

Abstract
The currently recognized two forms of “anabolic” protein aggregate myopathies, that is, defects in development, maturation and final formation of respective actin and myosin filaments encompass actinopathies and myosinopathies. The former are marked by mutations in the ACTA1 gene, largely of the de novo type. Aggregates of actin filaments are deposited within muscle fibers. Early clinical onset is often congenital; most patients run a rapidly progressive course and die during their first 2 years of life. Myosinopathies or myosin storage myopathies also commence in childhood, but show a much more protracted course owing to mutations in the myosin heavy chain gene MYH7. Protein aggregation consists of granular material in muscle fibers and few, if any, filaments.
Keywords: ACTA1 mutations, actinopathy, filament aggregation, hyaline body myopathy, myosin storage myopathy
INTRODUCTION
In the introduction, the term protein aggregate myopathies (PAMs) of the “anabolic” type has been used, in contrast to the “catabolic” forms, the myofibrillar myopathies (MFMs), the latter being defined by impaired extralysosomal protein degradation of hereditary or acquired background. The “anabolic” PAMs differ from the “catabolic” ones by: (i) frequently affecting children, sometimes even at birth; (ii) in recently described observations being associated with mutations different from those in the “catabolic” types; and (iii) demonstrating accumulation of the wild‐type and mutant proteins, but hardly any additional proteins.
At present, two anabolic forms of PAMs have been identified: the actin filament aggregate myopathies (AFAMs) marked by aggregation of actin filaments, and the myosinopathies, also called myosin storage myopathies, marked by accumulation of myosin. Nosographically, the AFAMs belong to the nemaline myopathy kindred (17) because they share mutations in the ACTA1 gene with nemaline myopathies marked by sarcoplasmic and/or intranuclear rods. In the premolecular era, aggregates of actin filaments in muscle fibers had only been mentioned in passing in patients with nemaline myopathy (24). Because of peculiar morphological features, they had also been emphasized in a textbook case report (13) as a separate condition and, then, as an unclassified myopathy. The filamentous body, also consisting of aggregated actin filaments, is a nonspecific feature within muscle fibers in many neuromuscular disorders as—to our knowledge—no respective report exists that describes filamentous bodies as a disease‐specific myopathological phenomenon in a patient's biopsied muscle or neuromuscular condition.
The second “anabolic” PAM, myosinopathy or myosin storage myopathy (MSM) 41, 42, was first recorded as a congenital myopathy marked by “probable lysis of myofibrils”(7). Subsequent reports emphasized the same myopathological features, that is, circumscribed patches (of granular material, by electron microscopy) within muscle fibers, which demonstrated ATPase activity. These were then called hyaline bodies and, thus, hyaline body myopathy (HBM) became a nomenclatorial predecessor of myosinopathy or MSM. Hence, nosographically, myosinopathy has been well‐defined, although under different names, as a distinct neuromuscular entity since its first observation. Identification of a Leu1793Pro mutation in the MYH7 gene (14) in post‐mortem tissue of one of the originally described siblings (7) attests to the nosographic, although not nomenclatorial, continuity of myosinopathy. Hyaline bodies as defined in HBM/MSM have not been described as a nonspecific myopathological feature, although the term “hyaline” has been used in different contexts as in “hyaline” or opaque hypercontracted muscle fibers in myopathology, especially of Duchenne muscular dystrophy; “hyaline structures” in MFM 11, 30; or as “hyaline” masses in a nosologically and, particularly, genetically unresolved “familial cardioneuromyopathy”(37). To date, no convincing forms of acquired AFAM or myosinopathy have been described.
It is interesting to note that AFAM and myosinopathy are defined by mutation‐associated protein aggregates of the two most important functional proteins of sarcomeres, actin and myosin, while other mutant sarcomeric proteins, usually associated with the Z‐disk, are linked to certain MFM. However, why mutations in the ACTA1 gene give rise to filamentous protein aggregates, while mutations in the MYH7 gene result in the aggregated protein slow myosin largely in a granular and not in a filamentous fashion, remains to be clarified.
AFAM
Clinical considerations
AFAM is a congenital disorder marked by muscle weakness and hypotonia. Patients may have feeding problems and may develop respiratory distress even requiring artificial ventilation. Other features may encompass facial diplegia, a dysmorphic face and club feet (10). Contractures and arthrogryposis may also be encountered (6). Electrophysiological studies may be normal or electromyography may reveal myopathic changes. Likewise, creatine kinase values are within normal limits or mildly elevated. Rarely, electrocardiographic data suggested cardiomyopathy (18) but, in general, cardiac function is not impaired in these patients. Most patients affected by AFAM die in early childhood of cardiorespiratory insufficiency, and few have been reported to survive into the second decade of life 18, 21. One boy who recovered from delayed motor development and poor head control is now able to walk but is, nevertheless, impaired in his motor activities (18) (Goebel, personal observation, December 2008). Notwithstanding a less dire prognosis in a small number of children, AFAM is a severe condition, often fatal in early life. So far, it appears that familial occurrence has not been reported.
Myopathology
The hallmark of AFAM, aggregation of actin filaments (Figure 1A–D) appears as a rather monotonous feature, varying only in quantity within muscle fibers. The actin filaments form in a haphazard fashion and, even in large areas of aggregation, are not mixed with other filamentous or nonfilamentous material but only an occasional mitochondrion. Aggregates of actin filaments may be encountered in the subsarcolemmal space as well as among myofibrils, although sometimes occupying a major part of a cross‐sectioned muscle fiber. The actin filament accumulations are sharply demarcated from sarcomeres and nuclei. At the light microscopic level, they appear as rather homogenous patches, sometimes resembling hyaline or opaque inclusions owing to the lack of the sarcotubular system and mitochondria. They are bluish in the hematoxylin–eosin preparation, and bluish‐reddish in the modified Gomori's trichrome preparation. These patches of actin filaments lack activities of oxidative enzymes and ATPases, the latter in contrast to ATPase activity in hyaline bodies of HBM. Acid phosphatase activity is not increased in patches of actin filaments. Sometimes, aggregates of filaments are apparently minute and only detected by electron microscopy without further light microscopic evidence (22). This corroborates the earlier textbook information, that aggregates of filaments were sometimes seen in nemaline myopathy, when these patches were actually described as lacking thick or myosin filaments rather than as showing aggregates of thin filaments (24).
Figure 1.
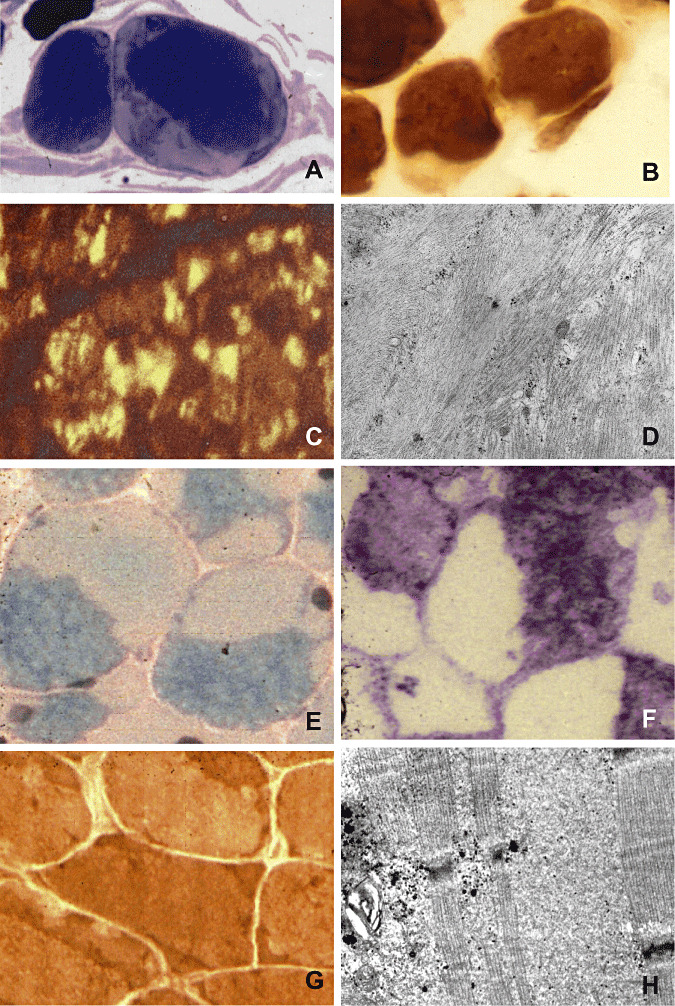
Actinopathy: (A) Large subsarcolemmal areas of actin filament aggregation (light) in two muscle fibers, semi‐thin section, methylene blue‐Azur II (Richardson). (B) In the ATPase preparation, subsarcolemmal areas of actin filament aggregation are devoid of enzyme histochemical activity. (C) An antibody against sarcomeric actin labels actin filament aggregates within muscle fibers (courtesy of C. Bönnemann, Göttingen/Philadelphia). (D) Ultrastructurally, there are disorganized sarcomeres (on the right), sharply separated from aggregated actin filaments (on the left). Myosinopathy: (E) Muscle fibers display very light opaque areas, the hyaline bodies, sharply demarcated from darker, greenish sarcomeric regions, modified Gomori's trichrome stain. (F) The hyaline bodies are devoid of oxidative enzyme histochemical activity, menadione‐linked α‐glycerophosphate dehydrogenase. (G) Hyaline bodies show enzyme histochemical activity of ATPase [contrary to actin filament aggregates, see (B)]. (H) Ultrastructurally, hyaline bodies consist of finely granular material, here seen among preserved sarcomeres.
AFAM belongs to the group of nemaline myopathies because nemaline bodies or rods may be encountered in addition to actin filament aggregates. These rods may be located in the sarcoplasm (1), within myofiber nuclei (18) or both (36). Sometimes, the rods were rather small, so‐called minirods(1). However, sarcoplasmic rods and actin filament aggregates have not always been found together in muscle fibers, but often separate, suggesting separate formation of both features. Filaments, too, have not been reported to be situated in nuclei, not even in the presence of intranuclear rods.
Some patients lack rods in their biopsied muscles, showing actin filament aggregates only 6, 13, 18, 19, 20.
The myopathological differential diagnosis of actin filament aggregates entails hyaline bodies, to be described in the subsequent section “Myosinopathy” and “Caps.”“Caps” are subsarcolemmal areas which are present in a haphazard fashion. They consist of Z‐disks and bilateral actin or thin filaments lacking myosin or thick filaments. Hence, apart from the different ultrastructural pathology of actin filament aggregates, hyaline bodies and “caps,” the light microscopic features are apparent in enzyme histochemical preparations in that actin filament aggegates lack oxidative enzymes and ATPase activities, hyaline bodies lack oxidative enzyme activities but display ATPase activities while “caps” show oxidative enzyme activities but lack ATPases 15, 16.
Immunohistochemically, the antibody against sarcomeric actin identifies actin filament aggregates 19, 20. The filaments can also be immunolabeled by electron microscopy with an antibody against α‐actinin (18). Desmin and vimentin have not been identified in these actin filament aggregates (6), although their presence was enhanced in many other muscle fibers, probably owing to their immaturity (6). A comparably large number of proteins, tested but not found by immunohistochemistry in MSM (Table 1), has not yet been studied in AFAM.
Table 1.
Immunohistochemistry in myosin aggregates/hyaline bodies.
| Proteins | Results | References |
|---|---|---|
| Myosin heavy chain slow | +/+/+/+ | 4, 27, 41, 43 |
| Myosin heavy chain fast | −/−/+/+ | 4, 8, 27, 38 |
| Myosin heavy chain neonatal | −/− | 4, 27 |
| Myosin heavy chain developmental | −/− | 4, 27 |
| Dystrophin | −/−/−/−/−/− | 2, 4, 8, 27, 38, 43 |
| Spectrin | −/−/−/− | 4, 8, 27, 38 |
| Desmin | −/−/−/−/− | 4, 8, 27, 38, 41 |
| α‐Actinin | −/−/− | 8, 27, 42 |
| Vimentin | −/− | 8, 27 |
| Ubiquitin | − | (27) |
| Sarcomeric actin | −/−/− | 4, 8, 42 |
| Tropomyosin | −/− | 4, 8 |
| Troponin T | −/− | 4, 8 |
| Dysferlin | −/− | 4, 8 |
| α‐, β‐, γ‐, δ‐Sarcoglycans | −/− | 4, 43 |
| α‐, β‐Dystroglycans | − | (4) |
| α‐B Crystallin | − | (4) |
| Nebulin | −/− | 4, 8, 42 |
| Titin | − | (42) |
| α‐Tubulin | − | (8) |
Genetics
To date, 10 different mutations in the skeletal muscle α‐actin gene leading to nine amino acid changes (Table 2) have been associated with the diagnosis of AFAM. All of the mutations are dominant; four have been determined in at least one instance to have arisen de novo in the affected child.
Table 2.
ACTA1 mutations associated with actin filament aggregate myopathies.*
| Gly15Arg |
| Leu142Phe |
| Ala144Ser145 duplication |
| Gly146Ser |
| Gly146Asp |
| Arg147Lys |
| Asp154Asn |
| Val163Leu |
| Ser348Leu |
Amino acids numbered according to the mature protein sequence, with the two N‐terminal amino acids deleted.
A previous analysis of the then six known AFAM amino acid changes concluded that the AFAM variant amino acids were clustered around the nucleotide binding cleft and the hinge at the bottom of the cleft (Figure 2) (40).
Figure 2.
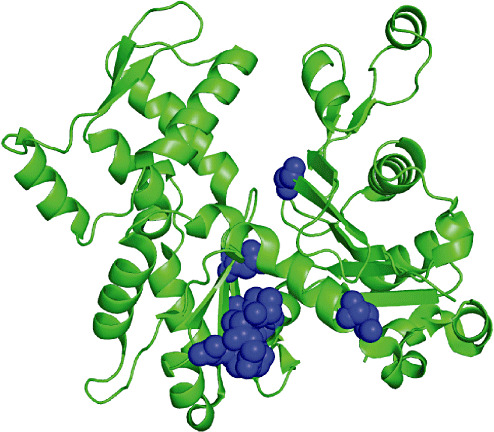
Location of all actin filament aggregate myopathy mutations within the ACTA1 monomer. Figure constructed using the Polyview website http://polyview.cchmc.org/polyview3d.html based on the 1j6z.pdb file for actin.
The AFAM variants discovered since then have continued this trend, and it is almost possible to predict where AFAM mutations will be found in the actin molecule. The AFAM mutations should theoretically affect nucleotide binding and in turn actin polymerization.
Two of the AFAM variants (Leu142Phe, Val163Leu) are also associated with intranuclear nemaline bodies, while one (Asp154Asn) is associated with both intranuclear and sarcoplasmic nemaline bodies (36).
Examined in tissue culture models, the Val163Leu mutant gave rise mainly to intranuclear aggregates 9, 12, 23, with some cytoplasmic aggregates. The suggestion was also made (12) that the intranuculear aggregates, which form within the nucleus, do so when there is an increase in the expression of the mutant in the cytoplasm, but the mutant cannot incorporate in normal thin filaments and migrates to the nucleus.
The G15R mutation studied in tissue culture did give rise to cytoplasmic aggregates 3, 9.
MYOSINOPATHY
Clinical features
Clinically, HBM or myosinopathy may become apparent in early childhood (7). Alternatively, it may be identified in adult patients in whom muscle weakness may be a predominant clinical symptom and dates back to childhood 2, 35, 38, 41, or in adult patients in whom weakness emerges between the fourth and sixth decades of life 4, 27. A number of patients have been reported without family history [2, 8, 35, 41 (patient 2)]. Occasionally, exercise‐associated myalgia may develop (38). Electromyography may reveal “myopathic” features. Creatine kinase may be normal or mildly elevated, occasionally even approaching 1000 U/L (38). The disease may progress to death in adulthood (14), or may progress at a much slower pace, even when commencing in childhood 2, 38.
Autosomal‐dominant inheritance has been documented 4, 27 and autosomal recessive inheritance inferred 33, 42. The originally described siblings (7) apparently had only a heterozygous mutation in the MYH7 gene (14). The parents of these siblings presented without muscle disease during the early childhood of their children (7), and they were neither clinically nor genetically further investigated, although the older affected daughter had died (14). Thus, the genetic and clinical affection status of the parents is still unresolved. Some patients remain without reported molecular results 2, 35. In most cases, the parents of the isolated affected patients were not tested for the same mutation, and it thus remains unclear whether these patients had de novo mutations. On the other hand, convincing de novo mutation in an HBM patient with a MYH7 gene mutation has been demonstrated in the isolated case described by Tajsharghi et al (41).
In a British family, three affected sibs with MSM presented with hypertrophic cardiomyopathy and respiratory muscle weakness, which resulted in premature death, in addition to symmetric limb girdle weakness (42). In this group of sibs, hyaline bodies were not only seen in biopsied type‐I fibers of skeletal muscle, but also at autopsy in cardiomyocytes remaining among areas of cardiac fibrosis (42). It was only possible to analyze the MYH7 gene in one of the affected sibs in this family, but a homozygous Glu1883Lys mutation was identified (42). This, along with the fact that the parents of the affected sibs were unaffected second cousins, suggests autosomal recessive inheritance in this family. However, as it was not possible to genetically analyze the other two affected sibs, it remains possible that the other two affected sibs had heterozygous mutations (42). Clinical, genetic and morphological observations in this British family, thus present a link between MSM and hypertrophic cardiomyopathy which is the most frequent disease associated with MYH7 gene mutations (31).
Severe cardiac and respiratory involvement was also noted in the male patient with apparent recessive MSM localized to the 3p22.2‐21.32 locus, the mutated protein of which remains unidentified 33, 44.
Myopathology
The morphological hallmark of MSM/myosinopathy is subsarcolemmal or intersarcomeric aggregation of granular material (Figure 1E–H), forming hyaline bodies, although they represent patches or plaques of granular material rather than true structured bodies, such as nemaline bodies in nemaline myopathy, reducing bodies in reducing body myopathy or crystalline inclusions. The hyaline bodies are observed in type I fibers only. Originally, in the first publication (7) and later, these plaques were highlighted by absent activities of oxidative enzymes and distinct activity of ATPase. In some reports, the ATPase activity is stated to be of the alkaline (pH 9.4) form 2, 7, 8, 38, while in others either both the alkaline and acid ATPase forms or exclusively the acid form 2, 4, 8, 38, 41 had been present. Ultrastructurally, the plaques of granular material (38) hardly contain any other organelles or structures, and are not membrane bound, although they are occasionally mixed with filamentous profiles (8). Adjacent sarcomeres appear largely intact, only rarely showing streaming of the Z‐band or multiminicore‐like lesions (7). Nuclei sometimes float in the lakes of granular material. Mitochondria always appeared unremarkable as to number, size and intrinsic structure. Occasionally (43), rimmed vacuoles and intranuclear tubulofilamentous 15–20 nm‐thick profiles were encountered. Few patients with an Arg1875Trp mutation lacked hyaline bodies in their biopsied muscle tissue (34). Increase in acid phosphatase activity indicating activation of the lysosomal compartment in muscle fibers was not conspicuous.
The enzyme histochemical demonstration of ATPase, recently especially after acid pre‐incubation in type I fibers suggested accumulation of slow myosin 4, 27, 41 and sometimes also fast myosin 8, 38 which, by immunohistochemistry, could be verified (in different specimens by different observers), while other isoforms of myosin, that is, neonatal and developmental, failed to be demonstrated by respective antibodies (Table 1). Furthermore, a rather large gamut of proteins, both sarcolemmal and sarcomeric, were tested and likewise failed to give reactions with the respective antibodies (Table 1). Occasionally, desmin, α‐B crystallin or tropomyosin could be found overexpressed at the margin of the hyaline bodies 4, 27, 38. Dearth or absence of other proteins but myosins, slow and fast, suggests that formation of sarcomeric myosin or thick filaments is blocked in MSM. Whether myosin‐associated proteins, such as myosin binding protein C, M‐protein or myomesin, accrue in these hyaline bodies has not been documented.
While Laing early‐onset distal myopathy is also related to mutations in the MYH7 gene, its myopathology appears nonspecific with variation in muscle fiber diameters and fiber‐type distributions and rimmed vacuoles in some biopsies, but without apparent hyaline bodies 26, 29.
Genetics
To date, only four different mutations in MYH7 have been described in association with MSM. These mutations are Leu1793Pro, Arg1845Trp, Glu1883Lys and His1901Leu (the His1901Leu mutation was originally published as His1904Leu, but this numbering of the amino acids is not standard (32)).
The Leu1793Pro (14), Arg1845Trp (41) and His1901Leu (5) mutations cause dominant MSM, while the Glu1883Lys mutation apparently causes recessive disease (42).
The dominant Leu1793Pro and His1901Leu mutations, and the recessive Glu1883Lys mutation are private mutations, only identified in single families to date 14, 42. On the other hand, the Arg1845Trp mutation has been identified in multiple families in different parts of the world 25, 34, 38, 41 and has arisen more than once in human history, as one incidence of definite de novo mutation has been identified for this mutation (41). This is a similar mutation pattern to the MYH7 mutations which cause Laing distal myopathy, where the Lys1617del mutation has been identified multiple times, but the other mutations are private mutations in individual families 26, 29 (Laing, unpub. obs.).
The tail of the myosin heavy chain in which the MSM mutations are found forms a coiled coil dimer. In order for a part of a protein to make a coiled coil, that part of the protein has to consist of a seven amino acid (heptad) repeat. The amino acid residues are labeled a–g, with hydrophilic and hydrophobic amino acid residues occupying certain of the positions which form the outer surface and inner core of the repeat. The Arg1845Trp and His1901Leu mutations are both at position f of the heptad repeat, while mutation Leu1793Pro is at position d, and Glu1883Lys is at position b (28).
Positions b and f are on the outside of the coiled coil, and the mutations may therefore alter interactions with myosin interacting proteins, while position d is internal within the core.
The MSM mutations are within or close to the 29 amino acid assembly competence domain close to the C‐terminal tail of myosin which stretches from residue 1871 to residue 1889 in MYH7 (39).
The MSM mutations may thus interfere with dimerization of the myosin monomers.
Genotype–phenotype correlations
Although only few mutations have been identified in the MYH7 gene, three types of genotype–clinicophenotype correlations have, so far, been recognized, but the consistency of such correlations needs to be corroborated by future observations: (i) Patients with the Arg1845Trp mutation having been identified in nine separate families, seven with MSM and two (34) with scapuloperoneal myopathy, have a mild form of myosinopathy, marked by slow progression and mild cardiac abnormalities, such as atrial fibrillation, septum hypertrophy or ventricular hypertrophy. Patients with this Arg1845Trp mutation may also have large calves. (ii) In patients with the Leu1793Pro and Glu1883Lys mutations, respectively, severely reduced life span and premature death in the third or fourth decades caused by respiratory and cardiac failure with cardiomyopathy are the major familial features. (iii) Intrafamilial clinical heterogeneity occurs in patients with the His1901Leu mutation (Table 3).
Table 3.
Genotype–clinicophenotype correlations in MYH7 mutations.
| Mutation | Mild/Severe course | Special clinical features | Familial (references) |
|---|---|---|---|
| Leu1793Pro Exon 37 | Severe Weak from birth, ventilatory assistance, postoperative death at age 27 years (one patient) | None | 7, 14 |
| Moderate Mother: Proximal weakness at age 30 years, ventilatory support at age 58 years; | Mild hypertrophic cardiomyopathy | (43) | |
| daughter: distal leg atrophy | At age 3 months, cardiomyopathy | ||
| Arg1845Trp Exon 37 | Family 1: mild Slow progression | Calf pseudohypertrophy Atrial fibrillation | (41) |
| Family 2: mild | Calf pseudohypertrophy | ||
| Patient 1: mild Slow progression | Minor dysmorphic features, mild calf hypertrophy | (8) | |
| Patient 2: mild Slow progression | None | (25) | |
| Mild | Slight ptosis | (38) | |
| Moderate/mild Middle age or later onset, scapulo‐peroneal syndrome or early‐onset proximal weakness | None | (34) | |
| Glu1883Lys Exon 38 | Severe Reduced life span: death at ages 32 and 57 years, respectively | Cardiomyopathy, respiratory failure | (42) |
| His1901Leu Exon 39 | Mild or severe Slow or rapidly progressive | Intrafamilial heterogeneity, loss of subcutaneous fat, dysarthria, contractures | 4, 5 |
| Chromosome 3p22.2‐21.32 Unknown mutation | Severe | Cardiac and respiratory insufficiencies | 33, 44 |
As the electron microscopic features of hyaline bodies appear uniformly finely granular and sharply demarcated from surrounding intact constituents of the muscle fiber throughout reported morphological findings, differences in genotype–morphotype correlations do not appear to exist.
ACKNOWLEDGMENTS
Editorial assistance by Astrid Wöber and photographic support by Walther Wagner are gratefully acknowledged.
REFERENCES
- 1. Agrawal PB, Strickland CD, Midgett C, Morales A, Newburger DE, Poulos MA et al (2004) Heterogeneity of nemaline myopathy cases with skeletal muscle α‐actin gene mutations. Ann Neurol 56:86–96. [DOI] [PubMed] [Google Scholar]
- 2. Barohn RJ, Brumback RA, Mendell JR (1994) Hyaline body myopathy. Neuromuscul Disord 4:257–262. [DOI] [PubMed] [Google Scholar]
- 3. Bathe FS, Rommelaere H, Machesky LM (2007) Phenotypes of myopathy‐related actin mutants in differentiated C2C12 myotubes. BMC Cell Biol 8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bohlega S, Lach B, Meyer BF, Al Said Y, Kambouris M, Al Homsi M, Cupler EJ (2003) Autosomal dominant hyaline body myopathy: clinical variability and pathologic findings. Neurology 61:1519–1523. [DOI] [PubMed] [Google Scholar]
- 5. Bohlega S, Abu‐Amero SN, Wakil SM, Carroll P, Al‐Amr R, Lach B et al (2004) Mutation of the slow myosin heavy chain rod domain underlies hyaline body myopathy. Neurology 62:1518–1521. [DOI] [PubMed] [Google Scholar]
- 6. Bornemann A, Bloch P, Petersen M, Schmalbruch H (1996) Fatal congenital myopathy with actin filament deposits. Acta Neuropathol 92:104–108. [DOI] [PubMed] [Google Scholar]
- 7. Cancilla PA, Kalyanaraman K, Verity MA, Munsat T, Pearson CM (1971) Familial myopathy with probable lysis of myofibrils in type I fibres. Neurology 21:579–585. [DOI] [PubMed] [Google Scholar]
- 8. Ceuterick C, Martin J‐J, Martens C (1993) Hyaline bodies in skeletal muscle of a patient with a mild chronic nonprogressive congenital myopathy. Clin Neuropathol 12:79–83. [PubMed] [Google Scholar]
- 9. Costa CF, Rommelaere H, Waterschoot D, Sethi KK, Nowak KJ, Laing NG et al (2004) Myopathy mutations in alpha‐skeletal‐muscle actin cause a range of molecular defects. J Cell Sci 117:3367–3377. [DOI] [PubMed] [Google Scholar]
- 10. Danon MJ, Carpenter S, Karpati G, Sherbany A, Stripathi N, Holland P (1995) A congenital myopathy with excessive actin filament deposition (693P). Neurology 45:A354. [Google Scholar]
- 11. De Bleecker JL, Engel AG, Ertl BB (1996) Myofibrillar myopathy with abnormal foci of desmin positivity. II. Immunocytochemical analysis reveals accumulation of multiple other proteins. J Neuropathol Exp Neurol 55:563–577. [DOI] [PubMed] [Google Scholar]
- 12. Domazetovska A, Ilkovski B, Cooper ST, Ghoddusi M, Hardeman EC, Minamide LS et al (2007) Mechanisms underlying intranuclear rod formation. Brain 130:3275–3284. [DOI] [PubMed] [Google Scholar]
- 13. Dubowitz V (1985) Muscle Biopsy: A Practical Approach, pp. 664–670. Baillière Tindall: London. [Google Scholar]
- 14. Dye DE, Azzarelli B, Goebel HH, Laing NG (2006) Novel slow‐skeletal myosin (MYH7) mutation in the original myosin storage myopathy kindred. Neuromuscul Disord 16:357–360. [DOI] [PubMed] [Google Scholar]
- 15. Fidzianska A (2002) “Cap disease”—a failure in the correct muscle fibre formation. J Neurol Sci 201:27–31. [DOI] [PubMed] [Google Scholar]
- 16. Fidzianska A, Badurska B, Ryniewicz B, Dembek I (1981) “Cap disease”: new congenital myopathy. Neurology 31:1113–1120. [DOI] [PubMed] [Google Scholar]
- 17. Goebel HH (2007) Cap disease uncapped. Neuromuscul Disord 17:429–432. [DOI] [PubMed] [Google Scholar]
- 18. Goebel HH, Anderson JR, Hübner C, Oexle K, Warlo I (1997) Congenital myopathy with excess of thin myofilaments. Neuromuscul Disord 7:160–168. [DOI] [PubMed] [Google Scholar]
- 19. Goebel HH, Brockmann K, Bönnemann C, Warlo IAP, Hanefeld F, Labeit S, Durling HJ (2004) Actin‐related myopathy without any missense mutation in the ACTA1 gene. J Child Neurol 19:149–153. [DOI] [PubMed] [Google Scholar]
- 20. Goebel HH, Brockmann K, Bönnemann C, Warlo IAP, Hanefeld F, Labeit S et al (2006) Patient with actin aggregate myopathy and not formerly identified ACTA1 mutation is heterozygous for the Gly15Arg mutation of ACTA1, which has previously been associated with actinopathy. J Child Neurol 21:545 [letter. [DOI] [PubMed] [Google Scholar]
- 21. Goez H, Sira LB, Jossiphov J, Borochowitz Z, Durling H, Laing NG, Nevo Y (2005) Predominantly upper limb weakness, enlarged cisterna magna, and borderline intelligence in a child with de novo mutation of the skeletal muscle alpha‐actin gene. J Child Neurol 20:236–239. [PubMed] [Google Scholar]
- 22. Hutchinson DO, Charlton A, Laing NG, Ilkovski B, North KN (2006) Autosomal dominant nemaline myopathy with intranuclear rods due to mutation of the skeletal muscle ACTA1 gene: clinical and pathological variability within a kindred. Neuromuscul Disord 16:113–121. [DOI] [PubMed] [Google Scholar]
- 23. Ilkovski B, Nowak KJ, Domazetovska A, Maxwell AL, Clement S, Davies KE et al (2004) Evidence for a dominant‐negative effect in ACTA1 nemaline myopathy caused by abnormal folding, aggregation and altered polymerization of mutant actin isoforms. Hum Mol Genet 13:1727–1743. [DOI] [PubMed] [Google Scholar]
- 24. Karpati G, Carpenter S (1992) Skeletal muscle pathology in neuromuscular diseases. In: Myopathies—Handbook of ClinicalNeurology, Vol. 18/62. Rowland LP, DiMauro S (eds), pp. 1–48. Elsevier Science Publishers B.V.: Amsterdam. [Google Scholar]
- 25. Laing NG, Ceuterick‐de Groote C, Dye DE, Liyanage K, Duff RM, Dubois B et al (2005) Myosin storage myopathy: slow skeletal myosin (MYH7) mutation in two isolated cases. Neurology 64:527–529. [DOI] [PubMed] [Google Scholar]
- 26. Lamont PJ, Udd B, Mastaglia FL, De Visser M, Hedera P, Voit T et al (2006) Laing early onset distal myopathy: slow myosin defect with variable abnormalities on muscle biopsy. J Neurol Neurosurg Psychiatry 77:208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Masuzugawa S, Kuzuhara S, Narita Y, Naito Y, Taniguchi A, Ibi T (1997) Autosomal dominant hyaline body myopathy presenting as scapuloperoneal syndrome: clinical features and muscle pathology. Neurology 48:253–257. [DOI] [PubMed] [Google Scholar]
- 28. McLachlan AD, Karn J (1982) Periodic charge distributions in the myosin rod amino acid sequence match cross‐bridge spacings in muscle. Nature 299:226–231. [DOI] [PubMed] [Google Scholar]
- 29. Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ et al (2004) Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause Laing early‐onset distal myopathy (MPD1). Am J Hum Genet 75:703–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakano S, Engel AG, Waclawik AJ, Emslie‐Smith AM, Busis NA (1996) Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol 55:549–562. [DOI] [PubMed] [Google Scholar]
- 31. Oldfors A (2007) Hereditary myosin myopathies. Neuromuscul Disord 17:355–367. [DOI] [PubMed] [Google Scholar]
- 32. Oldfors A, Tajsharghi H, Thornell LE (2005) Mutation of the slow myosin heavy chain rod domain underlies hyaline body myopathy. Neurology 64:580–581. [DOI] [PubMed] [Google Scholar]
- 33. Önengüt S, Ugur SA, Karasoy H, Yüceyar N, Tolun A (2004) Identification of a locus for an autosomal recessive hyaline body myopathy at chromosome 3p22.2‐p21.32. Neuromuscul Disord 14:4–9. [DOI] [PubMed] [Google Scholar]
- 34. Pegoraro E, Gavassini BF, Borsato C, Melacini P, Vianello A, Stramare R et al (2007) MYH7 gene mutation in myosin storage myopathy and scapulo‐peroneal myopathy. Neuromuscul Disord 17:321–329. [DOI] [PubMed] [Google Scholar]
- 35. Sahgal V, Sahgal S (1977) A new congenital myopathy: a morphological, cytochemical and histochemical study. Acta Neuropathol 37:225–230. 193343 [Google Scholar]
- 36. Schröder JM, Durling HJ, Laing NG (2004) Actin myopathy with nemaline bodies, intranuclear rods, and a heterozygous mutation in ACTA1 (Asp154Asn). Acta Neuropathol 108:250–256. [DOI] [PubMed] [Google Scholar]
- 37. Selcen D, Krueger BR, Engel AG (2002) Familial cardioneuromyopathy with hyaline masses and nemaline rods: a novel phenotype. Ann Neurol 51:224–234. [DOI] [PubMed] [Google Scholar]
- 38. Shingde MV, Spring PJ, Maxwell A, Wills EJ, Harper CG, Dye DE et al (2006) Myosin storage (hyaline body) myopathy: a case report. Neuromuscul Disord 16:882–886. [DOI] [PubMed] [Google Scholar]
- 39. Sohn RL, Vikstrom KL, Strauss M, Cohen C, Szent‐Gyorgyi AG, Leinwand LA (1997) A 29 residue region of the sarcomeric myosin rod is necessary for filament formation. J Mol Biol 266:317–330. [DOI] [PubMed] [Google Scholar]
- 40. Sparrow JC, Nowak KJ, Durling HJ, Beggs AH, Wallgren‐Pettersson C, Romero NB et al (2003) Muscle disease caused by mutations in the skeletal muscle alpha‐actin gene (ACTA1). Neuromuscul Disord 13:519–531. [DOI] [PubMed] [Google Scholar]
- 41. Tajsharghi H, Thornell L‐E, Lindberg C, Lindvall B, Henriksson KG, Oldfors A (2003) Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol 54: 494–500. [DOI] [PubMed] [Google Scholar]
- 42. Tajsharghi H, Oldfors A, Macleod DP, Swash M (2007) Homozygous mutation in MYH7 in myosin storage myopathy and cardiomyopathy. Neurology 68:962. [DOI] [PubMed] [Google Scholar]
- 43. Uro‐Coste E, Arne‐Bes MC, Pellissier JF, Richard P, Levade T, Heitz F et al (2009) Striking phenotypic variability in two familial cases of myosin storage myopathy with a MYH7 Leu1793Pro mutation. Neuromuscul Disord 19:165–166. [DOI] [PubMed] [Google Scholar]
- 44. Yuceyar AN, Baysal L, Ozbay OE, Kocaman AS, Karasoy H (2008) Severe cardiomyopathy in hyaline body myopathy: ten years of follow‐up (abstract G.P.9.08). Neuromuscul Disord 18:788. [Google Scholar]