

CLINICAL HISTORY
The patient was a 10‐year‐old African‐American female with a 1 year history of intermittent pain when she turned her neck, as well as pain in her shoulder and back. She also had a persistent rightward head tilt and progressive right hand weakness with paresthesias, as well as difficulty with ambulation secondary to progressive leg weakness. A full physical and neurological examination was significant for profound right hand weakness and sensory impairment, and bilateral lower extremity weakness and proprioceptive impairment.
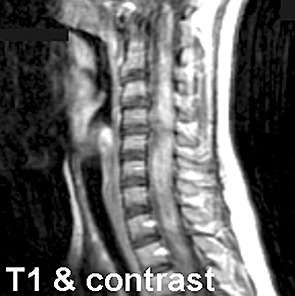
Magnetic resonance imaging showed widening of the cervical spine with hypointensity on T1 (Fig. 1), increased intradural intramedullary T2 signal (Fig. 2), and contrast enhancement (Fig. 3). The abnormal T2 signal extended from the craniocervical junction to T5, and the abnormal enhancement extended from the craniocervical junction to T3. No abnormalities were seen in the remainder of the brain.
Figure 1.
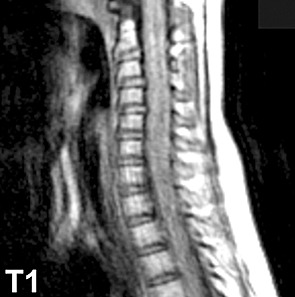
Figure 2.
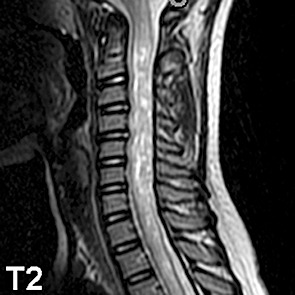
Figure 3.
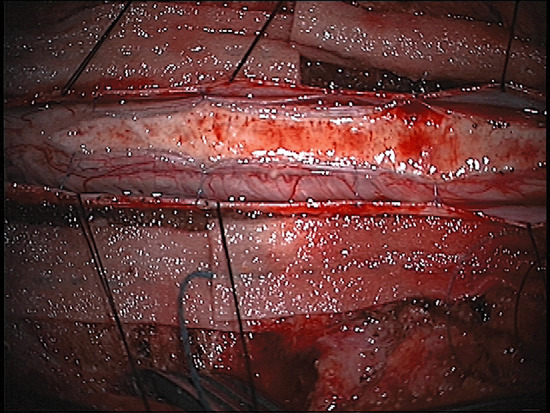
Corticosteroid therapy with dexamethasone was instituted, although the patient showed no improvement in either her symptoms or clinical findings. The spinal cord was thus decompressed via a C1 laminectomy and C2 through T3 osteoplastic laminotomy was then performed. The dura was tense from the underlying mass, which upon exposure was found to be brownish gray and well demarcated from the surrounding spinal cord, splaying the posterior columns laterally on each side (Fig. 4). A distinct plane between the mass and the surrounding cord was apparent circumferentially. Gross total resection was achieved, confirmed by ultrasound guidance during the resection.
Figure 4.
Postoperatively, the patient had only transient left upper extremity paresis and recovered extremely well, regaining full strength in both upper extremities, with resolution of her preoperative symptoms and neurological deficits. The patient was transferred to an inpatient rehabilitation facility on postoperative day 6. Her impressive recovery persisted, with no evidence of lesion recurrence on a follow‐up MRI obtained 2 months after surgery.
PATHOLOGICAL FINDINGS
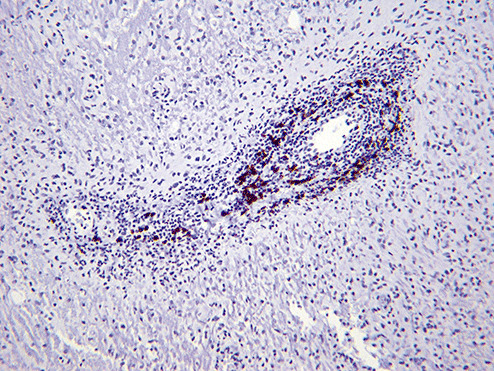
Sections of the excised lesion showed oblique profiles of abnormal spinal cord elements with hypercellularity in the parenchymal and perivascular regions (5, 6, 7). An LFB‐PAS stain (Fig. 8) showed sharply demarcated zones of myelinated and demyelinated axon fascicles. NF1 immunostain (Fig. 9) confirmed that the long processes were axons, and were focally swollen and dystrophic. GFAP immunostain showed widespread reactive piloid astrocytic processes and occasional gemistocytic reactive astrocytes in the parenchyma. CD68 immunostain highlighted columns of foamy macrophages interdigitating between the dystrophic and focally swollen neurofilament immunopositive axons. There were perivascular cuffs composed predominantly of CD3 positive T‐lymphocytes (Fig. 10) with some CD20 positive B‐cells (Fig. 11). What is the diagnosis?
Figure 5.
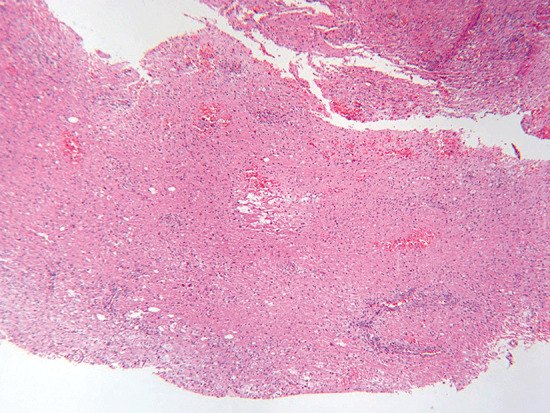
Figure 6.
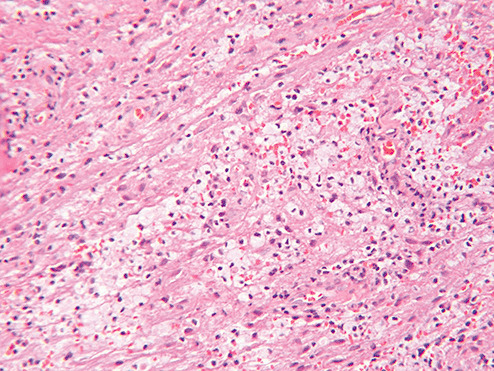
Figure 7.
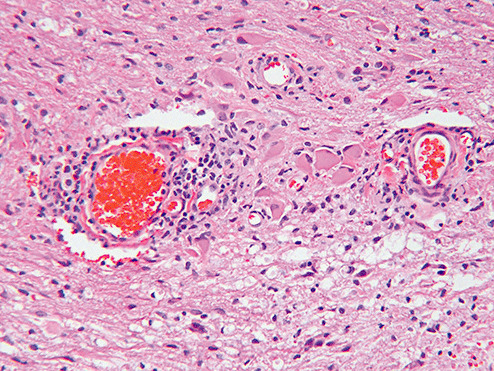
Figure 8.
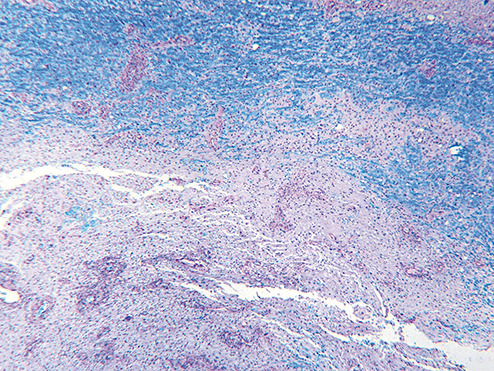
Figure 9.
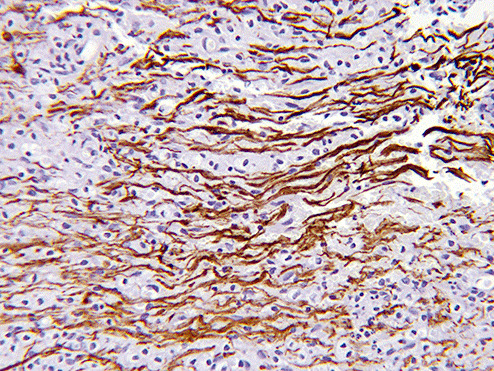
Figure 10.
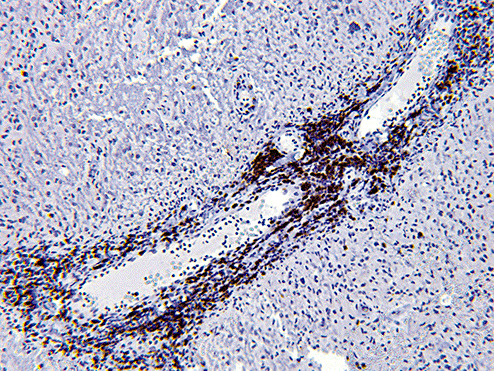
Figure 11.
FINAL DIAGNOSIS: NEUROMYELITIS OPTICA (NMO)
An extensive work‐up was conducted for potential etiologies of an inflammatory pseudotumor. A full serologic panel was positive only for anti‐aquaporin IgG antibody, consistent with the NMO spectrum of disorders. On further questioning, the family recalled that the patient had been evaluated several years ago for a “bad eye.” Ophthalmological examination confirmed visual impairment in the right eye.
NMO was once considered an aggressive variant of multiple sclerosis (MS), but with some differences (4). For example, unlike MS, NMO is relatively common in sub‐Saharan Africa, Asia, India, and the Caribbean, and interferon‐beta is not an effective therapy (6). Furthermore, detection of anti‐aquaporin IgG (a.k.a. NMO‐IgG) has been shown to be >90% specific and 75% sensitive for NMO 1, 3. These findings led to the modified NMO diagnostic criteria, which included optic neuritis and acute myelitis as major criteria plus either a spinal MRI lesion longer than 3 vertebral segments or the presence of anti‐aquaporin‐4 (AQP4) antibody (7). The mean age of NMO is around 40 years, but it is known to occur in children. Relapses with severe sequelae have been reported, especially if the patient is NMO‐IgG positive at initial presentation (1). Other risk factors for a relapsing clinical course include female gender and severe motor weakness at presentation. Each recurrent attack during the first 2 years of disease increases risk of early death (8).
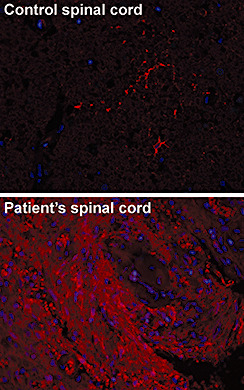
Long‐standing NMO lesions typically show reduced AQP4 immunostaining as do ischemic lesions and inactive MS plaques (5). Fluorescent microscopy showed increased AQP4 immunoreactivity in our case (Fig. 12, red). This increased staining may be due to the acute phase of the disease. Aquaporin channels increase plasma membrane osmotic permeability by facilitating the bidirectional movement of water. AQP4 is the main aquaphorin subtype found in the brain and is primarily expressed at key parenchymal‐fluid interfaces (2) Proposed pathologic mechanisms of action by the anti‐AQP4 antibody include endocytosis and degradation of membrane‐bound AQP4, disrupting the blood brain barrier, destroying AQP4‐producing astrocytes via complement fixation, and/or by indirect killing of nearby oligodendroglial cells via glutamate excitotoxicity (go to http://path.upmc.edu/divisions/neuropath/bpath/cases/case201/dx.html for references).
Figure 12.
POSTSCRIPT
The patient was treated with low‐dose oral corticosteroids and azathioprine but she had relapses at 4, 7, and 11 months, requiring high‐dose IV Solu‐Medrol each time. Plasmapheresis during the second relapse produced temporary remission. A fourth minor relapse 15 months after diagnosis prompted replacement of azathioprine with mycophenolate. While the patient has not yet demonstrated additional deficits, she has developed side effects to the steroid therapy, including Cushingoid features and diabetes.
ABSTRACT
Neuromyelitis optica (NMO), (Devic's disease), is a relatively uncommon autoimmune disease predominantly involving the spinal cord and optic nerves. We present a 10 year‐old female with intermittent neck pain, progressive right upper and bilateral lower extremity weakness. MR imaging and intraoperative findings were strongly suggestive of a neoplastic process. However, pathologic examination showed an inflammatory demyelinating lesion and serological studies were positive for NMO‐IgG. The patient improved dramatically following resection of the compressive “pseudotumor”, with resolution of her preoperative deficits. This case underscores the diverse clinical presentation of neuromyelitis optica and the importance of maintaining a broad differential diagnosis in pediatric lesions resembling neoplasms.
Affiliations: CH was supported by a Callie Rohr American Brain Tumor Association Fellowship
REFERENCES
- 1. Banwell B, Tenembaum S, Lennon VA, Ursell E, Kennedy J, Bar‐Or A, Weinshenker BG, Lucchinetti CF, Pittock SJ (2008) Neuromyelitis optica‐IgG in childhood inflammatory demyelinating CNS disorders. Neurology 70(5):344–52. [DOI] [PubMed] [Google Scholar]
- 2. Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, Lennon VA (2007) Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 69(24):2221–31. [DOI] [PubMed] [Google Scholar]
- 3. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364(9451):2106–12. [DOI] [PubMed] [Google Scholar]
- 4. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, et al. (2002) A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 125(Pt 7):1450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, et al (2007) Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 130(Pt 5):1224–34. [DOI] [PubMed] [Google Scholar]
- 6. Papeix C, Vidal JS, De Seze J, Pierrot‐Deseilligny C, Tourbah A, Stankoff B, et al. (2007) Immuno‐suppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler 13(2):256–9. [DOI] [PubMed] [Google Scholar]
- 7. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66(10):1485–9. [DOI] [PubMed] [Google Scholar]
- 8. Wingerchuk DM, Weinshenker BG (2003) Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology 60(5):848–53. [DOI] [PubMed] [Google Scholar]