

Abstract
Central nervous system neoplasms with combined features of malignant glioma and primitive neuroectodermal tumor (MG‐PNET) are rare, poorly characterized, and pose diagnostic as well as treatment dilemmas. We studied 53 MG‐PNETs in patients from 12 to 80 years of age (median = 54 years). The PNET‐like component consisted of sharply demarcated hypercellular nodules with evidence of neuronal differentiation. Anaplasia, as seen in medulloblastomas, was noted in 70%. Within the primitive element, N‐myc or c‐myc gene amplifications were seen in 43%. In contrast, glioma‐associated alterations involved both components, 10q loss (50%) being most common. Therapy included radiation (78%), temozolomide (63%) and platinum‐based chemotherapy (31%). Cerebrospinal fluid (CSF) dissemination developed in eight patients, with response to PNET‐like therapy occurring in at least three. At last follow‐up, 27 patients died, their median survival being 9.1 months. We conclude that the primitive component of the MG‐PNET: (i) arises within a pre‐existing MG, most often a secondary glioblastoma; (ii) may represent a metaplastic process or expansion of a tumor stem/progenitor cell clone; (iii) often shows histologic anaplasia and N‐myc (or c‐myc) amplification; (iv) has the capacity to seed the CSF; and (v) may respond to platinum‐based chemotherapy regimens.
Keywords: genetics, glioblastoma, gliosarcoma, metastases, MYC, neuroblastoma, oligodendroglioma, primitive neuroectodermal tumor, prognosis, small cell, stem cell
INTRODUCTION
Histogenetic models of brain tumor classification date back to the time of Bailey and Cushing in the 1920s (2). Although such models have been useful, our understanding of normal embryology, much less the development of neoplastic cell types, remains limited, and tumors do not always fall neatly within our conceptual models of histogenesis. For example, current views consider glioblastoma (GBM) to be the most malignant form of astrocytoma and as such, it is considered a purely astrocytic neoplasm. Nonetheless, GBMs with mesenchymal [gliosarcoma (GS)] and epithelial metaplasia are well recognized 1, 4, 5, 9, 15, 26, 27, 29, 33. GBM and GS present most often in adulthood, extensively permeate the central nervous system (CNS) parenchyma, and are treated with local radiation therapy and alkylating chemotherapeutic agents, such as temozolomide (20). Despite these features, however, the majority are resistant to therapy and the average survival is only about a year.
In contrast to GBM and its variants, primitive neuroectodermal tumor (PNET) of the CNS (CNS PNET) is a predominantly neuronal, but multipotential, non‐cerebellar embryonal neoplasm with medulloblastoma‐like histology (23). It affects primarily infants and children (mean age = 5.5 years), features a very high proliferation index, often undergoes cerebrospinal fluid (CSF) dissemination, and typically requires craniospinal radiation combined with platinum‐based chemotherapy 17, 23, 32, 39. As in GBM, the prognosis for CNS PNET is poor, although response to therapy is more frequent and long‐term survival is somewhat better for PNET. For example, a recent Canadian survey of 48 patients with CNS PNET showed therapeutic response rates of 52% and an estimated 4‐year survival time of 38% (17). Based on these differences and in current nosologic terms, GBM and PNET are considered non‐overlapping entities with distinct biologic behaviors and therapeutic responses.
Only rare brain tumors with combined features of GBM or GS and CNS PNET have previously been reported, usually as single case reports 10, 16, 18, 19, 22, 24, 37, 40. When encountered, they pose a diagnostic challenge, given pressures to place them into a single diagnostic category. As such, they are most often diagnosed as: (i) unusual variants of GBM/GS; (ii) CNS PNETs with prominent glial differentiation; or (iii) malignant or high‐grade glioneuronal neoplasms, not otherwise specified. Only rarely are they identified as combined GBM/GS and PNET. All of these designations are in fact, biologically plausible. However, the clinicopathologic and genetic features of the current series support the concept of an underlying high‐grade glioma with secondary development of PNET‐like foci. In most of our cases, patient survival was short and did not differ significantly from that of standard GBM. In a subset, however, PNET‐like clinical behavior, including CSF dissemination and responsiveness to platinum‐based therapeutic regimens, was also encountered. Possible explanations for the observation of a PNET‐like component include: (i) neuronal/neuroblastic metaplasia; and (ii) nodular expansion of the cancer stem cell/progenitor cell populations recently identified in conventional GBMs (13).
MATERIALS AND METHODS
Patient/tumor cohort
Tumors with combined features of high‐grade glioma and CNS PNET were prospectively collected from the in‐house clinical and consultation files of the authors between July 2004 and September 2007. Additionally, one 1998 case was identified retrospectively from a search of the archival files at the Mayo Clinic Tissue Registry, and three Washington University cases were identified upon a search of high‐grade gliomas diagnosed between 1990 and 2005 (25). Clinical data were obtained by review of medical records and were supplemented by dates of death derived from the Social Security Death Index (http://ssdi.genealogy.rootsweb.com/cgi‐bin/ssdi.cgi).
Ancillary stains and fluorescence in situ hybridization (FISH)
The majority of immunohistochemical and all FISH studies were performed and interpreted as previously described 11, 18, 25. The antibodies and DNA probes utilized are summarized in 1, 2, respectively. In consult cases, immunohistochemistry had often been performed in part by the referring pathologists using standard techniques. A reticulin histochemical stain was performed by either the referring or consultant pathologist, using standard techniques, in the cases showing either sarcoma‐like or desmoplastic/nodular features on routine histology.
Table 1.
Primary antibodies used for immunohistochemistry. Abbreviations: GFAP = glial fibrillary protein; NSE = neuron specific enolase.
| Immunostain | Source | Clone | Dilution |
|---|---|---|---|
| GFAP | Dako | 6F2 | 1:10000 |
| Synaptophysin | Dako | SY38 | 1:40 |
| Neu‐N | Chemicon | MAB 377 | 1:1000 |
| Neurofilament | Dako | 2F11 | 1:2000 |
| NSE | Dako | A0587 | 1:2000 |
| Chromogranin | Ventana | LK2H10 | Prediluted |
| p53 protein | BioGenex | 1801 | 1:200 |
| Ki‐67 | Dako | 1A4 | 1:100 |
Table 2.
Sources of DNA probes used for FISH. Abbreviation: FISH = fluorescence in situ hybridization; PNET = primitive neuroectodermal tumor.
| Probe name (clone) | Fluorescent Label | Chromosomal locus | Source |
|---|---|---|---|
| MYCN/CEP2 | SpectrumGreen/SpectrumOrange | 2p24.1/centromere 2 | Vysis, Downers Grove, IL |
| c‐MYC/CEP8 | SpectrumOrange/SpectrumGreen | 8q24.12‐q24.13/centromere 8 | Vysis |
| CEP7 | SpectrumGreen | centromere 7 | Vysis |
| EGFR (RPCI‐11 BAC 184P17) | Rhodamine | 7p12 | WU Sequencing Center, St. Louis, MO |
| PTEN | FITC | 10q23 | Dr Robert Jenkins, Mayo Clinic |
| DMBT1 | Rhodamine | 10q25.3‐q26.1 | Dr Robert Jenkins, Mayo Clinic |
| RPCI‐11 BAC 260I23 | FITC | 1p32 | Invitrogen, Huntsville, AL |
| RPCI‐11 BAC 184E11 | Rhodamine | 1q42 | Invitrogen |
| RPCI‐11 BAC 575H1 | FITC | 19p13 | Invitrogen |
| RPCI‐11 BAC 426G3 | Rhodamine | 19q13.3 | Invitrogen |
Statistics
All statistical analyses were performed using Intercooled Stata, version 9.2 (College Station, TX) as previously described (25). Chi‐square or Fisher exact tests were used for comparisons of proportions and Student's t‐test for mean comparisons of continuous variables such as age. All statistical tests were two‐sided and P < 0.05 was considered significant unless otherwise stated. Kaplan–Meier survival curves were plotted and log‐rank tests and Cox proportional hazards were used for univariate and multivariate comparisons of median overall survival (OS).
RESULTS
Study cohort
The study consisted of 30 male and 23 female patients (Table 3). With the exception of two pediatric patients (ages 12 and 17 years), the tumors arose in adults ranging in age from 21 to 80 years (median = 54 years). A majority of cases were derived from neuropathology consults submitted to one of four authors (AP, BWS, PCB and MKR). The three Washington University cases of malignant glioma and PNET (MG‐PNET) were identified among 581 recently reviewed GBMs (25), suggesting an overall frequency of PNET‐like components in roughly 0.5% of GBMs. One GS‐PNET was the subject of a prior case report (18).
Table 3.
Clinical features in 53 MG‐PNETs. Abbreviation: FU = followup; MG‐PNET = malignant glioma‐primitive neuroectodermal tumor.
| Age: | 12–80 years (median = 54 years) |
| Sex: | 30 male : 23 female (ratio = 1.3) |
| Survival Time (FU in 39; 27 dead) | <1 month–3.3 years (median = 9.1 months) |
| Length of Symptoms (n = 29) | 10 days–10 years (median = 3 months) |
| Gross Total Resection | 12 of 19 (63%) |
| Adjuvant Radiation | 18 of 23 (78%) |
| Adjuvant ChemoRx (n = 16) | Temozolomide (63%), Platinum‐based (31%) |
| Prior Glioma Dx (n = 13; 25%) | 8 months–10 years prior (median = 4 years) |
| Astrocytoma, World Health Organization grade II (n = 2) | |
| Oligoastrocytoma, World Health Organization grade II (n = 4) | |
| “Low‐grade glioma” (n = 1) | |
| Anaplastic oligoastrocytoma, World Health Organization grade III (n = 1) | |
| Glioblastoma with oligo features, World Health Organization grade IV (n = 2) | |
| Glioblastoma, World Health Organization grade IV (n = 3) | |
| Metastasis | eight of 20 cases (40%): eight CSF, one bone marrow |
Clinical and radiologic features
The clinical features are summarized in Table 3. Both presenting symptoms/signs and neuroimaging features were non‐specific and generally indistinguishable from those of either GBM or CNS PNET. Presentations were known in 29 patients and included headaches, seizures, dizziness, nausea/vomiting, blurred vision, language deficits, gait abnormalities, weakness and sensory abnormalities of 10 days to 10 years (median = 3 months) duration prior to the initial diagnosis of MG‐PNET. Neuroimaging studies revealed heterogeneously enhancing or ring‐enhancing cerebral masses in all instances wherein they were available.
Twenty‐seven patients (51%) were followed until death. Survival times ranged from less than 1 month to 3.3 years (median = 9.1 months) from the first diagnosis of MG‐PNET. An additional 12 patients (23%) were alive at last follow‐up, 1 month to 2.4 years (median = 10 months) from the initial diagnosis of MG‐PNET. The remaining 14 patients (26%) were either only recently diagnosed or lost to follow‐up. Only two patients were autopsied and both showed locally progressive disease. OS times did not differ significantly from those of patients with conventional GBM (Figure 1, log‐rank P = 0.4).
Figure 1.

Kaplan‐Meier curves comparing the survival of 53 MG‐PNET patients (blue curve) with 718 conventional GBMs (red curve). No statistically significant difference in median survival (9.1 vs. 10.3 months, respectively) was evident (log‐rank P = 0.39). Abbreviations: GBM = glioblastoma; MG = malignant glioma; PNET = primitive neuroectodermal tumor.
Details regarding adjuvant therapy were known in 23 patients. Three patients received none, with survival times being 2 and 5 months in the two that were followed until death. Eighteen patients (78%) received radiation therapy (17 local and one craniospinal), of which 14 were treated with concomitant chemotherapy (survival ranging from 1 month to 3.3 years, median = 12 months). Sixteen patients (70%) received GBM type adjuvant chemotherapy, including temozolomide in 10 and BCNU in one (survival ranging from 1 month to 3.3 years, median = 8 months). Three patients received “PNET‐like” platinum‐based chemotherapy regimens up front; survival times were 10 and 20 months in the two patients followed until death. In 13 patients receiving gross total resection, it was not possible to assess radiographic response, as no detectable residual tumor remained. In six patients having undergone subtotal resection, the tumor progressed despite therapy in five and it was not assessed in the sixth. In three instances, therapy was changed from temozolomide to platinum‐based regimens after signs of progression; on neuroimaging, all three showed subsequent signs of therapeutic response. In two other patients, platinum‐based regimens were changed to temozolomide; the tumor continued to progress in one case wherein subsequent imaging was obtained.
Although tumor progression from local disease was virtually universal, distant tumor spread was also common. This included cerebrospinal fluid (CSF) dissemination with ependymal seeding, intracranial meningeal deposits and drop metastases in eight, bone marrow metastasis in one that also had CSF spread, plus one instance of scalp recurrence at the prior incision site. Therefore, leptomeningeal metastases were encountered in 40% of 20 patients assessed for this complication and in 21% of the 39 patients with follow‐up. In two patients, CSF dissemination was detected at the time of initial diagnosis. In the other six, it was noted 1–15 months post‐operatively.
Histopathology
Histopathologic features are summarized in Table 4 and illustrated in 2, 3, 4, 5. The high‐grade glioma component resembled GBM in 47 (89%), GS in five (9%) and anaplastic oligodendroglioma in one (2%) (2, 3). The growth pattern was infiltrative and in areas of cortical involvement, secondary structures of Scherer were common. At low magnification, the PNET‐like component typically appeared as demarcated, markedly hypercellular nodules (2, 3). PNET‐like features included high nuclear to cytoplasmic ratios, hyperchromatic oval to carrot shaped nuclei, high mitotic‐karryorhectic indices, Homer Wright (neuroblastic) rosettes (62%; 2, 3), desmoplastic/nodular growth pattern (28%; 3, 4) and linear streaming of neoplastic cells (19%). Large cell and/or anaplastic cytologic features, such as those encountered in medulloblastomas, were noted in 37 cases (70%), including 25 (47%) cases featuring cell wrapping (2, 3). It is of note that the presence of anaplasia was not associated with further reductions in survival time.
Table 4.
Pathologic and genetic features in 53 MG‐PNETs.
| Histology Feature | Percent | Protein Expression/Genetics in neuroblastic component | Percent (median) |
|---|---|---|---|
| Foci of lower‐grade glioma* | 89 | GFAP** | 89 |
| Fibrillary astrocytoma | 62 | Synaptophysin | 87 |
| Gemistocytic astrocytoma | 40 | Neu‐N (n = 38) | 24 |
| Giant cell astrocytoma | 23 | Neurofilament (n = 36) | 11 |
| Oligoastrocytoma | 19 | NSE (n = 34) | 91 |
| Oligodendroglioma | 2 | Chromogranin (n = 34) | 29 |
| Pseudopalisading Necrosis | 60 | Ki‐67 LI (n = 41) | 30–100 (70) |
| Sarcoma (i.e. GS‐PNET) | 9 | p53 LI (n = 40) | 0–100 (80) |
| Homer Wright Rosettes | 62 | MYCN amplification (n = 40) | 33 |
| Perivascular pseudorosettes | 38 | MYCC amplification (n = 40) | 10 |
| Nodularity +/− desmoplasia | 28 | EGFR amplification (n = 40) | 10 |
| Streaming | 19 | Chromosome 10q deletion (n = 40) | 50 |
| Cell Wrapping | 47 | Chromosome 19q deletion (n = 12) | 42 |
| Large cell/Anaplastic PNET | 70 | 1p/19q codeletion (n = 12) | 8 |
These represented foci of lower‐grade appearing glial elements by histology, although a relatively bland‐appearing infiltrative component of a GBM could not always be entirely excluded;
GFAP expression was usually focal to patchy in the neuroblastic component and was much more extensive in the glioma component. Abbreviations: GBM = glioblastoma; GS = gliosarcoma; MG = malignant glioma; PNET = primitive neuroectodermal tumor.
Figure 2.
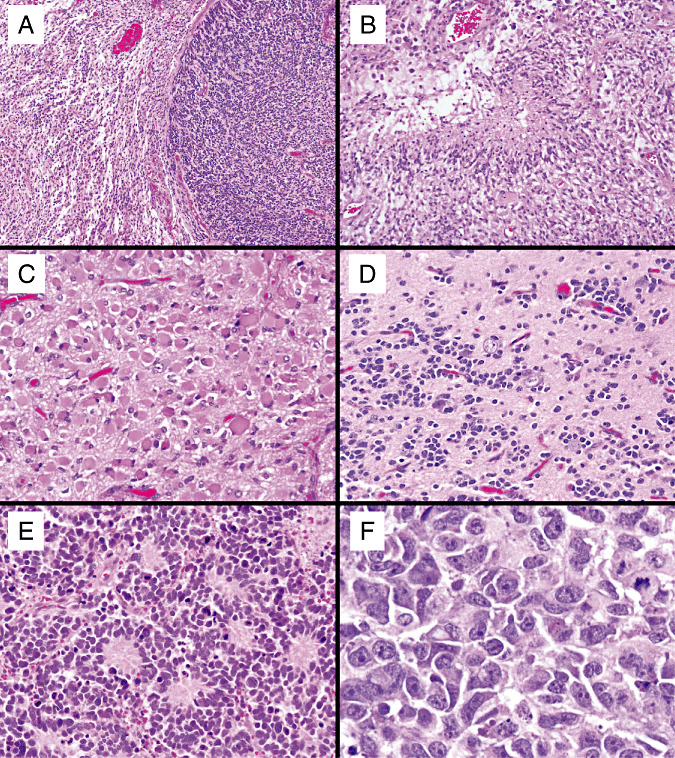
Examples of high‐grade gliomas with neuroblastic (PNET‐like) foci, including (A): a GBM‐PNET with the diffuse astrocytoma component on the left and the neuroblastic component on the right. The diffuse glioma components of MG‐PNET included: GBM with pseudopalisading necrosis (B), gemistocytic astrocytoma (C) and oligodendroglioma (D). The neuroblastic components resembled classic medulloblastomas with Homer Wright rosettes (E), often with anaplastic/large cell features including enlarged cells, vesicular nuclei, prominent nucleoli and cell wrapping (F). Abbreviations: GBM = glioblastoma; MG = malignant glioma; PNET = primitive neuroectodermal tumor.
Figure 3.
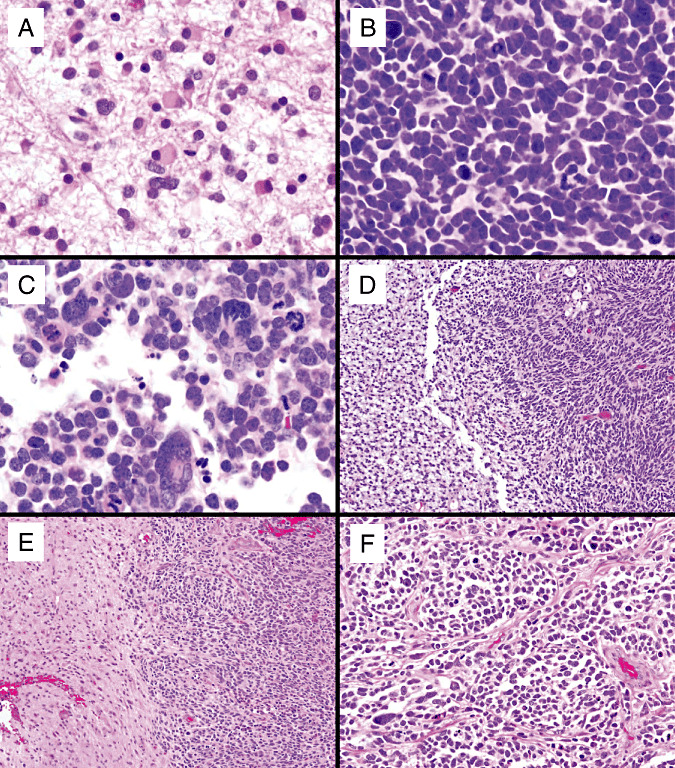
Additional representative high‐grade gliomas with neuroblastic (PNET‐like) foci, including (A–C): a GBM‐PNET that developed from progression of a diffuse astrocytoma. A. Diffuse astrocytoma, World Health Organization grade II diagnosed in a 30‐yo woman. B. A recurrence 4 years later showed the emergence of a PNET‐like component with neuroblastic rosettes comprising the majority of the tumor. C. Anaplastic foci were also noted. Two additional examples of the relatively sharp interface between the malignant glioma (left) and the PNET‐like nodules (right) are shown in (D) and (E). Medulloblastoma‐like nodularity from another GBM‐PNET is shown in (F). Abbreviations: GBM = glioblastoma; PNET = primitive neuroectodermal tumor.
Figure 4.

Representative ancillary stains: GFAP positivity within a minor fraction of the PNET‐like cells (A). Strong synaptophysin positivity in the neuroblastic component, but a lack of staining in the entrapped islands of glioma (B). Nuclear Neu‐N positivity in a subset of the PNET‐like cells (C). Nearly diffuse immunoreactivity for Ki‐67 (D) and p53 protein in primitive cells, including those forming Homer Wright rosettes (E). Reticulin shows a similar pattern to that of desmoplastic medulloblastoma in this example of GS‐PNET (F). Abbreviations: GFAP = glial fibrillary acidic protein; GS = gliosarcoma; PNET = primitive neuroectodermal tumor.
Figure 5.

Representative FISH images. Chromosome 10q deletion was seen in both the glioma and PNET‐like regions of a GBM‐PNET (A. one green PTEN and one red DMBT1 signal per cell). Another GBM‐PNET showed c‐myc gene amplification relatively restricted to the PNET‐like component (B. 2–4 green centromere 8 and numerous red c‐myc signals per cell). Another example showed N‐myc gene amplification in the PNET‐like component (C. centromere 2 in red and N‐myc gene in green), whereas the adjacent glioma (D) showed polysomy 2 (chromosomal gain), but no gene amplification. In the AO‐PNET case, codeletion of chromosome 1p (E. one green 1p32 and two red 1q42 signals in most nuclei) and 19q (F. one red 19q13 and two green19p13 signals in most nuclei) was seen in both components. In contrast, N‐myc gene amplification (G) was confined to the PNET‐like nodules. Abbreviations: GBM = glioblastoma; PNET = primitive neuroectodermal tumor.
Immunohistochemically (Table 4), the majority of the gliomas showed widespread GFAP expression. In the primitive component, GFAP staining was noted in 89%, although typically with far fewer positive cells than in the adjacent glioma (Figure 4A). Neuronal markers were relatively restricted to the PNET‐like foci, the exception being NSE. Synaptophysin staining was present in 87% of cases, reactivity being patchy to diffuse in primitive foci (Figure 4B); neuropil in the centers of neuroblastic rosettes was often highlighted. Although considerably less sensitive, Neu‐N expression was also limited to PNET‐like foci, and was easier to interpret because of its nuclear pattern of immunoreactivity (Figure 4C). NSE was highly sensitive with 91% reactivity, but was relatively non‐specific given that obviously glial elements were commonly positive as well. Nonetheless, the finding of strong and diffuse positivity within primitive nodules was supportive of the diagnosis. Ki‐67 labeling indices ranged from 30% to 100% within PNET‐like foci (Figure 4D), where it was always increased, in comparison with adjacent foci of glioma. Nuclear p53 expression was seen in 81% of cases, reactivity involving 5%–100% of tumor nuclei (Figure 4E); furthermore, it was always seen in both the glioma and the PNET‐like components.
In terms of the high‐grade glioma component, most tumors showed histopathologic features in support of the secondary, rather than primary or de novo type of GBM (28). For example, a prior diagnosis of pure glioma was rendered 8 months to 7 years previously in 13 cases (25%), eight of which (15%) were World Health Organization grades II or III (Table 3). In comparison, only 5.4% of all GBMs diagnosed at Washington University had a prior history of low‐grade glioma (P < 0.001; chi‐square). Furthermore, foci resembling diffuse low‐grade glioma were found in 47 cases (89%) of the MG‐PNET specimens under study. They included pure astrocytoma (fibrillary and/or gemistocytic) in 36 (77%), mixed oligoastrocytoma in 10 (21%) and pure oligodendroglioma in one (2%). Providing further support for the “secondary GBM” hypothesis, strong nuclear p53 immunoreactivity was found in 33 of 40 cases tested (83%), 22 of which (55%) showed staining in over 70% of tumor cells (Figure 4E). Nevertheless, a number of the cases presented with no prior history of glioma and EGFR amplification was found in a small subset of cases. As such, the development of PNET‐like foci can likely occur in either primary or secondary types of GBM.
The pathology of distant metastases was examined in three cases; two showed a pure PNET‐like pattern and one showed combined glioma and PNET‐like patterns. One case that recurred showed only the PNET‐like histopathology on re‐resection.
Fluorescence in situ hybridization (FISH)
FISH was employed in 40 cases; the results are summarized in Table 4. The most common GBM‐associated alteration was chromosome 10q deletion, a feature of 20 cases (50%); this was detected in both glial and PNET‐like elements (Figure 5A). One of these cases showed homozygous PTEN deletion and hemizygous DMBT1 loss; the rest harbored hemizygous loss for both markers. EGFR amplification was found in four tumors (10%) and was similarly seen in both components. Only one of these 4 tumors showed strong p53 immunoreactivity in >50% of cells. Chromosome 1 and 19 analyses were restricted to 12 tumors with suspected oligodendroglial features. Only the case of PNET arising from a pure anaplastic oligodendroglioma showed combined 1p and 19q codeletion in both glial and PNET‐like foci (Figure 5E–F). N‐myc gene amplification was also found, but was limited to the primitive element (Figure 5G). Clinically, this patient showed evidence of CSF dissemination at presentation and died 1 month after diagnosis. An additional four tumors showed 19q deletion alone, i.e. without concomitant 1p loss, in both components. Overall, PNET‐associated MYC gene amplifications (mostly N‐myc; Table 2) were identified in 17 (43%) cases; the alterations were relatively restricted to the primitive nodules (Figure 5B–D). Although MYC amplifications were usually diffuse in the nodules, scattered cells within the adjacent glioma‐like elements were also amplified in some cases, perhaps reflecting limited invasion of PNET‐like cells into the adjacent tissue. These amplifications were more commonly associated with large cell/anaplastic (15 of 28, 54%) than with non‐anaplastic (2 of 12, 17%; P = 0.041; Fisher's exact test) cytology. Combined “glioma‐like” (10q deletion, EGFR amplification and/or 1p/19q deletions) and “PNET‐like” (N‐myc or c‐myc amplifications) genetic alterations were found in nine (23%) cases. None of the genetic alterations noted above were clearly associated with patient survival.
DISCUSSION
Nomenclature and the origin of MG‐PNET
Although tumors with combined features of MG and CNS PNET (MG‐PNET) are rare, a motivating factor for the current study was the challenges they pose, both to the pathologist applying diagnostic nomenclature and to the oncologist in terms of patient management. To our knowledge, this is the first large series reported. In this predominantly adult cohort, the data strongly argue against either a collision tumor or simply a CNS PNET with extensive glial differentiation. Rather, they suggest that the PNET‐like nodules so typical of this tumor arise in a pre‐existing glioma, most often GBM. A number of findings support this interpretation. For example, the median patient age of 54 years is well above the average age for CNS PNET (5.5 years), the demographics being much more in line with high‐grade gliomas in general. Furthermore, aside from the relatively discrete hypercellular PNET‐like nodules, the classic glial morphology, extensive GFAP positivity and the pattern of infiltrative tumor growth, replete with secondary structure formation, were typical of diffuse glioma, often with both low‐grade and high‐grade features in the same tumor. Lastly, the fact that glioma‐like genetic alterations were seen in both components suggests a monoclonal process, the glioma arising first. In contrast, the relative restriction of MYC gene amplifications to the neuroblastic nodules suggests that it is a later or secondary alteration. Interestingly, these amplifications were often, but not invariably associated with large cell/anaplastic features as are seen in medulloblastoma (12). In this regard, the data of Lassman and colleagues are of particular interest in that they demonstrated that over‐expression of c‐myc protein in cultured GFAP‐positive astrocytes converted them into primitive, nestin‐positive, GFAP‐negative progenitor‐like cells (21). Therefore, based on the current data, diagnostic terms such as “malignant glioma with a PNET‐like component, i.e. MG‐PNET” seem appropriate and biologically accurate. In individual tumors, alternate terms such as GBM‐PNET, GS‐PNET and anaplastic oligodendroglioma (AO)‐PNET can be used to identify the specific glial components present. This is similar to the approach of McLendon and colleagues who previously reported two examples referred to as “cerebral neuroblastoma‐anaplastic astrocytoma” and “cerebral ganglioneuroblastoma‐glioblastoma”, respectively (22).
Evidence supporting the secondary (as opposed to primary or de novo) GBM concept for most, but not all of our MG‐PNET cases includes: (i) the slightly younger average age of onset [determined to be 45 years for secondary and 62 years for primary GBMs (28)]; (ii) the frequent history of a prior low‐grade glioma and/or histologic features of a lower‐grade precursor; (iii) often strong and diffuse p53 immunoreactivity; and (iv) the rarity of EGFR gene amplification. Confirming the PNET‐like or neuroblastic nature of the second tumoral component are: (i) cytologic features resembling those of classic, desmoplastic/nodular and large cell/anaplastic variants of medulloblastoma; (ii) the finding of Homer Wright rosettes; (iii) immunoreactivity for synaptophysin and other neuronal markers; (iv) extremely high proliferation indices; and (vi) the frequent occurrence of MYC gene amplifications.
The concurrence of PNET‐like elements in association with diffuse gliomas prompts at least two hypotheses. The first invokes neuroblastic/neuronal metaplasia. In support of this mechanism is the fact that GBMs are well known to undergo mesenchymal (GS) and/or epithelial (adenoid GBM/GS or GBM with true epithelial differentiation) metaplasia 1, 4, 5, 9, 15, 26, 27, 29, 33. The former was encountered in five of our study cases. Interestingly, sarcomatous metaplasia is being increasingly recognized in other gliomas; examples include “oligosarcoma” and “ependymosarcoma”6, 34, 35. Although “neuroblastic metaplasia” per se has not been previously described in CNS or extracranial tumors, neuronal or neurocytic differentiation is being increasingly reported in a variety of glial tumors, including diffuse gliomas 3, 8, 31, 36. It is possible that neuronal transformation in poorly differentiated regions of high‐grade gliomas, such as GBM or GS, might appear more PNET‐like than that manifesting as more mature forms in lower grade gliomas.
A second hypothesis, that the neuroblastic nodules represent clonal expansions of the tumor stem cell or progenitor cell populations, has its basis in the recent finding of such cells in high‐grade gliomas (13). It is of note that although the most dramatic examples of MG‐PNET were selected for our present study, small clusters of primitive‐appearing cells are common even in conventional GBMs. Whether they represent tumor stem/progenitor cells or merely foci of histologic anaplasia is unclear and will require further study. For the time being, we suggest that only those gliomas with distinct nodules showing both morphologically and immunohistochemically convincing neuroblastic features, be designated as MG‐PNET.
Distinction of MG‐PNET from small cell GBM
A potential source of diagnostic confusion is in the distinction of MG‐PNET from the small cell pattern of GBM (SC GBM) 7, 30, in that the latter terminology similarly suggests a primitive element. Although both tumor types feature cells with high nuclear to cytoplasmic ratios and marked proliferative activity, SC GBM is characterized by a diffuse parenchymal infiltrate of monomorphic, deceptively bland oval nuclei more likely to be confused with oligodendroglioma than with PNET (Figure 6A–C) . SC‐GBM often displays numerous thin GFAP‐positive processes (Figure 6B), rather than showing a predominantly neuronal pattern of differentiation. Furthermore, they lack the hypercellular nodules, marked hyperchromasia (Figure 6D), Homer Wright rosettes, large cell/anaplastic features and the extensive synaptophysin positivity that characterizes MG‐PNET. Clinically, SC GBMs is almost always of the primary or de novo type, rather than arising from lower grade precursors. In addition, SC GBM is associated with EGFR, rather than MYC amplifications. Lastly, to our knowledge, SC GBM has the same risk of CSF seeding as conventional GBMs.
Figure 6.

In contrast to malignant gliomas with PNET‐like foci, the small cell variant of GBM is characterized by a more infiltrative growth pattern with bland, monomorphic nuclei that superficially resemble oligodendroglioma, but display numerous mitotic figures (A) and abundant, thin GFAP‐positive processes (B). On intraoperative smears, the bland oval nuclei of SC‐GBM (C) contrast with the more hyperchromatic and irregular medulloblastoma‐like nuclei of GBM‐PNET (D), the latter example also suggesting the possibility of a Homer Wright rosette. Abbreviations: GBM = glioblastoma; GFAP = glial fibrillary protein; PNET = primitive neuroectodermal tumor; SC = small cell.
Implications for prognosis and therapy
In that most cases in our study originated as consults, it was often difficult to get complete data on therapy, radiologic response and clinical follow‐up. Nevertheless, the number was sufficient to make initial observations and conclusions. Not surprisingly, the biology was highly aggressive, the median survival time being only 9.1 months, a figure not statistically different from that of classic GBMs. Indeed, as in ordinary GBM, most patients experienced local failures. Nonetheless, metastases were also common, being seen in up to 40% of cases where it was specifically assessed or in 21% of all cases with follow‐up. Even the latter figure contrasts sharply with the 1.1% frequency of CSF spread seen in one large series of 267 adult GBMs (P < 0.001; chi‐square) (38). Our numbers are much closer to the 37% frequency of meningeal metastases in medulloblastomas and CNS PNETs (14). Additionally, three patients who switched to platinum‐based chemotherapy after failing radiation therapy and temozolomide subsequently showed radiographic responses. Therefore, although PNET‐like behavior may be seen in MG‐PNET, there is insufficient data to provide conclusive therapeutic recommendations. However, CSF dissemination is a substantial risk and platinum‐based chemotherapy should be considered, particularly in patients that fail an initial trial with temozolomide.
ACKNOWLEDGMENTS
The authors thank Drs. Alice Ahlgren, Leonard Berg, Leslie Bruch, Dario Caccamo, Geeta Chako, Abhik Chaudhury, Timothy Cole, Roger Crawford, Paul Dulai, James Eagan, Rajyasree Emmadi, Jonathan Finley, Ann Foyle, Cathryn Goldberg, Keith Kaplan, John Kunesh, Delphine Loussouarn, Robert Macaulay, Steve Moore, Dan Mornin, Jean‐Francois Mosnier, Peter Philpott, C. Harker Rhodes, Cheryl Rimmer, Christopher Robinson, Steve Rostad, Imran Shahab, Heidi Shappell, Eugene Vlodavsky, Hannes Vogel and John Yamashita, as well as to Mr. Joseph Friedman for sharing their cases with us and/or providing us with clinical follow‐up. We are also grateful to Diane Robirds, Julie Branson and Ruma Banerjee for expert technical support with the FISH assays.
REFERENCES
- 1. Actor B, Cobbers JM, Buschges R, Wolter M, Knobbe CB, Lichter P et al (2002) Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes Chromosomes Cancer 34:416–427. [DOI] [PubMed] [Google Scholar]
- 2. Bailey P, Cushing H (1926) A Classification of Tumors of the Glioma Group on a Histogenetic Basis With a Correlation Study of Prognosis. Lippincott: Philadelphia, PA. [Google Scholar]
- 3. Barbashina V, Salazar P, Ladanyi M, Rosenblum MK, Edgar MA (2007) Glioneuronal tumor with neuropil‐like islands (GTNI): a report of 8 cases with chromosome 1p/19q deletion analysis. Am J Surg Pathol 31:1196–1202. [DOI] [PubMed] [Google Scholar]
- 4. Biernat W, Aguzzi A, Sure U, Grant JW, Kleihues P, Hegi ME (1995) Identical mutations of the p53 tumour suppressor gene in the glial and sarcomatous part of gliosarcomas suggest a common origin from glial cells. J Neuropathol Exp Neurol 54:651–656. [DOI] [PubMed] [Google Scholar]
- 5. Boerman RH, Anderl K, Herath J et al (1996) The glial and mesenchymal elements of gliosarcomas share similar genetic alterations. J Neuropathol Exp Neurol 55:973–981. [DOI] [PubMed] [Google Scholar]
- 6. Boldrini L, Pistolesi S, Gisfredi S, Ursino S, Ali G, Pieracci N et al (2006) Telomerase activity and hTERT mRNA expression in glial tumors. Int J Oncol 28:1555–1560. [DOI] [PubMed] [Google Scholar]
- 7. Burger PC, Pearl DK, Aldape K, Yates AJ, Scheithauer BW, Passe SM et al (2001) Small cell architecture—a histological equivalent of EGFR amplification in glioblastoma multiforme? J Neuropathol Exp Neurol 60:1099–1104. [DOI] [PubMed] [Google Scholar]
- 8. Cenacchi G, Giangaspero F (2004) Emerging tumor entities and variants of CNS neoplasms. J Neuropathol Exp Neurol 63:185–192. [DOI] [PubMed] [Google Scholar]
- 9. Du Plessis DG, Rutherfoord GS, Joyce KA, Walker C (2004) Phenotypic and genotypic characterization of glioblastoma multiforme with epithelial differentiation and adenoid formations. Clin Neuropathol 23:141–148. [PubMed] [Google Scholar]
- 10. Dulai MS, Bosanko CM, Wang AM, Horoupian DS, Boodin S, Chen PY, Wilson JD (2004) Mixed cystic gliosarcoma and primitive neuroectodermal tumor: a case report. Clin Neuropathol 23:218–222. [PubMed] [Google Scholar]
- 11. Dunham C, Sugo E, Tobias V, Wills E, Perry A (2007) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): report of a case with prominent neurocytic differentiation. J Neurooncol 84:91–98. [DOI] [PubMed] [Google Scholar]
- 12. Giangaspero F, Eberhart CG, Haapasalo H, Pietsch T, Wiestler OD, Ellison DW (2007) Medulloblastoma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 132–140. IARC: Lyon. [Google Scholar]
- 13. Gilbertson RJ, Rich JN (2007) Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7:733–736. [DOI] [PubMed] [Google Scholar]
- 14. Hong TS, Mehta MP, Boyett JM, Donahue B, Rorke LB, Zeltzer PM (2005) Patterns of treatment failure in infants with primitive neuroectodermal tumors who were treated on CCG‐921: a phase III combined modality study. Pediatr Blood Cancer 45:676–682. [DOI] [PubMed] [Google Scholar]
- 15. Horiguchi H, Hirose T, Kannuki S, Nagahiro S, Sano T (1998) Gliosarcoma: an immunohistochemical, ultrastructural and fluorescence in situ hybridization study. Pathol Int 48:595–602. [DOI] [PubMed] [Google Scholar]
- 16. Ishizawa K, Kan‐nuki S, Kumagai H, Komori T, Hirose T (2002) Lipomatous primitive neuroectodermal tumor with a glioblastoma component: a case report. Acta Neuropathol (Berl) 103:193–198. [DOI] [PubMed] [Google Scholar]
- 17. Johnston DL, Keene DL, Lafay‐Cousin L, Steinbok P, Sung L, Carret AS et al (2008) Supratentorial primitive neuroectodermal tumors: a Canadian pediatric brain tumor consortium report. J Neurooncol 86:101–108. [DOI] [PubMed] [Google Scholar]
- 18. Kaplan KJ, Perry A (2007) Gliosarcoma with primitive neuroectodermal differentiation: case report and review of the literature. J Neurooncol 83:313–318. [DOI] [PubMed] [Google Scholar]
- 19. Kepes JJ (2002) Gliosarcoma with areas of primitive neuroepithelial differentiation and extracranial metastasis. Clin Neuropathol 21:193–195. author reply 5–6. [PubMed] [Google Scholar]
- 20. Kleihues P, Burger PC, Aldape KD, Brat DJ, Biernat W, Bigner DD et al (2007) Glioblastoma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 33–49. IARC: Lyon. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lassman AB, Dai C, Fuller GN, Vickers AJ, Holland EC (2004) Overexpression of c‐MYC promotes an undifferentiated phenotype in cultured astrocytes and allows elevated Ras and Akt signaling to induce gliomas from GFAP‐expressing cells in mice. Neuron Glia Biol 1:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McLendon RE, Bentley RC, Parisi JE, Tien RD, Harrison JC, Tarbell NJ et al (1997) Malignant supratentorial glial‐neuronal neoplasms: report of two cases and review of the literature. Arch Pathol Lab Med 121:485–492. [PubMed] [Google Scholar]
- 23. McLendon RE, Judkins AR, Eberhart CG, Fuller GN, Sarkar C, Ng HK (2007) Central nervous system primitive neuroectodermal tumours. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 141–146. IARC: Lyon. [Google Scholar]
- 24. McLendon RE, Provenzale J (2002) Glioneuronal tumors of the central nervous system. Brain Tumor Pathol 19:51–58. [DOI] [PubMed] [Google Scholar]
- 25. Miller CR, Dunham CP, Scheithauer BW, Perry A (2006) Significance of necrosis in grading of oligodendroglial neoplasms: a clinicopathologic and genetic study of newly diagnosed high‐grade gliomas. J Clin Oncol 24:5419–5426. [DOI] [PubMed] [Google Scholar]
- 26. Mork SJ, Rubinstein LJ, Kepes JJ, Perentes E, Uphoff DF (1988) Patterns of epithelial metaplasia in malignant gliomas. II. Squamous differentiation of epithelial‐like formations in gliosarcomas and glioblastomas. J Neuropathol Exp Neurol 47:101–118. [DOI] [PubMed] [Google Scholar]
- 27. Mueller W, Lass U, Herms J, Kuchelmeister K, Bergmann M, Von Deimling A (2001) Clonal analysis in glioblastoma with epithelial differentiation. Brain Pathol 11:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170:1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paulus W, Bayas A, Ott G, Roggendorf W (1994) Interphase cytogenetics of glioblastoma and gliosarcoma. Acta Neuropathol (Berl) 88:420–425. [DOI] [PubMed] [Google Scholar]
- 30. Perry A, Aldape KD, George DH, Burger PC (2004) Small cell astrocytoma: an aggressive variant that is clinicopathologically and genetically distinct from anaplastic oligodendroglioma. Cancer 101:2318–2326. [DOI] [PubMed] [Google Scholar]
- 31. Perry A, Scheithauer BW, Macaulay RJ, Raffel C, Roth KA, Kros JM (2002) Oligodendrogliomas with neurocytic differentiation. A report of 4 cases with diagnostic and histogenetic implications. J Neuropathol Exp Neurol 61:947–955. [DOI] [PubMed] [Google Scholar]
- 32. Reddy AT, Janss AJ, Phillips PC, Weiss HL, Packer RJ (2000) Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer 88:2189–2193. [DOI] [PubMed] [Google Scholar]
- 33. Reis RM, Konu‐Lebleblicioglu D, Lopes JM, Kleihues P, Ohgaki H (2000) Genetic profile of gliosarcomas. Am J Pathol 156:425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodriguez FJ, Scheithauer BW, Jenkins R, Burger PC, Rudzinskiy P, Vlodavsky E et al (2007) Gliosarcoma arising in oligodendroglial tumors (“oligosarcoma”): a clinicopathologic study. Am J Surg Pathol 31:351–362. [DOI] [PubMed] [Google Scholar]
- 35. Rodriguez FJ, Scheithauer BW, Perry A, Oliveira AM, Jenkins RB, Oviedo A et al (in press) Ependymal tumors with sarcomatous change (“ependymosarcoma”): a clinicopathologic and molecular cytogenetic study. Am J Surg Pathol. [DOI] [PubMed] [Google Scholar]
- 36. Rodriguez FJ, Scheithauer BW, Robbins PD, Burger PC, Hessler RB, Perry A et al (2007) Ependymomas with neuronal differentiation: a morphologic and immunohistochemical spectrum. Acta Neuropathol (Berl) 113:313–324. [DOI] [PubMed] [Google Scholar]
- 37. Shibahara J, Fukayama M (2005) Secondary glioblastoma with advanced neuronal immunophenotype. Virchows Arch 447:665–668. [DOI] [PubMed] [Google Scholar]
- 38. Stark AM, Nabavi A, Mehdorn HM, Blomer U (2005) Glioblastoma multiforme‐report of 267 cases treated at a single institution. Surg Neurol 63:162–169. discussion 9. [DOI] [PubMed] [Google Scholar]
- 39. Visee S, Soltner C, Rialland X, Machet MC, Loussouarn D, Milinkevitch S et al (2005) Supratentorial primitive neuroectodermal tumours of the brain: multidirectional differentiation does not influence prognosis. A clinicopathological report of 18 patients. Histopathology 46:403–412. [DOI] [PubMed] [Google Scholar]
- 40. Wharton SB, Whittle IR, Collie DA, Bell HS, Ironside JW (2001) Gliosarcoma with areas of primitive neuroepithelial differentiation and extracranial metastasis. Clin Neuropathol 20:212–218. [PubMed] [Google Scholar]