

CLINICAL HISTORY
A 22‐year‐old girl presented with convulsive status epilepticus and a previous history of recurrent seizures, myoclonus, ataxia and impaired cognitive functions. Testing revealed severe metabolic acidosis, elevated transaminases and creatine kinase, and respiratory insufficiency. After intubation and ventilation, thiopental was introduced but the patient's condition worsened dramatically with death after a few hours. The parents gave permission for autopsy. Born of healthy unrelated parents the patient had developed normally until the age of 16 years, when she suffered tonic‐clonic seizures and visual hallucinations. Some months prior to seizure onset, her parents had noted erratic jerks of the upper limbs, personality changes, and decline in school performance. An electroencephalographic (EEG) recording had shown spikes over the occipital regions and photosensitivity. Therapy with valproate had decreased the frequency of seizures. However, progressive worsening of cognitive and motor functions occurred within a few months. In addition, sudden and brief episodes of loss of contact were reported. Neurological examination had shown ataxia due to severe myoclonus at rest and action‐induced myoclonus, pyramidal signs, and opposition hypertonia. She could not tandem walk, and deep tendon reflexes were barely detectable. EEG showed diffusely slow background activity with superimposed generalized, high‐ voltage, paroxysmal abnormalities. Brain MRI displayed diffuse cerebral and cerebellar atrophy. Neuropsychological testing demonstrated a full‐scale IQ of 70 (verbal 75; performance 60). Adjunctive therapy with clonazepam and zonisamide significantly reduced the intensity of myoclonus. In the following years, mental decline continued unabated resulting in severe dementia, disorientation, akinesia, marked rigidity with opposition hypertonia, and inability to feed, which was her clinical state prior to her admission for status epilepticus and death.
MICROSCOPIC AND ULTRASTRUCTURAL FINDINGS
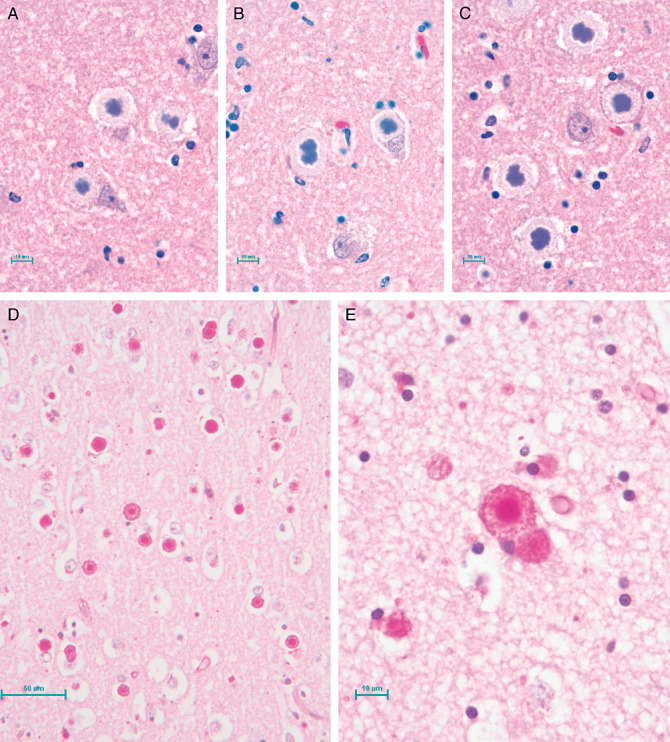
Histochemical analysis revealed that the CNS was diffusely replete with Intracellular inclusions (1, 2) which were highlighted by positive periodic acid–Schiff (PAS) staining (figure 2d e). The inclusions were most plentiful in the thalamus, cerebellum, and brainstem, and in lesser numbers in the frontal and occipital cortices. The inclusions were absent in subcortical white matter but were abundant in the spinal cord gray matter. Two types of inclusions could be distinguished: type I, punctate structures present in neuronal processes (Fig 2d;bar = 50 µm, and type II, large structures found in neuronal cell bodies usually apposed to the nucleus and often so large as to occupy the entire soma (Figure 2e; bar = 10 µm). All sections were ubiquitin‐negative.
Figure 1.
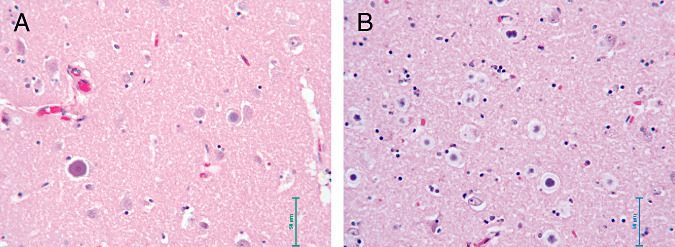
Figure 2.
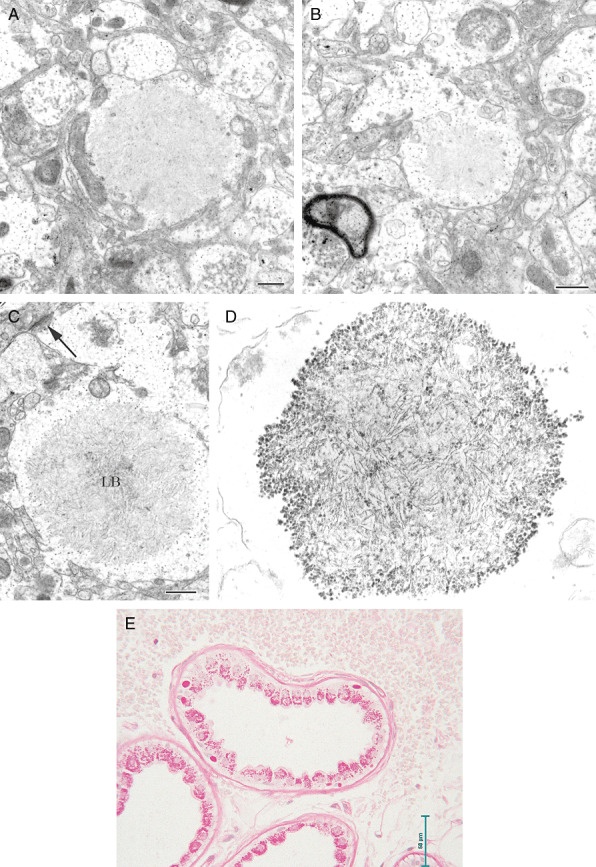
Electron microscopy showed that the inclusions consisted of accumulations of a fibrillar material in the dendrites but not in the axon terminals, recognized through their synaptic vesicles (SV) (Figures 3a, 3b; bar = 500 nm). Additional ultrastructural images show the filamentous nature (Figures 3c, 3d) In addition, the same inclusions were present in clinically unaffected organs too, especially skeletal and cardiac muscle, and the liver. In the apocrine sweat gland of skin (Fig 3e; bar = 50 µm) pathological inclusions (round bodies at base of cell) were clearly distinguishable from normal PAS‐positive, diastase‐resistant secretory material of the gland (granular material on luminal side).
Figure 3.
DIAGNOSIS
Progressive myoclonus epilepsy Lafora‐type (LD, OMIM# 254780), confirmed also by genetic testing, revealing a homozygous missense mutation (c.205C > G, P69A) in the EPM2B (NHLRC1) gene.
DISCUSSION
LD is the most common and severe form of adolescent‐onset progressive myoclonus epilepsies (PMEs), a group of devastating inherited neurodegenerative disorders characterized by progressively worsening myoclonus, epilepsy, early dementia and death (5). LD, first described by Lafora in 1911, is particularly frequent in Mediterranean countries (Spain, Italy, France), Northern Africa, the Middle East, and in some regions of Southern India where a high rate of consanguinity is present 3, 5. LD classically starts in adolescence in otherwise neurologically normal individuals, usually with action and stimulus‐sensitive myoclonus as well as tonic–clonic, absence, atonic, and visual seizures. Neuropsychiatric symptoms such as behavioral changes, depression, apathy, are also often present. Initial symptoms are followed by rapidly progressive dementia, refractory status epilepticus, psychosis, cerebellar ataxia, dysarthria, mutism, and respiratory failure which lead to death within about a decade 3, 5.
The main differential diagnosis is with four other forms of PMEs: Unverricht‐Lundborg disease (EPM1), the neuronal ceroid lipofuscinoses, myoclonic epilepsy with ragged red fibers (MERRF), and sialidosis 3, 5. The age of onset, presenting symptoms, occurrence of occipital seizures and the progressive and rapid course, together with the EEG features suggest LD, until the diagnosis is definitively confirmed by skin biopsy or genetic analysis. The pathologic hallmark of LD is the presence of the typical PAS‐positive intraneuronal inclusions (Lafora bodies, LBs). LBs are also found in other tissues such as heart, liver, muscle and skin composed of an abnormal form of glycogen 5, 6. Importantly, neuronal LB exclusively localize in perikarya and dendrites but not in axons. LB may be conveniently identified in the eccrine ducts or apocrine myoepithelia of sweat glands obtained from skin biopsy (1). LD is caused by mutations in the EPM2A (4) or the EPM2B (NHLRC1) (2) genes, encoding the laforin dual specificity protein phosphatase and the malin E3 ubiquitin ligase respectively, both involved in a complex poorly understood pathway regulating glycogen metabolism. At present, LD treatment remains palliative.
ABSTRACT
A 22‐year‐old girl presented with convulsive status epilepticus and a previous history of recurrent seizures, myoclonus, ataxia and impaired cognitive functions. Neurological examination revealed rest and action‐induced myoclonus, pyramidal signs and opposition hypertonia. Testing revealed severe metabolic acidosis, elevated transaminases and creatine kinase, and respiratory insufficiency. After intubation and ventilation, thiopental was introduced but the patient's condition worsened dramatically with death in a few hours. Autopsy showed profuse periodic acid–Schiff (PAS) positive intracellular inclusions in the CNS (Lafora bodies), most abundant in thalamus, cerebellum, and brainstem, as well as in other organs. Genetic testing revealed a homozygous missense mutation (c.205C > G, P69A) in the EPM2B (NHLRC1) gene, confirming the diagnosis of progressive myoclonic epilepsy Lafora‐type.
REFERENCES
- 1. Andrade DM, Ackerley CA, Minett TS, Teive HA, Bohlega S, Scherer SW, Minassian BA (2003) Skin biopsy in Lafora disease: genotype‐phenotype correlations and diagnostic pitfalls. Neurology 61:1611–4. [DOI] [PubMed] [Google Scholar]
- 2. Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, Avanzini G, Elia M, Ackerley CA, Jovic NJ, Bohlega S, Andermann E, Rouleau GA, Delgado‐Escueta AV, Minassian BA, Scherer SW (2003) Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet 35:125–127. [DOI] [PubMed] [Google Scholar]
- 3. Ganesh S, Puri R, Singh S, Mittal S, Dubey D (2006) Recent advances in the molecular basis of Lafora's progressive myoclonus epilepsy. J Hum Genet 51:1–8. [DOI] [PubMed] [Google Scholar]
- 4. Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, Dunham I, Gardner R, Fong CY, Carpenter S, Jardim L, Satishchandra P, Andermann E, Snead OC, 3rd , Lopes‐Cendes I, Tsui LC, Delgado‐Escueta AV, Rouleau GA, Scherer SW (1998) Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet 20:171–174. [DOI] [PubMed] [Google Scholar]
- 5. Minassian BA (2001) Lafora's disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol 25:21–29. [DOI] [PubMed] [Google Scholar]
- 6. Van Heycop Ten Ham MW (1975) Lafora disease, a form of progressive myoclonus epilepsy. In The Epilepsies. Handbook of Clinical Neurology, vol. 15, 382–422 (Eds. Vinken PJ and Bruyn GW), North‐Holland, Amsterdam. [Google Scholar]