

While brain tumors are fortunately uncommon (2% of all cancers), they are unfortunately among of the most debilitating and deadly disorders, with mean survival times typically less than 1 year for adults with malignant brain tumors (5). In this respect, brain tumors are the most common cause of cancer‐related death in children and the fourth leading cause in adults. In an effort to develop improved strategies for treating these tumors in both children and adults, numerous laboratories worldwide have investigated the seminal genetic changes important for human brain tumor development and growth, examined the various different cell types that comprise human tumors and searched for surrogate markers that might predict or reflect human tumor growth. The lessons learned from these studies have been instrumental in the design of new approaches to brain tumor therapeutics; however, they have provided only limited insights into the cellular and molecular changes essential for tumor formation and continued growth, and do not distinguish between causative changes and alterations resulting from progressive genomic instability. Moreover, research using human tumor specimens does not provide a tractable platform to adequately define the individual factors that limit the successful treatment of these complex tumors.
Improved outcomes following human brain tumor therapy will depend on surmounting several important barriers (Figure 1). First, we must target the correct cell type(s) within the tumor. Work from many laboratories has shown that brain tumors are composed of numerous different cell types, including stem/progenitor cells, immunologic elements (microglia), reactive astrocytes, angiogenic constituents and neoplastic cells (10, 14, 19, 27, 31). Each of these cell types has a critical role in tumorigenesis and growth, and may need to be individually targeted in order to achieve optimal tumor growth arrest. Similarly, it is important to inhibit the dominant growth regulatory pathways important for cancer cell proliferation, survival and invasion. While several signaling pathways contribute to inappropriate cell proliferation or survival, it is necessary to develop therapeutic strategies that block the activity of those molecules most responsible for conferring these cancer phenotypes. Along these lines, therapies that inhibit key signaling pathways may also allow the cancer cells to acquire secondary genetic or molecular changes that facilitate their resistance to treatment. Excellent examples abound in the literature, including secondary mutations that facilitate tumor recurrence following Imatinib treatment (9, 24). In addition to secondary changes that culminate in treatment resistance, other molecular or genetic factors may enhance treatment, including individual differences in the ability to metabolize antineoplastic agents (pharmacogenetics and pharmacokinetics) and the presence of “modifier” genes that influence tumor development, growth and response to therapy. Another consideration germane to the treatment of brain tumors is the effect of chemotherapy or radiation therapy on the normal brain. This is especially important when treating children whose brains are still maturing and are exquisitely sensitive to agents that impair normal cell growth or differentiation. Lastly, since it is often difficult to accurately assess tumor response to treatment by conventional imaging, the identification of serum or radiologic biomarkers of disease activity would greatly augment our therapeutic armamentarium.
Figure 1.
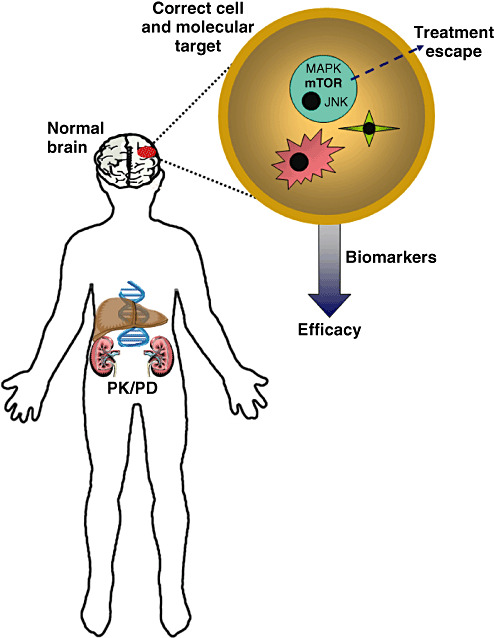
Improving the treatment of brain tumors. There are numerous variables that impact on successful brain tumor treatment. First, how the chemotherapeutic agents are metabolized and distributed influences drug availability (pharmacogenetics/pharmacodynamics; PK/PD). Second, genomic influences may partly determine who is most likely to develop a brain tumor and how that brain tumor will grow or respond to therapy. Third, the impact of treatment on the normal brain must be considered, especially in young children whose brains are still maturing. Fourth, we must identify the correct cell type(s) in the tumor for drug targeting as well as the optimal molecular pathways that drive tumor growth. Fifth, it is important to uncover potential escape mechanisms (ie, drug resistance) that undermine the continued effectiveness of anti‐cancer drug therapy. Lastly, the availability of surrogate markers of disease activity might serve as essential indicators of treatment efficacy early in the course of therapy. These “biomarkers” may be serum or radiologic surrogates with predictive value.
WHY MAKE MOUSE MODELS?
With the advent of mouse genetic engineering, it is now possible to generate animal models that develop brain tumors that closely resemble their human counterparts. As we move into an era of targeted therapeutics, it is important to properly exploit these models to provide insights that ultimately improve the care of brain tumor patients. While the study of human tumors has offered some valuable clues regarding the cellular and molecular composition of a mature neoplasm, it is extremely difficult to define the individual contributions of the various cell types to tumor growth and to establish cause‐and‐effect relationships between specific molecular alterations and tumor growth. In contrast, small animal models provide unprecedented opportunities to determine which molecular changes drive tumor growth. In addition, mouse models can be used to identify critical targets for therapeutic drug design, predict which tumors are most likely to respond to specific treatments, and define mechanisms that underlie tumor recurrence on therapy.
In this mini‐symposium, four leading experts in the field of small‐animal brain tumor modeling will discuss how we can best employ mouse models to decipher the basic cell and molecular biology of tumorigenesis (Dr Joshua Rubin), define targetable growth control pathways (Dr William Weiss), identify genetic risk factors for brain tumor formation and growth (Dr Karlyne Reilly) and inform human brain tumor clinical trials (Dr Eric Holland).
WHAT IS A MODEL?
When considering mouse models of human brain tumors, it is important to define what we mean by a “model”. One could define an animal model as any non‐human organism with either a natural or inducible disease similar to a known human condition. In addition, the modeled disease in small animals should be etiologically similar and recapitulate many of the seminal features of the human disorder (eg, proliferation, invasion, metastasis, histology, presence of a similar microenvironment, location and natural history). While it is preferable that the small animal model fully resembles all aspects of the human disease, models that recapitulate only select features of the disorder may also be instructive (eg, brain location, angiogenesis, histology and necrosis).
SIMPLE MODELS, COMPLEX ANSWERS
Over the past several years, it has become increasingly evident that brain tumorigenesis reflects complex interactions between different cell types within the tumor, diverse growth control pathways in participating cell types, the tumor microenvironment and the influence of genomic/genetic factors (Figure 2). Mouse brain tumor models have been incredibly instructive in identifying and dissecting these complexities.
Figure 2.
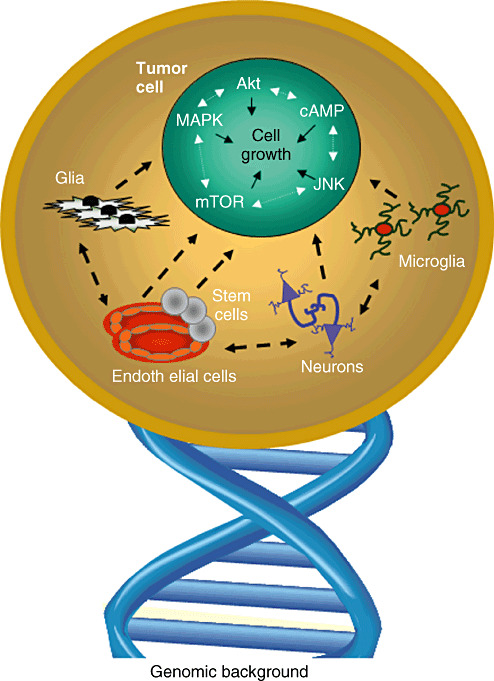
Brain tumors are complex biological systems. There are numerous different cell types present in human and mouse brain tumors that each impact on tumor development, growth and response to therapy. In addition, there is cross talk between the various signaling pathways within cancer cells and their microenvironment that may limit the utility of biologically targeted monotherapies. Lastly, there are genomic influences on tumor formation, continued growth and therapeutic response.
While there is considerable evidence that growth control pathways can intersect and influence each other, tumor cells can change their dependence on any particular signaling pathway. This can create a moving target, with tumor cells adapting to inhibition of a previously critical growth control pathway through positive and negative feedback loops (6, 29). Understanding the complicated cross talk between signaling pathways is essential to the design of pharmacologic inhibitors that can minimize tumor escape from treatment.
Work from a number of laboratories, including our own research team, has shown that brain tumor formation and growth requires signals from the tumor microenvironment. This is particularly evident in the case of pediatric low‐grade gliomas arising in the neurofibromatosis‐1 (NF1) inherited cancer syndrome: For instance, genetically engineered mice lacking Nf1 gene expression in glial cells do not develop brain tumors when the surrounding non‐glial cells are otherwise normal (1). In contrast, targeted Nf1 loss in glial cells of Nf1+/− mice (mice with reduced Nf1 gene expression in every cell in their body) predictably leads to the formation of optic gliomas, similar to those observed in children with NF1 (2, 3). This finding suggested that the presence of Nf1+/− non‐glial cells in the tumor microenvironment are necessary for optic glioma formation and growth (22). Using this Nf1 optic glioma mouse model, we have shown that Nf1+/− brain microglia produce chemokines and growth factors that uniquely promote the proliferation and survival of Nf1−/− glia (7, 30). These observations have led to the identification of potential drugs that target tumor microenvironment‐derived signals as adjuvants to therapies that specifically block Nf1‐deficient glial cell growth.
In addition to immunologic cells, other non‐neoplastic constituents within gliomas are important for tumor maintenance. For example, studies from several laboratories have shown that endothelial cells produce cytokines and growth factors that contribute to neoplastic cell growth (23, 26, 28). Moreover, the tumor vasculature may provide a specialized cellular niche for brain tumor‐associated stem cells (12). These “cancer stem cells” have been hypothesized to function as a cellular reservoir for neoplastic cells within the tumor and may be most critical for tumor maintenance (8, 15, 17). Numerous experiments support the notion that cancer stem cells have unique properties relevant to tumor growth and respond differently to chemotherapy and radiation therapy (13, 21), suggesting that effective treatments for brain tumors must consider these unique stem‐like cells.
Investigations using mouse models for epilepsy have demonstrated that specific inbred mouse strains have different susceptibilities to seizures (4, 11, 25), suggesting the presence of “modifier” genetic loci that contribute to disease expression and severity. The recent identification of “modifier” loci in mouse astrocytoma models has begun to provide similar insights into the genetic risk factors that may predispose to brain tumor formation (20). In addition, these specific genes may explain disparities in immune surveillance, microenvironmental contributions, and cellular responses that underlie differences in brain tumor predisposition, continued tumor growth and variable responses to therapy in humans.
Finally, mouse models may uncover tumor states for which human corollaries have yet to be discovered. In this regard, human brain tumors are not typically examined at their earliest, often asymptomatic stages of development, precluding an appreciation of the preneoplastic state. Similarly, we have few cellular and molecular insights into the dramatic cognitive and neuroendocrine effects of brain tumor therapy. The use of small animal models of brain tumors is likely to provide opportunities to further study these and other important issues in the intact animal.
WHEN GOOD MODELS GO BAD
While it is clear that mice are not just “furry little humans”, it is not often sufficiently appreciated that there are significant differences between mice and men with respect to innate immune responses, brain complexity and pharmacokinetic/pharmacodynamic responses. In this light, it is important to consider the implications of both positive and negative results to ensure that these findings are truly applicable to human brain tumors. When we fail to generate brain tumors in mice or do not observe a positive effect of drug treatment, we need to consider that our negative results do not necessarily detract from their relevance to the human condition, but rather, may reflect the limitations of our model systems. For example, these “failures” could result from differences in genetic modifiers or the immune system inherent to the strain of mice we have employed. Similarly, when treatments fail to show efficacy in preclinical studies, these findings might reflect differences in drug effects between humans and rodents. For example, penicillin is fatal for guinea pigs, aspirin is teratogenic in mice, and thalidomide does not cause birth defects in rats (16, 18). These considerations will have to be factored into our ultimate conclusions.
Despite these caveats and limitations, the future of mouse modeling of human brain tumors remains bright and exciting. We have already learned a great deal about the human condition by studying murine brain tumors and gained unique insights that only were possible using experimentally tractable systems. As our models become more and more sophisticated, it is highly likely that even greater rewards will be forthcoming.
Symposium Editor: David H. Gutmann, MD, PhD
REFERENCES
- 1. Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH (2002) Astrocyte‐specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol 22:5100–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, Gutmann DH (2003) Optic nerve glioma in mice requires astrocyte nf1 gene inactivation and nf1 brain heterozygosity. Cancer Res 63:8573–8577. [PubMed] [Google Scholar]
- 3. Bajenaru ML, Garbow JR, Perry A, Hernandez MR, Gutmann DH (2005) Natural history of neurofibromatosis 1‐associated optic nerve glioma in mice. Ann Neurol 57:119–127. [DOI] [PubMed] [Google Scholar]
- 4. Baraban SC (2007) Emerging epilepsy models: insights from mice, flies, worms and fish. Curr Opin Neurol 20:164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Central Brain Tumor Registry of the United States (2008) Statistical Report: Primary Brain Tumors in the United States, 2004. CBTRUS: Hinsdale, Illinois. [Google Scholar]
- 6. Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S et al (2008) Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN‐deficient glioblastoma. PLoS Med 5:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Daginakatte GC, Gutmann DH (2007) Neurofibromatosis‐1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1‐deficient astrocyte and glioma growth. Hum Mol Genet 16:1098–1112. [DOI] [PubMed] [Google Scholar]
- 8. Dirks PB (2008) Brain tumor stem cells: bringing order to the chaos of brain cancer. J Clin Oncol 26:2916–2924. [DOI] [PubMed] [Google Scholar]
- 9. Druker BJ (2006) Circumventing resistance to kinase‐inhibitor therapy. N Engl J Med 354:2594–2596. [DOI] [PubMed] [Google Scholar]
- 10. Fischer I, Gagner JP, Law M, Newcomb EW, Zagzag D (2005) Angiogenesis in gliomas: biology and molecular pathophysiology. Brain Pathol 15:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frankel WN, Taylor L, Beyer B, Tempel BL, White HS (2001) Electroconvulsive thresholds of inbred mouse strains. Genomics 74:306–312. [DOI] [PubMed] [Google Scholar]
- 12. Gilbertson RJ, Rich JN (2007) Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7:733–736. [DOI] [PubMed] [Google Scholar]
- 13. Hambardzumyan D, Squatrito M, Holland EC (2006) Radiation resistance and stem‐like cells in brain tumors. Cancer Cell 10:454–456. [DOI] [PubMed] [Google Scholar]
- 14. Hoelzinger DB, Demuth T, Berens ME (2007) Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst 99:1583–1593. [DOI] [PubMed] [Google Scholar]
- 15. Jandial R, UH, Levy ML, Snyder EY (2008) Brain tumor stem cells and the tumor microenvironment. Neurosurg Focus 24:E27. [DOI] [PubMed] [Google Scholar]
- 16. Koppanyi T, Avery MA (1966) Species differences and the clinical trial of new drugs: a review. Clin Pharmacol Ther 7:250–270. [DOI] [PubMed] [Google Scholar]
- 17. Lee Da Yong, Gutmann, DH (2007) Cancer stem cells and brain tumors: uprooting the bad seeds. Expert Rev Anticancer Ther 7:1581–1590. [DOI] [PubMed] [Google Scholar]
- 18. Mann RD (1984) Modern Drug Use. An Enquiry on Historical Principles, MTP Press: Lancaster. [Google Scholar]
- 19. Masson F, Calzascia T, Di Berardino‐Besson W, De Tribolet N, Dietrich PY, Walker PR (2007) Brain microenvironment promotes the final functional maturation of tumor‐specific effector CD8+ T cells. J Immunol 179:845–853. [DOI] [PubMed] [Google Scholar]
- 20. Reilly KM, Tuskan RG, Christy E, Loisel DA, Ledger J, Bronson RT et al (2004) Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc Natl Acad Sci USA 101:13008–13013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rich JN (2007) Cancer stem cells in radiation resistance. Cancer Res 67:8980–8984. [DOI] [PubMed] [Google Scholar]
- 22. Rubin JB, Gutmann DH (2005) Neurofibromatosis type 1—a model for nervous system tumour formation? Nat Rev Cancer 5:557–564. [DOI] [PubMed] [Google Scholar]
- 23. Salmaggi A, Gelati M, Pollo B, Frigerio S, Eoli M, Silvani A et al (2004) CXCL12 in malignant glial tumors: a possible role in angiogenesis and cross‐talk between endothelial and tumoral cells. J Neurooncol 67:305–317. [DOI] [PubMed] [Google Scholar]
- 24. Sawyers CL (2007) Where lies the blame for resistance–tumor or host? Nat Med 13:1144–1145. [DOI] [PubMed] [Google Scholar]
- 25. Schauwecker PE (2002) Complications associated with genetic background effects in models of experimental epilepsy. Prog Brain Res 135:139–148. [DOI] [PubMed] [Google Scholar]
- 26. Sheu BC, Chang WC, Cheng CY, Lin HH, Chang DY, Huang SC (2008) Cytokine regulation networks in the cancer microenvironment. Front Biosci 13:6255–6268. [DOI] [PubMed] [Google Scholar]
- 27. Stiles CD, Rowitch DH (2008) Glioma stem cells: a midterm exam. Neuron 58:832–846. [DOI] [PubMed] [Google Scholar]
- 28. Stiver SI (2004) Angiogenesis and its role in the behavior of astrocytic brain tumors. Front Biosci 9:3105–3123. [DOI] [PubMed] [Google Scholar]
- 29. Vogt PK, Kang S (2006) Kinase inhibitors: vice becomes virtue. Cancer Cell 9:327–328. [DOI] [PubMed] [Google Scholar]
- 30. Warrington NM, Woerner BM, Daginakatte GC, Dasgupta B, Perry A, Gutmann DH, Rubin JB (2007) Spatiotemporal differences in CXCL12 expression and cyclic AMP underlie the unique pattern of optic glioma growth in neurofibromatosis type 1. Cancer Res 67:8588–8595. [DOI] [PubMed] [Google Scholar]
- 31. Watters JJ, Schartner JM, Badie B (2005) Microglia function in brain tumors. J Neurosci Res 81:447–455. [DOI] [PubMed] [Google Scholar]