

Abstract
Adherence, invasion and translocation to and of the intestinal epithelium are important drivers of disease for many enteric bacteria. However, most work has been limited to transformed intestinal cell lines or murine models that often do not faithfully recapitulate key elements associated with human disease. The recent technological advances in organotypic culture of tissues and cells is providing unparalleled access to systems with human physiology and complexity. Human intestinal enteroids (HIEs), derived from patient biopsy or surgical specimens of intestinal tissues, are organotypic cultures now being adapted to the study of enteric infections. HIEs are comprised of the dominant cell types of the human gastrointestinal epithelium, can be grown in two or three-dimensional structures, form a crypt-villus axis with defined apical and basolateral compartments, and undergo physiologic responses to many different stimuli. Here, we describe a series of protocols that encompass the use of human enteroids for the measurement of the adherence, invasion and translocation of E. coli to and through the intestinal epithelium, outline the steps needed to grow and prepare enteroids for this purpose, and highlight some common problems to troubleshoot.
Keywords: Human Intestinal Enteroids, Bacteria, Adhesion, Invasion, Translocation, coli
INTRODUCTION
The gastrointestinal (GI) tract functions in food digestion, nutrient absorption, waste excretion, and is a barrier against bacteria that chronically or acutely inhabit this tissue. The recent explosion of studies associating the resident microbial flora with human health is further testament to the importance of studying the interaction between bacteria and the human intestinal epithelium. In addition, the GI tract represents a portal of entry for pathogenic bacteria, especially those that specifically induce the release of host fluid as a mechanism to spread progeny into the environment. Pathogen-induced diarrhea, perhaps the most common GI pathophysiology in humans, is responsible for approximately 2–4 billion episodes annually, especially in children [1]. Diarrhea is the second leading cause of death in children under the age of 5 years in developing countries [2]. Chronic diarrhea is associated with nutritional deficiency, stunted growth, and cognitive impairment [3]. Some GI bacteria do not induce diarrhea; instead, they are highly invasive of the intestinal epithelium and enter the underlying lymphoid tissue or even translocate into the blood. This may lead to bacteremia and dissemination to critical organs often resulting in life-threatening sepsis.
The molecular processes that lead to these pathophysiological effects are largely unidentified. Many rodent animal models do not recapitulate the hallmark features of human diarrhea. Larger animal species such as pigs resemble more closely the human condition, but their use is technically challenging and expensive. In vitro organ cultures of biopsies are short lived and transformed culture cells often have aberrant physiology. Recent advances in organotypic culturing methods of untransformed human tissue may be a viable alternative to animal models for some of the critical steps in the pathophysiology of these enteric infections. Of special significance is the progress made in the cultivation and growth of human intestinal enteroids, abbreviated to HIEs. These “mini guts” are derived from LGR5+ stem cells of the small intestine and colon [4–8] and are generated by modulating the presence and levels of key stem cell responsive growth factors, including Wnt3a, R-Spondin, and Noggin [6, 7, 9]. Upon removal of Wnt, stem cells differentiate into all major cell types of the intestinal epithelium, including entero-endocrine, Paneth, and Goblet cells, and obtain three-dimensional architecture with an enclosed lumen and a crypt/villus axis [10]. HIEs have now been generated from stomach, duodenum, jejunum, ileum, and colon and from many different patient donors [11]. Ongoing studies from various groups are examining the incorporation of immune cells, vasculature, stretch, and flow into enteroid cultures, as well as “gut-on-a-chip” approaches to model drug metabolism and multi-organ connections [12–14]. Here, we describe how to isolate, grow, maintain, store and infect human intestinal enteroids to model various interactions with enteric bacteria.
BASIC PROTOCOL 1
The cell culture system described below is often referred as enteroid or organoid culture system. The technology is novel owing to the use of non-transformed cells and an art by itself since the culturing methodology requires technical nuances and expertise that are vastly different from the traditional cell culture system (cancer cell lines). In brief, intestinal crypts containing stem cells are obtained from human biopsy/surgery samples and are propagated as 3-D enteroids embedded inside the matrigel. The enteroids are allowed to grow for a period of 7–10 days before making monolayers or further passaged. The preparation and growth of enteroid culture system are described in detail below.
Materials
Human 3D enteroids (isolated from intestinal crypts and based upon previously established protocol (4); about 100 3D enteroids in solid matrigel or at desired confluency after 7 days of growth
Matrigel, growth factor reduced, phenol free. (Corning, cat. no. 356231)
GlutaMAX-1 with final concentration 2 mM and stock solution at 200 mM (Invitrogen, cat. no. 35050–061)
Penicillin/Streptomycin at final concentration 1X and stock solution at 100X (Invitrogen , cat. no. 15140–122)
N2 supplement at final concentration 1X and stock solution at 100X (Invitrogen , cat. no. 17502–048)
B27 supplement at final concentration 1X and stock solution at 50X (Invitrogen , cat. no. 17504–044)
N-Acetylcysteine dissolved in ddH20 to a final concentration of 1X and stock solution at 500 mM (Sigma-Aldrich, cat. no. A9165–5G)
Mouse Epidermal Growth Factor (EGF) dissolved in PBS to a final concentration of 50 ng/mL and stock solution at 50 μg/mL (Invitrogen, cat. no. PMG8043)
A-83–01 dissolved in DMSO to a final concentration of 50 ng/mL and stock solution at 500 μM (Torcis, cat. no. 2939)
SB202190 dissolved in DMSO to a final concentration of 10 μM and stock solution at 10 mM (Sigma-Aldrich, cat. no. S7067)
Nicotinamide dissolved in ddH20 to a final concentration of 10 mM and stock solution at 1 M (Sigma-Aldrich, cat. no. N0636)
[Leu-15]- Gastrin I dissolved in PBS to a final concentration of 10nM and stock solution at 10 μM (Sigma-Aldrich, cat. no. G9145)
HEPES 1 M at final concentration 10 mM (Invitrogen, cat. no. 15630–080)
Advanced DMEM/F12 (Invitrogen, cat. no. 12634–028)
Recovery Cell Freezing Media (Invitrogen, cat. no. 12648–010)
24-well Nunclon delta surface tissue culture dish (ThermoScientific, cat. no. 142475)
Refrigerated centrifuge with swing rotor
Sterilized filter pipette tips
BD 1 mL TB syringe (cat. no. 309626)
Collagen IV solution (1 mg/mL in 100 mM cell culture grade Acetic acid), stored at 4°C (Sigma Aldrich, cat. no. C5533)
0.5 mM EDTA
0.05% Trypsin/ 0.5 mM EDTA (Gibco, cat. no. 25300–062)
10 μM Y-27632
CMGF− containing 10%FBS (See Reagents and Solutions)
40 μm cell strainer (Falcon, cat. no. 352340)
Differentiation Media (See Reagents and Solutions)
All stock solutions and aliquots can be stored at −20°C
Details of enteroid media used are listed in Table 1 and 2.
Table 1:
Complete media without growth factor (CMGF−)
| S. No | Components | Volume |
|---|---|---|
| 1 | Advanced DMEM/F12 | 500 mL |
| 2 | Glutamax 100X | 5 mL |
| 3 | HEPES 1M | 5 mL |
| 4 | Penicillin and Streptomycin | 5 mL |
Store at 4°C and can be used up to 4 weeks. Discard after 4 weeks.
Table 2:
Complete media with growth factor (CMGF+)
| S. No | Components | Volume (10 mL) |
|---|---|---|
| 1 | CMGF− | 1.5 mL |
| 2 | Wnt 3 conditioned media (ATCC L-Wnt3a cell line Cat #CRL-2647) | 5 mL |
| 3 | B27 (50X) | 200 μL |
| 4 | N2 (100X) | 100 μL |
| 5 | n-Acetylcysteine (500 mM) | 20 μL |
| 6 | Rspo-1 conditioned medium (R-Spondin HEK 293 cells) | 2 mL |
| 7 | Noggin (293-Noggin cells) | 1 mL |
| 8 | EGF (1000X final conc. 50 ng/mL) | 10 μL |
| 9 | Gastrin (1000X final conc. 10 nM) | 10 μL |
| 10 | Nicotinamide (final conc. 10 mM) | 100 μL |
| 11 | A83 (TGFb type I receptor inhibitor) (1000X final conc. 500 nM) | 10 μL |
| 12 | SB202190 (P38 inhibitor) (1000X final conc. 10 μM) | 10 μL |
Store at 4°C and can be used up to 2 weeks. Discard after 2 weeks.
Differentiation media: CMGF+ without Wnt 3A, Nicotinamide and SB202190, reduce R-Spondin and Noggin conditioned medium to half of the concentration.
Stage 1: Growth and maintenance of 3D enteroids
NOTE: Perform all steps in sterile cell culture cabinet.
-
1
Take 10 mL of CMGF− into 15 mL corning tube and keep on ice.
-
2
Transfer contents of frozen vial containing enteroids (after thawing) to 15 mL tube containing 10 mL ice cold CMGF− using 2 mL pipette.
-
3
Spin down the tube at 80xg for 5 minutes at 4°C.
-
4
Remove the supernatant and re-suspend pellet in 120 μL matrigel (enough to seed 4 wells, 30 μL/well) using cold P200 pipet tips (store tip boxes in −20°C to keep them cold).
-
5
Plate enteroids as droplets without air bubbles in 4 wells of 24 well plate and transfer plate to 37°C incubator.
-
6
Allow matrigel to solidify for 5–10 minutes, add 500 μL of room temperature CMGF+ to each well and culture in 37°C incubator.
-
7
Refresh culture with CMGF+ every other day.
-
8
Typically, enteroids are ready to be passaged or make monolayers after 7–10 days of culturing.
Stage 2: Passage of enteroids
NOTE: Passage ratio is 1:5 if growth density is about 100 3D enteroids per well, this protocol is calibrated for one well only, repeat with other wells
-
9
After 7–10 days of culturing, enteroids are ready to be passaged.
-
10
Remove medium from wells (around the solid Matrigel).
-
11
Add 500 μL of ice cold CMGF− to well and mechanically break up Matrigel by pipetting with P1000 up and down couple of times.
-
12
Aspirate the solution with 1mL syringe containing 25Gx5/8 needle, syringe up and down 2–3 times per well.
-
13
Transfer the whole contents into 15 mL tube.
-
14
Add equal volume of ice cold CMGF−.
-
15
Spin down at 80xg or 0.6 rpm for 5 minutes at 4°C.
-
16
Remove the supernatant or medium and keep the tube on ice.
-
17
Re-suspend enteroid pellet in Matrigel (calculate the amount of matrigel that will be needed, 30 μL/well) using cold P200 pipet tips.
-
18
Pipette 30 μL/well of enteroids matrigel mixture as droplet into 24 well plate using cold P200 pipet tips. Repeat step 6–7 from revival of enteroids.
Stage 3: Human intestinal enteroid monolayers (HIEM)
-
19
For coating wells, both collagen and matrigel can be used. For collagen, prepare 1:30 dilution of collagen IV solution in water and for matrigel, prepare 1:40 dilution of matrigel in ice cold PBS.
-
20
Coating with collagen: Add 100 μL of the diluted collagen solution and incubate at 37°C for 2 hours. Coating with Matrigel: Add 100 μL of diluted matrigel solution and incubate at 37°C for 20 minutes.
-
21
After 7–9 days of culturing 3D enteroids in CMGF+, collect desired number of 3D enteroids wells and wash with ice cold 500 μL of 0.5 mM PBS-EDTA.
-
22
Spin at 1000 rpm for 5 minutes at 4°C.
-
23
Remove the wash medium and dissociate the enteroids by adding 1mL of 0.05%Trypsin/0.5 mM EDTA and incubate at 37°C for 4 minutes.
-
24
Inactivate trypsin by adding 1 mL of CMGF− containing 10% FBS.
-
25
Dissociate enteroids by vigorously pipetting up and down using P1000 Pipette for 50 times.
-
26
Then pass the dissociate enteroids to 50 mL falcon tube containing 40 μM cell strainer. (Pre-wet the strainer with addition of CMGF− containing 10% FBS and rinse 2 times after passing the enteroids)
-
27
Spin at 1500 rpm for 5 minutes at 4°C.
-
28
Re-suspend the pellet in required volume of CMGF+ media containing 10 μM Y-27632.
-
29
Add 100 μL of the pellet containing CMGF+ media containing 10uM Y-27632 to one well of 96 well plate or chambered slide or to the upper compartment of Transwell (Only for the transwell system, add 500 mL of just the CMGF+ media containing 10 μM Y-27632 (no pellet) to lower compartment).
-
30
Next day or the day after, refresh the medium with differentiation media to allow proliferation of different types of intestinal epithelial cells.
Support protocol 1: Freezing and storing of enteroids
Introduction:
While enteroid culture system can be continuously grown for several passages, it is always a good practice to freeze down and store some early passage line of enteroids. This will be useful in case of contamination or failure to grow well at later passages for unknown reasons or even just as a good laboratory practice.
Materials:
Human 3D enteroids grown for 7–10 days
GlutaMAX-1 with final concentration 2 mM and stock solution at 200 mM (Invitrogen, cat. no. 35050–061)
Penicillin/Streptomycin at final concentration 1X and stock solution at 100X (Invitrogen , cat. no. 15140–122)
HEPES 1 M at final concentration 10 mM (Invitrogen, cat. no. 15630–080)
Advanced DMEM/F12 (Invitrogen, cat. no. 12634–028)
Recovery Cell Freezing Media (Invitrogen, cat. no. 12648–010)
Corning cryo-vial (Corning, cat. No. 430659)
Refrigerated centrifuge with swing rotor
Sterilized filter pipette tips
CMGF− containing 10%FBS (See Reagents and Solutions)
Protocol Steps
After 7 days of enteroid growth (until Step 8, Basic Protocol 1)), enteroids are ready to be freeze down.
Repeat Step 2–3 from Basic Protocol 1, Stage 2: Passage of enteroids.
Spin down at 80xg or 0.6 rpm for 5 minutes at 4°C.
Remove the supernatant or medium and keep the tube on ice.
Re-suspend enteroids into 1 mL freezing medium at the ratio of 2 wells of enteroid into 1 vial.
Keep cryo-vial into cell freezing container and store at −80°C for overnight. Next day transfer cryo-vials into liquid nitrogen.
BASIC PROTOCOL 2
ADHERENCE TO HUMAN INTESTINAL ENTEROID MONOLAYERS
It is now well known and appreciated that most commensal and pathogenic bacteria interact with host cells by expressing adhesive molecules on their surfaces that engage host cell receptors or soluble macromolecules. This host-microbe interaction, referred to as bacterial adherence, is often the first step of pathogenesis and thereby making adhesins, proteins that promote adhesion, a target for therapeutic development. E. coli is a very large and diverse group of bacteria with different pathotypes producing multiple adhesins that associate with the host cell. The following protocol uses HIEM to evaluate E. coli adherence to intestinal epithelial cells. For adherence, dilute and wash E. coli cells grown normally in suitable bacteriologic media. Re-suspend washed bacteria in enteroid CMGF− medium, add desired multiplicity of infection (MOI) to the differentiated HIEM, and incubate for (1–24 hours) at 37°C in the presence of 5% CO2 in a humidified incubator. Next, wash monolayers, lyse or scrape cells, and enumerate bacteria by plating serial dilutions on Luria-Bertani (LB) agar plates without or with antibiotic supplementation.
Materials
E. coli cultures with or without antibiotic resistance cassettes
37°C incubator with shaker
Micro-centrifuge
Disposable inoculating loops (Fisher Scientific, cat. no. 22363-600)
Sterile Luria-Bertani (LB) broth and agar plates without or with antibiotic supplementation
Spectrophotometer
1X Phosphate Buffered Saline (PBS)
CMGF− medium (See Reagents and Solutions)
Differentiation medium (See Reagents and Solutions)
Differentiated (4 day) HIEM in 96- well plate Basic Protocol 1 Stage 3
Differentiated (4 day) HIEM in chambered slides, coverslips or transwells Basic Protocol 1 Stage 3
Hemacytometer
0.25% trypsin/0.2% EDTA (trypsin) (Mediatech-Corning, cat. no. 25–053-CI)
1.5 mL micro-centrifuge tubes (Fischer Scientific, cat. no. 05-408-129)
Ca2+/Mg2+ free Hanks balanced salt solution (HBSS) (Mediatech-Corning, cat. no. 20–021-CV)
0.1% Triton X-100 (Sigma Aldrich, cat. no. T-8787) in PBS
Adhesion using traditional plating methods
Stage 1: Bacterial culture preparation
-
1
Grow E. coli strains from a single colony in LB broth without or with antibiotic supplementation at 37°C with aeration overnight (16–18 hours) [15] If experiment requires bacteria in log phase, sub-culture the overnight samples and grow accordingly.
-
2
Measure the optical density of bacterial cultures using a spectrophotometer and estimate the number of colony forming units (CFU) based on bacterial growth correlation curve.
-
3
Wash cultures in an equal volume of PBS 1 time and re-suspend in an equal volume of CMGF− from Basic Protocol 1 Stage 1. (Use micro-centrifuge 13,000 rpm to pellet bacteria)
Stage 2: HIEM infection
-
4
Remove differentiation media from HIEM cultured in 96 well plates (Basic Protocol 1 Stage 3) and replace with 100 μL fresh CMGF− (if infection is only 1–2 hours) or differentiation media (if infection is 3–24 hours) from Basic Protocol 1 Stage 1.
-
5
Determine the number of cells for MOI calculation by removing 1 HIEM in the 96-well plate. Add 200 μL of trypsin to the monolayer and incubate at 37°C in the presence of 5% CO2 in a humidified incubator for 5 minutes. Stop trypsinization by transferring cells to a micro-centrifuge tube containing 800 μL of HBSS and mix by pipetting. Next, load 10 μL of cell suspension to a hemocytometer, count the cells, and calculate the number of cells/mL according to manufacturer’s instructions. Since the volume of the seeded cell suspension is 100 μL per well, calculate the number of cells for the monolayer by dividing the number of cells/mL by 10. The number of cells for the monolayer ranges from 50,000–100,000. Calculate the desired MOI (MOI 1–200 for 1-hour infection, MO1 1–10 for 3–6-hour infection, MO1 0.01–0.1 for overnight infection).
-
6
Add each strain at the calculated MOI to the assigned wells and incubate for the desired time at 37°C in the presence of 5% CO2 in a humidified incubator.
-
7
Wash monolayers 3 times with PBS, lyse with 100 μl cold PBS containing 0.1% Triton X-100 or remove monolayer by scraping using only PBS, and enumerate the bacteria by plating serial dilutions (10−2-10−4) on LB agar plates with or without antibiotics.
Stage 3: Adhesion using staining methods
-
8
Use HIEM cultured on chambered slides, coverslips or Transwells from Basic Protocol 1 Stage 3 for studying adherence by staining methods.
-
9
Repeat steps 1–6 as mentioned in Basic Protocol 2 for performing bacterial infection to evaluate adherence.
-
10
To visualize adhered bacteria, various staining methods such as Giemsa-Wright staining, Immunofluorescence (IF) and Electron Microscopy techniques can be used.
-
11
HIEM can be fixed at room temperature using methanol for 5 minutes, or paraformaldehyde for 30 minutes or glutaraldehyde for 20 minutes or at 4°C overnight for all the three fixatives methanol, paraformaldehyde and glutaraldehyde.
BASIC PROTOCOL 3
BACTERIAL INVASION OF HUMAN INTESTINAL ENTEROID MONOLAYERS
Invasive E. coli strains are categorized according to their clinical manifestation as non- diarrheagenic or diarrhea-causing. Determining how these strains penetrate the intestinal epithelium after host colonization is crucial to understanding the initial steps of infection. The methods applied here are also applicable to other invasive species, including shigella, salmonella, and listeria. The following protocol describes how to use HIEM to evaluate invasion. This protocol was modified from Pízarro-Cerdá et al. (2002) where transformed Caco-2 cells were used as the model system to test Listeria monocytogenes invasion [16]. Briefly, as in Poole et al. (2017), dilute and wash E. coli cultures. Re-suspend washed bacteria in enteroid CMGF− medium, add desired MOI to the differentiated HIEM, and incubate for (1–24 hours) at 37°C in the presence of 5% CO2 in a humidified incubator [17]. Next, wash monolayers, add gentamicin for 2 hours to kill extracellular bacteria, lyse cells, and enumerate bacteria by plating serial dilutions on Luria-Bertani (LB) agar plates without or with antibiotic supplementation.
Materials
E. coli cultures of with or without antibiotic resistance cassettes
37°C incubator with shaker
Micro-centrifuge
Disposable inoculating loops (Fisher Scientific, cat. no. 22-363-600)
Sterile Luria-Bertani (LB) broth and agar plates without or with antibiotic supplementation
Spectrophotometer
1X Phosphate Buffered Saline (PBS)
CMGF− medium (See Reagents and Solutions)
Differentiation medium (See Reagents and Solutions)
Differentiated (4 day) HIEM in 96- well plate Basic Protocol 1 Stage 3
Hemacytometer
0.25% trypsin/0.2% EDTA (trypsin) (Mediatech-Corning, cat. no. 25–053-CI)
1.5 mL micro-centrifuge tubes (Fischer Scientific, cat. no. 05-408-129)
Ca2+/Mg2+ free Hanks balanced salt solution (HBSS) (Mediatech-Corning, cat. no. 20–021-CV)
0.1% Triton X-100 (Sigma Aldrich, cat. no. T-8787) in PBS
500X (50 mg/mL) Gentamicin sulfate stock (Santa Cruz Biotechnology, cat. no. sc-29066A)
Repeat steps 1–6 from Basic Protocol 2 Stages 1 (Bacterial culture preparation) and 2 (HIEM infection).
Make a working concentration of gentamicin sulfate 50 μg/mL by adding 1 μL/mL of manufacturer’s stock to CMGF− medium. The manufacturer recommends working concentrations of gentamicin sulfate range from (0.5 to 50 μg/mL). According to the Clinical and Laboratory Standards Institute (CLSI) the minimum inhibitory concentration interpretive standard for most Enterobacteriaceae for gentamicin is ≤4 μg/mL.
After the infection (1–24 hours), wash the HIEM 3 times with PBS, add 100 μL gentamicin sulfate, and incubate for an additional 2 hours to kill extracellular bacteria.
Wash monolayers 1 time with PBS to remove antibiotic and lyse with 100 μL cold PBS containing 0.1% Triton X-100 and enumerate by plating undiluted sample and serial dilutions (10−1-10−2) on LB agar plates with or without antibiotics.
BASIC PROTOCOL 4
BACTERIAL TRANSLOCATION THROUGH HUMAN INTESTINAL ENTEROID MONOLAYERS
E. coli can cause infections distal to the intestine and is the most frequently isolated bacterium in Gram-negative associated bacteremia and sepsis. The mechanism by which certain E. coli strains breach the intestinal epithelial barrier and disseminate to extrainestinal sites is poorly understood. The translocation assay using HIEM was modified from Kortman et al. where Salmonella typhimurium and other enteric pathogens passage across polarized Caco-2 cell monolayers were evaluated [18]. Briefly, check polarization of differentiated HIEM seeded on Transwell inserts in a 24-well plate by measuring the trans-epithelial electrical resistance (TEER). Dilute, wash, and re-suspend cultures of E. coli with or without antibiotic resistance cassettes in enteroid differentiation media. Apically infect HIEM with a MOI of 1–100 and determine the number of translocated bacteria after 1, 2, 4, and 6 hours by removing media from the bottom chamber. Enumerate bacteria by plating serial dilutions on Luria-Bertani (LB) agar plates without or with antibiotic supplementation. Monitor the effect of bacteria on monolayer integrity by measuring TEER at 0, 3, and 6 hours.
Materials
E. coli cultures of with or without antibiotic resistance cassettes
37°C incubator with shaker
Micro-centrifuge
Disposable inoculating loops (Fisher Scientific, cat. no. 22-363-600)
Sterile Luria-Bertani (LB) broth and agar plates without or with antibiotic supplementation
Spectrophotometer
1X Phosphate Buffered Saline (PBS)
CMGF− medium (See Reagents and Solutions)
Differentiation medium (See Reagents and Solutions)
Millicell® ERS-2 Voltohmmeter (Millipore Sigma, cat. no. MERS00002)
Hemacytometer
0.25% trypsin/0.2% EDTA (trypsin) (Mediatech-Corning, cat. no. 25–053-CI)
1.5 mL micro-centrifuge tubes (Fischer Scientific, cat. no. 05-408-129)
Ca2+/Mg2+ free Hanks balanced salt solution (HBSS) (Mediatech-Corning, cat. no. 20–021-CV)
0.1% Triton X-100 (Sigma Aldrich, cat. no. T-8787) in PBS
Day 5 differentiated and polarized HIEM seeded on Transwell inserts (filter pore size 3.0 μm in diameter, BD Biosciences, San Jose, CA, USA) in a 24-well plate from Basic Protocol 1 Stage 3
Sterile razor blade
Repeat steps 1–3 from Basic Protocol 2 Stage 1 (Bacterial culture preparation).
On day 5 of differentiation, replace differentiation medium with fresh media in upper and lower compartment and measure the TEER of HIEM seeded on Transwell inserts in a 24-well plate from Basic Protocol 1 Stage 3. The seeding volume for HIEM on a Transwell insert is 100 μL, however, increase the volume to 200 μL in upper compartment to measure TEER. Use the Millicell® ERS-2 Voltohmmeter to determine TEER values which measures membrane potential and resistance of epithelial cells in culture following manufacturer’s instructions. Polarized monolayers have a TEER greater than or equal to (800 Ω). If the TEER of monolayers does not reach this value, do not use the monolayers to measure translocation in HIEM.
To determine the number of cells for MOI calculation, remove 1 HIEM by using a razor blade to cut filter off the Transwell insert. Place cut filter in a micro-centrifuge tube containing 200 μl of trypsin and incubate at 37°C in the presence of 5% CO2 in a humidified incubator for 5 minutes. Dislodge cells from filter by pipetting and stop trypsinization by adding 800 μl of HBSS and mix by pipetting. As previously mentioned in Basic Protocol 2, use a hemocytometer to count the cells and calculate the desired MOI (For this system a MOI ranging from 1–100 has been used).
Add each E. coli strain at the calculated MOI to the upper compartment of the assigned Transwells and incubate for 1, 2, 4, and 6 hours at 37°C in the presence of 5% CO2 in a humidified incubator.
To determine the number of translocated bacteria, remove media from the bottom chamber at each time point and enumerate by plating undiluted samples and serial dilutions (10−1-10−4) on Luria-Bertani (LB) agar plates without or with antibiotic supplementation. During the infection, monitor the effect of bacteria on monolayer integrity by taking TER measurements at time 0, 3, and 6 hours.
REAGENTS AND SOLUTIONS
COMMENTARY
BACKGROUND INFORMATION
Caco-2 cells and other transformed cancer cell line models have been used for several years to determine and evaluate the virulence factors and steps in pathogenesis of different enteric pathogens such as: E. coli, Listeria, Salmonella, and Shigella. Even though these models have been instrumental in identifying key components of the pathogenic process, the data collected for these experiments do not always translate to what happens in vivo. Therefore, the ex vivo model describe in this unit can serve as a human surrogate and a bridge to fill the gap between in vitro and in vivo models currently used to evaluate gastrointestinal infections.
CRITICAL PARAMETERS AND TROUBLESHOOTING
Growth of 3D enteroids
Enteroid cultures are typically grown in very low or no antibiotic media and this makes them prone to contamination. To prevent contamination, perform all steps in a sterile environment. If absolutely required, Fungizone and Gentamicin can be added to prevent contamination. However, this can reduce the growth of enteroids and is usually not recommended. During passage of enteroids, if you observe more dead cells (usually appears as dark patch of 3D enteroids), incorporating an additional wash (Step 6–8 in passage of enteroids) may help in removal of dead cells, and survival of enteroids can be increased by adding more Roc inhibitor. A dedicated enteroid culture room equipped with biosafety hoods, separate incubators and with entry only allowed for those culturing the enteroids significantly cuts down on the level of culture contamination.
Ratio of 3D enteroids to monolayers
If the density of 3D enteroids is about 100/well, then use the following recommended ratio for making monolayers: (1) use one 3D well to make 3 wells of monolayers on plastic 96 well plate (2) use 2.5 3D wells to make 1 Transwell (3) use one 3D well to make 2 wells of monolayers on plastic chambered slides (4) use one 3D well to make one monolayer on cell culture grade modified glass chambered slides or coverslips.
Choice of Collagen or Matrigel
For plastic surfaces both collagen and matrigel provide optimal condition for seeding and growth of enteroids. For glass surfaces, matrigel works better and helps in formation of uniform monolayer. While working with matrigel, using previously chilled pipette tips allows for easy aliquot of matrigel without the formation of air bubbles.
Trypsinization of enteroids
For trypsinizing 3D enteroids, collect 8–12 wells into one 15mL falcon tube and incubate with 1 mL trypsin. If there are more 3D wells, use a different tube. Do not combine more than 12 well of 3D enteroids in one 15mL falcon tube, it can result in poor or improper digestion. Timing of trypsinization varies for every segment of intestine used. Use the following recommended timing as a guideline for trypsin digestion: (1) for jejunum and colon incubate at 37°C for 4 minutes (2) for duodenum incubate at 37°C for 4.5 minutes (3) for ileum incubate at 37°C for 5 minutes. Longer incubation duration can result in death of the enteroid cells.
Preparation of monolayers
Failure to form a uniform monolayer could be due to many reasons. Careful consideration of parameters like spin speed, air bubble formation, and trypsin digestion timing can help in troubleshooting. Do not increase the spin speed and strictly follow the suggested timing and temperature. The enteroid cells are very sensitive to centrifugal forces and higher speed and inappropriate temperature can reduce viability and increase the number of dead cells. If under visual examination using a microscope one observes dead cells in the 3D format (dead cells are marked by presence of dark and denser enteroid and typically undifferentiated 3D enteroid appears clear and globular), addition of 10 μM Y-27632 to growth media may help in rescuing the culture. Avoid formation of air bubbles by pipetting inside the liquid volume available. Air bubbles can result in loss of total available cells. If the number of cells is less while seeding, a uniform monolayer is less likely to be formed.
Differentiation of enteroids
Differentiated enteroid monolayers provide a great platform to study host–pathogen interactions in a physiologically relevant model system. Typically, after plating the enteroid monolayers, they can settle and grow to 80%−90% confluency for a day or two days if required. The day differentiation medium is changed or introduced is considered day zero and day four or five is considered as an ideal day for infection. However, the cells are ready for infection between day three and day five of differentiation process.
TIME CONSIDERATIONS
Basic Protocol 1:
Plating of enteroid monolayers will be completed over a period of two weeks, depending upon the growth of the cells. For a researcher or technician who has previous cell culturing experience and handling enteroid cultures for the first time, it is recommended to begin with 4 wells of 3D enteroids until comfortable with all the procedures. Seeding and growing 3D enteroids (Stage 1): After thawing the frozen enteroid vials, it will take about 15 minutes to seed and feed the enteroids. For passaging of enteroids (Stage 2), it can take about 30 minutes - 60 minutes depending on the number of wells handled. Preparation of enteroid monolayers (Stage 3): After coating the wells with matrigel (20 minutes) or collagen (2 hours), it can take about 30 minutes-50 minutes to plate enteroid monolayers depending on the number of 3D wells used.
Basic Protocol 2:
Bacterial adherence as determined by a plating method will be completed over approximately 48–72 hours. The time for this protocol does not include the number of days for HIEM preparation and differentiation (see Time Considerations for Basic Protocol 1) on 96-well tissue culture dishes. This protocol was adapted to E. coli but can be optimized for other bacteria. Step 1 growth of overnight E. coli strains will be for 16–18 hours. If the experiment requires bacteria in log phase, growth of sub-cultures the day of the experiment takes 2–3 hours. Steps 2–4, including the washing and re-suspending the bacteria, counting the numbers of cells in the HIEM, and changing the HIEM media will take 1–2 hours depending on the number of samples. Step 5 infecting the HIEM ranges for 1–24 hours depending on MOI used. Step 6 washing, dissociating or lysing the HIEM, and plating serial dilutions will take 30 minutes to 1 hour depending on the number of samples. The plates will be incubated overnight and counted the next day.
Bacterial adherence: The bacterial adherence assay has the same time considerations as Basic Protocol 2; however, histological staining methods may involve additional 1–2 hours depending on the reagents used, immunofluorescence may take additional 24–48 hours depending on the antibody used and electron microscopy may involve additional 72 hours based on the drying method chosen.
Basic Protocol 3:
The measurement of bacterial invasion has the same time consideration as adherence in Basic Protocol 2; however, invasion requires an additional 2-hour incubation with gentamicin to kill extracellular bacteria after infection.
Basic Protocol 4:
Bacterial translocation will be completed over 48 hours. The time for this protocol does not include the number of days for HIEM preparation and differentiation on Transwell inserts (see Time Considerations for Basic Protocol 1). The growth of overnight and sub-cultures, determination of the CFU number, preparation of bacteria, removing and counting the numbers of cells in the HIEM on the Transwell insert, and changing the HIEM media will take 1–2 hours depending on the number of samples. Measuring the TER value for each HIEM will take approximately 15 minutes to 1 hour depending on the number samples and Transwells. Apical HIEM infection and plating serial dilutions of media from the bottom chamber of each sample to determine the number of translocated bacteria will be approximately 7 hours. The plates will be incubated overnight and counted the next day.
Figure 1.
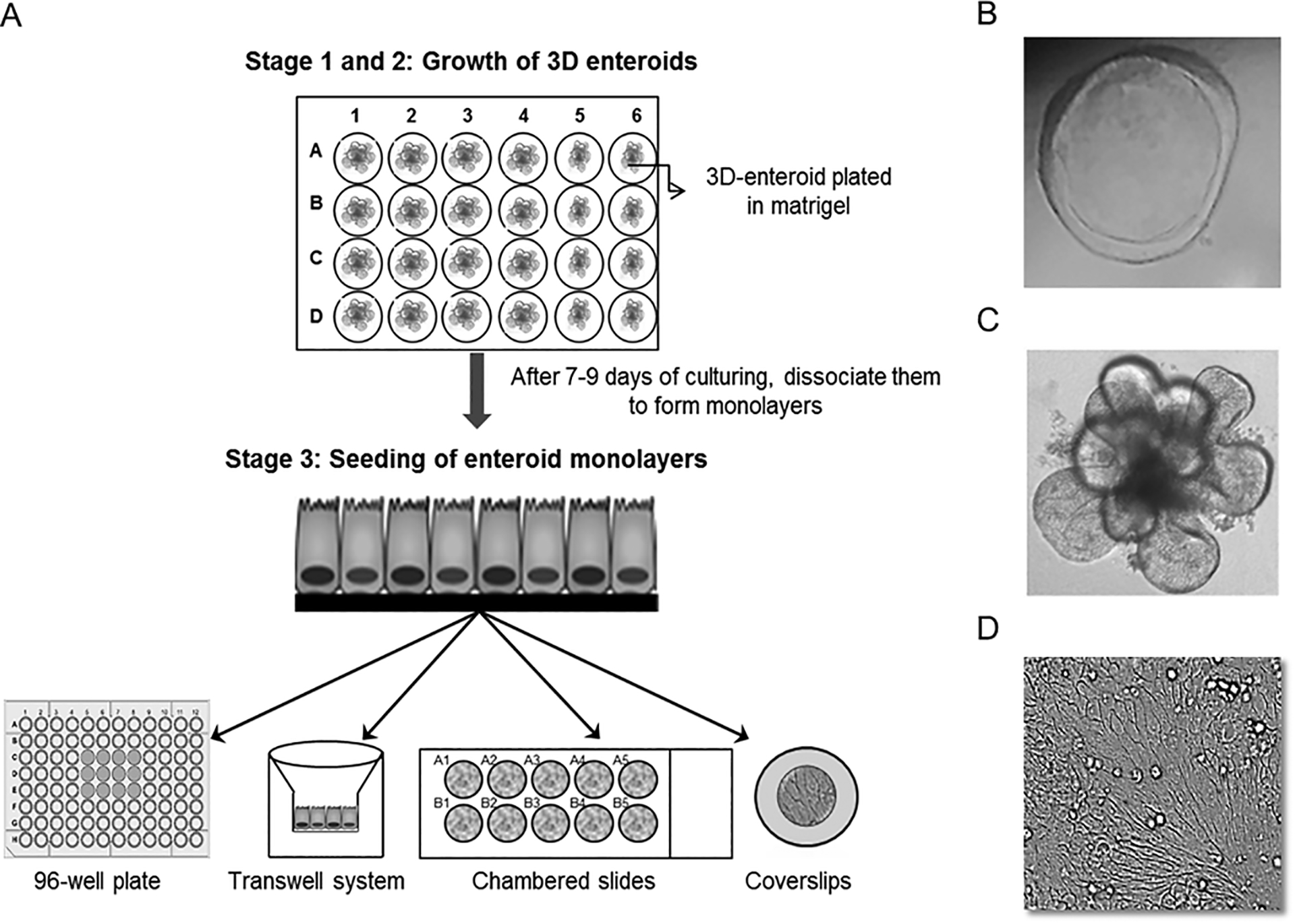
(A) Shows a schematic of enteroid monolayer preparation. Culturing of enteroids is broken down into three stages. In stage 1 and 2, 3D enteroids are seeded on matrigel support in 24 well plates. The 3D enteroids are maintained in undifferentiated growth condition (CMGF+ media) for 7–9 days before either passing them or making monolayers. In Stage 3, the monolayers are made from 3D enteroids and can be seeded on either collagen or matrigel coated 96 well plastic plates and Transwells or Cell-culture grade chambered slides and cover slips. After reaching 80%−90% confluency (usually within 24–48 hours), the undifferentiated enteroid monolayers are differentiated by addition of differentiation media to form a monolayer with the different intestinal epithelial cell types. The differentiation media are refreshed every alternate day and the infections are carried out after 4 days of differentiation. See Basic Protocol 1 for specifics. (B) Shows undifferentiated healthy 3D enteroid obtained from jejunum. (C) Shows differentiated 3D enteroid and the presence of dead cells (dark zone) in the center of enteroid can be observed. Dead cells in the center of 3D enteroid culture after 8 days are common. This is due to lack of penetration of growth media into the matrigel. However, if the 3D enteroid appears darker in most parts seen, that would indicate the enteroids are dying and not suitable for further proliferation or experiments. (D) Shows differentiated enteroid monolayer on 96 well plate.
SIGNIFICANCE STATEMENT.
Infectious diseases of the gastrointestinal (GI) tract are a substantial medical and economic burden worldwide. A complete understanding of how enteric pathogens cause illness in their human host is hampered by the lack of model systems that accurately recapitulate human physiology and disease. This article presents methods for the use of human intestinal enteroids, derived from the expansion of crypt stem cells, for the study of the adherence, invasion, and translocation of pathogenic bacteria such as E. coli. The utilization of “mini gut” enteroid systems as a human surrogate in the study of enteric pathogens may yield novel insights into mechanisms of pathogenesis and the development of anti-infective approaches.
ACKNOWLEDGEMENTS
This work was supported by the following: US National Institutes of Health grants U19 AI11497, T32 AI055413-14, and K12 GM084897.
LITERATURE CITED
- 1.Hodges K and Gill R, Infectious diarrhea: Cellular and molecular mechanisms. Gut Microbes, 2010. 1(1): p. 4–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wardlaw T, et al. , Diarrhoea: why children are still dying and what can be done. Lancet, 2010. 375(9718): p. 870–2. [DOI] [PubMed] [Google Scholar]
- 3.Steiner TS, et al. , Enteroaggregative Escherichia coli produce intestinal inflammation and growth impairment and cause interleukin-8 release from intestinal epithelial cells. J Infect Dis, 1998. 177(1): p. 88–96. [DOI] [PubMed] [Google Scholar]
- 4.McCracken KW, et al. , Generating human intestinal tissue from pluripotent stem cells in vitro. Nat Protoc, 2011. 6(12): p. 1920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato T and Clevers H, Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science, 2013. 340(6137): p. 1190–4. [DOI] [PubMed] [Google Scholar]
- 6.Sato T, et al. , Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology, 2011. 141(5): p. 1762–72. [DOI] [PubMed] [Google Scholar]
- 7.Sato T, et al. , Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature, 2011. 469(7330): p. 415–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato T, et al. , Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature, 2009. 459(7244): p. 262–5. [DOI] [PubMed] [Google Scholar]
- 9.Zachos NC, et al. , Human Enteroids/Colonoids and Intestinal Organoids Functionally Recapitulate Normal Intestinal Physiology and Pathophysiology. J Biol Chem, 2016. 291(8): p. 3759–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.In JG, et al. , Human mini-guts: new insights into intestinal physiology and host-pathogen interactions. Nat Rev Gastroenterol Hepatol, 2016. 13(11): p. 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahe MM, et al. , Establishment of human epithelial enteroids and colonoids from whole tissue and biopsy. J Vis Exp, 2015(97). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noel G, et al. , Erratum: A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions . Sci Rep, 2017. 7: p. 46790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee J, Choi JH, and Kim HJ, Human gut-on-a-chip technology: will this revolutionize our understanding of IBD and future treatments? Expert Rev Gastroenterol Hepatol, 2016. 10(8): p. 883–5. [DOI] [PubMed] [Google Scholar]
- 14.Dutta D and Clevers H, Organoid culture systems to study host-pathogen interactions. Curr Opin Immunol, 2017. 48: p. 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Son MS and Taylor RK, Growth and maintenance of Escherichia coli laboratory strains. Curr Protoc Microbiol, 2012. Chapter 5: p. Unit 5A.4. [DOI] [PubMed] [Google Scholar]
- 16.Pizarro-Cerdá J, Lecuit M, and Cossart P, 8 Measuring and analysing invasion of mammalian cells by bacterial pathogens: The Listeria monocytogenes system, in Methods in Microbiology. 2002, Academic Press. p. 161–177. [Google Scholar]
- 17.Poole NM, et al. , Role for FimH in Extraintestinal Pathogenic Escherichia coli Invasion and Translocation through the Intestinal Epithelium. Infect Immun, 2017. 85(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kortman GA, et al., Iron availability increases the pathogenic potential of Salmonella typhimurium and other enteric pathogens at the intestinal epithelial interface. PLoS One, 2012. 7(1): p. e29968. [DOI] [PMC free article] [PubMed] [Google Scholar]