

Abstract
Purple sweet potato color (PSPC), a class of naturally occurring anthocyanins, protects brain function against oxidative stress induced by D‐galactose (D‐gal) (Sigma‐Aldrich, St. Louis, MO, USA). Our data showed that PSPC enhanced open‐field activity, decreased step‐through latency, and improved spatial learning and memory ability in D‐gal‐treated old mice by decreasing advanced glycation end‐products' (AGEs) formation and the AGE receptor (RAGE) expression, and by elevating Cu,Zn‐superoxide dismutase (Cu,Zn‐SOD) (Sigma‐Aldrich) and catalase (CAT) expression and activity. Cleavage of caspase‐3 and increased terminal deoxynucleotidyl transferase (TdT)‐mediated deoxyuridine triphosphate (dUTP) nick‐end‐labeling (TUNEL)‐positive cells in D‐gal‐treated old mice were inhibited by PSPC, which might be attributed to its antioxidant property. PSPC also suppressed the activation of c‐Jun NH2‐terminal kinase (JNK) and the release of cytochrome c from mitochondria that counteracted the onset of neuronal apoptosis in D‐gal‐treated old mice. Furthermore, it was demonstrated that phosphoinositide 3‐kinase (PI3K) activation was required for PSPC to promote the neuronal survival accompanied with phosphorylation and activation of Akt and p44/42 mitogen‐activated protein kinase (MAPK) by using PI3K inhibitor LY294002 (Cell Signaling Technology, Inc., Beverly, MA, USA), implicating a neuronal survival mechanism. The present results suggest that neuronal survival promoted by PSPC may be a potentially effective method to enhance resistance of neurons to age‐related disease.
Keywords: Akt pathway, apoptosis, ERK pathway, neuronal survival, oxidative stress, PI3K, purple sweet potato color
INTRODUCTION
Brain aging refers to a multidimensional process including molecular, cellular and functional changes characterized by pathological features such as oxidative stress, alterations in cell metabolism, accumulation of misfolded proteins and nucleic acid damage. Increased oxidative stress and accumulation of oxidatively damaged molecules (proteins, nucleic acids and lipids) in the brain during aging promote dysfunction of various metabolic and signaling pathways 12, 20, 27. Along with realizing the adverse effects of oxidative stress on the brain, recent studies suggest advanced glycation end‐products (AGEs) are one of the risk factors in brain aging 30, 49. AGEs are the products of glycation reaction between a protein's primary amino group and a carbohydrate‐derived aldehyde group, and increase during aging 2, 29. Many studies have shown that D‐galactose (D‐gal) (Sigma‐Aldrich, St. Louis, MO, USA) induced mimetic aging in experimental animals 5, 45. D‐gal is a reducing sugar that can form AGEs in vivo and result in oxidative stress, which contributes to making clear the mechanism of this aging model to some extent 5, 7, 43. More evidence indicates that neuronal death induced by oxidative stress could exacerbate the deficits of learning and memory 6, 28, 55. Using this stimulus, we investigated whether D‐gal could induce apoptosis that contributed to the memory deficits in old mice.
Recently, interests in anthocyanin pigments have been intensified because of their physiological function, such as antioxidant, anti‐inflammatory and hepatoprotective activities 4, 39, 46. Purple sweet potato color (PSPC) is a kind of anthocyanins that is abundant in the tuber of the Okinawa purple sweet potato (Ipomoea batatas) 19, 31, 57. Our previous study has shown that PSPC could improve the ability of learning and memory in D‐gal‐injected mice via regulating the expression of synaptic proteins (54). On the basis of existing literature data, the primary aim of this research is to evaluate whether PSPC helps to promote survival of neurons to improve the ability of learning and memory in D‐gal‐treated old mice, and to investigate the molecular mechanism of its action in the brain of D‐gal‐treated old mice.
MATERIALS AND METHODS
Animals and treatments
Forty 15‐month‐old male Kunming strain mice were purchased from the Branch of National Breeder Center of Rodents (Shanghai). Prior to experiments, mice had free access to food and water, and were kept under constant conditions of temperature (23°C ± 1°C) and humidity (60%). Eight mice were housed per cage on a 12‐h light/dark schedule (lights on 8:30 am–8:30 pm). After a week of adaptation, mice were divided randomly into six groups. Groups 1, 5 and 6 served as vehicle control with injection of saline (0.9%) only, and the other three groups of mice (groups 2, 3 and 4) received daily subcutaneous injection of D‐gal at a dose of 500 mg/(kg/day) for 8 weeks, respectively.
After 8 weeks, mice in groups 3, 4 and 5 received PSPC (purity >90%; Qingdao Pengyuan Natural Pigment Research Institute) of 100 mg/(kg/day) in distilled water containing 0.1% Tween 80 (Sigma‐Aldrich, St. Louis, MO, USA) by oral gavage for another 4 weeks, and the mice of groups 1, 2 and 6 were given distilled water containing 0.1% Tween 80 orally at the same dose. Thirty minutes after PSPC treatment, 40 µg of LY294002 (Cell Signaling Technology, Inc., Beverly, MA, USA), dissolved in 10 µL of the solvent consisting of 99% sterile saline/1% dimethyl sulfoxide, was given for 4 minutes in groups 4 and 6 by means of intracerebroventricular infusion. The other four groups (groups 1, 2, 3 and 5) were infused with an equal volume of the solvent, respectively. Drug infusion was daily performed using a microinjector (KD scientific Inc., Holliston, MA, USA) through both cerebral ventricles (from the bregma: anteroposterior, 0.5 mm; mediolateral, 1.0 mm; depth, 2.0 mm) at a rate of 2.5 µL/minute for 4 weeks (35). All experiments were performed in compliance with the Chinese legislation on the use and care of laboratory animals, and were approved by the respective university committees for animal experiments. After the behavioral testing, mice were sacrificed, and brain tissues were immediately collected for experiments or stored at −70°C for later use.
Behavioral tests
Open‐field test
Open‐field test was performed as described previously 23, 24, 25. The open‐field apparatus was a circular arena (50 cm in diameter, 30 cm in height) made of wood laminated with light grey resin and illuminated with a single 40‐W bulb (3000 lux) placed 2.8 m above the center of it. The bottom of the arena was divided into 21 equal‐area grid cells by white lines. Tests were performed in the breeding room from 8:30 am to 4 pm. The individual mouse was placed in the middle of the chamber for each trial. After 1‐minute adaptation, the behavior of each mouse was recorded for 5 minutes by two observers 1 m away from the open‐field area. Between trials, the mouse was returned to its home cage in the same room, and the open field was wiped clean with a slightly damp cloth. The behavioral parameters were analyzed as the following: (i) ambulation: the number of grids crossed in the arena during the observation period; (ii) rearing: the number of times the mouse stood on its hind legs; (iii) leaning: the number of times the mouse placed one or two forelimbs on the wall of the arena; (iv) grooming: the number of times the mouse “washed” itself by licking, wiping, combing or scratching any part of the body.
Step‐through test
The step‐through passive avoidance apparatus consisted of an illuminated chamber (11.5 cm × 9.5 cm × 11 cm) attached to a darkened chamber (23.5 cm × 9.5 cm × 11 cm) containing a metal floor that could deliver footshocks. The two compartments were separated by a guillotine door. The illuminated chamber was lit with a 25‐W lamp. The step‐through test was performed as described previously 23, 24.
Morris water maze (MWM) test
The MWM test was performed as described previously 23, 24, 25. The experimental apparatus consisted of a circular water tank (100 cm in diameter, 35 cm in height), containing water (23°C ± 1°C) to a depth of 15.5 cm, which was rendered opaque by adding ink. A platform (4.5 cm in diameter, 14.5 cm in height) was submerged 1 cm below the water surface and placed at the midpoint of one quadrant. The pool was located in a test room that contained various prominent visual cues. Each mouse received four training trials per day for 5 consecutive days. Latency to escape from the water maze (finding the submerged escape platform) was calculated for each trial. On day 6, the probe test was carried out by removing the platform and allowing each mouse to swim freely for 60 s. The time that mice spent swimming in the target quadrant (where the platform was located during hidden‐platform training), and in the three nontarget quadrants (right, left and opposite quadrants), was measured, respectively. For the probe trials, the number of times each mouse crossed over the platform site was also measured and calculated. All data were recorded with a computerized video system.
Preparation of tissue samples
Tissue homogenates
For biochemical studies, animals were deeply anesthetized and sacrificed (25). Brains were promptly dissected and perfused with 50‐mM (pH 7.4) ice‐cold phosphate‐buffered saline (PBS) solution. Brains were homogenized in 1/5 (w/v) PBS containing a protease inhibitor cocktail (Sigma‐Aldrich) with 10 strokes at 1200 rpm in a Teflon‐glass Potter‐Elvehjem homogenizer (Kontes, Vineland, NJ, USA). Homogenates were divided into two portions, and one part was directly centrifuged at 8000 × g for 10 minutes to obtain the supernatant. Supernatant aliquots were used to determine brain catalase (CAT) and protein contents. The second part of homogenates was sonicated four times for 30 s with 20‐s intervals using a VWR Bronson Scientific sonicator (VWR Int. Ltd. Merch House Pool, UK), centrifuged at 5000 × g for 10 minutes at 4°C, and the supernatants were collected and stored at −70°C for the determination of Cu,Zn superoxide dismutase (Cu,Zn‐SOD) (Sigma‐Aldrich) enzyme activities.
For Western blot analysis, brains were homogenized in 1/3 (w/v) ice‐cold Radioimmunoprecipitation (RIPA) lysis buffer [1 × Tris‐buffered saline (TBS), 1% Nonidet P‐40 (NP‐40), 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS) and 0.004% sodium azide] combining 30 µL of 10 mg/mL phenylmethylsulfonyl fluoride (PMSF) solution, 30 µL of Na3VO4, 30 µL of NaF and 30 µL of protease inhibitor cocktails per gram of tissue 25, 54. The homogenates were sonicated four times for 30 s with 20‐s intervals using a VWR Bronson Scientific sonicator, centrifuged at 15 000 × g for 10 minutes at 4°C, and the supernatant was collected and stored at −70°C for Western blot studies. Protein levels in the supernatants were determined using the BCA™ assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA).
When necessary, brain tissues were immediately used to prepare mitochondrial fractions at 4°C. Nonfrozen brain tissues were homogenized in 1/3 (w/v) ice‐cold homogenization buffer [50‐mM 4‐morpholinepropanesulfonic acid (MOPS), pH 7.4 (Sigma‐Aldrich), 100‐mM KCl, 320‐mM sucrose, 50‐mM NaF, 0.5‐mM MgCl2, 0.2‐mM 1,4‐dithiothreitol, 1‐mM ethylenediaminetetraacetic acid (EDTA), 1‐mM ethylene glycol‐bis(2‐aminoethylether)‐N,N,N′,N′‐tetraacetic acid (EGTA), 1‐mM Na3VO4 (Sigma‐Aldrich), 20‐mM sodium pyrophosphate, 20‐mM β‐phosphoglycerol, 1‐mM p‐nitrophenyl phosphate, 1‐mM benzamidine, 1‐mM phenylmethylsulfonyl fluoride and 5 mg/mL each of leupeptin, aprotinin and pepstatin A]. The homogenates were centrifuged at 1000 × g for 10 minutes at 4°C. The pellets were discarded, and supernatants were centrifuged at 17 000 × g for 20 minutes at 4°C to get the cytosolic fraction in the supernatants and the crude mitochondrial fraction in the pellets. The protein concentrations were determined by the method of Lowry et al (22).
Collection of brain slice
The mice were perfused transcardially with 25 mL of normal saline (0.9%). The brain tissues were fixed in a fresh solution of 4% paraformaldehyde (pH 7.4) at 4°C for 4 h, incubated overnight at 4°C in 100‐mM sodium phosphate buffer (pH 7.4) containing 30% sucrose and embedded in optimal cutting temperature compound (OCT, Leica Microsystems, Nussloch, Germany). Coronal sections (12 µm) from cryofixed tissues were collected on 3‐aminopropyl‐trimethoxysilane‐coated slides (Sigma‐Aldrich) and stored at −70°C.
Biochemical assays
Assay of Cu,Zn‐SOD activity
Chemicals used in the assay, including xanthine, xanthine oxidase, cytochrome c, bovine serum albumin (BSA) and SOD, were purchased from Sigma‐Aldrich. Cu,Zn‐SOD activity was measured according to the method described by Lu et al 23, 25. Solution A was prepared by mixing 100 mL of 50‐mM PBS (pH 7.4) containing 0.1‐mM EDTA and 2 µmol of cytochrome c with 10 mL of 0.001 N NaOH solution containing 5 µmol of xanthine. Solution B contained 0.2‐U xanthine oxidase/mL and 0.1‐mM EDTA. Fifty microliters of a tissue supernatant was mixed with 2.9 mL of solution A, and the reaction was started by adding 50 µL of solution B. Change in absorbance at 550 nm was monitored in a spectrophotometer (Shimadzu UV‐2501PC, Shimadzu Corp., Kyoto, Japan). A blank was run by replacing the supernatant with 50 µL of ultra pure water. Cu,Zn‐SOD level was expressed as units per milligram protein, with reference to the activity of a standard curve of bovine copper‐zinc SOD under the same conditions.
Assay of CAT activity
CAT activity in brain tissue homogenates was determined using the method of Lu et al 23, 25. In brief, to a quartz cuvette (Shimadzu Corp., Kyoto, Japan), 0.65 mL of the phosphate buffer (50 mmol/L; pH 7.0) and a 50 µL sample were added, and the reaction was started by the addition of 0.3 mL of 30‐mM hydrogen peroxide (H2O2). The decomposition of H2O2 was monitored at 240 nm at 25°C. CAT activity was calculated in terms of nM H2O2 consumed per minute per milligram of protein in the samples.
Western blot analysis
Samples (80‐µg protein each) were separated by denaturing SDS‐polyacrylamide gel electrophoresis (PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane (Roche Diagnostics Corporation, Indianapolis, IN, USA) by electrophoretic transfer. The membranes were blocked with 5% BSA and 0.1% Tween 20 in TBS, incubated overnight with polyclonal anti‐AGE antiserum and raised against glyoxylic acid‐treated keyhole limpet hemocyanin (1:1000, Wako, Commercial Rafer, Zaragoza, Spain), monoclonal anti‐N‐Carboxymethyl‐lysine (CML) antibody (1:2000, Transgenic Inc., Kumamoto, Japan), a monoclonal anti‐N‐carboxyethyl‐lysine (CEL) antibody (1:2000, Transgenic Inc.) and polyclonal rabbit anti‐AGE receptor antibody (1:1000, Abcam, Cambridge, UK), polyclonal rabbit anti‐CAT antibody (1:4000, Abcam), polyclonal rabbit anti‐Cu,Zn‐SOD antibody (1:1000, Abcam), polyclonal rabbit anti‐cleaved caspase‐3 and anti‐caspase‐3 antibody (1:1000, Cell Signaling Technology, Inc.), monoclonal rabbit anti‐stress‐activated protein kinase/c‐Jun NH2‐terminal kinase (SAPK/JNK) (pThr183 or pTyr185) antibody (1:1000, Cell Signaling Technology, Inc.), monoclonal rabbit anti‐SAPK/JNK antibody (1:1000, Cell Signaling Technology, Inc.), monoclonal mouse anti‐cytochrome c antibody (1:2500, BD PharMingen, San Diego, CA, USA), monoclonal rabbit anti‐Akt (pThr308 or pSer473) antibody (1:1000, Cell Signaling Technology, Inc.), monoclonal rabbit anti‐Akt antibody (1:1000, Cell Signaling Technology, Inc.), monoclonal rabbit anti‐p44/p42 mitogen‐activated protein kinase (MAPK) [anti‐extracellular signal‐related kinases (ERK1/2)] (pThr202/Tyr204) antibody (1:1000, Cell Signaling Technology, Inc.), monoclonal rabbit anti‐ERK1/2 antibody (1:1000, Cell Signaling Technology, Inc.), polyclonal rabbit anti‐Bcl‐2‐associated death protein (Bad) (pSer112 or pSer136) antibody (1:1000, Cell Signaling Technology, Inc.), and polyclonal rabbit anti‐Bad antibody (1:1000, Cell Signaling Technology, Inc.), respectively. Quantitation of detected bands was performed with the Scion Image™ analysis software (Scion Corp., Frederick, MD, USA). The data were normalized using monoclonal mouse anti‐β‐actin (1:8000, Chemicon International Inc., Temecula, CA, USA), monoclonal mouse anti‐cytochrome c oxidase (COX) IV antibody (1:1000, Abcam), anti‐total SAPK/JNK, anti‐total‐Akt, anti‐total‐ERK1/2 or anti‐total‐Bad antibody as an internal control, and standardized with the vehicle control as 1.0.
Deoxyribonucleotidyl transferase (TdT)‐mediated dUTP‐fluorescein isothiocyanate (FITC) nick‐end labeling (TUNEL) assay
For the TUNEL staining, the standard protocol for frozen sections was followed (BD ApoAlert™ DNA Fragmentation assay kit, BD Biosciences Clontech, Palo Alto, CA, USA). The sections were immersed in a Coplin jar (VWR International, Aurora, CO, USA) containing fresh 4% formaldehyde/PBS, and incubated at room temperature for 5 minutes. The sections were washed twice with PBS for 5 minutes. The liquid was allowed to drain thoroughly, and the slides were placed on a flat surface. Each section was covered with 100 µL of 20 µg/mL Proteinase K solution (section III) and incubated at 37°C for 5 minutes. After two washes of 5 minutes each with PBS, the sections were transferred into a Coplin jar containing 4% formaldehyde/PBS and then washed in PBS again. The cells were covered in equilibration buffer (from the kit) and equilibrated at room temperature for 5 minutes. The equilibration buffer was drained, and TdT incubation buffer was added to the tissue sections. To perform the tailing reaction, the slice was placed in a dark and humidified 37°C incubator for 1 h. The tailing reaction was terminated by immersing the samples in 2 × saline‐sodium citrate (SSC) at room temperature for 15 minutes. Samples were washed three times with PBS for 5 minutes to remove unincorporated fluorescein‐dUTP. Finally, strong, nuclear green fluorescence in apoptotic cells was observed on a fluorescent microscopy equipped with a standard fluorescein filter (520 nm ± 20 nm). All cells stained with propidium iodide exhibit strong red cytoplasmic fluorescence when viewed at 620 nm. Specimens were analyzed with a Zeiss Axioskop 40™ microscope equipped for light microscopy (Carl Zeiss, Oberkochen, Germany). The images were taken with a CCD camera (CoolSNAP™ Color, Photometrics, Roper Scientific Inc., Trenton, NJ, USA) and processed with Image‐Pro® Plus 6.0 software (Media Cybernetics Inc., Newburyport, MA, USA). For analysis; plaque areas were excluded, and the number of stained cells in 0.01 mm2 was estimated by blind manual counting of seven regions located at a consistent position per section.
Statistical analysis
All statistical analyses were performed using the SPSS software, version 11.5 (SPSS Inc., Chicago, IL, USA). Group differences in the escape latency in the MWM training task were analyzed using two‐way analysis of variance (ANOVA) with repeated measures, the factors being treatment and training day. The other data were analyzed with one‐way ANOVA followed by Newman‐Keuls or Tukey's Honestly Significant Differences (HSD) post hoc test. Data were expressed as means ± standard deviation. Statistical significance was set at P < 0.05.
RESULTS
Effect of PSPC on the behavior of D‐gal‐treated old mice
Open‐field test
The effect of PSPC on the behavior of D‐gal‐treated old mice in open field is shown in Figure 1. D‐gal treatment reduced the locomotor activity, exploratory behavior and anxiety‐related behavior such as grid crossing, rearing/leaning [F(4,35) = 14.349, P < 0.001] and grooming [F(4,35) = 12.169, P < 0.001]. PSPC treatment produced a significant increase in open‐field activity in D‐gal‐treated old mice (P < 0.01). However, LY294002, a selective phosphatidylinositol 3‐kinase inhibitor, produced a decrease in open‐field activity in the old mice cotreated with D‐gal and PSPC. And there was no significant difference in open‐field activity between the D‐gal/PSPC/LY294002 group and the D‐gal group.
Figure 1.
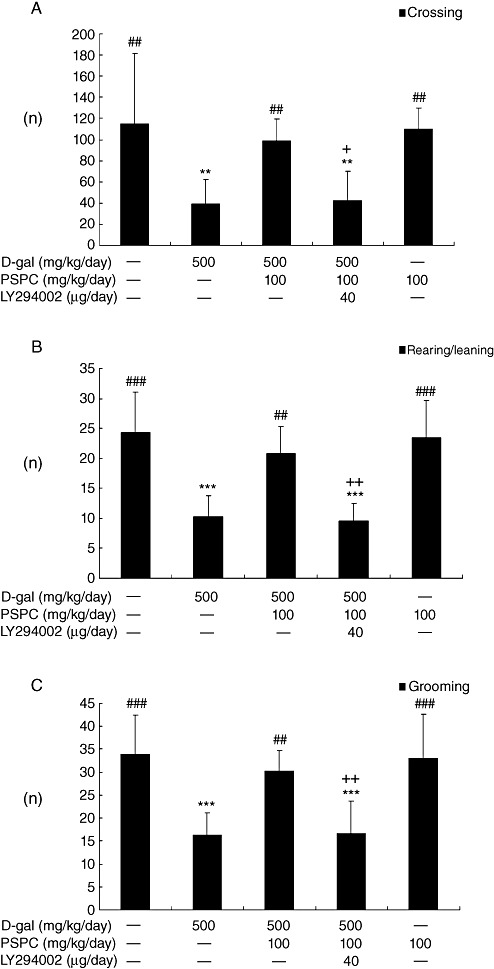
Effect of purple sweet potato color (PSPC) on the behavior of D‐galactose (D‐gal)‐treated old mice in the open field (n = 8). All values are expressed as means ± standard deviation. A. Comparison of crossing numbers (within 5 minutes). B. Comparison of rearing/leaning numbers (within 5 minutes). C. Comparison of grooming numbers (within 5 minutes). **P < 0.01, ***P < 0.01 vs. control group; ##P < 0.01, ###P < 0.001 vs. D‐gal group; +P < 0.05, ++P < 0.01 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 group.
Step‐through test
In the acquisition trial, the step‐through latencies did not differ among the five groups [F(4,35) = 1.510, P > 0.05] (Figure 2). The step‐through latency in the 24‐h retention trial was significantly decreased in D‐gal‐treated old mice [F(4,35) = 5.741, P < 0.05]. Post hoc analysis showed that PSPC significantly inhibited the amnesic effect of D‐gal on latency time for the step‐through trial (P < 0.05); however, the latency time in the D‐gal/PSPC group was markedly decreased by LY294002. And there was no significant difference in the latency time between the D‐gal/PSPC/LY294002 group and the D‐gal group.
Figure 2.
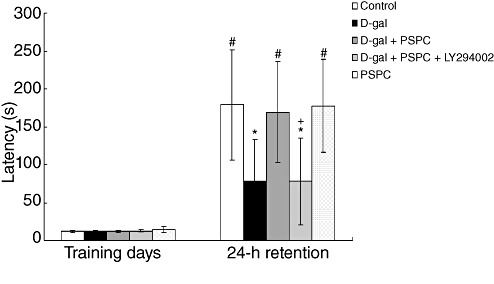
Effect of purple sweet potato color (PSPC) on the step‐through latency in D‐galactose (D‐gal)‐treated old mice (n = 8). All values are expressed as means ± standard deviation. *P < 0.05 vs. control group; #P < 0.05 vs. D‐gal group; +P < 0.05 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 group.
MWM test
Statistical analysis for the escape latency in the MWM test was performed using two‐way ANOVA for repeated measures, with day and treatment as sources of variation (Figure 3). Mice showed significant difference in mean latency to reach the platform between training days [F(4,175) = 54.747, P < 0.001] and between treatments [F(4,175) = 17.871, P < 0.001], but no interaction between the factors day and treatment [F(16,175) = 0.964, P > 0.05].
Figure 3.
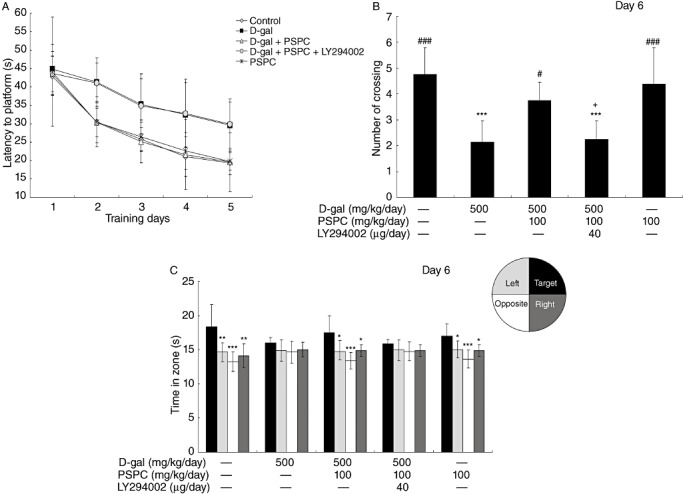
Effect of purple sweet potato color (PSPC) on the behavior of D‐galactose (D‐gal)‐treated old mice in Morris water maze (n = 8). All values are expressed as means ± standard deviation. A. Comparison of latency to platform during 5 training days. B. Comparison of numbers of crossing over platform site on day 6. ***P < 0.001 vs. control group; #P < 0.05, ###P < 0.001 vs. D‐gal group; +P < 0.05 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 group. C. Comparison of the time spent in target quadrant with other quadrants on day 6. *P < 0.05, **P < 0.01, ***P < 0.01 compared with any other quadrant.
During the hidden‐platform acquisition, the performance was improved as indicated by the decreased escape latencies across successive days (Figure 3A). In the D‐gal group, the latency was prolonged significantly as compared with that in the vehicle controls (P < 0.001), and remained unchanged in the D‐gal group cotreated with PSPC (P > 0.05). However, the D‐gal/PSPC group remained with prolonged latency caused by LY294002. During the probe trial, the spatial accuracy of old mice was determined by the number of times they crossed the former platform area (Figure 3B). A significantly decreased number of crossing was observed in the D‐gal group [F(4,35) = 12.276, P < 0.001], which was reversed by PSPC treatment. In contrast, no significant difference was found in the number of crossings between the D‐gal group and the D‐gal/PSPC/LY294002 group. Probe trial performance can also be represented as time spent in different quadrants, and the well‐trained mouse will show high preference for the target quadrant where the platform used to be (Figure 3C). The preference, which was not significant in both D‐gal group and D‐gal/PSPC/LY294002 group, approached significance in the control, D‐gal/PSPC and PSPC groups [control group: F(3,28) = 9.369, P < 0.01; D‐gal group: F(3,28) = 1.747, P > 0.05; D‐gal/PSPC group: F(3,28) = 8.956, P < 0.05; D‐gal/PSPC/LY294002 group: F(3,28) = 1.655, P > 0.05; PSPC group: F(3,28) = 8.598, P < 0.05 vs. target quardrant].
Effect of PSPC on AGEs, CML, CEL formation and RAGE expression in D‐gal‐treated old mice
The antibodies to AGEs, CML and CEL were used as markers of glycoxidation and carbonyl production. The formation of AGEs, CML and CEL (ranging from 30 kDa to 200 kDa) was analyzed by Western blotting (Figure 4). The intensity of AGEs, CML and CEL was significantly increased in the brain of D‐gal‐treated mice as compared with vehicle controls [AGEs: F(4,10) = 80.714, P < 0.001; CML: F(4,10) = 56.097, P < 0.001; CEL: F(4,10) = 67.233, P < 0.001]. PSPC significantly reduced the formation of AGEs, CML and CEL in the brain of D‐gal‐treated mice [AGEs: P < 0.001; CML: P < 0.01; CEL: P < 0.05 vs. D‐gal group]. However, LY294002 significantly inhibited the reduced AGEs, CML and CEL formation in the brain of old mice cotreated with D‐gal and PSPC [AGEs: P < 0.001; CML: P < 0.01; CEL: P < 0.01 vs. D‐gal/PSPC group].
Figure 4.
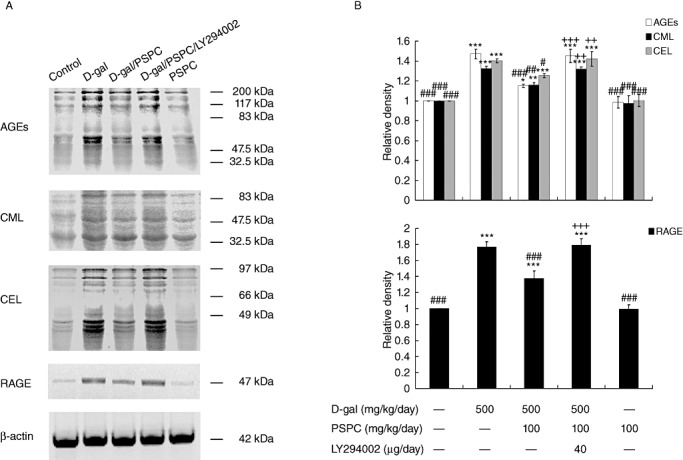
Effect of purple sweet potato color (PSPC) on the expression of advanced glycation end‐products (AGEs), Carboxymethyl‐lysine (CML), carboxyethyl‐lysine (CEL) and AGE receptor (RAGE) in the brain of D‐galactose (D‐gal)‐treated old mice (n = 3). A. Immunoreactivity of AGEs, CML, CEL, RAGE and β‐actin in all treated groups. B. Relative density analysis of the AGEs, CML, CEL and RAGE protein bands. β‐Actin was probed as an internal control. The relative density is expressed as the ratio (AGEs/β‐actin, CML/β‐actin, CEL/β‐actin, RAGE/β‐actin), and the vehicle control is set as 1.0. Values are averages from three independent experiments. Each value is the mean ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. D‐gal group; ++P < 0.01, +++P < 0.001 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 groups.
RAGE, the receptor for AGEs, is a member of the immunoglobulin superfamily of cell surface molecules; its interaction with ligands enhances receptor expression 34, 38. D‐gal markedly increased the expression of RAGE in the brain of mice [RAGE: F(4,10) = 104.291, P < 0.001] (Figure 4). PSPC significantly inhibited the overexpression of RAGE in the brain of D‐gal‐treated mice (P < 0.001). LY294002 markedly induced the expression of RAGE (P < 0.001), which was consistent with the changes in the formation of AGEs. No significant difference was found in the expression of AGEs, CML, CEL and RAGE between mice treated with PSPC alone and vehicle controls.
Effect of PSPC on the expression and activity of Cu,Zn‐SOD and CAT in D‐gal‐treated old mice
We had previously found that mice, when treated with D‐gal, underwent age‐related changes via redox balance alterations 23, 25. To determine whether antioxidative effect of PSPC was responsible for the improved learning and memory performance in the same model, we examined the changes in the expression and activities of enzymes Cu,Zn‐SOD and CAT. As shown in 5, 6, the expressions of Cu,Zn‐SOD and CAT in D‐gal‐treated old mice were significantly decreased as compared with vehicle controls [Cu,Zn‐SOD: F(4,10) = 198.972, P < 0.001; CAT: F(4,10) = 270.723, P < 0.001]. The expression levels of Cu,Zn‐SOD and CAT were significantly higher in old mice cotreated with PSPC and D‐gal than that in D‐gal treated old mice (Cu,Zn‐SOD: P < 0.001; CAT: P < 0.001). Correspondingly, the decreased activities of Cu,Zn‐SOD and CAT in D‐gal‐treated old mice were significantly elevated by PSPC [Cu,Zn‐SOD: F(4,20) = 29.601, P = 0.01; CAT: F(4,20) = 21.006, P < 0.05 vs. D‐gal group]. LY294002 markedly inhibited the increased expression and activities of Cu,Zn‐SOD and CAT in old mice cotreated with D‐gal and PSPC.
Figure 5.
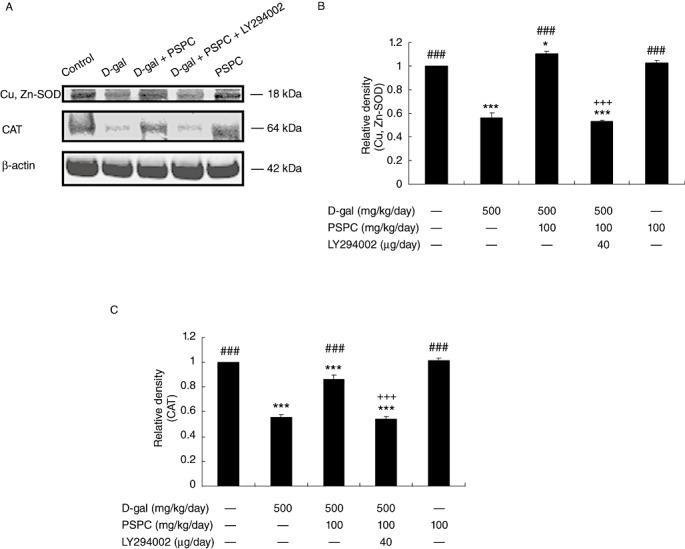
Effect of purple sweet potato color (PSPC) on the expression of Cu,Zn‐superoxide dismutase (Cu,Zn‐SOD) and catalase (CAT) in the brain of D‐galactose (D‐gal)‐treated old mice (n = 3). A. Immunoreactivity of Cu,Zn‐SOD, CAT and β‐actin in all treated groups. B. Relative density analysis of the Cu,Zn‐SOD protein bands. C. Relative density analysis of the CAT protein bands. β‐Actin was probed as an internal control. The relative density is expressed as the ratio (Cu,Zn‐SOD/β‐actin, CAT/β‐actin), and the vehicle control is set as 1.0. Values are averages from three independent experiments. Each value is the mean±standard deviation. *P < 0.05, ***P < 0.001 vs. control group; ###P < 0.001 vs. D‐gal group; +++P < 0.001 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 group.
Figure 6.
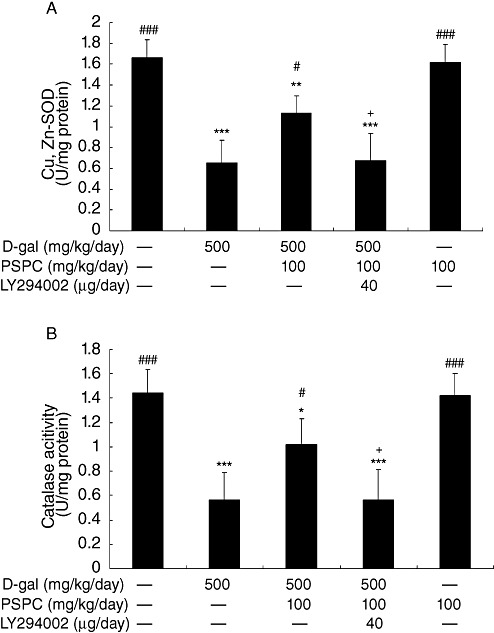
Effect of purple sweet potato color (PSPC) on the activity of Cu,Zn‐superoxide dismutase (Cu,Zn‐SOD) and catalase (CAT) in the brain of D‐galactose (D‐gal)‐treated old mice (n = 5). Values are averages from five independent experiments. Each value is the mean ± standard deviation. A. Cu,Zn‐SOD. B. CAT. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05, ###P < 0.001 vs. D‐gal group; +P < 0.05 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 group.
Effect of PSPC on the D‐gal‐induced caspase‐3‐dependent apoptosis in mouse brain
We used the TUNEL assay to investigate the effect of PSPC on the D‐gal‐induced apoptosis (7, 8). The number of TUNEL‐positive cells in the hippocampus and cerebral cortex of D‐gal‐treated old mice was significantly increased [hippocampus: F(4,30) = 45.566, P < 0.001; cerebral cortex: F(4,30) = 37.602, P < 0.001]. There were significantly fewer TUNEL‐positive cells in the hippocampus and cerebral cortex of old mice cotreated with D‐gal and PSPC as compared with those treated with D‐gal (hippocampus: P < 0.001; cerebral cortex: P < 0.001). However, the significantly increased TUNEL‐positive cells were observed in the hippocampus and cerebral cortex of old mice cotreated with D‐gal and PSPC group caused by LY294002 (hippocampus: P < 0.001; cerebral cortex: P < 0.001).
Figure 7.
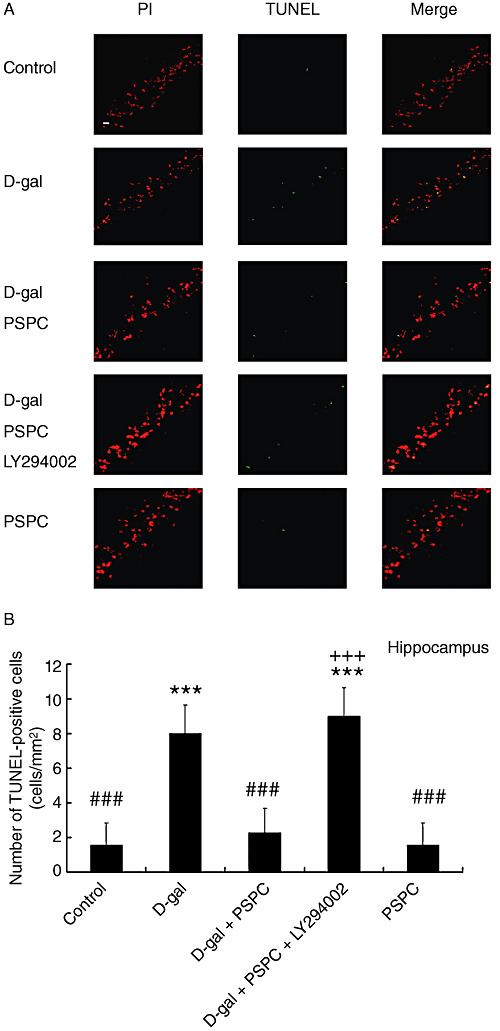
A. In situ detection of fragmented DNA [deoxyribonucleotidyl transferase‐mediated dUTP‐FITC nick‐end labeling (TUNEL) assay] in the hippocampus of old mice. The brain tissues were processed for TUNEL and photographed using a fluorescence microscope with either a propidium iodide (PI) filter alone (left) or an FITC filter alone (middle). The merged images show that apoptotic cells appear yellow and non‐apoptotic cells appear red (right). Scale bars = 100 µm. B. The histogram showed the relative proportion of TUNEL‐positive cells in the hippocampus of old mice. Data are means ± standard deviation (n = 7). ***P < 0.001 vs. control group; ###P < 0.001 vs. D‐galactose (D‐gal) group; +++P < 0.001 D‐gal/purple sweet potato color (PSPC) group vs. D‐gal/PSPC/LY294002 group.
Figure 8.
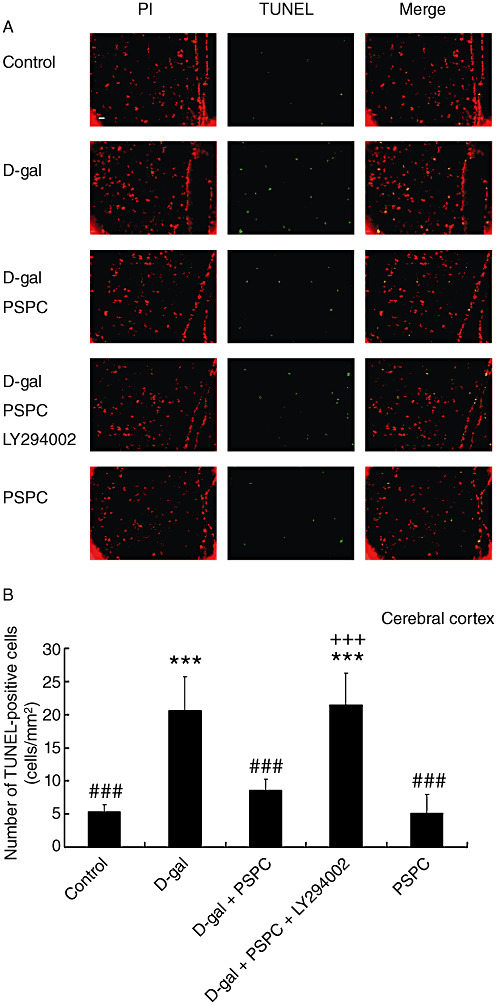
A. In situ detection of fragmented DNA [deoxyribonucleotidyl transferase‐mediated dUTP‐(FITC) nick‐end labeling (TUNEL) assay] in the cerebral cortex of old mice. The brain tissues were processed for TUNEL and photographed using a fluorescence microscope with either a propidium iodide (PI) filter alone (left) or an FITC filter alone (middle). The merged images show that apoptotic cells appear yellow and non‐apoptotic cells appear red (right). Scale bars = 100 µm. B. The histogram showed the relative proportion of TUNEL‐positive cells in the cerebral cortex of old mice. Data are means ± standard deviation (n = 7). ***P < 0.001 vs. control group; ###P < 0.001 vs. D‐galactose (D‐gal) group; +++P < 0.001 D‐gal/purple sweet potato color (PSPC) group vs. D‐gal/PSPC/LY294002 group.
The cleavage and activation of caspase‐3 in the brain of old mice were also examined by Western blotting (Figure 9). As reported previously, D‐gal increased the activation of caspase‐3 in the brain of mice (25). In the present study, we found that PSPC could markedly inhibit the activation of caspase‐3 in D‐gal‐treated old mice [F(4,10) = 383.581, P < 0.001 vs. D‐gal group]. The activation of caspase‐3 was markedly increased in the brain of D‐gal/PSPC‐cotreated old mice by treatment with LY294002 (P < 0.001 vs. D‐gal/PSPC group).
Figure 9.
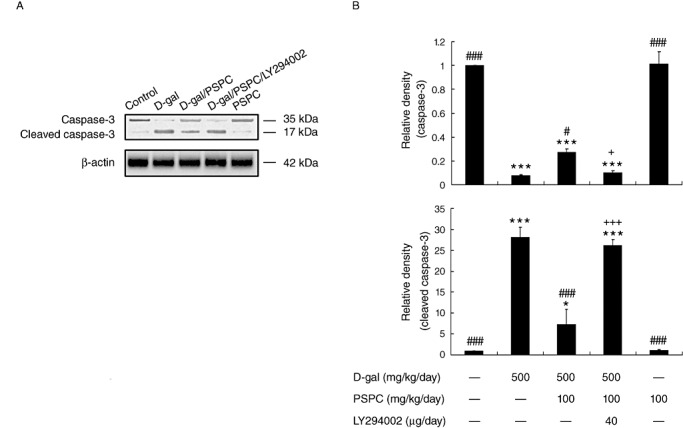
Effect of purple sweet potato color (PSPC) on the cleavage of caspase‐3 in the brain of D‐galactose (D‐gal)‐treated old mice (n = 3). A. Immunoreactivity of cleaved caspase‐3 and β‐actin in all treated groups. B. Relative density analysis of the cleaved caspase‐3 protein bands. β‐Actin was probed as an internal control. The relative density is expressed as the ratio of cleaved caspase‐3 to β‐actin, and the vehicle control is set as 1.0. Values are averages from three independent experiments. Each value is the mean ± standard deviation. *P < 0.05, ***P < 0.001 vs. control group; #P < 0.05, ###P < 0.001 vs. D‐gal group; +P < 0.05, +++P < 0.001 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 group.
Effect of PSPC on the activation of JNK and the release of cytochrome c in the brain of D‐gal‐treated old mice
In Figure 10, we examined the activation of JNK by Western blot analysis. D‐gal induced the activation of JNK significantly in the brain of old mice [p‐p46 JNK: F(4,10) = 66.735, P < 0.001; p‐p54 JNK: F(4,10) = 27.679, P < 0.001], which was markedly inhibited by PSPC treatment (p‐p46 JNK: P < 0.01; p‐p54 JNK: P < 0.05 vs. D‐gal group). The activation of JNK in the brain of old mice cotreated with D‐gal and PSPC was significantly increased by LY294002 (p‐p46 JNK: P < 0.01; p‐p54 JNK: P < 0.05 vs. D‐gal/PSPC group).
Figure 10.
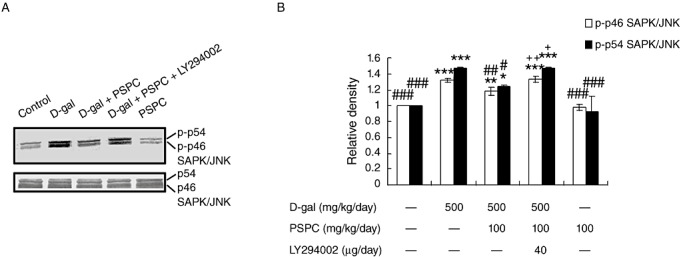
Effect of purple sweet potato color (PSPC) on the phosphorylation of stress‐activated protein kinase (SAPK)/c‐Jun NH2‐terminal kinase (JNK) in the brain of D‐galactose (D‐gal)‐treated old mice. A. Immunoreactivity of phospho‐JNK (p‐JNK) and β‐actin in all treated groups. B. Relative density analysis of the p‐JNK protein bands. β‐Actin was probed as an internal control. The relative density is expressed as the ratio of p‐JNK to β‐actin, and the vehicle control is set as 1.0. Values are averages from three independent experiments. Each value is the mean ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. D‐gal group; +P < 0.05, ++P < 0.01 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 group.
It has been demonstrated that the activation of JNK is responsible for the cytochrome c release, which leads to the cleavage of caspase‐3 and the induction of apoptosis. In the present study, we then measured the mitochondrial and cytosolic cytochrome c in the brain of old mice by Western blot analysis (Figure 11). Without D‐gal treatment, most of the detectable cytochrome c in the brain of old mice was in the mitochondria. Cytochrome c in the cytosol of D‐gal‐treated old mice significantly increased [F(4,10) = 314.371, P < 0.001], which was markedly inhibited by cotreatment with PSPC (P < 0.001 vs. D‐gal group). The amount of cytochrome c in mitochondria showed a corresponding increase in the brain of old mice cotreated with D‐gal and PSPC [F(4,10) = 166.061, P < 0.001 vs. D‐gal group]. Interestingly, the amount of cytosolic cytochrome c in the brain of old mice cotreated with D‐gal and PSPC was significantly increased by LY294002 (P < 0.001 vs. D‐gal/PSPC group).
Figure 11.
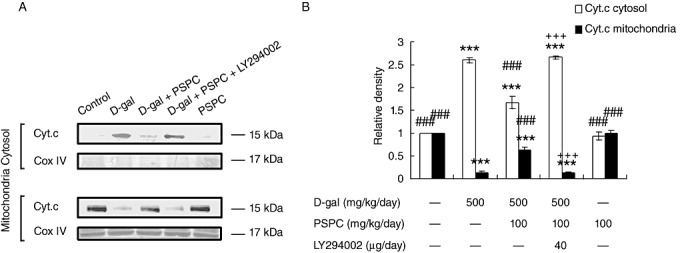
Effect of purple sweet potato color (PSPC) on the release of cytochrome c (Cyt.c) from cytochondria in the brain of D‐galactose (D‐gal)‐treated old mice. A. Immunoreactivity of cytosolic and mitochondrial Cyt.c and COX IV in all treated groups. B. Relative density analysis of the mitochondrial and cytosolic Cyt.c protein bands. COX IV was probed as an internal control. The relative density is expressed as the ratio of mitochondrial and cytosolic Cyt.c to β‐actin, and the vehicle control is set as 1.0. Values are averages from three independent experiments. Each value is the mean ± standard deviation. ***P < 0.001 vs. control group; ###P < 0.001 vs. D‐gal group; +++P < 0.05 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 group.
Effect of PSPC on the phosphorylation of Akt and p44/42 MAPK in the brain of D‐gal‐treated old mice
Both Akt and p44/42 MAPK (ERK1/2) are serine/threonine kinases that are invoked by numerous growth and survival factors. We investigated the effect of PSPC on phosphorylation of Akt and p44/42 MAPK by Western blot analysis (Figure 12). Akt activation requires phosphorylation on both Thr308 and Ser473 (26). The phosphorylation of Akt (Thr308/Ser473) was significantly decreased by D‐gal treatment [Akt (Thr308): F(4,10) = 287.585, P < 0.001; Akt (Ser473): F(4,10) = 298.725, P < 0.001]. PSPC remarkably attenuated the decrease of pAkt (Thr308/Ser473) in the brain of D‐gal‐treated old mice [Akt (Thr308/Ser473): P < 0.001 vs. D‐gal group]. LY294002 significantly decreased the phosphorylation of Akt (Thr308/Ser473) in the brain of old mice cotreated with D‐gal and PSPC [Akt (Thr308/Ser473): P < 0.001 vs. D‐gal/PSPC group].
Figure 12.
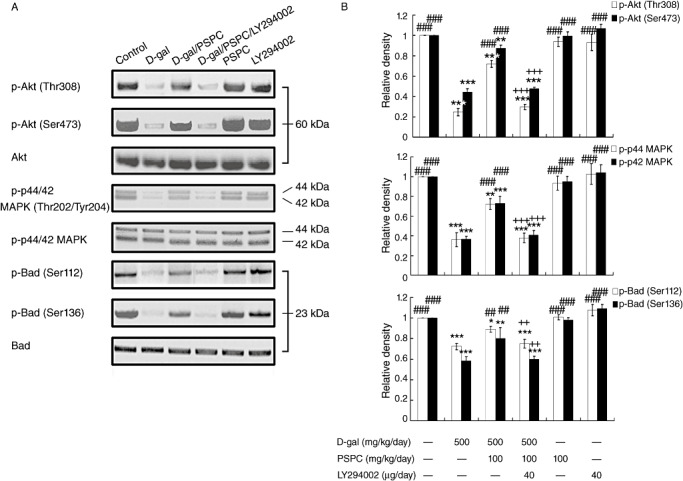
Effect of purple sweet potato color (PSPC) on the phosphorylation of Akt, p44/42 MAPK and Bad in the brain of D‐gal‐treated old mice. A. Immunoreactivity of p‐Akt(Thr308/Ser473), p‐p44/42 mitogen‐activated protein kinase (MAPK)(Thr202/Tyr204), p‐Bcl‐2‐associated death protein (Bad)(Ser112/Ser136), total‐Akt (t‐Akt), t‐p44/42 MAPK and t‐Bad in all treated groups. B. Relative density analysis of the p‐Akt, p‐p44/42 MAPK and p‐Bad protein bands. t‐Akt, t‐p44/42 MAPK and t‐Bad were probed as internal controls, respectively. The relative density is expressed as the ratio (p‐Akt/t‐Akt; p‐p44/42/t‐p44/42; p‐Bad/t‐Bad), and the vehicle control is set as 1.0. Values are averages from three independent experiments. Each value is the mean ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group; ##P < 0.01, ###P < 0.001 vs. D‐gal group; ++P < 0.01, +++P < 0.001 D‐gal/PSPC group vs. D‐gal/PSPC/LY294002 group.
The p44/42 MAPK phosphorylation status was also analyzed by Western blot. Similarly, the analysis of Western blot showed that D‐gal markedly decreased the phosphorylation of p44/42 MAPK [p‐p44 MAPK: F(4,10) = 79.357, P < 0.001; Akt (Ser473): F(4,10) = 117.092, P < 0.001]. Figure 12 also revealed that the PSPC induced phosphorylation of p44/42 MAPK (Thr202/Tyr204) in the brain of D‐gal‐treated old mice (P < 0.001 vs. D‐gal group), which was inhibited by LY294002 (P < 0.001 vs. D‐gal/PSPC group).
Effect of PSPC on the phosphorylation of Bad in the brain of D‐gal‐treated old mice
Bad, a proapoptotic Bcl‐2 family protein, is modulated by phosphorylation at two sites, serine 112 (Ser‐112) and serine 136 (Ser‐136). The analysis of Western blot showed that D‐gal significantly reduced the phosphorylation of Bad at Ser‐112 and Ser‐136 [pBad (Ser112): F(4,10) = 53.169, P < 0.001; pBad (Ser136): F(4,10) = 39.689, P < 0.001] (Figure 12). However, PSPC significantly increased the phosphorylation of Bad at these two sites in the brain of D‐gal‐treated old mice (P < 0.01 vs. D‐gal), which would promote the neuronal survival by the association of Bad with 14‐3‐3. There was no significant effect of PSPC on the Bad phosphorylaton of mice without D‐gal treatment. LY294002 markedly inhibited the PSPC‐induced phosphorylation of Bad in the brain of D‐gal‐treated old mice (P < 0.01 vs. D‐gal/PSPC group).
DISCUSSION
Previous studies have revealed that D‐gal has a neurotoxic effect in mice 23, 54. D‐gal is a reducing sugar that can form AGEs in vivo and result in oxidative stress. It is also demonstrated that AGEs exacerbate and accelerate aging change and contribute to the development of age‐related diseases, including atherosclerosis, neurodegenerative disease and age‐related macular degeneration 3, 49. Our present study showed that D‐gal induced the oxidative stress, including decreased expression and activity of Cu,Zn‐SOD and CAT, which might result from the increased expression of AGEs, CML, CEL and RAGE, (4, 5, 6). In the open‐field tests, the D‐gal model group showed a significantly decreased number of crossing, rearing/leaning and grooming as compared with the vehicle group. These results indicated that spontaneous behavior and explorative response were decreased by the D‐gal‐induced oxidative stress. Furthermore, D‐gal‐treated old mice in the step‐through test and MWM test also showed an impaired learning and memory ability.
PSPC, a class of naturally occurring anthocyanins, possesses potent anti‐inflammatory and antioxidant properties 32, 36. It has been reported to play a useful role in improving the learning and memory ability by increasing the expression of synaptic proteins such as GAP43 and PSD95 (54). Dietary supplementation with PSPC improved the learning and memory ability of D‐gal‐treated old mice, accompanied with increased expression and activity of Cu,Zn‐SOD and CAT (5, 6). Cu,Zn‐SOD converts superoxide enzymically into hydrogen peroxide that can be converted nonenzymically into the highly reactive hydroxyl radical (OH) in the presence of reduced transition metals (eg, ferrous or cuprous ions) or converted into water by the enzyme CAT, thereby promoting the clearance of reactive oxygen species (ROS) (10). The present results indicate that PSPC plays an effective role in protecting brain function from D‐gal‐induced oxidative insults.
High ROS concentrations contribute to the apoptotic cell death whenever they are generated in the context of the apoptotic process (44). The TUNEL method was used to analyze apoptotic cells in situ, based on labeling of DNA strand breaks in the cerebral cortex and the hippocampus of mice. Previous reports show that anthocyanins play neuroprotective effects as assessed in vitro by reducing oxidative stress‐induced apoptosis 14, 15, 48. As shown in 7, 8, D‐gal increased the number of TUNEL‐positive cells in the cerebral cortex and the hippocampus of old mice. The activation of caspase‐3 was involved in the apoptosis pathway induced by D‐gal (Figure 9). We found that PSPC decreased the number of TUNEL‐positive cells and inhibited the activation of caspase‐3 in old mice treated with D‐gal. Thus, it suggests that PSPC has a potential anti‐apoptotic effect by its antioxidant activity.
MAPKs are proposed to participate in various cellular processes, including cell proliferation, differentiation and apoptosis. Numerous studies indicate that JNK, also known as SAPK, can be activated by oxidative stress and involved in apoptosis signaling pathways (Figure 13) 21, 51. The release of cytochrome c is tightly linked to the presence and activation of JNK, and the JNK‐mediated cytochrome c release contributes to caspase‐3 activation and onset of apoptosis 11, 51. The activation of JNK and the release of cytochrome c from mitochondria into cytosol were significantly increased by the treatment with D‐gal (10, 11). Our results further revealed that PSPC markedly prevented the activation of JNK and the release of cytochrome c induced by D‐gal. Suppressing apoptosis using PSPC that targets key proteins in the cell death process, such as cytochrome c and caspase‐3, might be a potent method to enhance resistance of neurons to age‐related disease.
Figure 13.
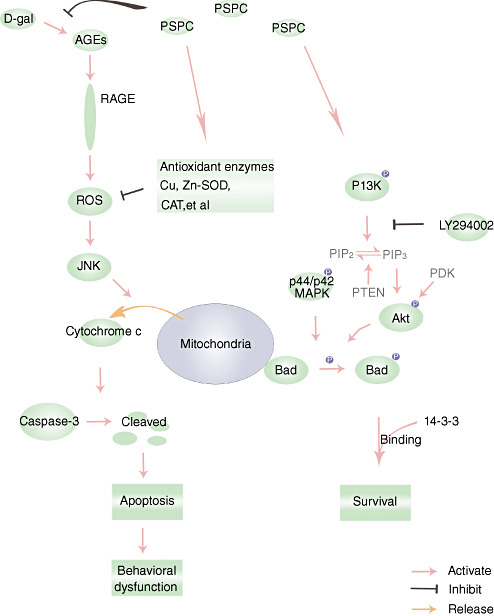
Schematic diagram for the protective effects of purple sweet potato color (PSPC) against D‐galactose (D‐gal)‐induced neurotoxicity. Abbreviations: AGEs = advanced glycation end‐products; RAGE = AGE receptor; CAT = catalase; Bad = Bcl‐2‐associated death protein; Cu,Zn‐SOD = Cu,Zn‐superoxide dismutase; PI3K = phosphoinositide 3‐kinase; ROS = reactive oxygen species; JNK = c‐Jun NH2‐terminal kinase; MAPK = mitogen‐activated protein kinase.
It has been reported that Bad, a proapoptotic Bcl‐2 family protein, may be an integrated checkpoint of phosphoinositide 3‐kinase (PI3K)/Akt‐mediated survival signaling and JNK‐mediated death signaling. JNK reduces the affinity of Bad for 14‐3‐3 and promotes the association of Bad with Bcl‐XL or Bcl‐2 via phosphorylation of Bad (Ser128) or 14‐3‐3 8, 47. Bad promotes an apoptotic cascade by binding to Bcl‐XL and Bcl‐2. Previous reports suggest that the decreased activation of JNK maycontribute to neuronal survival via the formation of heterodimers of Bad with 14‐3‐3.
PI3K/Akt pathway may lead to Bad phosphorylation and may thereby suppress cell death and promote cell survival. Akt is a potent Bad Ser‐136 kinase in vitro (9). The decreased phosphorylation of Akt at Thr308 and Ser473 was found in the D‐gal‐treated old mice accompanied with the decreased phosphorylation of Bad at Ser136 (Figure 12). PSPC treatment caused the phosphorylation of Bad (Ser136) associated with the phosphorylation of Akt (Thr308/Ser473) in the brain of D‐gal‐treated old mice.
The p44/42 MAPK, also called the ERK1/2, plays an important role in promoting cell survival by phosphorylation of Bad at Ser112 41, 58. Similarly, the decreased phosphorylation of Bad at Ser112 was observed in the D‐gal‐treated old mice accompanied with the decreased phosphorylation of p44/42 MAPK at Thr202 and Tyr204 (Figure 12). PSPC induced the phosphorylation and activation of p44/42 MAPK in the D‐gal‐treated old mice, which resulted in the concomitant phosphorylation of Bad (Ser112). PSPC‐induced phosphorylation of Bad at Ser136 and Ser112 sites promoted the neuronal survival in the D‐gal‐treated old mice. Phosphorylation of these two sites has been associated with binding of Bad with 14‐3‐3 protein, which may sequester Bad from Bcl‐XL or Bcl‐2, thus promoting the neuronal survival.
LY294002 was used to further investigate whether PI3K‐dependent survival pathways were required for PSPC to promote neuronal survival (Figure 13). LY294002, a flavonoid derivative, is a competitive and reversible inhibitor of the adenosine‐5′‐triphosphate (ATP) binding site of PI3K 42, 52. Akt pleckstrin homology (PH) domain with 3′‐phosphoinositides is thought to provoke conformational changes in Akt, resulting in its phosphorylation at Thr308 and Ser473 respectively by PDK1 and PDK2 50, 53. As shown in Figure 12, LY294002 significantly inhibited the phosphorylation of Akt (Thr308/Ser473) and Bad (Ser136) in the brain of old mice cotreated with D‐gal and PSPC. Many investigations indicate that there is a strong cross talk between the PI3K pathway and the MEK (mitogen‐activated protein kinase kinase) pathway 1, 40. Inhibition of the PI3K has been shown to inhibit the ERK pathway in a cell type‐ and ligand‐specific manner 1, 37. Our results also showed that LY294002 significantly inhibited the phosphorylation of p44/42 MAPK and Bad (Ser112) in the brain of old mice cotreated with D‐gal and PSPC. Accompanied with the inhibition of pBad (Ser112/Ser136), LY294002 resulted in the onset of apoptosis in the hippocampus and cerebral cortex of old mice cotreated with D‐gal and PSPC. As shown in 7, 8, 9, LY294002 significantly induced the cleavage of caspase‐3 and increased the number of TUNEL‐positive cells in the old mice cotreated with D‐gal and PSPC.
In addition, the phosphorylation and activation of JNK have been found to be inhibited via the activation of PI3K/Akt pathways in response to various stress 16, 37. This may help to explain in part why PSPC significantly decreased the phosphorylation of JNK in the D‐gal‐treated old mice. On the contrary, LY294002 induced the phosphorylation of JNK in the brain of old mice cotreated with D‐gal and PSPC, which might result from the inhibition of pAkt (Figure 10). Consequently, LY294002 also induced the release of cytochrome c from the mitochondria in the old mice cotreated with D‐gal and PSPC, which contributed to the initiation of apoptosis (Figure 11). Furthermore, our present study concluded that the release of cytochrome c from mitochondria probably resulted from the reduction of pBad induced by LY294002. It was likely that the reduction of pBad induced the efflux of cytochrome c via the formation of heterodimers with, and inactivation of, anti‐apoptotic proteins Bcl‐2 and Bcl‐XL 13, 17, 18, 33, 56. As a result, LY294002 induced obvious deficits in learning and memory in old mice cotreated with D‐gal and PSPC, which was associated with the neuronal apoptosis.
In conclusion, our present data suggest that D‐gal induces multiple molecular and functional changes in the brain similar to the natural aging. In the D‐gal‐treated old mice, PSPC promotes neuronal survival against oxidative stress, ultimately resulting in the enhanced cognitive performance of D‐gal‐treated old mice. Further research was carried out to identify the mechanism that promoted neuronal survival. The survival signaling pathways involve the concomitant phosphorylation of Bad at Ser‐112 and Ser‐136 by simultaneous activation of PI3K/Akt pathways and ERK pathway, respectively.
DISCLOSURE STATEMENT
There are no actual or potential conflicts of interest in this work. All experiments were performed in compliance with the Chinese legislation on the use and care of laboratory animals, and were approved by the respective university committees for animal experiments.
ACKNOWLEDGMENTS
This work is supported by the National Natural Science Fundation of China (30950031), the Major Fundamental Research Program of Natural Science Foundation of the Jiangsu Higher Education Institutions of China (07KJA36029), grants from the Key Laboratory of Jiangsu Province, grants from Qing Lan Project of Jiangsu Province, China, grants from the Natural Science Foundation by Xuzhou Normal University (08XLR09) and grants from the Natural Science Foundation for Colleges and Universities in Jiangsu Province (09KJB180009).
REFERENCES
- 1. Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Graos MM et al (2005) Neuroprotection by BDNF against glutamate‐induced apoptotic cell death is mediated by ERK and PI3‐kinase pathways. Cell Death Differ 12:1329–1343. [DOI] [PubMed] [Google Scholar]
- 2. Asif M, Egan J, Vasan S, Jyothirmayi GN, Masurekar MR, Lopez S et al (2000) An advanced glycation endproduct cross‐link breaker can reverse age‐related increases in myocardial stiffness. Proc Natl Acad Sci U S A 97:2809–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brownlee M (1995) Advanced protein glycosylation in diabetes and aging. Annu Rev Med 46:223–234. [DOI] [PubMed] [Google Scholar]
- 4. Chen CM, Li SC, Lin YL, Hsu CY, Shieh MJ, Liu JF (2005) Consumption of purple sweet potato leaves modulates human immune response: T‐lymphocyte functions, lytic activity of natural killer cell and antibody production. World J Gastroenterol 11:5777–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cui X, Zuo P, Zhang Q, Li X, Hu Y, Long J et al (2006) Chronic systemic D‐galactose exposure induces memory loss, neurodegeneration, and oxidative damage in mice: protective effects of R‐alpha‐lipoic acid. J Neurosci Res 84:647–654. [DOI] [PubMed] [Google Scholar]
- 6. Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K et al (2004) Involvement of oxidative stress‐induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A 101:2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deng HB, Cui DP, Jiang JM, Feng YC, Cai NS, Li DD (2003) Inhibiting effects of Achyranthes bidentata polysaccharide and Lycium barbarum polysaccharide on nonenzyme glycation in D‐galactose induced mouse aging model. Biomed Environ Sci 16:267–275. [PubMed] [Google Scholar]
- 8. Donovan N, Becker EBE, Konishi Y, Bonni A (2002) JNK phosphorylation and activation of BAD couples the stress‐activated signaling pathway to the cell death machinery. J Biol Chem 277:40944–40949. [DOI] [PubMed] [Google Scholar]
- 9. Downward J (2004) PI 3‐kinase, Akt and cell survival. Semin Cell Dev Biol 15:177–182. [DOI] [PubMed] [Google Scholar]
- 10. Dröge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82:47–95. [DOI] [PubMed] [Google Scholar]
- 11. Eminel S, Klettner A, Roemer L, Herdegen T, Waetzig V (2004) JNK 2 translocates to the mitochondria and mediates cytochrome c release in PC 12 cells in response to 6‐hydroxydopamine. J Biol Chem 279:55385–55392. [DOI] [PubMed] [Google Scholar]
- 12. Floyd RA, Hensley K (2002) Oxidative stress in brain aging: implications for therapeutics of neurodegenerative diseases. Neurobiol Aging 23:795–807. [DOI] [PubMed] [Google Scholar]
- 13. Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C et al (1999) Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL‐XL prevents this release but not tumor necrosis factor‐R1/Fas death. J Biol Chem 274:1156–1163. [DOI] [PubMed] [Google Scholar]
- 14. Heo HJ, Lee CY (2005) Strawberry and its anthocyanins reduce oxidative stress‐induced apoptosis in PC12 cells. J Agric Food Chem 53:1984–1989. [DOI] [PubMed] [Google Scholar]
- 15. Huber A, Bürkle A, Münch G (2007) Neuroprotective mechanisms: oxidative stress as a target for neuroprotective therapies in Alzheimer's and Parkinson's disease. In: Handbook of Neurochemistry and Molecular Neurobiology. Abel L, Moussa BHY, Peter R, Sylvia AM, Leontino B (eds), pp. 77–102. Springer: Springer‐Verlag, Berlin Heidelberg, Germany. [Google Scholar]
- 16. Hui L, Pei DS, Zhang QG, Guan QH, Zhang GY (2005) The neuroprotection of insulin on ischemic brain injury in rat hippocampus through negative regulation of JNK signaling pathway by PI3K/Akt activation. Brain Res 1052:1–9. [DOI] [PubMed] [Google Scholar]
- 17. Kelekar A, Chang BS, Harlan JE, Fesik SW, Thompson CB (1997) Bad is a BH3 domain‐containing protein that forms an inactivating dimer with Bcl‐XL. Mol Cell Biol 17:7040–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kluck RM, Bossy‐Wetzel E, Green DR, Newmeyer DD (1997) The release of cytochrome c from mitochondria: a primary site for Bcl‐2 regulation of apoptosis. Science 275:1132–1136. [DOI] [PubMed] [Google Scholar]
- 19. Konczak‐Islam I, Yoshimoto M, Hou D‐X, Terahara N, Yamakawa O (2003) Potential chemopreventive properties of anthocyanin‐rich aqueous extracts from in vitro produced tissue of sweetpotato (Ipomoea batatas L.). J Agric Food Chem 51:5916–5922. [DOI] [PubMed] [Google Scholar]
- 20. Li XM, Ma YL, Liu XJ (2007) Effect of the Lycium barbarum polysaccharides on age‐related oxidative stress in aged mice. J Ethnopharmacol 111:504–511. [DOI] [PubMed] [Google Scholar]
- 21. Liu J, Lin A (2005) Role of JNK activation in apoptosis: a double‐edged sword. Cell Res 15:36–42. [DOI] [PubMed] [Google Scholar]
- 22. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275. [PubMed] [Google Scholar]
- 23. Lu J, Zheng YL, Luo L, Wu DM, Sun DX, Feng YJ (2006) Quercetin reverses D‐galactose induced neurotoxicity in mouse brain. Behav Brain Res 171:251–260. [DOI] [PubMed] [Google Scholar]
- 24. Lu J, Zheng YL, Wu DM, Sun DX, Shan Q, Fan SH (2006) Trace amounts of copper induce neurotoxicity in the cholesterol‐fed mice through apoptosis. FEBS Lett 580:6730–6740. [DOI] [PubMed] [Google Scholar]
- 25. Lu J, Zheng YL, Wu DM, Luo L, Sun DX, Shan Q (2007) Ursolic acid ameliorates cognition deficits and attenuates oxidative damage in the brain of senescent mice induced by D‐galactose. Biochem Pharmacol 74:1078–1090. [DOI] [PubMed] [Google Scholar]
- 26. Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mattson MP, Duan W, Chan SL, Cheng A, Haughey N, Gary DS et al (2002) Neuroprotective and neurorestorative signal transduction mechanisms in brain aging: modification by genes, diet and behavior. Neurobiol Aging 23:695–705. [DOI] [PubMed] [Google Scholar]
- 28. Morrison JH, Hof PR (1997) Life and death of neurons in the aging brain. Science 278:412–419. [DOI] [PubMed] [Google Scholar]
- 29. Münch G, Thome J, Foley P, Schinzel R, Riederer P (1997) Advanced glycation endproducts in ageing and Alzheimer's disease. Brain Res Brain Res Rev 23:134–143. [DOI] [PubMed] [Google Scholar]
- 30. Münch G, Schinzel R, Loske C, Wong A, Durany N, Li JJ et al (1998) Alzheimer's disease—synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Transm 105:439–461. [DOI] [PubMed] [Google Scholar]
- 31. Odake K, Terahara N, Saito N, Toki K, Honda T (1992) Chemical structures of two anthocyanins from purple sweet potato, Ipomoea batatas. Phytochemistry 31:2127–2130. [Google Scholar]
- 32. Oki T, Masuda M, Furuta S, Nishiba Y, Terahara N, Suda I (2002) Involvement of anthocyanins and other phenolic compounds in radical‐scavenging activity of purple‐fleshed sweet potato cultivars. J Food Sci 67:1752–1756. [Google Scholar]
- 33. Ottilie S, Diaz J‐L, Horne W, Chang J, Wang Y, Wilson G et al (1997) Dimerization properties of human BAD. Identification of a BH‐3 domain and analysis of its binding to mutant BCL‐2 and BCL‐XL proteins. J Biol Chem 272:30866–30872. [DOI] [PubMed] [Google Scholar]
- 34. Pamplona R, Dalfo E, Ayala V, Bellmunt M, Prat J, Ferrer I, Portero‐Otin M (2005) Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation: effects of Alzheimer disease and identification of lipoxidation targets. J Biol Chem 280:21522–21530. [DOI] [PubMed] [Google Scholar]
- 35. Paxinos G, Frank KB (2001) The Mouse Brain in Sterotaxic Coordinates. San Diego: Academic Press. [Google Scholar]
- 36. Philpott M, Gould KS, Lim C, Ferguson LR (2004) In situ and in vitro antioxidant activity of sweetpotato anthocyanins. J Agric Food Chem 52:1511–1513. [DOI] [PubMed] [Google Scholar]
- 37. Qiao M, Shapiro P, Kumar R, Passaniti A (2004) Insulin‐like growth factor‐1 regulates endogenous RUNX2 activity in endothelial cells through a phosphatidylinositol 3‐kinase/ERK‐dependent and Akt‐independent signaling pathway. J Biol Chem 279:42709–42718. [DOI] [PubMed] [Google Scholar]
- 38. Ramasamy R, Vannucci SJ, Yan SSD, Herold K, Yan SF, Schmidt AM (2005) Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15:16–28. [DOI] [PubMed] [Google Scholar]
- 39. Sakatani M, Suda I, Oki T, Kobayashi S, Kobayashi S, Takahashi M (2007) Effects of purple sweet potato anthocyanins on development and intracellular redox status of bovine preimplantation embryos exposed to heat shock. J Reprod Dev 53:605–614. [DOI] [PubMed] [Google Scholar]
- 40. Sato S, Fujita N, Tsuruo T (2004) Involvement of 3‐phosphoinositide‐dependent protein kinase‐1 in the MEK/MAPK signal transduction pathway. J Biol Chem 279:33759–33767. [DOI] [PubMed] [Google Scholar]
- 41. Scheid MP, Schubert KM, Duronio V (1999) Regulation of Bad phosphorylation and association with Bcl‐xL by the MAPK/Erk kinase. J Biol Chem 274:31108–31113. [DOI] [PubMed] [Google Scholar]
- 42. Semba S, Itoh N, Ito M, Harada M, Yamakawa M (2002) The in vitro and in vivo effects of 2‐(4‐Morpholinyl)‐8‐phenyl‐chromone (LY294002), a specific inhibitor of phosphatidylinositol 3′‐kinase, in human colon cancer cells. Clin Cancer Res 8:1957–1963. [PubMed] [Google Scholar]
- 43. Shen YX, Xu SY, Wei W, Sun XX, Yang J, Liu LH, Dong C (2002) Melatonin reduces memory changes and neural oxidative damage in mice treated with D‐galactose. J Pineal Res 32:173–178. [DOI] [PubMed] [Google Scholar]
- 44. Slater AFG, Stefan C, Nobel I, Van Den Dobbelsteen DJ, Orrenius S (1995) Signalling mechanisms and oxidative stress in apoptosis. Toxicol Lett 82:149–153. [DOI] [PubMed] [Google Scholar]
- 45. Song X, Bao M, Li D, Li YM (1999) Advanced glycation in D‐galactose induced mouse aging model. Mech Ageing Dev 108:239–251. [DOI] [PubMed] [Google Scholar]
- 46. Suda I, Ishikawa F, Hatakeyama M, Miyawaki M, Kudo T, Hirano K et al (2008) Intake of purple sweet potato beverage affects on serum hepatic biomarker levels of healthy adult men with borderline hepatitis. Eur J Clin Nutr 62:60–67. [DOI] [PubMed] [Google Scholar]
- 47. Sunayama J, Tsuruta F, Masuyama N, Gotoh Y (2005) JNK antagonizes Akt‐mediated survival signals by phosphorylating 14‐3‐3. J Cell Biol 170:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tarozzi A, Morroni F, Hrelia S, Angeloni C, Marchesi A, Cantelli‐Forti G, Hrelia P (2007) Neuroprotective effects of anthocyanins and their in vivo metabolites in SH‐SY5Y cells. Neurosci Lett 424:36–40. [DOI] [PubMed] [Google Scholar]
- 49. Tian J, Ishibashi K, Ishibashi K, Reiser K, Grebe R, Biswal S et al (2005) Advanced glycation endproduct‐induced aging of the retinal pigment epithelium and choroid: a comprehensive transcriptional response. Proc Natl Acad Sci U S A 102:11846–11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toker A, Newton AC (2000) Cellular signaling: pivoting around PDK‐1. Cell 103:185–188. [DOI] [PubMed] [Google Scholar]
- 51. Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A et al (2000) Requirement of JNK for stress‐induced activation of the cytochrome c‐mediated death pathway. Science 288:870–874. [DOI] [PubMed] [Google Scholar]
- 52. Vlahos CJ, Matter WF, Hui KY, Brown RF (1994) A specific inhibitor of phosphatidylinositol 3‐kinase, 2‐(4‐morpholinyl)‐8‐phenyl‐4H‐1‐benzopyran‐4‐one (LY294002). J Biol Chem 269:5241–5248. [PubMed] [Google Scholar]
- 53. West KA, Castillo SS, Dennis PA (2002) Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist Updat 5:234–248. [DOI] [PubMed] [Google Scholar]
- 54. Wu DM, Lu J, Zheng YL, Zhou Z, Shan Q, Ma DF (2008) Purple sweet potato color repairs d‐galactose‐induced spatial learning and memory impairment by regulating the expression of synaptic proteins. Neurobiol Learn Mem 90:19–27. [DOI] [PubMed] [Google Scholar]
- 55. Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B et al (2004) Increased oxidative stress is associated with chronic intermittent hypoxia‐mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience 126:313–323. [DOI] [PubMed] [Google Scholar]
- 56. Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J et al (1997) Prevention of apoptosis by Bcl‐2: release of cytochrome c from mitochondria blocked. Science 275:1129–1132. [DOI] [PubMed] [Google Scholar]
- 57. Yoshinaga M, Tanaka M, Nakatani M (2000) Changes in anthocyanin content and composition of developing storage root of purple‐fleshed sweet potato (Ipomoea batatas (L.) Lam). Breed Sci 50:59–64. [Google Scholar]
- 58. Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che XM, Culmsee C et al (2002) Transforming growth factor‐beta 1 increases Bad phosphorylation and protects neurons against damage. J Neurosci 22:3898–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]