

Abstract
X‐adrenoleukodystrophy (X‐ALD) is a metabolic, peroxisomal disease affecting the nervous system, adrenal cortex and testis resulting from inactivating mutations in ABCD1 gene which encodes a peroxisomal membrane half‐adenosine triphosphate (ATP)‐binding cassette transporter, ABCD1 (or ALDP), whose defect is associated with impaired peroxisomal β‐oxidation and accumulation of saturated very long‐chain fatty acids (VLCFA) in tissues and body fluids. Several phenotypes are recognized in male patients including cerebral ALD in childhood, adolescence or adulthood, adrenomyeloneuropathy (AMN), Addison's disease and, eventually, gonadal insufficiency. Female carriers might present with mild to severe myeloneuropathy that resembles AMN. There is a lack of phenotype–genotype correlations, as the same ABCD1 gene mutation may be associated with different phenotypes in the same family, suggesting that genetic, epigenetic, environmental and stochastic factors are probably contributory to the development and course of the disease. Degenerative changes, like those seen in pure AMN without cerebral demyelination, are characterized by loss of axons and secondary myelin in the long tracts of the spinal cord, possibly related to the impaired lipid metabolism of VLCFAs and the associated alterations (ie, oxidative damage). Similar lesions are encountered following inactivation of ABCD1 in mice (ABCD1 ‐). A different and more aggressive phenotype is secondary to cerebral demyelination, very often accompanied by inflammatory changes in the white matter of the brain and associated with activation of T lymphocytes, CD1 presentation and increased levels of cytokines, γ‐interferon, interleukin (IL)‐1α, IL‐2 and IL‐6, Granulocyte macrophage colony‐stimulating factor (GM‐CSF), tumor necrosis factor‐α, chemokines and chemokine receptors.
Keywords: adrenoleukodystrophy, adrenomyeloneuropathy, autoimmunity, gene therapy, hematopoietic stem cell transplantation, neurometabolic disease, peroxisome, very long‐chain fatty acids, X‐ALD
INTRODUCTION
X‐adrenoleukodystrophy (X‐ALD, ALD, [Mendelian inheritance in Man (MIM) 300100]) is a metabolic, peroxisomal monogenic disease affecting the nervous system, adrenal cortex and testis resulting from inactivating mutations in ABCD1 gene located in chromosome Xq28. ABCD1 encodes a peroxisomal membrane half‐ATP‐binding cassette (ABC) transporter, ABCD1 (or ALDP), whose defect is related to abnormal peroxisomal β‐oxidation and accumulation of saturated very long‐chain fatty acids (VLCFA) in tissues and body fluids 2, 7, 9, 35, 97, 99, 107, 147.
The estimated frequency is about 1:17 000 in the USA (13) and 1:20 000 in France (99), whereas the estimated frequency for heterozygous women is 1:14 000 (13).
X‐ALD is a very enigmatic disorder as several phenotypes might occur in male patients within the same family: cerebral childhood adrenoleukodystrophy (cALD), cerebral juvenile, cerebral adult form, adrenomyeloneuropathy (AMN) and isolated Addison's disease. Female carriers present frequently with moderate to severe myelopathy, usually without cerebral involvement or adrenal insufficiency (96). Frontotemporal dementia is a common presentation of cerebral ALD in adults 75, 90, 137. Importantly, the different phenotypes do not depend on the type of the mutation (10). In contrast, clinical symptoms are closely related with neuropathological features, either inflammatory or degenerative or combined. Moreover, members of the same family may suffer from different phenotypes, and a phenotype can be modified in a particular individual during his life. Different clinical phenotypes have been reported in monozygotic twins (156), suggesting that genetic, epigenetic, environmental and stochastic factors are probably contributory to the development and course of the disease 1, 55, 76, 130.
The history of the discoveries related with X‐ALD is fascinating in its complexity, and it is a good example of endeavor, tremendously rigorous work and serendipity in science. It is worth reading the experiences of major contributors in this field to have an idea of the evolution from what was formerly called Schilder's disease, a fatal inflammatory leukodystrophy of childhood probably due to different causes, to the present concept of one of the more common phenotypes of X‐ALD encompassing a broader conception of the disease 97, 108. Several aspects are still obscure, such as the relationship between the inactivation of a peroxisomal ALDP protein and the abnormal metabolism of lipids, the causes and effects of the severe inflammatory response in most cases.
In parallel with these events, several strategies have emerged aimed at finding a cure or alleviation of the disease. Importantly, efforts in the field of allogeneic hematopoietic cell transplant have yielded significant help to a subgroup of patients at the very early stages of cALD. Also, more than 15 adults with cerebral ALD have successfully been transplanted in Germany and France and have encouraged new approaches to increase understanding about the pathogenesis of the disorder and the rational use of therapeutic tools.
Here, we will review some aspects of the neuropathology of the most common forms of X‐ALD and possible mechanisms involved in their molecular neuropathogenesis. We will also summarize the principal therapeutic approaches. Excellent studies and reviews of adrenal gland and testicular pathology in X‐ALD can be found elsewhere 112, 113, 115, 116, 117.
GENETICS
Adrenoleukodystrophy was first linked to the chromosome Xq28 and later demonstrated to be caused by mutations in the ABCD1 gene 88, 102, 103. ABCD1 contains 10 exons and encodes for an mRNA of 4.3 kb that translates a protein of 745 amino acids.
More than 460 different ALD mutations have been reported including missense, frameshift, nonsense and large deletions (X‐linked Adrenoleukodystrophy Database: http://www.x‐ald.nl). Missense mutations account for more than 50% of the total (67).
A closely related gene is ABCD2 in chromosome 12q1 that codes for an ALD‐related protein (ALDRP) with high sequence homology and overlapping functions to ABCD1 41, 45, 59, 60, 118, 125. Although X‐ALD phenotype is independent of ABCD2 genotype (81), exploiting the functional redundancy of these transporters might have some therapeutic implications (see below).
BIOCHEMISTRY
As detailed elsewhere (69), the principal and pathognomonic biochemical alteration in X‐ALD is the accumulation of VLCFA, especially hexacosanoic (C26:0) and tetracosanoic (C24:0) in brain and other tissues and body fluids (63). VLCFA content in X‐ALD is increased in cholesterol esters and complex lipids such as gangliosides, phosphatidylcholine, sphingomyelin, cerebrosides and sulfatides. A main component of myelin sheaths, the proteolipid protein of myelin, PLP, is enriched in VLCFA in X‐ALD 14, 48, 63, 97, 138.
ABCD1 encodes an ABC transporter that harbors a putative lipid‐binding domain, belonging to the lipid transporters subclass. Of 48 transcriptionally active ABC transporters, at least 13 are expressed in the brain and participate in brain lipid transport across membranes and in homeostasis. Three of the ABCD transporters are components of peroxisomal membranes; ABCD1 is known as adrenoleukodystrophy protein, ABCD2 is the ALDRP, ABCD3 is named peroxisomal membrane protein (PMP70). For the fourth member of the ABCD transporters, ABCD4 is also known as 69 kDa PMP (PMP69) 71, 155. A recent study doubted the peroxisomal localization (66). The tree peroxisomal ABCDs may work as homo‐ or heterodimers, demonstrated in vivo and in vitro respectively, and ABCD1 might transport acyl coenzyme A esters across the peroxisomal membrane 57, 68, 78, 151. This transport is energy dependent (123).
NEUROPATHOLOGY
X‐linked adrenoleukodystrophy, childhood cerebral form (cALD)
Postmortem studies in childhood cerebral ALD show that the cortex is grossly intact but that myelin is replaced by gray‐to‐brown tissue (128).The neuropathology of cALD is characterized by the progressive degeneration of the cerebral white matter, with demyelinating lesions that are confluent, generally symmetric and show either a caudorostral progression starting from initial parieto‐occipital involvement (65%) or a rostrocaudal progression starting frontally (35%). The corpus callosum, optic pathways and posterior limbs of the internal capsules are characteristically involved in the occipital forms, the arcuate U fibers being often spared. In this parieto‐occipital forms of cALD, demyelination usually starts in the splenium of corpus callosum and then extend into the parieto‐occipital white matter. In the rostrocaudal progressing forms, demyelination either starts in the genu of corpus callosum and then extends, or it starts in pyramidal tracts in the internal capsules and extends to the centrum ovale (frontal), progressing to the anterior parts of the brain. The white matter of the cerebellum is also affected, although less extensively than the white matter of the cerebrum.
System atrophy affects the pyramidal tracts in the brain stem and the spinal cord is seen at the later stage of the disease. In contrast to the severe involvement of the white matter, the cerebral cortex, cerebellar cortex and deep cerebellar nuclei, and grey matter of the brain stem are largely spared (Figure 1).
Figure 1.
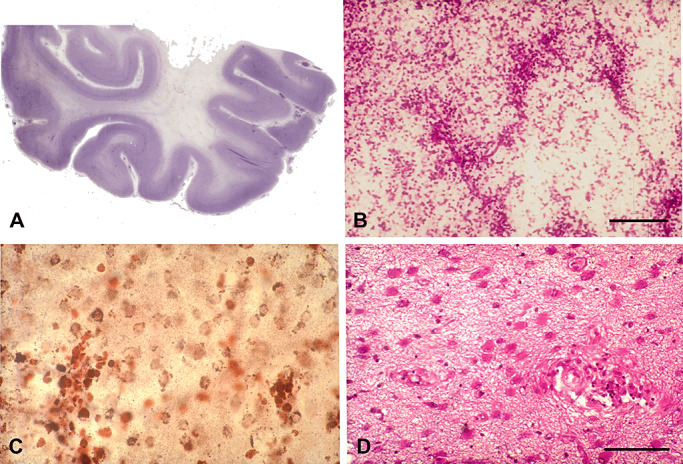
X‐ALD, childhood cerebral form (cALD). A. Loss of myelin and axons in the white matter of the temporal lobe. B. Inflammatory infiltrates in the white matter at the edge of the demyelinating lesion. C. Infiltrating reactive CD68+ cells in the intermediate zone between the edge of demyelination and the demyelinated area. D. Hypertrophic astrocytes in the demyelinated area. Paraffin sections: A, B, D. hematoxylin and eosin; C. CD68 immunohistochemistry. B, bar = 200 µm; C, D, bar = 50 µm.
Three histopathological zones with a spatiotemporal sequence can be defined in the demyelinating lesions of cerebral ALD (128). The first zone or peripheral edge, in which destruction of myelin occurs initially, contains scattered Periodic Acid Schiff (PAS)‐positive and sudanophilic macrophages, but axons are spared. It is closely followed by a second zone in which there are many lipid‐laden macrophages with lamellar inclusions and some surviving myelinated axons. The third zone in the center of the lesion consists of a dense mesh of glial fibrils with absence of oligodendroglia, myelin sheaths and very often axons. Cavitated areas and calcium deposits can be present in the third zone, reflecting usually an advanced state of the lesion, although it can also be a relative early event based on magnetic resonance imaging (MRI), reflecting that a long initial non‐inflammatory period, usually asymptomatic, that entered an active phase of neuroinflammation becoming symptomatic. Very recently, beyond the leading edge of peripheral demyelination, a zone lacking microglia has been described (40).
Myelin and axon loss is accompanied by perivascular and infiltrating macrophages containing PAS‐positive granular and lamellar inclusions which are more abundant at the sites of active degeneration than in older degenerating areas. Infiltration and perivascular accumulation of macrophages and mononuclear cells, mostly lymphocytes, is seen between the first and the second zone. This infiltrate resembles that observed in multiple sclerosis (MS) and experimental allergic encephalomyelitis (EAE). The perivascular cuffs in ALD disclose a T‐ and B‐cell response typical of a cellular immune response, such as in active MS or EAE 12, 53. In contrast to MS or EAE, the inflammation in ALD tends to be found behind the active edge of demyelination. The B‐cell component of the perivascular cuff is also not prominent, although in view of novel findings, the participation of B lymphocytes might have been underestimated.
Myelin and axon stains reveal loss of myelin, myelin varicosities, loss of axons and occasional small axonal enlargements containing Amyloid precursor protein (APP) and synaptophysin, suggesting impaired axonal transport. A few macrophages may also be seen in apparently spared zones of the white matter, indicating early degeneration of myelin and axons in these zones. Reactive microglia is present in active demyelinating lesions with infiltration of B and T lymphocytes. Loss of oligodendrocytes and reactive astrocytes is found in the demyelinated areas of the white matter (Figure 1). Little is known, however, about the mechanism of oligodendroglial death.
A most striking change is the presence of inflammatory infiltrates at the border of the lesions of the white matter, just behind the active demyelinating front (Figure 1). A few inflammatory infiltrates can also be encountered in the deep demyelinated areas. Inflammatory cells are CD4+ and CD8+ T lymphocytes, some of them containing granzyme B, and rare B lymphocytes 53, 62, 64, 111, 122. Plasma cells are rarely encountered. Importantly, CD8+ lymphocytes are seen in white matter areas distant from the lesions (64).
Ocular anomalies have been reported in cALD. These consist of primary degeneration of the photoreceptors and optic pathways 23, 50, 139.
In spite of a biochemically abnormal myelin, with excess of fatty acids as components of complex lipids and proteins, X‐ALD cannot be considered a dismyelinating disease, as is the case for other leukodystrophies such as Pelizaeus‐Merzbacher disease. The ultrastructural aspect of myelin sheaths is strictly normal during development and adulthood in AMN patients, and the onset of disease is most frequently in adulthood, years after myelination has ended.
Although targeted on myelin, primary axon damage in cALD cannot be ruled out on the basis of our current knowledge (39).
AMN
AMN was first recognized in patients with progressive spastic paraparesis and corticoadrenal insufficiency, due to predominant spinal cord and peripheral nerve involvement together with specific changes in the cortex of the adrenal gland 18, 54, 126, 127. Yet not all patients develop adrenal insufficiency, whereas nearly half of them show abnormalities consistent with variable demyelination in the cerebral white matter. About one‐third of them develop a rapid progressive cerebral form similar to that seen in cALD form 110, 148.
The neuropathology of AMN is characterized by loss of axons in the long tracts of the spinal cord, mainly dorsal fascicles and pyramidal tracts, and secondary loss of myelin. There is no accompanying infiltration of hematogenous mononuclear cells, with inflammatory changes restricted to activated microglia. Macrophages filled with granular or lamellar inclusions are extremely rare in the pure AMN forms with only spinal cord (and brain stem) involvement. Therefore, the typical lesions in the spinal cord in AMN are manifested as a dying‐back myelopathy. Spinal motor neurons do not appear to be grossly affected, whereas neurons in the dorsal ganglia, although apparently preserved in number, show atrophy and abnormal lipid inclusions in mitochondria (109).
Inflammatory and non‐inflammatory features are combined in AMN with cerebral involvement (110), representing a double pathology dependent on separate pathogenic mechanisms. Cerebral lesions in AMN with cerebral involvement are similar to those seen in cALD, as are the symptoms and prognosis, with only a more protracted course initially.
Peripheral neuropathy is a common feature in AMN although usually surpassed by spinal cord symptoms. As in the spinal cord, the primary pathology in peripheral nerves appears to be axonopathy 22, 32, 110, 149, but electrophysiology contradicts often this affirmation; while some patients have electrophysiological signs of axonal neuropathy, others suffer of real demyelinating neuropathy. Trilammelar inclusions are also observed in Schwann cells. Optic nerve atrophy, manifested as impaired visual function, optic disk pallor and demyelination of the optic nerves, as seen by neuroimaging methods, may occur in AMN (51).
Adult cerebral X‐ALD
Pure adult cerebral X‐ALD is a rare condition, although symptomatic cerebral involvement occurs in a significant percentage of cases with AMN 96, 110, 148. Pure cerebral forms in adulthood are manifested initially with subacute cognitive decline. Neuroimaging and analysis of VLCFA in serum may help to orient the diagnosis, the indication of genetic study and the genetic counseling for other family members (31). Typical cerebral lesions in adult‐onset cerebral X‐ALD are shown in 2, 3.
Figure 2.
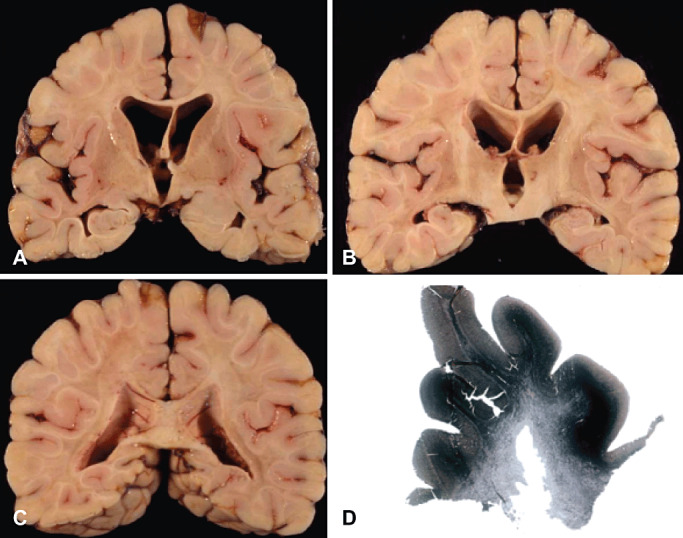
Pure adult cerebral X‐ALD. Gross neuropathological examination of the cerebral hemispheres at the levels of the striatum and globus pallidus (A), mid‐thalamus (B) and posterior part of the parietal lobes (C) show demyelination of the cerebral white matter largely sparing the subcortical fibers. Note the thinning of the anterior part of the corpus callosum and internal capsule. Demyelination is better seen by using myelin stains (Spielmeyer‐Vogt) (D).
Figure 3.
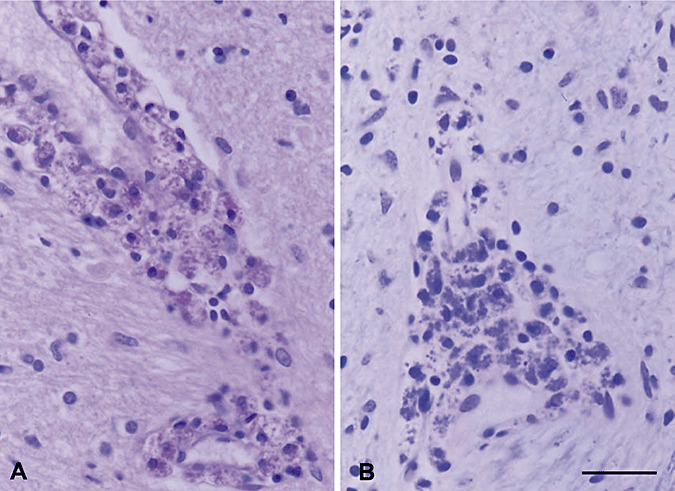
Pure adult cerebral X‐ALD. A, B. Perivascular infiltrates of round cells and macrophages filled with granular material that is stained with hematoxylin and eosin (A) and luxol fast blue (B). Bar = 50 µm.
NEURORADIOLOGY
Differential diagnosis with other myelin disorders can easily be made using radiological means, as the white matter abnormalities of central nervous system (CNS) myelin density observed are typically localized.
Computed tomography (CT) scanning was used initially to diagnose demyelinating lesions of ALD, but only at an advanced stage 6, 17, 30, 34, 36, 38, 52, 82, 84, 159. Nowadays, MRI has supplanted CT 3, 8, 124, 143, 146. Brain MRI demonstrates demyelination earlier and allows correlating more precisely structure–function relationships. The magnetic resonance (MR) signal changes of affected white matter result in prolongation of T1 and T2 relaxation times. Early involvement of corpus callosum (splenium or genu), pyramidal tracts within brain stem or internal capsules, is thus detected. The localization of the demyelinating lesions within the parieto‐occipital junctions and the frontal lobes can be precisely evaluated and make easier correlation with the neuropsychological deficits. Volume measurement (4) and scoring systems (79) of demyelinating lesions have been developed to evaluate the effects of the therapeutic interventions.
As the disease progresses, MRI defines the zones of demyelination precisely: (i) the primary visual pathways (optic radiations, lateral geniculate body, Meyer's loop); (ii) the auditory pathway (medial geniculate body, brachium of inferior colliculus, white matter of transverse temporal gyri); (iii) the pyramidal tracts within the brain stem and internal capsules on successive coronal sections; (iv) the external capsules, putamen and thalamus; (v) the lateral lemnisci, fronto‐pontine and ponto‐cerebellar tracts in brain stem and pons; and (vi) the white matter of cerebellar hemispheres. Abnormal zones reflecting focal disruption of the blood‐brain barrier can be identified with intravenous injection of gadolinium on T1 sequences. The region showing contrast accumulation is located between two demyelinating regions that have different T2 relaxation times. The first zone located in front of enhanced region has lower hyperintensity than the second zone located behind. These MRI findings are in agreement with pathological data showing that accumulation of perivascular lymphocytes are behind and not in front of the edge of the initial demyelinating lesions. Today, brain MRI is the only means to detect early signs of cerebral demyelination in the absence of markers predicting phenotype.
In AMN, MRI shows spinal cord atrophy but without correlation with the degree of neurological disability (3). T2‐weighted images commonly reveal a hyperintensity of the pyramidal tracts that reaches the posterior arms of the internal capsules. This abnormal MR finding can easily be differentiated from the normal T2 relaxation time variations seen in the posterior internal capsules (89).
Interestingly, diffusion tensor‐based three‐dimensional imaging fiber tracking has detected corticospinal tract abnormalities in genu of corpus callosum, pyramids and posterior columns in pure AMN patients, indicating that pathology might not be limited to spinal cord long tracts alone, thus reflecting what is seen on neuropathology (33).
Multislice proton magnetic resonance spectroscopy (MRSI) is very helpful as it allows to analyze brain metabolites with concentrations above 1 mmol. They include N‐acetyl aspartate (NAA), choline‐containing compounds (Cho), glutamate, creatinine and phosphocreatine (Cr) and lactate. In the white matter, X‐ALD induces an increase in Cho and then a reduction in NAA 24, 73, 141. Cerebral regions with advanced demyelination show increased lactate content. These abnormalities are not specific for ALD and their significance remains unclear. They are, however, more widespread than those detected by conventional MRI. An elevated content of Cho can also be detected in white matter apparently normal on MRI. Thus, MRSI may help to identify patients with early neurological involvement.
Recently, a study using MRSI imaging at 7 Tesla has allowed better visualization of X‐ALD lesion architecture, white matter tracts and gray–white matter distinction compared with 1.5 Tesla. The myo‐inositol/creatine ratio was 46% higher and Cho/Cr was 21% higher in non‐affected white matter of adult cALD than in AMN (P < 0.05). There were no significant differences between AMN patients and female heterozygotes. This global myo‐inositol/creatine ratio demonstrates a significant association with the severity of the symptoms measured with Expanded Disability Status Scale score, and deserves further testing to be validated as a biomarker in adult X‐ALD (121).
DIAGNOSIS
Early diagnosis is compulsory in X‐ALD as treatment and genetic counseling and follow‐up of family members is crucial in the prognosis. High concentrations of C26:0 and abnormal ratios of C26:0 and C24:0 to C22:0 in plasma are very suggestive of X‐ALD and are usually found at birth of affected males and in most heterozygous females 94, 100, 145. Monounsaturated VLCFA levels are also increased in ALD, although at a less extent than saturated VLCFA. For diagnosis purposes, the marked increase of C26:0 in the lysophosphatidylcholine fraction is of interest. Currently, neonatal screening is being validated using this parameter in Maryland State (USA) (61).
Combined liquid chromatography‐tandem mass spectrometric methods, electrospray ionisation tandem mass spectrometry (ESI‐MS‐MS) and High Performance Liquid Chromatography – electrospray tandem mass spectrometry (HPLC‐ESI‐MS/MS), are highly accurate methods for diagnosis (61). Biochemical diagnosis of affected males is nearly 100% accurate, provided that plasma has been taken in fasting conditions. This is not the case for women as VLCFA analysis can detect between 85% and 95% of heterozygous (Hz) women only. It is therefore mandatory to establish the status of women at risk to be heterozygous by searching ABCD1 gene mutation.
Analysis of VLCFA in cultured amniocytes and chorionic cells has been used for the prenatal diagnosis with high sensitivity and accuracy (95), although gene mutation analysis is currently frequently performed for this purpose. Yet genetic studies are mandatory in every case to confirm the status of women at risk to be heterozygous (99). Neuroradiological changes appear early in the course of the disease and its monitoring is mandatory in the evaluation of the disease progression and indication of aggressive treatment, such as allogeneic hematopoietic transplantation. Clinical tracking is also important, as early signs of impaired visual perception and altered evoked potentials, and early neuropsychological signs, may draw attention to the neuroradiology changes 25, 47, 65, 122. Newborn screening has been initiated in parts of the USA which will allow an identification of all X‐ALD patients presymptomatically 61, 122.
ANIMAL MODELS
Inactivation of ABCD1 in mice (ABCD ‐) results in decreased VLCFA β‐oxidation in fibroblasts, accumulation of VLCFA in target organs, such as adrenal gland, testis, nervous tissue 42, 72, 80, 157 and neurological phenotype at about 18–20 months with moderate axonal degeneration in the long tracts of the spinal cord but no inflammatory infiltrates and white matter alterations in brain 37, 119. These characteristics are reminiscent of AMN (119). cALD has not been reproduced in mice so far, indicating that additional factors which do not operate in mice are required to trigger demyelination and then neuroinflammation. The lack of a demyelinating mouse model for ALD certainly hampers the comprehension of the molecular pathology and slows the rate of progress in the development of therapeutic approaches. Attempts to place the mutation in a genetic background more prone to neuroinflammation have been made, including the strains SJL and Swiss, finding little success (S Kemp and P Aubourg, personal communication).
Inactivation of ABCD2 results in late‐onset cerebellar and sensory ataxia associated with loss of Purkinje cells, degeneration of dorsal ganglion cells and VLCFA accumulation and axonal degeneration in the dorsal and lateral columns of the spinal cord (41). A double mutant ABCD1/ABCD2 exhibits an earlier onset and more severe phenotype, starting at 12 months of age, a fact that can facilitate experimental therapeutic strategies 83, 118. Axonal damage, as evidenced using synaptophysin, APP and ubiquitin accumulation as markers of swellings, occurs as the first pathological event in this model, followed by myelin degeneration (Figure 4). Additionally, inflammatory infiltrates of T lymphocytes can be found, which are absent from ABCD1‐ or ABCD2‐single knockouts (118).
Figure 4.
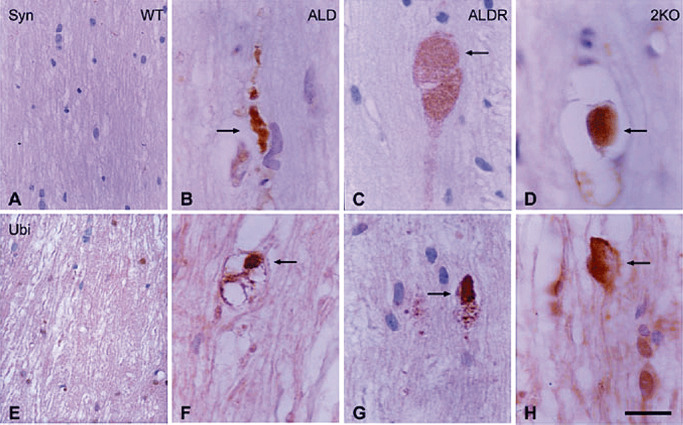
The dorsal column of the spinal cord in wild type: WT (A, E); ABCD1‐, ALD (B, F); ABCD2‐, ALDR (C, G); and double mutant ABCD1 ‐ /ABCD2 ‐, 2KO (D, H) aged 20 months immunostained with anti‐synaptophysin (A–D) and anti‐ubiquitin (E–H) antibodies. Axonal swellings (arrows) are seen in ALD, ALDR and 2KO mice. Paraffin sections slightly counterstained with haematoxylin. Bar = 20 µm.
MOLECULAR NEUROPATHOLOGY
ALD and the role of ABC transporters in lipid transport
ABC transporters regulate lipid transport across membranes. Of 48 transcriptionally active ABC transporters, at least 13 are expressed in the brain and participate in brain lipid transport and homeostasis. At least three of the four ABCD transporters are components of peroxisomal membranes; ABCD1 is known as adrenoleukodystrophy protein, ABCD2 is the ALDRP, ABCD3 is named peroxisomal membrane protein (PMP70). Peroxisomal ABCDs may work as homo‐ or heterodimers, demonstrated in vivo and in vitro respectively, and ABCD1 might transport acyl coenzyme A esters across the peroxisomal membrane 68, 71, 78, 118, 140, 151, 155. This transport is energy dependent (123).
ALDP in the central nervous system is localized in endothelial cells, astrocytes, microglia and in oligodendrocytes, mostly those of the corpus callosum, anterior commissure and internal capsule, whereas ALDP is very low or absent in the neurons (43). More recent studies have shown that ALDP‐encoding mRNA is conspicuous in heart, muscle, liver, kidney and endocrine organs. ALDP is expressed in certain neuron populations (hypothalamus and basal nucleus of Meynert), adrenal gland (zona reticularis and fasciculata), testis (Sertoli and Leydig cells), in addition to skin, colon and other organs (58). Although these studies are not contradictory, it is clear that further work is needed to refine the cellular distribution of ALDP and ALDRP in the nervous system on the basis of a possible heterodimerization of these proteins under appropriate conditions. Curiously, ALDRP expression mirrors ALDP distribution in the nervous system, where it is more abundant in the neurons (140). ALDRP is expressed in oligo and microglia, not endothelial cells in contrast to ALDP. Better understanding of ABCD1 and ABCD2 cellular localization is particularly important as ABCD2 compensates for ABCD1 deficits in cultured cells (104) and in ABCD1‐ mice (118), a feature with potential therapeutic implications.
Impaired lipid peroxisomal transport and impaired oxidation of VLCFAs in peroxisomes
Peroxisomal β‐oxidation of fatty acids starts with the formation of coenzyme A ester catalyzed by VLCFA‐Coenzyme A (CoA) synthetase 136, 142. This reaction is altered in skin fibroblasts of X‐ALD 74, 134, 153, 154. It is assumed that impaired enzymatic activity results in VLCFA accumulation. In this line, overexpression of ABCD1 cDNA in X‐ALD fibroblasts increases VLCFA oxidation and reduces the accumulation of VLCFAs 15, 21, 132. These observations suggest a link between abnormal ALDP and VLCFA accumulation in X‐ALD. This is further supported by the observation that impaired VLCFA β‐oxidation also occurs in ABCD2 knockouts (45), most likely as a secondary consequence caused by the defective transport. Thus, ABCD1 is proposed to play a crucial role in transporting VLCFAs, or their CoA derivatives, across the peroxisome membrane in humans. This is based on the accumulation of VLCFA in patients with X‐ALD and the role of the ABCD transporter orthologs in the peroxisomal importation of fatty acids in yeast (57). Recent studies support this hypothesis, as the overexpression of human ABCD1 in Saccharomyces cerevisiae cells allows the import of several long and VLCFA‐CoA, such as C18:1n‐9, C16:0, C22:0 and C24:6n‐3 into yeast peroxisomes (151). In mice, deficiency of ABCD2 leads to impairment of C24:6n‐3 and C26:0 beta‐oxidation and accumulation of C20:1 and C22:1 in various tissues 45, 77.
However, the link between impaired fatty acid transport into the peroxisome and decreased activity of very long‐chain fatty acyl‐CoA synthetase activity is not clearly understood. Decreased very long‐chain acyl‐CoA synthetase activity is accompanied by decreased peroxisomal VLCFA β‐oxidation in a very long‐chain acyl‐CoA synthetase knockout mice, but these animals do not have accumulation of VLCFAs and do not exhibit an X‐ALD phenotype (56).
Oxidative damage and mitochondria involvement
Increased oxidative stress has been reported in X‐ALD as manifested by increased expression levels of thiobarbituric acid‐reactive substances (TBA‐RS), indicating a stimulation of lipid peroxidation, and decreased total antioxidant reactivity (TAR) implying reduced capacity to buffer augmented reactive species, in plasma, together with increased catalase and superoxide dismutase (SOD) activity in fibroblasts (152). Interestingly, total antioxidant status (TAS) is decreased in symptomatic but not in asymptomatic X‐ALD cases (27). Moreover, high levels of VLCFA, and NO and cytokines are found in X‐ALD lymphoblasts (144).
Oxidative stress also occurs in asymptomatic obligatory female carriers (mothers of X‐ALD patients) as manifested by increased expression of TBA‐RS and decreased TAR in plasma, whereas TAS is not altered (28). Interestingly, no correlation was observed between oxidative stress markers and VLCFA (C26:0 and C26:0/C22:0) levels in plasma (28).
Increased expression of inducible nitric oxide synthase has been observed in astrocytes and microglia, together with increased nitration of proteins in the center of inflammatory demyelinating lesions in X‐ALD (49). Subsequent studies have shown nitrotyrosylated proteins in astrocytes, malondialdehyde, 4‐hydroxynonenal and hemoxygenase‐1 immunoreacticvity in astrocytes and microglia, and increased manganese‐SOD (MnSOD) in astrocytes in juvenile and adult ALD and AMN cases (114). Importantly, increased oxidative markers, although variable from one region to another and from one case to the next, are mainly associated with demyelinating lesions and myelin debris but are also present beyond the demyelinating edge (114).
Preliminary studies in KO ABCD1 (ABCD1 ‐) mice were not conclusive as no relevant increase in oxidative stress markers was found in the brain of ALD mice when compared with wild type excepting the increase of MnSOD in a variety of tissues (114). However, later studies using more sensitive methods (gas chromatography/mass spectrometry) have shown oxidative damage to proteins in ABCD1 ‐ mice aged of 3.5 months, long before the first neuropathological lesions in the spinal cord at around 16 months of age and clinical signs at 20 months of age (44). Parallel studies have shown activation of oxidative responses following VLCFA in organotypic spinal cord slices of ABCD1 ‐ mice and in human ALD fibroblasts that were reversed by Trolox, an analogue of α‐tocopherol (44).
Ultrastructurally, altered mitochondria have been described in the adrenal cortex in X‐ALD (117) and ABCD1 ‐ mice (85). Altered mitochondria have also been observed in neurons of spinal cords of ABCD1 and ABCD2 ‐ mice (41). Functional studies have shown altered rates of mitochondrial LCFA β‐oxidation suggesting a role of mitochondria in fatty acid metabolism that may have implications in X‐ALD 85, 87. As mitochondria are the major subcellular source of reactive oxygen species (ROS), and play a role in several major neurological disorders, studies aimed at analyzing mitochondria function in X‐ALD are warranted and will be discussed elsewhere in this symposium (120). Interestingly, it has been shown that the accumulation of VLCFA per se is not the cause of the mitochondrial abnormalities, and vice versa, mitochondrial abnormalities are not responsible for the accumulation of VLCFA in X‐ALD mice (105).
Inflammation and immunity
Inflammation and altered immune responses are present in a percentage of ALD cases with cerebral involvement but not in pure AMN, in the majority of heterozygous women and in ALD mouse models. Inflammation in X‐ALD is characteristic of cerebral forms, and it is manifested with perivascular infiltrates of CD8+, CD4+ lymphocytes, lowered numbers of B lymphocytes, rare plasma cells and macrophages located immediately beyond the front of myelin damage (64). These areas with infiltrates are characterized by breakdown of the blood‐brain barrier as revealed with MRI sequences using gadolinium contrast. In addition, activated microglia and hypertrophic astrocytes are found in demyelinated areas. CD1b immunoreactivity is observed in perivascular lymphocytes, microglia, macrophages and astrocytes (64). As CD1 is an antigen‐presenting molecule that can present lipid antigens to immunocompetent cells, it has been suggested that CD1 expression may trigger T‐cell inflammatory responses in ALD (64) as already found in other settings (92). Yet the mechanism that mediates oligodendroglial cell death following inflammation is not known.
Powers et al (111) have suggested that excess of VLCFA stimulates nearby astrocytes, perivascular cells and macrophages to initiate a tumor necrosis factor‐α (TNF)‐α‐dependent cytokine cascade that leads to demyelination mediated primarily by cytokines with superimposed destruction of myelin by T cells and to a minimal degree by complement and B cells. Further work has reported increased mRNA and protein levels of cytokines, γ‐interferon, interleukin (IL)‐1α, IL‐2 and IL‐6, GM‐CSF, TNF‐α, chemokines and chemokine receptors have been reported in chronic and acute lesions 86, 106. This is accompanied by increased activation of peripheral blood mononuclear cells following stimulation with phytohemoagglutinin or lipopolysaccharide (29).
Although the inflammatory demyelination in X‐ALD resembles MS in that inflammatory cells are mostly macrophages and T lymphocytes, with infrequent B cells, the occurrence of cell‐mediated auto‐antibody immune responses is not clear in ALD (129).
LESSONS DERIVED FROM THERAPEUTIC PROCEDURES
Understanding of fragmentary aspects of the pathogenesis of X‐ALD has permitted the implementation of distinct strategies. One of these consists of the replacement therapy with adrenal steroids which is mandatory in patients with Addison's disease. Another is based on the dietary administration of glyceroltrioleate/glyceroltrierucate (Lorenzo's oil: LO) combined with a VLCFA‐poor diet which is aimed at reducing the accumulation of VLCFAs. Early administration of LO may delay the appearance of neurological symptoms when given to asymptomatic children before 6 years of age 26, 101. Finally, hematopoietic stem cell transplantation, either derived from human leukocyte antigen (HLA)‐compatible bone marrow or umbilical cord, has been successfully used since 20 years to arrest inflammatory demyelination cerebral ALD 5, 98, 131. The mechanism by which hematopoietic stem cell transplantation is able to arrest the cerebral disease is not known. In the absence of any effect of myeloablative conditioning per se, it raises the possibility that replacement of ALD microglia by normal microglia is sufficient to arrest the demyelinating process. Work is in progress to determine whether microglia with ABCD1 gene mutation have impaired function. Very recently, a combined approach using gene therapy to correct the patient hematopoietic stem cells have been tried in two cerebral childhood ALD cases. The hematopoietic stem cells were genetically corrected ex vivo with a lentiviral vector encoding wild‐type ABCD1 and reinfused into the patients after full myeloablation. Arrest of cerebral demyelination was observed in the two treated patients with effectiveness similar to that observed in successful allogeneic hematopoieitic transplantation (20). Delivery of neurotrophic factors through gene therapy has been efficacious in mice (83). Pharmacological therapies include the use of lovastatine (133) and, potentially, the use of histone deacetylase (HDAC) inhibitors to upregulate the ABCD2 gene (46). The different therapeutic strategies will be discussed in depth in other chapters in this symposium 11, 19.
CONCLUDING COMMENTS
Increased VLCFA levels are the biomarker per excellence of the disease, and its excess produces oxidative stress and oxidative damage to proteins. Accumulation of VLCFA, in particular, complex lipid subsets, or in subsets of acylated proteins, most likely impairs metabolic and signal transduction pathways. However, increased VLCFA levels observed at birth in X‐ALD are not accompanied by neurological symptoms; certain ALD‐carriers never develop the disease in spite of high VLCFA levels. Human patients may present variegated phenotypes starting at variable ages, while ALD mice do not develop clinical symptoms until about 18–20 months. These observations indicate that accumulation of VLCFAs is necessary but not sufficient to develop the whole spectrum of disease. A balance between VLCFA and other lipids seems may play a role in the alteration of lipid homeostasis in ALD. In spite of increased VLCFAs, plasmalogens are deficient in the nervous system in ALD (70). Whether plasmalogens participate in the preservation of tissues with high VLCFA levels in X‐ALD, as suggested by the results of ABCD1 ‐ mice models (16), deserves further attention, as these ether‐phospholipids seem to have an antioxidant role.
It seems clear that the development of the severe cerebral phenotypes is mediated by immune responses that target oligodendrocytes and myelin, and results in demyelination and reactive gliosis (62). CD1‐mediated presentation of lipids to T cells is a known mechanism of T‐cell activation by lipopeptide antigens 91, 93, 150, 158. This mechanism may have a crucial role in cALD pathogenesis. In addition, recent studies have shown that silencing ABCD1 and ABCD2 sensitizes mice astrocytes in primary cultures, increases the levels of VLCFA and induces the expression of cytokines mediated by transcription factors nuclear factor kappa B (NFk‐B), AP‐1 and CCAAT/enhancer binding protein (C/EBP)(135). These findings demonstrate a link between accumulation of VLCFA and induction of inflammatory mediators in vitro (135). The possibility that VLCFA accumulation and neuroinflammation are the only causative factors in cerebral form of ALD (106) is attractive but still speculative.
Work is still needed to understand on an individual patient basis which environmental and/or genetic vulnerability factors contribute to the development of so various clinical phenotypes in ALD. Although great progress has been made in the last few years, we are still far from understanding fully the mechanisms that trigger axonal damage, cerebral demyelination and neuroinflammation. Fortunately, early diagnosis and sophisticated therapies administered at appropriate times have curbed the fateful natural progression of cerebral forms of ALD for a significant fraction of ALD patients.
ACKNOWLEDGMENTS
Personal work was supported by grants from the European Commission (FP7‐241622), the European Leukodystrophy Association (ELA 2006‐043I4), the Spanish Institute for Health Carlos III (FIS PI080991), the Autonomous Government of Catalonia (2009SGR85) and the COST action BM0604 to AP, and the European Brain bank Network (Brain Net II: EU 2004‐CT‐2004‐503039) to IF. Reference National Centers CIBERNED (Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas) and CIBERER (Centro de Investigación Biomédica en Red de Enfermedades Raras) are research branches of the Instituto Carlos III (ISCIII). We thank T. Yohannan for editorial assistance.
REFERENCES
- 1. Asheuer M, Bieche I, Laurendeau I, Moser A, Hainque B, Vidaud M, Aubourg P (2005) Decreased expression of ABCD4 and BG1 genes early in the pathogenesis of X‐linked adrenoleukodystrophy. Hum Mol Genet 14:1293–1303. [DOI] [PubMed] [Google Scholar]
- 2. Aubourg P (1996) X‐linked adrenoleukodystrophy. In: Handbook of Clinical Neurology, Vinken PJ, Bruyn GW, Moser HW (eds), pp. 447–483. Elsevier: Amsterdam. [Google Scholar]
- 3. Aubourg P, Adamsbaum C, Lavallard‐Rousseau MC, Lemaitre A, Boureau F, Mayer M, Kalifa G (1992) Brain MRI and electrophysiologic abnormalities in preclinical and clinical adrenomyeloneuropathy. Neurology 42:85–91. [DOI] [PubMed] [Google Scholar]
- 4. Aubourg P, Adamsbaum C, Lavallard‐Rousseau MC, Rocchiccioli F, Cartier N, Jambaque I et al (1993) A two‐year trial of oleic and erucic acids (“Lorenzo's oil”) as treatment for adrenomyeloneuropathy. N Engl J Med 329:745–752. [DOI] [PubMed] [Google Scholar]
- 5. Aubourg P, Blanche S, Jambaque I, Rocchiccioli F, Kalifa G, Naud‐Saudreau C et al (1990) Reversal of early neurologic and neuroradiologic manifestations of X‐linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med 322:1860–1806. [DOI] [PubMed] [Google Scholar]
- 6. Aubourg P, Diebler C (1982) Adrenoleukodystrophy—its diverse CT appearances and an evolutive or phenotypic variant: the leukodystrophy without adrenal insufficiency. Neuroradiology 24:33–42. [DOI] [PubMed] [Google Scholar]
- 7. Aubourg P, Mandel JL (1996) X‐linked adrenoleukodystrophy. Ann N Y Acad Sci 804:461–476. [DOI] [PubMed] [Google Scholar]
- 8. Aubourg P, Sellier N, Chaussain JL, Kalifa G (1989) MRI detects cerebral involvement in neurologically asymptomatic patients with adrenoleukodystrophy. Neurology 39:1619–1621. [DOI] [PubMed] [Google Scholar]
- 9. Berger J, Gartner J (2006) X‐linked adrenoleukodystrophy: clinical, biochemical and pathogenetic aspects. Biochim Biophys Acta 1763:1721–1732. [DOI] [PubMed] [Google Scholar]
- 10. Berger J, Molzer B, Fae I, Bernheimer H (1994) X‐linked adrenoleukodystrophy (ALD): a novel mutation of the ALD gene in 6 members of a family presenting with 5 different phenotypes. Biochem Biophys Res Commun 205:1638–1643. [DOI] [PubMed] [Google Scholar]
- 11. Berger J, Pujol A, Aubourg P, Forss‐Petter S (2010) Current and future pharmacological treatment strategies in X‐linked adrenoleukodystrophy. Brain Pathol 20:845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bernheimer H, Budka H, Muller P (1983) Brain tissue immunoglobulins in adrenoleukodystrophy: a comparison with multiple sclerosis and systemic lupus erythematosus. Acta Neuropathol 59:95–102. [DOI] [PubMed] [Google Scholar]
- 13. Bezman L, Moser AB, Raymond GV, Rinaldo P, Watkins PA, Smith KD et al (2001) Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol 49:512–517. [PubMed] [Google Scholar]
- 14. Bizzozero OA, Zuniga G, Lees MB (1991) Fatty acid composition of human myelin proteolipid protein in peroxisomal disorders. J Neurochem 56:872–878. [DOI] [PubMed] [Google Scholar]
- 15. Braiterman LT, Zheng S, Watkins PA, Geraghty MT, Johnson G, McGuinness MC et al (1998) Suppression of peroxisomal membrane protein defects by peroxisomal ATP binding cassette (ABC) proteins. Hum Mol Genet 7:239–247. [DOI] [PubMed] [Google Scholar]
- 16. Brites P, Mooyer PA, El Mrabet L, Waterham HR, Wanders RJ (2009) Plasmalogens participate in very‐long‐chain fatty acid‐induced pathology. Brain 132:482–492. [DOI] [PubMed] [Google Scholar]
- 17. Brooks BS, El Gammal T (1982) Case report. An additional case of adrenoleukodystrophy with both type I and type II CT features. J Comput Assist Tomogr 6:385–388. [DOI] [PubMed] [Google Scholar]
- 18. Budka H, Sluga E, Heiss WD (1976) Spastic paraplegia associated with Addison's disease: adult variant of adreno‐leukodystrophy. J Neurol 213:237–250. [DOI] [PubMed] [Google Scholar]
- 19. Cartier N, Aubourg P (2010) Hematopoietic stem cell transplant and gene therapy in X‐linked adrenoleukodystrophy. Brain Pathol 20:857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cartier N, Hacein‐Bey‐Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I et al (2009) Hematopoietic stem cell gene therapy with a lentiviral vector in X‐linked adrenoleukodystrophy. Science 326:818–823. [DOI] [PubMed] [Google Scholar]
- 21. Cartier N, Lopez J, Moullier P, Rocchiccioli F, Rolland MO, Jorge P et al (1995) Retroviral‐mediated gene transfer corrects very‐long‐chain fatty acid metabolism in adrenoleukodystrophy fibroblasts. Proc Natl Acad Sci U S A 92:1674–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chaudhry V, Moser HW, Cornblath DR (1996) Nerve conduction studies in adrenomyeloneuropathy. J Neurol Neurosurg Psychiatry 61:181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cohen SM, Green WR, De La Cruz ZC, Brown FR 3rd, Moser HW, Luckenbach MW et al (1983) Ocular histopathologic studies of neonatal and childhood adrenoleukodystrophy. Am J Ophthalmol 95:82–96. [DOI] [PubMed] [Google Scholar]
- 24. Confort‐Gouny S, Vion‐Dury J, Chabrol B, Nicoli F, Cozzone PJ (1995) Localised proton magnetic resonance spectroscopy in X‐linked adrenoleukodystrophy. Neuroradiology 37:568–575. [DOI] [PubMed] [Google Scholar]
- 25. Cox CS, Dubey P, Raymond GV, Mahmood A, Moser AB, Moser HW (2006) Cognitive evaluation of neurologically asymptomatic boys with X‐linked adrenoleukodystrophy. Arch Neurol 63:69–73. [DOI] [PubMed] [Google Scholar]
- 26. Deon M, Garcia MP, Sitta A, Barschak AG, Coelho DM, Schimit GO et al (2008) Hexacosanoic and docosanoic acids plasma levels in patients with cerebral childhood and asymptomatic X‐linked adrenoleukodystrophy: Lorenzo's oil effect. Metab Brain Dis 23:43–49. [DOI] [PubMed] [Google Scholar]
- 27. Deon M, Sitta A, Barschak AG, Coelho DM, Pigatto M, Schmitt GO et al (2007) Induction of lipid peroxidation and decrease of antioxidant defenses in symptomatic and asymptomatic patients with X‐linked adrenoleukodystrophy. Int J Dev Neurosci 25:441–444. [DOI] [PubMed] [Google Scholar]
- 28. Deon M, Sitta A, Barschak AG, Coelho DM, Terroso T, Schmitt GO et al (2008) Oxidative stress is induced in female carriers of X‐linked adrenoleukodystrophy. J Neurol Sci 266:79–83. [DOI] [PubMed] [Google Scholar]
- 29. Di Biase A, Merendino N, Avellino C, Cappa M, Salvati S (2001) Th 1 cytokine production by peripheral blood mononuclear cells in X‐linked adrenoleukodystrophy. J Neurol Sci 182:161–165. [DOI] [PubMed] [Google Scholar]
- 30. Di Chiro G, Eiben RM, Manz HJ, Jacobs IB, Schellinger D (1980) A new CT pattern in adrenoleukodystrophy. Radiology 137:687–692. [DOI] [PubMed] [Google Scholar]
- 31. Dohle CI, Bannykh SI, Hisama FM, Baehring JM (2009) Leukoencephalopathy in adults: is it adrenoleukodystrophy? A case report and molecular analysis. J Neurol Sci 285:235–237. [DOI] [PubMed] [Google Scholar]
- 32. Dubey P, Fatemi A, Barker PB, Degaonkar M, Troeger M, Zackowski K et al (2005) Spectroscopic evidence of cerebral axonopathy in patients with “pure” adrenomyeloneuropathy. Neurology 64:304–310. [DOI] [PubMed] [Google Scholar]
- 33. Dubey P, Fatemi A, Huang H, Nagae‐Poetscher L, Wakana S, Barker PB et al (2005) Diffusion tensor‐based imaging reveals occult abnormalities in adrenomyeloneuropathy. Ann Neurol 58:758–766. [DOI] [PubMed] [Google Scholar]
- 34. Dubois PJ, Freemark M, Lewis D, Drayer BP, Heinz ER, Osborne D (1981) Case report. Atypical findings in adrenoleukodystrophy. J Comput Assist Tomogr 5:888–891. [DOI] [PubMed] [Google Scholar]
- 35. Dubois‐Dalcq M, Feigenbaum V, Aubourg P (1999) The neurobiology of X‐linked adrenoleukodystrophy, a demyelinating peroxisomal disorder. Trends Neurosci 22:4–12. [DOI] [PubMed] [Google Scholar]
- 36. Duda EE, Huttenlocher PR (1976) Computed tomography in adrenoleukodystrophy. Correlation of radiological and histological findings. Radiology 120:349–350. [DOI] [PubMed] [Google Scholar]
- 37. Dumser M, Bauer J, Lassmann H, Berger J, Forss‐Petter S (2007) Lack of adrenoleukodystrophy protein enhances oligodendrocyte disturbance and microglia activation in mice with combined Abcd1/Mag deficiency. Acta Neuropathol 114:573–586. [DOI] [PubMed] [Google Scholar]
- 38. Eiben RM, Di Chiro G (1977) Computer assisted tomography in adrenoleukodystrophy. J Comput Assist Tomogr 1:308–314. [DOI] [PubMed] [Google Scholar]
- 39. Eichler FS, Barker PB, Cox C, Edwin D, Ulug AM, Moser HW, Raymond GV (2002) Proton MR spectroscopic imaging predicts lesion progression on MRI in X‐linked adrenoleukodystrophy. Neurology 58:901–907. [DOI] [PubMed] [Google Scholar]
- 40. Eichler FS, Ren JQ, Cossoy M, Rietsch AM, Nagpal S, Moser AB et al (2008) Is microglial apoptosis an early pathogenic change in cerebral X‐linked adrenoleukodystrophy? Ann Neurol 63:729–742. [DOI] [PubMed] [Google Scholar]
- 41. Ferrer I, Kapfhammer JP, Hindelang C, Kemp S, Troffer‐Charlier N, Broccoli V et al (2005) Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late‐onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum Mol Genet 14:3565–3577. [DOI] [PubMed] [Google Scholar]
- 42. Forss‐Petter S, Werner H, Berger J, Lassmann H, Molzer B, Schwab MH et al (1997) Targeted inactivation of the X‐linked adrenoleukodystrophy gene in mice. J Neurosci Res 50:829–843. [DOI] [PubMed] [Google Scholar]
- 43. Fouquet F, Zhou JM, Ralston E, Murray K, Troalen F, Magal E et al (1997) Expression of the adrenoleukodystrophy protein in the human and mouse central nervous system. Neurobiol Dis 3:271–285. [DOI] [PubMed] [Google Scholar]
- 44. Fourcade S, Lopez‐Erauskin J, Galino J, Duval C, Naudi A, Jove M et al (2008) Early oxidative damage underlying neurodegeneration in X‐adrenoleukodystrophy. Hum Mol Genet 17:1762–1773. [DOI] [PubMed] [Google Scholar]
- 45. Fourcade S, Ruiz M, Camps C, Schluter A, Houten SM, Mooyer PA et al (2009) A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am J Physiol Endocrinol Metab 296:E211–E221. [DOI] [PubMed] [Google Scholar]
- 46. Fourcade S, Ruiz M, Guilera C, Hahnen E, Brichta L, Naudi A et al (2010) Valproic acid induces antioxidant effects in X‐linked adrenoleukodystrophy. Hum Mol Genet [Epub ahead of print] PMID:20179078. [DOI] [PubMed] [Google Scholar]
- 47. Furushima W, Inagaki M, Gunji A, Inoue Y, Kaga M, Mizutani S (2009) Early signs of visual perception and evoked potentials in radiologically asymptomatic boys with X‐linked adrenoleukodystrophy. J Child Neurol 24:927–935. [DOI] [PubMed] [Google Scholar]
- 48. Garashi M, Belchis D, Suzuki K (1976) Brain gangliosides in adrenoleukodystrophy. J Neurochem 27:327–328. [DOI] [PubMed] [Google Scholar]
- 49. Gilg AG, Singh AK, Singh I (2000) Inducible nitric oxide synthase in the central nervous system of patients with X‐adrenoleukodystrophy. J Neuropathol Exp Neurol 59:1063–1069. [DOI] [PubMed] [Google Scholar]
- 50. Glasgow BJ, Brown HH, Hannah JB, Foos RY (1987) Ocular pathologic findings in neonatal adrenoleukodystrophy. Ophthalmology 94:1054–1060. [DOI] [PubMed] [Google Scholar]
- 51. Grainger BT, Papchenko TL, Danesh‐Meyer HV (2010) Optic nerve atrophy in adrenoleukodystrophy detectable by optic coherence tomography. J Clin Neurosci 17:122–124. [DOI] [PubMed] [Google Scholar]
- 52. Greenberg HS, Halverson D, Lane B (1977) CT scanning and diagnosis of adrenoleukodystrophy. Neurology 27:884–886. [DOI] [PubMed] [Google Scholar]
- 53. Griffin DE, Moser HW, Mendoza Q, Moench TR, O'Toole S, Moser AB (1985) Identification of the inflammatory cells in the central nervous system of patients with adrenoleukodystrophy. Ann Neurol 18:660–664. [DOI] [PubMed] [Google Scholar]
- 54. Griffin JW, Goren E, Schaumburg H, Engel WK, Loriaux L (1977) Adrenomyeloneuropathy: a probable variant of adrenoleukodystrophy. I. Clinical and endocrinologic aspects. Neurology 27:1107–1113. [DOI] [PubMed] [Google Scholar]
- 55. Heinzer AK, McGuinness MC, Lu JF, Stine OC, Wei H, Van der Vlies M et al (2003) Mouse models and genetic modifiers in X‐linked adrenoleukodystrophy. Adv Exp Med Biol 544:75–93. [DOI] [PubMed] [Google Scholar]
- 56. Heinzer AK, Watkins PA, Lu JF, Kemp S, Moser AB, Li YY et al (2003) A very long‐chain acyl‐CoA synthetase‐deficient mouse and its relevance to X‐linked adrenoleukodystrophy. Hum Mol Genet 12:1145–1154. [DOI] [PubMed] [Google Scholar]
- 57. Hettema EH, Van Roermund CW, Distel B, Van Den Berg M, Vilela C, Rodrigues‐Pousada C et al (1996) The ABC transporter proteins Pat1 and Pat2 are required for import of long‐chain fatty acids into peroxisomes of Saccharomyces cerevisiae . EMBO J 15:3813–3822. [PMC free article] [PubMed] [Google Scholar]
- 58. Hoftberger R, Kunze M, Weinhofer I, Aboul‐Enein F, Voigtlander T, Oezen I et al (2007) Distribution and cellular localization of adrenoleukodystrophy protein in human tissues: implications for X‐linked adrenoleukodystrophy. Neurobiol Dis 28:165–174. [DOI] [PubMed] [Google Scholar]
- 59. Holzinger A, Kammerer S, Berger J, Roscher AA (1997) cDNA cloning and mRNA expression of the human adrenoleukodystrophy related protein (ALDRP), a peroxisomal ABC transporter. Biochem Biophys Res Commun 239:261–264. [DOI] [PubMed] [Google Scholar]
- 60. Holzinger A, Mayerhofer P, Berger J, Lichtner P, Kammerer S, Roscher AA (1999) Full length cDNA cloning, promoter sequence, and genomic organization of the human adrenoleukodystrophy related (ALDR) gene functionally redundant to the gene responsible for X‐linked adrenoleukodystrophy. Biochem Biophys Res Commun 258:436–442. [DOI] [PubMed] [Google Scholar]
- 61. Hubbard WC, Moser AB, Liu AC, Jones RO, Steinberg SJ, Lorey F et al (2009) Newborn screening for X‐linked adrenoleukodystrophy (X‐ALD): validation of a combined liquid chromatography‐tandem mass spectrometric (LC‐MS/MS) method. Mol Genet Metab 97:212–220. [DOI] [PubMed] [Google Scholar]
- 62. Hudspeth MP, Raymond GV (2007) Immunopathogenesis of adrenoleukodystrophy: current understanding. J Neuroimmunol 182:5–12. [DOI] [PubMed] [Google Scholar]
- 63. Igarashi M, Schaumburg HH, Powers J, Kishmoto Y, Kolodny E, Suzuki K (1976) Fatty acid abnormality in adrenoleukodystrophy. J Neurochem 26:851–860. [DOI] [PubMed] [Google Scholar]
- 64. Ito M, Blumberg BM, Mock DJ, Goodman AD, Moser AB, Moser HW et al (2001) Potential environmental and host participants in the early white matter lesion of adreno‐leukodystrophy: morphologic evidence for CD8 cytotoxic T cells, cytolysis of oligodendrocytes, and CD1‐mediated lipid antigen presentation. J Neuropathol Exp Neurol 60:1004–1019. [DOI] [PubMed] [Google Scholar]
- 65. Kaga M, Furushima W, Inagaki M, Nakamura M (2009) Early neuropsychological signs of childhood adrenoleukodystrophy (ALD). Brain Dev 31:558–561. [DOI] [PubMed] [Google Scholar]
- 66. Kashiwayama Y, Seki M, Yasui A, Murasaki Y, Morita M, Yamashita Y et al (2009) 70‐kDa peroxisomal membrane protein related protein (P70R/ABCD4) localizes to endoplasmic reticulum not peroxisomes, and NH2‐terminal hydrophobic property determines the subcellular localization of ABC subfamily D proteins. Exp Cell Res 315:190–205. [DOI] [PubMed] [Google Scholar]
- 67. Kemp S, Pujol A, Waterham HR, Van Geel BM, Boehm CD, Raymond GV et al (2001) ABCD1 mutations and the X‐linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum Mutat 18:499–515. [DOI] [PubMed] [Google Scholar]
- 68. Kemp S, Wanders RJ (2007) X‐linked adrenoleukodystrophy: very long‐chain fatty acid metabolism, ABC half‐transporters and the complicated route to treatment. Mol Genet Metab 90:268–276. [DOI] [PubMed] [Google Scholar]
- 69. Kemp S, Wanders RJ (2010) Biochemical aspects of X‐adrenoleukodystrophy. Brain Pathol 20:831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Khan M, Singh J, Singh I (2008) Plasmalogen deficiency in cerebral adrenoleukodystrophy and its modulation by lovastatin. J Neurochem 106:1766–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim WS, Weickert CS, Garner B (2008) Role of ATP‐binding cassette transporters in brain lipid transport and neurological disease. J Neurochem 104:1145–1166. [DOI] [PubMed] [Google Scholar]
- 72. Kobayashi T, Shinnoh N, Kondo A, Yamada T (1997) Adrenoleukodystrophy protein‐deficient mice represent abnormality of very long chain fatty acid metabolism. Biochem Biophys Res Commun 232:631–636. [DOI] [PubMed] [Google Scholar]
- 73. Kruse B, Barker PB, Van Zijl PC, Duyn JH, Moonen CT, Moser HW (1994) Multislice proton magnetic resonance spectroscopic imaging in X‐linked adrenoleukodystrophy. Ann Neurol 36:595–608. [DOI] [PubMed] [Google Scholar]
- 74. Lazo O, Contreras M, Hashmi M, Stanley W, Irazu C, Singh I (1988) Peroxisomal lignoceroyl‐CoA ligase deficiency in childhood adrenoleukodystrophy and adrenomyeloneuropathy. Proc Natl Acad Sci U S A 85:7647–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li JY, Hsu CC, Tsai CR (2010) Spinocerebellar variant of adrenoleukodystrophy with a novel ABCD1 gene mutation. J Neurol Sci 290:163–165. [DOI] [PubMed] [Google Scholar]
- 76. Linnebank M, Kemp S, Wanders RJ, Kleijer WJ, Van Der Sterre ML, Gartner J et al (2006) Methionine metabolism and phenotypic variability in X‐linked adrenoleukodystrophy. Neurology 66:442–443. [DOI] [PubMed] [Google Scholar]
- 77. Liu J, Sabeva NS, Bhatnagar S, Li XA, Pujol A, Graf GA (2010) ABCD2 is abundant in adipose tissue and opposes the accumulation of dietary erucic acid (C22:1) in fat. J Lipid Res 51:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Liu LX, Janvier K, Berteaux‐Lecellier V, Cartier N, Benarous R, Aubourg P (1999) Homo‐ and heterodimerization of peroxisomal ATP‐binding cassette half‐transporters. J Biol Chem 274:32738–32743. [DOI] [PubMed] [Google Scholar]
- 79. Loes DJ, Hite S, Moser H, Stillman AE, Shapiro E, Lockman L et al (1994) Adrenoleukodystrophy: a scoring method for brain MR observations. Am J Neuroradiol 15:1761–1766. [PMC free article] [PubMed] [Google Scholar]
- 80. Lu JF, Lawler AM, Watkins PA, Powers JM, Moser AB, Moser HW, Smith KD (1997) A mouse model for X‐linked adrenoleukodystrophy. Proc Natl Acad Sci U S A 94:9366–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Maier EM, Mayerhofer PU, Asheuer M, Kohler W, Rothe M, Muntau AC et al (2008) X‐linked adrenoleukodystrophy phenotype is independent of ABCD2 genotype. Biochem Biophys Res Commun 377:176–180. [DOI] [PubMed] [Google Scholar]
- 82. Marler JR, O'Neill BP, Forbes GS, Moser HW (1983) Adrenoleukodystrophy (ALD): clinical and CT features of a childhood variant. Neurology 33:1203–1205. [DOI] [PubMed] [Google Scholar]
- 83. Mastroeni R, Bensadoun JC, Charvin D, Aebischer P, Pujol A, Raoul C (2009) Insulin‐like growth factor‐1 and neurotrophin‐3 gene therapy prevents motor decline in an X‐linked adrenoleukodystrophy mouse model. Ann Neurol 66:117–122. [DOI] [PubMed] [Google Scholar]
- 84. McDonald MI, Corey GR, Gallis HA, Durack DT (1984) Single and multiple pyogenic liver abscesses. Natural history, diagnosis and treatment, with emphasis on percutaneous drainage. Medicine 63:291–302. [DOI] [PubMed] [Google Scholar]
- 85. McGuinness MC, Lu JF, Zhang HP, Dong GX, Heinzer AK, Watkins PA et al (2003) Role of ALDP (ABCD1) and mitochondria in X‐linked adrenoleukodystrophy. Mol Cell Biol 23:744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. McGuinness MC, Powers JM, Bias WB, Schmeckpeper BJ, Segal AH, Gowda VC et al (1997) Human leukocyte antigens and cytokine expression in cerebral inflammatory demyelinative lesions of X‐linked adrenoleukodystrophy and multiple sclerosis. J Neuroimmunol 75:174–182. [DOI] [PubMed] [Google Scholar]
- 87. McGuinness MC, Zhang HP, Smith KD (2001) Evaluation of pharmacological induction of fatty acid beta‐oxidation in X‐linked adrenoleukodystrophy. Mol Genet Metab 74:256–263. [DOI] [PubMed] [Google Scholar]
- 88. Migeon BR, Moser HW, Moser AB, Axelman J, Sillence D, Norum RA (1981) Adrenoleukodystrophy: evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci U S A 78:5066–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mirowitz S, Sartor K, Gado M, Torack R (1989) Focal signal‐intensity variations in the posterior internal capsule: normal MR findings and distinction from pathologic findings. Radiology 172:535–539. [DOI] [PubMed] [Google Scholar]
- 90. Mishra S, Modi M, Das CP, Prabhakar S (2006) Adrenoleukodystrophy manifesting as spinocerebellar degeneration. Neurol India 54:195–196. [PubMed] [Google Scholar]
- 91. Moody DB (2006) The surprising diversity of lipid antigens for CD1‐restricted T cells. Adv Immunol 89:87–139. [DOI] [PubMed] [Google Scholar]
- 92. Moody DB, Besra GS, Wilson IA, Porcelli SA (1999) The molecular basis of CD1‐mediated presentation of lipid antigens. Immunol Rev 172:285–296. [DOI] [PubMed] [Google Scholar]
- 93. Moody DB, Young DC, Cheng TY, Rosat JP, Roura‐Mir C, O'Connor PB et al (2004) T cell activation by lipopeptide antigens. Science 303:527–531. [DOI] [PubMed] [Google Scholar]
- 94. Moser AB, Kreiter N, Bezman L, Lu S, Raymond GV, Naidu S, Moser HW (1999) Plasma very long chain fatty acids in 3000 peroxisome disease patients and 29 000 controls. Ann Neurol 45:100–110. [DOI] [PubMed] [Google Scholar]
- 95. Moser AB, Moser HW (1999) The prenatal diagnosis of X‐linked adrenoleukodystrophy. Prenat Diagn 19:46–48. [DOI] [PubMed] [Google Scholar]
- 96. Moser H, Smith KD, Watkins PA, Powers J, Moser AB (2001) X‐linked adrenoleukodystrophy. In: The Metabolic and Molecular Bases of Inherited Disease, Scriver C (ed.) pp. 3257–3301. McGraw‐Hill: New‐York. [Google Scholar]
- 97. Moser HW (1997) Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain 120:1485–1508. [DOI] [PubMed] [Google Scholar]
- 98. Moser HW (2006) Therapy of X‐linked adrenoleukodystrophy. NeuroRx 3:246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Moser HW, Mahmood A, Raymond GV (2007) X‐linked adrenoleukodystrophy. Nat Clin Pract Neurol 3:140–151. [DOI] [PubMed] [Google Scholar]
- 100. Moser HW, Moser AE, Trojak JE, Supplee SW (1983) Identification of female carriers of adrenoleukodystrophy. J Pediatr 103:54–59. [DOI] [PubMed] [Google Scholar]
- 101. Moser HW, Raymond GV, Lu SE, Muenz LR, Moser AB, Xu J et al (2005) Follow‐up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo's oil. Arch Neurol 62:1073–1080. [DOI] [PubMed] [Google Scholar]
- 102. Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H et al (1993) Putative X‐linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361:726–730. [DOI] [PubMed] [Google Scholar]
- 103. Mosser J, Lutz Y, Stoeckel ME, Sarde CO, Kretz C, Douar AM et al (1994) The gene responsible for adrenoleukodystrophy encodes a peroxisomal membrane protein. Hum Mol Genet 3:265–271. [DOI] [PubMed] [Google Scholar]
- 104. Netik A, Forss‐Petter S, Holzinger A, Molzer B, Unterrainer G, Berger J (1999) Adrenoleukodystrophy‐related protein can compensate functionally for adrenoleukodystrophy protein deficiency (X‐ALD): implications for therapy. Hum Mol Genet 8:907–913. [DOI] [PubMed] [Google Scholar]
- 105. Oezen I, Rossmanith W, Forss‐Petter S, Kemp S, Voigtlander T, Moser‐Thier K et al (2005) Accumulation of very long‐chain fatty acids does not affect mitochondrial function in adrenoleukodystrophy protein deficiency. Hum Mol Genet 14:1127–1137. [DOI] [PubMed] [Google Scholar]
- 106. Paintlia AS, Gilg AG, Khan M, Singh AK, Barbosa E, Singh I (2003) Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X‐ALD: implications for potential therapies. Neurobiol Dis 14:425–439. [DOI] [PubMed] [Google Scholar]
- 107. Powers J (2008) Peroxisomal disorders. In: Greenfield's Neuropathology, Love S, Louis D, Ellison D (eds), pp. 643–673. London: Hodder Arnold. [Google Scholar]
- 108. Powers JM (2005) Adreno‐leukodystrophy: a personal historical note. Acta Neuropathol 109:124–127. [DOI] [PubMed] [Google Scholar]
- 109. Powers JM, DeCiero DP, Cox C, Richfield EK, Ito M, Moser AB, Moser HW (2001) The dorsal root ganglia in adrenomyeloneuropathy: neuronal atrophy and abnormal mitochondria. J Neuropathol Exp Neurol 60:493–501. [DOI] [PubMed] [Google Scholar]
- 110. Powers JM, DeCiero DP, Ito M, Moser AB, Moser HW (2000) Adrenomyeloneuropathy: a neuropathologic review featuring its noninflammatory myelopathy. J Neuropathol Exp Neurol 59:89–102. [DOI] [PubMed] [Google Scholar]
- 111. Powers JM, Liu Y, Moser AB, Moser HW (1992) The inflammatory myelinopathy of adreno‐leukodystrophy: cells, effector molecules, and pathogenetic implications. J Neuropathol Exp Neurol 51:630–643. [DOI] [PubMed] [Google Scholar]
- 112. Powers JM, Moser HW, Moser AB, Ma CK, Elias SB, Norum RA (1987) Pathologic findings in adrenoleukodystrophy heterozygotes. Arch Pathol Lab Med 111:151–153. [PubMed] [Google Scholar]
- 113. Powers JM, Moser HW, Moser AB, Schaumburg HH (1982) Fetal adrenoleukodystrophy: the significance of pathologic lesions in adrenal gland and testis. Hum Pathol 13:1013–1019. [DOI] [PubMed] [Google Scholar]
- 114. Powers JM, Pei Z, Heinzer AK, Deering R, Moser AB, Moser HW et al (2005) Adreno‐leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol 64:1067–1079. [DOI] [PubMed] [Google Scholar]
- 115. Powers JM, Schaumberg HH (1974) Adreno‐leukodystrophy. Similar ultrastructural changes in adrenal cortical and Schwann cells. Arch Neurol 30:406–408. [DOI] [PubMed] [Google Scholar]
- 116. Powers JM, Schaumburg HH (1981) The testis in adreno‐leukodystrophy. Am J Pathol 102:90–98. [PMC free article] [PubMed] [Google Scholar]
- 117. Powers JM, Schaumburg HH, Johnson AB, Raine CS (1980) A correlative study of the adrenal cortex in adreno‐leukodystrophy—evidence for a fatal intoxication with very long chain saturated fatty acids. Invest Cell Pathol 3:353–376. [PubMed] [Google Scholar]
- 118. Pujol A, Ferrer I, Camps C, Metzger E, Hindelang C, Callizot N et al (2004) Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X‐adrenoleukodystrophy. Hum Mol Genet 13:2997–3006. [DOI] [PubMed] [Google Scholar]
- 119. Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL (2002) Late onset neurological phenotype of the X‐ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet 11:499–505. [DOI] [PubMed] [Google Scholar]
- 120. Pujol A, Singh I (2010) Pathomechanisms of X‐adrenoleukodystrophy: an hypothesis. Brain Pathol 20:838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ratai E, Kok T, Wiggins C, Wiggins G, Grant E, Gagoski B et al (2008) Seven‐Tesla proton magnetic resonance spectroscopic imaging in adult X‐linked adrenoleukodystrophy. Arch Neurol 65:1488–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Raymond GV, Jones RO, Moser AB (2007) Newborn screening for adrenoleukodystrophy: implications for therapy. Mol Diagn Ther 11:381–384. [DOI] [PubMed] [Google Scholar]
- 123. Roerig P, Mayerhofer P, Holzinger A, Gartner J (2001) Characterization and functional analysis of the nucleotide binding fold in human peroxisomal ATP binding cassette transporters. FEBS Lett 492:66–72. [DOI] [PubMed] [Google Scholar]
- 124. Romero C, Dietemann JL, Kurtz D, Bataillard M, Christmann D (1990) Adrenoleukodystrophy. Value of contrast‐enhanced MR imaging. J Neuroradiol 17:267–276. [PubMed] [Google Scholar]
- 125. Savary S, Troffer‐Charlier N, Gyapay G, Mattei MG, Chimini G (1997) Chromosomal localization of the adrenoleukodystrophy‐related gene in man and mice. Eur J Hum Genet 5:99–101. [PubMed] [Google Scholar]
- 126. Schaumburg HH, Powers JM, Raine CS, Johnson AB, Kolodny EH, Kishmoto Y et al (1976) The myeloneuropathy variant of adrenoleukodystrophy. J Neuropathol Exp Neurol 35:312. [Google Scholar]
- 127. Schaumburg HH, Powers JM, Raine CS, Spencer PS, Griffin JW, Prineas JW, Boehme DM (1977) Adrenomyeloneuropathy: a probable variant of adrenoleukodystrophy. II. General pathologic, neuropathologic, and biochemical aspects. Neurology 27:1114–1119. [DOI] [PubMed] [Google Scholar]
- 128. Schaumburg HH, Powers JM, Raine CS, Suzuki K, Richardson EP Jr (1975) Adrenoleukodystrophy. A clinical and pathological study of 17 cases. Arch Neurol 32:577–591. [DOI] [PubMed] [Google Scholar]
- 129. Schmidt S, Haase CG, Bezman L, Moser H, Schmidt M, Kohler W et al (2001) Serum autoantibody responses to myelin oligodendrocyte glycoprotein and myelin basic protein in X‐linked adrenoleukodystrophy and multiple sclerosis. J Neuroimmunol 119:88–94. [DOI] [PubMed] [Google Scholar]
- 130. Semmler A, Bao X, Cao G, Kohler W, Weller M, Aubourg P, Linnebank M (2009) Genetic variants of methionine metabolism and X‐ALD phenotype generation: results of a new study sample. J Neurol 256:1277–1280. [DOI] [PubMed] [Google Scholar]
- 131. Semmler A, Kohler W, Jung HH, Weller M, Linnebank M (2008) Therapy of X‐linked adrenoleukodystrophy. Expert Rev Neurother 8:1367–1379. [DOI] [PubMed] [Google Scholar]
- 132. Shinnoh N, Yamada T, Yoshimura T, Furuya H, Yoshida Y, Suzuki Y et al (1995) Adrenoleukodystrophy: the restoration of peroxisomal beta‐oxidation by transfection of normal cDNA. Biochem Biophys Res Commun 210:830–836. [DOI] [PubMed] [Google Scholar]
- 133. Singh I, Khan M, Key L, Pai S (1998) Lovastatin for X‐linked adrenoleukodystrophy. N Engl J Med 339:702–703. [DOI] [PubMed] [Google Scholar]
- 134. Singh I, Moser AE, Moser HW, Kishimoto Y (1984) Adrenoleukodystrophy: impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts, and amniocytes. Pediatr Res 18:286–290. [DOI] [PubMed] [Google Scholar]
- 135. Singh J, Khan M, Singh I (2009) Silencing of Abcd1 and Abcd2 genes sensitizes astrocytes for inflammation: implication for X‐adrenoleukodystrophy. J Lipid Res 50:135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Smith BT, Sengupta TK, Singh I (2000) Intraperoxisomal localization of very‐long‐chain fatty acyl‐CoA synthetase: implication in X‐adrenoleukodystrophy. Exp Cell Res 254:309–320. [DOI] [PubMed] [Google Scholar]
- 137. Sutovsky S, Petrovic R, Chandoga J, Turcani P (2007) Adult onset cerebral form of X‐linked adrenoleukodystrophy with dementia of frontal lobe type with new L160P mutation in ABCD1 gene. J Neurol Sci 263:149–153. [DOI] [PubMed] [Google Scholar]
- 138. Theda C, Moser AB, Powers JM, Moser HW (1992) Phospholipids in X‐linked adrenoleukodystrophy white matter: fatty acid abnormalities before the onset of demyelination. J Neurol Sci 110:195–204. [DOI] [PubMed] [Google Scholar]
- 139. Traboulsi EI, Maumenee IH (1987) Ophthalmologic manifestations of X‐linked childhood adrenoleukodystrophy. Ophthalmology 94:47–52. [DOI] [PubMed] [Google Scholar]
- 140. Troffer‐Charlier N, Doerflinger N, Metzger E, Fouquet F, Mandel JL, Aubourg P (1998) Mirror expression of adrenoleukodystrophy and adrenoleukodystrophy related genes in mouse tissues and human cell lines. Eur J Cell Biol 75:254–264. [DOI] [PubMed] [Google Scholar]
- 141. Tzika AA, Ball WS Jr, Vigneron DB, Dunn RS, Nelson SJ, Kirks DR (1993) Childhood adrenoleukodystrophy: assessment with proton MR spectroscopy. Radiology 189:467–480. [DOI] [PubMed] [Google Scholar]
- 142. Uchiyama A, Aoyama T, Kamijo K, Uchida Y, Kondo N, Orii T, Hashimoto T (1996) Molecular cloning of cDNA encoding rat very long‐chain acyl‐CoA synthetase. J Biol Chem 271:30360–30365. [DOI] [PubMed] [Google Scholar]
- 143. Uchiyama M, Hata Y, Tada S (1991) MR imaging of adrenoleukodystrophy. Neuroradiology 33:25–29. [DOI] [PubMed] [Google Scholar]
- 144. Uto T, Contreras MA, Gilg AG, Singh I (2008) Oxidative imbalance in nonstimulated X‐adrenoleukodystrophy‐derived lymphoblasts. Dev Neurosci 30:410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Valianpour F, Selhorst JJ, Van Lint LE, Van Gennip AH, Wanders RJ, Kemp S (2003) Analysis of very long‐chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab 79:189–196. [DOI] [PubMed] [Google Scholar]
- 146. Van Der Knaap MS, Valk J (1989) MR of adrenoleukodystrophy: histopathologic correlations. Am J Neuroradiol 10:S12–S14. [PMC free article] [PubMed] [Google Scholar]
- 147. Van Geel BM, Assies J, Wanders RJ, Barth PG (1997) X linked adrenoleukodystrophy: clinical presentation, diagnosis, and therapy. J Neurol Neurosurg Psychiatry 63:4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Van Geel BM, Bezman L, Loes DJ, Moser HW, Raymond GV (2001) Evolution of phenotypes in adult male patients with X‐linked adrenoleukodystrophy. Ann Neurol 49:186–194. [DOI] [PubMed] [Google Scholar]
- 149. Van Geel BM, Koelman JH, Barth PG, Ongerboer de Visser BW (1996) Peripheral nerve abnormalities in adrenomyeloneuropathy: a clinical and electrodiagnostic study. Neurology 46:112–118. [DOI] [PubMed] [Google Scholar]
- 150. Van Rhijn I, Zajonc DM, Wilson IA, Moody DB (2005) T‐cell activation by lipopeptide antigens. Curr Opin Immunol 17:222–229. [DOI] [PubMed] [Google Scholar]
- 151. Van Roermund CW, Visser WF, Ijlst L, Van Cruchten A, Boek M, Kulik W et al (2008) The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl‐CoA esters. FASEB J 22:4201–4208. [DOI] [PubMed] [Google Scholar]
- 152. Vargas CR, Wajner M, Sirtori LR, Goulart L, Chiochetta M, Coelho D et al (2004) Evidence that oxidative stress is increased in patients with X‐linked adrenoleukodystrophy. Biochim Biophys Acta 1688:26–32. [DOI] [PubMed] [Google Scholar]
- 153. Wanders RJ, Van Roermund CW, Van Wijland MJ, Schutgens RB, Schram AW, Tager JM et al (1988) X‐linked adrenoleukodystrophy: identification of the primary defect at the level of a deficient peroxisomal very long chain fatty acyl‐CoA synthetase using a newly developed method for the isolation of peroxisomes from skin fibroblasts. J Inherit Metab Dis 11(Suppl. 2):173–177. [DOI] [PubMed] [Google Scholar]
- 154. Wanders RJ, Van Roermund CW, Van Wijland MJ, Schutgens RB, Van Den Bosch H, Schram AW, Tager JM (1988) Direct demonstration that the deficient oxidation of very long chain fatty acids in X‐linked adrenoleukodystrophy is due to an impaired ability of peroxisomes to activate very long chain fatty acids. Biochem Biophys Res Commun 153:618–624. [DOI] [PubMed] [Google Scholar]
- 155. Wanders RJ, Visser WF, Van Roermund CW, Kemp S, Waterham HR (2007) The peroxisomal ABC transporter family. Pflugers Arch 453:719–734. [DOI] [PubMed] [Google Scholar]
- 156. Wilichowski E, Ohlenbusch A, Korenke GC, Hunneman DH, Hanefeld F (1998) Identical mitochondrial DNA in monozygotic twins with discordant adrenoleukodystrophy phenotype. Ann Neurol 43:835–836. [DOI] [PubMed] [Google Scholar]
- 157. Yamada T, Shinnoh N, Kondo A, Uchiyama A, Shimozawa N, Kira J, Kobayashi T (2000) Very‐long‐chain fatty acid metabolism in adrenoleukodystrophy protein‐deficient mice. Cell Biochem Biophys 3:239–246. [DOI] [PubMed] [Google Scholar]
- 158. Young DC, Moody DB (2006) T‐cell recognition of glycolipids presented by CD1 proteins. Glycobiology 16: 103R–112R. [DOI] [PubMed] [Google Scholar]
- 159. Young RS, Ramer JC, Towfighi J, Weidner W, Lehman R, Moser HW (1982) Adrenoleukodystrophy: unusual computed tomographic appearance. Arch Neurol 39:782–783. [DOI] [PubMed] [Google Scholar]