

Abstract
Experimental autoimmune neuritis (EAN) is a well‐known animal model of Guillain‐Barré Syndrome. In this study, we studied the spatiotemporal expression of interleukin‐16 (IL‐16) in the nervous system of EAN rats and pharmacological effects of minocycline on IL‐16 expressions in EAN rats. In sciatic nerves and dorsal/ventral roots of EAN rats, IL‐16+ cells, identified as macrophages and T cells, were mainly found to concentrate around blood vessels. However, in spinal cords, IL‐16+ microglial cells were mainly found in lumbar dorsal horns. Massive IL‐16+ cell accumulation in sciatic nerves and spinal roots was temporally correlated with severity of neurological signs of EAN. Furthermore, a strong correlation of IL‐16+ cell accumulation with local demyelination in perivascular areas of sciatic nerves, and significant reduction of IL‐16+ cell numbers in sciatic nerves and spinal cords by minocycline suggested a pathological contribution of IL‐16+ cells in EAN. Taken together, robust IL‐16+ cell accumulation in the nervous system and its temporal correlation with severity of neurological signs in EAN might suggest a pathological role of IL‐16 in EAN, which makes IL‐16 a potential pharmacological target.
Keywords: EAN, IL‐16, macrophage, T lymphocyte
INTRODUCTION
Experimental autoimmune neuritis (EAN) is an autoantigen‐specific T cell‐mediated animal model of human demyelinating inflammatory diseases of the peripheral nervous system (PNS), like Guillain‐Barré Syndrome (GBS), which is pathogenetically considered an inflammatory reaction directed against specific components of the myelin sheath of peripheral nerves. EAN can be actively induced by immunization with autoantigen (purified peptide P0 or P2), and is pathologically characterized by the breakdown of the blood‐nerve barrier (BNB), severe demyelination in the PNS, and accumulation of infiltrated T cells and macrophages. As a monophasic disease, EAN is clinically characterized by weight loss, ascending paraparesis/paralysis and spontaneous recovery (6).
Activated autoreactive T cells, which can recognize peripheral nerve autoantigens on antigen presenting cells, like monocytes or Schwann cells, are of importance for the initiation of EAN. Following activation, autoreactive T cells attach to the venular endothelium in the PNS, penetrate the BNB, and generate an immune reaction within the PNS that orchestrates the invasion of autoreactive T cells and monocytes and local inflammation. Activated macrophages cause demyelination by direct phagocytic attack or secreted inflammatory mediators. Depletion of macrophages and inhibition of their activity have been shown to suppress the development of EAN. In EAN, the local inflammatory reactions are further enhanced by autoantibodies. Altogether, activation and accumulation of T cells and macrophages in the PNS are critical to the EAN development (19).
Interleukin‐16 (IL‐16), initially named lymphocyte chemoattractant factor, is a cytokine with chemotactic properties, but lacks the classic structural motifs that would classify it as a chemokine (4). IL‐16 can be produced by a variety of immune cells, like granulocytes, dendritic cells, T cells, monocytes/macrophages, and microglial cells (40). IL‐16 was found to exert a wide range of effects on cells, including induction of progression to the G1 phase, upregulation of CD25, and inhibition of antigen‐specific proliferation, but with retained antigen nonspecific proliferative properties (5). Through its interaction with CD4, IL‐16 acts as a chemoattractant for lymphocytes, monocytes and eosinophils, which express CD4 (29). IL‐16 regulates the proliferation, activation and recruitment of these cells, and the secretion of proinflammatory cytokines, including IL‐1β, IL‐6, IL‐15 and tumor necrosis factor‐α (TNF‐α) from monocytes (18). Recently, IL‐16's diverse immunoregulatory functions have been implicated in several infectious, immune and autoimmune inflammatory processes and diseases, like asthma, rheumatoid arthritis, multiple sclerosis, viral infections, astrocytic brain tumors, spinal cord injury and experimental autoimmune encephalomyelitis (EAE) 5, 8, 15, 34).
Minocycline is a member of the tetracycline class, and, in addition to its antimicrobial activity, has been shown to possess anti‐inflammatory effects caused by modulating microglial activation, cytokine and chemokine release, nitric oxide (NO) production, cellular apoptosis and so on (39). Our investigation demonstrated that minocycline greatly decreased the severity and duration of neurological signs and mechanical allodynia in EAN (Zhang et al, submitted).
In the present investigation, we analyzed the spatiotemporal expression of IL‐16 in sciatic nerves, dorsal/ventral roots and spinal cords from EAN rats, its correlation with pathological progress of EAN, and whether minocycline treatment could alter the expression of IL‐16.
MATERIALS AND METHODS
Animal experiments
Male Lewis rats (8–10 weeks old, 200–250 g, Charles River, Sulzfeld, Germany) were housed with equal daily periods of light and dark and free access to food and water. All procedures were performed in accordance with the published International Health Guidelines under a protocol approved by the local Administration District Official Committee.
EAN was induced by immunization using subcutaneous injection of 100 µg of synthetic neuritogenic P2 peptide into both hindpaws of anesthetized rats as described previously (32). Rats were scored daily for the development of EAN. The severity of EAN was scored as follows: 0—normal, 1—reduced tonus of tail, 2—limp tail, impaired righting, 3—absent righting, 4—gait ataxia, 5—mild paresis of the hind limbs, 6—moderate paraparesis, 7—severe paraparesis or paraplegia of the hind limbs, 8—tetraparesis, 9—moribund, 10—death.
Three rats in each group were sacrificed 7, 11, 13, 15, 17, 22 and 41 days after immunization for the study of sciatic nerves, and other five normal rats were sacrificed as the control group. Rats were deeply anaesthetized with ether and perfused intracardially with 4°C, 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS). Sciatic nerves were quickly removed and postfixed in 4% paraformaldehyde overnight at 4°C. Sciatic nerves were cut to two equal long segments, embedded in paraffin and sectioned serially (3 µm).
To study IL‐16 expression in EAN spinal cords and dorsa/ventral roots, a previously reported EAN spinal cord library (1), containing tissues at days 6, 10, 12, 15, 18, 24 and 48 (three rats each time point) was used.
Sciatic nerve and spinal cord library for EAN rats treated with minocycline or vehicle (PBS) was established in our lab recently (Zhang et al, submitted). Briefly, EAN rats were intraperitoneally injected with minocycline (Sigma, Steinheim, Germany; 45 mg/kg body weight in 1 mL PBS) once daily after immunization, from day 0 to the end of the experiment, day 17. Another group of rats were injected with the same volume of PBS as control. At day 17, all rats were sacrificed and tissues were quickly removed for immunohistochemistry (IHC).
IHC
IHC was performed on 3‐µm paraffin‐embedded sections using purified mouse monoclonal antibody directed against IL‐16 (1:100; BMA, Augst, Switzerland), whose binding site is localized to the mature processed IL‐16 C‐terminal part. The specificity of this antibody has been demonstrated by ELISA, Western blotting and peptide absorbing experiments by our group previously 31, 34). After dewaxing, sections were boiled (in an 850 W microwave oven) for 15 minutes in citrate buffer (2.1 g citric acid monohydrate/L, pH 6) (Carl Roth, Karlsruhe, Germany). Endogenous peroxidase was inhibited by 1% H2O2 in pure methanol (Merck, Darmstadt, Germany) for 15 minutes. Sections were incubated with 10% normal pig serum (Biochrom, Berlin, Germany) to block nonspecific binding of immunoglobulins and then with the IL‐16 antibody overnight at 4°C. Antibody binding to tissue sections was visualized with a biotinylated rabbit antimouse IgG antibody (1:400; DAKO, Hamburg, Germany). Subsequently, sections were incubated with a horseradish peroxidase‐conjugated Streptavidin complex for 30 minutes (1:100; DAKO), followed by development with diaminobenzidine (DAB) substrate (Fluka, Neu‐Ulm, Germany). Finally, sections were counterstained with hemalum. As negative controls for EAN diseased sciatic nerves, spinal roots and spinal cords, the primary antibodies were omitted.
Double staining
In double‐staining experiments, spinal cord and sciatic nerve sections were immunolabeled as described above. Then, they were once more irradiated in a microwave for 15 minutes in citrate buffer and were incubated with 10% normal pig serum (Biochrom). Subsequently, the sections were incubated with the appropriate second primary monoclonal and polyclonal antibodies for 1 h at room temperature. The following antibodies were used: ED1 (1:100; Serotec, Oxford, UK) for activated microglia/macrophages, W3/13 (1:50; Serotec) for T lymphocytes, OX‐62 (1:50; Serotec) for dendritic cells and S100 (1:500; Dako) for Schwann cells in the PNS. Consecutively, visualization was achieved by adding secondary antibody at a dilution of 1:400 in tris buffered saline with bovine serum albumin (TBS‐BSA) for 30 minutes, and then alkaline phosphatase‐conjugated Avidin complex diluted 1:100 in TBS‐BSA for another 30 minutes. Finally, immunostaining was developed with Fast Blue BB salt chromogen‐substrate solution, but by omission of counterstaining with hemalum. The double staining with monoclonal antibody against MBP (myelin basic protein; 1:200; Serotec) for myelinated axon was performed separately. The coloration of this antibody was developed with Histo‐Green (green), and the nuclei were shown by counterstaining with hemalum (blue).
Evaluation and statistical analysis
After immunostaining, sections of each time point were examined by light microscopy. IL‐16 expression was evaluated at the transections of inflamed sciatic nerves and spinal cord and in dorsal/ventral roots. To semiquantify IL‐16 expression, the numbers of IL‐16+ cells were counted. Positive cell counting based on IHC results has been well developed to semiquantify protein expression (13). IL‐16+ cells were counted manually by independent observers unaware of the experimental time points. To evaluate IL‐16+ cell numbers in sciatic nerves, four cross‐sections for each rat were evaluated. Microphotos of the whole sciatic nerve cross‐sections were taken under 100× magnification using Nikon Coolscope® (Nikon, Düsseldorf, Germany). IL‐16+ cells were counted manually on these photos, and only positive cells with the nucleus at the focal plane were counted. Areas of sciatic nerve cross‐sections were measured on the same pictures using software MetaMorph® Offline 7.1 (Molecular Devices, Toronto, Canada). Results were calculated as arithmetic means of IL‐16+ cells per square millimeter and standard errors of means (SEM).
To evaluate IL‐16+ cell numbers in dorsal/ventral roots, sections were scanned by light microscop, and 10 dorsal/ventral roots that have the maximum numbers of IL‐16+ cells were selected for each slide for cell counting. Microphotos were taken for each selected dorsal/ventral root. IL‐16+ cells were counted, and areas of dorsal/ventral roots were measured using the method described above. Results were calculated as arithmetic means of IL‐16+ cells per square millimeter and SEM.
To evaluate IL‐16+ cell numbers in dorsal horns of lumbar spinal cord, sections were first examined under dark field microscopy to determine the lumbar segmental level according to the method of Molander et al (22). Microphotos were taken for lumbar dorsal horns selected. IL‐16+ cells were counted, and areas of dorsal horns were measured as described above. For each rat, three coronal lumbar spinal cord sections were analyzed. Both left and right dorsal horns were counted for each of the section. Results were calculated as arithmetic means of IL‐16+ cells per square millimeter and SEM.
The correlation between axon demyelination and IL‐16 expression was analyzed in sciatic nerves from day 10 and 17 EAN rats. Cross‐sections of sciatic nerve were double‐stained for IL‐16 (brown) and MBP (for myelin, green). Cell nuclei were counterstained by hemalum as blue. For each rat, four cross‐sections were analyzed. Stained cross‐sections were evaluated by light microscopy. Under 400× magnifications, all perivascular areas present in cross‐sections were analyzed. Numbers of myelinated axons and IL‐16+ cells were counted for each perivascular area by two independent observers. Arithmetic means were calculated and expressed as numbers of myelinated axons or IL‐16+ cells per perivascular area. Correlation between numbers of myelinated axons and IL‐16+ cells in all these observation fields was analyzed subsequently.
Difference of IL‐16+ cell counting among different time points was analyzed by one‐way ANOVA followed by Dunnett's multiple comparison test (GraphPad PrismTM 4.0 software, GraphPad Software, San Diego, USA). Correlation analysis was evaluated by checking Spearman's correlation coefficient (for nonparametric correlation) (GraphPad PrismTM 4.0 for Windows). For all statistical analysis, significance levels were set at P < 0.05.
RESULTS
Development of EAN
Rats developed the first neurological signs of EAN (reduced tonus of tail) 12 days after immunization. Severity of neurological signs was maximal at day 15 (arithmetic mean of neurological scores: 5.3 ± 0.3). Soon after that, rats recovered fast from EAN and no neurological signs were observed by day 22 (Figure 1A).
Figure 1.
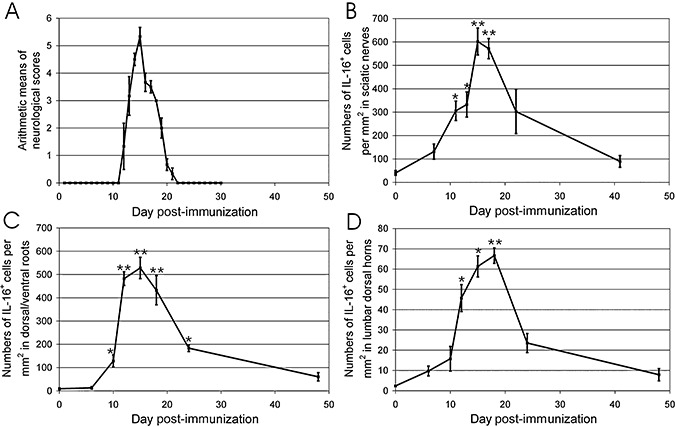
Time courses of neurological signs and IL‐16 + cell accumulation in EAN. (A) Score of EAN severity. Rats were immunized with synthetic neuritogenic P2 peptide and monitored for development of EAN. Severity of the disease was graded as described in Experimental Procedures. Results are given as arithmetic means of neurological scores ± standard errors of means (SEM). (B–D) The time course of lesional IL‐16+ cell numbers in sciatic nerves (B), dorsal/ventral roots (C) and spinal cord (D) of EAN rats. The numbers of IL‐16+ cells were counted in whole cross section of sciatic nerves, dorsal/ventral roots of each rat, and lumbar dorsal horns as described in Experimental Procedures. Data were calculated as arithmetic means of IL‐16+ cells per square millimeter and SEM. Statistical analysis was performed by one‐way ANOVA followed by Dunnett's multiple comparison test (GraphPad PrismTM 4.0 software, GraphPad Software, San Diego, USA). Abbreviations: IL‐16 = interleukin‐16; EAN = experimental autoimmune neuritis.
IL‐16 expression in rat sciatic nerves following EAN
IL‐16 expression in sciatic nerves of normal and EAN rats was studied using IHC. No immunoreactivity was detected in control staining for EAN‐diseased sciatic nerves without primary IL‐16 antibody (Figure 2A). In sciatic nerves of normal rats, immunoreactivity of IL‐16 was observed in single cells around vessels (40.5 ± 10.1 IL‐16+ cells per mm2) (1, 2). Following immunization, slight accumulation of IL‐16+ cells was observed at day 7 (131.5 ± 32.3 per mm2, n = 3, P > 0.05, compared with normal control). Accumulation of IL‐16+ cells became significant at day 11 (305.8 ± 41.5 per mm2, n = 3, P < 0.05, compared with normal control), and maximal cell numbers were recorded at day 15 (601.7 ± 57.6 per mm2, n = 3, P < 0.01, compared with normal control) (Figure 2C). Subsequently, a considerable decrease of numbers of IL‐16+ cells was observed at day 22 (302.8 ± 94.3 per mm2, n = 3, P > 0.05, compared with normal control) (Figure 2D) and finally returned to normal control level by day 41 (89.7 ± 25.1 per mm2, n = 3, P > 0.05, compared with normal control) (Figure 1B).
Figure 2.
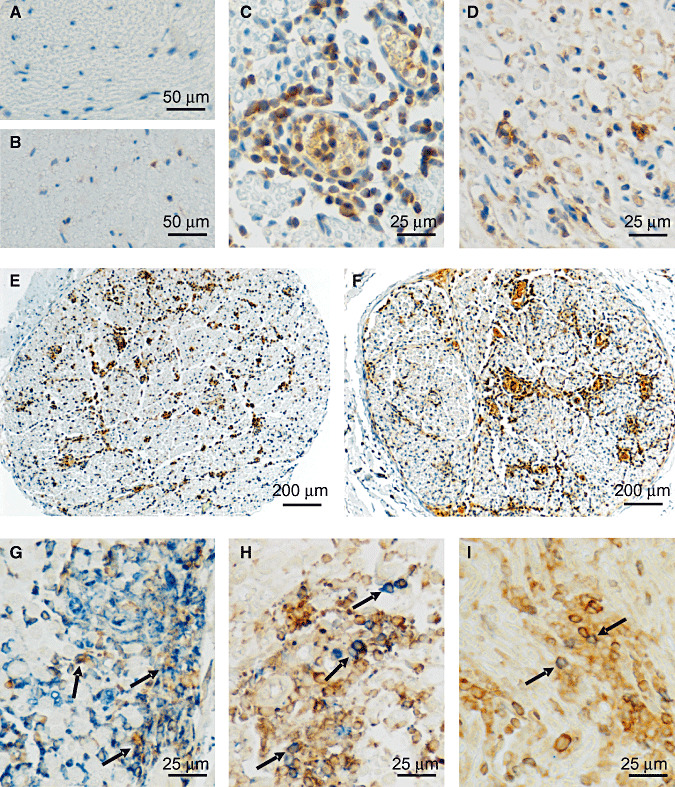
Immunohistochemical labelling of IL‐16 in sciatic nerves of EAN rats. No immunoreactivity was seen in control staining for EAN‐diseased sciatic nerves without primary IL‐16 antibody (A). In normal rat sciatic nerves, IL‐16 expression was only occasionally observed in single cells (B). At day 15, the cellular accumulation of IL‐16+ cells was maximal and seen mainly around vessels (C). Until day 22, the accumulation of IL‐16+ cells decreased rapidly (D). At low magnifications, the distributions of accumulated IL‐16+ cells can be clearly observed throughout the entire cross‐sections of sciatic nerves. IL‐16+ cells were mainly observed in perineurium and endoneurium, and concentrated around vessels, especially during the early (day 11) (E) and acute (day 15) (F) phase of EAN. Further double‐labeling experiments showed that over 70% IL‐16+ (brown) cells (arrow) coexpressed ED‐1 (blue), (G) representing active macrophages, and about 20% IL‐16+ (brown) cells (arrow) coexpressed pan‐T‐cell marker W3/13 (blue) (H) at day 15. Only a small population (about 3%) of IL‐16+ cells (brown) (arrow) were found to coexpress OX‐62 (Blue) (I). Abbreviations: IL‐16 = interleukin‐16; EAN = experimental autoimmune neuritis.
In sciatic nerves, IL‐16+ cells were not uniformly distributed. IL‐16+ cells were mainly observed in perineurium and concentrated around vessels, but infiltration of IL‐16+ cells could be seen in endoneurium of sciatic nerves as well (Figure 2E,F).
Accumulated IL‐16+ cells in sciatic nerves were further characterized by double staining with monoclonal antibodies directed against activated macrophages (ED1), pan‐T cells (W3/13), dendritic cells (OX‐62) and polyclonal antibody against Schwann cells (S100). Three representative time points, days 7, 15 and 22, were selected for double‐staining experiments. Most IL‐16+ cells were identified to be macrophages (ED‐1+) (Figure 2G). At day 7, about 60% of IL‐16+ cells coexpressed CD68 (ED‐1 staining), and this fraction increased to 70% at day 15 and to about 80% at day 22. About 35% ED‐1+ cells were observed IL‐16 positive at day 7 and more than 55% at day 15. However, only less than 15% macrophages (ED‐1+) expressed IL‐16 at day 22. Another major cellular sources of IL‐16 were T cells (W3/13+ cells) (Figure 2H). At day 7, about 30% IL‐16+ cells were T cells, and this fraction decreased to 20% and 10% at day 15 and 22, respectively. Nearly 70% W3/13+ cells were double stained with IL‐16 at day 7, and this percentage increased slightly up to about 80% at day 15. But at day 22, only the half of T cells coexpressed IL‐16. Further, only a small population (about 3%) of IL‐16+ cells were found to coexpress OX‐62, indicating that dendritic cells expressed IL‐16 as well (Figure 2I). But no IL‐16+ cells double stained with S100 were observed in our study. Therefore, the major cellular sources of IL‐16 in sciatic nerves of EAN rats were macrophages and T cells.
IL‐16 expression in rat dorsal/ventral roots following EAN
No immunoreactivity was seen in control staining for EAN‐diseased spinal roots (Figure 3F) without primary IL‐16 antibody. In dorsal and ventral roots of normal rats, IL‐16+ cells were only occasionally seen (9.0 ± 3.8 IL‐16+ cells per mm2, 1, 3), and their numbers were lower than in sciatic nerves. Significant accumulation of IL‐16+ cells was observed as early as day 10 (128.5 ± 26.3 per mm2, n = 3, P < 0.05, compared with normal control), was maximal at day 15 (527.9 ± 45.9 per mm2, n = 3, P < 0.01, compared with normal control; Figure 3B) and then gradually decreased over time (at day 24: 183.4 ± 14.4 per mm2, n = 3, P < 0.05, compared with normal control, Figure 3C) until the end of our observation period (day 48). Similar to sciatic nerves, IL‐16+ cells were mainly detected to concentrate around vessels in perineurium and endoneurium.
Figure 3.
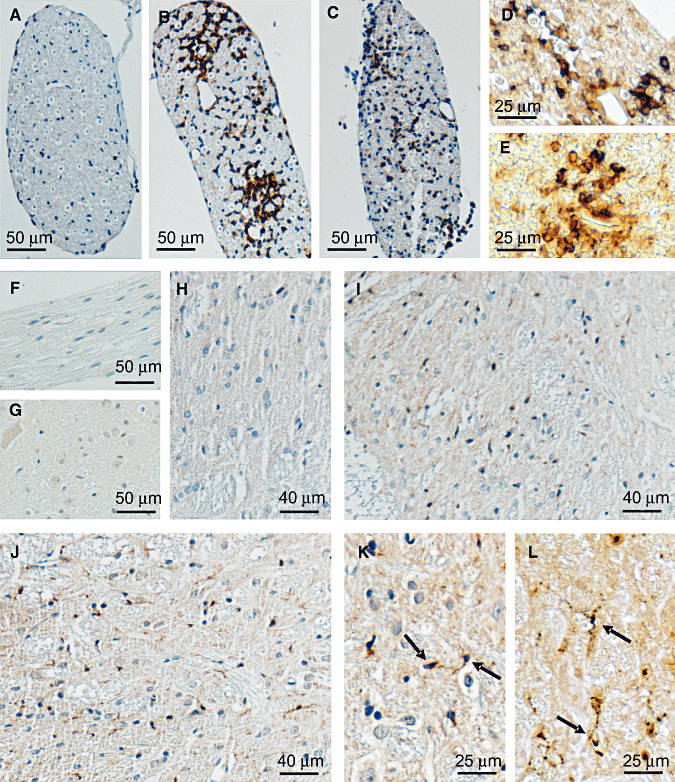
Immunohistochemical labeling of IL‐16 in dorsal/ventral roots and spinal cords of EAN rats. No immunoreactivity was seen in control staining for EAN diseased spinal roots (F) or spinal cords (G) without primary IL‐16 antibody. In dorsal/ventral roots of normal rats, IL‐16+ cells could be rarely seen (A). Larger numbers of IL‐16+ cells were observed around day 15. Similar to in sciatic nerves, IL‐16+ cells were mainly detected to concentrate around vessels in perineurium and endoneurium (B). At day 24, the accumulations of IL‐16+ cells were obviously decreased, but the cellular distribution pattern remained the same (C). Double labeling experiments showed that about 65% IL‐16+ (brown) cells (arrow) coexpressed ED1 (blue) (D), and more than 25% IL‐16+ (brown) cells (arrow) were double stained with W3/13 (blue) (E) at day 15. In spinal cords of normal rats, IL‐16+ cells were rare (H). A significant accumulation of IL‐16+ microglia was observed in the areas of lumbar dorsal horn of spinal cord as early as 12 days after immunization, (I) and the strongest accumulation was seen at day 18 (J). More details of the cellular accumulation and microglial hypertrophic morphology (arrow) are represented in (K) with a higher resolution. Further double staining with ED‐1 (blue) also proved that microglia (arrow) were the only cellular resource of IL‐16+ cells in EAN spinal cords (L). Abbreviations: IL‐16 = interleukin‐16; EAN = experimental autoimmune neuritis.
Double immunostaining revealed that in dorsal/ventral roots, about 65% IL‐16+ cells were macrophages (ED1+) (Figure 3D), and about 25% IL‐16+ cells were T cells (W3/13+) during the acute phase of EAN (day 15) (Figure 3E).
IL‐16 expression in rat spinal cords following EAN
No immunoreactivity was seen in the control staining for EAN‐diseased spinal cords (Figure 3G) without primary IL‐16 antibody. In spinal cords of normal rats, IL‐16+ cells were rarely seen (2.4 ± 0.4 per mm2) (1, 3). Significant accumulation of IL‐16+ cells was detected at day 12 (Figure 3I) after immunization, and the strongest accumulation was seen at day 18 (Figure 3J). The numbers of IL‐16+ cells were more pronounced in sections of lumbar in comparison with cervical or thoracic levels, and relatively higher numbers were seen in grey matter than in white matter, particularly in the dorsal horns. In spinal cords, the IL‐16+ cells exhibited a hypertrophic morphology characterized by enlarged, darkened soma and shorter, thicker and less branched processes (Figure 3K). Double immunostaining experiments with ED‐1 antibody revealed that almost all IL‐16+ cells were ED‐1+ (Figure 3L). ED‐1 labeling together with their morphological characteristics of reactive microglia indicated that the major IL‐16+ cells in the spinal cord of EAN rat were reactive microglia.
As IL‐16+ cells were relatively more abundant in dorsal horns, we further semiquantified numbers of IL‐16+ cells in dorsal horns of EAN rats at different time points. Some accumulation of IL‐16+ cells were detected in the dorsal horns as early as 6 days after immunization (9.7 ± 2.4 per mm2, n = 3, P > 0.05, compared with normal control), and a significant accumulation was observed at day 12 (45.7 ± 6.7 per mm2, n = 3, P < 0.05, compared with normal control) (Figure 1D). The maximal accumulation of IL‐16+ cells was seen at day 18 (66.7 ± 4.8 per mm2, n = 3, P < 0.01, compared with normal control) (1, 3). Thereafter, numbers of IL‐16+ cells decreased rapidly and returned to normal level at day 48 (day 24: 23.6 ± 4.7 per mm2, n = 3, P > 0.05; day 48: 7.9 ± 3.1 per mm2, n = 3, P > 0.05, compared with normal control). The pattern of appearance of IL‐16+ cells in lumbar dorsal horns was different from the patterns described above, as a slight cellular accumulation could be observed already at day 6, but maximal cell numbers were seen 3 days after maximal disease severity.
Correlation of IL‐16 expression with EAN severity
In sciatic nerves, the accumulation of IL‐16+ cells appeared earlier and persisted longer compared with the development of neurological signs. However, the maximum levels of IL‐16+ cells and clinical scores were found at the same day (day 15), and the significant accumulations of IL‐16+ cells were observed in accordance with the severity of EAN. Further correlation analysis proved a significant positive correlation of the time course of IL‐16+ cell accumulation in sciatic nerves with the time course of neurological scores of EAN rats in our observation (r = 0.87; P < 0.01) (Figure 4A).
Figure 4.
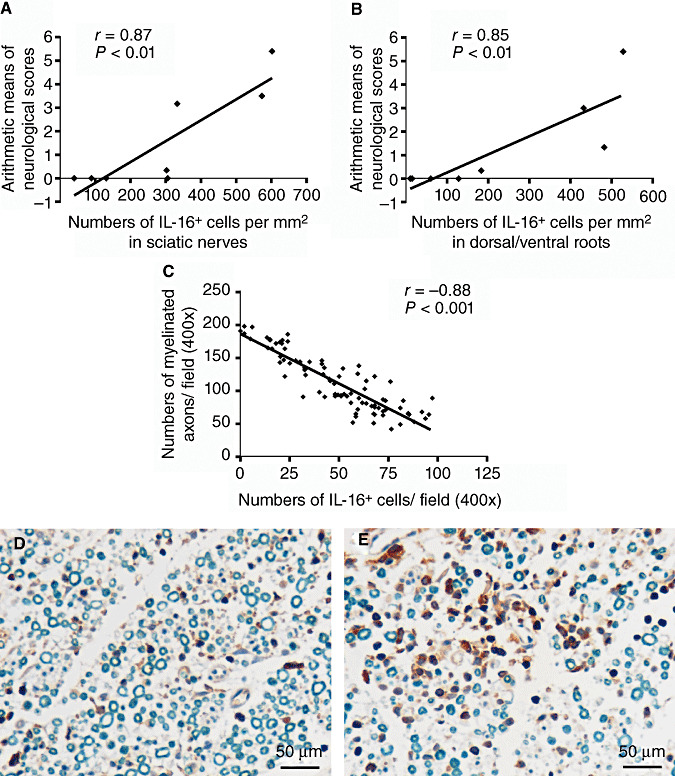
Correlation of IL‐16 + cell accumulations with the pathology of EAN. (A,B) Time course of IL‐16+ cell accumulation in sciatic nerves (A) and dorsal/ventral roots (B) was positively correlated with that of the neurological scores in EAN rats. IL‐16+ cells were counted as described in Experimental Procedures. Correlation analysis was evaluated by checking Spearman's correlation coefficient (for nonparametric correlation) (GraphPad PrismTM 4.0 for Windows, GraphPad Software, San Diego, USA). (C) A significant negative correlation between numbers of IL16+ cells with numbers of myelinated axons in perivascular areas of sciatic nerves from days 10 and 17 EAN rats was observed. The counting of IL‐16+ cells and myelinated axons was described in Experimental Procedures. Representative microphotos of two observation fields are presented to show that high IL‐16 expression was related with low numbers of myelinated axons (E), and weak IL‐16 expression was associated with high numbers of myelinated axons (F) in perivascular areas. Abbreviations: IL‐16 = interleukin‐16; EAN = experimental autoimmune neuritis.
In the dorsal/ventral roots of EAN rats, even after the disappearance of neurological signs by day 22, minor IL‐16+ cell infiltrations could still be observed at days 24 and 48, but in general, the time course of IL‐16+ cell accumulation was similar to that of neurologic severity of EAN. Further, correlation analysis showed that a significant positive correlation existed between the time course of IL‐16+ cell accumulation in dorsal/ventral roots and the time course of neurological scores of EAN (r = 0.85; P < 0.01) (Figure 4B).
In addition, a negative correlation between numbers of infiltrated IL‐16+ cells in perivascular areas—where there are hotspots of demyelination in EAN—with local myelinated axons, was found in sciatic nerves of EAN rats (Figure 4C, r = −0.83; P < 0.001), indicating more IL‐16+ cells in perivascular areas with less myelinated axons (Figure 4D,E).
IL‐16 expression following minocycline treatment
Minocycline has been shown to reduce the severity and duration of neurological signs and mechanical allodynia in EAN by our group (Zhang et al, submitted). Here we analyzed the effects of minocycline on IL‐16 expression.
IL‐16 expression in sciatic nerves and lumbar dorsal horns was analyzed in day 17 EAN rats treated with minocycline or PBS by IHC. In sciatic nerves of minocycline‐treated EAN rats, numbers of IL‐16+ cells were significantly reduced compared with PBS‐injected controls at day 17 (126.0 ± 28.1 per mm2, n = 5, P < 0.01, compared with PBS group 486.6 ± 50.9 per mm2; Figure 5A). In lumbar dorsal horns of day 17 EAN rats, numbers of IL‐16+ cells were also significantly decreased after treatment with minocycline (11.2 ± 1.1 per mm2, n = 5, P < 0.01, compared with PBS group 62.2 ± 3.0 per mm2; Figure 5B).
Figure 5.
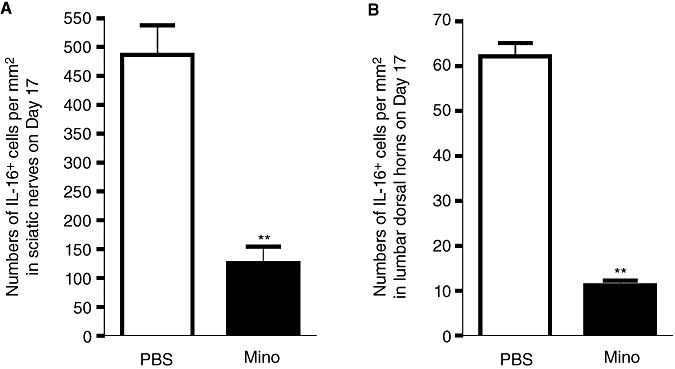
IL‐16 expression following minocycline‐treatment in EAN. (A,B) IL‐16 expression in sciatic nerves and spinal cords was analyzed in EAN rats treated with minocycline (50 mg/kg in 1 mL PBS) or PBS (1 mL) by IHC. Minocycline or PBS was intraperitoneally injected once per day following EAN induction. Rats were sacrificed at day 17. Bar graphs showing that numbers of IL‐16+ cells in sciatic nerves (A) and in lumbar dorsal horns (B) of minocycline‐treated EAN rats were significantly lower that of PBS‐treated EAN rats. Abbreviations: IL‐16 = interleukin‐16; EAN = experimental autoimmune neuritis; PBS = phosphate buffered saline; IHC = immunohistochemistry.
DISCUSSION
Here we have analyzed the expression of IL‐16 in sciatic nerves, dorsal/ventral roots and spinal cords of EAN rats. The major IL‐16+ cells were identified as infiltrated macrophages and T cells in sciatic nerves and dorsal/ventral roots, and as reactive microglia in the spinal cord. The time course of IL‐16+ cell accumulation in sciatic nerves and spinal roots was in accordance with the severity of neurological signs of EAN and infiltrated IL‐16+ cells negatively correlated with myelinated axons demyelination in perivascular areas of sciatic nerves. Further, administration of minocycline, which was known to decrease the severity and duration of neurological signs and mechanical allodynia in EAN, greatly reduced the accumulation of IL‐16+ cells in sciatic nerves and lumbar dorsal horns.
IL‐16 is a pleiotropic cytokine and a key mediator of inflammation. Upregulation of IL‐16 has been reported in a variety of inflammatory and autoimmune diseases, such as asthma, inflammatory bowel disease, atopic dermatitis, systemic lupus erythematosus, rheumatoid arthritis, graves disease, Whipple's disease, delayed‐type hypersensitivity, HIV infection in central nervous system (CNS), multiple sclerosis, EAE, human focal cerebral infarctions and rat spinal cord injury 2, 5, 8, 11, 12, 16, 17, 24, 34). More importantly, neutralization of IL‐16 in several of these disorders has been shown to suppress inflammatory cell infiltration and greatly improve clinical recovery, like EAE, the central nervous system analogous disease of EAN (35), trinitrobenzene sulfonic acid (TNBS)‐induced colitis (11) and delayed‐type hypersensitivity reaction (38), which confirms that IL‐16 plays an important role in the pathogenesis of these inflammatory disorders. Here we observed a robust accumulation of IL‐16+ cells in sciatic nerves and spinal roots in EAN, and the time course of cell accumulations positively correlated with EAN severity. This observation, together with pathological roles of IL‐16 in autoimmune/inflammatory disorders, may suggest a pathological role of IL‐16 in EAN.
IL‐16 is expressed by a variety of immune cells, like granulocytes, dendritic cells, CD4+ and CD8+ T cells, monocytes/macrophages, and microglial cells (40). In our study, IL‐16 was mainly seen in T cells and macrophages. It is interesting to find that the percentage of IL‐16+ T cells to total T cells and IL‐16+ macrophages to total macrophages differed at different time points in EAN sciatic nerves. These observations showed that the expression of IL‐16 in T cells and macrophages varied during EAN, indicating that alteration of IL‐16 production was induced in EAN.
In sciatic nerves and dorsal/ventral roots of EAN rats, immunoreactivity of IL‐16 was mainly detected in infiltrated macrophages and T cells. It has been reported that IL‐16 has chemotactic activities and plays an important role in a variety of inflammatory and immunomodulatory processes through CD4‐dependent and independent signaling pathways (34). In lymphocytes and monocytes, cross‐linking of CD4 by IL‐16 results in phosphorylation of CD4, increase of cellular Ca2+ concentrations and activation of inositol 1,4,5‐trisphosphate (IP3) (3). IL‐16 can cause T cell migration, proliferation and surface expression of the high‐affinity IL‐2 receptor (CD25) and MHC‐II molecules. In macrophages, IL‐16 also promotes cell migration and proliferation, and induces expression of several important proinflammatory cytokines by monocytes, like IL‐1β, IL‐6, IL‐15 and TNF‐α4, 5, 18). Therefore, as a pleiotropic cytokine, IL‐16 may regulate migration and proliferation of T cells and macrophages, and promotes the release of several important inflammatory cytokines from these cells. Activation and infiltration of T cells and macrophages into PNS lesions are critical to the development of EAN (30). Autoreactive T cell activation is the initial force for the pathological process of EAN. Macrophages are considered to be the predominant cell population, which infiltrate into EAN lesions and function as effectors destroying myelin (33). Therefore, IL‐16 might be involved also in the progression of EAN through regulating infiltration and cytokine secretion of T cells and macrophages.
Negative correlation of infiltrated IL‐16+ cells with local myelinated axons in perivascular areas of sciatic nerves might suggest an involvement of IL‐16 in pathology of demyelination. In EAN, macrophages are considered to be the predominant effector destroying myelin (33). In sciatic nerves of EAN, IL‐16+ cells were mainly found in perivascular spaces, which are of particular neuroimmunological interest, as they are prime portals of monocyte transmigration and lymphocyte drainage (7). IL‐16 is known to induce migration of macrophages and indirectly increases vascular permeability by induction of TNF‐α and IL‐1β expression 15, 40). Therefore, it is reasonable to speculate that IL‐16 might facilitate macrophage infiltration and indirectly contribute to axon demyelination in EAN.
We also observed an upregulation of IL‐16 in spinal cord microglia. While EAN mainly affects the PNS, impacts of EAN on spinal cord were occasionally reported as well, particularly on spinal microglia, which are very sensitive to changes in the CNS microenvironment 25, 28). In the spinal cord, microglia are activated in response to a variety of peripheral stimuli, resulting from degeneration of central terminals of dying sensory neurons or through the release of substances by incoming sensory afferents or pain‐responsive neurons in the dorsal horn (37). In EAN spinal cords, microglia activation is known (1). Our study here further showed significantly enhanced immunoreactivity of IL‐16 in spinal microglia.
In EAN spinal cords, IL‐16+ microglia were most pronounced in the dorsal horn, an area closely related with nociceptive signaling. IL‐16 is known to stimulate macrophages to secrete proinflammatory cytokines, like IL‐6, TNF‐α and IL‐1β(18), which are reported to contribute to central sensitization of neuropathic pain (10). Neuropathic pain, which is caused by the injury and/or inflammation of the peripheral nerves, is reported in EAN recently (21), and is a common symptom of GBS, occurring in 55%–85% of cases 23, 27). Recently, in our group, significant mechanical allodynia in EAN rats was observed at day 9, reaching a maximal level around day 17 and then slowly recovered until day 37 postimmunization (Zhang et al, submitted). A correlation analysis was performed, and a significant positive correlation between the time course of the accumulations of IL‐16+ cells in lumbar dorsal horns with that of mechanical allodynia was established (r = −0.95, P < 0.001). Therefore, the accumulation of IL‐16+ microglial in dorsal horns might facilitate pain hypersensitivity in EAN.
Minocycline has been reported to possess anti‐inflammatory properties, and is known to inhibit neuropathic pain in a variety of models 9, 14, 20). Its anti‐inflammatory actions were suggested to be caused by modulating immune cell activation and subsequent release of cytokines, chemokines, lipid mediators of inflammation, matrix metalloproteases and nitric oxide (36). And minocycline's anti‐neuropathic pain activity has been related to microglia deactivation (26). Recently, we proved that minocycline in EAN rats could reduce severity and duration of EAN neurological signs and neuropathic pain (Zhang et al, submitted). In this study, we found that in parallel with the improved recovery from EAN, immunoreactivity of IL‐16 in sciatic nerves and spinal cords was also greatly reduced by minocycline, indicating a potential pathological involvement of IL‐16 in EAN.
In summary, our observations demonstrated a robust accumulation of IL‐16+ cells in sciatic nerves, dorsal/ventral roots and spinal cords of EAN, which temporally correlated with the severity of neurological signs and mechanical allodynia of EAN, respectively. A negative correlation of infiltrated IL‐16+ cells with local myelinated axons in perivascular areas of sciatic nerves may suggest a contribution of IL‐16 to pathology of demyelination. Furthermore, minocycline, which proved to reduce severity and duration of EAN neurological signs and neuropathic pain, decreased numbers of IL‐16+ cells in sciatic nerves and spinal cords as well. Our data here suggests that IL‐16 might play a role in the EAN pathology and might be considered as a potential target of pharmacological therapy.
ACKNOWLEDGMENTS
This work has been supported in part by a grant of the BMBF (03134298). The authors are solely responsible for the content of the article.
REFERENCES
- 1. Beiter T, Artelt MR, Trautmann K, Schluesener HJ (2005) Experimental autoimmune neuritis induces differential microglia activation in the rat spinal cord. J Neuroimmunol 160:25–31. [DOI] [PubMed] [Google Scholar]
- 2. Benoit M, Fenollar F, Raoult D, Mege JL (2007) Increased levels of circulating IL‐16 and apoptosis markers are related to the activity of Whipple's disease. PLoS ONE 2:e494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cruikshank WW, Berman JS, Theodore AC, Bernardo J, Center DM (1987) Lymphokine activation of T4+ T lymphocytes and monocytes. J Immunol 138:3817–3823. [PubMed] [Google Scholar]
- 4. Cruikshank WW, Kornfeld H, Center DM (2000) Interleukin‐16. J Leukoc Biol 67:757–766. [DOI] [PubMed] [Google Scholar]
- 5. Glass WG, Sarisky RT, Vecchio AM (2006) Not‐so‐sweet sixteen: the role of IL‐16 in infectious and immune‐mediated inflammatory diseases. J Interferon Cytokine Res 26:511–520. [DOI] [PubMed] [Google Scholar]
- 6. Gold R, Hartung HP, Toyka KV (2000) Animal models for autoimmune demyelinating disorders of the nervous system. Mol Med Today 6:88–91. [DOI] [PubMed] [Google Scholar]
- 7. Guillemin GJ, Brew BJ (2004) Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol 75:388–397. [DOI] [PubMed] [Google Scholar]
- 8. Guo LH, Mittelbronn M, Brabeck C, Mueller CA, Schluesener HJ (2004) Expression of interleukin‐16 by microglial cells in inflammatory, autoimmune, and degenerative lesions of the rat brain. J Neuroimmunol 146:39–45. [DOI] [PubMed] [Google Scholar]
- 9. Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M (2005) Intrathecal minocycline attenuates peripheral inflammation‐induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci 22:2431–2440. [DOI] [PubMed] [Google Scholar]
- 10. Inoue K (2006) ATP receptors of microglia involved in pain. Novartis Found Symp 276:263–272. [PubMed] [Google Scholar]
- 11. Keates AC, Castagliuolo I, Cruickshank WW, Qiu B, Arseneau KO, Brazer W et al (2000) Interleukin 16 is up‐regulated in Crohn's disease and participates in TNBS colitis in mice. Gastroenterology 119:972–982. [DOI] [PubMed] [Google Scholar]
- 12. Klimiuk PA, Goronzy JJ, Weyand CM (1999) IL‐16 as an anti‐inflammatory cytokine in rheumatoid synovitis. J Immunol 162:4293–4299. [PubMed] [Google Scholar]
- 13. Leifeld L, Fielenbach M, Dumoulin FL, Speidel N, Sauerbruch T, Spengler U (2002) Inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS) expression in fulminant hepatic failure. J Hepatol 37:613–619. [DOI] [PubMed] [Google Scholar]
- 14. Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF et al (2005) Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain 115:71–83. [DOI] [PubMed] [Google Scholar]
- 15. Liebrich M, Guo LH, Schluesener HJ, Schwab JM, Dietz K, Will BE et al (2007) Expression of interleukin‐16 by tumor‐associated macrophages/activated microglia in high‐grade astrocytic brain tumors. Arch Immunol Ther Exp 55:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Little FF, Cruikshank WW (2004) Interleukin‐16 and peptide derivatives as immunomodulatory therapy in allergic lung disease. Expert Opin Biol Ther 4:837–846. [DOI] [PubMed] [Google Scholar]
- 17. Masuda K, Katoh N, Soga F, Kishimoto S (2005) The role of interleukin‐16 in murine contact hypersensitivity. Clin Exp Immunol 140:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mathy NL, Scheuer W, Lanzendorfer M, Honold K, Ambrosius D, Norley S et al (2000) Interleukin‐16 stimulates the expression and production of pro‐inflammatory cytokines by human monocytes. Immunology 100:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maurer M, Toyka KV, Gold R (2002) Cellular immunity in inflammatory autoimmune neuropathies. Rev Neurol 158:S7–15. [PubMed] [Google Scholar]
- 20. Mika J, Osikowicz M, Makuch W, Przewlocka B (2007) Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur J Pharmacol 560:142–149. [DOI] [PubMed] [Google Scholar]
- 21. Moalem‐Taylor G, Allbutt HN, Iordanova MD, Tracey DJ (2007) Pain hypersensitivity in rats with experimental autoimmune neuritis, an animal model of human inflammatory demyelinating neuropathy. Brain Behav Immun 21:699–710. [DOI] [PubMed] [Google Scholar]
- 22. Molander C, Xu Q, Grant G (1984) The cytoarchitectonic organization of the spinal cord in the rat. I. The lower thoracic and lumbosacral cord. J Comp Neurol 230:133–141. [DOI] [PubMed] [Google Scholar]
- 23. Moulin DE, Hagen N, Feasby TE, Amireh R, Hahn A (1997) Pain in Guillain‐Barre syndrome. Neurology 48:328–331. [DOI] [PubMed] [Google Scholar]
- 24. Mueller CA, Schluesener HJ, Conrad S, Pietsch T, Schwab JM (2006) Spinal cord injury‐induced expression of the immune‐regulatory chemokine interleukin‐16 caused by activated microglia/macrophages and CD8+ cells. J Neurosurg Spine 4:233–240. [DOI] [PubMed] [Google Scholar]
- 25. Nakamura Y (2002) Regulating factors for microglial activation. Biol Pharm Bull 25:945–953. [DOI] [PubMed] [Google Scholar]
- 26. Narita M, Yoshida T, Nakajima M, Narita M, Miyatake M, Takagi T et al (2006) Direct evidence for spinal cord microglia in the development of a neuropathic pain‐like state in mice. J Neurochem 97:1337–1348. [DOI] [PubMed] [Google Scholar]
- 27. Pentland B, Donald SM (1994) Pain in the Guillain‐Barre syndrome: a clinical review. Pain 59:159–164. [DOI] [PubMed] [Google Scholar]
- 28. Piehl F, Lidman O (2001) Neuroinflammation in the rat—CNS cells and their role in the regulation of immune reactions. Immunol Rev 184:212–225. [DOI] [PubMed] [Google Scholar]
- 29. Qi JC, Wang J, Mandadi S, Tanaka K, Roufogalis BD, Madigan MC et al (2006) Human and mouse mast cells use the tetraspanin CD9 as an alternate interleukin‐16 receptor. Blood 107:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schabet M, Whitaker JN, Schott K, Stevens A, Zurn A, Buhler R et al (1991) The use of protease inhibitors in experimental allergic neuritis. J Neuroimmunol 31:265–272. [DOI] [PubMed] [Google Scholar]
- 31. Schluesener HJ, Seid K, Kretzschmar J, Meyermann R (1996) Leukocyte chemotactic factor, a natural ligand to CD4, is expressed by lymphocytes and microglial cells of the MS plaque. J Neurosci Res 44:606–611. [DOI] [PubMed] [Google Scholar]
- 32. Schluesener HJ, Seid K, Kretzschmar J, Meyermann R (1998) Allograft‐inflammatory factor‐1 in rat experimental autoimmune encephalomyelitis, neuritis, and uveitis: expression by activated macrophages and microglial cells. Glia 24:244–251. [DOI] [PubMed] [Google Scholar]
- 33. Schmidt B, Stoll G, Van Der Meide P, Jung S, Hartung HP (1992) Transient cellular expression of gamma‐interferon in myelin‐induced and T‐cell line‐mediated experimental autoimmune neuritis. Brain 115:1633–1646. [DOI] [PubMed] [Google Scholar]
- 34. Schwab JM, Nguyen TD, Meyermann R, Schluesener HJ (2001) Human focal cerebral infarctions induce differential lesional interleukin‐16 (IL‐16) expression confined to infiltrating granulocytes, CD8+ T‐lymphocytes and activated microglia/macrophages. J Neuroimmunol 114:232–241. [DOI] [PubMed] [Google Scholar]
- 35. Skundric DS, Dai R, Zakarian VL, Bessert D, Skoff RP, Cruikshank WW et al (2005) Anti‐IL‐16 therapy reduces CD4+ T‐cell infiltration and improves paralysis and histopathology of relapsing EAE. J Neurosci Res 79:680–693. [DOI] [PubMed] [Google Scholar]
- 36. Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W (2005) Minocycline as a neuroprotective agent. Neuroscientist 11:308–322. [DOI] [PubMed] [Google Scholar]
- 37. Watkins LR, Maier SF (2002) Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev 82:981–1011. [DOI] [PubMed] [Google Scholar]
- 38. Yoshimoto T, Wang CR, Yoneto T, Matsuzawa A, Cruikshank WW, Nariuchi H (2000) Role of IL‐16 in delayed‐type hypersensitivity reaction. Blood 95:2869–2874. [PubMed] [Google Scholar]
- 39. Zemke D, Majid A (2004) The potential of minocycline for neuroprotection in human neurologic disease. Clin Neuropharmacol 27:293–298. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Z, Fauser U, Schluesener HJ (2007) Early attenuation of lesional interleukin‐16 up‐regulation by dexamethasone and FTY720 in experimental traumatic brain injury. Neuropathol Appl Neurobiol (e‐published ahead of print). [DOI] [PubMed] [Google Scholar]