

Abstract
Sporadic inclusion body myositis (s‐IBM), the most common muscle disease of older persons, is of unknown cause, and there is no enduring treatment. Abnormal accumulation of intracellular multi‐protein inclusions is a characteristic feature of the s‐IBM phenotype, and as such s‐IBM can be considered a “conformational disorder,” caused by protein unfolding/misfolding combined with the formation of inclusion bodies. Abnormal intracellular accumulation of unfolded proteins may lead to their aggregation and inclusion body formation.
The present article is focusing on the multiple proteins that are accumulated in the form of aggregates within s‐IBM muscle fibers, and it explores the most recent research advances directed toward a better understanding of mechanisms causing their impaired degradation and abnormal aggregation. We illustrate that, among other factors, abnormal misfolding, accumulation and aggregation of proteins are associated with their inadequate disposal—and these factors are combined with, and perhaps provoked by, an aging intracellular milieu. Other concurrent and possibly provocative phenomena known within s‐IBM muscle fibers are: endoplasmic reticulum stress and unfolded protein response, mitochondrial abnormalities, proteasome inhibition, lysosome abnormality and endodissolution. Together, these appear to lead to the s‐IBM‐specific vacuolar degeneration, and muscle fiber atrophy, concluding with muscle fiber death.
Keywords: α‐synuclein, amyloid‐β, autophagy, heat shock proteins, inclusion body myositis, inclusions, phosphorylated tau, proteasome inhibition, protein aggregation, protein misfolding
INTRODUCTION
Sporadic inclusion body myositis (s‐IBM) is the most common muscle disease of persons 50 years and older. s‐IBM muscle tissue shares several phenotypic similarities with brain tissue of Alzheimer's disease (AD) and Parkinson's disease Lewy bodies (recently reviewed in 7). The progressive course of s‐IBM leads to pronounced muscle weakness and wasting, resulting in severe disability, while brain function is unaffected. There is no enduring treatment 7, 34, 40. Clinical features of s‐IBM, pathologic muscle diagnostic criteria and various treatment approaches were recently summarized (40). Pathologically, two processes—vacuolar degeneration and atrophy of muscle fibers, and mononuclear cell inflammation—are characteristic features of s‐IBM muscle biopsies 5, 6, 7, 34. The vacuolar degeneration of muscle fibers is accompanied by accumulations within muscle fibers, mainly in their non‐vacuolated cytoplasm, of ubiquitinated, multi‐protein aggregates containing amyloid‐β (Aβ), phosphorylated tau (p‐tau) in the form of paired helical filaments (PHFs) and multiple other proteins 3, 5, 6, 7. The multi‐protein, intra‐muscle fiber aggregates contain proteins in the β‐pleated sheet conformation of amyloid 3, 5, 6, 7, 12, 69, indicating their unfolded/misfolded status. Because accumulation of these intracellular congophilic proteinaceous aggregates (inclusions) is a characteristic feature of the s‐IBM phenotype, s‐IBM has been considered a “conformational disorder”(5), characterized by the inclusion bodies containing aggregated unfolded/misfolded proteins. Unfolded/Misfolded proteins especially as oligomers are considered to be very toxic to cells 42, 61, 119. Intracellular abnormalities occurring in s‐IBM muscle fibers are illustrated in Figure 1. Aβ and/or its toxic oligomers are considered to play a key upstream pathogenic role in the s‐IBM pathogenesis leading to the demonstrated proteasome inhibition, oxidative stress, mitochondrial abnormalities and possibly to the inhibition of autophagy (details in 6, 7, and below).
Figure 1.
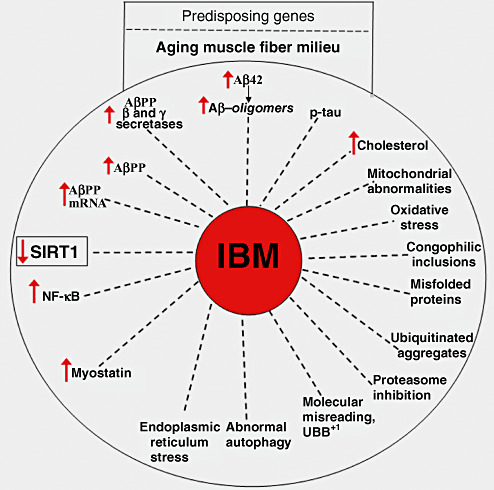
Intracellular abnormalities present in sporadic inclusion body myositis (s‐IBM) muscle fibers. We propose that predisposing genes and an aging muscle fiber milieu contribute to the muscle fiber abnormalities typical of s‐IBM (details in the text). Decreased SIRT1 activity might play a central, age‐related role in the s‐IBM pathogenic cascade.
We have recently demonstrated that the activity of SIRT1, an NAD‐dependent deacetylase, is inhibited in s‐IBM muscle fibers—this might be the cause of the detrimental activation of NF‐κB and contribute to other important abnormalities, including Aβ accumulation 78, 79, 80.
The present article focuses on the multiple proteins that are accumulated in the form of aggregates within s‐IBM muscle fibers, and it explores the most recent research advances directed toward better understanding of mechanisms causing their impaired degradation and abnormal aggregation. We illustrate that, among other factors, abnormal misfolding, accumulation and aggregation of proteins are associated with their inadequate disposal—and these factors are combined with, and perhaps provoked by, an aging intracellular milieu. Together, these appear to lead to the s‐IBM‐specific vacuolar degeneration and muscle fiber atrophy, concluding with muscle fiber death. The mechanism of muscle fiber death in s‐IBM is not well understood. There is compelling evidence that apoptosis does not participate in this process 46, 65, 72. Also, muscle fiber necrosis is very rare in s‐IBM muscle biopsy. The most likely process involves a recently described “autophagic cell death”(62 and below.), a mechanism described much earlier by one of us as the “endodissolusion” of muscle fibers (39).
The virtually identical complex of numerous abnormalities occurring within the thousands of long multinucleated non‐mitotic muscle fibers of each patient, in a consistently preferential topographical distribution in the musculature, and the similarity in essentially all s‐IBM patients suggest a similar initial driving mechanism (such as an integrated virus, self‐replicating protein or molecular deficiency or toxicity) that affects each individual muscle fiber de novo. This may be occurring possibly: (i) from an extra‐muscular source; or (ii) triggered by spontaneous disturbance of intra‐fiber age‐fragilized molecules. Each initiating mechanism presents a golden therapeutic opportunity, if it can be identified. Alternatively, a step in the ensuing self‐perpetuating, currently invincible pathogenic cascade involving the now‐known molecular abnormalities discussed below might prove easier to treat.
CHARACTERISTIC FEATURES OF MULTI‐PROTEIN AGGREGATES IN s‐IBM MUSCLE FIBERS
Multinucleated muscle fibers are usually several centimeters long. On a given 10 µm section of an s‐IBM muscle biopsy, the aggregates are present mainly in vacuole‐free regions of vacuolated muscle fiber cytoplasm and in cytoplasm of “non‐vacuolated” fibers—the latter can have vacuoles located farther along the fiber. Thus, in a given region of a fiber, aggregates seem to precede vacuole formation. The vacuoles themselves usually do not contain the IBM characteristic inclusions. The IBM autophagic vacuoles, which typically do contain membranous debris, appear to be lysosomal, and to be an end result of muscle fiber destruction (Figure 2A–D). While some of the vacuoles appear “rimmed” by a trichrome reddish material [that color indicating lipoprotein membranous material (41)], often the vacuoles do not have an obvious rim and appear “empty” (those must be differentiated from freezing artifact holes). By electron microscopy, s‐IBM vacuoles are often filled with myelin‐like bodies and other lysosomal‐like structures (Figure 2E). The autophagic nature of s‐IBM vacuoles is also demonstrated by their increased immunoreactivity of some of lysosomal enzymes 63, 100, and see below).
Figure 2.
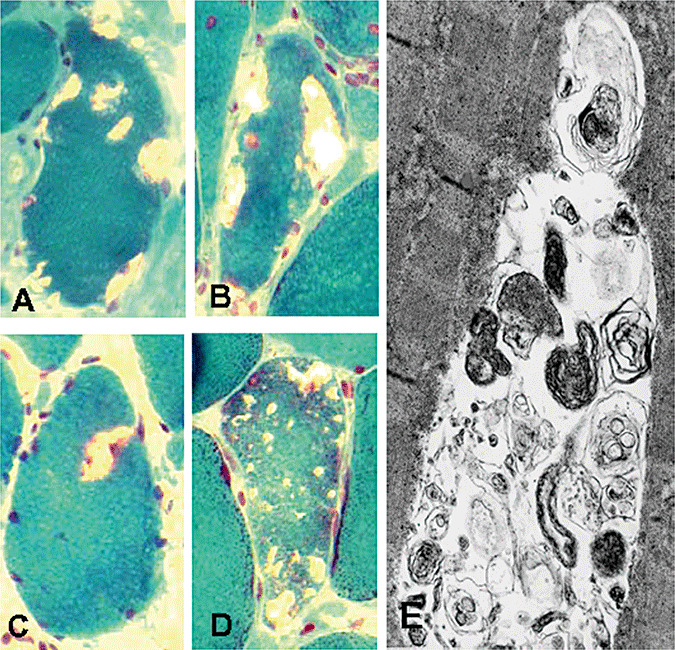
Light and electron microscopic (EM) features of sporadic inclusion body myositis vacuolated muscle fibers. Engel trichrome staining demonstrating typical vacuolated muscle fibers (A–D). On a given 10 µm transverse section, vacuoles are of various sizes; some contain floccular pinkish material, and some appear empty. Only one vacuole, in (C), appears to have a slight rim. (E) Transmission EM shows a vacuole containing inclusions consisting of numerous, various‐sized membranous whorls of autophagosomal/lysosomal debris. A–D, *2100; E, ×50 000.
Intra‐muscle fiber protein aggregates, identified by immunocytochemical staining with an antibody recognizing a specific protein accumulated within a given aggregate, are of two major types: larger rounded “plaque‐like” inclusions, containing Aβ (Figure 3A), and delicate squiggly inclusions containing p‐tau (Figure 3B). As both Aβ and p‐tau have a tendency to form β‐pleated sheet amyloid, accumulation of amyloid identified by fluorescence‐enhanced Congo red visualized through Texas red filters (12) typically has a similar pattern as the Aβ and p‐tau (Figure 3C). (Note: “Aβ” refers to one specific protein, whereas “amyloid” designates congophilic β‐pleated sheet configuration of any one of many proteins that can aggregate into this rather insoluble, abnormal three‐dimensional shape. We emphasize this because these similar terms are sometimes confused in the literature.) Multiple or single foci of amyloid are evident within about 60%–80% of the s‐IBM abnormal muscle fibers in a given transverse section.
Figure 3.
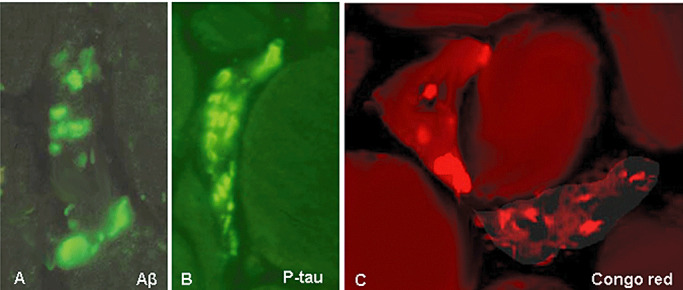
Immunohistochemistry and Congo red staining of sporadic inclusion body myositis muscle fibers. Inclusions composed of amyloid‐β (Aβ) appear as roundish, large plaque‐like aggregates (A), while those composed of phosphorylated tau (p‐tau) are more delicate and squiggly (B). Two different types of typical congophilic amyloid deposits by Congo red staining, visualized through Texas red filters and epifluorescence illumination (12); the fiber on the left has round, plaque‐like deposits, while the one at lower right, has more delicate, linear and squiggly inclusions (C). A–C, ×2300.
In our hands, positive crystal violet metachromasia staining, which specifically identifies β‐pleated sheet amyloid, is positive within the same muscle fibers that contain Congo red‐positive amyloid (Figure 4). In hereditary IBM caused by the GNE gene mutation, amyloid is usually not present, but in some older patients it is within rare muscle fibers. In hereditary IBM caused by VCP gene mutation, large clumps of amyloid deposits are present in muscle fiber nuclei, and occasionally within the cytoplasm (1). In myofibrillar myopathy, although abnormal muscle fibers were previously reported to contain congophilic amyloid by Congo red staining (93), by our studies they are crystal violet negative (not shown) and, therefore, presumably do not contain true amyloid in β‐pleated sheet conformation.
Figure 4.
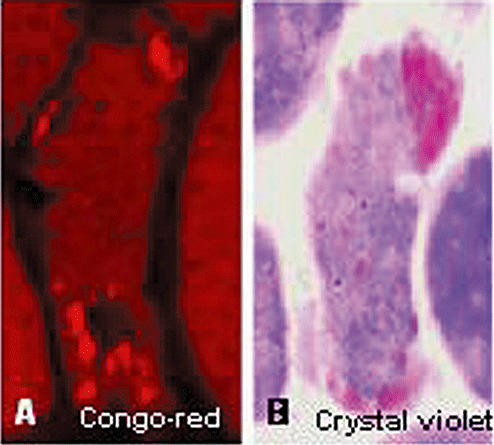
Congo red and crystal violet stainings of the same sporadic inclusion body myositis muscle fiber. Congo red staining, visualized through Texas red filters and epifluorescence illumination (A), shows a small congophilic amyloid deposit at the top right, and several deposits below the vacuole in the lower part of the fiber. Crystal violet staining of the same fiber, but several sections away, illustrates a large pink amyloid deposit at the top, and small deposits at the bottom of the fiber. As the sections are not closely adjacent, the fiber section in (B) does not have a vacuole in the lower part. A and B, ×2100.
PROTEINS AGGREGATED WITHIN s‐IBM MUSCLE FIBERS, AND THEIR PUTATIVE PATHOGENIC ROLE: A BRIEF DESCRIPTION
Both plaque‐like and squiggly aggregates contain, in addition to Aβ and p‐tau, several other proteins that also have the propensity to unfold/misfold and form β‐pleated sheet amyloid, for example, α‐synuclein (α‐syn), presenilin1 and cellular prion protein (11), as well as a number of other proteins having various functions and significance including: (i) markers of oxidative stress; (ii) endoplasmic reticulum (ER) chaperones indicative of the unfolded protein response (UPR); (iii) 26S proteasome components and the proteasome shuttle protein p62; (iv) mutated ubiquitin (UBB+1); (v) heat shock proteins; (vi) various transduction and transcription factors; and (vii) several other proteins (Table 1 and references therein, and reviewed in 3, 4, 5, 6, 7).
Table 1.
Protein components of the amyloid‐β (Aβ) and phosphorylated tau (p‐tau) intracellular inclusions and their possible functions in sporadic inclusion body myositis muscle fibers, as identified by immunohistochemical and immuno‐electron microscopical studies (see references therein). Abbreviations: NK = not known; Aβ = amyloid‐β; BACE = β‐amyloid‐converting enzyme; UBCH7 = ubiquitin conjugating enzyme H7; RNF5 = RING finger protein 5; HSP = heat‐shock protein; CHIP = carboxyl terminus of HSP70‐ interacting protein; BiP/GRP78 = immunoglobulin heavy chain‐binding protein/glucose‐regulated protein 78; ERP72 = endoplasmic reticulum protein 72 kDa; HERP = homocysteine‐induced endoplasmic reticulum protein; ERK = extracellular signal‐regulated kinase; CDK5 = cyclin‐dependent kinase 5; GSK‐3β = glycogen synthase kinase 3β; SOD = superoxide dismutase; NF‐κB = nuclear factor‐ κB; Ref1 = redox factor 1; NOS = nitric oxide synthase; SMN = survival motor‐neuron protein; PPARγ = peroxisome proliferator‐activated receptorγ; VCP = valosin containing protein; TDP‐43 = TAR DNA binding protein; LC3 = microtubule‐associated protein 1 light chain 3; RNA = ribonucleic acid.
| Aβ‐aggregates | p‐tau aggregates | References | |||
|---|---|---|---|---|---|
| Light microscopy | Electron microscopy | Light microscopy | Electron microscopy | ||
| Morphology, typical | Plaque‐like, rounded various size inclusions | 6–10 nm filaments, floccular and amorphous material | Squiggly | 15–21 nm paired‐helical filaments | 9, 10, 13, 70 |
| β‐Pleated sheet amyloid (Congo‐red+, crystal violet+) | + | + | 4, 5, 12, 69 | ||
| Proteins, various | |||||
| Aggregate‐prone proteins | |||||
| Aβ | + | + | − | − | 9, 10 |
| α‐Synuclein | + | + | − | − | 16, 85 |
| p‐Tau | − | − | + | + | 13, 70 |
| Prion protein, cellular | + | + | + | + | (11) |
| AβPP processing/Aβ deposition | |||||
| BACE1 and BACE2 | + | + | − | − | 102, 104 |
| Nicastrin | + | + | + | + | (105) |
| Presenilin1 | + | + | + | + | (15) |
| Neprilysin | + | + | NK | NK | (28) |
| NOGO B | + | + | − | − | (123) |
| Cystatin C | + | + | − | − | (103) |
| Transglutaminase 1 & 2 | + | NK | NK | NK | (30) |
| Ubiquitin–proteasome system | |||||
| Ubiquitin | + | + | + | + | (8) |
| Proteasome subunits | + | + | + | + | (45) |
| Parkin | + | + | NK | NK | (85) |
| UbcH7 | + | + | − | − | (85) |
| UBB+1 | + | + | + | + | (44) |
| RNF5 | + | NK | − | NK | (35) |
| Heat shock proteins | |||||
| Hsp70 and its cofactors | + | + | + | + | (84) |
| Hsp40 | + | + | + | + | (84) |
| CHIP | + | + | + | + | (Paciello and Askanas, unpub. obs.) |
| ER chaperones | |||||
| BiP/GRP78 | + | + | − | − | (107) |
| GRP94 | + | + | − | − | (107) |
| Calnexin | + | + | − | − | (107) |
| Calreticulin | + | + | − | − | (107) |
| ERP72 | + | + | − | − | (107) |
| HERP | + | + | − | − | (76) |
| Signal transduction components | |||||
| ERK | − | − | + | + | (115) |
| CDK5 | − | − | + | + | (116) |
| GSK‐3β | NK | NK | + | + | (117) |
| Casein kinase 1α | NK | NK | + | NK | (57) |
| Markers of oxidative stress | |||||
| Nitrotyrosine | + | + | + | + | (125) |
| SOD1 | NK | NK | NK | NK | (14) |
| Malondialdehyde | + | + | + | + | (22) |
| α1‐ Antichymotrypsin | + | NK | NK | NK | (19) |
| NFκB | NK | NK | + | + | (126) |
| Ref‐1 | + | + | + | + | (26) |
| iNOS, eNOS | − | − | + | + | (125) |
| Seleno‐glutathione peroxidase‐1 | NK | NK | NK | NK | (24) |
| Transcription components | |||||
| RNA polymeraseII | − | − | + | + | (118) |
| RNA | − | NK | + | NK | (25) |
| SMN | − | − | + | + | (23) |
| c‐Jun | + | + | + | + | (27) |
| NFκB | + | NK | + | + | (126) |
| Ref‐1 | + | + | + | + | (26) |
| PPARγ | + | + | NK | − | (77) |
| Other proteins | |||||
| Apolipoprotein E | + | + | + | + | (71) |
| Myostatin | + | + | − | − | (121) |
| VCP | NK | NK | NK | NK | (112) |
| TDP‐43 | NK | NK | NK | NK | 64, 114 |
| LC3 | + | NK | NK | NK | (67) |
| p62 | − | − | + | + | (81) |
Below, we briefly describe properties of some of the proteins that accumulate, concentrating on their possible pathogenic roles; details of other accumulated proteins are available in the references cited in Table 1.
Aβ precursor protein (AβPP) and Aβ
The first intracellular accumulation of Aβ in any disease was identified in s‐IBM muscle fibers (9)—an important role of intracellular Aβ toxicity was therefore postulated for s‐IBM muscle and, analogously, for AD neurons (2), in contrast to the then‐widespread view that Aβ toxicity in AD is extracellular. Subsequently, intraneuronal Aβ42 was demonstrated, and its intracellular toxicity also proposed (50, and reviewed in 48). Several recent experimental studies provide strong evidence that overexpression of AβPP and its proteolytic product Aβ plays an upstream role in the s‐IBM pathogenesis (reviewed in detail 6).
Increased accumulation of both AβPP and Aβ are identifiable early in s‐IBM abnormal muscle fibers (10). In addition, there are abnormalities of the AβPP processing machinery. For example, BACE1 and BACE2, which are transmembrane β‐secretases that cleave AβPP at the N‐terminal of Aβ, as well as nicastrin and presenilins, which are two of the components of the γ‐secretase system that cleaves AβPP at the C‐terminal of Aβ to generate either Aβ40 or Aβ42 (reviewed in 109), are increased in s‐IBM muscle fibers where they accumulate in aggregates colocalizing with Aβ102, 104. By electron microscopy, there are large clusters of densely (Figure 5B), or loosely packed 6–10 nm amyloid‐like fibrils, which are mainly composed of Aβ42 (Figure 5E). In s‐IBM muscle fibers, there is preferential accumulation of the Aβ42 fragment 7, 106, which is known to be more hydrophobic and more prone to self‐association and oligomerization, and is much more cytotoxic than Aβ40 42, 48, 111. There are also several factors acting in s‐IBM muscle fibers that might contribute to Aβ production, deposition and oligomerization (Table 1 and references therein).
Figure 5.
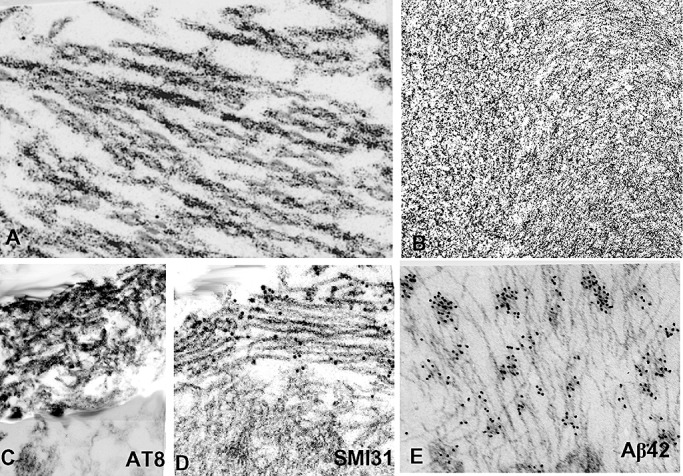
Transmission and immuno‐electron microscopy (EM) of sporadic inclusion body myositis (s‐IBM) abnormal muscle fibers. (A,B) Transmission EM. (A) A bundle of typical s‐IBM paired helical filaments. (B) A tightly packed cluster of 6–10 nm amyloid‐like filaments. (C–E) Immuno‐EM. (C) Horseradish peroxidase immunolocalization of phosphorylated tau (p‐tau) using AT8 antibody, shows that only a cluster of paired helical filaments (PHFs) in the upper left is immunostained, while the unaffected cytoplasm (below) is not immunoreactive. (D) Gold‐immuno‐EM using SMI31 antibody, shows gold particles, indicating p‐tau, only on the cluster of PHFs, while the unaffected cytoplasm (below) does not have any gold particles. (E) Gold‐immuno‐EM with a specific antibody recognizing Aβ42 showing gold particles on 6–10 nm amyloid‐like filaments. A, ×83 000; B,D,C×50 000; E, ×65 000.
p‐tau
As in AD brain, in s‐IBM muscle fibers p‐tau is accumulated intracellularly in the form of congophilic aggregates of delicate squiggly or linear inclusions (70 and Figure 3C), which by electron microscopy appear as PHFs (Figure 5A,D). Various antibodies recognizing several epitopes of p‐tau localize to those inclusions by light microscopic immunohistochemistry (Figure 3B), and by immunoelectron microscopy they are exclusively associated with the clusters of PHFs (Figure 5C,D) (70). Occasionally, accumulations of p‐tau occur within muscle fiber nuclei, but most of the p‐tau‐immunoreactive inclusions are cytoplasmic (Figure 6A,B). Several kinases known to phosphorylate tau are also accumulated within s‐IBM muscle fibers where they colocalize with p‐tau‐positive inclusions. Those include extracellular signal‐regulated kinase (115), cyclin‐dependent kinase 5 (116), glycogen synthase kinase 3β(117) and casein kinase 1 (57). s‐IBM–PHFs also contain RNA and the RNA‐binding protein survival motor neuron, and both were proposed to contribute to PHF formation (25). New studies related to neurodegeneration strongly suggest that accumulation of p‐tau could be cytotoxic to neurons (reviewed in 53, 56, 96). In contrast to Aβ exerting an intra‐muscle fiber cytotoxicity, there is no direct evidence yet that p‐tau might be toxic to s‐IBM muscle fibers; however, this possibility should be explored. Conceivably, the large masses of aggregated PHFs composed of p‐tau (3, 5) could severely impair muscle fiber integrity and function by: (i) physically disturbing contraction; (ii) hypothetical invisible tau oligomers sticking to and impairing various normal cellular components such as mitochondria and ER; and (iii) depriving the muscle fiber of its normal tau function.
Figure 6.
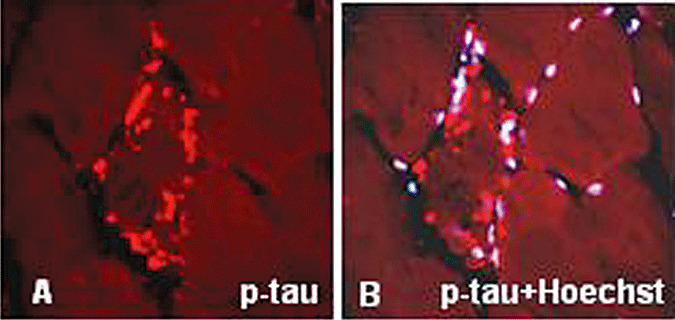
Immunohistochemistry of phosphorylated tau (p‐tau) in sporadic inclusion body myositis. (A) Several bundles of paired helical filaments immunostained with SMI‐31 antibody, which recognizes p‐tau, are present in an abnormal muscle fiber. (B) The same preparation as in (A) counterstained with a nuclei‐marker Hoechst, illustrates that most of the p‐tau immunoreactive aggregates are not associated with the nuclei. A,B, ×1250.
α‐Syn and parkin
α‐Syn has been implicated in the pathogenesis of several neurodegenerative diseases 18, 21, 32, and its overexpression has been associated with oxidative stress, impairment of proteasome and mitochondrial functions and other abnormalities 18, 32, 54, 66. Several years ago, we showed immunocytochemically that α‐syn is accumulated in s‐IBM muscle fibers in aggregates colocalizing with Aβ(16). More recently, we have shown by immunoblots that in s‐IBM human muscle fibers, the 22 kDa form of α‐syn, which is O‐glycosylated, is more expressed than its native 16 kDa form (85). The 22 kDa form was shown by others to be a target of ubiquitination by parkin (95). The preferential increase of the 22 kDa O‐glycosylated form of α‐syn in s‐IBM muscle fibers might be caused by proteasome inhibition, which has been demonstrated in s‐IBM fibers (45 and see below). α‐Syn is degraded by both the 26S proteasome and by lysosomal autophagy 68, 113. [Whether inhibition of lysosomal activity, as recently demonstrated in s‐IBM muscle fibers (81 and see below), also contributes to α‐syn increase, and accumulation is not yet known.]
Accordingly, a putative toxicity of α‐syn, in addition to the demonstrated cytotoxicity of Aβ, may contribute to the muscle fiber degeneration in s‐IBM. Such toxicity might not be related to the α‐syn and Aβ in the insoluble aggregates, but rather to an intracellular toxicity of their soluble oligomers and protofibrils 5, 85, 92.
Parkin is an E3–ubiquitin ligase that ubiquitinates α‐syn (91). Parkin is increased in s‐IBM muscle fibers, where it accumulates in the form of intra‐muscle fiber aggregates, which closely colocalize with α‐syn (85 and Figure 7A,B). In brains of sporadic Parkinson's disease patients, parkin and α‐syn accumulate in Lewy bodies, which are considered aggresomes (91). Parkin, in addition to ubiquitinating several proteins, is also considered to protect cells against toxicity induced by α‐syn, ER and other stresses, perhaps by helping to aggregate toxic α‐syn oligomers and promote their degradation 55, 99. Accordingly, we propose that increase of parkin in s‐IBM muscle fibers may represent a cellular defense mechanism against toxicity induced by α‐syn, ER and other stresses. However, the 2.7‐fold increase of parkin in s‐IBM muscle fibers might not be sufficient to overcome a sixfold increase of α‐syn (85), or to protect against other continuing stresses.
Figure 7.
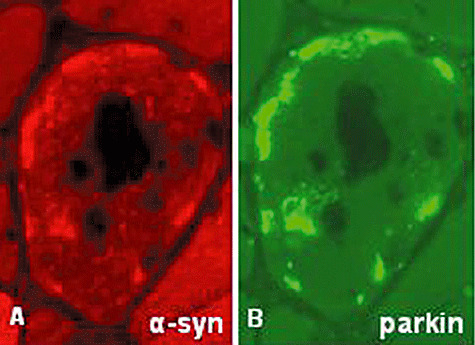
Immunohistochemistry of α‐synuclein (α‐syn) and parkin in sporadic inclusion body myositis (s‐IBM). (A,B) While there is a close localization of α‐syn‐ and parkin‐immunoreactive aggregates in an s‐IBM vacuolated muscle fiber, α‐syn appears to be increased somewhat more diffusely (this could be a real difference in distribution or an effect of different sensitivities of two antibodies). A,B, ×2100.
Accumulation of mutated ubiquitin (UBB+1)
Accumulation of UBB+1 within s‐IBM muscle fibers reflects the phenomenon of “molecular misreading.” This term designates acquired, non‐DNA‐encoded dinucleotide deletions occurring within mRNAs, resulting in production of potentially toxic mutant proteins (recently reviewed in 101). The aberrant transcripts are formed during or after transcription, and they can be translated from the deletion onward into the +1 reading frame to produce abnormal proteins, that is, mutant ubiquitin, termed UBB+1 (101). UBB+1 protein was shown accumulated in the plaques (containing Aβ) and neurofibrillary tangles (containing p‐tau) of AD brain (101). It was also found in brains of other neurodegenerative disorders in which inhibition of the proteasome has been proposed to play a pathogenetic role (101). UBB+1 itself can become ubiquitinated, and that form inhibits the proteasome (101). Accordingly, accumulation of UBB+1 was proposed to be a marker for proteasomal dysfunction in brain (101).
In s‐IBM muscle fibers, UBB+1 is accumulated in the form of aggregates, which can also contain wild‐type ubiquitin, Aβ and p‐tau (44). Those associations raise a possibility that UBB+1 might promote formation of those aggregates.
Our study showing accumulation of UBB+1 in muscle fibers of s‐IBM demonstrated for the first time that molecular misreading can occur in diseased human muscle (44). We proposed that the aging cellular environment of s‐IBM muscle fibers, combined with factors such as oxidative stress and perhaps other detrimental molecular events, leads to abnormal production and accumulation of UBB+1 (44), which might contribute to proteasome inhibition 44, 45 and see below). Moreover, if those aspects have led to the one example of molecular misreading we tested for, there may be yet undiscovered, and possibly pathogenic, examples of similar mutations on other proteins.
Accumulation of other proteins
The scope of this article does not permit a detailed description of several other proteins aggregated in s‐IBM muscle fibers that are illustrated and referenced in Table 1, such as myostatin, which was recently discussed 7, 121.
POSSIBLE MECHANISMS UNDERLYING PROTEIN CROWDING, MISFOLDING AND AGGREGATION IN s‐IBM MUSCLE FIBERS
General comments regarding protein misfolding and aggregation
In general, protein aggregation is considered to be caused by binding of partly unfolded or misfolded polypeptides induced by interaction between their inappropriately exposed hydrophobic surfaces 38, 42, 61, 94. Those interactions are highly specific (87). Normal cellular proteins folded correctly are soluble, structural or associated with cell membranes (reviewed in 94). In s‐IBM, insoluble aggregates of improperly folded proteins are usually cytoplasmic, occasionally nuclear. Although fully formed amyloid fibrils, which are insoluble, were previously considered to be cytotoxic, they may not be. There is current experimental evidence that pre‐amyloid oligomeric complexes or aggregates, either diffuse or in a protofibril stage, can be very cytotoxic 38, 42, 49. Unfolding or misfolding of proteins can occur in vivo and in vitro under several circumstances, including macromolecular crowding, oxidative stress, impaired disposal, exposure to toxins and “aging”38, 42, 47, 94. Increased transcription of several proteins and markers of oxidative stress occur in s‐IBM muscle fibers (reviewed in 3, 4, 5, 6, 7)—this mechanism, plus impaired disposal of proteins (see below) might contribute to unfolding or misfolding of IBM proteins. Aggregations of proteins into insoluble intracellular complexes/inclusion bodies have been proposed to be importantly related to several neurodegenerative disorders, including AD, Parkinson's disease, Huntington disease and amyotrophic lateral sclerosis (reviewed in 21, 29, 68, 94, 119).
Cellular mechanisms to eliminate misfolded/unfolded proteins
To eliminate misfolded proteins, a cell recruits mainly the following mechanisms: (i) protein refolding through the ER chaperones; (ii) protein refolding through heat shock proteins; (iii) protein degradation through the 26S ubiquitin–proteasome system (UPS); and (iv) protein degradation through autophagy, which involves formation of autophagosomes, their fusion with lysosomes and degradation of proteins by the lysosomal catabolic enzymes (reviewed in detail in 31, 33, 43, 68, 73, 82, 86, 128, 129). Below we describe abnormalities of these systems in s‐IBM muscle fibers.
ER Stress (ERS) and the UPR
The ER is an intracellular compartment having a critical role in the processing, folding and exporting of newly synthesized proteins into the secretory pathway (reviewed in 128, 129). In the ER, molecular chaperones are required to assure proper folding of unfolded or misfolded proteins 128, 129. Unfolded proteins accumulating in the ER cause ERS 128, 129. This elicits the UPR, a functional mechanism by which a cell attempts to protect itself against ERS 128, 129. In s‐IBM muscle fibers, we have previously reported evidence of ERS and the UPR 76, 107. As misfolded proteins continue to accumulate and aggregate in s‐IBM muscle fibers, we propose that in them the UPR is not adequate, because it is overwhelmed and/or impaired by the misfolded proteins. Our most recent studies have shown that ERS might be detrimental to the muscle fiber because, in cultured normal human muscle fibers, experimentally produced ERS: (i) induced myostatin, a negative regulator of muscle mass, through an NF‐κB‐related mechanism; and (ii) decreased SIRT1 deacetylase activity 78, 79, reviewed in detail in 7). Accordingly, ERS may importantly contribute to the s‐IBM pathogenesis.
Heat shock proteins (HSPs) and other chaperones
Chaperones of the HSP70 family constitute a very important group of molecular chaperones that play a role in protein folding and refolding, and disaggregation of partially unfolded proteins (reviewed in 43, 86). Their induction occurs in response to detrimental cellular conditions causing unfolded proteins. HSP70 binds exposed hydrophobic regions in proteins, preventing their aggregation. HSP70 function depends on ATP binding and hydrolysis, and it is linked to HSP70 cofactors. HSP40 increases the HSP70 folding function by enhancing ATPase activity of HSP70 43, 86.
s‐IBM‐vacuolated muscle fibers and some non‐vacuolated fibers (at a given cross‐sectional level) have strongly HSP70‐immunoreactive inclusions, which colocalize with Aβ and p‐tau immunoreactive inclusions (84 and Figure 8A,B). By immunoblots, HSP70 is 4.5‐fold increased as compared to control muscle samples, and it associates with Aβ by immunoprecipitation (84 and Figure 8C,D). Accordingly, HSP70 may participate in Aβ refolding, and may facilitate the refolding of other proteins in s‐IBM muscle fibers.
Figure 8.
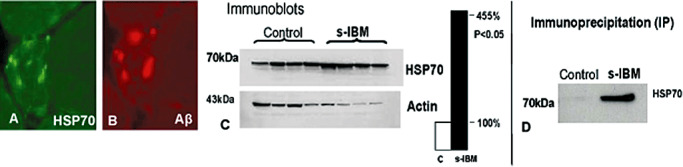
Heat shock protein 70 (HSP70) in sporadic inclusion body myositis (s‐IBM). (A,B) A close immuno‐colocalization of HSP70 and amyloid‐β (Aβ)‐immunoreactive aggregates in an abnormal muscle fiber. Both × 700. (C) Immunoblot showing that HSp70 is more than 4.5‐fold increased in muscle of s‐IBM as compared to normal controls. (D) Immunoprecipitation; s‐IBM and control muscle biopsies were immunoprecipitated with an antibody against Aβ and the membrane was then immunoprobed with an antibody against HSP70. In s‐IBM, HSP70 physically associates with Aβ, while control is negative.
αB‐crystallin (αBC) was shown to be accumulated in s‐IBM abnormal muscle fibers and in several other muscle diseases (17). αBC specifically recognizes and stabilizes proteins that have a propensity to aggregate and precipitate 36, 98. In cell‐free systems, αBC binds Aβ, prevents Aβ aggregation and prevents Aβ spontaneous fibril formation and fibril growth (88). However, when applied to cultured rat neurons extracellularly concomitantly with Aβ, αBC increases Aβ cytotoxicity (97), possibly because of an αBC influence in maintaining Aβ in its soluble oligomeric, highly cytotoxic form 88, 97. Our study (122) provided a novel demonstration that αBC physically associates with AβPP and Aβ oligomers both in s‐IBM‐biopsied muscle fibers and in AβPP‐overexpressing cultured human muscle fibers (122). We suggested that the binding of αBC to Aβ oligomers might be detrimental by retarding and diminishing their fibrillization and aggregation into visible, putatively nontoxic aggregates, thereby prolonging their existence as toxic oligomers (122).
The 26S proteasome
The UPS is a major degradation mechanism for normal, short‐lived proteins, and for misfolded proteins exported from the ER through a ubiquitin‐mediated ATP‐independent process 31, 82, 110. The 26S proteasome, composed of a catalytic 20S core and a 19S regulatory complex, is a ∼700 kDa multi‐subunit protease complex present in the cytoplasm and nucleus of eukaryotic cells.
In s‐IBM muscle fibers, we have demonstrated reduced activity of the three major proteasomal proteolytic enzymes (45), which probably contributes strongly to the abnormal accumulation of the ubiquitinated proteins aggregated in s‐IBM muscle fibers. Additional evidence of proteasome impairment in s‐IBM muscle fibers that we have described is formation of aggresomes (45), which experimentally form when proteasome function is inhibited (60). Whether aggresomes, in general, contribute to cellular death or protect cells from toxic effects of misfolded proteins remains uncertain. We also demonstrated that the AβPP/Aβ/proteasome interrelationship appears to be important in inducing proteasome abnormalities in s‐IBM muscle fibers because: (i) Aβ colocalized with proteasome subunits at the light‐microscopy level and they were both associated electron microscopically with the same structures; (ii) there was a physical association of Aβ/AβPP and proteasome protein by immunoprecipitation studies; and (iii) proteasome activity was inhibited in cultured human muscle fibers overexpressing Aβ/AβPP (45). Other factors present in s‐IBM muscle fibers that might contribute to inhibiting proteasome function include: an aging muscle fiber environment; protein over‐crowding; oxidative stress; and accumulated p‐tau, α‐syn and UBB+1 (reviewed in 6, 7)—all of these have been shown to be capable of inhibiting proteasome activity in other systems 32, 58, 66, 82, 101. The possible factors leading to proteasome inhibition in s‐IBM muscle fibers are illustrated in Table 1, (6). Whether proteasomal abnormalities participate in antigen presentation and T‐cell inflammation in s‐IBM muscle fibers remains to be studied.
Autophagy
“Autophagy” is used to describe catabolism of proteins and cellular components, such as mitochondria and other membranous structures, through lysosomal degradation (reviewed in 33, 68, 73, 75, 120). Autophagy has been closely linked to the protein quality control system. Disturbance of autophagy has been associated with several neurodegenerative disorders, including AD, Parkinson's and Huntington diseases 33, 68, 73, 75, 89, 120. The lysosome is an intracellular organelle whose main function is degradation and recycling of proteins originating both intracellularly and extracellularly (review in 33, 68, 73, 75, 120). Because of the ability of autophagy to remove unwanted or damaged proteins, this system, together with the 26S proteasome, constitutes the most important mechanism of unwanted protein disposal. The process of autophagy is generally divided into three different types: macroautophagy, microautophagy and chaperone‐mediated autophagy 33, 37, 68, 73, all three culminating in the final step of degradation of proteins by lysosomes. Macroautophagy relates to the formation and maturation of autophagosomes, which in mammalian cells are identified on immunoblots by the presence of LC3‐II protein 75, 120. Autophagosomes carry the proteins destined for degradation by lysosomes. After an autophagosome fuses with the lysosomal membrane, it disposes proteins into the lysosome, and then the final degradation of proteins by lysosomal enzymes occurs 33, 68, 73, 75, 120. In normal cells, the autophagosomal–lysosomal system (A‐L‐S) functions properly, and it assures proper protein quality and quantity. When misfolded or damage proteins accumulate and increase in a cell, either because of the proteasome inhibition, oxidative stress or other stressors, the need for autophagic degradation increases, which is followed by the increased formation of autophagosomes, often leading to the formation of autophagic vacuoles 37, 62, 75. This phenomenon is dramatically increased when the lysosomal activity is partially impaired, or the amount of material to be degraded exceeds lysosomal capabilities. s‐IBM vacuoles are considered autophagic, as they often contain: (i) lysosomal membranous debris by light and electron microscopy (Figure 2A–E), which is considered a result of partial damage within a still‐living muscle fiber; and (ii) increased immunoreactivity of some of the lysosomal enzymes 63, 100. In contrast to neurodegenerative disorders in which the role of autophagy has been intensively studied 33, 68, 73, 75, 89, 120, relatively little is known about how this process might be contributing to protein aggregation in s‐IBM muscle fibers. And, the A‐L‐S function and formation, indicated by autophagosome maturation and lysosomal activity, have not been adequately studied in s‐IBM, to our knowledge. Our most recent studies showed that in s‐IBM muscle fibers, in contrast to controls: (i) LC3‐II, which indicates increased autophagosome formation and maturation 75, 120, becomes evident (81); and (ii) the activity of lysosomal enzyme cathepsin D was decreased 50% despite actual increase of its protein (81). Together, these data suggest that in s‐IBM muscle fibers, there is: (i) impaired autophagy by reducing some key lysosomal functions; and (ii) an attempted compensation by excessive proliferation of lysosomes containing acid phosphatase (evident histochemically) and probably other hydrolytic enzymes that spill into the cytoplasm and gradually dissolve cellular contents, a phenomenon termed “endodissolution”(39).
In addition, p62/sequestosome, a ubiquitin‐binding protein shuttling ubiquitinated proteins for their degradation by both proteasome and A‐L‐S, is accumulated in aggregates and increased by immunoblots in s‐IBM muscle fibers (81, Nogalska et al, unpub. obs.). Immunopositive p62 inclusions were demonstrated in brains of neurodegenerative disorders 37, 124, 127. Experimentally, when functions of either the 26S proteasome or lysosomes are inhibited, LC3‐II, a marker of autophagosome maturation, and p62‐immunoreactive inclusions are present 20, 124, 127. The accumulation of p62‐immunopositive inclusions in s‐IBM muscle fibers suggests that p62 is recruited to carry ubiquitinated proteins for degradation, but its effort is not successful because of impaired lysosomal and proteasomal functions. s‐IBM muscle appears to suffer a “double‐hit” crippling protein disposal, namely inhibition of both the proteasome and impaired autophagy.
Muscle fiber aging
s‐IBM typically becomes clinically manifest in patients after age 50, and more often in their 60s and 70s. Among sporadic “inflammatory myopathy” patients, the described specific degenerative accumulations and aggregations of proteins within muscle fibers occur only in s‐IBM, not in polymyositis or dermatomyositis. We suggest that these accumulations are related to an aging‐based degenerative pathogenic cascade. For example, cellular aging was shown to promote accumulation of abnormal proteins and slow the degradation rate of normal and abnormal proteins 33, 51, 52, 59, 108. Playing a critical role in cellular aging is unsatisfactory removal of damaged cellular components caused by various factors, including oxidative stress and other stressors 33, 108. Understanding the interrelated phenomena of aging‐related protein misfolding and aggregation of proteins may clarify mechanisms relevant to s‐IBM. In the cellular milieu of the aging muscle fiber, there may be diminished homeostatic mechanisms caused by either: (i) diminished expression of “youthful” genes encoding beneficial cellular factors; or (ii) overexpression of yet unknown “aging” genes encoding toxic cellular factors. Premature aging of s‐IBM myoblasts in culture has recently been demonstrated (74). Several aspects of aging of human muscle, for example, those associated with mitochondrial abnormalities, such as ragged‐red fibers, cytochrome oxidase‐negative muscle fibers and multiple mitochondrial DNA deletions, are more common in s‐IBM muscle 83, 90. Aging changes within muscle mitochondria, or elsewhere in the muscle fiber, may predispose to the s‐IBM mitochondrial abnormalities, potentially provoking a vicious circle between mitochondrial malfunctions, oxidative stress and protein aggregation/accumulation.
CONCLUSION
We have presented our current understanding of mechanisms leading to the abnormal protein unfolding/misfolding and aggregation within s‐IBM muscle fibers, and their putative cytotoxic consequences. Improving the impaired protein disposal machinery and a better understanding of the mechanisms and consequences of human muscle fiber aging might provide new avenues for therapy in s‐IBM.
ACKNOWLEDGMENTS
Our studies described in this review were supported by the National Institutes of Health (NINDS grant NS34103 and NIA Merit Award AG16768), the Muscular Dystrophy Association, The Myositis Association and the Helen Lewis Research Funds. We thank our many research team colleagues who participated over the years in the studies described herein. Dr Nogalska is on leave from Department of Biochemistry, Medical University of Gdansk, Gdansk, Poland.
REFERENCES
- 1. Alvarez RB, Simmons Z, Askanas V (1998) New autosomal dominant inclusion‐body myopathy with many congophilic muscle nuclei that contained paired‐helical filaments composed of phosphorylated tau. Neurology 50:A204. [Google Scholar]
- 2. Askanas V, Engel WK (1998) Does overexpression of betaAPP in aging muscle have a pathogenic role and a relevance to Alzheimer's disease? Clues from inclusion body myositis, cultured human muscle, and transgenic mice. Am J Pathol 153:1673–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Askanas V, Engel WK (2001) Inclusion‐body myositis: newest concepts of pathogenesis and relation to aging and Alzheimer disease. J Neuropathol Exp Neurol 60:1–14. [DOI] [PubMed] [Google Scholar]
- 4. Askanas V, Engel WK (2003) Proposed pathogenetic cascade of inclusion‐body myositis: importance of amyloid‐beta, misfolded proteins, predisposing genes, and aging. Curr Opin Rheumatol 15:737–744. [DOI] [PubMed] [Google Scholar]
- 5. Askanas V, Engel WK (2006) Inclusion‐body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology 66:S39–S48. [DOI] [PubMed] [Google Scholar]
- 6. Askanas V, Engel WK (2007) Inclusion‐body myositis, a multifactorial muscle disease associated with aging: current concepts of pathogenesis. Curr Opin Rheumatol 19:550–559. [DOI] [PubMed] [Google Scholar]
- 7. Askanas V, Engel WK (2008) Inclusion‐body myositis: muscle‐fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer's and Parkinson's disease brains. Acta Neuropathol 116:583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Askanas V, Serdaroglu P, Engel WK, Alvarez RB (1991) Immunolocalization of ubiquitin in muscle biopsies of patients with inclusion body myositis and oculopharyngeal muscular dystrophy. Neurosci Lett 130:73–76. [DOI] [PubMed] [Google Scholar]
- 9. Askanas V, Engel WK, Alvarez RB (1992) Light and electron microscopic localization of beta‐amyloid protein in muscle biopsies of patients with inclusion‐body myositis. Am J Pathol 141:31–36. [PMC free article] [PubMed] [Google Scholar]
- 10. Askanas V, Alvarez RB, Engel WK (1993) Beta‐amyloid precursor epitopes in muscle fibers of inclusion body myositis. Ann Neurol 34:551–560. [DOI] [PubMed] [Google Scholar]
- 11. Askanas V, Bilak M, Engel WK, Alvarez RB, Tome F, Leclerc A (1993) Prion protein is abnormally accumulated in inclusion‐body myositis. Neuroreport 5:25–28. [DOI] [PubMed] [Google Scholar]
- 12. Askanas V, Engel WK, Alvarez RB (1993) Enhanced detection of congo‐red‐positive amyloid deposits in muscle fibers of inclusion body myositis and brain of Alzheimer's disease using fluorescence technique. Neurology 43:1265–1267. [DOI] [PubMed] [Google Scholar]
- 13. Askanas V, Engel WK, Bilak M, Alvarez RB, Selkoe DJ (1994) Twisted tubulofilaments of inclusion body myositis muscle resemble paired helical filaments of Alzheimer brain and contain hyperphosphorylated tau. Am J Pathol 144:177–187. [PMC free article] [PubMed] [Google Scholar]
- 14. Askanas V, Sarkozi E, Alvarez RB, McFerrin J, Engel WK, Siddique T (1996) Superoxide dismutase‐1 gene and protein in vacuolated muscle fibers of sporadic inclusion‐body myositis, hereditary inclusion‐body myopathy, and cultured human muscle after β‐amyloid precursor protein gene transfer. Neurology 46:A487. [Google Scholar]
- 15. Askanas V, Engel WK, Yang CC, Alvarez RB, Lee VM, Wisniewski T (1998) Light and electron microscopic immunolocalization of presenilin 1 in abnormal muscle fibers of patients with sporadic inclusion‐body myositis and autosomal‐recessive inclusion‐body myopathy. Am J Pathol 152:889–895. [PMC free article] [PubMed] [Google Scholar]
- 16. Askanas V, Engel WK, Alvarez RB, McFerrin J, Broccolini A (2000) Novel immunolocalization of alpha‐synuclein in human muscle of inclusion‐body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. J Neuropathol Exp Neurol 59:592–598. [DOI] [PubMed] [Google Scholar]
- 17. Banwell BL, Engel AG (2000) AlphaB‐crystallin immunolocalization yields new insights into inclusion body myositis. Neurology 54:1033–1041. [DOI] [PubMed] [Google Scholar]
- 18. Bennett MC (2005) The role of alpha‐synuclein in neurodegenerative diseases. Pharmacol Ther 105:311–331. [DOI] [PubMed] [Google Scholar]
- 19. Bilak M, Askanas V, Engel WK (1993) Strong immunoreactivity of alpha 1‐antichymotrypsin co‐localizes with beta‐amyloid protein and ubiquitin in vacuolated muscle fibers of inclusion‐body myositis. Acta Neuropathol 85:378–382. [DOI] [PubMed] [Google Scholar]
- 20. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A et al (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bossy‐Wetzel E, Schwarzenbacher R, Lipton SA (2004) Molecular pathways to neurodegeneration. Nat Med 10:S2–S9. [DOI] [PubMed] [Google Scholar]
- 22. Broccolini A, Engel WK, Alvarez RB, Thomas C, Yang CC, Askanas V (1998) Possible pathogenic role of malondialdehyde, a toxic product of lipid peroxidation, in sporadic inclusion‐body myositis. Neurology 50:A367–A368. [Google Scholar]
- 23. Broccolini A, Engel WK, Askanas V (1999) Localization of survival motor neuron protein in human apoptotic‐like and regenerating muscle fibers, and neuromuscular junctions. Neuroreport 10:1637–1641. [DOI] [PubMed] [Google Scholar]
- 24. Broccolini A, Mirault ME, Engel WK, Askanas V (1999) Abnormal accumulation of seleno‐glutathion peroxidase‐1 and catalase and their mRNAs in sporadic inclusion‐body myositis. Neurology 52(Suppl. 2):A333. [Google Scholar]
- 25. Broccolini A, Engel WK, Alvarez RB, Askanas V (2000) Paired helical filaments of inclusion‐body myositis muscle contain RNA and survival motor neuron protein. Am J Pathol 156:1151–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Broccolini A, Engel WK, Alvarez RB, Askanas V (2000) Redox factor‐1 in muscle biopsies of patients with inclusion‐body myositis. Neurosci Lett 287:1–4. [DOI] [PubMed] [Google Scholar]
- 27. Broccolini A, Engel WK, Askanas V (2000) Possible pathogenic role of redox factor 1 and AP‐1 transcription factor complex in sporadic inclusion‐body myositis. Neurology 54(Suppl. 3):A464. [Google Scholar]
- 28. Broccolini A, Gidaro T, Morosetti R, Gliubizzi C, Servidei T, Pescatori M et al (2006) Neprilysin participates in skeletal muscle regeneration and is accumulated in abnormal muscle fibres of inclusion body myositis. J Neurochem 96:777–789. [DOI] [PubMed] [Google Scholar]
- 29. Chiesa R, Piccardo P, Biasini E, Ghetti B, Harris DA (2008) Aggregated, wild‐type prion protein causes neurological dysfunction and synaptic abnormalities 10.1523/JNEUROSCI.3109‐08.2008. J Neurosci 28:13258–13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choi YC, Park GT, Kim TS, Sunwoo IN, Steinert PM, Kim SY (2000) Sporadic inclusion body myositis correlates with increased expression and cross‐linking by transglutaminases 1 and 2. J Biol Chem 275:8703–8710. [DOI] [PubMed] [Google Scholar]
- 31. Ciechanover A (2006) The ubiquitin proteolytic system: from a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology 66(2 Suppl. 1):S7–S19. [DOI] [PubMed] [Google Scholar]
- 32. Cookson MR (2005) The biochemistry of Parkinson's disease. Annu Rev Biochem 383:225–230. [DOI] [PubMed] [Google Scholar]
- 33. Cuervo AM (2008) Autophagy and aging: keeping that old broom working. Trends Genet 24:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dalakas MC (2006) Sporadic inclusion‐body myositis—diagnosis, pathogenesis and therapeutic strategies. Nat Clin Pract Neurol 2:437–447. [DOI] [PubMed] [Google Scholar]
- 35. Delaunay A, Bromberg KD, Hayashi Y, Mirabella M, Burch D, Kirkwood B et al (2008) The ER‐bound RING finger protein 5 (RNF5/RMA1) causes degenerative myopathy in transgenic mice and is deregulated in inclusion body myositis. PLoS ONE 3:e1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Derham BK, Harding JJ (1999) Alpha‐B crystallin as a molecular chaperone. Prog Retin Eye Res 18:463–509. [DOI] [PubMed] [Google Scholar]
- 37. Ding WX, Yin XM (2008) Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 4:141–150. [DOI] [PubMed] [Google Scholar]
- 38. Ellis RJ, Pinheiro TJ (2002) Medicine: danger—misfolding proteins. Nature 416:483–484. [DOI] [PubMed] [Google Scholar]
- 39. Engel WK (1979) Dagen des oordeels. Pathokinetic mechanisms and molecular messengers (a dramatic view). Arch Neurol 36:329–339. [DOI] [PubMed] [Google Scholar]
- 40. Engel WK, Askanas V (2006) Inclusion‐body myositis: clinical, diagnostic, and pathologic aspects. Neurology 66:S20–S29. [DOI] [PubMed] [Google Scholar]
- 41. Engel WK, Cunningham GG (1963) Rapid examination of muscle tissue and improved trichrome method for fresh‐frozen biopsy sections. Neurology 13:919–923. [DOI] [PubMed] [Google Scholar]
- 42. Ferreira ST, Vieira MN, De Felice FG (2007) Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB Life 59:332–345. [DOI] [PubMed] [Google Scholar]
- 43. Fink AL (1999) Chaperone‐mediated protein folding. Physiol Rev 79:425–449. [DOI] [PubMed] [Google Scholar]
- 44. Fratta P, Engel WK, Van Leeuwen FW, Hol EM, Vattemi G, Askanas V (2004) Mutant ubiquitin UBB+1 is accumulated in sporadic inclusion‐body myositis muscle fibers. Neurology 63:1114–1117. [DOI] [PubMed] [Google Scholar]
- 45. Fratta P, Engel WK, McFerrin J, Davies KJ, Lin SW, Askanas V (2005) Proteasome inhibition and aggresome formation in sporadic inclusion‐body myositis and in amyloid‐{beta} precursor protein‐overexpressing cultured human muscle fibers. Am J Pathol 167:517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fyhr IM, Lindberg C, Oldfors A (2002) Expression of Bcl‐2 in inclusion body myositis. Acta Neurol Scand 105:403–407. [DOI] [PubMed] [Google Scholar]
- 47. Garcia‐Mata R, Gao YS, Sztul E (2002) Hassles with taking out the garbage: aggravating aggresomes. Traffic 3:388–396. [DOI] [PubMed] [Google Scholar]
- 48. Glabe C (2001) Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer's disease. J Mol Neurosci 17:137–145. [DOI] [PubMed] [Google Scholar]
- 49. Glabe CG, Kayed R (2006) Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 66:S74–S78. [DOI] [PubMed] [Google Scholar]
- 50. Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F et al (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guarente L, Kenyon C (2000) Genetic pathways that regulate ageing in model organisms. Nature 408:255–262. [DOI] [PubMed] [Google Scholar]
- 52. Hekimi S, Guarente L (2003) Genetics and the specificity of the aging process. Science 299:1351–1354. [DOI] [PubMed] [Google Scholar]
- 53. Honson NS, Kuret J (2008) Tau aggregation and toxicity in tauopathic neurodegenerative diseases. J Alzheimers Dis 14:417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M et al (2000) Alpha‐synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol 157:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105:891–902. [DOI] [PubMed] [Google Scholar]
- 56. Iqbal K, Liu F, Gong CX, Alonso AD, Grundke‐Iqbal I (2009) Mechanisms of tau‐induced neurodegeneration. Acta Neuropathol Doi:10.00401‐009‐0486‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kannanayakal TJ, Mendell JR, Kuret J (2008) Casein kinase 1 alpha associates with the tau‐bearing lesions of inclusion body myositis. Neurosci Lett 431:141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keck S, Nitsch R, Grune T, Ullrich O (2003) Proteasome inhibition by paired helical filament‐tau in brains of patients with Alzheimer's disease. J Neurochem 85:115–122. [DOI] [PubMed] [Google Scholar]
- 59. Kirkwood TBL, Austad SN (2000) Why do we age? Nature 408:233–238. [DOI] [PubMed] [Google Scholar]
- 60. Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10:524–530. [DOI] [PubMed] [Google Scholar]
- 61. Kopito RR, Ron D (2000) Conformational disease. Nat Cell Biol 2:E207–E209. [DOI] [PubMed] [Google Scholar]
- 62. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kumamoto T, Ueyama H, Tsumura H, Toyoshima I, Tsuda T (2004) Expression of lysosome‐related proteins and genes in the skeletal muscles of inclusion body myositis. Acta Neuropathol 107:59–65. [DOI] [PubMed] [Google Scholar]
- 64. Kusters B, Van Hoeve BJA, Schelhaas HJ, Ter Laak H, Van Engelen BGM, Lammens M (2008) TDP‐43 accumulation is common in myopathies with rimmes vacuoles. Acta Neuropathol 117: 209–211. [DOI] [PubMed] [Google Scholar]
- 65. Li M, Dalakas MC (2000) Expression of human IAP‐like protein in skeletal muscle: a possible explanation for the rare incidence of muscle fiber apoptosis in T‐cell mediated inflammatory myopathies. J Neuroimmunol 106:1–5. [DOI] [PubMed] [Google Scholar]
- 66. Lindersson E, Beedholm R, Hojrup P, Moos T, Gai W, Hendil KB, Jensen PH (2004) Proteasomal inhibition by alpha‐synuclein filaments and oligomers. J Biol Chem 279:12924–12934. [DOI] [PubMed] [Google Scholar]
- 67. Lunemann JD, Schmidt J, Schmid D, Barthel K, Wrede A, Dalakas MC, Munz C (2007) Beta‐amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol 61:476–483. [DOI] [PubMed] [Google Scholar]
- 68. Martinez‐Vicente M, Cuervo AM (2007) Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol 6:352–361. [DOI] [PubMed] [Google Scholar]
- 69. Mendell JR, Sahenk Z, Gales T, Paul L (1991) Amyloid filaments in inclusion body myositis. Novel findings provide insight into nature of filaments. Arch Neurol 48:1229–1234. [DOI] [PubMed] [Google Scholar]
- 70. Mirabella M, Alvarez RB, Bilak M, Engel WK, Askanas V (1996) Difference in expression of phosphorylated tau epitopes between sporadic inclusion‐body myositis and hereditary inclusion‐body myopathies. J Neuropathol Exp Neurol 55:774–786. [DOI] [PubMed] [Google Scholar]
- 71. Mirabella M, Alvarez RB, Engel WK, Weisgraber KH, Askanas V (1996) Apolipoprotein E and apolipoprotein E messenger RNA in muscle of inclusion body myositis and myopathies. Ann Neurol 40:864–872. [DOI] [PubMed] [Google Scholar]
- 72. Mirabella M, Engel WK, Passinetti G, Finch CE, Askanas V (1996) Denervation of adult human muscle fibers induces apoptosis, evidenced by fragmentation of nuclear DNA, and increased expression of the clusterin (ApoJ) gene. Neurology 46:A270. [Google Scholar]
- 73. Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self‐digestion. Nature 451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Morosetti R, Broccolini A, Sancricca C, Gliubizzi C, Gidaro T, Tonali PA et al (2008) Increased aging in primary muscle cultures of sporadic inclusion‐body myositis. Neurobiol Aging Doi:10.1016/j.neurobiolaging.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 75. Nixon RA (2007) Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120:4081–4091. [DOI] [PubMed] [Google Scholar]
- 76. Nogalska A, Engel WK, McFerrin J, Kokame K, Komano H, Askanas V (2006) Homocysteine‐induced endoplasmic reticulum protein (Herp) is up‐regulated in sporadic inclusion‐body myositis and in endoplasmic reticulum stress‐induced cultured human muscle fibers. J Neurochem 96:1491–1499. [DOI] [PubMed] [Google Scholar]
- 77. Nogalska A, Engel WK, Askanas V (2007) Abnormalities of peroxisome proliferator‐activated receptor γ in sporadic inclusion‐body myositis muscle fibers. Ann Neurol 62:S13. [Google Scholar]
- 78. Nogalska A, Wojcik S, Engel WK, McFerrin J, Askanas V (2007) Endoplasmic reticulum stress induces myostatin precursor protein and NF‐kappaB in cultured human muscle fibers: relevance to inclusion body myositis. Exp Neurol 204:610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nogalska A, D'Agostino C, Engel WK, Davies KJ, Askanas V (2008) Decreased SIRT1 deacetylase activity in sporadic inclusion‐body myositis muscle fibers. Neurobiol Aging Doi:10.1016/j.neurobiolaging.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 80. Nogalska A, D'Agostino C, Engel WK, Askanas V (2009) Treatment with resveratrol, trans‐3,4′,5‐trihydroxystilbene polyphenol decreases amyloid β percursor protein, amyloid‐β oligomerization and myostatin in AβPP‐overexpressing cultured human muscle fibers (AβPP+‐CHMF‐IBM Model): relevance to treatment of sporadic‐IBM patients. Neurology 72:A103. [Google Scholar]
- 81. Nogalska A, Terracciano C, D'Agostino C, Engel WK, Askanas V (2009) Inhibited lysosomal activity in sporadic inclusion‐body myositis muscle fibers is associated with increased maturation of autophagososmes and accumulation of sequestososm1/p62 immunopositive inclusions. Neurology 72:A103. [Google Scholar]
- 82. Oddo S (2008) The ubiquitin‐proteasome system in Alzheimer's disease. J Cell Mol Med 12:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Oldfors A, Moslemi AR, Jonasson L, Ohlsson M, Kollberg G, Lindberg C (2006) Mitochondrial abnormalities in inclusion‐body myositis. Neurology 66:S49–S55. [DOI] [PubMed] [Google Scholar]
- 84. Ozturk A, McFerrin J, Engel WK, Askanas V (2004) HSP70 chaperone machinery in sporadic inclusion‐body myositis muscle fibers. Neurology 62:A168. [Google Scholar]
- 85. Paciello O, Wojcik S, Engel WK, McFerrin J, Askanas V (2006) Parkin and its association with alpha‐synuclein and AbetaPP in inclusion‐body myositis and AbetaPP‐overexpressing cultured human muscle fibers. Acta Myologica 25:13–22. [PubMed] [Google Scholar]
- 86. Pockley AG (2001) Heat shock proteins in health and disease: therapeutic targets or therapeutic agents? Expert Rev Mol Med 3:1–21. [DOI] [PubMed] [Google Scholar]
- 87. Rajan RS, Illing ME, Bence NF, Kopito RR (2001) Specificity in intracellular protein aggregation and inclusion body formation. Proc Natl Acad Sci U S A 98:13060–13065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Raman B, Ban T, Sakai M, Pasta SY, Ramakrishna T, Naiki H et al (2005) AlphaB‐crystallin, a small heat‐shock protein, prevents the amyloid fibril growth of an amyloid beta‐peptide and beta2‐microglobulin. Biochem J 392:573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rubinsztein DC (2006) The roles of intracellular protein‐degradation pathways in neurodegeneration. Nature 443:780–786. [DOI] [PubMed] [Google Scholar]
- 90. Santorelli FM, Sciacco M, Tanji K, Shanske S, Vu TH, Golzi V et al (1996) Multiple mitochondrial DNA deletions in sporadic inclusion body myositis: a study of 56 patients. Ann Neurol 39:789–795. [DOI] [PubMed] [Google Scholar]
- 91. Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L et al (2002) Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol 160:1655–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schulz B Jr (2007) Mechanisms of neurodegeneration in idiopathic Parkinson's disease. Parkinsonism Relat Disord 13:S306–S308. [DOI] [PubMed] [Google Scholar]
- 93. Selcen D, Ohno K, Engel AG (2004) Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain 127:439–451. [DOI] [PubMed] [Google Scholar]
- 94. Sherman MY, Goldberg AL (2001) Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29:15–32. [DOI] [PubMed] [Google Scholar]
- 95. Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R et al (2001) Ubiquitination of a new form of alpha‐synuclein by parkin from human brain: implications for Parkinson's disease. Science 293:263–269. [DOI] [PubMed] [Google Scholar]
- 96. Spires‐Jones TL, Stoothoff WH, De Calignon A, Jones PB, Hyman BT (2009) Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci 32:150–159. [DOI] [PubMed] [Google Scholar]
- 97. Stege GJ, Renkawek K, Overkamp PS, Verschuure P, Van Rijk AF, Reijnen‐Aalbers A et al (1999) The molecular chaperone alphaB‐crystallin enhances amyloid beta neurotoxicity. Biochem Biophys Res Commun 262:152–156. [DOI] [PubMed] [Google Scholar]
- 98. Sun Y, MacRae TH (2005) The small heat shock proteins and their role in human disease. FEBS J 272:2613–2627. [DOI] [PubMed] [Google Scholar]
- 99. Tsai YC, Fishman PS, Thakor NV, Oyler GA (2003) Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem 278:22044–22055. [DOI] [PubMed] [Google Scholar]
- 100. Tsuruta Y, Furuta A, Furuta K, Yamada T, Kira J, Iwaki T (2001) Expression of the lysosome‐associated membrane proteins in myopathies with rimmed vacuoles. Acta Neuropathol 101: 579–584. [DOI] [PubMed] [Google Scholar]
- 101. Van Leeuwen FW, Hol EM, Fischer DF (2006) Frameshift proteins in Alzheimer's disease and in other conformational disorders: time for the ubiquitin‐proteasome system. J Alzheimers Dis 9:319–325. [DOI] [PubMed] [Google Scholar]
- 102. Vattemi G, Engel WK, McFerrin J, Buxbaum JD, Pastorino L, Askanas V (2001) Presence of BACE1 and BACE2 in muscle fibres of patients with sporadic inclusion‐body myositis. Lancet 358:1962–1964. [DOI] [PubMed] [Google Scholar]
- 103. Vattemi G, Engel WK, McFerrin J, Askanas V (2003) Cystatin C colocalizes with amyloid‐beta and coimmunoprecipitates with amyloid‐beta precursor protein in sporadic inclusion‐body myositis muscles. J Neurochem 85:1539–1546. [DOI] [PubMed] [Google Scholar]
- 104. Vattemi G, Engel WK, McFerrin J, Pastorino L, Buxbaum JD, Askanas V (2003) BACE1 and BACE2 in pathologic and normal human muscle. Exp Neurol 179:150–158. [DOI] [PubMed] [Google Scholar]
- 105. Vattemi G, Kefi M, Engel WK, Askanas V (2003) Nicastrin, a novel protein participating in amyloid‐β production, is overexpressed in sporadic inclusion‐body myositis muscle. Neurology 60:A315. [Google Scholar]
- 106. Vattemi G, Nogalska A, Engel WK, D'Agostino C, Checler F, Askanas V (2009) Amyloid‐β42 is preferentially deposited in muscle biopsies of patients with sporadic inclusion‐body myositis (s‐IBM). Acta Neuropathol 117:569–574. [DOI] [PubMed] [Google Scholar]
- 107. Vattemi G, Engel WK, McFerrin J, Askanas V (2004) Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol 164:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Vellai T (2008) Autophagy genes and ageing. Cell Death Differ 16:94–102. [DOI] [PubMed] [Google Scholar]
- 109. Vetrivel KS, Thinakaran G (2006) Amyloidogenic processing of beta‐amyloid precursor protein in intracellular compartments. Neurology 66:S69–S73. [DOI] [PubMed] [Google Scholar]
- 110. Voges D, Zwickl P, Baumeister W (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem 68:1015–1068. [DOI] [PubMed] [Google Scholar]
- 111. Walsh DM, Selkoe DJ (2007) A beta oligomers—a decade of discovery. J Neurochem 101:1172–1184. [DOI] [PubMed] [Google Scholar]
- 112. Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D et al (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat Genet 36:377–381. [DOI] [PubMed] [Google Scholar]
- 113. Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003) Alpha‐synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278:25009–25013. [DOI] [PubMed] [Google Scholar]
- 114. Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M et al (2008) TDP‐43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry 79:1186–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wilczynski GM, Engel WK, Askanas V (2000) Association of active extracellular signal‐regulated protein kinase with paired helical filaments of inclusion‐body myositis muscle suggests its role in inclusion‐body myositis tau phosphorylation. Am J Pathol 156:1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wilczynski GM, Engel WK, Askanas V (2000) Cyclin‐dependent kinase 5 colocalizes with phosphorylated tau in human inclusion‐body myositis paired‐helical filaments and may play a role in tau phosphorylation. Neurosci Lett 293:33–36. [DOI] [PubMed] [Google Scholar]
- 117. Wilczynski GM, Broccolini A, Engel WK, Askanas V (2001) Novel proposed role of glycogen synthase kinase 3β in the pathogenesis of inclusion‐body myositis. Neurology 56(Suppl. 3):A464. [Google Scholar]
- 118. Wilczynski GM, Engel WK, Askanas V (2001) Novel cytoplasmic immunolocalization of RNA polymerase II in inclusion‐body myositis muscle. Neuroreport 12:1809–1814. [DOI] [PubMed] [Google Scholar]
- 119. Williams AJ, Paulson HL (2008) Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 31:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Winslow AR, Rubinsztein DC (2008) Autophagy in neurodegeneration and development. Biochim Biophys Acta—Mol Basis Dis 1782:723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wojcik S, Engel WK, McFerrin J, Askanas V (2005) Myostatin is increased and complexes with amyloid‐beta within sporadic inclusion‐body myositis muscle fibers. Acta Neuropathol 110:173–177. [DOI] [PubMed] [Google Scholar]
- 122. Wojcik S, Engel WK, McFerrin J, Paciello O, Askanas V (2006) AbetaPP‐overexpression and proteasome inhibition increase alphaB‐crystallin in cultured human muscle: relevance to inclusion‐body myositis. Neuromuscul Disord 16:839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wojcik S, Engel WK, Yan R, McFerrin J, Askanas V (2007) NOGO is increased and binds to BACE1 in sporadic inclusion‐body myositis and in A beta PP‐overexpressing cultured human muscle fibers. Acta Neuropathol 114:517–526. [DOI] [PubMed] [Google Scholar]
- 124. Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG, Wooten MC (2006) Signaling, polyubiquitination, trafficking, and inclusions: sequestosome 1/p62's role in neurodegenerative disease. J Biomed Biotechnol 62079:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yang CC, Alvarez RB, Engel WK, Askanas V (1996) Increase of nitric oxide synthases and nitrotyrosine in inclusion‐body myositis. Neuroreport 8:153–158. [DOI] [PubMed] [Google Scholar]
- 126. Yang CC, Askanas V, Engel WK, Alvarez RB (1998) Immunolocalization of transcription factor NF‐kappaB in inclusion‐body myositis muscle and at normal human neuromuscular junctions. Neurosci Lett 254:77–80. [DOI] [PubMed] [Google Scholar]
- 127. Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L et al (2002) p62 is a component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 160:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zhang K, Kaufman RJ (2006) The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 66:S102–S109. [DOI] [PubMed] [Google Scholar]
- 129. Zhang K, Kaufman RJ (2008) From endoplasmic‐reticulum stress to the inflammatory response. Nature 454:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]