

Abstract
Polyamines are small cationic molecules that in adult brain are connected to neuronal signaling by regulating inward‐rectifier K+‐channels and different glutamate receptors. Antizyme inhibitors (AZINs) regulate the cellular uptake of polyamines and activate ornithine decarboxylase (ODC), the rate‐limiting enzyme of polyamine synthesis. Elevated levels of ODC activity and polyamines are detected in various brain disorders including stroke and Alzheimer's disease (AD).
We originally reported a novel brain‐ and testis‐specific AZIN, called AZIN2, the distribution of which we have now studied in normal and diseased human brain by in situ hybridization and immunohistochemistry. We found the highest accumulation of AZIN2 in a pearl‐on‐the‐string‐like distribution along the axons in both the white and gray matter. AZIN2 was also detected in a vesicle‐like distribution in the somas of selected cortical pyramidal neurons. Double‐immunofluorescence staining revealed co‐localization of AZIN2 and N‐methyl D‐aspartate‐type glutamate receptors (NMDARs) in pyramidal neurons of the cortex. Moreover, we found accumulation of AZIN2 in brains affected by AD, but not by other neurodegenerative disorders (CADASIL or Lewy body disease). ODC activity is mostly linked to cell proliferation, whereas its regulation by AZIN2 in post‐mitotically differentiated neurons of the brain apparently serves different purposes. The subcellular distribution of AZIN2 suggests a role in vesicular trafficking.
Keywords: Alzheimer's disease, antizyme inhibitor, NMDA, ornithine decarboxylase, polyamine
INTRODUCTION
The activity of ornithine decarboxylase (ODC), the rate‐limiting enzyme of polyamine synthesis, is intimately linked to cell growth, transformation and differentiation (10). In the brain, high concentrations of polyamines (putrescine, spermidine and spermine) are present both intra‐ and extracellularly. The adult brain also contains large amounts of ODC protein but its enzymatic activity is very low. This indicates that most of the ODC in the central nervous system (CNS) is in complex with its inhibitors, the antizymes (AZ) (28). AZ sequesters monomeric ODC and prevents the formation of the catalytically active dimeric enzyme (37). AZ inhibitors (AZINs) are regulator proteins that display a high degree of sequence homology with ODC. Through their higher affinity for AZ, the AZINs liberate ODC from the AZ–ODC complex, leading to the emergence of catalytically active ODC 21, 25, 30, 50. In addition, AZINs also enhance the cellular uptake of polyamines 23, 31. Two isoforms of AZIN are known, AZIN1 and AZIN2. AZIN1 regulates cell growth and is ubiquitously expressed in most cells (23). We originally identified and cloned a second AZIN, now called AZIN2, and reported its high expression in testis and the CNS (46). The cellular distribution and the functional role of AZIN2 are still to be established.
The polyamines and ODC are related to the functional regulation of ion channels in the CNS. The inward‐rectifier K+ channels that regulate membrane‐resting potential and excitability are blocked by polyamines 14, 15, 29. Polyamines also inhibit the alfa‐amino‐3‐hydroxyl‐5‐methyl‐4‐isoxazole‐propionate (AMPA)‐ and kainate‐dependent fast depolarization of glutaminergic synapses, and thereby modulate neurotransmitter signals 8, 20. The N‐methyl D‐aspartate (NMDA)‐type excitatory glutamate receptors (NMDAR) form glutamate‐gated ion channels in the brain, which are regulated in multiple ways by polyamines. NMDARs mediate synaptic plasticity, which is pivotal for cognitive functions such as memory and learning (6). The local regulation of ODC activity and polyamine concentrations is obviously important for the channel functioning and neurotransmitter signaling. The molecular details of the local regulation of ODC activity are mostly unknown, however.
An increased amount and/or activity of ODC has been found in the brain in neurodegerative disorders like Alzheimer's disease (AD) 5, 45. Beta‐amyloid, which is implicated in the neurotoxicity in AD, accelerates the generation of reactive oxygen species (ROS), the accumulation of which induces ODC activity and the production of polyamines (54). The generation of ROS is enhanced by the binding of β‐amyloid oligomers to NMDARs and is dependent on NMDAR activity (11). The activation of NMDAR upregulates the expression of ODC in cultured neuronal cells (43), and this might reflect a physiological role of NMDAR signalling in the regulation of polyamine synthesis in vivo.
In this study, we investigated the expression and localization of AZIN2 in normal and diseased brain tissue, using immunohistochemistry and in situ hybridization. As NMDAR has been functionally implemented in both polyamine metabolism and neurotoxicity, we also studied the co‐distribution of AZIN2 and NMDAR1.
MATERIALS AND METHODS
The production of AZIN2 antibodies
Two antisera were raised in rabbits against synthetic peptides STRDLLKELTLGASQATTDEVA (antiserum 2) and STRDLLKELTLGASQATT (antiserum 3), corresponding to amino acids 18–39 and 18–35 of AZIN2 sequence (RefSeq Accession NM_052998). This N‐terminal region of AZIN2 has low homology to ODC and AZIN1 (14% and 5%, respectively). The longer peptide spans from exon one to exon three, leaving out exon two that is not contained in the splicing variants (SVs) 1–7 and 9. However, the shorter peptide is encoded only by exon 1, thus being capable of recognizing all splicing variants, including variants 8 and 10 (46) (see Supporting Information Figure S1). Artificially branched peptides for the immunization were synthesized with automated peptide synthesizer 433A (Applied Biosystems, Foster City, CA, USA) or Multipep (Intavis Ag, Koeln, Germany) using Fmoc chemistry and purified by reverse‐phase chromatography (Vydac C18, The Nest Group Inc., Southborough, MA, USA). Peptide purity was determined by matrix‐assisted laser desorption ionization‐time of flight mass spectrometry. The antibodies were produced by the Viikki Laboratory Animal Centre, University of Helsinki, Finland (permission no. HY176‐02 obtained from the Animal Experiment Board of the State Provincial Office of Southern Finland).
Specificity testing of AZIN2 antibodies
The antisera were tested against total cell lysates of COS‐7 cells transfected with flag‐tagged ODC, AZIN1 or AZIN2 (splicing variant 1), and empty vector (p3XFLAG‐CMV10, Sigma, St. Louis, MO, USA) by immunofluorescence stainings and western blotting. Immunofluorescent stainings were performed with antiserum 2 and mouse monoclonal M2 flag antibody (3 µg/mL, Sigma) followed by fluorophore‐labeled secondary antibodies. The stainings were visualized with an immunofluorescence microscope (Axiophot2, Zeiss, Jena, Germany, and SensiCam, PCO CCD Imaging, Kelheim, Germany).
In western blotting, 20 µg of proteins from empty vector, ODC‐, AZIN1‐ and AZIN2‐transfected cells were separated in 12% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE). After transferring the proteins to nitrocellulose membrane (Bio‐Rad Laboratories, Hercules, CA, USA) they were immunoblotted with antiserum 3 for AZIN2 (1:200 dilution in 1:1 Odyssey blocking buffer: Tris‐buffered saline (Li‐Cor, Lincoln, NE, USA) for 2 h at room temperature (RT). In antigen absorption testing, 150 µg of peptide used for immunization were incubated with antiserum dilution for 1.5 h, RT prior to immunoblotting. The proteins were visualized by the Odyssey infrared imaging system (Li‐Cor) after labeling with Alexa Fluor 680 goat anti‐rabbit IgG (1:10 000 dilution, Invitrogen, Carlsbad, CA, USA). The specificity of AZIN2 antiserum was further validated by immunoblotting the same membrane with mouse monoclonal M2 flag antibody (Sigma). Digital image processing was performed with Li‐Cor software and Adobe Photoshop CS2, version 9.0.2 (Adobe Systems Incorporated, San Jose, California, USA).
Tissue samples
Tissue samples from human brain taken for diagnostic purposes were used for immunohistochemistry. Seven control brain samples and five AD samples were obtained from the archives of the Department of Pathology, University of Helsinki, Finland. In addition, five AD samples and five matching controls belong to the Vantaa 85+ material, University of Helsinki, which is previously described in detail (47). The cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) and Lewy body dementia (DLB) samples, and the corresponding control (n = 4) and AD (n = 5) cases were provided by professor Hannu Kalimo, University of Helsinki, Finland. All dementia samples (AD, CADASIL and DLB) and 10 of the 17 control brain samples were taken post‐mortem. The other seven control samples were taken for diagnostic purposes during epilepsy surgery. All diagnoses were based on clinical assessment and histopathological evaluation by experienced neuropathologists.
The control samples obtained from autopsies (n = 10, mean age 78 years, range 56–95 years) were immunostained for beta‐amyloid, phospho‐tau and alpha‐synuclein and some of them with modified Bielchowsky silver stain as well. Tau tangles, if any, were detected only in entorhinal cortex in autopsied control samples; additionally Braak staging (9) 2–3 was available for five of control cases. Brain tissue from the temporal or frontal cortex of seven patients at a mean age of 13 years (range 1–38 years) surgically removed for epilepsy (n = 6) or astrocytoma G2 (n = 1) was used as control. The sections included our study were histologically normal or contained mild atrophia or dysplasia.
The AD diagnosis was based on neuropathological examination in which a specialist neuropathologist estimated the frequency of senile plaques in entorhinal cortex, hippocampus, and neocortex according to CERAD and NIA‐Reagan criteria including assessment of the Braak staging of the AD according to the immunohistochemistry‐based scheme (9) using AT8 phospho‐tau antibody. Braak staging of the samples was 5–6. The AD samples used in this study were collected from the frontal cortex and hippocampus.
The CADASIL diagnosis was verified by restriction enzyme analysis to identify the c.397C > T (p.Arg133Cys) or c.3206A > G (p.Tyr1069Cys) mutation in Notch3 gene (the two predominant mutations in the Finnish CADASIL population) as well as by electron microscopy of skin biopsy to identify the presence of the CADASIL‐specific granular osmiophilic material (GOM) as presented by Tikka et al (51). The CADASIL samples used in our study were from frontal cortex and hippocampus.
DLB diagnosis was based on neuropathological examination by a specialist neuropathologist using alpha‐synuclein immunostaining as recommended in the 2005 consensus criteria (36). PD Braak was available but was not specifically applied to these cases. Alpha‐synuclein pathology was present in all three regions, gyrus cinguli, frontal cortex and hippocampus. Variable extent of AD pathology was seen in all DLB cases.
Permission for using human material was obtained in accordance to the Finnish legislation, following the guidelines of the Declaration of Helsinki.
Immunohistochemistry
Sections of formalin‐fixed, paraffin‐embedded human brain tissue were air‐dried overnight at 37°C, deparaffinized in xylene and rehydrated in a graded ethanol series. Deparaffinized sections were boiled in a microwave oven (900 W) for 20 minutes in 10 mM citrate buffer, pH 6, for antigen retrieval, and blocked with CAS‐block (Zymed, South San Francisco, CA, USA) for 20 minutes at RT. Tissue sections were incubated with fresh aliquots of antiserum 2 or antiserum 3 for AZIN2 (diluted 1:300–600 in Chemmate, Dako, Glostrup, Denmark) overnight at +4°C. A Vectastain Elite Rabbit IgG kit (Vector Laboratories Inc, CA, USA) with 3‐amino‐9‐ethylcarbazole (Sigma) as a chromogenic substrate was used. Nuclei were visualized with hematoxylin counter‐staining. A parallel section stained with preimmune serum was included in all immunohistochemistry as a negative control. Light microscopic images were obtained with an Olympus BX51 microscope (Olympus Optical, Tokyo, Japan) and a Nikon Digital Sight DS‐5M camera (Nikon Corporation, Tokyo, Japan) using NIS‐Elements F 2.30 software (Nikon Corporation). Digital image processing was performed with Adobe Photoshop CS2, version 9.0.2 (Adobe Systems Incorporated).
Immunofluorescence staining
For double‐immunofluorescence staining, deparaffinized brain sections were boiled for 10 minutes in citrate buffer and avidin‐biotin blocked before sequential incubations, first with antibodies against AZIN2 (dilution 1:20) followed by biotinylated anti‐rabbit secondary antibody (dilution 1:30) and streptavidin : TRITC Star3B (dilution 1:30, Serotec ltd, Oxford, UK). After the first staining cycle with AZIN2 antibodies and fluorophore labeled‐streptavidin, avidin‐biotin block was performed. The samples were then stained with NMDAR1 antibodies (dilution 1:50, mouse monoclonal, BD Pharmingen, Franklin Lakes, NJ, USA) followed by biotinylated anti‐mouse secondary antibody (dilution 1:30) and streptavidin‐fluorescein isothiocyanate (FITC; dilution 1:30, Dako). Finally, autofluorescence was blocked with 0.3% Sudan Black B (Allied Chemical, NY, USA) treatment for 10 minutes. Antibodies, streptavidin‐labels and CAS‐block were diluted in 5% horse serum. 1% bovine serum albumin (Sigma), 0.1% Tween20 (Sigma) in phosphate‐buffered saline (PBS) was used for washing the samples. Microscopic images were obtained with an Axiophot2 immunofluorescence microscope (Zeiss) and SensiCam (PCO CCD Imaging).
In situ hybridization
Parallel tissue sections from human brain used in immunohistochemistry were hybridized with antisense and sense cRNA probes of AZIN2. The AZIN2 cDNA was amplified by polymerase chain reaction (PCR) using the upstream primer 5′‐CACTCCCTGAGCTGC‐3′ and the downstream primer 5′‐CTGCTCCGTGGATGGT‐3′, and subcloned in both orientations into the pCR2.1‐TOPO vector. cRNA probes were labeled with digoxigenin‐UTP by in vitro transcription with T7 polymerase using a DIG RNA Labelling Kit (Roche Diagnostics GmbH, Mannheim, Germany), and 200 ng of a probe were used for each hybridization. A Ventana Discovery Staining Platform was used for automated hybridization. The immunological detection of the DIG label was carried out with monoclonal biotinylated anti‐digoxin antibody (Jackson ImmunoResearch Laboratories Inc., PA, USA) at 1:2000 dilution, and using a BlueMap kit (Ventana Medical Systems Inc., Tucson, Arizona, USA). Finally the slides were counterstained with Nucleofast Red (Ventana). The probes were tested by Southern blotting not to react with ODC nor with AZIN1 mRNA.
Western blotting
Equal amounts of human neocortical gray and white matter (post‐mortem delay <48 h) were homogenized in 2% SDS supplemented with 1x Complete Protease Inhibitor Cocktail (Roche) and incubated for 10 minutes at +70°C. Cooled samples were boiled for 5 minutes with 2× Laemmli sample buffer, after which proteins were separated in 12% SDS‐PAGE. Nitrocellulose membranes with transferred proteins were immunoblotted with antibody 3 against AZIN2 (1:200 dilution) and mouse monoclonal anti‐β‐actin antibody (1:5000, Sigma). Primary antibodies were labeled with 1:10 000 dilutions of Alexa Fluor 680 goat anti‐rabbit IgG (Invitrogen) and Odyssey donkey anti‐mouse IRdye 800 (Li‐Cor). The detection was carried out using the Odyssey infrared imaging system (Li‐Cor).
Reverse transcription PCR (RT–PCR)
Two hundred nanograms of human brain Poly A+ RNA (Clontech Laboratories, Inc., Palo Alto, CA, USA) was used as a template in the RT‐PCR reaction (RobusT I RT‐PCR Kit, Finnzymes, Espoo, Finland) with primers to 5′‐ and 3′‐ends of the open reading frame of AZIN2 (5′‐TCAGCTCCTCCTGCAAGGCATGG‐3′ and 5′‐GAGGCCCACTCACATGATGCTCGCT‐3′). DNA was separated in 1.5% agarose‐gel and visualized after staining with SYBR Green I Nucleic Acid Gel Stain (Cambrex, Rockland, ME, USA) by AlphaImager HP (Alpha Innotech, San Leandro, CA, USA). A positive control was included from the RobusT RT‐PCR Kit.
RESULTS
AZIN2 is expressed in neurons
Different parts of the human brain were studied immunohistochemically with two peptide antibodies for the expression of AZIN2 (Table 1). Interestingly, the neuronal tracts were strongly visualized in all studied areas. Axon bundles emerging from lamina V to VI were strongly positive in neocortex and white matter (Figure 1B). Also dendrites in the subpial plexus were visualized. In the hippocampus, antiserum 3 detected AZIN2 in axons of the dentate gyrus, of the perforant pathway, alveus and entorhinal cortex (Figure 1C,D). In the cerebellum, the axons of basket and Purkinje cells expressed AZIN2 (Figure 1E–H). Also the tracts of the thalamus (Figure 1I) and medulla oblongata (Figure 1K) showed reactivity with antiserum 3.
Table 1.
The distribution of antizyme inhibitor 2 (AZIN2) in human brain. The distribution was investigated from immunohistochemical stainings.
| Area of central nervous system | AZIN2 expressing cells | Subcellular distribution of AZIN2 |
|---|---|---|
| Frontal cortex | Large pyramidal | Soma, apical dendrite and axon |
| Temporal cortex | Large pyramidal | Soma, apical dendrite and axon |
| Cerebellum | Purkinje and basket cells | Axon |
| Hippocampus | Dentate gyrus, perforant pathway, alveus, entorhinal ctx | Axon |
| Basal ganglia | Neurons | Axon |
| Thalamus | Neurons | Axon |
| Medulla oblongata | Neurons | Soma and axon |
Figure 1.
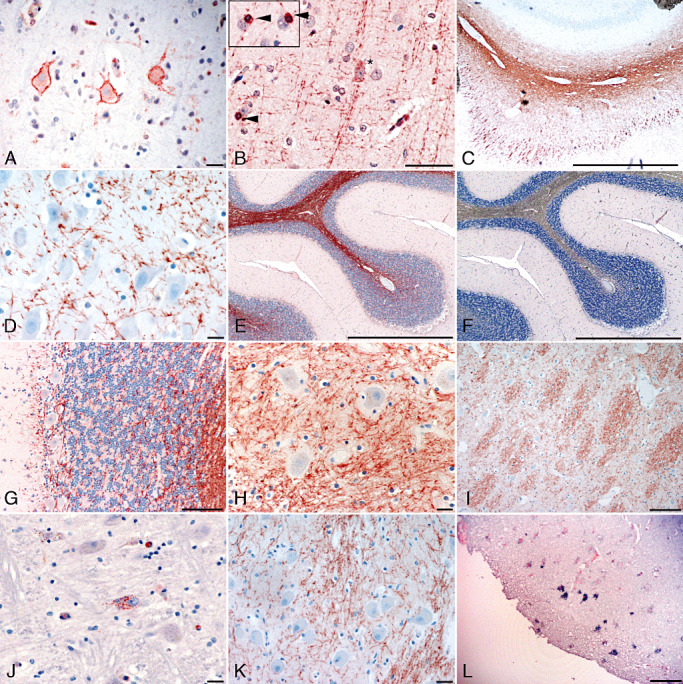
The expression of antizyme inhibitor 2 (AZIN2) in the human brain. Neuron‐specific expression of AZIN2 is detected with immunohistochemistry (A–K) and in situ hybridization (L). Immunohistochemistry with antibody 2 (A and J) against AZIN2 detects mainly neuronal bodies. Axons are strongly stained with antibody 3 (B–I, and K). (A–B) Neocortex, a pyramidal neuron (asterisk) and occasional oligodendrocytes (arrowhead), (C–D) hippocampus, (E–G) cerebellum, (H–I) thalamus, (J–K) medulla oblongata and (L) neocortex. Scale bar = 50 µm.
Staining of the temporal and frontal lobe visualized AZIN2 also in the soma of medium and large pyramidal neurons in lamina III to V (Figure 1A). However, the somas of all large pyramidal cells did not contain AZIN2. The expression was regional and involved most of the pyramidal neurons in selected areas. In addition to the neocortex, somas of some neurons in the medulla oblongata (Figure 1J) and Purkinje and basket cells in the cerebellum also expressed AZIN2. Neurons of the hippocampus, basal ganglia and thalamus expressed only minor reactivity with antiserum 2. In the sections from the temporal lobe, AZIN2 was occasionally detected in oligodendrocytes (Figure 1B, insert). No immunoreactivity was observed in parallel sections stained with preimmune serum (Figure 1F). The staining intensity and spatial distribution of AZIN2 was the same both in the surgically removed samples and in samples obtained from autopsies.
In situ hybridization was used to verify the immunohistochemical staining in the neocortex. An anti‐sense cRNA probe showed reactivity with large pyramidal neurons in the same neocortical areas where AZIN2 was detected with antisera (Figure 1L). In situ hybridization further demonstrated the regional distribution of cells expressing AZIN2 mRNA. No reactivity was detected with the sense probe.
Variable expression of the SVs of AZIN2
Our two peptide antibodies differed in their affinity for AZIN2 in the different subcellular compartments. Antiserum 3 detected a strong expression of AZIN2 in axons, and with a lower intensity it also stained pyramidal neurons of the neocortex. Antiserum 2, in contrast, recognized AZIN2 in the soma and apical dendrites of neurons but not axon‐localized AZIN2. We surmise that this variation in staining pattern was caused by the selective specificity of our antisera for different SVs of AZIN2. We have previously reported the existence of eight alternatively spliced forms of AZIN2 (46), and have further sequenced two additional SVs. Both of these variants have the same initiation‐ and stop‐codons as SV1. SV9 lacks 84‐bp from the 5′‐end of exon V, and SV10 has a 93‐bp extension in the 5′‐end of exon II. These variants, like all the others, were cloned from human adult brain QUICKclone cDNA library (Clontech). As illustrated in Figure 2A, RT‐PCR with primers to exon I and XI from human brain demonstrates multiple SVs, of which two predominate. Antibody 3 detected two strong bands of AZIN2 in western blotting from the white matter, whereas an additional band of apparently larger size dominated in the blottings from the gray matter (Figure 2B). Taken together, these results indicate that differently spliced variants of AZIN2 occur in the CNS.
Figure 2.
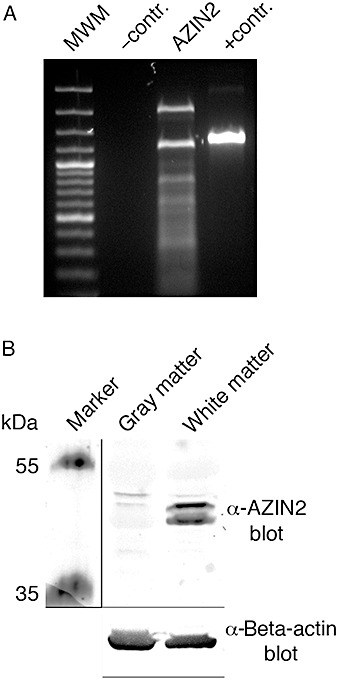
Alternative splicing of antizyme inhibitor 2 (AZIN2) in human brain. A. Reverse transcription polymerase chain reaction with primers to AZIN2 shows various splicing variants from pooled brain material. B. Antibody to AZIN2 (antibody 3) has a higher affinity for the splicing variants that are expressed in cerebral white matter, detected by immunoblotting, compared with apparently larger splicing variants in the gray matter.
The subcellular localization of AZIN2
AZIN2 was visualized in granular or vesicle‐like structures in the soma close to the plasma membrane of pyramidal neurons in the frontal cortex (Figure 3A). In axons, accumulation of AZIN2 was also observed in granular or vesicle‐like structures along the processus (Figure 3B). As in the somal area, AZIN2 was localized close to the inside of the plasma membrane following the outer boundaries of the neurite.
Figure 3.
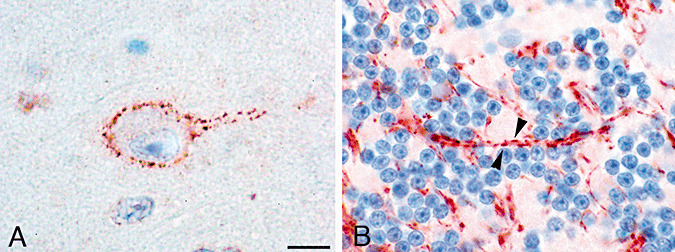
The subcellular localization of antizyme inhibitor 2 (AZIN2). A. In the somal area of neurons, AZIN2 is located close to the plasma membrane in a granular or vesicular distribution (staining with antibody 2). B. Patch‐like accumulations (arrowheads) of AZIN2 are detected in axons (antibody 3). Scale bar = 10 µm.
A more diffuse staining for AZIN2 was seen in the Purkinje cells, and in neurons of the hypoglossal nucleus and the pyramid of the medulla oblongata, although vesicle‐like distribution of the staining was also detected. A distinct granular distribution of AZIN2 was found in the lateral neurons of the medulla oblongata (area of spinal trigeminal and ambiguous nuclei) (Figure 1J). This staining was not restricted to the vicinity of the plasma membrane, but was rather dispersed throughout the cytoplasm and along the apical dendrite. Nuclear staining was occasionally observed in immunofluorescent and immunohistochemical specimens with both antisera to AZIN2.
AZIN2 co‐localizes with the NMDA receptor in neurons
As polyamines regulate NMDARs, we wanted to investigate the interrelationship between AZIN2 and NMDAR. The distribution of these proteins was studied by double immunofluorescence. NMDAR1 was detected in most of the pyramidal cells, whereas AZIN2 showed a more restricted expression. However, all the cells expressing AZIN2 were also positive for NMDAR1. This indicates an overlapping localization of AZIN2 and NMDAR1 in the cytoplasm of large pyramidal cells in the neocortex of normal brain (Figure 4).
Figure 4.
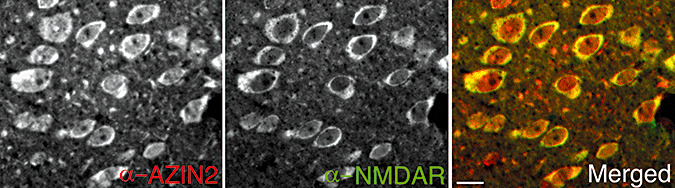
Co‐localization of antizyme inhibitor 2 (AZIN2) and N‐methyl D‐aspartate‐type glutamate receptor (NMDAR)1. Double‐immunofluorescence staining of neurons from the temporal cortex; yellow indicates co‐localization. Scale bar = 10 µm.
AD alters the expression of AZIN2
The expression of AZIN2 was studied in sections from the frontal lobe and the hippocampus of 15 brains affected by AD (Table 2). Virtually all the pyramidal cells in the frontal cortex stained positively for AZIN2, lacking the regional variation seen in normal brain. The individual cells of the AD brains also appeared to contain greater amounts of AZIN2. In the pyramidal cells, AZIN2 was detected in large aggregates or in vacuoles (Figure 5A,B). AZIN2 was also accumulated in the axons of AD patients (Figure 5C). In the samples stained with antiserum 3, the axons appeared swollen and AZIN2 was located in vacuole‐like aggregates that were larger than those seen in the axons of normal brain tissue.
Table 2.
The expression of AZIN2 in neurodegenerative diseases. The table sums up the basic information of the samples studied. The expression, investigated in human brain samples by immunohistochemistry, was strongest and most widely distributed in the cases of Alzheimer's disease (+++), followed by normal brain samples. In CADASIL and Lewy body dementias, only minor amounts (+, +/−) of AZI2 were detected.
| Diagnosis | Level of AZI2 expression | Number of cases (n) | Age, mean; range (years) | Gender (male : female) | Dementia |
|---|---|---|---|---|---|
| Lewy body dementia | +/− | 4 | 75; 63–81 | 2:2 | 4/4 |
| CADASIL‐dementia | + | 4 | 62; 58–64 | 3:1 | 4/4 |
| Alzheimer's disease | +++ | 15 | 81; 63–98 | 6:9 | 15/15 |
| Normal | +/++ | 17 | 41; 1–95 | 13:4 | 2/17 |
Figure 5.
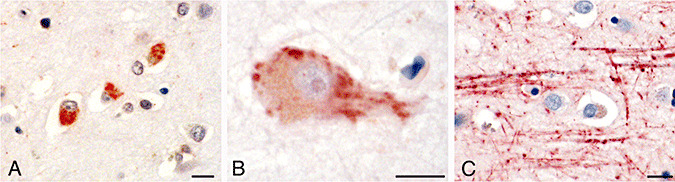
In Alzheimer's disease, antizyme inhibitor 2 (AZIN2) accumulates in the neuronal bodies and axons. Immunohistochemical stainings with antibodies to AZIN2 of neurons of hippocampus (A), and neurons and axons from the frontal cortex (B and C). Scale bar = 10 µm.
The neurons in hippocampal regions' subdivisions 1‐3 of Ammon's horn/cornu Ammonis (CA1‐3) of AD brains displayed a robust expression of AZIN2, contrary to the case in normal brain samples. Other hippocampal neurons in AD brains showed interindividual differences in their expression of AZIN2. Staining with preimmune serum remained negative.
The expression of AZIN2 was also studied in brain samples from patients with CADASIL dementia, characterized by a thickening of arterioles, and from patients with DLB in which intraneural Lewy bodies are detected in the neocortex and brain stem. The expression of AZIN2 in DLB and CADASIL dementia was mainly similar to that seen in samples from the corresponding areas of normal brain (Table 2).
Validation of the specificity of antibodies
Validation of the antibody specificity was carried out by western blotting of cell lysates expressing flag‐tagged AZIN2, AZIN1, ODC or empty vector. Antiserum 3 detected three bands of approximately 51–55 kDa in size in lysates from AZIN2 overexpressing cells (Supporting Information, Figure S2). No signals from ODC‐ or AZIN1‐expressing cells were observed. All transfected proteins were visualized with an antibody against the flag‐tag. Our AZIN2 antibody detected no bands after antigen absorption, thus indicating its specificity for AZIN2 (Supporting Information, Figure 2A).
As antiserum 2 did not work in western blotting, its specificity was tested with these same COS‐7 transfectants by immunofluorescence staining. Positive signals for AZIN2 were received from stainings of AZIN2‐ and ODC‐transfected cells. At the subcellular level, the localizations of AZIN2 and flag‐tag overlapped only in the AZIN2‐transfectants.
DISCUSSION
Polyamines and ODC activity, which AZIN2 is assumed to regulate, are strongly associated with cell growth and transformation. The expression of AZIN2 in non‐proliferating neurons, however, implicates other functions. In addition to neural cells, we have detected the expression of AZIN2 in Leydig cells and in mast cells 56, 57 both of which are terminally differentiated cells. Most of the ODC in the brain is bound to AZ that blocks its enzymatic activity 26, 27, 28. However, various stimuli, including electrical and chemical stimulation, and traumatic injuries, activate ODC 2, 7, 12, 35, 44.
AZIN1 is distributed ubiquitously and its expression is stimulated by mitotic signaling (41) leading to upregulated ODC activity and elevated polyamine concentrations 23, 24. AZIN1 is also highly expressed in certain cancers, indicating that they play a role in cell transformation and malignant growth 19, 23, 49. We have earlier found a low or absent expression of AZIN2 in cancer cell lines (46). The ultimate role of AZIN2 in regulating ODC activity and the concentration of polyamines is still incompletely understood, but the activity of AZIN2 is evidently not connected with cell proliferation, and it appears that AZIN1 and AZIN2 regulate ODC for different purposes.
The axon bundles in neocortex, thalamus, cerebellum and medulla oblongata stained strongly positively for AZIN2. AZIN2 was distributed in vesicle‐like or granular structures along the edges of the axons. A similar vesicle‐like distribution was detected under the plasma membrane in the somal area. The vivid traffic of neurosecretory granules along the axonal cytoskeleton depends greatly on the activation of G‐proteins [for a review, see (40)]. We have recently demonstrated that RhoA, a G‐protein regulating cytoskeleton dynamics, is activated via polyamination, which is dependent on the activity and translocation of ODC (33). It is tempting to speculate that AZIN2 might regulate ODC‐dependent local polyamination of also other G‐proteins involved in the traffic of neurosecretory vesicles.
Both of our two rabbit antibodies raised against AZIN2 peptides showed specificity in western blotting and immunofluorescence staining against full‐length AZIN2. However, the antibodies detected partly different distributions of AZIN2 in the brain. Only antibody 3 showed robust staining of AZIN2 in axons, whereas antibody 2 detected AZIN2 mainly in the somas of selected pyramidal neurons. This suggests an anatomically divergent distribution of the different SVs of the protein. Indeed, by RT‐PCR we detected the expression of several SVs in human brain and western blotting revealed differently spliced species of AZIN2 in the gray and white matter. The functional role of these SVs of AZIN2 remains to be elucidated, but it is possible that the variants target their expression to specific cellular or sub‐cellular distribution.
The expression of AZIN2 in the somas of pyramidal neurons differed between adjacent areas in the neocortex, giving an impression of spatial and/or temporal regulation of AZIN2 expression. Furthermore, AZIN2 co‐localized with NMDAR in pyramidal neurons, although the positive staining for NMDAR was more widely distributed. NMDARs constantly circulate between the synaptic plasma membrane and cytoplasmic vesicles upon neuronal activation and sensory experience, contributing to long‐term potentiation 17, 48. Vesicles containing NMDARs are transported along the microtubules to postsynaptic density and extrasynaptic areas 52, 53. Furhermore, the binding of polyamines from the extra‐ or intracellular side to NMDAR can either enhance or inhibit the influx of Ca2+ 1, 4, 13. Pretreatment of cultured mouse cortical neurons with difluoromethylornithine (DFMO), a specific inhibitor of ODC, abolished NMDA‐induced neurotoxicity (34). This indicates that intracellular levels of polyamines are involved in the function of NMDAR. Given that polyamines regulate NMDAR, AZIN2 might influence glutamate‐mediated signaling by controlling the local synthesis of polyamines.
Elevated ODC activity and putrescine concentrations have been detected in the brain in pathological conditions such as ischemia and AD 5, 22, 32, 38, 39. In AD, the pathological accumulation of β‐amyloid induces activation of ODC, in addition to its ability to provoke the generation of neurotoxic ROS 3, 18, 43. Putrescine functions as a scavenger of free radicals and thereby buffers the cytotoxic effects of ROS (55). Yatin et al have further shown that β‐amyloid, in addition to the induction of ODC activity, also stimulates polyamine uptake (54). The cellular uptake of polyamines and the induction of ODC activity are regulated by AZINs. The accumulation of AZIN2 in AD could display neuroprotective functions by enhancing the production and uptake of putrescine stimulated by β‐amyloid. Nilsson et al have compared the localization of ODC in normal brain and AD brain (42). They found evidence for translocation of ODC from nucleus to the cytoplasm of neocortical pyramidal cells early in AD. In addition, elevated expression of ODC was found in cerebellar Purkinje cells and in hippocampus of AD brains. We detected an elevated expression of AZIN2 also in Purkinje cells and the hippocampus in AD. Earlier reports on the immunohistochemical location of ODC in the brain may nevertheless require reinterpretation. The antibodies used were raised before the identification of AZIN2 and may therefore have resulted in substantial cross‐reactivity because of the high degree of homology between ODC and AZIN2.
In DLB and in Parkinson's disease, the aggregation of α‐synuclein is considered to be a major pathological event. The binding of spermine to α‐synuclein provokes folding changes and leads to the formation of aggregates (16). We, however, did not detect any increased expression of AZIN2 in Lewy body disease, indicating that the accumulation of AZIN2 is merely a specific feature of AD.
As polyamines participate in many functions of a cell, their concentration has to be locally regulated. This would require translocation of ODC to the different cell compartments, with local differences in the regulation and regulators. AZIN2 apparently acts as a local regulator of ODC, providing a rapidly mobilized tool for the fine adjustment of polyamine generation.
Supporting information
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
We thank Tiiu Arumäe, Anu Harju, Jussi Hepojoki, Hannu Kalimo, Irina Suomalainen and Anna Wilenius for technical assistance. This work was supported by the Sigrid Juselius Foundation, the Academy of Finland, and Finska Läkaresällskapet.
REFERENCES
- 1. Araneda RC, Lan JY, Zheng X, Zukin RS, Bennett MV (1999) Spermine and arcaine block and permeate N‐methyl‐D‐aspartate receptor channels. Biophys J 76:2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baskaya MK, Rao AM, Prasad MR, Dempsey RJ (1996) Regional activity of ornithine decarboxylase and edema formation after traumatic brain injury. Neurosurgery 38:140–145. [DOI] [PubMed] [Google Scholar]
- 3. Behl C, Davis JB, Lesley R, Schubert D (1994) Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77:817–827. [DOI] [PubMed] [Google Scholar]
- 4. Benveniste M, Mayer ML (1993) Multiple effects of spermine on N‐methyl‐D‐aspartic acid receptor responses of rat cultured hippocampal neurones. J Physiol 464:131–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bernstein HG, Muller M (1995) Increased immunostaining for L‐ornithine decarboxylase occurs in neocortical neurons of Alzheimer's disease patients. Neurosci Lett 186:123–126. [DOI] [PubMed] [Google Scholar]
- 6. Bliss TV, Collingridge GL (1993) A synaptic model of memory: long‐term potentiation in the hippocampus. Nature 361:31–39. [DOI] [PubMed] [Google Scholar]
- 7. Bondy SC, Mitchell CL, Rahmaan S, Mason G (1987) Regional variation in the response of cerebral ornithine decarboxylase to electroconvulsive shock. Neurochem Pathol 7:129–141. [DOI] [PubMed] [Google Scholar]
- 8. Bowie D, Mayer ML (1995) Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine‐mediated ion channel block. Neuron 15:453–462. [DOI] [PubMed] [Google Scholar]
- 9. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davis RH, Morris DR, Coffino P (1992) Sequestered end products and enzyme regulation: the case of ornithine decarboxylase. Microbiol Rev 56:280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST et al (2007) Abeta oligomers induce neuronal oxidative stress through an N‐methyl‐D‐aspartate receptor‐dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 282:11590–11601. [DOI] [PubMed] [Google Scholar]
- 12. Dienel GA, Cruz NF (1984) Induction of brain ornithine decarboxylase during recovery from metabolic, mechanical, thermal, or chemical injury. J Neurochem 42:1053–1061. [DOI] [PubMed] [Google Scholar]
- 13. Durand GM, Bennett MV, Zukin RS (1993) Splice variants of the N‐methyl‐D‐aspartate receptor NR1 identify domains involved in regulation by polyamines and protein kinase C. Proc Natl Acad Sci U S A 90:6731–6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fakler B, Brandle U, Bond C, Glowatzki E, Konig C, Adelman JP et al (1994) A structural determinant of differential sensitivity of cloned inward rectifier K+ channels to intracellular spermine. FEBS Lett 356:199–203. [DOI] [PubMed] [Google Scholar]
- 15. Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM (1994) Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science 266:1068–1072. [DOI] [PubMed] [Google Scholar]
- 16. Grabenauer M, Bernstein SL, Lee JC, Wyttenbach T, Dupuis NF, Gray HB et al (2008) Spermine binding to Parkinson's protein alpha‐synuclein and its disease‐related A30P and A53T mutants. J Phys Chem B 112:11147–11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grosshans DR, Clayton DA, Coultrap SJ, Browning MD (2002) LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci 5:27–33. [DOI] [PubMed] [Google Scholar]
- 18. Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF et al (1994) A model for beta‐amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A 91:3270–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jung MH, Kim SC, Jeon GA, Kim SH, Kim Y, Choi KS et al (2000) Identification of differentially expressed genes in normal and tumor human gastric tissue. Genomics 69:281–286. [DOI] [PubMed] [Google Scholar]
- 20. Kamboj SK, Swanson GT, Cull‐Candy SG (1995) Intracellular spermine confers rectification on rat calcium‐permeable AMPA and kainate receptors. J Physiol 486(Pt 2):297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kanerva K, Makitie LT, Pelander A, Heiskala M, Andersson LC (2008) Human ornithine decarboxylase paralogue (ODCp) is an antizyme inhibitor but not an arginine decarboxylase. Biochem J 409:187–192. [DOI] [PubMed] [Google Scholar]
- 22. Keinanen R, Miettinen S, Yrjanheikki J, Koistinaho J (1997) Induction of ornithine decarboxylase mRNA in transient focal cerebral ischemia in the rat. Neurosci Lett 239:69–72. [DOI] [PubMed] [Google Scholar]
- 23. Keren‐Paz A, Bercovich Z, Porat Z, Erez O, Brener O, Kahana C (2006) Overexpression of antizyme‐inhibitor in NIH3T3 fibroblasts provides growth advantage through neutralization of antizyme functions. Oncogene 25:5163–5172. [DOI] [PubMed] [Google Scholar]
- 24. Kim SW, Mangold U, Waghorne C, Mobascher A, Shantz L, Banyard J et al (2006) Regulation of cell proliferation by the antizyme inhibitor: evidence for an antizyme‐independent mechanism. J Cell Sci 119:2583–2591. [DOI] [PubMed] [Google Scholar]
- 25. Kitani T, Fujisawa H (1989) Purification and characterization of antizyme inhibitor of ornithine decarboxylase from rat liver. Biochim Biophys Acta 991:44–49. [DOI] [PubMed] [Google Scholar]
- 26. Laitinen PH (1985) Involvement of an “antizyme” in the inactivation of ornithine decarboxylase. J Neurochem 45:1303–1307. [DOI] [PubMed] [Google Scholar]
- 27. Laitinen PH, Huhtinen RL, Hietala OA, Pajunen AE (1985) Ornithine decarboxylase activity in brain regulated by a specific macromolecule, the antizyme. J Neurochem 44:1885–1891. [DOI] [PubMed] [Google Scholar]
- 28. Laitinen PH, Hietala OA, Pulkka AE, Pajunen AE (1986) Purification of mouse brain ornithine decarboxylase reveals its presence as an inactive complex with antizyme. Biochem J 236:613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lopatin AN, Makhina EN, Nichols CG (1994) Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372:366–369. [DOI] [PubMed] [Google Scholar]
- 30. Lopez‐Contreras AJ, Lopez‐Garcia C, Jimenez‐Cervantes C, Cremades A, Penafiel R (2006) Mouse ornithine decarboxylase‐like gene encodes an antizyme inhibitor devoid of ornithine and arginine decarboxylating activity. J Biol Chem 281:30896–30906. [DOI] [PubMed] [Google Scholar]
- 31. Lopez‐Contreras AJ, Ramos‐Molina B, Cremades A, Penafiel R (2008) Antizyme inhibitor 2 (AZIN2/ODCP) stimulates polyamine uptake in mammalian cells. J Biol Chem 283:20761–20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lukkarinen JA, Grohn OH, Alhonen LI, Janne J, Kauppinen RA (1999) Enhanced ornithine decarboxylase activity is associated with attenuated rate of damage evolution and reduction of infarct volume in transient middle cerebral artery occlusion in the rat. Brain Res 826:325–329. [DOI] [PubMed] [Google Scholar]
- 33. Makitie LT, Kanerva K, Andersson LC (2009) Ornithine decarboxylase regulates the activity and localization of rhoA via polyamination. Exp Cell Res 315:1008–1014. [DOI] [PubMed] [Google Scholar]
- 34. Markwell MA, Berger SP, Paul SM (1990) The polyamine synthesis inhibitor alpha‐difluoromethylornithine blocks NMDA‐induced neurotoxicity. Eur J Pharmacol 182:607–609. [DOI] [PubMed] [Google Scholar]
- 35. Martinez E, De Vera N, Artigas F (1991) Differential response of rat brain polyamines to convulsant agents. Life Sci 48:77–84. [DOI] [PubMed] [Google Scholar]
- 36. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 37. Mitchell JL, Chen HJ (1990) Conformational changes in ornithine decarboxylase enable recognition by antizyme. Biochim Biophys Acta 1037:115–121. [DOI] [PubMed] [Google Scholar]
- 38. Morrison LD, Kish SJ (1995) Brain polyamine levels are altered in Alzheimer's disease. Neurosci Lett 197:5–8. [DOI] [PubMed] [Google Scholar]
- 39. Morrison LD, Cao XC, Kish SJ (1998) Ornithine decarboxylase in human brain: influence of aging, regional distribution, and Alzheimer's disease. J Neurochem 71:288–294. [DOI] [PubMed] [Google Scholar]
- 40. Ng EL, Tang BL (2008) Rab GTPases and their roles in brain neurons and glia. Brain Res Rev 58:236–246. [DOI] [PubMed] [Google Scholar]
- 41. Nilsson J, Grahn B, Heby O (2000) Antizyme inhibitor is rapidly induced in growth‐stimulated mouse fibroblasts and releases ornithine decarboxylase from antizyme suppression. Biochem J 346:699–704. [PMC free article] [PubMed] [Google Scholar]
- 42. Nilsson T, Bogdanovic N, Volkman I, Winblad B, Folkesson R, Benedikz E (2006) Altered subcellular localization of ornithine decarboxylase in Alzheimer's disease brain. Biochem Biophys Res Commun 344:640–646. [DOI] [PubMed] [Google Scholar]
- 43. Nilsson T, Malkiewicz K, Gabrielsson M, Folkesson R, Winblad B, Benedikz E (2006) Antibody‐bound amyloid precursor protein upregulates ornithine decarboxylase expression. Biochem Biophys Res Commun 341:1294–1299. [DOI] [PubMed] [Google Scholar]
- 44. Pajunen AE, Hietala OA, Virransalo EL, Piha RS (1978) Ornithine decarboxylase and adenosylmethionine decarboxylase in mouse brain–effect of electrical stimulation. J Neurochem 30:281–283. [DOI] [PubMed] [Google Scholar]
- 45. Paschen W, Csiba L, Rohn G, Bereczki D (1991) Polyamine metabolism in transient focal ischemia of rat brain. Brain Res 566:354–357. [DOI] [PubMed] [Google Scholar]
- 46. Pitkanen LT, Heiskala M, Andersson LC (2001) Expression of a novel human ornithine decarboxylase‐like protein in the central nervous system and testes. Biochem Biophys Res Commun 287:1051–1057. [DOI] [PubMed] [Google Scholar]
- 47. Polvikoski T, Sulkava R, Myllykangas L, Notkola IL, Niinisto L, Verkkoniemi A et al (2001) Prevalence of Alzheimer's disease in very elderly people: a prospective neuropathological study. Neurology 56:1690–1696. [DOI] [PubMed] [Google Scholar]
- 48. Quinlan EM, Philpot BD, Huganir RL, Bear MF (1999) Rapid, experience‐dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci 2:352–357. [DOI] [PubMed] [Google Scholar]
- 49. Schaner ME, Davidson B, Skrede M, Reich R, Florenes VA, Risberg B et al (2005) Variation in gene expression patterns in effusions and primary tumors from serous ovarian cancer patients. Mol Cancer 4:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Snapir Z, Keren‐Paz A, Bercovich Z, Kahana C (2008) ODCp, a brain‐ and testis‐specific ornithine decarboxylase paralogue, functions as an antizyme inhibitor, although less efficiently than AzI1. Biochem J 410:613–619. [DOI] [PubMed] [Google Scholar]
- 51. Tikka S, Mykkanen K, Ruchoux MM, Bergholm R, Junna M, Poyhonen M et al (2009) Congruence between NOTCH3 mutations and GOM in 131 CADASIL patients. Brain 132:933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tovar KR, Westbrook GL (1999) The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci 19:4180–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Washbourne P, Bennett JE, McAllister AK (2002) Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci 5:751–759. [DOI] [PubMed] [Google Scholar]
- 54. Yatin SM, Yatin M, Aulick T, Ain KB, Butterfield DA (1999) Alzheimer's amyloid beta‐peptide associated free radicals increase rat embryonic neuronal polyamine uptake and ornithine decarboxylase activity: protective effect of vitamin E. Neurosci Lett 263:17–20. [DOI] [PubMed] [Google Scholar]
- 55. Yatin SM, Yatin M, Varadarajan S, Ain KB, Butterfield DA (2001) Role of spermine in amyloid beta‐peptide‐associated free radical‐induced neurotoxicity. J Neurosci Res 63:395–401. [DOI] [PubMed] [Google Scholar]
- 56. Mäkitie LT, Kanerva K, Sankila A, Andersson LC (2009) High expression of antizyme inhibitor 2, an activator of ornithine decarboxylase in steroidogenic cells of human gonads. Histochem Cell Biol DOI 10.1007/s00418‐009‐0636‐ [DOI] [PubMed] [Google Scholar]
- 57. Kanerva K, Lappalainen J, Mäkitie LT, Virolainen S, Kovanen PT, Andersson LC (2009) Expression of antizyme inhibitor 2 in mast cells and role of polyamines as selective regulators of serotonin secretion. PLoS One 4:e6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item