [DOI] [PubMed] [Google Scholar]
- 41. Wisniewski HM, Ghetti B, Terry RD (1973) Neuritic (senile) plaques and filamentous changes in aged rhesus monkeys. J Neuropathol Exp Neurol 32:566–584. [DOI] [PubMed] [Google Scholar]
- 42. Woltjer RL, Sonnen JA, Sokal I, Rung LG, Yang W, Kjerulf JD et al (2008) Quantitation and Mapping of Cerebral Detergent‐Insoluble Proteins in the Elderly. Brain Pathol 19:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of demographic and neuropathological characteristics of study subjects.
Table S2. Causes of death in controls.
Table S3. Cause of death in AD patients.
Table S4. Summary of relationships between Aβ species.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item